
Submitted:
28 March 2023
Posted:
30 March 2023
You are already at the latest version
Abstract
It is important to add second- or third-phase materials to improve the physical, mechanical, and biodegradable properties of polypropylene (PP) nanocomposites. A unique hybrid bio-nanocomposite was fabricated using microparticle cellulose powder from wastepaper with a polypropylene (PP) polymeric matrix using a low-shear chaotic mixing method. A small amount (2-5%) of layered micro clay was also added to investigate the hybrid effect of nanocomposite fabrication on the mechanical properties. A low-shear chaotic melt mixing method induces the formation of nano cellulose structures and layered clay nanoparticles by via exfoliation. The nanoparticles were characterized using Fourier transform infrared (FTIR) spectroscopy. Mechanical properties such as the flexural strength (FS) and tensile strength (TS) were investigated using an Izod impact tester (IZ), and a dynamic mechanical testing analyzer (DMTA). Further, scanning electron microscopy (SEM) was used to observe the microstructure of the nanocomposite. In addition, differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were conducted to investigate the crystallinity and degradation of the samples, respectively. The addition of wastepaper cellulose powder below 2.5 wt. % did not improve the mechanical properties, however, improvements were observed when more than 5wt.% wastepaper cellulose powder was added. With the addition of a small amount of nano-clay, the mechanical properties were significantly improved, and a synergistic effect could be noticed. The observed results were then compared with PP-clay nanocomposites used in automotive industries to replace cellulose nanocomposites. The application areas can also cover lead production in paper cups and vacuum foaming.
Keywords:
nano-cellulose
; nanocomposites
; mechanical properties
; biopolymers
Introduction
Bio nanocomposites are cutting-edge materials with many benefits having potential in biomedical applications like bone fillers, antimicrobial agents, drug delivery and tissue engineering. To date, the primary challenge for scientists to solve expanding environmental consciousness, is the development of bio-based nanomaterials and nanocomposites that contribute to a cleaner and more sustainable environment [1,2,3,4,5]. Currently, cellulose fibers derived from natural sources have received significant attention as potential alternatives to the large-scale production of biodegradable plastics. Additionally, cellulose fibers or powders can be used to enhance the properties of plastics by incorporating them as a secondary phase. Cellulosic raw materials offer different options in the production of biodegradable polymers, such as sawdust, rice husks, natural cotton, sisal fibers, and wood cellulose. [6,7].
Many scientists have researched the structures and morphology of cellulose composites and discovered that cellulose is a long-chain polymer made by the linkage of smaller polymers. The link in the cellulose chain consists of poly-(1,4)-D-glucose [8], and the glucose unit residue has the potential to link through condensation to form a polydisperse linear polymer combining the -H and -OH groups [9]. Typically, cellulose chains have a high potential for polymerization owing to the presence of numerous hydroxyl groups. However, it can be challenging to measure the chain length by analyzing the mechanical and degradation properties because of molecular weight distribution and variety of degradation mechanisms. Additionally, the presence of a hydrophilic group such as a hydroxyl functional group renders cellulose compounds more hydrophilic and limits their ability to bond with the polymeric matrix. Using cellulose particulates as a secondary component with non-biodegradable polymeric materials can result in a biodegradable composite material [10,11,12,13].
At optimal processing conditions, cellulose materials derived from agriculture have been widely reported by many researchers to be effective replacements for inorganic/mineral-based fillers, such as Talc, TiO2, CaCO3, MgO, and Mg2(OH)3, in thermoplastics [14,15,16]. Incorporating long natural fibers, such as jute, as reinforcements in thermoplastics has environmental advantages, improved mechanical properties, lower weight, and higher cost effectiveness. Recycling cellulose-filled thermoplastic composites is also simpler and more cost-efficient than recycling composites reinforced with inorganic fillers [17,18]. However, cellulose materials have limitations in mass production and industrial use because of the poor bonding between the cellulose fiber and the polymeric matrix. To overcome this, the use of surface treatments and compatibilizers is crucial. [19,20,21,22,23,24].
