Submitted:
23 March 2023
Posted:
24 March 2023
You are already at the latest version
Abstract
Cancers have been interpreted either as somatic evolution of cheater cells that escape replication regulation or alternatively as non-healing wounds. Both the interpretations have substantial support as well as glaring anomalies but the two have not been put together to make a coherent synthesis. We argue here that mechanisms and pathways to escape the normal regulation of cell proliferation do not need to evolve de novo. Mechanisms to override the normal regulation have already evolved for wound healing and tissue regeneration. Almost all of the hallmarks of cancer are also seen in the wound healing process. This suggests that cancer develops not by any de novo gain of function but by exaptation of pre-evolved wound healing functions. Somatic evolution that makes the wound healing triggers constitutive is not mutation limited but selection limited and the selective forces are dependent on the tissue microenvironment. Some mechanisms for such selection have been suggested. Many more need to be investigated. A series of mechanisms have evolved to minimize the risk of cancers which may fail in an altered lifestyle context. We support our synthesis with multiple lines of evidence and also make differential testable predictions. This evolutionary perspective suggests novel lines of research and also has translatable implications for cancer prevention.
Keywords:
Cancer
; wound healing
; exaptation
; somatic evolution
Introduction
Cancer is a phenomenon of uncontrolled growth of certain cells defying the regulation mechanisms of the body. Development of cancer is perceived as a process of somatic evolution (Casás-Selves and DeGregori 2011, Rozhok and DeGregori 2017).Conceptually there are multiple ways of interpreting cancer generically. One school of thought views cancer cells as ‘cheater’ cells (Matapurkar and Watve 1997, Aktipis and Nesse 2013, Aktipis et al. 2015, Aktipis 2020). Another independent perspective is that of cancer as a non-healing wound (Schäfer and Werner 2008, Dvorak 2015, Sundaram et al. 2018, Hua and Bergers 2019, MacCarthy-Morrogh, and Martin 2020, Deyell et al. 2021,). Somatic mutations are commonly assumed to be necessary for transformation of a normal cell into malignant cell, but the dynamics of mutation accumulation is complex. The classical view of somatic evolution of cancer has been that of accumulation of a series of mutations. Since most differentiated cells undergo senescence and die, mutations in them are most unlikely to be relevant to tumorigenesis. Mutations in the population of adult stem cells (ASC) are the ones under focus (White and Lowry 2015, Tomasetti and Vogelstein 2015, Tomasetti et al. 2017). A single mutation is generally not sufficient and collection of mutations specific to each type of canceris required for the transformation. One view is that of a purely chance driven accumulation of the required set of mutations (Tomasetti et al. 2017). Since the probability of co-occurrence of multiple mutations is very small, a process such as clonal selection was thought necessary (Nowell 1976, Greaves and Maley 2012).On the other hand is the idea of chromothripsis, in which a large number chromosomal changes happen simultaneously and explosively in a single shot (Forment et al. 2012, Voronina et al. 2020). The thinking in the field has been largely mutation centered. The extensively debated Peto’s paradox (Nagy et al. 2007, Noble et al. 2015, Tollis et al. 2017), the presumption of Chromothripsis as well as the idea of cancer as “bad luck” arises from the assumption that somatic evolution of cancer is mutation limited. A nuanced view, supported by multiple lines of evidence is that somatic evolution of cancer is selection limited rather than mutation limited (Casás-Selves and DeGregori 2011, Rozhok, A. I. &DeGregori 2017, Vibishan and Watve 2020). Further rapidly accumulating molecular and cellular details reveal that cancer is much more than mutation accumulation. Cells with a set of mutations that might develop into a tumor in one set of conditions, fail to do so in another (Cao et al. 2010). Several components of the tissue microenvironment are crucial in the development of cancer (Hu and Polyak 2007, Medema and Vermeulen 2011, Lu et al. 2012, Quail and Joyce 2013, Pickup et al. 2014, Schulz et al. 2019, Liu et al. 2020, Elgundi et al. 2020). The malignant tissue also exhibits a number of complex “ecological” or “social” phenomena including cell-cell cooperation, cross talk between different cells, co-option of blood vessels, interaction with the immune system, responsiveness to the microenvironment etc (Tabassum and Polyak 2015, Adler andGordon 2019, Reynolds et al. 2020, Paczkowski et al. 2021, Somarelli 2021).Organs of future metastasis are not passive receivers of circulating tumor cells. A very intricate and sophisticated level of communication is involved in metastasis (Peinado et al. 2017). The hallmarks and enabling characteristics of cancer have kept on increasing with increasing research inputs (Hanahan and Weinberg 2000, 2011, Hanahan 2022). Of particular interest is the involvement of neurons in contributing to the hallmarks and enabling characteristics (Faulkner et al. 2019, Zahalka and Frenette 2020, Ayala 2023, Hanahan and Monje 2023). If cancers arise by de novo gain of function and are cheater cells, why should the nervous system take a pro-active role in their growth? If some cells arise as cheater cells by a set of mutations, why should other normal cells proactively promote their growth? All these apparently well coordinated phenomena are difficult to arise de novo so frequently and reproducibly in the population.
At a different level lies the question of possible evolution of mechanisms to prevent cancer (Trivedi et al. 2023). A number of tumor suppressor genes (Chen et al. 2020, Sherr et al. 2004, Kontomanolis et al. 2020, Joyce et al. 2022) have been identified and they are believed to have evolved as mechanisms of cancer defense. However, each one of them has one or more normal physiological functions independent of cancer. Therefore whether they evolved for preventing cancers or for their normal physiological functions is questionable. Since barring a few exceptions, most cancers appear at later ages, selection for mechanisms of prevention is likely to be weak by the Peter Medawar principle (Turan et al. 2019). Nevertheless, some mechanisms for arresting cancer can be expected and are claimed to have evolved and the dynamics of their evolution also needs to be interpreted carefully.
Limitations and criticism of existing perspectives of cancer: No single theory so far has the ability to accommodate all the well demonstrated phenomena observed during the development of cancer. The simple view of cancer as accumulation of chance mutations serially or at once (Tomasetti and Vogelstein 2015, Tomasetti et al. 2017, Forment et al. 2012, Voronina et al. 2020), which imply that cancer is only bad luck (Tomasetti and Vogelstein 2015, Tomasetti et al. 2017) is not compatible with the observed epidemiological patterns (Vibishan and Watve 2020). Furthermore attempts to prevent DNA damage have not succeeded in preventing cancers (Cockfield and Schafer 2019). The population level predictions of clonal expansion theory are also not compatible with the epidemiological picture (Vibishan and Watve 2020). The main limitation of the “cheater cell” paradigm (Aktipis and Nesse 2013, Aktipis et al. 2015, Aktipis 2020) is that cancer development and metastasis has extremely complex dynamics involving a number of characteristics and complex cell-cell cooperation processes. Whether the entire complexity arises de novo by mutations is questionable. Secondly unlike its implicit assumption, experiments do not always demonstrate the selective advantage of the intermediate mutants. In vitro, the IGF-II concentration in culture media was found to markedly alter the selective advantage of an IFG-II over-expressing mutant in cell competition (Archetti et al. 2015). Also this hypothesis does not account for the remarkable similarity in cancer and wound healing pathways. The perspective of cancer as a non-healing wound (Schäfer and Werner 2008, Dvorak 2015, Sundaram et al. 2018, Hua and Bergers 2019, Deyell et al. 2021) does not explain what makes the wound healing pathways derail and give rise to cancer. Based on the epidemiological as well as physiological patterns some studies show that the somatic evolution of cancers in not mutation limited but is selection limited (Casás-Selves and DeGregori 2011, Rozhok, A. I. &DeGregori 2017, Vibishan and Watve 2020). The mutants get a selective advantage only under certain microenvironmental contexts. The microenvironment is variable across individuals based on their genetic, developmental, life style and age related factors. The life time number of adult stem cell division is large enough so that the probability of each type of mutations is sufficiently large, but whether the mutant gets selected in competition with normal ASCs is the critical question that decides the development of cancer. Although this view is largely compatible with the epidemiological patterns, the details of the selective forces required for this hypothesis are still hazy.
The New Synthesis
In multicellular organisms with differentiated tissues, a control on cell proliferation is needed at two distinct levels. One is the level of normal healthy tissue maintenance. The other is the occasional requirement of healing a wound or making up for the tissue lost or damaged for any reason. The latter needs increased dynamics of replication starting from ASCs at a different level of coordination and differentiation. Therefore the mechanisms for surpassing the normal regulation on cell division are already present in the body. They are highly complex but very well coordinated with stage specific mechanisms of regulation. All mechanisms of shifting between the two levels of coordination have evolved and preexist in the cell and can be activated by a set of triggers. A different set of triggers down-regulates these mechanisms when the healing is more or less complete. By the new synthesis, cancer is not about escaping the regulation mechanisms by some novel mechanisms acquired by mutations. It is about wrongly triggering the healing and regeneration process without a genuine need. Normally the signals coming from injured tissue provide the triggers for starting the process. Consequently when healing is near completion a different set of signals coming from the healed tissue downregulates the process. In cancers since there is no real wound, the signals that control the process after healing are not generated at all. Therefore the process of making new cells to replace the perceived damaged tissue continues without a full stop. Thus cancer cells are not cheater cells, but are “cheated” or misled cells that are made to “believe” that there is a wound when in reality there may not be any.
The crucial question now is what constitutes the misleading signal to start wound healing and cell replacement protocol. The wound healing process needs not a single but multitudes of signals to get started. Therefore a misled trigger would also need multiple signals. One possibility is that a set of mutations can make some of the inducible pathways of wound healing constitutive. We will exemplify by the EGF signaling pathway. EGF signaling is one of the crucial mechanisms in the regulation of cell dynamics at both normal and wound healing levels of regulation. A basic level of EGF signaling is required for normal ASC and tissue dynamics (). The damaged tissue generates higher than normal levels of EGF which is one of the many triggers to start the wound healing process. Three types of mutations related to EGF signaling are known to occur in different types of cancers. One leads to overexpression of EGF receptor (EGFR), another leads to internal synthesis of EGF and the third makes pathways downstream of EGF signaling constitutive. In all the three, the cell becomes independent or oversensitive to external EGF signaling. Therefore in such a cell the EGF signaling might be misread as injury signaling even when it is normal. The crucial question may not be whether one of the three types of mutations arises, but whether a cell with one of the mutations will survive and outcompete a normal cell. Differential selection acting on one of the EGF related mutants, or more generally any growth factor signaling related mutants can be hypothesized as follows.
Since the growth factors are synthesized and regulated centrally, individual tissues or cells do not invest in their synthesis but receive the signal free of individual cost. However if there is chronic deficiency of a growth factor, a mutant that overexpresses or auto-activates a growth factor receptor, itself synthesizes the growth factor or makes the downstream pathway constitutive, would be at a selective advantage. Since the cell has to pay some energy cost to do so (Oña and Lachmann 2020), it would be at a disadvantage when external supply of the growth factor is adequate (Figure 1). However, when the external supply is deficient, normal cell replication would be suboptimal and the mutant would get a selective advantage in competition. The investment in say overexpressing the receptor may be overcompensated by the unique benefit. The important point to realize is that the selective advantage to the growth factor independent mutant cell is not an all time advantage, but only a conditional advantage under long term growth factor deficiency.
