
Submitted:
24 March 2023
Posted:
27 March 2023
You are already at the latest version
Abstract
Hexokinases (HKs) convert hexose sugars to hexose-6-phosphate, thus trapping them inside cells to meet the synthetic and energetic demands. HKs participate in various standard and altered physiological processes, including cancer, primarily through the reprogramming of cellular metabolism. Four canonical HKs have been identified with different expression patterns across tissues. HKs 1-3 play a role in glucose utilization, whereas HK 4 (glucokinase, GCK) also acts as a glucose sensor. Recently, a novel 5th HK, hexokinase domain containing 1 (HKDC1), has been identified, which plays a role in whole-body glucose utilization and insulin sensitivity. Beyond the metabolic functions, HKDC1 is differentially expressed in many forms of human cancer. This review focuses on the role of HKs, particularly HKDC1, in metabolic reprogramming and cancer progression.
Keywords:
Cancer metabolism
; HKDC1
; Hexokinases
; glucose metabolism
; metabolic reprogramming
1. Introduction
First observed by Otto Warburg in 1924, one of the hallmarks of cancer cells is reprogrammed glucose metabolism, where glucose uptake and lactate production are enhanced regardless of oxygen concentrations, popularly known as the “Warburg effect.” Initially, this phenomenon was thought to be due to mitochondrial dysfunction in cancer cells [1,2]. However, research has now established that enhanced glucose metabolism coupled with altered mitochondrial metabolism supplies increased energy needs and provides metabolites for biosynthetic pathways, such as nucleotides, fatty acids, and amino acids needed by proliferating cancer cells [3]. The first step of glucose metabolism is catalyzed by hexokinases (HKs). HKs are a family of phosphotransferase enzymes with different kinetic properties, expression profiles, and subcellular localization that initiate glucose metabolism [4,5,6]. Four canonical isoforms of the HK family have been well characterized: HKs 1-3 have a broad range of expression, and the fourth isoform, more commonly known as glucokinase (GCK), is expressed mainly in the liver and pancreas. [4,5,6,7]. Although specific roles have been described for each HK, the existence of multiple isozymes catalyzing the same reaction within the same cell or tissue is a pressing question. Glucose-6-phosphate (G6P) is the first stable intracellular intermediate of glucose metabolism; therefore, its generation is tightly regulated by the selective expression of different HK isoforms in normal and pathophysiological scenarios [8]. For the same reason, HKs vary in cellular distribution, expression patterns, and substrate affinity levels depending on the cell’s physiological state. This review describes the role, distribution, and regulation of different isoforms and their metabolic functions in cancer.
1.1. General characteristics and distribution
Genes that code for HK protein isoforms are conserved in bacteria to humans [4,6]. However, bacterial and lower vertebrate genes code for smaller proteins (about 50 kDa), while mammalian HKs 1-3 and HKDC1 are ≅ 100 kDa in size. HK isoforms possess high sequence similarities (Supplementary Figure S1) at the ‘N’ and ‘C’ terminal domains referred to as “Hemi domains” (Figure 1) [4,5,6,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. These hemi domains are thought to have evolved because of a gene duplication event from the bacterial HK enzyme [23,24]. Upon subsequent evolutionary divergence, the N-terminal Hemi domain acquired different properties in each isoform (Figure 1) [9,10,11,12,13,14,15,16,17,18,19,20,21,22,25,26]. One characteristic feature of mammalian HKs is allosteric inhibition by G6P, which supports the “gene duplication event theory,” suggesting that the duplication event led to the formation of a hemi-domain that evolved into a regulatory binding site [27]. Amino acid sequence comparisons of hexokinases from lower to higher vertebrates are available to support the gene duplication hypothesis. Various comparison analysis studies suggest that high similarities in sequence exist not only between the hemi-domains but HKs from lower to higher vertebrates, indicating a slow rate of amino acid substitution (rate of mutation through evolution) at homologous HK genes across species [27,28,29]. Some of the common characteristics of each isozyme are described and listed in Table 1. HK1 gene encodes a protein of 100 kDa, and only the C-terminal domain is catalytically active [4,6]. It is ubiquitously expressed inside all cells with a granular cytoplasmic expression pattern. The enzyme is also localized to the mitochondrial outer membrane [4,5,6]. HK1 has the highest level of tissue expression in the brain, followed by the urinary bladder, thyroid gland, colon, and bone marrow [4,6]. HK2 is the most well-characterized isoform of the HK family, primarily expressed in insulin-sensitive tissues like the adipose and skeletal muscle [30]. It undergoes significant changes in expression in different cancers and is the most well-studied HK in cancer biology [31,32,33,34,35,36,37,38,39,40,41]. It is the only identified HK with both N and C terminal domains catalytically active and is the most highly regulated isoform [4,5,6,10,11,12]. Like HK1, HK2 has been shown to localize to the mitochondria [6]. HK3 is a less well-characterized 100 kDa isoform of the hexokinase family, which lacks the N-terminal mitochondrial binding domain of HK1 and 2 (Figure 1). HK3 is expressed in lung, kidney, and liver tissue at levels low compared to HK1 & 2. It is also the predominant isozyme in granulocytes [11,12,42] (Table 1). HK4, or glucokinase (GCK), is a unique 50 kDa enzyme mainly expressed in the liver and pancreas and closely resembling the ancestral bacterial enzyme [43,44]. The enzyme is also expressed in enteroendocrine cells and the brain [45,46]. The distinguishing feature of GCK in metabolic regulation is its role as the body’s primary glucose sensor. Small fluctuations in GCK activity alter the threshold for glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells, which is not observed with other hexokinases [47,48,49]. Mutations in the GCK gene lead to two different diseases of blood glucose regulation: maturity-onset diabetes of the young type 2 (MODY-2), and persistent hyperinsulinemic hypoglycemia of infancy (PHHI) [50,51,52]. GCK is localized in the cytoplasm, but reports have also suggested that GCK forms a heteropentameric complex at the mitochondria with BCL 2-associated death promoter (BAD), protein kinase A (PKA, cAMP-dependent protein kinase), protein phosphatase 1 (PP1, dual-specificity serine/threonine phosphatase), and Wiskott-Aldrich family member (WAVE1) under certain conditions to integrate glycolysis and apoptosis [53,54].
Phylogenetic analyses carried out in the middle of the 2000s to comprehend the diversification of the HKs and the evolution of GCK [20,21] led to the discovery of a novel HK-like gene known as the hexokinase domain containing-1 (HKDC1). The gene that codes for HKDC1 lies on chromosome 10 in humans near the HK1 gene, and the two share more than 70% sequence similarity [21]. The enzyme has been shown to play a role in modulating glucose tolerance during pregnancy by identifying its genetic variants in a genome-wide association study (GWAS) [14,19]. Like HK1-2, it also contains an ‘N’ and a ‘C’ terminal domain, which are the regions predicted to bind glucose and ATP, respectively, and includes amino acid residues, which remain conserved with those of the other HKs (Figure 1). [20,21]. HKDC1 is broadly expressed in the retina of the eyes, kidneys, brain, small intestine, duodenum, pharynx, esophagus, and thyroid gland [21]. We [15,17,55] and others [56] have shown that HKDC1, like HK1-2, associates with the outer mitochondria membrane via its interaction with the voltage-dependent anion channel (VDAC).
1.2. Regulation of Hexokinase expression
The primary metabolic role of HKs is the phosphorylation of glucose, thus trapping it inside cells and initiating glucose metabolism [4,5,6,57]. HKs therefore dictate the direction of glucose flux within the cells. However, if phosphorylation of glucose were the only role of HKs, the presence of a single HK would seem reasonable. The existence of different isoforms raises a question about the non-redundant role played by each enzyme. It also suggests that different rates of glucose phosphorylation are needed depending on the cell/tissue requirements. This allows “metabolic plasticity” to the cells to allow better regulation and channeling of glucose metabolism, and the role of GCK in this context has been defined most elaborately [7]. Therefore the expression of HKs is profoundly altered in cancer cells, and it varies widely among different cancer types (Supplementary Figure S2).
Transcriptional Control: This can be illustrated by highlighting differences in transcriptional regulation between different isoforms. Promoter regions of different hexokinases have been analyzed to contain several regulatory elements governing different transcription profiles under varied conditions [58]. The isoforms have also been observed to bind multiple transcriptional factors [59,60,61,62,63,64,65,66]. The promoter of HK1 in rats has been shown to contain transcriptional start site elements, lack a TATA sequence, and lie within a CpG island that extends into the translational start site [67]. These features are similar to promoter element features of housekeeping genes, tailored for their ubiquitous expression [67]. HK1 promoter also contains regulatory sites known as sp sites within the P2 BOX which are essential for promoter activity and binding of protein factors in lower vertebrates to humans [60,68,69]. The role of non-coding elements in regulating HK1 and their association with congenital hyperinsulinism has also been reported [68]. Alternative splicing forms multiple HK1 isoforms, i.e., HK1 (ubiquitous), HKR (erythrocytes), HK-TA/TB (testis-A/B), HK-TB (testis-B), and HK-TD (testis-D). HK1 and HKR isoforms differ only in exon 1 and share the remaining 17 exons, while HK-TA, HK-TB, HK-TC, and HK-TD have different 5′ UTR exons and share the 17 exons with all other isoforms of HK1 [67]. On the other hand, the promoter region of HK2 contains a single transcriptional start insulin-binding element, leading to transcriptional upregulation of HK2 by insulin [59]. However, the promoter of HKII contains a binding motif for hypoxia-inducible factor 1α (HIF1α). HIF1α is upregulated due to hypoxia brought about by the tumor microenvironment, which results in the upregulation of HKII, making HKII the most highly expressed HK in multiple tumors. Recently, it has been reported that HIF1α is negatively regulated by the long non-coding RNA LINC00365 in breast cancer cell lines, resulting in a decline in HKII levels and cellular proliferation [70]. Enhanced expression of HKII in hepatocellular carcinoma in rat models has also been reported to be induced by loss of DNA methylation on the CpG island on the HK2 promoter [71]. Post-transcriptional regulation of HKII activity by MicroRNAs has also been documented. Anti-tumorigenic microRNA miR 143 acts on HKII mRNA, leading to its degradation and decreased stability. Upregulation of oncogenic microRNA miR155 takes place in multiple tumors, leading to repression of miR 143, thereby stabilizing HKII mRNA [72,73]. HK3 contains a binding site for the basic leucine zipper transcription factor CCAAT/enhancer binding protein alpha (CEBPA) which leads to its transcriptional upregulation during all-trans retinoic acid (ATRA) mediated neutrophil differentiation [74]. Interestingly, the HKDC1 promoter contains high levels of epigenetic marks like H3K4me1 and H3K27ac in multiple human cell lines. The regulatory roles of these marks on their expression in normal and cancer cells need further exploration [22].
