
Submitted:
27 March 2023
Posted:
28 March 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
There have been significant collaborative efforts over the past three years to develop therapies against COVID-19. During this journey, there has also been a lot of focus on understanding at-risk groups of patients who either have pre-existing conditions or have developed concomitant health conditions due to the impact of COVID-19 on immune system. There was a high incidence of COVID-19 induced pulmonary fibrosis (PF) observed in patients. PF can cause significant morbidity, long-term disability, and lead to death in the long run. Additionally, being a progressive disease, PF can also impact the patient for a long time after COVID infection and affect the overall quality of life. Although current therapies are being used as the mainstay for treating PF, there is no therapy specifically for COVID-induced PF. As observed in the treatment of other diseases, nanomedicine can show a significant promise in overcoming the limitations of current anti-PF therapies. In this review, we summarize the efforts reported by various groups to develop nanomedicine therapeutics to treat COVID-induced PF.
Keywords:
SARS-CoV-2
; COVID-19
; pulmonary fibrosis
; coronavirus
; nanoparticles
; nanomedicine
; pandemic
; acute respiratory syndrome
1. Background
Pulmonary Fibrosis (PF) is a disease characterized by decline in lung function which eventually leads to respiratory failure, if untreated. PF is a progressive disease with lung transplant being the only option in late stages [1]. In some cases, PF has histopathologic and radiologic indications that are like other non-PF forms of interstitial lung disease, making it difficult to distinguish for accurate diagnosis [2]. Histologically, it is characterized by permanent scarring and extracellular matrix deposition in lung parenchyma [3,4]. Recent advances have determined certain genomic as well as natural history factors that increase the risk of PF. It has been reported that single nucleotide polymorphism on the p-terminus of chromosome 11, which is located within a highly conserved area of the promoter region for the mucin 5B (MUC5B) gene is associated with the high risk of PF [5]. Other polymorphisms and loci have also been identified that are associated with high risk of PF. Interestingly, members of extended families with variant abnormalities of the same gene were at high risks of different fibrotic forms of lung injury [6]. Genomic factors and epigenetic mechanisms such as histone modification and microRNA expression are also found to be responsible for high risk of fibrosis [7,8]. Moreover, advanced age is likely to increase the risk of PF, males are at high risk compared to females, and occupational hazards such as silica exposure, material dust exposure, livestock exposure have shown to contribute to the pathogenesis of PF as well [9,10]. However, it should be noted that in many cases, patients with PF do not necessarily have prevalence of previously mentioned risk factors, however, these risk factors do require genetic predisposition to develop PF. The key histologic indications of PF are characteristic fibroblastic foci accompanied with distortion and fibrosis of lung tissue. Primary symptoms of PF include persistent dry cough, shortness of breath, tiredness, and weight loss. If undiagnosed the disease may worsen causing pneumonia, bronchitis, followed by chest pain, and cardiac failure in the later stages of disease progression. According to the World Health Organization (WHO), about 100,000 people are affected by IPF in the United States, and 30,000 to 40,000 new cases are diagnosed each year. Moreover, the global prevalence of IPF is 13 to 20 cases per 100,000 people.
In the post-COVID era, the PF is also viewed as an aftereffect of COVID-19 infection. PF was not only a major cause of death worldwide in COVID-19 infected patients but also a cause of permanent lung tissue damage in recovered patients [11,12]. COVID-19 and PF also share common risk factors such as old age, male sex, and comorbidities like diabetes and hypertension. Current treatment options for PF include antifibrotic agents such as pirfenidone and nintedanib. Although these therapies are shown to improve survival expectancy by as much as 2 years, they have been effective in controlling the rate of lung function decline by only about 50% [13,14,15]. Additionally, these therapies are available only in the form of oral dosage forms and hence administration is difficult for patients in coma or patients that are intubated/mechanically ventilated in critical care units. In this review, we discuss various nanotherapeutic approaches that can potentially be used in the control, maintenance, and treatment of PF. They can be designed to facilitate targeted delivery of therapeutic agent to the lungs and avoid drug dumping.
2. Pathophysiology of COVID-induced PF
While the complex molecular mechanisms that precipitate the grave pathophysiological outcomes in the lung are being elucidated, the significant extent of overlap in the progression and genetic etiology of idiopathic pulmonary fibrosis (IPF) and COVID induced PF has been demonstrated [16,17]. The heterogeneity of the severity of the disease progression for COVID-19 has led to wide spectrum of health outcomes ranging from asymptomatic to needing mechanical ventilation, lung damage, and death [18]. The downstream pulmonary effects produced by COVID overlap uniquely with the signaling pathways shown by IPF. The macrophagial infiltration, progressive fibroblast proliferation, and metaplasia have been reported in patients with COVID, while not correlated with H1N1 influenza or bacterial infections [19,20]. In approximately 84% of COVID-19 patients, signs and symptoms of PF follow a clinical cure and are seen in all patients with severe or critical SARS-CoV-2 infection [12]. Fadista and co-workers reported a positive genetic correlation of IPF and the severity of COVID-19 symptoms in an age stratified Mendelian randomization analysis. However, there is also parallel evidence signifying the divergence in etiology in the PF caused by COVID-19 and IPF. Flaifel and co-workers reported the incidence of a non-specific interstitial pneumonia (NSIP) and bronchiectasis in COVID-19 patients instead of the interstitial pneumonia pattern of fibrosis seen in IPF, and this finding was also corroborated by Parimon and co-workers [21].
Figure 1.
Pathophysiology of COVID-induced PF. The aberrant interaction of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) with the alveolar epithelium (Type I and II) leads to cellular death, inflammation, and keratinization. Disruption of respiration is a grave cardinal symptom of PF and has been linked to patients affected severely by COVID-19. The molecular mechanisms behind the positive feedback loop of inflammatory activity have been linked to a phenomenon of ‘cytokine storm’ mediated through monocytes and myeloid cells. A more complex mechanism of cellular metaplasia of krt+ basal cells and fibroblasts to the alveoli has been linked to decreased lung function, the furtherance of aberrant inflammatory responses, and reduced overall survival.
Figure 1.
Pathophysiology of COVID-induced PF. The aberrant interaction of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) with the alveolar epithelium (Type I and II) leads to cellular death, inflammation, and keratinization. Disruption of respiration is a grave cardinal symptom of PF and has been linked to patients affected severely by COVID-19. The molecular mechanisms behind the positive feedback loop of inflammatory activity have been linked to a phenomenon of ‘cytokine storm’ mediated through monocytes and myeloid cells. A more complex mechanism of cellular metaplasia of krt+ basal cells and fibroblasts to the alveoli has been linked to decreased lung function, the furtherance of aberrant inflammatory responses, and reduced overall survival.