Owing to the tissue cell dynamics in higher animals where somatic cells have a definite life span and inevitable senescence and death, any mutation in the differentiated tissue is unlikely to sustain over a long time. However since growth factors are involved in the maintenance and self-renewal of adult stem cells (Shi et al. 2008, Discher et al. 2009, Coutu, D. L., &Galipeau2011), a stem cell mutant that becomes independent of growth factor signaling can rapidly invade the stem cell population when the normal growth factor levels are depleted. From the tissue dynamics point of view this is the most crucial phenomenon.
There are likely to be multiple mechanisms by which altered growth factor levels can create selective advantages for carcinogenic mutations. The involvement of EGF and P53 in the adult stem cell dynamics suggests that a deficiency of EGF can select for P53 mutant as well (Figure 2). The normal dynamics of stem cells crucially depends upon asymmetric division of stem cells with one of the daughter cells differentiating into tissue cells and the other being renewed as a stem cell. The asymmetry of division is under control of multiple signals including EGF and p53. While EGF signaling facilitates stem cell renewal (Tamama et al. 2010, Piryani et al. 2016, Wang et al. 2019), p53 facilitates differentiation and prevents dedifferentiation (Yu et al. 2014). If EGF signal is weak, the normal cell would have a reduced probability of renewal. This would lead to a gradual depletion of stem cell pool. Under these conditions a p53 mutant, which is less likely to differentiate can have an increased probability of contributing to the stem cell pool. Thus under EGF deficient conditions, EGF independent mutant and a p53 mutant or both get a selective advantage over a normal ASC. With normal EGF levels the normal cell has a good rate of getting renewed in the ASC population and the mutants would face a tough competition from normal cells. If the mutants have to pay some cost of the mutation, they are more likely to lose out in the competition.
If the new mutant has any selective advantage owing to altered levels of any other internal environmental factor, its population will rise making the stage available for any further spontaneous genomic change. This way the improbable nature of coming together of many chromosomal changes necessary to give rise to a cancerous cell alters substantially.
A natural question to follow is what causes chronic alterations in the levels of EGF and other growth factors that can give a selective advantage to mutants. A stone age ancestral hunter-gatherer is likely to have experienced minor injuries such as pricking, pinching, bruises, blunt impacts and the like at a high frequency. It is therefore not surprising that the injuries as well as anticipation of injuries stimulate a variety of growth factors (Roberts 1974, Nexo et al. 1981, 1984, Aloe et al. 1994, Wilson et al. 1999). The regulation of growth factor production would have evolved to suit the natural and inevitable frequency of injuries. As compared to the stone age or agricultural societies, modern urban life style has substantially lower frequency of cutaneous injuries. In addition acts of physical adventure which anticipate injuries are becoming increasingly deficient owing to multiple reinforced measures of safety and physical risk avoidance. Since in ancestral populations, the injury induced growth factor release would be frequent, the mean growth factor levels in normal stone age life would have been sufficient for all normal functions of growth factors. Therefore it is likely that no other mechanisms for maintaining healthy growth factor levels in the absence of injury stimulus would have evolved.
In a non-injury-prone and non-adventurous modern lifestyle, a cumulative deficiency of growth factors may develop. Current data on population levels of growth factors are scanty but available studies show that the levels of EGF, NGF and many other growth factors are altered in many lifestyle related disorders (Kasayama et al. 1989, Rasmussen 1995, Mraz et al. 2009).
The wound healing process needs multiple triggers and therefore making only one of the triggers constitutive does not make the mutant cell malignant. Until a minimum number of triggers are altered by mutation or epigenetic or physiological changes the process will not begin. Therefore either a series of mutations or a concerted genetic, epigenetic and physiological changes are required for malignancy (Miranda-Gonçalves et al. 2018, Saggese et al. 2020, Crispo et al. 2019, Izzo et al. 2021, Sun et al. 2022, Thakur and Chen 2019). It is also likely that when only some of the triggering pathways are constitutive, the presence of an actual wound provides the necessary stimuli and a tissue regeneration process begins. This is the likely reason why many tumors initiate only after a local injury or inflammation (Walter et al. 2011, Kuraishy et al. 2011, Arwert et al. 2012, Lee et al. 2018). However owing to the mutations, the down-regulation at the right time is disabled and as a result the cell process fails to stop leading to cancerous growth.
Evolution of Mechanisms for Cancer Prevention
Classically cancer is considered to be somatic evolution that needs to reinvent itself every time (Gatenby et al. 2010). All characteristics of cancer have to evolve de novo by this perspective. By this view a number of mechanisms have evolved to minimize the risk of cancer (Gatenby et al. 2010, Nedelcu and Caulin 2016). Our synthesis necessitates a rethinking of this perspective. The mechanisms and pathways that characterize cancers do not need to evolve de novo. Almost all of them have already evolved for wound healing and tissue regeneration process and somatic evolution only needs to make their expression constitutive or out of context.
The mutation limited view of cancers has emphasized on strategies to reduce the chances of mutations on the one hand and detect and eliminate the transformed cells by metabolic or by immune mechanisms on the other. The tumor suppressor genes that have been identified get a different interpretation by our synthesis. If the so called tumor suppressor genes and mechanisms are a normal part of regulation and closure of the wound healing process, they may not have evolved specifically as a defense against cancer. Instead their normal role in regulating and terminating the wound healing cascade will make them suppress cancers too. Our synthesis also identifies many more efficient mechanisms of preventing cancers and also identifies the conditions in which such mechanism can fail.
There are five classes of strategies to prevent cancers (A) strategies to ensure that potentially cancer causing mutants do not pass on to the next generation (B) strategies to reduce the chances of somatic mutations and (C) strategies to prevent erroneous triggering of wound healing (D) strategies to regulate and terminate the wound healing cascades (E) strategies to ensure that the mutants are at a selective disadvantage.
A: To prevent cancer related mutations from passing on to the next generation:
A combination of two strategies can ensure this almost completely (i) a single cell stage in the life-cycle and (ii) pleiotropy between adult tissue regulation and early embryonic development. In social cheating, individual isolation at some stage of lifecycle is known to arrest cheating (Matapurkar and Watve 1997). In multicellular organisms in which the life cycle needs to go through a single cell stage, if mechanisms in this cell are defective, it will simply not develop into an organism. The only possibility of passing on cancer related mutations to the next generation is if the mutants are individually neutral and become cancerous only in combination with others. However, if the genes are involved in the early developmental process, any mutation altering its expression will impair development itself. Evolution of pleiotropy between early embryonic developmental mechanisms and adult cell replication regulation mechanisms can ensure that cancer causing mutations do not pass on to the next generation. It is easy to see that the growth factors in particular have important roles in early embryonic development (Spanos et al. 2000, Teruel and Smith 2000, Guzeloglu-Kayisli et al. 2009, Llobat 2021, Allan et al. 2001) as well as in wound healing (Vaidyanathan 2021) and whose mutants are often related to cancers (Witsch et al. 2010).
B: Strategies to reduce mutational burden:
The stem cell and asymmetric division system by itself is efficient in reducing the chances of mutation accumulation. The adult stem cells undergo asymmetric division and thereby one of the daughter cells differentiates and the other adds back to the ASC pool. The differentiated cells may replicate, but ultimately undergo senescence and die. The mutation rate in the stem cell is substantially smaller than that is cells at other stages (Brazhnik 2020). Therefore any mutations accumulated in the differentiated cells vanish along with cell death. New lines start again from ASCs. This mechanism arrests mutation accumulation considerably if not completely. The other side of the coin, however is that a mutation the ASC will persist. Further in normal cell cycles there are check points and apoptosis that can trigger self destruction in a cell with DNA damage (Roos and Kaina 2006, Aitken and Koppers 2011).
C. Strategies to prevent erroneous triggering of wound healing: If the wound healing process was to get triggered by a single trigger, a single mutation could have been sufficient to cause cancer. But beginning of wound healing involves complex signaling including the ones released by cell lysis, degraded collagen, calcium signaling and growth factors released by platelets and cells (Niethammer2016, Shannon et al. 2017, Yamakawa and Hayashida 2019, Ghilardi et al. 2020). The advantage of multiple signaling pathways is that a single mutation making a pathway constitutive will not initiate cancer. Further the multiple triggers may also contain signatures of the site and type of tissue that needs regeneration. In the absence of selection, it is highly unlikely that a cell can have multiple mutations to internalize all the necessary triggers and start the wound healing process in the absence of a wound.
D. Strategies for effective termination of wound healing:
Equally critical for wound healing is to terminate proliferation, migration and other responses at the right stage. This process is also regulated by a number of well coordinated signals and pathways. Interestingly many so called “tumor suppressor genes” are involved at this stage of wound healing (supplementary Table S1). Evolution of multiple mechanisms of regulating the process simultaneously ensures cancer prevention, unless there is selection for one or more mutants affecting the regulatory mechanisms.
E. Strategies to ensure that the mutants are at a selective disadvantage: A mutant cell that can internalize a signal under normal circumstances pays a greater cost than cells responding to external growth factor signals as described earlier. Therefore mutants internalizing one of the multiple necessary wound healing signals are unlikely to gain a selective advantage and proliferate. They are more likely to get selected against owing to its higher cost.
The signaling pathways within a cell are often branched and the branches linked to different vital functions (Figure 3). We argued above that when the external signal is inadequate, any mutation making the pathway constitutive may get selected. Potentially any signal along the pathway towards triggered cell proliferation can mutate to become constitutive or overexpressed. However if it is downstream to the branching point, it will not upregulate the other vital function and as a result the mutant cell may die instead of proliferating. The structure of the signaling pathways, particularly its branching might have evolved to minimize the types of mutations that can lead to successful proliferation of the cell.
Optimization of the anti cancer defenses: The cancer defense mechanisms come at a cost and enforce certain constraints on cellular and physiological processes (Boutry et al. 2020). Therefore an optimization is expected to evolve in the cancer defense mechanisms. We discussed above that a subnormal level of growth factors can lead to selection of a signal internalizing mutant. However growth factors have a cost (Oña andLachmann 2020). Apart from the cost of making and maintaining the growth factor levels, the metabolic rate and rate of cell replacement also increases with growth factor levels. Therefore having higher levels of growth factors in normal conditions is energetically costly. There should be an optimum level of growth factors that keep the energy cost to a minimum but do not increase the risk of selecting mutants considerably. The net cost with varying levels of growth factors is likely to follow a cliff edge fitness function (Figure 4) (Nesse 2004). The optimum should ideally lie near the tip, but some variations being inevitable in biological systems, some individuals may drift towards a propensity to develop cancers normally. Because of the cliff edge fitness function a small probability of developing cancer may exist in a population growing in its natural habitat. But if growth factor expressions are suboptimum due to a mismatched lifestyle, the risk can increase substantially. Growth factor independent mutant is only an example. Similar function can be expected for selective factors for other cancer causing mutations. There is also likely to be a trade-off of adult stem cell numbers and maintenance mechanisms with growth rate and accordingly the ASC number could have been optimized by evolution.