Most cancers shift their HK expression profiles in favor of HKII, a consequence of metabolic re-programming in cancer. This could be illustrated by the induction of gene expression of HKII and silencing of GCK in liver and pancreatic cancers [75,76,77,78,79]. Also, in humans, progression from the normal brain to low-grade gliomas and finally to glioblastoma multiforme (N) occurs with a progressive shift from HKI to HKII with a concomitant decrease in prognosis. HK-II expression levels are closely associated with tumor grade and mortality in hepatocellular carcinoma and breast metastasis [80,81]). One of the reasons for this response is the catalytic activity in both domains in HKII, favoring greater utilization of glucose and maintaining a downhill gradient for glucose phosphorylation. Also, HKII expression in cancers is stimulated by the insulin signaling pathway Akt/mTORC1, which is upregulated in tumor cells to regulate glucose metabolism, cellular growth, and survival through the phosphorylation of target molecules. Akt phosphorylates HKII at Thr 473, which lies within the Akt consensus binding motif RARQKT*, stabilizing HKII protein. This motif is conserved from mice through humans [82]. For the same reason, expression of HKII is decreased in type I diabetes mellitus (T1DM) due to reduced insulin signaling and is recovered upon insulin treatment in T1DM [83,84,85,86,87]. Although HKI and HKDC1 share the mitochondrial localization property with HKII, they lack the Akt consensus motif, making them more susceptible to degradation through apoptosis. However, we have previously reported that in mouse models overexpressing HKDC1 enhances Akt phosphorylation (15,16).
1.3. Regulation of hexokinase activity
HK isoforms have different catalytic and regulatory properties. HK1 is activated by high inorganic phosphate levels (Pi) and inhibited by the product G-6-P. Therefore, a cellular milieu with a high ratio of Pi/G6P because of high rates of ATP utilization favors glycolysis through HK1 activity for the generation of ATP [6]. One of the best examples to illustrate this is the reversal of G6P-induced inhibition of HK1 by inorganic phosphate (Pi), which leads to the evasion of G6P-induced feedback inhibition of glucose phosphorylation and favoring its ubiquitous expression since glycolysis is a primary requirement of all mammalian cells [4,5,6].
On the other hand, HK2 lacks this antagonizing response by Pi, and instead, Pi adds to the inhibition caused by G-6-P in the case of HK2 [46]. This feature favors HK2 activity in metabolically active tissues like skeletal muscles for replenishment of glycogen synthesis following muscle contraction, and a body of existing literature suggests an anabolic role for HK2 [46,48]. Additionally, a wealth of literature agrees with an anabolic role for HK2, funneling G-6-P to synthesize NADPH for lipid biosynthesis via the pentose phosphate pathway (PPP) in the liver and mammary [88,89].
HK3 is known to be inhibited by glucose at high concentrations in the 1mmol l -1(substrate inhibition) but is less sensitive to inhibition by G-6-P. Interestingly, HK3 has a similar response towards G-6-P and Pi similar to HK2 (Table 1), which supports an anabolic role for HK3, but further research is needed to answer this question [46,49]. It also has the lowest affinity for the second substrate, ATP, among all HKs, but the physiological role of this property remains elusive [46].
GCK has the highest Km (lowest affinity) for glucose among all HKs, allowing the liver and pancreas to serve as a “glucose buffer” and a “glucose sensor,” respectively. It is not inhibited by G-6-P and has a 50-fold lower affinity for glucose than other isoforms. Within the liver, the low affinity is tailored to ensure the availability of glucose to physiologically sensitive tissues like the brain under starvation and its utilization only when glucose is abundantly available. Within the pancreas, this feature allows GCK to act as a “glucose sensor” to regulate insulin release. Mutations in the glucokinase (GCK) gene lead to maturity-onset diabetes of the young, type 2 (MODY-2), and persistent hyperinsulinemic hypoglycemia of infancy (PHHI) [50,52,90]. MODY-2 is a mild type 2 diabetes resulting from a defect in glucose-induced insulin secretion [50,51,52]. Mutations in the GCK leading to MODY-2 are arguably the most common cause of monogenic diabetes due to these specific mutations. More than 40 mutations have been linked to MODY-2, including frameshifts, nonsense, missense, and splice-site variants [1,2,3,4,5,6,7,8]. The proposed role of GCK as a “glucose sensor” in pancreatic β-cells [3,12,13] is consistent with the MODY-2 phenotype wherein small reductions in β-cell activity increase the threshold for glucose-induced insulin secretion resulting in the phenotype. However, a report by Postic et al. suggests that hepatic GCK also plays a role in MODY-2. Alterations in GCK activity are also associated with many other diseases that have been reviewed elsewhere in detail [13,43]. Owing to its unique role, GCK regulation is complex, and several regulatory mechanisms have been discovered. Alternative and tissue-specific promoters drive GCK transcription and gene expression to varying degrees [91,92,93,94,95,96,97]. Several metabolites, including insulin, glucose, and hormones, regulate GCK expression at the transcriptional level [98,99,100,101,102]. Regulation of GCK has been recently reviewed elsewhere in more detail [103].
Not much is known about the kinetic and regulatory properties of HKDC1, and it needs further exploration. However, the genetic locus near HKDC1 is a “hot spot” for various “histone modifications,” and it is believed that HKDC1 is subject to different levels of regulation under different physiological and pathophysiological conditions [19,22]. Although HKDC1 has two kinase domains like HK1, there have been contrasting reports on its catalytic potential. An early study suggests that HKDC1 possesses hexokinase activity, where experiments on INS-1 rat pancreatic cells with HKDC1 overexpression showed changes in HK activity [22]. Interestingly, the hexokinase activity of the other HKs was unaffected by the expression of HKDC1 [22]. Going further, our group has recently shown that the hexokinase activity of HKDC1 is quite low, and the principal function of the protein may be more related to binding to mitochondria and modulating glucose flux [17].
1.4. Differences in subcellular localization
This feature allows the utilization of different HK isoforms for channeling G6P to pathways dictated by the cell’s metabolic state. Under normal conditions, GCK is primarily cytosolic [104], while HK3 is mostly perinuclear in localization [105]. The subcellular localization of HK1 and 2 has important influences on their metabolic, antioxidant, and anti-apoptotic effects. HK1 localizes to the mitochondrial membrane, and HK2 is localized to the outer mitochondrial membrane through a voltage-dependent anion channel (VDAC) [4,6]. However, HK2 binds to mitochondria with less affinity than HK1 and can translocate between cytoplasm and mitochondria depending on glucose and glucose 6-phosphate [8]. Mitochondrial-bound HK1 promotes efficient glucose catabolism by coupling glycolysis with oxidative phosphorylation. This feature makes it the ideal HK isoform for brain cells. On the other hand, HK2 in normal cells is mostly cytosolic and promotes anabolic functions like glycogen synthesis through PPP, making it ideal for muscle and cardiac cells. Also, PPP leads to the generation of reduced glutathione from NADPH which is essential for the anti-oxidant activity of HK2. Although the mitochondrial binding property of HK2 appears to be in tune with the metabolic demands of cancer cells, allowing them to couple glucose consumption with energy (ATP) production, its role in mediating glucose consumption and anabolic processes under normal conditions remains elusive [4,5,6,31,32,33,34,35,36,37]. Studies, however, show that HK2 dynamically shuttles between the mitochondria and cytoplasm in response to changes in intracellular G6P, pH, and Akt signaling pathways [106].
As a result of low-grade inflammation (aging and diabetes), HK1 has been shown to predominantly localize in cytoplasm and favor an inflammatory phenotype [107,108]. In a landmark study conducted by De Jesus et al., it was observed that mice lacking the N-terminal mitochondrial binding domain (MBD) on HK1 produced an inflammatory response when challenged with lipopolysaccharide (LPS), increased glucose flux through the PPP but decreased flux below the level of glyceraldehyde phosphate dehydrogenase (GAPDH) brought about by nitrosylation of GAPDH which leads to reduced GAPDH activity [109]. HK3 has also been shown to be associated with mitochondrial-associated membranes (MAMs) in normal mice brains through unknown mechanisms. This effect is abolished due to chronic stress in mice [110]. It has been reported that hexokinases are differentially translocated within cells depending upon the physiological conditions and the mechanisms through which HKs migrate between cellular compartments; however, they remain unidentified and warrant more investigation in this area [111]. Recently, HKDC1 has also been shown to bind with mitochondria via interaction with VDAC [16,17,18,19]. More research is needed in this area to understand better the significance of differential localization of hexokinases under different conditions.
1.5. Roles of hexokinases in cancer-mediated metabolic reprogramming
One of the characteristic features of cancer is unabated cell division. For this reason, neoplastic cells preferentially obtain energy and biomolecules through glycolysis through metabolic reprogramming. Metabolic reprogramming refers to the ability of cancer cells to alter their metabolism to support their enhanced metabolic requirements of high ATP and intermediates for biosynthetic processes. This requirement brings about extensive changes in the expression of different hexokinase enzymes.