Because polymeric materials are widely used in packaging, textile and in medical devices, incorporating cellulose materials as a secondary phase to improve biodegradability can be a practical and effective solution. Using recycled cellulose from paper waste contributes to creating a cleaner and more sustainable environment. Developing multi-layered silicates modified with organic molecules and polymeric materials is a new approach to creating high-performance nano polymers. [25,26,27,28,29]. Layered silicate is commonly blended with various polymers, primarily polyethylene and polypropylene nylon, using traditional melt-mixing extruder machines. Researchers have suggested ways to increase the interlayer distance of clay minerals containing silicate using treated organic molecules to make it easier for monomers to penetrate the interlayer space within the silicate clay minerals [30].
There are three common methods for creating polymer-clay nanocomposites: melt compounding, solution processing, and in situ intercalation polymerization. In the typical melt compounding technique, polymer pellets and clay are extruded to produce polymer-clay nanocomposites via high-shear mixing. Owing to the high shear and brief mixing time, the clay particles are not detached from their bonded layered structure, resulting in intercalated clay composites which typically exhibit poor mechanical properties. A low-shear mix melting method is required to achieve a high level of nano-clay dispersal into the polymeric matrix and exfoliate the clay layers [31,32]. The main goal of this study was to fabricate hybrid biodegradable polymeric nanocomposites using the low-shear chaotic melt compounding method and microcellulose recycled powder from used paper with the addition of a small amount of nano-clay for a synergistic effect. Thus, the use of recycled paper can also reduce the threat of global pollution. The mechanical properties of the nanocomposites were investigated, evaluated, and discussed. The chemical structure of the cellulose fiber is shown in Figure 1. A few hydroxyl functional groups are observed in the cellulose structure, which are responsible for polymeric bonding.
2. Experimental Section
2.1. Materials
The cellulose powder was collected from Eco Research Institute (ERI), Japan. Micron-sized paper powder was fabricated using the super cryogenic technique, as mentioned by ERI. Polypropylene BX-3900 was purchased from SK Chemical, Korea, and nano-clay 93A was obtained from Shinil Chemical Korea. The toughening agent EG 8407 was purchased from Dow chemicals. Other supporting materials such Ethanox and EV were used as antioxidants and thermal stabilizers, respectively, and used as is.
2.2. Composite fabrication
Cellulose nanocomposites were fabricated by the polymer melt chaotic mixing method using a twin extruder. All raw materials with defined weight percentages were premixed and added to the extruder hopper. Table 1 shows the composite component ratios and their respective nomenclature. Ethanox and EV were used as antioxidants and thermal stabilizers, respectively. The extruder temperature was kept at 220°C–240°C, which allowed the polymer matrix, cellulose, and clay to be homogeneous. The strands were cooled in water and cut using a pelletizer. A 5–10 mm composite pellet was dried at 80°C for 4 h and injected into an injection machine for the test specimen.
2.2. Characterization
INSTRON USA was used to measure the flexural strength, tensile strength, and modulus. Thermogravimetric analysis (TGA) was performed on a thermal analysis (TA) instrument. The samples were heated to 120°C and maintained isothermally for 1 h to normalize the moisture content. After air-cooling, thermal scans were performed from 40°C to 600°C at a heating rate of 10°C/min. Differential scanning calorimetry (DSC) was performed using a TA instrument (DSC Q2000). The dynamical mechanical properties of the composites were measured by DMTA FTIR and were performed using aby TA FTIR analyzer. The sample morphologies were studied using scanning electron microscopy (SEM; JEOL JSM-6330F, Japan). The impact strength of the composites was measured using an Izod impact tester (IZ). For DSC, DMTA and FT-IRm 10-15 gm composite pellets were used for analysis. SEM samples were prepared from injection-molded parts, and the upper surfaces were polished.
3. Results and discussion
The FTIR spectra of the nanocomposites are shown in Figure 2. The spectra of the modified composites showed distinct peaks at 2917, 1690, 1450, and 1375 cm-1. The characteristic peak of the OH group in cellulose and the presence of water molecules can be easily detected by the presence of broad peaks between 3325-3340 cm-1 [33]. OH, stretching in the region of 2900-3000 cm-1 has also been confirmed by various researchers [34]. In the SP-1 composite, no distinct peak was observed at 3400-3500 cm-1 for the pure PP virgin polymer. However, a small peak was observed for all other nanocomposites. Although the peaks were very small, it indicates the presence of a small amount of OH or water molecules. The presence of a small peak might be due to the minute amount of water molecules present in the cellulose powder as they were fully dried. The distinct peaks at 2917 cm-1 can be attributed to the C-H symmetrical stretching of the PP polymeric material. The presence of a small peak at 1640 cm-1 in SP-2-5 confirms the presence of the OH group in the cellulose backbone, whereas no such peak was observed in the PP matrix. The wide peak at 1000-1050 cm-1 in SP-3 composites confirms the presence of inorganic clay; however, it decreased in SP-4 and SP-5 as the amount of clay was reduced from 5.5% to 2%. No characteristic hydrogen bonding peaks were observed in the FTIR analysis.