Evidence for the Synthesis
Our synthesis accounts for a number of well known patterns and phenomena in cancer that have been unexplained or paradoxical with prior perspectives. This can be treated as existing support to the synthesis, before any novel testable predictions can be made.
- The parallels between wound healing and cancer:
Many authors have pointed out the similarities between mechanisms and pathways involved in cancer and in wound healing (Sundaram et al. 2018, Foster et al. 2018, MacCarthy-Morrogh and Martin 2020, Deyell et al. 2021) (Supporting information Table S1–S3). In vitro wound healing assays have been commonly used to assess cell proliferation migration and metastasis in cancer (Wang et al. 2019, Freitas et al. 2021, Kauanova et al. 2021) as well as in wound healing (Stamm et al. 2016). But the parallels between the two are not restricted to what is reflected in wound healing assays. All of the hallmarks and enabling characteristics of cancers (Hanahan and Weinberg 2000, 2011, Hanahan 2022, Hanahan and Monje 2023) namely proliferative signaling, evading growth suppressors, replicative immortality, inducing angiogenesis, altered cellular metabolism avoiding immune destruction, genome instability or increased rates of mutations and tumour promoting inflammation are demonstrable in some or the other stages of the wound healing process as described under and in supplementary information.
The process of wound healing and tissue regeneration has been divided into six phases namely (i) hemostasis (ii) inflammatory phase (iii) growth and neovascularization phase (iv) re-epithelization (v) tissue maturation and remodeling (Rodrigues et al. 2019). When a wound causes serious damage or loss of tissue, first defense from invading pathogens and removal of damaged cells and debris is needed. This involves inflammatory cells, oxidative burst and other mechanisms of defense. Then certain cells from the surrounding tissues need to proliferate more and make up for the loss. This requires proliferation beyond the normal regulated levels by evading growth suppressors, dedifferentiation, epithelial to mesenchymal transition (EMT), altered dynamics of ASCs, coordinated and directed migration to the site of damage, cell-cell communication, and cooperation between different cell types, co-opting blood vessels and enhancement of angiogenesis, rapid metabolic burst needed to supply energy for the proliferation and repair process. Thus these phenomena are not unique to cancer cells. All the mechanisms witnessed in the development of cancer have evolved for a normal healing, regeneration, repair and tissue homeostasis process (Supplementary Table S1). Out of the 264 genes listed by the cosmic catalogue of genes whose mutations are causally implicated in cancers, 243 genes have some or the other role in wound healing (Supplementary Table S2). Conversely out of the 554 genes differentially expressed in wound healing, 462 were evidently involved in tumorigenesis, only 16% were not. (Supplementary Table S3).
Mechanisms to prevent ectopic growth of cells such as anoikis as well as mechanisms to become anoikis resistant are required for normal tissue dynamics and wound healing processes (Malagobadan and Nagoor 2019, Yin et al. 2022). Therefore the phenomenon of anoikis resistance (Weems et al. 2023) is not unique to cancer cells.
But since the coordination of wound healing is dependent upon gradients of locale specific signals which are absent in cancer, the cancer proliferation lacks the superb site specific coordination and regulation seen in wound healing. For example migration of cells to the site of damage is well directed by chemoattractant gradients in wound healing (Brubaker et al. 2013, Ansorge et al. 2016, Ridiandries et al. 2018). Some of these chemoattractants are active in cancers as well but the absence of site specificity in these signals can result into metastasis to unrelated tissues. Further as the healing is near completion another coordinated cascade for terminating the proliferation, remodeling the tissue, destruction of the now unwanted cells, cleaning and wound closure is activated. It can be noted from Table 1 that many of the important genes and mechanisms involved in the later phases of wound healing are also known to be tumor suppressors. This offers a new interpretation to the evolution of tumor suppressor genes. They might have primarily evolved to regulate and appropriately terminate the wound healing process and not as tumor suppressors.
A wound healing process requires coordination and cross talk between different types of cells, tissues, blood vessels and nerves. A number of such complex interplays have been demonstrated (Ashrafi et al. 2016, Brazil et al. 2019, Zhou et al. 2020, Bird 2021, Gupta et al. 2022, Beura et al. 2022). It’s no wonder therefore that such complex cross-talks are seen in cancer development too (Pascut et al. 2020, Su et al. 2021). Injuries and behavior have a necessary two way relationship. Injuries potentially affect social hierarchies and foraging behavior and therefore an injured animal needs to fine tune his behavior according to the severity of injury. The behavior in turn feeds back to the process of healing. Therefore complex neuronal interaction with the healing tissue are expected to have evolved normally (Beura et al. 2022) and they contribute to the tumorigenic process as well (Hanahan and Monje 2023).
At the level of chromosomes and nucleic acids, some of the mechanisms are adaptive and functionally important in wound healing. A few others appear to be inevitable effects of rapid proliferation. For example RNA splicing has specific role in wound healing and there is considerable overlap between the altered splicing agents in wound healing and cancers (Jensen et al. 2014, Anczuków and Krainer 2016, She et al. 2021). Involvement of circular RNA and specifically circ-Amotl1 is common to wound healing and cancer (Yang et al. 2017, Li et al. 2022).
The speed of replication being the priority in wound healing, more mutations and chromosomal abnormalities may accumulate in the proliferating cells (Zhang and Xu 2017, Bielas and Heddle 2000). In addition some of the mechanisms involved in inflammation and proliferation are known to be mutagenic (Kiraly et al. 2015, Zhang and Xu 2017, Kay et al. 2019). Genomic instability also may result from inflammatory mechanisms (Butin-Israeli et al. 2019). Therefore the rate of mutation and chromosomal abnormalities is expected to be high in the healing tissue. Therefore active apoptotic mechanisms are needed in the later phases of wound healing to detect and eliminate defective cells. However, since differentiated cells are subject to senescence and death, normally there would be a reasonable amount of mutation tolerance in this process. Further mutations may also generate neoantigens and such cells will be cleared by the immune system normally. Therefore increased rates of mutation and chromosomal alterations may not cause long term problems in wound healing. However, if mechanisms including apoptosis and macrophage mediated clearance are impaired, accumulation of mutation and chromosomal anomalies may result. This appears to happen in chronic inflammation induced cancers ().
A plausible alternative interpretation also exists for the chromosomal alterations. Some of the phenomena have been speculated to have a functional significance. For example, polyploidization and cell fusion is suggested to have useful functions in wound healing (Alvarez-Dolado, and Martínez-Losa 2011, Losick et al. 2013, Dornen et al. 2020) and therefore they may be a normal part of the wound healing process and not unique to cancer.
Macrophages have a dual role in both wound healing and cancer. Although somewhat oversimplified, there is a macrophage polarization paradigm that classifies them into M1 and M2 types. During wound healing M1 macrophages clear invading pathogens as well as apoptotic or effete cells. Conversely M2 macrophages suppress inflammation and promote cell proliferation during repair and regeneration (Krzyszczyk et al. 2018). Similarly in tumor development M2 promote proliferation (Cassetta and Pollard 2023) and M1 appear to attack and clear tumor cells (Boutilier, A. J., & Elsawa 2021).
The expected high rate of mutations in inflammation and healing processes raises the possibility that neoantigens will be created in the process. This is a possible threat or hurdle at certain stages of wound healing. It is possible therefore that the PD-1, PD-L1, CTLA-4/B7-1/B7-2 driven immune checkpoints (Hua and Bergers 2019, Hohnson et al. 2022) evolved for this purpose. They regulate the immune mechanisms during critical phases of cell proliferation for tissue regeneration. In some of the healing associated disorders, macrophages appear to be activated by antigens (Weyand et al. 1994, Watanabe et al. 2017), but the origin of these antigens is not known. Activating immune checkpoints facilitates wound healing (Su et al. 2019, Wang et al. 2022). Given the probability of generating new antigens during wound healing, mechanisms for contextual and short time suppression of immune response to new antigens are needed. Therefore these mechanisms may have evolved to accompany wound healing. If cancers were only invasion by cheater cells, it is difficult to explain why immune checkpoint mechanisms are turned on in cancers.
Owing to multiple similarities between wound healing and tumor growth most cancer therapies interfere in wound healing mechanisms (Haubner et al. 2012, Deptula et al. 2019) as expected. This is not a “side effect” of cancer therapy, it is the main effect according to our perspective.
1. Epidemiological patterns: Vibishan and Watve (2020), using multiple lines of epidemiological evidence argue that cancers are not mutation limited, but selection limited and a selection limited view explains the epidemiological patterns better. Our synthesis makes further suggestions on how the selection works. This explains why there can be non-mutagenic carcinogens, why the relationship between number of stem cell divisions and cancer incidence is non-linear, why the incidence of cancer reduces at late age and why cancer incidence does not increase with the size of the organism.
1. Importance of tissue microenvironment: The tissue microenvironment has dual importance. On the one hand it shapes the selective pressures on the mutants and on the other it influences the triggers for wound healing pathways. Therefore it is expected that to a large extent the tissue microenvironment will shape the history of developing cancer at all stages. This is compatible with empirical findings (Wang et al. 2017, Hu and Polyak 2008, Medema and Vermeulen 2011, Lu et al. 2012, Quail and Joyce 2013, Pickup et al. 2014, Liu et al. 2020, Elgundi et al. 2020).
Accounting for Peto’s paradox: Since the somatic evolution of cancer is not a mutation limited process, the Peto’s paradox (Nagy et al. 2007, Noble et al. 2015, Tollis et al. 2017) simply does not exist. The paradox was created by a mutation centric view. In our synthesis the perspective is radically different. Cancers are selection limited rather than being mutation limited and there is no reason why cancer incidence should increase with body size, or the number of cell divisions throughout the range.
Lifestyle factors and cancer incidence: Since life style factors can influence body’s internal environment, they can influence cancer incidence in a significant way. Many studies show the effects of several lifestyle factors (Anand et al. 2008, Katzke et al. 2015, Wolin et al. 2010, Avgerinos et al. 2019, Singh et al. 2022, Pati et al. 2023). But so far the detailed causal links have been underexplored. Our synthesis gives a theoretical platform on which the pathways and links between life style factors and tumorigenesis can be elucidated in detail.
Behaviorally rich environment suppresses implanted tumor growth: Perhaps potentially the most important and so far ignored life style factor is behavior. In our synthesis, behavior has a central role in shaping the tissue microenvironment. An important experiment highlighting the importance of behavior is that by Cao et al. (2010) which showed that by providing a behaviorally rich environment progression of an implanted tumor could be effectively suppressed. Many experiments could reproduce the results although they differ in the detail (Li et al. 2015, Takai et al. 2019, Watanabe et al. 2020, Xiao et al. 2021, de Sousa Fernandes et al. 2022). Our synthesis demands a revival of this line of work to trace the multiple molecular links between behavior and carcinogenesis.