Hexokinase 1: Expression of HK1 is amplified in some cancers where it is responsible for rewiring the metabolic state towards aerobic glycolysis to supply ATP and macromolecules (Figure 2) [112,113,114]. The observation that most normal cells express HK1 while cancer cells express HK1 and HK2 stimulated interest in reducing HK2 activity in cancers. However, studies demonstrated that the knockdown of HK2 alone doesn’t inhibit in vivo tumor progression with reduced glucose consumption, suggesting that HK1 compensates for the overall tumorigenic potential. In contrast, the knockdown of HK2 in HK1- HK2+ cancers reduced xenograft tumor progression [115,116,117,118]. These studies suggest a greater involvement of HK1 in tumor progression beyond its currently known role and possibly as a regulatory function in cancer cells. For example, in a study by Daniela et al., it has been observed to be involved in ovarian cancer in a glucose phosphorylation-independent fashion [119] that HKI also serves as the effector of KRAS4A, an isoform of the most frequently mutated oncogene KRAS, during tumorigenesis [120].
Hexokinase 2: HK2 is significantly overexpressed in treatment-resistant primary and metastatic breast cancer [38,39,40,41], bladder cancer [121], cervical squamous cell carcinoma [122], colorectal cancer [123], neuroendocrine tumor [112], ovarian epithelial tumors [113], glioblastoma [58,114, hepatocellular carcinoma [31], laryngeal squamous cell carcinoma [32], lung cancer [33], neuroblastoma [34], pancreatic cancer [35], and prostate cancer. HK2 expression in these cancers inversely correlates to overall patient survival rates [36]. Genetic ablation of HK2 is known to inhibit malignant growth in mouse models [37,115,116,117,118]. A landmark study on an adult tumor model of mice demonstrated the therapeutic effects of systemic deletion of HK2 [37,124,125,126,127]. In addition to its enzymatic activity, the mitochondrial binding ability of HK2 plays a role in inhibiting apoptosis and upregulation of synthetic pathways which support tumor growth (Figure 2). The mitochondrial-bound HK2 is therefore elevated in many forms of cancer [38,39,40]. The amplification of HK2 appears to be related to the expression of p53. Recent studies have shown that p53-inducible protein TIGAR (Tp53-induced Glycolysis and Apoptosis Regulator), Akt, and ER stress sensor kinase could regulate mitochondrial HK2 localization [128,129,130,131,132,133,134]. Interestingly, mitochondrial TIGAR–HK2 complex upregulated HK2 and hypoxia-inducible factor 1 (HIF1) activity, which limits reactive oxygen species (ROS) production and protects against tumor cell death under hypoxic conditions [135,136,137,138,139,140]. It is also observed that GCK to HK2 switch occurs in hepatocellular carcinoma (HCC), and the expression of HK2 is highest in HCC [138]. Additionally, HK2 is also regulated by epigenetic mediators, including long non-coding RNAs [39,40,115,116], microRNAs [132,135,136,137,138], histone, and DNA methylation [118]. HK2 is localized to the outer mitochondrial membrane through a voltage-dependent anion channel (VDAC) [137] (Figure 2). This association permits direct access to the ATP generated within the mitochondria [135]. This phenomenon is especially significant in malignant cells where rates of aerobic glycolysis go up tremendously to meet the energy demands of the transformed cell (Warburg effect) [114].
Hexokinase 3: HK3 is upregulated in several cancers, including acute myeloid leukemia (AML), where it plays a role as an anti-apoptotic protein to promote tumor cell survival alongside HK1 and 2 [141,142]. The previously identified functions of the enzyme include cell survival through attenuation of apoptosis and enhancement of mitochondrial biogenesis [3,143,144]. The latest research about the functions of HK3 in normal and cancer cells has uncovered previously unanticipated roles of this protein. A recent study by Seiler et al. has reported that Hexokinase 3 enhances myeloid cell survival via non-glycolytic functions [145], while another report by Xu et al. showed that HK3 dysfunction promotes tumorigenesis and immune escape by upregulating macrophage infiltration in renal cell carcinoma [146].
Glucokinase: Glucose phosphorylation activity for GCK has been observed in several cancer cell lines [147]. GCK is also known to interact with BAD (Bcl-2 agonist of cell death) to integrate glycolysis with apoptosis [148,149,150,151]. To date, 17 activating mutations targeted by multiple activators have been identified in the allosteric activator site of GCK [152,153,154,155]. The activating variations and their targeting by the activators lead to enhanced cellular proliferation, including the proliferation of cancer cell lines like INS, which indicates a putative pro-oncogenic role for GCK [156,157,158]. Although there is no direct evidence for the role of GCK as a pro-oncogene, recent reports exploring somatic variations of allosterically regulated proteins in cancer genomes suggest that somatic mutations of GCK could play a role in tumorigenesis [159]. Těšínský et al. provide the first direct evidence of the role of GCK in tumorigenesis by demonstrating a change in the kinetic properties of GCK which include an increased affinity for glucose and changes in cooperative binding [160].
Hexokinase domain containing 1: Studies performed over the past decade have linked HKDC1 to various functions (Figure 3). Much of the interest in HKDC1′s role in cancer stems from the fact that, like HK1 and 2, it localizes in the mitochondrial outer membrane (MOM) and binds with the voltage-dependent anion channel (VDAC) [15]. We were the first to identify the role of hepatic HKDC1 in glucose metabolism. Using a mouse model of HKDC1, we demonstrated that hepatic HKDC1 modulates glucose metabolism and insulin sensitivity in mice. While HKDC1 has nominal expression in normal hepatocytes [18], it is significantly upregulated in hepatocellular carcinoma (HCC) cells [161,162], implying that it plays an essential role in HCC. By using HKDC1 knockout models, we have shown that HK activity is not affected by HKDC1 ablation; however, there is a significant increase in glucose uptake, where the bulk of glucose carbons flow through the glycolytic shunt pathways PPP and HBP (Figure 3) [17]. We further show that HKDC1 interacts with the mitochondria, and its loss results in mitochondrial dysfunction [17]. Since cancer cells require ATP to prepare for cell division during the synthetic (S) phase of the cell cycle, a deficiency in ATP may cause cell cycle arrest. Others have shown that HKDC1 is also significantly increased in breast cancer cells, enhancing glucose uptake and mitochondrial membrane potential to encourage cell survival and growth. In agreement with this phenomenon, HKDC1 knockdown increased the production of reactive oxygen species (ROS), the activation of caspase 3, and apoptosis [55]. Li et al. [163] used RNA-seq data from The Cancer Genome Atlas to pinpoint genetically altered genes in a univariate survival analysis of patients with squamous cell lung carcinoma (SQCLC). Seven thousand two hundred twenty-two genetically modified genes were discovered by analysis of RNA-seq data from 550 SQCLC patients, and HKDC1 was one of 14 feature genes with more than 100 frequencies linked to a worse prognosis [163,164]. HKDC1 mRNA and protein levels also expressed higher in lung cancer cell lines than in healthy lung epithelial cells.
Additionally, there was a direct correlation between the degree of HKDC1 protein expression and histological differentiation, reduced survival, tumor size, pN (N refers to the number of nearby lymph nodes with cancer) stage, and poor prognosis. In agreement with these results, lung cancer cell lines stably overexpressing HKDC1 demonstrated increased glucose consumption and lactate generation and increased proliferation, migration, and invasion compared to healthy lung epithelial cells [163,164]. A comparison study on RNA sequencing (RNA-Seq) analysis of colorectal cancer (CRC) and matched standard tissue samples has observed significant splicing variations in nine genes in CRC. Interestingly, the authors discovered alternate regulation of the first exon in HKDC1 using exon sequencing (DEXSeq) to uncover variations in relative exon usage. HKDC1 E1a-E3a was elevated in CRC, suggesting a potential functional impact because of a projected change in the HKDC1 protein sequence [165,166,167]. Another study has reported a 13-h phase change in HKDC1 expression between SW480 cells and their metastatic counterpart SW620 (a core clock gene) that occurs in conjunction with a phase shift in aryl hydrocarbon receptor nuclear translocator-like protein-1 (BMAL1). In SW480 cells, silencing BMAL1 results in an elevation of HKDC1 expression, and this effect was eliminated in SW620 cells. These findings imply that HKDC1 and the circadian clock interact, as the circadian clock is altered in metastatic cells [168].
Eukaryotic cells adjust to cellular stress by phosphorylating eukaryotic translation initiation factor 2 alpha (eIF2), which results in the translation of specific transcripts that enable the cell to withstand stress [132,135,136,137,169,170]. Activating Transcription Factor 4 (ATF4) is a leucine zipper transcription factor that modulates the cellular integrated stress response to allow cells to adapt to and endure stressors [171,172,173]. The overexpression of ATF4 causes the HKDC1 gene transcription to increase significantly under cellular stress, changing hepatocyte mitochondrial dynamics [174]. HKDC1 is upregulated in response to the endoplasmic reticulum (ER) stress or mitochondrial respiratory chain inhibition; however, when these stressors are present in combination with RNA interference to decrease ATF4, HKDC1 gene expression is reduced [174].
2. Future Directions
Accelerated aerobic glycolysis is a hallmark of cancer cells which provides a rapid source of ATP and good metabolic intermediates for synthesizing nucleic acids, lipids, and proteins in the rapidly dividing cells [175,176]. The increased dependency of cancer cells on glucose metabolism sets them apart from their regular counterparts and could render them more vulnerable to disruption in glucose metabolism. Cancer cells could therefore be selectively targeted through disruption of glucose metabolism, and therapeutic targeting of HK enzymes in cancers has seemed a plausible strategy. However, considering the overarching redundancy in the catalytic activity of different isozymes, it seems reasonable to argue that one isoform could compensate for another under specified conditions. A lack of literature on the non-redundant functions of each isozyme further complicates this approach, and therapeutic targeting of HKs in cancer per se awaits more targeted approaches for effective outcomes. Identification of isoform-specific roles in cancer could reveal more selective targets that could be utilized for therapeutic purposes without compromising overall homeostasis.