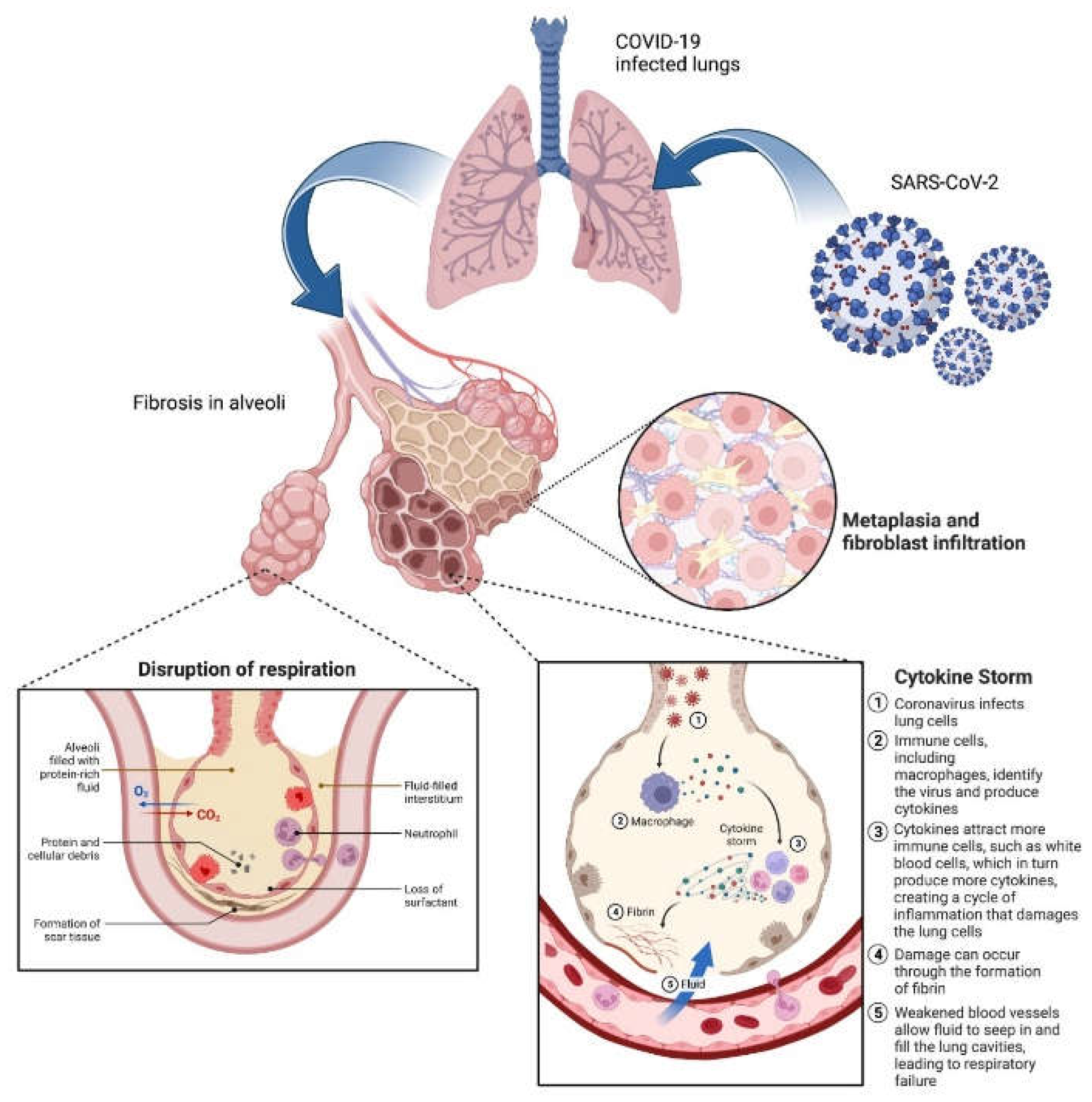
A key element of the etiology of COVID-19 induced PF is preferential infectivity that this virus has towards alveolar macrophages and the basal epithelial cells in the respiratory tract, owing to the attachment protein ACE2 [22]. The alveolar epithelial cells (AECs) play a key role in pulmonary homeostasis, with Type I AECs regulating gas exchange while the Type II AECs are responsible for the production of a protein-lipid surfactant containing surfactant protein C, sterols, and dipalmitoylphosphatidylcholine (DPPC), which is critical for lowering of the surface tension and alveolar function [23,24]. The epithelial-mesenchymal trophic units (EMTUs) are a functional element representing the feedback interactions between the epithelial and mesenchymal cells [25,26,27]. This signaling channel can be disrupted by cellular death, disruption, and inflammation in the type II AECs perpetuated by environmental factors like silica or metal dust, radiation, or infections like COVID-19. The surviving type II AECs have been shown to overexpress profibrotic markers like pSTAT3 and KIT, apoptotic markers like CASP3, and proinflammatory signaling molecules like IL-6.
The persistence of inflammation in these loci has direct links to increased levels of proinflammatory biomarkers like IFN-γ type 2, TGF-β, IL-1α, and IL-8 [28,29]. Monocytes, macrophages, and myeloid cells induced by these signaling markers trigger further trigger a ‘cytokine storm’ and thus perpetuate a positive feedback loop with further immune cell recruitment, ROS generation, and colocalization with lung tissue showing profibrotic histology [30,31]. The high degree of chronic inflammation found in COVID-19 patients with PF remains a key feature of the condition [32]. Aberrant activation of lung epithelial progenitor cells that leads to increased active myofibroblasts with associated fibroblast alveolar migration is a downstream effect of the disrupted cell signaling in PF. Indeed, proliferation of myofibroblasts with other elements of the mesenchyme have been reported in patients with late- or end-stage fibrotic COVID-19 [33]. Alveolar epithelial damage and the subsequent dysregulation of the paracrine cell signaling contribute to the aberrant cell proliferation which leads to metaplasia, a critical feature of PF [34,35,36,37]. The altered numbers and function of the type II AECs lead to metaplasic infiltration of krt+ basal cells into the alveoli, subsequent keratinization of the pulmonary tissue, and eventually decreased lung function [38,39,40]. This reduced lung function, overexpressed TGF-β signaling, and progression of PF is linked to decreased overall survival rates [41]. TGF-β plays a key role in the progression of PF by inducing recruitment of myofibroblasts, accumulation of extracellular matrix, and persistent induction of fibronectin and collagen [42,43,44]. The effects of TGF-β associated cascades are balanced by the activity of the bone morphogenetic protein (BMP4), which is responsible for inhibiting the proliferation of human pulmonary fibroblasts and induces the proliferation of pulmonary epithelial cells [45]. Koli and co-workers demonstrated elevated levels of Gremlin/Drm mRNA, an antagonist of BMP-4, in fibrotic tissue [42]. Tilting of the balance towards TGF-β cascades in antagonism to BMP-4 in fibrotic tissue has been demonstrated in murine models. There might be a diagnostically relevant unique expression profile for PF marked by lower levels of circulating IFN-β and higher IL-1α and TGF-β.
The overall pathophysiology of PF and the high extent of overlap with COVID-19 warrants further investigation into the metabolome and the interactome of fibroproliferative cell signaling, metaplasia, and immune activity. The covariables of history of exposure to tobacco smoke, environmental irritants, chronic obstructive pulmonary disorder (COPD), autoimmunity, as well as genetic predisposition may also contribute to the severity of the PF seen in COVID-19 patients.
Current management strategies for COVID-induced PF heavily rely on its pathophysiology. The next section in this review focuses on the current management strategies and efforts.
3. Current Status of COVID-induced PF therapy
Current therapy of PF is performed with either pirfenidone or nintedanib. Pirfenidone’s mechanism of action is not completely understood yet, however, it is believed to inhibit fibroblast activity and matrix deposition by disrupting TGF- β activity [46]. Pirfenidone underwent multiple clinical trials, however, three separate sets of trials granted it authorization to use in Japan, Europe, and the USA [47]. A randomized, double-blind, placebo-controlled phase III trial that included mild-to-moderate IPF patients was conducted in Japan over 52 weeks [48]. The progression-free survival was significantly longer in 1800mg/day dose arm compared to placebo arm leading to granting authorization in Japan. Moreover, two CAPACITY trials were conducted by enrolling mild-to-moderate progression IPF patients, of which one trial i.e., trial 004 met the primary endpoint [49]. Trial 006 on the other hand could not demonstrate difference between pirfenidone treated subjects as compared to placebo arm in terms of the % change in forced vital capacity (FVC) of the lungs. These trials granted pirfenidone an authorization in Europe. However, an additional study was required by the US FDA.