Testable Predictions and Suggested Lines of Experimental Research
A number of testable predictions emerge from our synthesis which may guide certain novel lines of research and lead to greater insights into the fundamental biology of cancer.
One cell stage in life cycle: Organisms in which cancers can be fatal should have a mandatory one cell stage in the life cycle. Organism that have other mechanisms due to which cancers are either very infrequent or not always fatal, may have single cell stage optional. For example, plants can shed a diseased part and grow new tissues. The cancer like conditions in plants are not necessarily fatal and therefore plants can have a single cell stage optionally in the life cycle. They may reproduce vegetatively. Animals like hydra reproduce vegetatively, but there is no report of cancer in vegetatively reproducing hydra. Whether this is a generalized statement needs to be evaluated across the diversity of life.
In vitro cell competition experiments: The concept of context dependent selective advantage to specific driver mutations involved in different types of cancers can be tested using in vitro competition experiments. One such demonstration comes from Archetti et al. (2015) studies. These experiments mark a line of work by which the selective conditions for different mutations thought to be drivers of carcinogenesis can be identified.
Extension of Cao et al. (2010) experiments: The in vivo counterpart of selection experiments should follow the experimental design exemplified by Cao et al. (2010) and replicated by many others (Li et al. 2015, Foglesong et al. 2019, Watanabe 2020). Although the findings were largely reproducible, the mechanisms by which behavior influences tumor growth remains underexplored. Our suggestion that behavior alters the selective landscape in tissues is testable in vivo on the background of such experiments.
Behavior centered lifestyle studies: The concept of lifestyle has been largely restricted to diet and physical activity. The nature of physical activity and behavior is an important component of lifestyle according to our synthesis. Many links between behavior and physiology that could potentially alter the microenvironment are reviewed by Watve (2013). Association between behavioral traits and cancer incidence is potentially testable using epidemiological designs similar to the earlier studies, but with data on behavioral traits. There are indications that metabolic diseases as well as cancers are rare in hunter-gatherer and agri-horticultural societies (Eaton et al. 1994). How far the behavioral component contributes to the difference is an open question that can be pursued through careful cross cultural studies.
Looking for cancer specific phenomena in wound healing: We have seen that a large number of molecular, cellular and system level phenomena, once thought to be unique to cancer have been detected during wound healing (Table 1). However, a few more are thought to be cancer specific and wound healing has not been explored for presence of them. We predict that most of such phenomena would be found to be operative during wound healing in some form. Hunting for such phenomena in the wound healing process with a functional relevance can serve as empirical tests for our synthesis.
A good example is the question whether neoantigens are generated during wound healing. Neoantigens in cancer is not only a result of mutations. There appear to be specific mechanisms to increase the chances of forming new epitopes on the cell surface. The phenomena of codon reassignment and frameshift polypeptides happening at the ribosomal level without any mutational change (Bartok et al. 2020, Pataskar et al. 2022) is evidently a context specific and highly regulated process. Its occurrence across many different types of tumors suggests that it is unlikely to arise de novo in somatic evolution of cancer but it may have evolved for some specific contextual function in normal physiology which expresses itself during cancer. We expect that this function would be crucial during some stage of the wound healing process. Macrophages need to remove damaged, exhausted and effete cells (Peng et al. 2007, Westman et al. 2020, Vignali et al. 2022) during wound healing and how they selectively identify such cells is an open question. The codon reassignment and frame shift can help generate novel peptide epitopes on cell surface which enable immune response to such cells in addition to some known “eat me” signals (Lemke 2019, Birkle and Brown 2021). What we already know is that the IDO1 pathway that triggers the codon reassignment in cancer (Pataskar et al. 2022) is active during wound healing and has a role in antimicrobial activity as well as immune regulation to avoid autoimmune complications, both being crucial in wound healing (Nino-Castro et al. 2014, Bandeira et al. 2015, Ito et al. 2015, Lemos et al. 2020, Bello et al. 2021).
There are other specialized mechanisms for generating neoantigens including mRNA splicing (Merlotti et al. 2023) that are currently thought to be unique to cancers. But the splicing protein SFRS6 (Jensen et al. 2014) or SFRS3 (Li-Korotky 2006) are already known to be active in wound healing. We speculate that such mechanisms of generating novel surface epitopes will help macrophage mediated clearance of exhausted, effete cells and cells who’s role in the healing process is over (Riwaldt et al. 2017) from the wound healing site. An experimental demonstration of one or more of such phenomena during wound healing is a testable prediction that would give strong empirical support to our synthesis.
The expected limitations of immunotherapy: Cancer immunotherapy has attracted much attention and research investment currently. Our synthesis predicts very limited success to this approach. If immune response is mediated by neoantigens generated by mutations, there will be strong selection acting against such mutants and in the rapid somatic evolution macrophage action will quickly select against cells carrying neoantigens or neoepitopes (Anagnostou et al. 2017, Nagel et al. 2022, Niknafs et al. 2023). Since most neoantigens originate in passenger, rather than driver mutations, there is likely to be rapid succession of different neoantigens but immunotherapy is less likely to clear the tumor entirely. Only if an essential or driver mutation is associated with a neoantigen, or if a back mutation from the neoantigen is lethal (Niknafs et al. 2023), therapy such as CAR-T can have sustained benefit.
If, on the other hand, antigens are generated by the built in process of ribosomal level codon reassignment and frame shifted polypeptides, these processes are presumably evolved for removing specific cells and they will continue to do so in tumors as well. They are unlikely to destroy the entire tumor. Compatible with our expectation is the extremely limited success of immunotherapy alone (Ventola 2017, Sambi et al. 2019, Taefehshokr 2022). At the most it may be useful in supporting other lines of treatment.
Relevance of Our Synthesis to Cancer Research and Clinical Applications
Although many lines of evidence point to the evolution of cancer being selection limited rather than mutation limited, so far we have very little understanding of microenvironmental factors that would select specific driver mutants. This needs to become a major focus of research in near future. Which lifestyle and environmental variables affect the microenvironmental factors is the next critical question that would follow. Normalizing the microenvironment to prevent selection for driver mutations can potentially prevent cancers to a large extent. Among the life style factors, behavioral environment is an interesting possibility that needs to be explored seriously (Cao et al. 2010, Watve 2013). If behavioral deficiencies are central contributors to cancer proneness, sports, exercise and activities that act as appropriate behavioral supplements should be able to prevent cancers to a significant extent. This is potentially an important public health implication of our synthesis.
There is a possible bad news for immunotherapy, but at the same time we predict that studying the pathways that regulate and terminate the wound healing process might give useful novel breakthroughs in controlling tumor growth.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgement
Discussions at an early stage of concept development with Ruby Singh, Ruchika kaul-Ghanekar, Preirna Raina, Vibishan B., Nazneen Gheewala were useful.
References
- Adler, F.R.; Gordon, D.M. Cancer ecology and evolution: Positive interactions and system vulnerability. Curr. Opin. Syst. Biol. 2019, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.; Koppers, A.J. Apoptosis and DNA damage in human spermatozoa. Asian J. Androl. 2011, 13, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Aktipis A. (2020). The Cheating Cell: How Evolution Helps Us Understand and Treat Cancer. Princeton University Press.
- Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS. 2015 Cancer across the tree of life: Cooperation and cheating in multicellularity. Phil. Trans. R. Soc. B 370: 20140219. [CrossRef]
- Aktipis, C.A.; Nesse, R.M. Evolutionary foundations for cancer biology. Evol. Appl. 2013, 6, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.; Flint, D.; Patel, K. Insulin-like growth factor axis during embryonic development. Reproduction 2001, 122, 31–39. [Google Scholar] [CrossRef]
- Aloe, L.; Bracci-Laudiero, L.; Alleva, E.; Lambiase, A.; Micera, A.; Tirassa, P. Emotional stress induced by parachute jumping enhances blood nerve growth factor levels and the distribution of nerve growth factor receptors in lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 10440–10444. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dolado, M., & Martínez-Losa, M. (2011). Cell fusion and tissue regeneration. Advances in experimental medicine and biology, 713, 161–175. [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef]
- Ansorge, M.; Rastig, N.; Steinborn, R.; König, T.; Baumann, L.; Möller, S.; Schnabelrauch, M.; Cross, M.; Werner, C.; Beck-Sickinger, A.G.; Pompe, T. Short-range cytokine gradients to mimic paracrine cell interactions in vitro. J. Control. Release Off. J. Control. Release Soc. 2016, 224, 59–68. [Google Scholar] [CrossRef]
- Archetti, M.; Ferraro, D.A.; Christofori, G. Heterogeneity for IGF-II production maintained by public goods dynamics in neuroendocrine pancreatic cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1833–1838. [Google Scholar] [CrossRef]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.; Baguneid, M.; Bayat, A. The Role of Neuromediators and Innervation in Cutaneous Wound Healing. Acta Dermato-Venereologica 2016, 96, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G. Neuroepithelial Interactions in Cancer. Annu. Rev. Pathol. 2023, 18, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, L.G.; Bortolot, B.S.; Cecatto, M.J.; Monte-Alto-Costa, A.; Romana-Souza, B. Exogenous Tryptophan Promotes Cutaneous Wound Healing of Chronically Stressed Mice through Inhibition of TNF-α and IDO Activation. PLoS ONE 2015, 10, e0128439. [Google Scholar] [CrossRef]
- Bello, C.; Heinisch, P.P.; Mihalj, M.; Carrel, T.; Luedi, M.M. Indoleamine-2,3-Dioxygenase as a Perioperative Marker of the Immune System. Front. Physiol. 2021, 12, 766511. [Google Scholar] [CrossRef]
- Beura, S.K.; Panigrahi, A.R.; Yadav, P.; Agrawal, S.; Singh, S.K. Role of Neurons and Glia Cells in Wound Healing as a Novel Perspective Considering Platelet as a Conventional Player. Mol. Neurobiol. 2022, 59, 137–160. [Google Scholar] [CrossRef]
- Bielas, J.H.; Heddle, J.A. Proliferation is necessary for both repair and mutation in transgenic mouse cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11391–11396. [Google Scholar] [CrossRef]
- Bird, L. Healing powers of neuron–macrophage crosstalk. Nat. Rev. Immunol. 2021, 21, 408–409. [Google Scholar] [CrossRef]
- Birkle, T.; Brown, G.C. I’m Infected, Eat Me! Innate Immunity Mediated by Live, Infected Cells Signaling To Be Phagocytosed. Infect. Immun. 2021, 89, e00476–20. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Boutry, J.; Dujon, A.M.; Gerard, A.-L.; Tissot, S.; Macdonald, N.; Schultz, A.; Biro, P.A.; Beckmann, C.; Hamede, R.; Hamilton, D.G.; et al. Ecological and Evolutionary Consequences of Anticancer Adaptations. iScience 2020, 23, 101716. [Google Scholar] [CrossRef] [PubMed]
- Brazhnik, K.; Sun, S.; Alani, O.; Kinkhabwala, M.; Wolkoff, A.W.; Maslov, A.Y.; Dong, X.; Vijg, J. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci. Adv. 2020, 6, eaax2659. [Google Scholar] [CrossRef] [PubMed]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell–epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, A.L.; Rendon, J.L.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J. Immunol. 2013, 190, 1746–1757. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726. [Google Scholar] [CrossRef]
- Cao, L.; Liu, X.; Lin, E.-J.D.; Wang, C.; Choi, E.Y.; Riban, V.; Lin, B.; During, M.J. Environmental and genetic activation of a brain-a BDNF/leptin axis causes cancer remission and inhibition. Cell 2010, 142, 52–64. [Google Scholar] [CrossRef]
- Casás-Selves, M.; DeGregori, J. How cancer shapes evolution and how evolution shapes cancer. Evol. Educ. Outreach 2011, 4, 624–634. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 2023, 23, 238–257. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef]
- Cockfield, J.A.; Schafer, Z.T. Antioxidant Defenses: A Context-Specific Vulnerability of Cancer Cells. Cancers 2019, 11, 1208. [Google Scholar] [CrossRef]
- Coutu, D.L.; Galipeau, J. Roles of FGF signaling in stem cell self-renewal, senescence and aging. Aging 2011, 3, 920–933. [Google Scholar] [CrossRef]
- Crispo, F.; Condelli, V.; Lepore, S.; Notarangelo, T.; Sgambato, A.; Esposito, F.; Maddalena, F.; Landriscina, M. Metabolic Dysregulations and Epigenetics: A Bidirectional Interplay that Drives Tumor Progression. Cells 2019, 8, 798. [Google Scholar] [CrossRef]
- Dawkins, Richard. (2006)The Blind Watchmaker. Penguin Books.