HK1-2 and HKDC1 contain a mitochondrial binding site in the N-terminal domain. This domain mediates HK1 activity in normal cells while it plays a role in tumorigenesis in HK2 and HKDC1 [22,177,178]. HK2 is known to inhibit apoptosis and regulate autophagy [28]. The recent identification of HK2 localization to contact points between mitochondria and endoplasmic reticulum, known as mitochondria, associated membranes (MAMs), has unveiled a novel role of HK2 in regulating Ca2+ flux within the cells [179,180]. HKDC1 is also postulated to bind to MAMs similarly and regulate Ca2+ flux. In the future, the binding of HK2 and HKDC1 could be specifically targeted as a promising therapeutic strategy for effective outcomes in cancer. Of particular interest, small molecular inhibitors which specifically target the binding of HK2 and HKDC1 to mitochondria and MAMs need further exploration [181]. Such inhibitors have recently been characterized for HK2, which specifically and selectively target HK2 without producing off-target effects. Evaluation of similar inhibitors for HKDC1 could prove to be an effective therapeutic avenue for cancer treatment in the future [181]. Evaluation of the role of HK2 on HKDC1 protein stabilization in HK2+ HKDC1+ cancers followed by independent and combined therapeutic targeting of these enzymes also holds promise for more effective treatment of cancers due to hexokinase targeting.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.WK. and ZF; writing—original draft preparation, ZF, HI, and SB.; writing—review and editing, MWK and BTL. All authors have read and agreed to the published version of the manuscript.
Funding
MWK is supported by DOD Career Development Grant (W81XWH2010650). B.T.L. is supported by a VA Merit Review Award (I01BX00382), NIH grants (R01 DK104927, R01 DK111848, U01 DK127378, and P30 DK020595).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data used in this manuscript is publicly available at https://www.cancer.gov/ccg/research/genome-sequencing/tcga.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Warburg O (1956). On the origin of cancer cells. Science 123: 309–314, 1956. [CrossRef]
- Warburg O. On the origin of cancer cells. Science 1956; 123:309-14. [CrossRef]
- Liberti, M. V., & Locasale, J. W. (2016). The Warburg effect: How does it benefit cancer cells? Trends in Biochemical Sciences, 41(3), 211–218.
- Katzen HM, Schimke RT (1965). Multiple forms of hexokinase in the rat: tissue distribution, age dependency, and properties. Proc Natl Acad Sci USA 54: 1218–1225. [CrossRef]
- Sebastian S, et al (1997). Assignment of hexokinase types 1,2,3 (Hk1,2,3) and glucokinase (Gck) to rat chromosome band 20q11, 4q34, 17q12 and 14q21 respectively, by in situ hybridization. Cytogenet Cell Genet 77: 266–267. [CrossRef]
- Wilson JE (2003). Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function J Exp Biol 206: 2049–2057.
- Franz M Matschinsky, David F Wilson. (2019). The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front Physiol . 2019. [CrossRef]
- Martínez-Reyes, I., Chandel, N.S. Cancer metabolism: looking forward. Nat Rev Cancer 21, 669–680 (2021). [CrossRef]
- Mathupala et al (2006). Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 7;25(34):4777-86.
- Scott John et al (2011). Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One. 2 9;6(3):e17674. [CrossRef]
- Wyatt E et al (2010). “Regulation and cytoprotective role of hexokinase III”. PLOS ONE. 5 (11): e13823. [CrossRef]
- Cárdenas ML et al (1998). “Evolution and regulatory role of the hexokinases”. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1401 (3): 242–64.
- Postic C et al (2001). Cell-specific roles of glucokinase in glucose homeostasis. Recent Prog Horm Res. ;56:195-217. [CrossRef]
- Ludvik AE et al (2016). HKDC1 Is a Novel Hexokinase Involved in Whole-Body Glucose Use. Endocrinology. 2016 Sep;157(9):3452-61. [CrossRef]
- Pusec CM et al (2019). Hepatic HKDC1 Expression Contributes to Liver Metabolism. Endocrinology.1;160(2):313-330. [CrossRef]
- Khan MW et al (2019). Hepatic hexokinase domain containing 1 (HKDC1) improves whole body glucose tolerance and insulin sensitivity in pregnant mice. Biochim Biophys Acta Mol Basis Dis.1;1865(3):678-687.
- Khan MW et al (2022). The hexokinase “HKDC1” interaction with the mitochondria is essential for liver cancer progression. Cell Death Dis.28;13(7):660.
- Khan MW et al (2018). Studies on the Tissue Localization of HKDC1, a Putative Novel Fifth Hexokinase, in Humans. J Histochem Cytochem.;66(5):385-392.
- Zapater JL et al (2022). Hexokinase domain-containing protein-1 in metabolic diseases and beyond. Trends Endocrinol Metab. 2022 Jan;33(1):72-84. [CrossRef]
- Hayes MG et al. (2013). “Identification of HKDC1 and BACE2 as genes influencing glycemic traits during pregnancy through genome-wide association studies”. Diabetes. 62 (9): 3282–91. [CrossRef]
- Irwin DM et al (2008). “Molecular evolution of the vertebrate hexokinase gene family: Identification of a conserved fifth vertebrate hexokinase gene”. Comparative Biochemistry and Physiology. Part D, Genomics & Proteomics. 3 (1): 96–107. [CrossRef]
- Guo, C et al (2015). Coordinated regulatory variation associated with gestational hyperglycemia regulates expression of the novel hexokinase HKDC1. Nature communications, 6(1), 1-8.
- Colowick SP (1973) The hexokinases. In: Boyer PD (ed) The enzymes, vol 9. Academic, New York, pp 1–48.
- Easterby JS, O’Brien MJ (1973) Purification and properties of pig-heart hexokinase. Eur J Biochem 38:201–211.
- White TK, Wilson JE (1989) Isolation and characterization of the discrete N-and C-terminal halves of rat brain hexokinase: retention of full catalytic activity in the isolated C-terminal half. Arch Biochem Biophys 274:375–393.
- Arora KK, Filburn CR, Pedersen PL (1993) Structure/function relationships in hexokinase. Site-directed mutational analyses and characterization of overexpressed fragments implicate different functions for the N-and C-terminal halves of the enzyme. J Biol Chem 268:18259–18266.
- T Ureta, C Medina, A Preller (1987). The evolution of hexokinases. Arch Biol Med Exp;20(3-4):343-57.
- H J Tsai (1999). Functional organization and evolution of mammalian hexokinases: mutations that caused the loss of catalytic activity in N-terminal halves of type I and type III isozymes. Arch Biochem Biophys; 369(1):149-56. [CrossRef]
- Shigeyuki Kawai (2005). Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J Biosci Bioeng ;99(4):320-30. [CrossRef]
- Heikkinen, S., Supploa, S., Malkki, M. and Deeb, S.(2000). Mouse hexokinase II gene: structure, cDNA, promoter analysis, and expression pattern. Mamm. Genome 11, 91-96. [CrossRef]
- Kwee SA et al (2012). Choline kinase alpha and hexokinase-2 protein expression in hepatocellular carcinoma: association with survival. PLoS ONE. 7:e46591. [CrossRef]
- Chen J et al (2014). Hexokinase 2 overexpression promotes the proliferation and survival of laryngeal squamous cell carcinoma. Tumor Biol. 35:3743–53.
- Wang H et al (2016). Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 16: 38.
- Botzer LE et al (2016). Hexokinase 2 is a determinant of neuroblastoma metastasis. Br J Cancer. 114:759–66. [CrossRef]
- Ogawa H et al (2015). The combination of the expression of hexokinase 2 and pyruvate kinase M2 is a prognostic marker in patients with pancreatic cancer. Mol Clin Oncol. 3:563–571. [CrossRef]
- He HC, et al (2009). “Real-time quantitative RT-PCR assessment of PIM-1 and hK2 mRNA expression in benign prostate hyperplasia and prostate cancer”. Medical Oncology. 26 (3): 303–8. [CrossRef]
- Patra KC et al (2013). Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell ;24:213–28.
- Bacci M et al. (2016). Morandi: miR-155 drives metabolic reprogramming of ER+ breast cancer cells following long-term estrogen deprivation and predicts clinical response to aromatase inhibitors. Cancer Res. 76:1615–26.
- van’t LJ et al (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415:530–6.
- Liu X et al (2019). Elevated hexokinase II expression confers acquired resistance to 4-hydroxytamoxifen in breast cancer cells. Mol Cell Proteomics. 18:2273–2284.
- Palmieri D et al (2009). Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol Cancer Res. 7:1438–45. [CrossRef]
- Federzoni EA et al (2014). CEBPA-dependent HK3 and KLF5 expression in primary AML and during AML differentiation. Scientific Reports. 4: 4261. [CrossRef]
- Franz M Matschinsky (2005). Glucokinase, glucose homeostasis, and diabetes mellitus. Curr Diab Rep;5(3):171-6. [CrossRef]
- Iynedjian PB (1993) Mammalian glucokinase and its gene. Biochem J 293:1–13.
- Ferre T, Riu E, Bosch F, Valera A, Evidence from transgenic mice that glucokinase is rate limiting for glucose utilization in the liver., FASEB J. 10 (1996) 1213–8. [CrossRef]
- Wilson JE, Hexokinases, Rev Physiol Biochem Pharmacol. 126 (1995) 65–198.
- Matschinsky FM, Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes, Diabetes. 39 (1990) 647–652. [CrossRef]
- Cárdenas ML, Cornish-Bowden A, Ureta T, Evolution and regulatory role of the hexokinases, Biochim. Biophys. Acta - Mol. Cell Res 1401 (1998) 242–264. [CrossRef]
- Kawai S, Mukai T, Mori S, Mikami B, Murata K, Hypothesis: Structures, evolution, and ancestor of glucose kinases in the hexokinase family, J. Biosci. Bioeng (2005). [CrossRef]
- Velho G, Froguel P, Clement K, Pueyo ME, Rakotoambinina B, Zouali H, Passa P, Cohen D, Robert JJ: Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. L a n c e t 340:444–448, 1992.