This ASCEND study enrolled patients with high risk of disease progression [15]. These trials demonstrated a 45% reduction in FVC decline compared to placebo arm satisfying the primary objective of this trial. Following this study FDA granted pirfenidone an authorization for use in IPF. The reported side effects of pirfenidone include nausea, vomiting, dyspepsia, anorexia, photosensitivity reaction, skin rash, and elevation of liver enzymes. Pirfenidone is metabolized primarily in the liver via the cytochrome system (CYP1A2 enzyme). Hence, co-administration of CYP1A2 inhibitors (e.g., amiodarone, ciprofloxacin, fluvoxamine) or inducers (e.g., omeprazole) that can alter pirfenidone bioavailability is avoided [50,51]. Pirfenidone is contraindicated in patients with severe hepatic dysfunction or patients with advanced renal dysfunction (glomerular filtration rate <30 ml/min). On the other hand, nintedanib inhibits multiple intracellular kinases by blocking receptors for vascular endothelial growth factor (VEGFR 1-3), platelet-derived growth factor (PDGFR α and β), and fibroblast growth factor (FGFR 1-3), and it also inhibits Fms-like tyrosine kinase-3. In INPULSIS-1 and 2 trials it showed reduced decline in FVC in nintedanib treated patients as compared to placebo arm [14]. The most common side effects are diarrhea and gastrointestinal discomfort. Nintedanib is mainly metabolized by hepatic ester cleavage and minor role is played by CYP3A4. It is also a substrate for p-glycoprotein. Hence, co-administration of inhibitors of both CYP3A4 and P-glycoprotein (e.g., erythromycin, imidazoles) or inducers (e.g., phenytoin, carbamazepine, rifampicin, St. John’s Wort) is avoided. Also, nintedanib is contraindicated in patients with moderate to severe hepatic dysfunction. Additionally, both these drugs hold potential to be explored for their use in COVID-19 induced IPF. However, the clinical studies to claim whether these drugs help prevent post-COVIID fibrotic injury have not taken place yet apart from the few case studies in combination with other drugs. Additionally, safety and specificity to lung tissue remain viable concerns for all the currently utilized therapies. Table 1 shows on-going clinical trials to develop therapies against COVID-induced PF.
As widely reported for other disease areas, nanomedicine can overcome the limitations of the current PF therapies [52,53]. Nanotherapies like nanoparticles, cell-derived vesicles, nanopolymers are more amenable to modification to enhance their safety, specificity, and stimuli-responsive drug release [54,55]. Additionally, there have been multiple reports of tailoring pharmacokinetics of drugs by using nanomedicine-based approaches [56]. Nanoparticle applications to deliver drug combinations have been widely reported [57,58]. In the following section, we report a few nanotherapeutic approaches that have been reported in the literature that look specifically at COVID-induced PF.
4. Nano-therapeutic approaches to treat COVID induced PF
4.1. Cell-mimicking nanodecoys
Reports which elucidated the mechanism of COVID indicated that angiotensin-converting enzyme 2 (ACE2) played pivotal role in viral entry into the host cell [59,60]. The role of ACE2 has been well characterized in multiple viral infections. In COVID, the spike protein of the virus specifically interacts with ACE2-presenting pneumocytes in the lungs and goblet secretory cells in the nasal mucosa [61]. Li and co-workers developed a cell therapy using a mixture of resident lung epithelial cells and mesenchymal cells which they termed lung spheroid cells (LSCs) [62]. Since these cells express ACE2, the authors utilized their cell membrane to fabricate ACE2 nanodecoys. The LSC-nanodecoys could act as cell mimics and bind to the SARS-CoV-2 spike (S) protein, consequently triggering a phagocytotic response from macrophages resulting in the elimination of the virus. This therapeutic system provided an advantage over spike protein targeting therapies like antiviral drugs and vaccines as they target human cells expressing ACE2 receptor and are independent of viral proteins. Consequently, the authors are of the view that this therapy could also be more effective in managing more aggressive variants associated with the mutations in the viral spike proteins.
Nanodecoys were generated by serial extrusion of LSCs through polycarbonate membranes with various pore sizes and characterized. The obtained nanodecoys exhibited an average size of 320nm and were spherical in shape. A yield of approximately 11,000 nanodecoys was obtained from one LSC. Control nanodecoys were also generated from HEK293 cells which do not express the ACE2 receptors. The authors also demonstrated the ability of nanodecoys to bind to S-protein in a dose responsive manner. While LSC nanodecoys could competitively bind within 4 hours, HEK293 nanodecoys did not show binding to S-protein. In addition, LSC-nanodecoys were internalized more in the macrophage cells as compared to lung cells, indicating that they could be potentially cleared out by the immune cells. Similar studies were also performed to show that the LSC nanodecoys could protect from SARS-CoV2 mimics. The biodistribution of the nanocdecoys was performed in CD1 mice. Fluorescent labeled nanodecoys were administered to mice via inhalation route and the biosidtribution analysis was performed as 24, 48 and 72h post administration. The authors reported that nanodecoys could still be found in the lungs 72h after a single inhalation treatment. In addition, nanodecoys were also found in spleen, liver and kidney indicating clearance via the reticuloendothelial system (RES). No impact was observed on CD68+ macrophage infiltration indicating biocompatibility with immune cells. Additionally, the authors also demonstrated that the LSC nanodecoys could significantly reduce the number of SARS-CoV2 mimics that were internalized by lung cells avoiding further infection. This effect was not observed for free form of recombinant ACE2 or HEK293 nanodecoys. Confocal micriscpy assessments showed that the nanodecoys could accelerate the clearance of the mimics. Cytokine and histopathological assessments of all major organs showed no adverse effects of either LSC or control nanodecoys.
To demonstrate therapeutic efficacy of the LSC nanodecoys, a pilot non-human primate (NHP) study was performed in cynomolgus macaques. This model could replicate many clinical symptoms of SARS-CoV-2 infection as well as show robust viral replication. The animals were challenged with SARS-CoV-2 using intranasal and intrathecal routes followed by random assignment into the LSC nanodecoy and PBS control groups. Nanodecoys were administered daily from days 2 to 5 post-challenge via inhalation using a nebulizer and fitted mask at the dose of 1010 particles/kg. 8 days post-challenge the animals were sacrificed for further assessments. Viral loads in bronchoalveolar lavage (BAL) and nasal swabs (NS) were assessed by using RT-PCR to measure genomic RNA which was indicative of viral replication. High levels of RNA were observed in control animals with a median peak of 6.243 log10RNA copies ml−1 in BAL and a median peak of 5.595 log10RNA copies per swab in NSs on day 2. On the contrary, RNA levels showed a dramatic reduction in nanodecoy treated animals with a <1.7log10 reduction of median peak RNA in both BAL and NS on day 8 post-challenge. Hisptopathological analysis of the lung tissue was performed at the end of the study. Multifocal regions of inflammation, monocellular cell infiltrates and edema were observed in the control animals. On the other hand, LSC-nanodecoy treatment significantly reduced the numbers of inflammatory cells infiltrating the lungs. Immunohistochemistry (IHC) staining was performed to detect the viral levels within the lung tissue. LSC-nanodecoy treated group showed significantly low levels of viral protein as well as viral replication within the lung tissue as compared to control groups.
Additionally, the authors also mentioned that in terms of drug development, the major pain points they wanted to address were off-target effects and undesired biodistribution. Based in this study, the authors were able to show that the nano decoys not only showed specificity towards lung accumulation but also exerted their effect only in the impacted tissue without eliciting toxicity in other systemic organs. Based on all these findings the authors believe that LSC-nanodecoys can serve as a potential therapeutic agent for treating COVID-19 and COVID-induced PF.
4.2. CD-24 exosomes
Shapira and co-workers tried to develop a therapy based on CD24, which is a small, heavily glycosylated membrane-anchored protein which acts as an immune checkpoint regulator [63]. CD24 allows immune differentiation between molecular patterns of damaged or dying cells versus the ones derived from pathogens like bacteria and viruses [64]. Binding of CD23 to damage associated molecular patterns (DAMPs) prevents binding to pathogen associated molecular patterns (PAMPs). Additionally, this also causes inhibition of DAMP-induced inflammatory cytokine activation. As a result, while CD24 dampens immune activation, it does not affect immune recognition, hence does not interfere with viral clearance. Exosomes are vesicles which play a role in intercellular communication [65]. They have been reported to be useful delivery vehicles as they can increase stability and extend bioavailability of therapeutic molecules as evidenced by multiple on-going clinical trials [66,67]. Additionally, a recent report showed that nebulized exosomes can help repair PF [68]. To combine the benefits of both CD24 and exosomes, the authors developed CD-24 enriched exosomes (EXO-CD24) as a targeted therapy against COVID-19 immune activation.
The authors initially have reported creation of a HEK293 cell line stably transfected with CD24 which they used for purification of exosomes displaying high levels of CD24 (EXO-CD24) [69]. The authors utilized flow cytometry and western blot analysis to demonstrate high expression of CD24 on the isolated exosomes. Further characterization studies showed thatEXO-CD24 had a particle size of approximately 100-200 nm. Additionally, the EXO-CD24 had a spherical morphology with clearly visible lipid bilayers and vesicular internal structures.