- Fernandes, M.S.d.S.; Santos, G.C.J.; Filgueira, T.O.; Gomes, D.A.; Barbosa, E.A.S.; dos Santos, T.M.; Câmara, N.O.S.; Castoldi, A.; Souto, F.O. Cytokines and Immune Cells Profile in Different Tissues of Rodents Induced by Environmental Enrichment: Systematic Review. Int. J. Mol. Sci. 2022, 23, 11986. [Google Scholar] [CrossRef]
- Deptuła, M.; Zieliński, J.; Wardowska, A.; Pikuła, M. Wound healing complications in oncological patients: Perspectives for cellular therapy. Adv. Dermatol. Allergol. 2019, 36, 139–146. [Google Scholar] [CrossRef]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth factors, matrices, and forces combine and control stem cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef]
- Dörnen, J.; Sieler, M.; Weiler, J.; Keil, S.; Dittmar, T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. Int. J. Mol. Sci. 2020, 21, 1811. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal—redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.B.; Pike, M.C.; Short, R.V.; Lee, N.C.; Trussell, J.; Hatcher, R.A.; Wood, J.W.; Worthman, C.M.; Konner, M.J.; Hill, K.R.; et al. Women’s reproductive cancers in evolutionary context. Q. Rev. Biol. 1994, 69, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Elgundi, Z.; Papanicolaou, M.; Major, G.; Cox, T.R.; Melrose, J.; Whitelock, J.M.; Farrugia, B.L. Cancer Metastasis: The Role of the Extracellular Matrix and the Heparan Sulfate Proteoglycan Perlecan. Front. Oncol. 2020, 9, 1482. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Foglesong, G.D.; Queen, N.J.; Huang, W.; Widstrom, K.J.; Cao, L. Enriched environment inhibits breast cancer progression in obese models with intact leptin signaling. Endocrine-Related Cancer 2019, 26, 483–495. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef]
- Freitas, J.T.; Jozic, I.; Bedogni, B. Wound Healing Assay for Melanoma Cell Migration. Methods Mol. Biol. 2021, 2265, 65–71. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J.; Brown, J.S. Evolutionary dynamics of cancer prevention. Nat. Rev. Cancer 2010, 10, 526–527. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, S.J.; O’Reilly, B.M.; Sgro, A.E. Intracellular signaling dynamics and their role in coordinating tissue repair. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1479. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Kaushik, D.; Mohan, V. Role of neurotransmitters in the regulation of cutaneous wound healing. Exp. Brain Res. 2022, 240, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Guzeloglu-Kayisli, O.; Kayisli, U.; Taylor, H. The role of growth factors and cytokines during implantation: Endocrine and paracrine interactions. Semin. Reprod. Med. 2009, 27, 062–079. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Monje, M. Cancer hallmarks intersect with neuroscience in the cancer microenvironment. Cancer Cell 2023, 41, 573–580. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Haubner, F.; Ohmann, E.; Pohl, F.; Strutz, J.; Gassner, H.G. Wound healing after radiation therapy: Review of the literature. Radiat. Oncol. 2012, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Polyak, K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008, 18, 27–34. [Google Scholar] [CrossRef]
- Hua, Y.; Bergers, G. Tumors vs. Chronic Wounds: An Immune Cell’s Perspective. Front. Immunol. 2019, 10, 2178. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Ando, T.; Ogiso, H.; Arioka, Y.; Saito, K.; Seishima, M. Inhibition of indoleamine 2,3-dioxygenase activity accelerates skin wound healing. Biomaterials 2015, 53, 221–228. [Google Scholar] [CrossRef]
- Izzo, L.T.; Affronti, H.C.; Wellen, K.E. The Bidirectional Relationship Between Cancer Epigenetics and Metabolism. Annu. Rev. Cancer Biol. 2021, 5, 235–257. [Google Scholar] [CrossRef] [PubMed]
- A Jensen, M.; E Wilkinson, J.; Krainer, A.R. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat. Struct. Mol. Biol. 2014, 21, 189–197. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Joyce, C., Rayi, A., & Kasi, A. (2022). Tumor-Suppressor Genes. In StatPearls. StatPearls Publishing.
- Kasayama, S.; Ohba, Y.; Oka, T. Epidermal growth factor deficiency associated with diabetes mellitus. Proc. Natl. Acad. Sci. USA 1989, 86, 7644–7648. [Google Scholar] [CrossRef]
- A Katzke, V.; Kaaks, R.; Kühn, T. Lifestyle and Cancer Risk. Cancer J. 2015, 21, 104–110. [Google Scholar] [CrossRef]
- Kauanova, S.; Urazbayev, A.; Vorobjev, I. The Frequent Sampling of Wound Scratch Assay Reveals the “Opportunity” Window for Quantitative Evaluation of Cell Motility-Impeding Drugs. Front. Cell Dev. Biol. 2021, 9, 640972. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, O.; Gong, G.; Olipitz, W.; Muthupalani, S.; Engelward, B.P. Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLOS Genet. 2015, 11, e1004901. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer. Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kollara, A.; May, T.; Brown, T.J. Wounding promotes ovarian cancer progression and decreases efficacy of cisplatin in a syngeneic mouse model. J. Ovarian Res. 2018, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef]
- Lemos, H.; Mohamed, E.; Ou, R.; McCardle, C.; Zheng, X.; McGuire, K.; Homer, N.Z.M.; Mole, D.J.; Huang, L.; Mellor, A.L. Co-treatments to Boost IDO Activity and Inhibit Production of Downstream Catabolites Induce Durable Suppression of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2020, 11, 1256. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gan, Y.; Fan, Y.; Wu, Y.; Lin, H.; Song, Y.; Cai, X.; Yu, X.; Pan, W.; Yao, M.; et al. Enriched environment inhibits mouse pancreatic cancer growth and down-regulates the expression of mitochondria-related genes in cancer cells. Sci. Rep. 2015, 5, srep07856. [Google Scholar] [CrossRef] [PubMed]
- Li-Korotky, H.-S.; Hebda, P.A.; Kelly, L.A.; Lo, C.-Y.; Dohar, J.E. Identification of a pre-mRNA splicing factor, arginine/serine-rich 3 (Sfrs3), and its co-expression with fibronectin in fetal and postnatal rabbit airway mucosal and skin wounds. Biochim. et Biophys. Acta 2006, 1762, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, J.; Chen, M.; Xu, J.; Zhu, D. Circular RNA in cancer development and immune regulation. J. Cell. Mol. Med. 2022, 26, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, Q.; Ju, Y.; Song, G. Role of the mechanical microenvironment in cancer development and progression. Cancer Biol. Med. 2020, 17, 282–292. [Google Scholar] [CrossRef]
- Llobat, L. Pluripotency and Growth Factors in Early Embryonic Development of Mammals: A Comparative Approach. Veter- Sci. 2021, 8, 78. [Google Scholar] [CrossRef]
- Losick, V.P.; Fox, D.T.; Spradling, A.C. Polyploidization and cell fusion contribute to wound healing in the adult Drosophila epithelium. Curr. Biol. 2013, 23, 2224–2232. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- MacCarthy-Morrogh, L.; Martin, P. The hallmarks of cancer are also the hallmarks of wound healing. Sci. Signal. 2020, 13, eaay8690. [Google Scholar] [CrossRef]
- Malagobadan Sharan, Noor Hasima Nagoor. Anoikis. in Encyclopedia of Cancer (Third Edition) Editor(s): Paolo Boffetta, Pierre Hainaut, Academic Press, 2019, Pages 75-84, ISBN 9780128124857.
- Matapurkar, A.K.; Watve, M.G. Altruist cheater dynamics in Dictyostelium: Aggregated distribution gives stable oscillations. Am. Nat. 1997, 150, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; Vermeulen, L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011, 474, 318–326. [Google Scholar] [CrossRef]
- Merlotti, A.; Sadacca, B.; Arribas, Y.A.; Ngoma, M.; Burbage, M.; Goudot, C.; Houy, A.; Rocañín-Arjó, A.; Lalanne, A.; Seguin-Givelet, A.; et al. Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci. Immunol. 2023, 8, eabm6359. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Mraz, M.; Bartlova, M.; Lacinova, Z.; Michalsky, D.; Kasalicky, M.; Haluzikova, D.; Matoulek, M.; Dostalova, I.; Humenanska, V.; Haluzik, M. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin. Endocrinol. 2009, 71, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.D.; Victor, E.M.; Cropper, J.H. Why don’t all whales have cancer? A novel hypothesis resolving Peto’s paradox. Integr. Comp. Biol. 2007, 47, 317–328. [Google Scholar] [CrossRef]
- Nedelcu, A.M., Caulin, A.F. (2016). The Evolution of Cancer Suppression Mechanisms. In: Maley, C., Greaves, M. (eds) Frontiers in Cancer Research. Springer, New York, NY.