- Byrne MM, Sturis J, Clement K, Vionnet N, Pueyo ME, Stoffel M, Takeda J, Passa P, Cohen D, Bell GI, Velho G, Froguel P, Polonsky KS: Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 93:1120–1130, 1994.
- Velho G, Petersen KF, Perseghin G, Hwang JH, Rothman DL, Pueyo ME, Cline GW, Froguel P, Shulman GI: Impaired hepatic glycogen synthesis in glucok i n a s e - d e ficient (MODY-2) subjects. J Clin Invest 98:1755–1761, 1996.
- Danial NN, Gramm CF, Scorrano L, Zhang C-Y, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ, BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis, Nature. 424 (2003) 952–956.
- Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJA, Datta SR, Pitter KL, Bird GH, Wikstrom JD, Deeney JT, Robertson K, Morash J, Kulkarni A, Neschen S, Kim S, Greenberg ME, Corkey BE, Shirihai OS, Shulman GI, Lowell BB, Korsmeyer SJ, Dual role of proapoptotic BAD in insulin secretion and beta cell survival, Nat. Med 14 (2008) 144–153.
- Chen, X et al. (2019). PGC1β regulates breast tumor growth and metastasis by SREBP1-mediated HKDC1 expression. Frontiers in oncology, 9, 290. [CrossRef]
- Chen, Q. et al (2020). HKDC1 C-terminal based peptides inhibit extranodal natural killer/T-cell lymphoma by modulation of mitochondrial function and EBV suppression. Leukemia, 34(10), 2736-2748. [CrossRef]
- Ahn KJ et al (2009). “Enzymatic properties of the N- and C-terminal halves of human hexokinase II”. BMB Reports. 42 (6): 350–5.
- Demetrius, L., & Tuszynski, J. A. (2010). Quantum metabolism explains the allometric scaling of metabolic rates. Journal of the Royal Society Interface, 7(44), 507–514. [CrossRef]
- R L Printz et al (1993). Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. J Biol Chem. 5;268(7):5209-19.
- White, J. A., Liu, W. and Wilson, J. E. (1996). Isolation of the promoter for Type I hexokinase from rat. Arch. Biochem. Biophys. 335,161-172. [CrossRef]
- Liu, W. and Wilson, J. E. (1997). Two Sp sites are important cis elements regulating the upstream promoter region of the gene for rat Type I hexokinase. Arch. Biochem. Biophys. 346,142-150.
- Mathupala, S. P., Rempel, A. and Pedersen, P. L.(1995). Glucose catabolism in cancer cells. Isolation, sequence,and activity of the promoter for Type II hexokinase. J. Biol. Chem. 270,16918-16925.
- Osawa, H., Robey, R. B., Printz, R. L. and Granner, D. K.(1996). Identification and characterization of basal and cyclic AMP response elements in the promoter of the rat hexokinase II gene. J. Biol. Chem. 271,17296-17303. [CrossRef]
- Sebastian, S., White, J. A. and Wilson, J. E.(1999). Characterization of the rat Type III hexokinase gene promoter. A functional octamer 1 motif is critical for basal promoter activity. J. Biol. Chem. 274,31700-31706.
- Sebastian, S., Edassery, S. and Wilson, J. E.(2001). The human gene for the Type III isozyme of hexokinase. Structure, basal promoter, and evolution. Arch. Biochem. Biophys. 395,113-120. [CrossRef]
- S Heikkinen et al (1999). Hexokinase II-deficient mice. Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J Biol Chem. 1999 Aug 6;274(32):22517-23.
- Koko Murakami et al (2002). Gene expression and biological significance of hexokinase in erythroid cells. Acta Haematol;108(4):204-9. [CrossRef]
- Matthew N. et al (2022). Non-coding variants disrupting a tissue-specific regulatory element in HK1 cause congenital hyperinsulinism. Nature Genetics. 54, 1615–1620.
- Feriotto G et al. Transcriptional activity and Sp 1/3 transcription factor binding to the P1 promoter sequences of the human AbetaH-J-J locus. FEBS J. 2007 Sep;274(17):4476-90.
- Buhan Liu et al (2023). LINC00365 functions as a tumor suppressor by inhibiting HIF-1α-mediated glucose metabolism reprogramming in breast cancer. Exp Cell Res. 2023 Apr 1;425(1):113514. [CrossRef]
- Kim HR et al (2013). p53 regulates glucose metabolism by miR-34a. Biochem Biophys Res Commun. Jul 26;437(2):225-31.
- Tong AW et al (2008). Modulation of miRNA activity in human cancer: a new paradigm for cancer gene therapy? Cancer Gene Ther;15:341–355.
- D J Roberts, S Miyamoto (2015). Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. Feb;22(2):248-57.
- Elena A Federzoni et al (2014). CEBPA-dependent HK3 and KLF5 expression in primary AML and during AML differentiation. Sci Rep. Mar 3;4:4261. [CrossRef]
- E Bustamante et al (1981). Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem. Aug 25;256(16):8699-704. [CrossRef]
- Rempel A et al (1996). Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res; 56:2468–2471.
- Mathupala SP et al (1997). Aberrant glycolytic metabolism of cancer cells: a remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J Bioenerg Biomembr;29:339–343. [CrossRef]
- Mayer D et al (1997). Hexokinase expression in liver preneoplasia and neoplasia. Biochem Soc Trans.; 25:122–127. [CrossRef]
- Pedersen PL et al (2002). Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. ;1555 :14–20. [CrossRef]
- Kwee SA et al (2012). Choline kinase alpha and hexokinase-2 protein expression in hepatocellular carcinoma: association with survival. PLoS One;7:e46591.
- Palmieri D et al (2009). Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol Cancer Res.7:1438–1445. [CrossRef]
- S Miyamoto et al (2008). Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. Mar;15(3):521-9.
- Katzen HM (1966). The effect of diabetes and insulin in vivo and in vitro on a low Km form of hexokinase from various rat tissues. Biochem Biophys Res Commun;24:531–536.
- Katzen HM et al (1970). Multiple forms of hexokinase. Activities associated with subcellular particulate and soluble fractions of normal and streptozotocin diabetic rat tissues. J Biol Chem.245:4081–4096.
- Burcelin R et al (1993). Regulation of glucose transporter and hexokinase II expression in tissues of diabetic rats. Am J Physiol;265:E392–E401. [CrossRef]
- Printz RL et al (1993). Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. J Biol Chem;268:5209–5219.
- Gurel E et al (2013). Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol Biol Rep; 40:4153–4160. [CrossRef]
- Sebastian, S. et al (2000). Anabolic function of the Type II isozyme of hexokinase in hepatic lipid synthesis. Biochem. Biophys. Res. Commun. 270,886-891. [CrossRef]
- Kaselonis G. L. et al (1999). Expression of hexokinase 1 and hexokinase 2 in mammary tissue of nonlactating and lactating rats: Evaluation by RT-PCR. Mol. Gen. Metab. 68,371-374. [CrossRef]
- Osbak KK et al (2009). Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia, Hum. Mutat 30: 1512–1526. [CrossRef]
- Iynedjian PB, Molecular physiology of mammalian glucokinase, Cell. Mol. Life Sci 66 (2009) 27–42. [CrossRef]
- Shelton KD et al (1992).Multiple elements in the upstream glucokinase promoter contribute to transcription in insulinoma cells, Mol Cell Biol. Oct;12(10):4578-89.
- Moates JM et al (2003). BETA2 activates transcription from the upstream glucokinase gene promoter in islet β-cells and gut endocrine cells, Diabetes. Feb;52(2):403-8. [CrossRef]
- Jetton TL et al (1994). Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut, J. Biol. Chem. Feb 4;269(5):3641-54. [CrossRef]
- Moates JM et al (2004). The Pal elements in the upstream glucokinase promoter exhibit dyad symmetry and display cell-specific enhancer activity when multimerised, Diabetologia. Sep;47(9):1632-40. [CrossRef]
- Shawn M et al(2019). Molecular and Cellular Regulation of Human Glucokinase. Arch Biochem Biophys. Mar 15; 663: 199–213.
- P. B. Iynedjian (2008). Molecular Physiology of Mammalian Glucokinase. Cell Mol Life Sci. 2009 Jan; 66(1): 27–42. [CrossRef]
- Andreas Peter et al(2011).Hepatic Glucokinase Expression Is Associated with Lipogenesis and Fatty Liver in Humans. The Journal of Clinical Endocrinology & Metabolism 96:7, 1 July;E1126–E1130.
- Iynedjian PB (1993). Mammalian glucokinase and its gene., Biochem. J 293; 1–13. [CrossRef]
- Iynedjian PB et al (1988). Stimulation by insulin of glucokinase gene transcription in liver of diabetic rats, J. Biol. Chem. Jan 15;263(2):740-4. [CrossRef]
- Wu C, et al (2004). A Potential Role for Fructose-2,6-Bisphosphate in the Stimulation of Hepatic Glucokinase Gene Expression, Endocrinology. Feb;145(2):650-8. [CrossRef]
- Roth U et al (2004). The Transcription Factors HIF-1 and HNF-4 and the Coactivator p300 Are Involved in Insulin-regulated Glucokinase Gene Expression via the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway, J. Biol. Chem. Jan 23;279(4):2623-31.