The authors also demonstrated inhibition of inflammatory cytokine/chemokine secretion in EXO-CD24 treated human monocyte cell line, U937. In vivo studies with mice were performed to assess the toxicity as well as efficacy. For safety assessment, A dose of either 5 × 108 or 1 × 109 EXO-CD24 was administered by inhalation, once daily, for 5 days. Animals were either sacrificed on day 6 or followed for an additional week. Saline, the carrier for the exosomes, was used as vehicle. The authors reported that even for the highest dose, no adverse effects or differences were observed between control and treated groups in behavior, food and water consumption, body weight, organ weight at the end of the study, nor in hematology, blood chemistry, and urine analyses. Additionally, histological evaluation of organs showed no toxicity concerns. In order to assess the ability of the EXO-CD24 in reducing inflammatory cytokines and lung inflammation, a mouse model of acute respiratory distress syndrome (ARDS) was used. ARDS affected mice were administered either 5 × 108 or 1 × 109 EXO-CD24 once daily for 3 days. After 3 days of treatment, the mice were sacrificed and assessed for histological differences as well as levels of various inflammatory markers. The authors reported that the animals treated with lower dose had moderate-to-severe lung injury while animals with the higher dose showed a marked reduction in lung injury. Additionally, a significant dose dependent reduction in cytokines was observed. Collectively, these findings suggested that EXO-CD24 was a safe and efficacious treatment for COVID-induced lung inflammation.
Following promising observations in animal studies, a Phase Ib/IIa clinical study was performed mainly with an intent to assess safety with an additional focus on pharmacokinetics. 35 patients were enrolled in a four dose escalation groups (1x108, 5x108, 1x109, 1x1010 EXO-CD24/dose). The decrease in serum cytokine arrays and inflammatory markers, in patients that participated in the study, in a time-dependent manner, suggested the efficacy of EXO-CD24. Additionally, an increase in respiratory rate and blood oxygen levels showed the promise of this therapy in treatment of lung injury. Overall, the results from this study showed that EXO-CD24 had a high tolerability and potential efficacy justified further clinical investigations.
4.3. Mannosylated albumin-siRNA NPs
It has been reported that PF involves activation of the innate and adaptive immune pathways that release inflammatory cytokines [70]. During this process a crucial role is played by resident alveolar macrophages are replaced by newly penetrating monocyte-derived macrophages (Mo-AMs) which transition into alveolar macrophages [71]. These newly arriving macrophages have an overexpression of mannose receptor CD206 [72]. Current therapies like Pirfenidone and Nintedanib only dampen immune activation [73]. However, there is no target specific approach to disease inducing macrophage population [74].
Singh and co-workers leveraged these mannose receptors by developing mannosylated albumin nanoparticles (MANPs) that could be internalized by CD206+ macrophages [75]. They utilized these nanoparticles to deliver a TGFβ1 siRNA that could reduce inflammatory cytokine secretion and prevent PF [76]. The MANPs were prepared by coacervation process and coated with D-mannose. Albumin based NPs have been widely reported to possess safety as well as relevance to clinical translation. Mannose coating was enabled by opening the aldehyde groups at low pH and high temperatures to allow reaction of the activated mannose with free amine groups of ANPs. Surface mannose concentration was optimized at 8mM which was the concentration at which the NP surface appeared to be saturated as evidence by quantification studies and 1NMR. Coating of ANPs with mannose resulted in almost two times increase in particle size from 60nm to 100nm. The cellular uptake of the synthesized MANPs in macrophages was assessed by flow cytometry. Additionally, the authors showed that the uptake was a result of CD206 targeting by incubating these cells with CD206 antibody which resulted in reduction in the macrophage uptake.
To assess the application of MANPs as delivery vehicles, MANPs encapsulating TGFβ1 siRNA were synthesized. The entrapment efficiency of siRNA was determined to be about 60% and about 50% release of siRNA was observed in 5 days in PBS pH 7.4 at 37 °C. Fluorescently labeled siRNA was encapsulated and macrophage uptake was assessed using flow cytometry and confocal microscopy. These studies showed that MANP-siRNA had significantly more internalization as compared to free siRNA.
To assess the in vivo efficacy of TGFβ1 siRNA loaded MANPs, the authors utilized a bleomycin-induced PF model which was 15 days long. TGFβ1 siRNA loaded MANPs were administered on day 5 and 10. On day 15, the mice were sacrificed, and histological assessments were performed. Marked reduction was observed in TGFβ1 siRNA loaded MANPs treated mice. Additionally, significant reduction in fibrotic mass as well as collagen deposition was observed. The treated group also showed a 3-fold reduction in macrophage proliferation in the lungs. Further assessment was performed to assess the levels of proinflammatory cytokines TGFβ1 and IL-1β in lungs by ELISA. It was observed that TGFβ1 siRNA loaded MANPs treated mice showed marked reduction in the cytokine levels. Lung function was assessed by measuring multiple mechanical properties of the respiratory system using an instrument called flexiVent. Marked improvements were observed in lung function of animals treated with TGFβ1 siRNA loaded MANPs. Overall, the authors demonstrated the therapeutic efficacy of MANPs to target a specific subset of lung macrophages and mitigate PF.
4.4. Nanostructured hydroxychloroquine
Hydroxychloroquine (HCQ) is an inexpensive drug which has been commonly indicated for the treatment of malaria [77]. Additionally, in recent times HCQ has been investigated for its anti-inflammatory effects in treatment of disorders like rheumatoid arthritis, lupus and inflammatory bowel disease [78]. There have been multiple reports of modifying HCQ to alter its pharmacokinetics so that its therapeutic action can be made more specific [79,80]. Following a similar rationale, Ali and co-workers investigated hydroxychloroquine nanostructured lipid carriers (HCQ-NLCs) as a pulmonary delivery system to treat COVID-induced PF [81]. HCQ-NLC was prepared using a hot emulsification-ultrasonication method. The final nanocarrier comprised of majorly of almond oil and Compritol 888ATO. Additionally, L-phosphatidylcholine was included as an amphiphilic surfactant to promote stability of NLCs. The total lipid content used in the synthesis was about 10% w/v while the surfactant was added at 2% w/v. Targeted HCQ content was 50 mg. Characterization of the obtained NLCs showed a particle size of approximately 250nm with a PDI of 0.3. In vitro release testing showed that 15% of HCQ was released in 30 minutes followed by a cumulative release of 80% over 12 hours. In order to evaluate in vivo efficacy, the authors investigated HCQ-NLCs in a murine bleomycin (BLM) – induced fibrosis model. Mice with fibrosis were divided into different treatment groups and administered with six doses of HCQ-NLC via intrathecal route, HCQ suspension via oral route and Dexamethasone (Dexa) administered via intraperitoneal route. Dexa was included as a positive control as it is used extensively in the management of PF. Post-sacrifice, lung tissue was analyzed for pro-inflammatory cytokine levels and histopathological evaluation. All the three treatment groups showed a reduction in TNFα, IL-6, IL-1β and NFK-β as compared to the untreated mice. Additionally, HCQ-NLCs administered mice reduced pro-inflammatory cytokine levels to base levels as compared to the HCQ treated group. Similar results were observed with histopathological evaluation. HCQ-NLC treated mice showed marked open alveoli free of any inflammatory cell infiltration. Slight improvement was observed for HCQ administered mice. This report lacked the depth in their study design in terms of explaining why the authors chose to evaluate the efficacy of different treatments via different routes of administration. In our opinion, this approach to study design does not provide adequate comparative information to confirm whether the HCQ-NLCs are significantly better than the other treatment groups. Nevertheless, this report highlights a potential nanotherapeutic approach where an existing small molecule drug was packaged into a nanocarrier and its application to PF was assessed.