- Nagel, R.; Pataskar, A.; Champagne, J.; Agami, R. Boosting Antitumor Immunity with an Expanded Neoepitope Landscape. Cancer Res 2022, 82, 3637–3649. [Google Scholar] [CrossRef]
- Nesse, R.M. Cliff-edged fitness functions and the persistence of schizophrenia. Behav. Brain Sci. 2004, 27, 862–863. [Google Scholar] [CrossRef]
- Nexø, E.; Hollenberg, M.D.; Bing, J. Aggressive behavior in mice provokes a marked increase in both plasma epidermal growth factor and renin. Acta Physiol. Scand. 1981, 111, 367–371. [Google Scholar] [CrossRef]
- Nexø, E.; Olsen, P.S.; Poulsen, K. Exocrine and endocrine secretion of renin and epidermal growth factor from the mouse submandibular glands. Regul. Pept. 1984, 8, 327–334. [Google Scholar] [CrossRef]
- Niethammer, P. The early wound signals. Curr. Opin. Genet. Dev. 2016, 40, 17–22. [Google Scholar] [CrossRef]
- Niknafs, N.; Balan, A.; Cherry, C.; Hummelink, K.; Monkhorst, K.; Shao, X.M.; Belcaid, Z.; Marrone, K.A.; Murray, J.; Smith, K.N.; et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat. Med. 2023, 29, 440–449. [Google Scholar] [CrossRef]
- Niño-Castro, A.; Abdullah, Z.; Popov, A.; Thabet, Y.; Beyer, M.; Knolle, P.; Domann, E.; Chakraborty, T.; Schmidt, S.V.; Schultze, J.L. The IDO1-induced kynurenines play a major role in the antimicrobial effect of human myeloid cells against Listeria monocytogenes. Innate Immun. 2014, 20, 401–411. [Google Scholar] [CrossRef]
- Noble, R.; Kaltz, O.; Hochberg, M.E. Peto’s paradox and human cancers. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2015, 370, 20150104. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Oña, L.; Lachmann, M. Signalling architectures can prevent cancer evolution. Sci. Rep. 2020, 10, 674. [Google Scholar] [CrossRef] [PubMed]
- Paczkowski, M.; Kretzschmar, W.W.; Markelc, B.; Liu, S.K.; Kunz-Schughart, L.A.; Harris, A.L.; Partridge, M.; Byrne, H.M.; Kannan, P. Reciprocal interactions between tumour cell populations enhance growth and reduce radiation sensitivity in prostate cancer. Commun. Biol. 2021, 4, 6. [Google Scholar] [CrossRef]
- Pascut, D.; Pratama, M.Y.; Vo, N.V.; Masadah, R.; Tiribelli, C. The Crosstalk between Tumor Cells and the Microenvironment in Hepatocellular Carcinoma: The Role of Exosomal microRNAs and Their Clinical Implications. Cancers 2020, 12, 823. [Google Scholar] [CrossRef]
- Pataskar, A.; Champagne, J.; Nagel, R.; Kenski, J.; Laos, M.; Michaux, J.; Pak, H.S.; Bleijerveld, O.B.; Mordente, K.; Navarro, J.M.; et al. Tryptophan depletion results in tryptophan-to-phenylalanine substitutants. Nature 2022, 603, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Pati, S.; Irfan, W.; Jameel, A.; Ahmed, S.; Shahid, R.K. Obesity and Cancer: A Current Overview of Epidemiology, Pathogenesis, Outcomes, and Management. Cancers 2023, 15, 485. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Martin, D.A.; Kenkel, J.; Zhang, K.; Ogden, C.A.; Elkon, K.B. Innate and adaptive immune response to apoptotic cells. J. Autoimmun. 2007, 29, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. Embo Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Piryani, S.O.; Kam, A.Y.; Kliassov, E.G.; Chen, B.J.; Spector, N.L.; Chute, J.P.; Chao, N.J.; Doan, P.L. EGF Accelerates Hematopoietic Stem Cell Regeneration Following 5-FU Chemotherapy Via G-CSF Receptor Signaling. Blood 2016, 128, 1490. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H.; Hvidberg, A.; Juul, A.; Main, K.M.; Gotfredsen, A.; E Skakkebaek, N.; Hilsted, J. Massive weight loss restores 24-hour growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J. Clin. Endocrinol. Metab. 1995, 80, 1407–1415. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Oli, M.W.; Oli, M.K. Eco-oncology: Applying ecological principles to understand and manage cancer. Ecol. Evol. 2020, 10, 8538–8553. [Google Scholar] [CrossRef]
- Ridiandries, A.; Tan JT, M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef]
- Riwaldt, S.; Monici, M.; Petersen, A.G.; Jensen, U.B.; Evert, K.; Pantalone, D.; Utpatel, K.; Evert, M.; Wehland, M.; Krüger, M.; et al. Preparation of A Spaceflight: Apoptosis Search in Sutured Wound Healing Models. Int. J. Mol. Sci. 2017, 18, 2604. [Google Scholar] [CrossRef]
- Roberts, M.L. Testosterone-induced accumulation of epidermal growth factor in the submandibular salivary glands of mice, assessed by radioimmunoassay. Biochem. Pharmacol. 1974, 23, 3305–3308. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Rozhok, A., & DeGregori, J. (2017). Aging, Somatic Evolution, and Cancer. On Human Nature, 193-209. [CrossRef]
- Saggese, P.; Sellitto, A.; Martinez, C.A.; Giurato, G.; Nassa, G.; Rizzo, F.; Tarallo, R.; Scafoglio, C. Metabolic Regulation of Epigenetic Modifications and Cell Differentiation in Cancer. Cancers 2020, 12, 3788. [Google Scholar] [CrossRef] [PubMed]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 4508794. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Schulz, M.; Salamero-Boix, A.; Niesel, K.; Alekseeva, T.; Sevenich, L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front. Immunol. 2019, 10, 1713. [Google Scholar] [CrossRef]
- Shannon, E.K.; Stevens, A.; Edrington, W.; Zhao, Y.; Jayasinghe, A.K.; Page-McCaw, A.; Hutson, M.S. Multiple Mechanisms Drive Calcium Signal Dynamics around Laser-Induced Epithelial Wounds. Biophys. J. 2017, 113, 1623–1635. [Google Scholar] [CrossRef]
- She, W.; Shao, J.; Jia, R. Targeting Splicing Factor SRSF6 for Cancer Therapy. Front. Cell Dev. Biol. 2021, 9, 780023. [Google Scholar] [CrossRef]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sun, G.; Zhao, C.; Stewart, R. Neural stem cell self-renewal. Crit. Rev. Oncol. /Hematol. 2008, 65, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mayengbam, S.S.; Yaduvanshi, H.; Wani, M.R.; Bhat, M.K. Obesity Programs Macrophages to Support Cancer Progression. Cancer Res 2022, 82, 4303–4312. [Google Scholar] [CrossRef]
- PeiSomarelli, J.A. The Hallmarks of Cancer as Ecologically Driven Phenotypes. Front. Ecol. Evol. 2021, 9, 661583. [Google Scholar] [CrossRef] [PubMed]
- Spanos, S.; Becker, D.L.; Winston, R.M.; Hardy, K. Anti-apoptotic action of insulin-like growth factor-I during human preimplantation embryo development. Biol. Reprod. 2000, 63, 1413–1420. [Google Scholar] [CrossRef]
- Stamm, A.; Reimers, K.; Strauß, S.; Vogt, P.; Scheper, T.; Pepelanova, I. In vitro wound healing assays—State of the art. BioNanoMaterials 2016, 17, 79–87. [Google Scholar] [CrossRef]
- Su, T.; Zhang, P.; Zhao, F.; Zhang, S. Exosomal MicroRNAs Mediating Crosstalk Between Cancer Cells With Cancer-Associated Fibroblasts and Tumor-Associated Macrophages in the Tumor Microenvironment. Front. Oncol. 2021, 11, 631703. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, G.M.; Quah, S.; Sampath, P. Cancer: The dark side of wound healing. FEBS J. 2018, 285, 4516–4534. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef]
- Taefehshokr, S.; Parhizkar, A.; Hayati, S.; Mousapour, M.; Mahmoudpour, A.; Eleid, L.; Rahmanpour, D.; Fattahi, S.; Shabani, H.; Taefehshokr, N. Cancer immunotherapy: Challenges and limitations. Pathol.—Res. Pract. 2022, 229, 153723. [Google Scholar] [CrossRef]
- Tamama, K.; Kawasaki, H.; Wells, A. Epidermal growth factor (EGF) treatment on multipotential stromal cells (MSCs). Possible enhancement of therapeutic potential of MSC. J. Biomed. Biotechnol. 2010, 2010, 795385. [Google Scholar] [CrossRef]
- Teruel, M., Smith, R., & Catalano, R. (2000). Growth factors and embryo development. Biocell : Official journal of the Sociedades Latinoamericanas de Microscopia Electronica ... et. al, 24(2), 107–122.
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Takai, D.; Abe, A.; Miura, H.; Tanaka, S.; Komura, J.-I. Minimum environmental enrichment is effective in activating antitumor immunity to transplanted tumor cells in mice. Exp. Anim. 2019, 68, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boddy, A.M.; Maley, C.C. Peto’s Paradox: How has evolution solved the problem of cancer prevention? BMC Biol. 2017, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Trivedi, D.D.; Dalai, S.K.; Bakshi, S.R. The Mystery of Cancer Resistance: A Revelation Within Nature. J. Mol. Evol. 2023, 91, 133–155. [Google Scholar] [CrossRef]
- Turan, Z.G.; Parvizi, P.; Dönertaş, H.M.; Tung, J.; Khaitovich, P.; Somel, M. Molecular footprint of Medawar’s mutation accumulation process in mammalian aging. Aging Cell 2019, 18, e12965. [Google Scholar] [CrossRef]
- Vaidyanathan, L. Growth Factors in Wound Healing—A Review. Biomed. Pharmacol. J. 2021, 14, 1469–1481. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Vibishan, B.; Watve, M.G. Context-dependent selection as the keystone in the somatic evolution of cancer. Sci. Rep. 2020, 10, 4223. [Google Scholar] [CrossRef]
- Vignali, P.D.A.; DePeaux, K.; Watson, M.J.; Ye, C.; Ford, B.R.; Lontos, K.; McGaa, N.K.; Scharping, N.E.; Menk, A.V.; Robson, S.C.; et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat. Immunol. 2022, 24, 267–279. [Google Scholar] [CrossRef]
- Voronina, N.; Wong, J.K.L.; Hübschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.D.; Rice, P.L.; Redente, E.F.; Kauvar, E.F.; Lemond, L.; Aly, T.; Wanebo, K.; Chan, E.D. Wound Healing after Trauma May Predispose to Lung Cancer Metastasis. Am. J. Respir. Cell Mol. Biol. 2011, 44, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Decker, C.C.; Zechner, L.; Krstin, S.; Wink, M. In vitro wound healing of tumor cells: Inhibition of cell migration by selected cytotoxic alkaloids. BMC Pharmacol. Toxicol. 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Feige, P.; Brun, C.E.; Hekmatnejad, B.; Dumont, N.A.; Renaud, J.-M.; Faulkes, S.; Guindon, D.E.; Rudnicki, M.A. EGFR-Aurka Signaling Rescues Polarity and Regeneration Defects in Dystrophin-Deficient Muscle Stem Cells by Increasing Asymmetric Divisions. Cell Stem Cell 2019, 24, 419–432. [Google Scholar] [CrossRef]
- Watanabe, J.; Kagami, N.; Kawazoe, M.; Arata, S. A simplified enriched environment increases body temperature and suppresses cancer progression in mice. Exp. Anim. 2020, 69, 207–218. [Google Scholar] [CrossRef]
- Watanabe, R.; Zhang, H.; Berry, G.; Goronzy, J.J.; Weyand, C.M. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am. J. Physiol. Circ. Physiol. 2017, 312, H1052–H1059. [Google Scholar] [CrossRef]
- Watve M. G. (2013) Doves, diplomats and diabetes: A Darwinian reinterpretation of type 2 diabetes and related disorders. Springer, New York, Heidelberg, London 2013.