Figure 3 (a, b) show images of rod-like cellulose particles with primary diameters of 20 µm. These SEM images also confirmed the powdered nature of the cellulose used in the experiment. Figure 3 c shows the microstructure of the composites; fractures were observed on the surface of the cellulose nanocomposites, and the white region indicates the presence of cellulose nanoparticles.
Figure 4 shows the typical flexural stress-strain curves of the PP polymer matrix and its composites with cellulose and cellulose inorganic clay hybrid. The results indicate that the flexural stress of the virgin PP was higher than that of the cellulose-added composites. However, the stress increased further when a small amount of inorganic clay was added. Maximum stress was observed in SP-5 composites when 9% of cellulose was added, demonstrating the synergetic effect of cellulose and the inorganic clay. The lower flexural strength of the 5.5% cellulose sample could be attributed to the inhomogeneous dispersion of cellulose into the polymeric matrix.
Moreover, hydrogen or chemical bonding did not occur between the cellulose and the PP matrix, which was proven by FTIR and SEM analyses. This was also consistent with the tensile strength measurements. Figure 5 shows the tensile stress-strain curve of the composites. The SP-1 sample of virgin PP composites had a higher tensile strength than the 5.5% cellulose dispersed composites. The cellulose-inorganic clay composite SP-5 sample showed the highest tensile strength and tensile modulus. Table 2 lists the average mechanical properties after tensile testing.
Figure 6 and Figure 7 show the storage moduli and Tan delta temperatures of all the samples. It was observed that the storage modulus of the PP was significantly lower than that of the cellulose and clay-dispersed composites. When 2.5% of cellulose was added, the modulus increased. Whereas when 2.5% of inorganic clay was added, the modulus increased significantly.
However, the addition of both cellulose and inorganic clay did not lead to any significant improvements. The improvement in the storage modulus by the addition of inorganic clay in sample SP-3 can be explained by the fact that inorganic clay is a rigid material. When dispersed into polymeric materials via intercalation or exfoliation, the rigidity of the polymeric matrix increases, resulting in a higher storage modulus. As the temperature increases, the clay has little influence in retaining the stiffness of PP after −60°C. D’ Angelo et al. [35] observed a similar phenomenon when he added Talc nano powder into the PP matrix.
The viscoelastic behavior of the polymeric materials was obtained by measuring the tan delta. The temperature dependence of the dynamic mechanical properties is shown in Figure 7. The spectra of the measured samples showed three relaxation peaks at −85°C, −45°C, and 50–75°C. The relaxation peak at −85°C corresponds to the motions of small chain groups [36]. The virgin PP peak was stronger and more predominant than those of the other composites, followed by the cellulose-dispersed composites. The SP-3 composites had the highest tan delta value of the composites near −85°C, followed by SP-2, SP-5 SP-4, and SP-1. A similar trend was also observed in the storage modulus measurements. In addition, the gamma peak was also found to be shifted at a higher temperature for filled composites, indicating reduced polymeric chain mobility with the addition of cellulose and inorganic clay.
The beta relation peak followed a similar trend as the gamma relation peak. A small shift at higher temperatures is evident from the graph. However, a larger shift at higher temperatures was observed in the alpha peaks. PP composites in SP-1 showed an alpha relation peak at 50°C, which shifted to 70°C, 75°C, and 100°C for SP-5, SP-3, and SP-2 composites, respectively. The tan delta peak of SP-5, SP-3, and SP-2 composites was also much higher than that of the SP-1 composites. This phenomenon indicates that fewer polymer chains participated in this transition. The tan delta peak at lower temperatures (<-50°C) corresponds to the presence of 20% rubbery polymer in the composition. The delta peak in this region is believed to be the glass transition temperature (Tg) of the rubbery polymer. The increase in storage modulus and shift in delta peak position suggests the physical interaction between the polymer and reinforcing agents limited the segmental polymer chain mobility in the vicinity of nano-reinforcements [37]. Similar results were observed by Okasan [38], who pointed out that the results from the viscoelastic behavior of the composites were consistent with the mechanical properties, suggesting that the reinforcing effect of nanofibers in the polymer restricted the molecular chain mobility. The highest restriction was observed when only cellulose was added to the PP polymer in the SP-2 composites. The delta 2 peak, which is the Tg of the PP polymer, was observed at approximately 80–90°C. The dynamic mechanical properties of the composites are shown in Table 3.