4.5. PLGA-PEG-G0-C14-IL11 siRNA NPs
Bai and co-workers reported the development of a novel lipid-polymer platform consisting of lipid like compound G0-C14 and PLGA-PEG [82]. The developed NPs encapsulated siRNA against IL11, which is a proinflammatory cytokine [83]. The developed NPs enabled pulmonary delivery of siRNA through bronchiolar and alveolar epithelium on inhalation. The authors reported that these NPs had good biocompatibility as well as could stand up to the shear stress generated during nebulization. In vitro studies were performed to exhibit effective gene silencing. In vivo studies in mice showed that these NPs did not elicit any immune reactions in lung or liver which showed their promise as an efficient inhaled RNA therapy.
Additionally, the authors assessed the therapeutic efficacy of these NPs in a bleomycin-induced PF mouse model which is clinically relevant and has been widely used for PF studies [84]. NP treated mice showed a marked reduction in inflammatory markers. Histologically, a significant reduction in thickness of alveolar septa was observed. Additionally, restoration of impaired alveolar barrier and reduced collagen deposition was observed. The authors indicate that while promising evaluation of this system in other models of PF would greatly help considering only one disease model does not cover all the characteristics of the disease. Nevertheless, the authors believe that this system can be used for simultaneous delivery of multiple siRNAs which could potentially provide synergistic benefits.
5. Conclusions
COVID-19 pandemic has been a major threat to human health. While the medical and pharmaceutical industry has been quick to develop an initial understanding of this disease and developing vaccines, there has not been a lot of focus on the long-term effects of COVID [85]. Partly, this is because the pandemic is about 3 years old so long-term effects are still being monitored [86,87]. However, a significant percentage of COVID affected individuals have shown a certain degree of lung damage [88]. In rare cases, few patients with existing lung conditions have developed PF [89,90,91]. Overall, it remains to be seen whether COVID affected patients exhibit progressive PF in the long term. Although PF is a well characterized disease, COVID induced PF might have a different pathophysiology and consequently its own treatment challenges. The current mainstay of therapies for COVID-induced PF are still drugs that are indicated for PF. However, recently there has been a shift in the field to evaluate these therapies specifically for COVID-induced PF. As a result, there are multiple clinical trials on-going to assess the efficacy of PF drugs in COVID-induced PF [92]. However, the major challenges include safety as well as specificity of these therapeutics. Most of these drugs function by silencing the immune system [93]. Lack of specificity results in non-specific immune silencing which can lead to a lot of concerning side effects. Nanotherapeutic approaches have shown benefits over similar concerns in context of other disease areas [94,95,96]. As a result, there are many reports of evaluations of various nanotherapeutic systems in COVID-induced PF [97,98,99].
In the future, the hope is that more studies would result in therapies with higher margin of safety and specificity towards COVID-induced PF [100,101,102,103]. However, the major challenges to clinical translation would be development of industrial processes to scale up these therapies to produce at scale large enough to supply phase III clinical trials and commercial production [104]. Additionally, processes capable of consistently producing uniform therapeutics will be a challenge. Better understanding of regulatory landscape especially surrounding lung-targeted therapies will be needed. Overall, we are headed in the right direction in terms of starting to understand lung-targeted therapies and patient needs. However, a lot of learning is still needed to allow for successful bedside translation of these nanotherapies [105].
Author Contributions
Conceptualization, S.K., T.K., S.D., and C.J.; writing—original draft preparation, review, and editing, S.K., T.K., S.D., and C.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
NA.
Informed Consent Statement
NA.
Data Availability Statement
NA.
Conflicts of interest: The authors declare no conflict of interest.
References
- P. M. George, C. M. Patterson, A. K. Reed, and M. Thillai, “Lung transplantation for idiopathic pulmonary fibrosis,” Lancet Respir. Med., vol. 7, no. 3, pp. 271–282, 2019. [CrossRef]
- K. C. Meyer, “Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis,” Expert Rev. Respir. Med., vol. 11, no. 5, pp. 343–359, May 2017. [CrossRef]
- T. E. King Jr, A. Pardo, and M. Selman, “Idiopathic pulmonary fibrosis,” Lancet, vol. 378, no. 9807, pp. 1949–1961, Dec. 2011. [CrossRef]
- W. D. Travis et al., “An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias,” Am. J. Respir. Crit. Care Med., vol. 188, no. 6, pp. 733–748, Sep. 2013. [CrossRef]
- M. A. Seibold et al., “A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis,” N. Engl. J. Med., vol. 364, no. 16, pp. 1503–1512, Apr. 2011. [CrossRef]
- C. A. Newton et al., “Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive,” Eur. Respir. J., vol. 48, no. 6, pp. 1710 LP – 1720, Dec. 2016. [CrossRef]
- V Yang, “Epigenomics of idiopathic pulmonary fibrosis,” Epigenomics, vol. 4, no. 2, pp. 195–203, Mar. 2012. [CrossRef]
- Tzouvelekis and N. Kaminski, “Epigenetics in idiopathic pulmonary fibrosis,” Biochem. Cell Biol., vol. 93, no. 2, pp. 159–170, Jan. 2015. [CrossRef]
- Caminati, F. Madotto, G. Cesana, S. Conti, and S. Harari, “Epidemiological studies in idiopathic pulmonary fibrosis: pitfalls in methodologies and data interpretation,” Eur. Respir. Rev., vol. 24, no. 137, pp. 436 LP – 444, Sep. 2015. [CrossRef]
- G. Raghu et al., “Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11,” Lancet Respir. Med., vol. 2, no. 7, pp. 566–572, Jul. 2014. [CrossRef]
- S. E. Tanni et al., “Pulmonary fibrosis secondary to COVID-19: a narrative review,” Expert Rev. Respir. Med., vol. 15, no. 6, pp. 791–803, Jun. 2021. [CrossRef]
- J.-N. Zou et al., “The characteristics and evolution of pulmonary fibrosis in COVID-19 patients as assessed by AI-assisted chest HRCT,” PLoS One, vol. 16, no. 3, p. e0248957, Mar. 2021, [Online]. [CrossRef]
- M. Fisher et al., “Predicting Life Expectancy for Pirfenidone in Idiopathic Pulmonary Fibrosis,” J. Manag. Care Spec. Pharm., vol. 23, no. 3-b Suppl, pp. S17–S24, Mar. 2017. [CrossRef]
- L. Richeldi et al., “Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis,” N. Engl. J. Med., vol. 370, no. 22, pp. 2071–2082, May 2014. [CrossRef]
- T. E. King et al., “A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis,” N. Engl. J. Med., vol. 370, no. 22, pp. 2083–2092, May 2014. [CrossRef]
- J. Fadista et al., “Shared genetic etiology between idiopathic pulmonary fibrosis and COVID-19 severity,” eBioMedicine, vol. 65, Mar. 2021. [CrossRef]