- Weems, A.D.; Welf, E.S.; Driscoll, M.K.; Zhou, F.Y.; Mazloom-Farsibaf, H.; Chang, B.-J.; Murali, V.S.; Gihana, G.M.; Weiss, B.G.; Chi, J.; et al. Blebs promote cell survival by assembling oncogenic signalling hubs. Nature 2023, 615, 517–525. [Google Scholar] [CrossRef]
- Westman, J.; Grinstein, S.; Marques, P.E. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front. Immunol. 2020, 10, 3030. [Google Scholar] [CrossRef]
- Weyand, C.M.; Schönberger, J.; Oppitz, U.; Hunder, N.N.; Hicok, K.C.; Goronzy, J.J. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J. Exp. Med. 1994, 179, 951–960. [Google Scholar] [CrossRef]
- White, A.C.; Lowry, W.E. Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol. 2015, 25, 11–20. [Google Scholar] [CrossRef]
- Wilson, S.E.; Chen, L.; Mohan, R.R.; Liang, Q.; Liu, J. Expression of HGF, KGF, EGF and receptor messenger RNAs following corneal epithelial wounding. Exp. Eye Res. 1999, 68, 377–397. [Google Scholar] [CrossRef]
- Witsch, E.; Sela, M.; Yarden, Y.; Li, T.Y.; Lin, S.-Y.; Lin, S.-C.; Staruschenko, A.; Palygin, O.; Ilatovskaya, D.V.; Pavlov, T.S. Roles for growth factors in cancer progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef]
- Wolin, K.Y.; Carson, K.; Colditz, G.A. Obesity and cancer. Oncologist 2010, 15, 556–565. [Google Scholar] [CrossRef]
- Xiao, R.; Ali, S.; Caligiuri, M.A.; Cao, L. Enhancing Effects of Environmental Enrichment on the Functions of Natural Killer Cells in Mice. Front. Immunol. 2021, 12, 695859. [Google Scholar] [CrossRef]
- Yang, Z.-G.; Awan, F.M.; Du, W.W.; Zeng, Y.; Lyu, J.; Wu, D.; Gupta, S.; Yang, W.; Yang, B.B. The circular RNA interacts with STAT3, increasing its nuclear translocation and wound repair by modulating Dnmt3a and miR-17 function. Mol. Ther. 2017, 25, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, S.; Hayashida, K. Advances in surgical applications of growth factors for wound healing. Burn. Trauma 2019, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, J.; Zhang, X.; Liao, Y.; Luo, W.; Wang, S.; Ding, J.; Huang, J.; Chen, M.; Wang, W.; et al. A missing piece of the puzzle in pulmonary fibrosis: Anoikis resistance promotes fibroblast activation. Cell Biosci. 2022, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Sa, C.; Xiaobing, F.; Andong, Z. p53: The Barrier or Guardian for Cell Dedifferentiation? BioScience 2014, 64, 883–892. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, X. Mutation-promoting molecular networks of uncontrolled inflammation. Tumor Biol. 2017, 39, 1010428317701310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Brown, B.A.; Siegel, A.P.; El Masry, M.S.; Zeng, X.; Song, W.; Das, A.; Khandelwal, P.; Clark, A.; Singh, K.; et al. Exosome-Mediated Crosstalk between Keratinocytes and Macrophages in Cutaneous Wound Healing. ACS Nano 2020, 14, 12732–12748. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Expected growth responses of normal (solid line) and receptor overexpressing (dashed line) mutant to different growth factor concentrations. At low growth factor concentrations, the mutant may gain a competitive advantage over the normal cell, but at high concentration they may lose it owing to the extra cost they pay for overexpressing the receptor.
Figure 1.
Expected growth responses of normal (solid line) and receptor overexpressing (dashed line) mutant to different growth factor concentrations. At low growth factor concentrations, the mutant may gain a competitive advantage over the normal cell, but at high concentration they may lose it owing to the extra cost they pay for overexpressing the receptor.
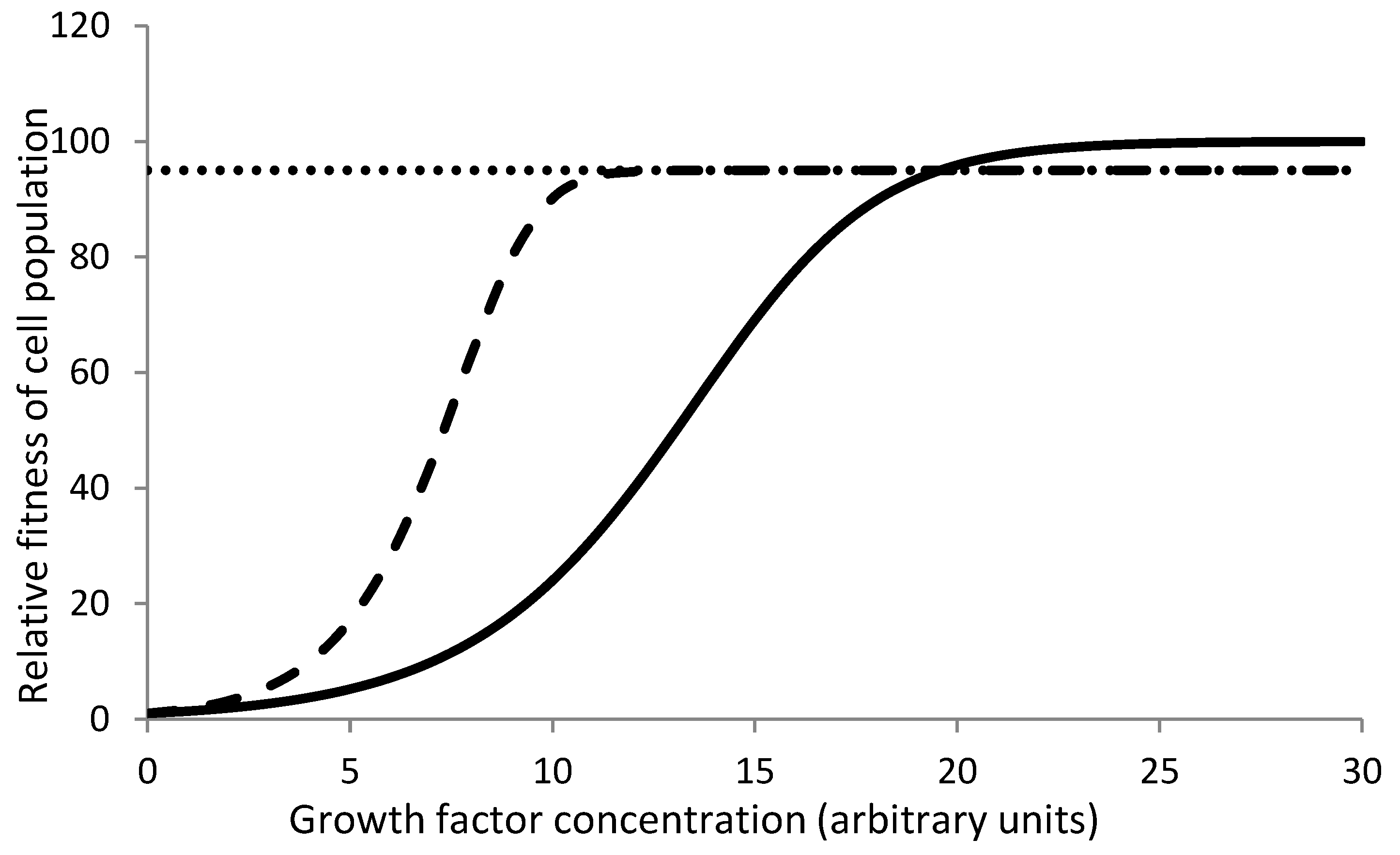
Figure 2.
Effects of EGF and p53 on adult stem cell renewal and differentiation dynamics. In this dynamics a weak input of EGF signal can give selective advantage to EGFR overexpressing or p53 mutants or both.
Figure 2.
Effects of EGF and p53 on adult stem cell renewal and differentiation dynamics. In this dynamics a weak input of EGF signal can give selective advantage to EGFR overexpressing or p53 mutants or both.
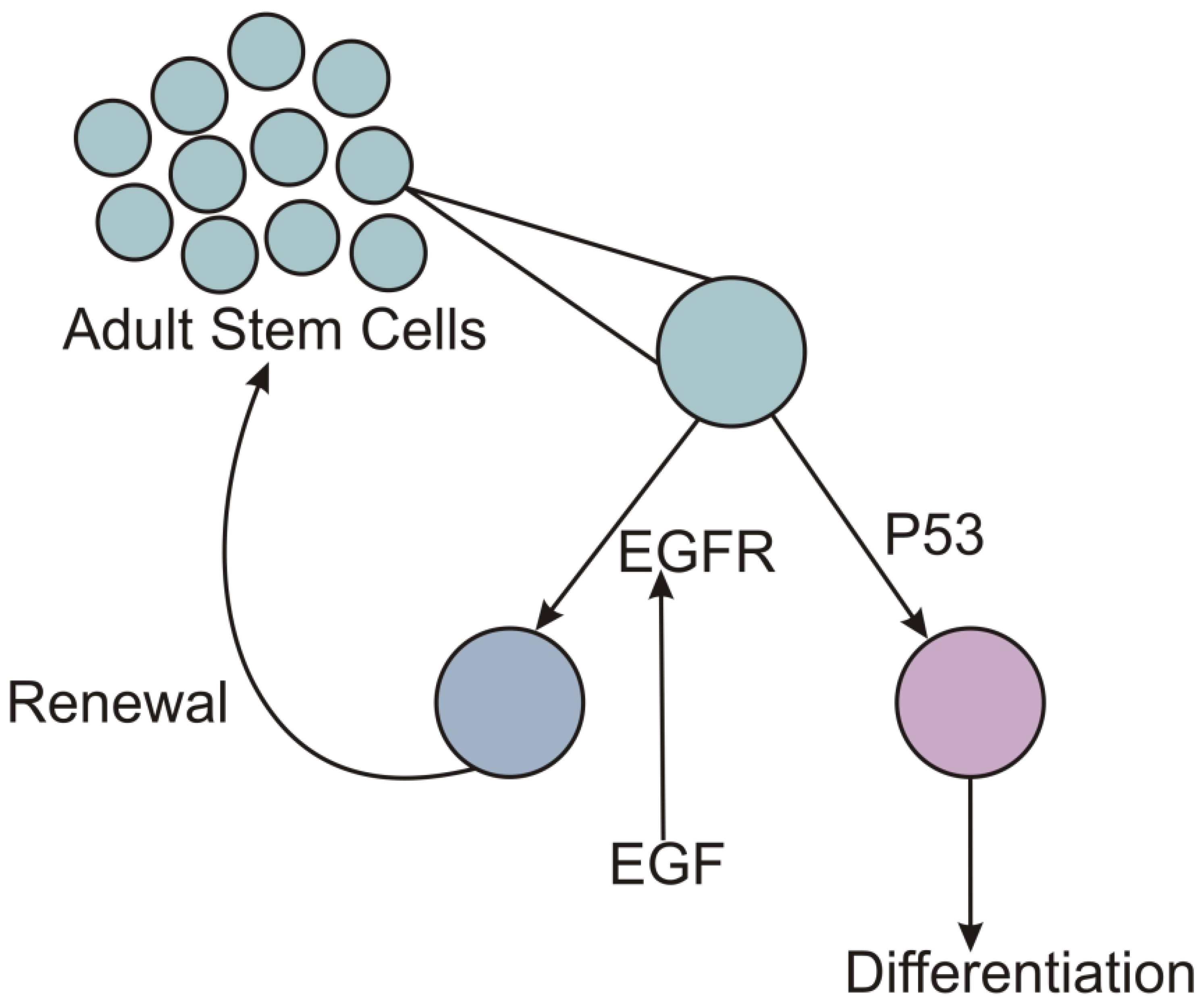
Figure 3.