- Iynedjian PB et al (1995). Glucokinase and cytosolic phosphoenolpyruvate carboxykinase (GTP) in the human liver. Regulation of gene expression in cultured hepatocytes., J. Clin. Invest 95; 1966–73. [CrossRef]
- Agius L (2016). Hormonal and Metabolite Regulation of Hepatic Glucokinase, Annu. Rev. Nutr 36; 389–415. [CrossRef]
- M Stubbs, S Aiston, L Agius (2000). Subcellular localization, mobility, and kinetic activity of glucokinase in glucose-responsive insulin-secreting cells. Diabetes.Dec;49(12):2048-55. [CrossRef]
- Preller A et al (1992). Localization of the type III isozyme of hexokinase at the nuclear periphery. Arch Biochem Biophys.;294:482–92. [CrossRef]
- Guillaume Calmettes et al (2015). Hexokinases and cardioprotection. J Mol Cell Cardiol . Jan;78:107-15.
- Tomohiro Nishizawa et al (2014). Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep.Apr 24;7(2):356-365. [CrossRef]
- 108 Jong-Seok Moon et al (2015). mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. Jul 7;12(1):102-115.
- Adam De Jesus et al (2022). Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol Cell. Apr 7;82(7):1261-1277.
- Michael A van der Kooij et al (2022). Chronic social stress disrupts the intracellular redistribution of brain hexokinase 3 induced by shifts in peripheral glucose levels. J Mol Med (Berl).Oct;100(10):1441-1453. [CrossRef]
- Ji-Biao Huang et al (2002). Hexokinase translocation during neutrophil activation, chemotaxis, and phagocytosis: disruption by cytochalasin D, dexamethasone, and indomethacin. Cell Immunol. Jul-Aug;218(1-2):95-106. [CrossRef]
- Mark P Labrecque et al (2021). Cabozantinib can block growth of neuroendocrine prostate cancer patient-derived xenografts by disrupting tumor vasculature. PLoS One 20;16(1):e0245602. [CrossRef]
- Lee HG et al (2016). Regulation of HK2 expression through alterations in CpG methylation of the HK2 promoter during progression of hepatocellular carcinoma. Oncotarget.7:41798–810.
- Sun L et al (2008). Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell. Biol. 28, 1007–1017.
- Wilson JE (1985) Regulation of mammalian hexokinase activity. In: Beitner R (ed) Regulation of carbohydrate metabolism, vol I. CRC, Boca Raton, pp 45–85.
- Yanqing Li et al.(2020) Prognostic Significance and Related Mechanisms of Hexokinase 1 in Ovarian Cancer. Onco Targets Ther; 13: 11583–11594.
- Xu S et al (2018). Hexokinase 2 is targetable for HK1 negative, HK2 positive tumors from a wide variety of tissues of origin. J Nucl Med. 2018 Jun 7;60(2):212-7.
- Xu S et al (2018). A precision therapeutic strategy for hexokinase 1-null, hexokinase 2-positive cancers.Cancer Metab. 2018 Jun 28;6:7.
- Daniela Šimčíková et al (2021). Loss of hexokinase 1 sensitizes ovarian cancer to high-dose metformin. Cancer & Metabolism volume 9, Article number: 41. [CrossRef]
- Amendola CR et al (2019). KRAS4 directly regulates HK1. Nature.;576(7787):482–6.
- Yang X et al (2015). A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour Biol 36:383–91.
- Huang X et al (2015). HK2 is a radiation resistant and independent negative prognostic factor for patients with locally advanced cervical squamous cell carcinoma. Int J Clin Exp Pathol. 8:4054–63.
- Iwamoto M et al (2014). Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J Nucl Med. 55:2038–44.
- Christofk, H. R., Vander Heiden, M. G., Harris, M. H., Ramanathan, A., Gerszten, R. E., Wei, R., Fleming, M. D., Schreiber, S. L., & Cantley, L. C. (2008). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature, 452(7184), 230–233. [CrossRef]
- Anderson M et al (2016). Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget ;8:56081–94. [CrossRef]
- Wang L et al (2014). Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-drven prostate cancer growth. Cell Rep;8:1461–74.
- Wolf A et al (2011). Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med;208:313–26.
- Semenza, G. L. (2010). HIF-1: Upstream and downstream of cancer metabolism. Current Opinion in Genetics & Development, 20(1), 51–56. [CrossRef]
- Cheung EC et al (2012). Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci USA. 109:20491–6. [CrossRef]
- Neary CL, Pastorino JG (2013). Akt inhibition promotes hexokinase 2 redistribution and glucose uptake in cancer cells. J Cell Physiol. 228:1943–8. [CrossRef]
- Gao F et al (2015). Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2mediated glycolysis. Oncol Rep. 33:1533–9.
- Hou X et al (2015). PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2′s mitochondria translocation. Sci Rep.5:9065.
- Wang H et al (2016). Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 16: 38.
- Wang L et al (2014). Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep;8:1461–74.
- Yoshino H et al (2013). Tumor-suppressive microRNA-143/145 cluster targets hexokinase-2 in renal cell carcinoma. Cancer Sci.104:1567–74.
- W et al (2015). MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology. 62:1132–44.
- Qin Y et al (2016). MiR-4458 suppresses glycolysis and lactate production by directly targeting hexokinase2 in colon cancer cells. Biochem Biophys Res Commun. 469:37–43.
- Jiang S et al (2012). A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 31:1985–98.
- Gregersen LH et al (2012). MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer. 12:232. [CrossRef]
- Weiqi Dai et al (2015). By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice. Oncotarget. 30; 6(15): 13703–13717. [CrossRef]
- Federzoni EA et al (2012). PU.1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood. 119 (21): 4963–70. [CrossRef]
- Gao HY et al (2015). Identification of key genes affecting disease free survival time of pediatric acute lymphoblastic leukemia based on bioinformatic analysis. Blood Cells, Molecules & Diseases. 54 (1): 38–43.
- Lu, J. (2019). The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Demetrius, L., & Tuszynski, J. A. (2010). Reviews, 38(1–2), 157–164.
- Jose, C., Bellance, N., & Rossignol, R. (2011). Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochimica et Biophysica Acta, 1807(6), 552–561.
- Kristina Seiler et al (2022). Hexokinase 3 enhances myeloid cell survival via non-glycolytic functions. Cell Death & Disease 13,: 448. [CrossRef]
- Wenhao Xu et al (2021). Hexokinase 3 dysfunction promotes tumorigenesis and immune escape by upregulating monocyte/macrophage infiltration into the clear cell renal cell carcinoma microenvironment. Int J Biol Sci. 2021; 17(9): 2205–2222. [CrossRef]
- M. Board, et al (1995). High Km glucose-phosphorylating (glucokinase) activities in a range of tumor cell lines and inhibition of rates of tumor growth by the specific enzyme inhibitor mannoheptulose. Cancer Res., 55pp. 3278-3285.
- N.N. Danial, et al (2003). BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature, 424 (2003), pp. 952-956. [CrossRef]
- N.-L.-C. Bui, et al (2018). Bad phosphorylation as a target of inhibition in oncology. Cancer Lett., 415, pp. 17-186. [CrossRef]
- N.N. Danial (2008). BAD: undertaker by night, candyman by day. Oncogene, 27, pp. S5-S70. [CrossRef]
- P. Jiang, et al (2006). The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol. Cell. Biol., 26, pp. 9071-9082. [CrossRef]
- M. Matschinsky, et al (2006).The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes, 55, pp. 1-12.
- J. Grimsby, et al (2003).Allosteric Activators of Glucokinase: potential Role in Diabetes Therapy. Science, 301, pp. 370-373. [CrossRef]
- F.M. Matschinsky (2009). Assessing the potential of glucokinase activators in diabetes therapy. Nat. Rev. Drug Discov., 8, pp. 399-416. [CrossRef]
- A. Nakamura et al (2015). Present status of clinical deployment of glucokinase activators. J. Diabetes Investig., 6, pp. 124-132. [CrossRef]
- Y.S. Oh, et al (2014). Treatment with glucokinase activator, YH-GKA, increases cell proliferation and decreases glucotoxic apoptosis in INS-1 cells. Eur. J. Pharm. Sci., 51, pp. 137-145. [CrossRef]
- S. Porat, et al (2011). Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab., 13, pp. 440-449. [CrossRef]
- S. Kassem, et al (2010). Large islets, beta-cell proliferation, and a glucokinase mutation. N. Engl. J. Med., 362, pp. 1348-1350.
- Q. Shen, et al (2017). Proteome-scale investigation of protein allosteric regulation perturbed by somatic mutations in 7,000 cancer genomes. Am. J. Hum. Genet., 100, pp. 5-20. [CrossRef]
- Miroslav Těšínský et al (2019). Biochim Biophys Acta Proteins Proteom. 1867(3):213-218.
- Orci, L. A et al (2021). Incidence of hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: a systematic review, meta-analysis, and meta-regression. Clinical Gastroenterology and Hepatology. [CrossRef]
- Nagaoki, Y. et al (2021). Increasing incidence of non-HBV-and non-HCV-related hepatocellular carcinoma: single-institution 20-year study. BMC gastroenterology, 21(1), 1-10. [CrossRef]
- Li, J. et al (2017). A prognostic 4-gene expression signature for squamous cell lung carcinoma. Journal of cellular physiology, 232(12), 3702-3713. [CrossRef]
- Zhu, Y et al (2016). Treatment of advanced squamous cell lung cancer. Zhongguo fei ai za zhi= Chinese Journal of Lung Cancer, 19(10), 687-691.
- Lian, H et al . (2020). Identification of novel alternative splicing isoform biomarkers and their association with overall survival in colorectal cancer. BMC gastroenterology, 20(1), 1-12. [CrossRef]
- García-Campelo, R. et al (2015). SEOM clinical guidelines for the treatment of non-small cell lung cancer (NSCLC) 2015. Clinical and Translational Oncology, 17(12), 1020-1029. [CrossRef]
- Anders, S. et al (2012). Detecting differential usage of exons from RNA-seq data. Nature Precedings, 1-1.
- Fuhr, L et al (2018). The circadian clock regulates metabolic phenotype rewiring via HKDC1 and modulates tumor progression and drug response in colorectal cancer. EBioMedicine, 33, 105-121. [CrossRef]
- Fan L et al (2018). Long noncoding RNA urothelial cancer associated 1 regulates radioresistance via the hexokinase 2/glycolytic pathway in cervical cancer. Int J Mol Med. 42:2247–59.