- D. Wendisch et al., “SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis,” Cell, vol. 184, no. 26, pp. 6243-6261.e27, Dec. 2021. [CrossRef]
- W. J. Wiersinga, A. Rhodes, A. C. Cheng, S. J. Peacock, and H. C. Prescott, “Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review,” JAMA, vol. 324, no. 8, pp. 782–793, Aug. 2020. [CrossRef]
- T. Parimon, M. Espindola, A. Marchevsky, R. Rampolla, P. Chen, and C. M. Hogaboam, “Potential mechanisms for lung fibrosis associated with COVID-19 infection,” QJM An Int. J. Med., p. hcac206, Aug. 2022. [CrossRef]
- F. Rendeiro et al., “The spatial landscape of lung pathology during COVID-19 progression,” Nature, vol. 593, no. 7860, pp. 564–569, 2021. [CrossRef]
- Flaifel et al., “Pulmonary Pathology of End-Stage COVID-19 Disease in Explanted Lungs and Outcomes After Lung Transplantation,” Am. J. Clin. Pathol., vol. 157, no. 6, pp. 908–926, Jun. 2022. [CrossRef]
- P. Cheresh, S.-J. Kim, S. Tulasiram, and D. W. Kamp, “Oxidative stress and pulmonary fibrosis,” Biochim. Biophys. Acta - Mol. Basis Dis., vol. 1832, no. 7, pp. 1028–1040, 2013. [CrossRef]
- S. Tran, A. Ksajikian, J. Overbey, P. Li, and Y. Li, “Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review,” Cells, vol. 11, no. 16. 2022. [CrossRef]
- K. Sehlmeyer, J. Ruwisch, N. Roldan, and E. Lopez-Rodriguez, “Corrigendum: Alveolar Dynamics and Beyond – the Importance of Surfactant Protein C and Cholesterol in Lung Homeostasis and Fibrosis ,” Frontiers in Physiology , vol. 11. 2020, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fphys.2020.00943. [CrossRef]
- M. A. Seibold et al., “The Idiopathic Pulmonary Fibrosis Honeycomb Cyst Contains A Mucocilary Pseudostratified Epithelium,” PLoS One, vol. 8, no. 3, p. e58658, Mar. 2013, [Online]. [CrossRef]
- F. Conforti et al., “Paracrine SPARC signaling dysregulates alveolar epithelial barrier integrity and function in lung fibrosis,” Cell Death Discov., vol. 6, no. 1, p. 54, 2020. [CrossRef]
- M. J. Evans, L. S. Van Winkle, M. V Fanucchi, and C. G. Plopper, “The Attenuated Fibroblast Sheath of the Respiratory Tract Epithelial–Mesenchymal Trophic Unit,” Am. J. Respir. Cell Mol. Biol., vol. 21, no. 6, pp. 655–657, Dec. 1999. [CrossRef]
- Ghazavi, A. Ganji, N. Keshavarzian, S. Rabiemajd, and G. Mosayebi, “Cytokine profile and disease severity in patients with COVID-19,” Cytokine, vol. 137, p. 155323, 2021. [CrossRef]
- Colarusso et al., “Post-COVID-19 Patients Who Develop Lung Fibrotic-like Changes Have Lower Circulating Levels of IFN-β but Higher Levels of IL-1α and TGF-β,” Biomedicines, vol. 9, no. 12. 2021. [CrossRef]
- J. C. Melms et al., “A molecular single-cell lung atlas of lethal COVID-19,” Nature, vol. 595, no. 7865, pp. 114–119, 2021. [CrossRef]
- Yao et al., “Cell-Type-Specific Immune Dysregulation in Severely Ill COVID-19 Patients,” Cell Rep., vol. 34, no. 1, Jan. 2021. [CrossRef]
- Bharat et al., “Lung transplantation for patients with severe COVID-19,” Sci. Transl. Med., vol. 12, no. 574, p. eabe4282, Dec. 2020. [CrossRef]
- S. W. Aesif et al., “Pulmonary Pathology of COVID-19 Following 8 Weeks to 4 Months of Severe Disease: A Report of Three Cases, Including One With Bilateral Lung Transplantation,” Am. J. Clin. Pathol., vol. 155, no. 4, pp. 506–514, Apr. 2021. [CrossRef]
- J. C. T. Horowitz Victor J, “Epithelial-Mesenchymal Interactions in Pulmonary Fibrosis,” Semin Respir Crit Care Med, vol. 27, no. 06, pp. 600–612, 2006. [CrossRef]
- T. M. Maher, A. U. Wells, and G. J. Laurent, “Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms?,” Eur. Respir. J., vol. 30, no. 5, pp. 835 LP – 839, Nov. 2007. [CrossRef]
- D. Uhal, I. Joshi, W. F. Hughes, C. Ramos, A. Pardo, and M. Selman, “Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung,” Am. J. Physiol. Cell. Mol. Physiol., vol. 275, no. 6, pp. L1192–L1199, Dec. 1998. [CrossRef]
- L. Yao et al., “Paracrine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblast differentiation in lung fibrosis,” Cell Death Differ., vol. 26, no. 5, pp. 943–957, 2019. [CrossRef]
- P. Jiang et al., “Ineffectual Type 2–to–Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8hi Transitional State,” Am. J. Respir. Crit. Care Med., vol. 201, no. 11, pp. 1443–1447, Feb. 2020. [CrossRef]
- M. Cassandras et al., “Gli1+ mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung,” Nat. Cell Biol., vol. 22, no. 11, pp. 1295–1306, 2020. [CrossRef]
- Prasse et al., “BAL Cell Gene Expression Is Indicative of Outcome and Airway Basal Cell Involvement in Idiopathic Pulmonary Fibrosis,” Am. J. Respir. Crit. Care Med., vol. 199, no. 5, pp. 622–630, Aug. 2018. [CrossRef]
- T. S. Blackwell et al., “Future Directions in Idiopathic Pulmonary Fibrosis Research. An NHLBI Workshop Report,” Am. J. Respir. Crit. Care Med., vol. 189, no. 2, pp. 214–222, Oct. 2013. [CrossRef]
- K. Koli et al., “Bone Morphogenetic Protein-4 Inhibitor Gremlin Is Overexpressed in Idiopathic Pulmonary Fibrosis,” Am. J. Pathol., vol. 169, no. 1, pp. 61–71, Jul. 2006. [CrossRef]
- J. K. Keski-Oja Katri; Lohi, Jouko; Laiho, Marikki, “Growth Factors in the Regulation of Plasminogen-Plasmin System in Tumor Cells,” Semin Thromb Hemost, vol. 17, no. 03, pp. 231–239, 1991. [CrossRef]
- R. Raghow, A. E. Postlethwaite, J. Keski-Oja, H. L. Moses, and A. H. Kang, “Transforming growth factor-beta increases steady state levels of type I procollagen and fibronectin messenger RNAs posttranscriptionally in cultured human dermal fibroblasts.,” J. Clin. Invest., vol. 79, no. 4, pp. 1285–1288, Apr. 1987. [CrossRef]
- J. M. Shannon and B. A. Hyatt, “Epithelial-Mesenchymal Interactions in the Developing Lung,” Annu. Rev. Physiol., vol. 66, no. 1, pp. 625–645, Feb. 2004. [CrossRef]
- K. C. Meyer, “Pulmonary fibrosis, part II: state-of-the-art patient management,” Expert Rev. Respir. Med., vol. 11, no. 5, pp. 361–376, May 2017. [CrossRef]
- M. Kreuter, “Pirfenidone: an update on clinical trial data and insights from everyday practice,” Eur. Respir. Rev., vol. 23, no. 131, pp. 111 LP – 117, Mar. 2014. [CrossRef]
- H. Taniguchi et al., “Pirfenidone in idiopathic pulmonary fibrosis,” Eur. Respir. J., vol. 35, no. 4, pp. 821 LP – 829, Apr. 2010. [CrossRef]
- P. W. Noble et al., “Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials,” Lancet, vol. 377, no. 9779, pp. 1760–1769, May 2011. [CrossRef]
- P. Spagnolo, T. M. Maher, and L. Richeldi, “Idiopathic pulmonary fibrosis: Recent advances on pharmacological therapy,” Pharmacol. Ther., vol. 152, pp. 18–27, 2015. [CrossRef]
- S. King and S. D. Nathan, “Practical considerations in the pharmacologic treatment of idiopathic pulmonary fibrosis,” Curr. Opin. Pulm. Med., vol. 21, no. 5, 2015, [Online]. Available: https://journals.lww.com/co-pulmonarymedicine/Fulltext/2015/09000/Practical_considerations_in_the_pharmacologic.10.aspx. [CrossRef]
- Wani et al., “Surface PEGylation of Mesoporous Silica Nanorods (MSNR): Effect on loading, release, and delivery of mitoxantrone in hypoxic cancer cells,” Sci. Rep., vol. 7, no. 1, p. 2274, 2017. [CrossRef]