The signaling pathways may have evolved to minimize the possible number mutations that can trigger uncontrolled cell proliferation. Linking a vital function of the cell as a branch of the proliferation signaling pathway is a potential preventive mechanism. In the hypothetical signaling pathway above, if the external signal is weak, constitutive or overexpressing mutation at the receptor or signal 1 level can lead to uncontrolled proliferation but mutation making signal 3 or signal 4 constitutive cannot. In reality the signaling pathways are often branched and link to many different functions of the cell.
Figure 3.
The signaling pathways may have evolved to minimize the possible number mutations that can trigger uncontrolled cell proliferation. Linking a vital function of the cell as a branch of the proliferation signaling pathway is a potential preventive mechanism. In the hypothetical signaling pathway above, if the external signal is weak, constitutive or overexpressing mutation at the receptor or signal 1 level can lead to uncontrolled proliferation but mutation making signal 3 or signal 4 constitutive cannot. In reality the signaling pathways are often branched and link to many different functions of the cell.
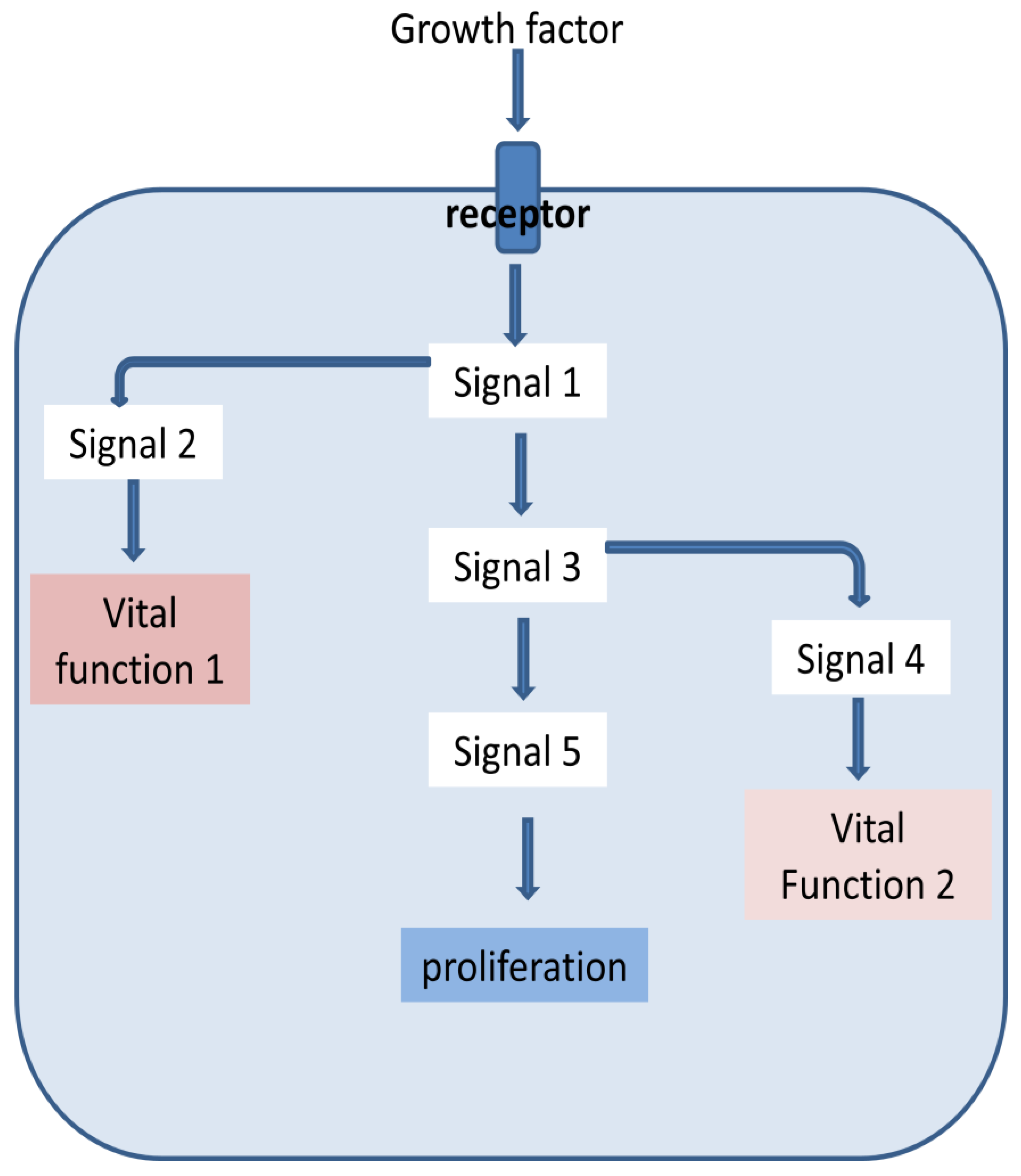
Figure 4.
Cliff edge fitness function in optimizing mechanisms of cancer prevention. Continuing with the example of growth factor independence mutations discussed above, at low basal GF levels, the probability of selecting mutant increases. On the other hand high basal level of GF comes with some cost. The peak fitness at the optimum is highly asymmetric with steep decline on the side of cancer risk. Since multiple factors result into individual phenotypic variability in any character, when the optimum is selected genetically, a small chance of cancer will remain. Subnormal GF expression due to life style factors can increase the chance rapidly.
Figure 4.
Cliff edge fitness function in optimizing mechanisms of cancer prevention. Continuing with the example of growth factor independence mutations discussed above, at low basal GF levels, the probability of selecting mutant increases. On the other hand high basal level of GF comes with some cost. The peak fitness at the optimum is highly asymmetric with steep decline on the side of cancer risk. Since multiple factors result into individual phenotypic variability in any character, when the optimum is selected genetically, a small chance of cancer will remain. Subnormal GF expression due to life style factors can increase the chance rapidly.
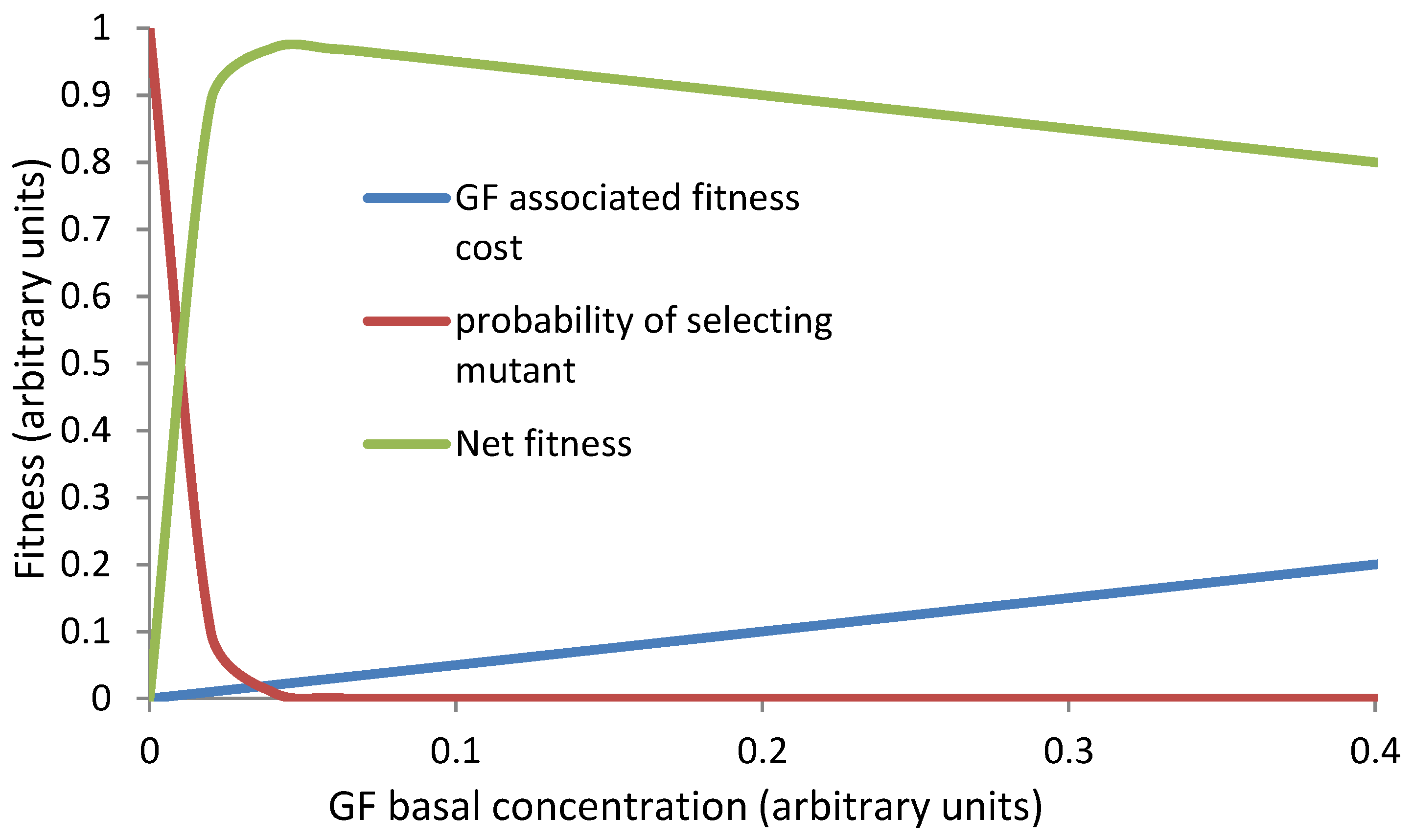
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