- Ma Y et al (2019). Long non-coding RNA HOTAIR promotes cancer cell energy metabolism in pancreatic adenocarcinoma by upregulating hexokinase-2. Oncol Lett. 18:2212–9.
- Nabi, K., & Le, A. (2021). The intratumoral heterogeneity of cancer metabolism. Advances in Experimental Medicine and Biology, 1311.
- Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311.
- Jose, C., Bellance, N., & Rossignol, R. (2011). Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochimica et Biophysica Acta, 1807(6), 552–561.
- Evstafieva, A. G.et al (2018). Implication of KRT16, FAM129A and HKDC1 genes as ATF4 regulated components of the integrated stress response. PloS one, 13(2), e0191107.
- Lunt SY, Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64. [CrossRef]
- Schulze A, Harris AL (2012). How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature.;491(7424):364-73.
- Kamarajugadda, S., et al. (2012). Glucose oxidation modulates anoikis and tumor metastasis. Molecular and Cellular Biology, 32(10), 1893–1907. [CrossRef]
- Iynedjian PB (1993) Mammalian glucokinase and its gene. Biochem J 293:1–13.
- Ciscato F et al (2021). Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int J Mol Sci.;22(9):4716. [CrossRef]
- Xue YN et al (2019). Zinc and p53 disrupt mitochondrial binding of HK2 by phosphorylating VDAC1. Exp Cell Res. 1;374(1):249-258.
- Shaopei Wang et al (2023). Advances in the Study of Hexokinase 2 (HK2) Inhibitors. Anticancer Agents Med Chem;23(7):736-746. [CrossRef]
Figure 1.
Schematic representation of the functional domains of the five hexokinase isoforms. The rust-colored cylinders represent domains with catalytic activity, and the blue cylinders have no catalytic activity. Both cylinders in HKDC1 are gray colored because this isoform has very low kinase activity. MLS = mitochondrial localization sequence (red-colored cylinder). Numbers represent amino acid sequences adapted from Uniprot.org.
Figure 1.
Schematic representation of the functional domains of the five hexokinase isoforms. The rust-colored cylinders represent domains with catalytic activity, and the blue cylinders have no catalytic activity. Both cylinders in HKDC1 are gray colored because this isoform has very low kinase activity. MLS = mitochondrial localization sequence (red-colored cylinder). Numbers represent amino acid sequences adapted from Uniprot.org.
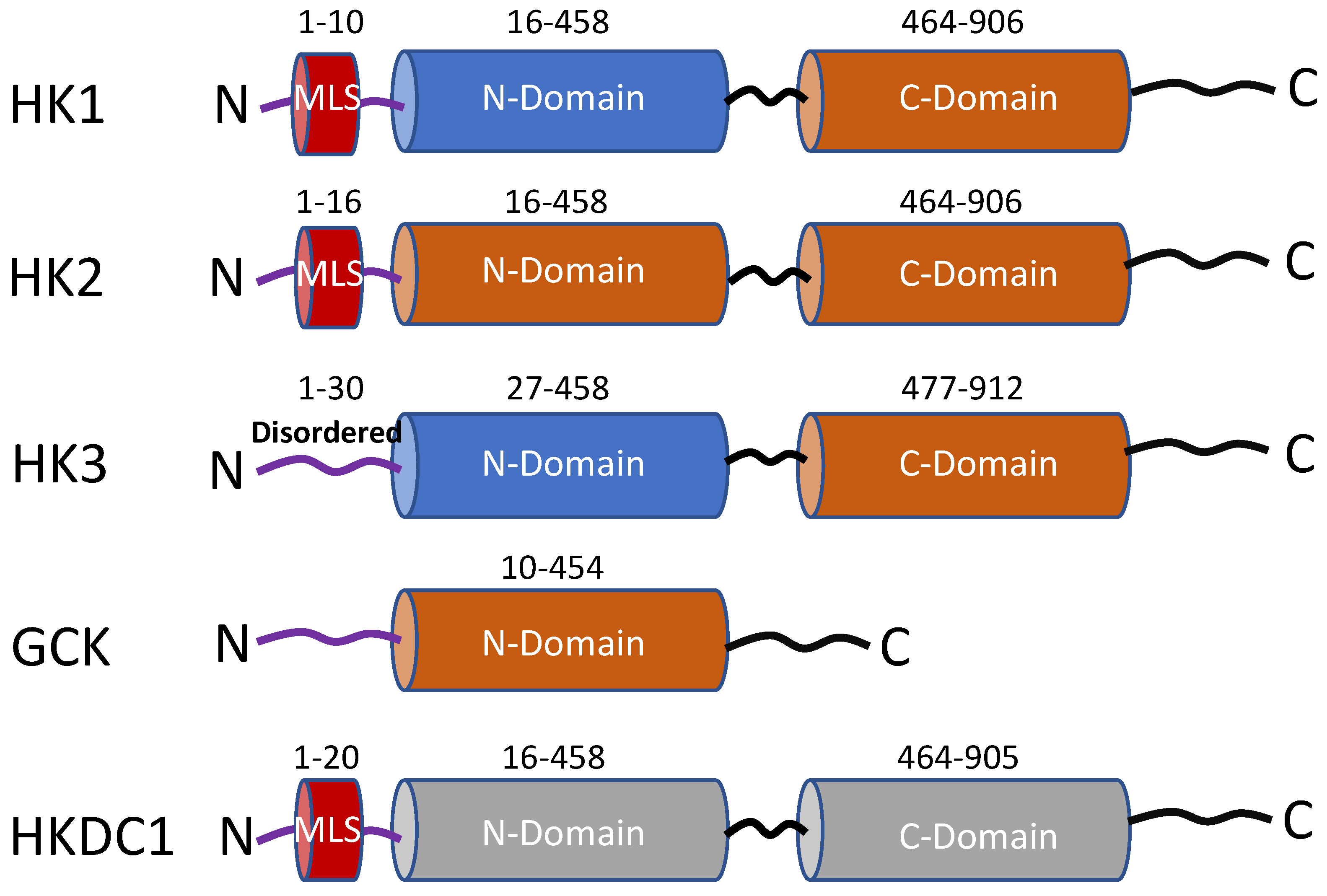
Figure 2.
Illustration of the delivery of glucose to membrane-bound HKs in malignant cells. Illustration of the delivery of Glucose to HKs 1, 2, and HKDC1 bound to the outer mitochondrial membrane (OMM) and metabolic fates of the glucose-6-phosphate (G6P) formed thereof within a malignant cell. Glucose transport across the plasma membrane by glucose transporters is phosphorylated by HKs (HK1, HK2, or HKDC1) bound to a voltage-dependent anion channel (VDAC) located on the outer mitochondrial membrane. VDAC allows direct access of ATP generated by the ATP synthase within the mitochondria to the HKs, which can be transported across the inner-mitochondrial membrane by the adenine nucleotide translocator. To maintain malignant cells’ highly glycolytic metabolic flux, the product G6P is rapidly distributed across key metabolic routes (see thick green arrows). The primary metabolic routes for G6P are (a) entry into the pentose-phosphate pathway for biosynthesis of nucleic-acid precursors and (b) conversion to pyruvate and lactate through glycolysis. In cancer cells, most lactate is transported out of the with the aid of lactate transporters. In contrast, small amounts of pyruvate are transported to mitochondria through the pyruvate transporters to supply intermediates to the tricarboxylic acid (TCA) cycle (thin red arrows). Citrate transporters transport citrate produced in the TCA cycle to aid in synthesizing membrane components like phospholipids and cholesterol, essential for tumor cell proliferation.
Figure 2.
Illustration of the delivery of glucose to membrane-bound HKs in malignant cells. Illustration of the delivery of Glucose to HKs 1, 2, and HKDC1 bound to the outer mitochondrial membrane (OMM) and metabolic fates of the glucose-6-phosphate (G6P) formed thereof within a malignant cell. Glucose transport across the plasma membrane by glucose transporters is phosphorylated by HKs (HK1, HK2, or HKDC1) bound to a voltage-dependent anion channel (VDAC) located on the outer mitochondrial membrane. VDAC allows direct access of ATP generated by the ATP synthase within the mitochondria to the HKs, which can be transported across the inner-mitochondrial membrane by the adenine nucleotide translocator. To maintain malignant cells’ highly glycolytic metabolic flux, the product G6P is rapidly distributed across key metabolic routes (see thick green arrows). The primary metabolic routes for G6P are (a) entry into the pentose-phosphate pathway for biosynthesis of nucleic-acid precursors and (b) conversion to pyruvate and lactate through glycolysis. In cancer cells, most lactate is transported out of the with the aid of lactate transporters. In contrast, small amounts of pyruvate are transported to mitochondria through the pyruvate transporters to supply intermediates to the tricarboxylic acid (TCA) cycle (thin red arrows). Citrate transporters transport citrate produced in the TCA cycle to aid in synthesizing membrane components like phospholipids and cholesterol, essential for tumor cell proliferation.