- R. Roggers, S. Kanvinde, S. Boonsith, and D. Oupický, “The practicality of mesoporous silica nanoparticles as drug delivery devices and progress toward this goal,” AAPS PharmSciTech, vol. 15, no. 5, pp. 1163–1171, Oct. 2014. [CrossRef]
- Y. Zhu et al., “Self-immolative polycations as gene delivery vectors and prodrugs targeting polyamine metabolism in cancer,” Mol. Pharm., vol. 12, no. 2, pp. 332–341, Feb. 2015. [CrossRef]
- S. Deodhar et al., “Transformation of dolutegravir into an ultra-long-acting parenteral prodrug formulation,” Nat. Commun., vol. 13, no. 1, p. 3226, 2022. [CrossRef]
- M. Moss and M. Siccardi, “Optimizing nanomedicine pharmacokinetics using physiologically based pharmacokinetics modelling,” Br. J. Pharmacol., vol. 171, no. 17, pp. 3963–3979, Sep. 2014. [CrossRef]
- N. Gautam et al., “Lipophilic nanocrystal prodrug-release defines the extended pharmacokinetic profiles of a year-long cabotegravir,” Nat. Commun., vol. 12, no. 1, p. 3453, 2021. [CrossRef]
- T. A. Kulkarni et al., “A year-long extended release nanoformulated cabotegravir prodrug,” Nat. Mater., vol. 19, no. 8, pp. 910–920, 2020. [CrossRef]
- S. Beyerstedt, E. B. Casaro, and É. B. Rangel, “COVID-19: angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection,” Eur. J. Clin. Microbiol. Infect. Dis., vol. 40, no. 5, pp. 905–919, 2021. [CrossRef]
- W. Ni et al., “Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19,” Crit. Care, vol. 24, no. 1, p. 422, 2020. [CrossRef]
- J. Yang et al., “Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor,” Nat. Commun., vol. 11, no. 1, p. 4541, 2020. [CrossRef]
- Z. Li et al., “Cell-mimicking nanodecoys neutralize SARS-CoV-2 and mitigate lung injury in a non-human primate model of COVID-19,” Nat. Nanotechnol., vol. 16, no. 8, pp. 942–951, 2021. [CrossRef]
- S. Shapira et al., “A novel platform for attenuating immune hyperactivity using EXO-CD24 in COVID-19 and beyond,” EMBO Mol. Med., vol. 14, no. 9, p. e15997, Sep. 2022. [CrossRef]
- X. Fang, P. Zheng, J. Tang, and Y. Liu, “CD24: from A to Z,” Cell. Mol. Immunol., vol. 7, no. 2, pp. 100–103, 2010. [CrossRef]
- S. Gurung, D. Perocheau, L. Touramanidou, and J. Baruteau, “The exosome journey: from biogenesis to uptake and intracellular signalling,” Cell Commun. Signal., vol. 19, no. 1, p. 47, 2021. [CrossRef]
- H. Chen et al., “Exosomes, a New Star for Targeted Delivery ,” Frontiers in Cell and Developmental Biology , vol. 9. 2021, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fcell.2021.751079. [CrossRef]
- K. Herrmann, M. J. A. Wood, and G. Fuhrmann, “Extracellular vesicles as a next-generation drug delivery platform,” Nat. Nanotechnol., vol. 16, no. 7, pp. 748–759, 2021. [CrossRef]
- P.-U. C. Dinh et al., “Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis,” Nat. Commun., vol. 11, no. 1, p. 1064, 2020. [CrossRef]
- S. Shapira, D. Kazanov, S. Weisblatt, A. Starr, N. Arber, and S. Kraus, “The CD24 Protein Inducible Expression System Is an Ideal Tool to Explore the Potential of CD24 as an Oncogene and a Target for Immunotherapy in vitro and in vivo,” J. Biol. Chem., vol. 286, no. 47, pp. 40548–40555, Nov. 2011. [CrossRef]
- Wick, A. Backovic, E. Rabensteiner, N. Plank, C. Schwentner, and R. Sgonc, “The immunology of fibrosis: innate and adaptive responses,” Trends Immunol., vol. 31, no. 3, pp. 110–119, Mar. 2010. [CrossRef]
- L. Zhang, Y. Wang, G. Wu, W. Xiong, W. Gu, and C.-Y. Wang, “Macrophages: friend or foe in idiopathic pulmonary fibrosis?,” Respir. Res., vol. 19, no. 1, p. 170, 2018. [CrossRef]
- K. Tsuchiya et al., “Macrophage Mannose Receptor CD206 Predicts Prognosis in Community-acquired Pneumonia,” Sci. Rep., vol. 9, no. 1, p. 18750, 2019. [CrossRef]
- K. M. Roach et al., “Evaluation of Pirfenidone and Nintedanib in a Human Lung Model of Fibrogenesis,” Frontiers in Pharmacology, vol. 12. 2021, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fphar.2021.679388. [CrossRef]
- J. A. Ardura, G. Rackov, E. Izquierdo, V. Alonso, A. R. Gortazar, and M. M. Escribese, “Targeting Macrophages: Friends or Foes in Disease? ,” Frontiers in Pharmacology , vol. 10. 2019, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fphar.2019.01255. [CrossRef]
- Singh et al., “Nanoparticle targeting of de novo profibrotic macrophages mitigates lung fibrosis,” Proc. Natl. Acad. Sci., vol. 119, no. 15, p. e2121098119, Apr. 2022. [CrossRef]
- M. Patel et al., “The Immunopathobiology of SARS-CoV-2 Infection,” FEMS Microbiol. Rev., vol. 45, no. 6, p. fuab035, Nov. 2021. [CrossRef]
- R. I. Fox, “Mechanism of action of hydroxychloroquine as an antirheumatic drug,” Semin. Arthritis Rheum., vol. 23, no. 2, Supplement 1, pp. 82–91, 1993. [CrossRef]
- Y. S. Chhonker, S. Kanvinde, R. Ahmad, A. B. Singh, D. Oupický, and D. J. Murry, “Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS,” Metabolites , vol. 11, no. 2. 2021. [CrossRef]
- S. S. Kesharwani et al., “Site-directed non-covalent polymer-drug complexes for inflammatory bowel disease (IBD): Formulation development, characterization and pharmacological evaluation,” J. Control. Release, vol. 290, pp. 165–179, 2018. [CrossRef]
- S. Kanvinde et al., “Pharmacokinetics and efficacy of orally administered polymeric chloroquine as macromolecular drug in the treatment of inflammatory bowel disease,” Acta Biomater., vol. 82, pp. 158–170, Dec. 2018. [CrossRef]
- S. Ali, M. G. Alrashedi, O. A. Ahmed, and I. M. Ibrahim, “Pulmonary Delivery of Hydroxychloroquine Nanostructured Lipid Carrier as a Potential Treatment of COVID-19,” Polymers, vol. 14, no. 13. 2022. [CrossRef]
- X. Bai et al., “Inhaled siRNA nanoparticles targeting IL11 inhibit lung fibrosis and improve pulmonary function post-bleomycin challenge,” Sci. Adv., vol. 8, no. 25, p. eabn7162, Feb. 2023. [CrossRef]
- K. Y. Fung et al., “Emerging roles for IL-11 in inflammatory diseases,” Cytokine, vol. 149, p. 155750, 2022. [CrossRef]
- D. M. Walters and S. R. Kleeberger, “Mouse Models of Bleomycin-Induced Pulmonary Fibrosis,” Curr. Protoc. Pharmacol., vol. 40, no. 1, pp. 5.46.1-5.46.17, Mar. 2008. [CrossRef]
- V Raveendran, R. Jayadevan, and S. Sashidharan, “Long COVID: An overview,” Diabetes Metab. Syndr. Clin. Res. Rev., vol. 15, no. 3, pp. 869–875, 2021. [CrossRef]
- S. Lopez-Leon et al., “More than 50 long-term effects of COVID-19: a systematic review and meta-analysis,” Sci. Rep., vol. 11, no. 1, p. 16144, 2021. [CrossRef]
- R. L. Levine, “Addressing the Long-term Effects of COVID-19,” JAMA, vol. 328, no. 9, pp. 823–824, Sep. 2022. [CrossRef]
- M. Suran, “Autopsies Reveal Lung Damage Patterns From COVID-19,” JAMA, vol. 326, no. 24, p. 2463, Dec. 2021. [CrossRef]
- F. McGroder et al., “Pulmonary fibrosis 4 months after COVID-19 is associated with severity of illness and blood leucocyte telomere length,” Thorax, vol. 76, no. 12, pp. 1242 LP – 1245, Dec. 2021. [CrossRef]