The glass-rubber transition temperature, Tg, of cellulose-filled polymer composites is an important parameter that controls different properties of the resulting composites, such as their mechanical behavior, matrix chain dynamics, and swelling behavior. Differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA) were used to evaluate the Tg values of the polymers and composites. In DSC experiments, Tg is generally taken as the inflection point of the specific heat increment at the glass-rubber transition. The DMA tests showed a relaxation process in this temperature range, instead of a transition process. The temperature of this relaxation process depends on both Tg and the measurement frequency, which can be taken as the temperature at the maximum peak of the internal friction factor (tanδ) or loss modulus (E’’), where E’ corresponds to the storage tensile modulus. Table 4 summarizes thed thermal properties of the composites and cellulose nanocomposites measured by DSC. All the materials exhibited single melting endotherms and various temperatures depending on the filler content. Notably, all composites showed a higher melting temperature (Tm) than the virgin PP composite. The PP composite had a Tm of 157.35°C, while all the cellulose and clay dispersed showed around 163°C. The melting temperature increased with the addition of clay and cellulose materials, resulting in better thermal stability. Increased nucleation caused by the addition of filler particles produced a shift to a higher temperature of peak temperature of crystallization exotherm on cooling from the melt.
The enthalpy (ΔHm) of the PP composite (SP-1) at the transition temperature was 62.37 J/g, however, in SP-2 ΔHm where only cellulose was added, the ΔHm decreased to 60.58. On the other hand, the addition of inorganic clay (SP-3) and both additives (SP-4) led to higher ΔHm values, which indicates increased thermal stability owing to the addition of inorganic clay. The SP-2 and SP-5 composites showed a lower ΔHm compared to the others, suggesting that the loading of cellulose absorbed more energy in the melting of composites, as the ΔHm of cellulose was much lower than that of virgin PP [39].
The crystalline enthalpy (-ΔHm) of the SP-1 composite was 69.92 J/g. When cellulose was added to SP-3, its -ΔHm decreased to 69.51. Whereas with the addition of inorganic clay to SP-3, it increased to 72.86 J/g. The addition of both cellulose and clay into SP-4 increased its -ΔHm to 76.45 J/g. Further, the -ΔHm in SP-5 decreased to 66.63 J/m. These results suggest that the addition of cellulose interfered with crystallization. It is assumed that the decrease in –ΔHm was caused by the decrease in the crystal size. Lee S. et al [40] also observed a similar behavior and suggested that clay and wood floors were responsible for crystallization, as they play a significant role as nucleating agents.
Table 5 lists the crystallinity (Xc) of the composites. Crystallinity was calculated using ΔH = 190 J/g of virgin PP and the enthalpy of fusion obtained from the DSC curves. The crystallinity of the PP matrix in SP-1 was 36%. Further, it increased by approximately 38.4% when inorganic clay was added. Meanwhile, a decrease in crystallinity was observed when only cellulose was added to the SP-3 sample. The crystallinity was further increased by the addition of both cellulose and clay in the SP-4 and SP-5 samples. These results suggest that the viscosity of the base PP was affected by the dispersion of cellulose particles and the surface chemistry of cellulose molecules [41].
Figure 8 shows the crystallization analysis of the samples determined using CRYSTAL (Polymer Char, Spain) conducted with an infrared (IR) detector in 1, 2, and 4-trichlorobenzene. Crystallization was carried out in a 50 mL stainless steel stirred vessel. Five crystallization vessels were installed in the main oven, a gas chromatography oven, and attached to a dual-channel optoelectronic IR detector via a rotary valve. The sample solution of 0.15% (w/w) concentration was prepared in 1, 2, 4-trichlorobenzene (TCB) at 150°C and stirred continuously for 60 min. The solution was then equilibrated at 95°C for 45 min, and subsequently crystallized at a cooling rate of 0.2°C/min from 95 to 30°C. The amount of polymer in the solutions was measured using a dual-wavelength optoelectronic IR detector. The qualitative differential composition distribution (dw/dT versus T) was obtained by numerical differentiation of the integral analog. The IR cell was heated isothermally during the entire experiment at 150°C.