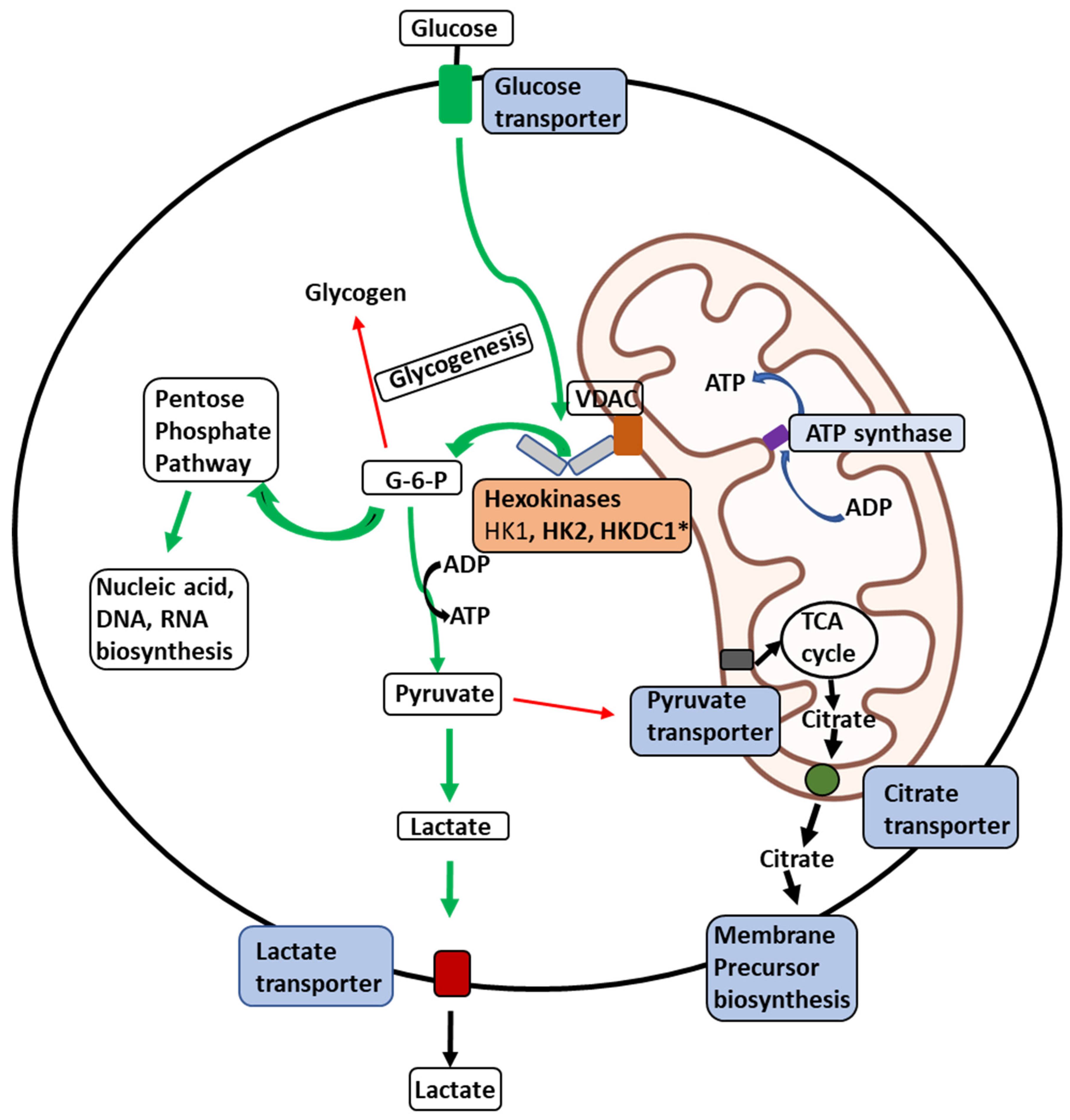
Figure 3.
Schematic representation of the effects of HKDC1 over-expression in cancer cells. The cell membrane glucose transporters (GLUT 1/3) mediate the glucose uptake, which is degraded to pyruvate by glycolysis. Upregulation of HKDC1 (and other HKs) in many cancer types leads to enhanced generation of glycolytic intermediate, which functions as precursors for numerous metabolic pathways necessary for the biosynthesis of cellular components; pentose phosphate pathway (marked with thick red arrows), cholesterol biosynthesis and fatty acid biosynthesis. Notably, HKDC1 upregulation leads to an increase in HKDC1-mitochondrial binding, which is responsible for the maintenance of glycolysis and TCA cycle and contributes to unabated cell proliferation through aversion of apoptosis and Endoplasmic reticulum (ER) mediated stress response mechanisms by reducing the number of physical contact points between ER and mitochondria.
Figure 3.
Schematic representation of the effects of HKDC1 over-expression in cancer cells. The cell membrane glucose transporters (GLUT 1/3) mediate the glucose uptake, which is degraded to pyruvate by glycolysis. Upregulation of HKDC1 (and other HKs) in many cancer types leads to enhanced generation of glycolytic intermediate, which functions as precursors for numerous metabolic pathways necessary for the biosynthesis of cellular components; pentose phosphate pathway (marked with thick red arrows), cholesterol biosynthesis and fatty acid biosynthesis. Notably, HKDC1 upregulation leads to an increase in HKDC1-mitochondrial binding, which is responsible for the maintenance of glycolysis and TCA cycle and contributes to unabated cell proliferation through aversion of apoptosis and Endoplasmic reticulum (ER) mediated stress response mechanisms by reducing the number of physical contact points between ER and mitochondria.
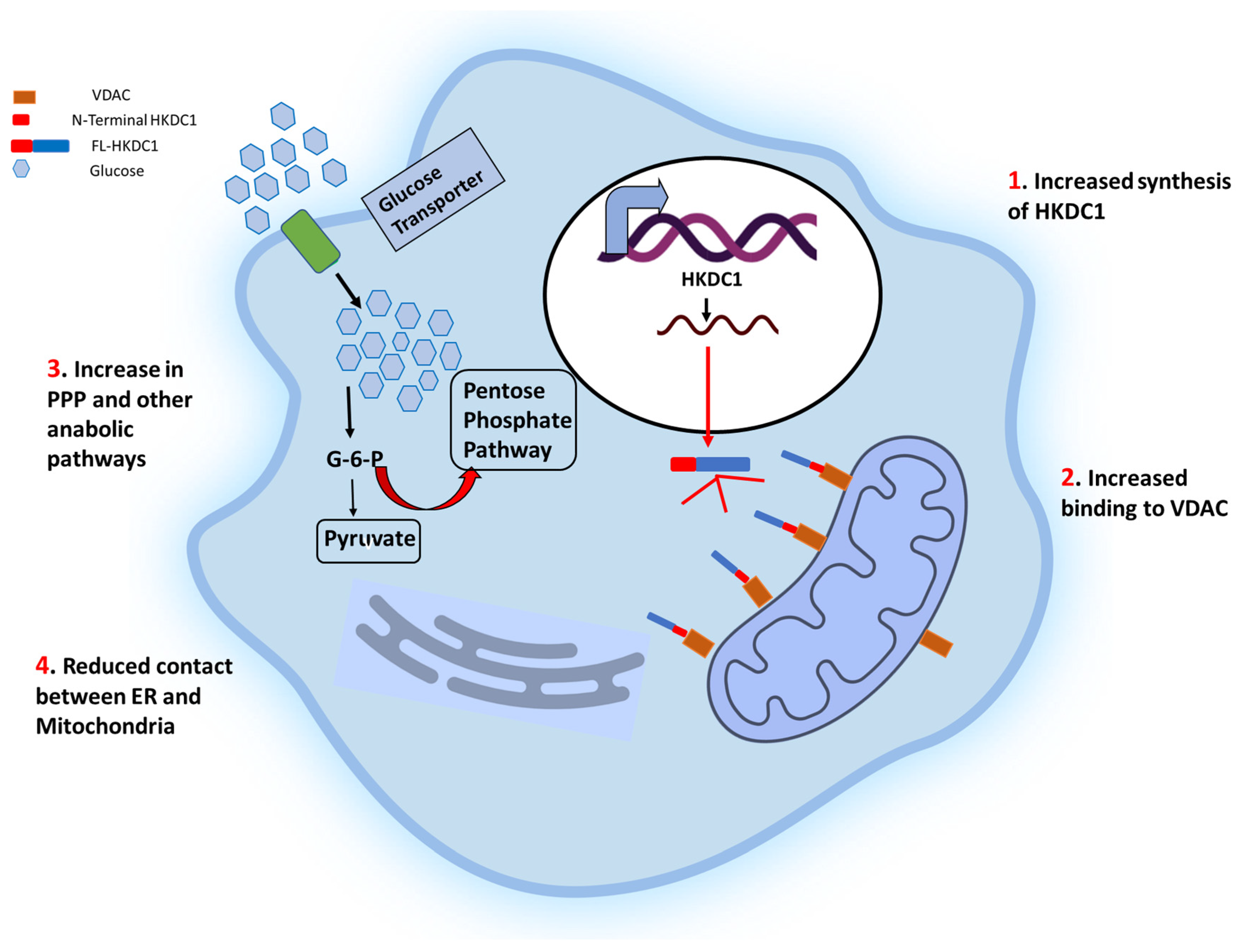

Table 1.
Characteristics of HK isoforms in humans.
| HKI | HKII | HKIII | GCK | HKDC1 | |
|---|---|---|---|---|---|
|
Gene location (Human) |
10q22 | 2p13 | 5q35.2 | 7p15.1 | 10q22 |
| MW (kDa) | ~100 | ~100 | ~100 | ~50 | ~100 |
| Number of catalytic domains | 1 | 2 | 1 | 1 | 1 |
| Km for glucose ( mmol 1-1) | 0.03 | 0.3 | 0.003 | 6 | - |
| Km for ATP ( mmol 1-1) | 0.5 | 0.7 | 1.0 | 0.6 | - |
| G6P inhibitionKi (mmol 1-1) | 0.02 | 0.02 | 0.10 | - | - |
| Effect of pi | Low conc counteracts G6P inhibition, but high conc is inhibitory | inhibitory | Inhibitory | - | - |
| Insulin regulation | - | + | * | + | * |
| Major tissue expression | Brain, Kidney | Muscle, adipose | Lung, spleen | Liver, pancreas | GI, Kidney, and Brain |
| Mitochondrial binding | ✓ | ✓ | ✕ | ✕ | ✓ |
|
+ = effect; - = NO effect; * = Sufficient data not available; pi = inorganic phosphate; ✓ = binding; ✕ = no binding | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