- E. Bazdyrev, P. Rusina, M. Panova, F. Novikov, I. Grishagin, and V. Nebolsin, “Lung Fibrosis after COVID-19: Treatment Prospects,” Pharmaceuticals, vol. 14, no. 8. 2021. [CrossRef]
- J. Hama Amin et al., “Post COVID-19 pulmonary fibrosis; a meta-analysis study,” Ann. Med. Surg., vol. 77, p. 103590, 2022. [CrossRef]
- Mohammadi et al., “Post-COVID-19 Pulmonary Fibrosis,” Cureus, vol. 14, no. 3, p. e22770, 2022. [CrossRef]
- T. A. Wynn, “Integrating mechanisms of pulmonary fibrosis,” J. Exp. Med., vol. 208, no. 7, pp. 1339–1350, Jul. 2011. [CrossRef]
- S. Kanvinde, T. Kulkarni, S. Deodhar, D. Bhattacharya, and A. Dasgupta, “Non-Viral Vectors for Delivery of Nucleic Acid Therapies for Cancer,” BioTech, vol. 11, no. 1. 2022. [CrossRef]
- S. Soares, J. Sousa, A. Pais, and C. Vitorino, “Nanomedicine: Principles, Properties, and Regulatory Issues ,” Frontiers in Chemistry , vol. 6. 2018, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fchem.2018.00360. [CrossRef]
- J. Ren et al., “Precision Nanomedicine Development Based on Specific Opsonization of Human Cancer Patient-Personalized Protein Coronas,” Nano Lett., vol. 19, no. 7, pp. 4692–4701, Jul. 2019. [CrossRef]
- C. Velino et al., “Nanomedicine Approaches for the Pulmonary Treatment of Cystic Fibrosis ,” Frontiers in Bioengineering and Biotechnology , vol. 7. 2019, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fbioe.2019.00406. [CrossRef]
- M. Skibba, A. Drelich, M. Poellmann, S. Hong, and A. R. Brasier, “Nanoapproaches to Modifying Epigenetics of Epithelial Mesenchymal Transition for Treatment of Pulmonary Fibrosis ,” Frontiers in Pharmacology , vol. 11. 2020, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fphar.2020.607689. [CrossRef]
- C.-Y. Loo and W.-H. Lee, “Nanotechnology-based therapeutics for targeting inflammatory lung diseases,” Nanomedicine, vol. 17, no. 12, pp. 865–879, Mar. 2022. [CrossRef]
- E. S. White, M. Thomas, S. Stowasser, and K. Tetzlaff, “Challenges for Clinical Drug Development in Pulmonary Fibrosis ,” Frontiers in Pharmacology , vol. 13. 2022, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fphar.2022.823085. [CrossRef]
- R. J. Kaner et al., “Design of Idiopathic Pulmonary Fibrosis Clinical Trials in the Era of Approved Therapies,” Am. J. Respir. Crit. Care Med., vol. 200, no. 2, pp. 133–139, Apr. 2019. [CrossRef]
- S. Harari and A. Caminati, “Idiopathic pulmonary fibrosis: from clinical trials to real-life experiences,” Eur. Respir. Rev., vol. 24, no. 137, pp. 420 LP – 427, Sep. 2015. 2015. [CrossRef]
- L. Lancaster et al., “Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials,” BMJ Open Respir. Res., vol. 6, no. 1, p. e000397, Mar. 2019. [CrossRef]
- S. Hua, M. B. C. de Matos, J. M. Metselaar, and G. Storm, “Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization ,” Frontiers in Pharmacology , vol. 9. 2018, [Online]. Available: https://www.frontiersin.org/articles/10.3389/fphar.2018.00790. [CrossRef]
- S. Đorđević et al., “Current hurdles to the translation of nanomedicines from bench to the clinic,” Drug Deliv. Transl. Res., vol. 12, no. 3, pp. 500–525, 2022. [CrossRef]

Table 1.
On-going clinical trials for COVID-induced PF therapy.
| Clinical Trial ID | Title | Intervention | Sponsor |
|---|---|---|---|
| NCT04607928 | Phase-II Randomized Clinical Trial to Evaluate the Effect of Pirfenidone Compared to Placebo in Post-COVID19 | Drug: Pirfenidone Drug: Placebo |
Institut d'Investigació Biomèdica de Bellvitge |
| NCT04818489 | Impact of Colchicine on the Clinical Outcome of COVID-19 and the Development of Post-COVID-19 Pulmonary Fibrosis: Randomized Controlled Clinical Trial | Drug: Colchicine 0.5 mg Other: the standard protocol only |
ClinAmygate |
| NCT04551781 | Short Term Low Dose Corticosteroids for Management of Post Covid-19 Pulmonary Fibrosis | Drug: 20 mg Prednisone for 14 days Drug: control |
South Valley University |
| NCT05648734 | Impact of Anti-Inflammatory and Anti-Fibrotic Drugs on Post-acute COVID-19 Pulmonary Fibrosis | Drug: Corticosteroids alone Drug: Corticosteroids + Colchicine Drug: Corticosteroids + Pirfenidone Drug: Corticosteroids + Colchicine + Pirfenidone for ≥ 14 day |
Mansoura University |
| NCT04279197 | Efficacy and Safety of Fuzheng Huayu Tablets in Post-COVID-19 Patients with Pulmonary Inflammation and Fibrosis: A Multicenter Double-blind Randomized Controlled Trial | Drug: Fuzheng Huayu Tablet Drug: Vitamin C tablets Drug: Placebo Other: respiratory function rehabilitation training |
ShuGuang Hospital |
| NCT04541680 | Nintedanib for the Treatment of SARS-Cov-2 Induced Pulmonary Fibrosis" | Drug: Nintedanib 150 mg Other: Placebo |
Assistance Publique - Hôpitaux de Paris |
| NCT04856111 | A Study of the Efficacy and Safety of Pirfenidone vs. Nintedanib in the Treatment of Fibrotic Lung Disease After Coronavirus Disease-19 |
Drug: Pirfenidone Drug: Nintedanib |
Postgraduate Institute of Medical Education and Research |
| NCT04948203 | SECOVID: A Multi-center, Randomized, Dose-ranging Parallel-group Trial Assessing the Efficacy of Sirolimus in Hospitalized Patients With COVID-19 Pneumonia for the Prevention of Post-COVID Fibrosis |
Drug: Sirolimus | University of Chicago |
| NCT05387239 | Safety and Effectiveness of EV-Pure + WJ-Pure Treatment on Pulmonary Fibrosis Secondary to Covid-19 |
Drug: EV-Pure™ and WJ-Pure™ plus standard care Drug: Placebo (Saline plus standard care) |
Vitti Labs, LLC |
| NCT04645368 | Multicenter, Open-label Prospective Cohort Study of the Efficacy and Safety of the Inclusion of Longidaze in the Prevention and Treatment of Post-inflammatory Pulmonary Fibrosis and Interstitial Lung Diseases Caused by COVID-19 | Drug: bovhyaluronidase azoxymer | NPO Petrovax |
| NCT04805086 | Phase I/II MONACO Cell Therapy Study: Monocytes as an Anti-fibrotic Treatment After COVID-19 | Biological: MON002 | Guy's and St Thomas' NHS Foundation Trust |
| NCT04912011 | The Use of a Mineralocorticoid Receptor Antagonist (Spironolactone) in the Treatment of Pulmonary Fibrosis Associated With SARS-CoV-2 Infection | Drug: Canrenoate Potassium Drug: Normal Saline |
Pomeranian Medical University Szczecin |
| NCT04338802 | Efficacy and Safety of Nintedanib Ethanesulfonate Soft Capsule in the Treatment of Pulmonary Fibrosis in Patients with Moderate to Severe COVID-9(COVID 19) : a Single-center, Randomized, Placebo-controlled Study | Drug: Nintedanib 150 mg Other: Placebo |
Tongji Hospital |
| NCT04619680 | Early Nintedanib Deployment in COVID-19 Interstitial Lung Disease | Drug: Nintedanib Drug: Placebo |
Icahn School of Medicine at Mount Sinai |
| NCT04482595 | A Phase 2 Study of BIO 300 Oral Suspension in Discharged COVID-19 Patients | Drug: BIO 300 Oral Suspension Drug: Placebo |
Humanetics Corporation |
| NCT04537130 | Phase Ib Controlled Exploratory Trial for Treatment of Fibrosing Interstitial Lung Disease Patients Secondary to SARS-CoV-2 Infection with IN01 Vaccine (COVINVAC) | Biological: IN01 vaccine | Instituto Oncológico |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