Figure 9 shows the TGA curves of all the prepared composites. The analyzed data are summarized in Table 5. The PP composite in SP-1 began degrading at approximately 400°C and gradually decreased with 0.76% residue at 460°C. However, the thermal stability (thermal degradation) of clay composites in SP-2 improved significantly from 400.00 °C to 428.00 °C (1st onset) but deteriorated at 450°C with 3.78% residue. Meanwhile, composites with only cellulose (SP-2) or clay and cellulose (SP-4,5) showed early degradation around 290–309°C but the 2nd onset temperature increased around 435–446°C. Hybrid composites SP-4 and SP-5 showed a higher 2nd onset temperature. The 1st decomposition onset temperature for the composites can be attributed to the volatile decomposition products associated with cellulose molecules. However, the increase in the 2nd onset temperature might be caused by the synergetic effect of clay and cellulose, in which cellulose acted as a temperature barrier along with inorganic clay.
Conclusion
In this study, hybrid bio-nanocomposites were fabricated using microparticle cellulose powder from wastepaper and a small amount of layered microclay with a polypropylene (PP) polymeric matrix using a low-shear chaotic mixing method. FTIR and SEM analyses confirmed the presence of cellulose in the PP polymeric matrix. The flexural stress-strain curves demonstrated the synergetic effect of the cellulose and inorganic clay in the composites. The maximum stress was obtained in SP-5 composites when 9% of cellulose was added. The cellulose-inorganic clay composite SP-5 also showed the highest tensile strength and tensile modulus. In addition, the gamma peak was also found to be shifted at a higher temperature for filled composites, which indicates reduced polymeric chain mobility owing to the addition of cellulose and inorganic clay. In the case of SP-3 and SP-4, the DSC results of the composites showed that with the addition of inorganic clay, the ΔHm values increased, indicating an increase in thermal stability owing to the addition of inorganic clay. SP-5 composites showed a lower ΔHm compared to the others, suggesting that the loading of cellulose absorbed more energy during the melting of composites, as the ΔHm of cellulose is much lower than that of virgin PP. The TGA results also confirmed the superior thermal properties of the composites. This study confirms that with the addition of cellulose powder and a small amount of nano-clay, the mechanical properties of the composites improved significantly, and a synergistic effect was observed. Therefore, these composites can be used in a variety of biodegradable applications.
References
- Cao, Y.; Wu, Y.Q. Evaluation of statistical strength of bamboo fiber and mechanical properties of fiber reinforced green composites. J. Cent. South Univ. Technol. 2008, 15, 564–567. [Google Scholar] [CrossRef]
- Li, X.; et al. Chemical treatments of natural fiber for use in natural fiber-reinforced composites: a review. J. Polym. Environ. 2007, 15, 25–33. [Google Scholar] [CrossRef]
- Lee, B.-H.; et al. Fabrication of long and discontinuous natural fiber reinforced polypropylene biocomposites and their mechanical properties. Fibers Polym. 2009, 10, 83–90. [Google Scholar] [CrossRef]
- Mehta, G.; et al. Novel biocomposites sheet molding compounds for low-cost housing panel applications. J. Polym. Environ. 2005, 13, 169–175. [Google Scholar] [CrossRef]
- Ioelovich, M.; Figovsky, O. Advanced environment-friendly polymer materials. Polym. Adv. Technol. 2002, 13, 1112–1115. [Google Scholar] [CrossRef]
- Lee, S.-Y.; et al. Creep behavior and manufacturing parameters of wood flour filled polypropylene composites. Compos. Struct. 2004, 65, 459–469. [Google Scholar] [CrossRef]
- Kim, H.-S.; et al. Thermogravimetric analysis of rice husk flour filled thermoplastic polymer composites. J. Therm. Anal. Calorim. 2004, 76, 395–404. [Google Scholar]
- Dorée. The Methods of Cellulose Chemistry, Chapman & Hall, London, UK, 1947.
- The Merck Index, Merck & Co, Rahway, NJ, USA, 8th edition, 1968.
- Chand, N.; Fahim, M. Natural fibers and their composites, in Tribology of Natural Fiber Polymer Composites (Second Edition), N. Chand and M. Fahim, Editors. 2021, Woodhead Publishing. p. 1-59.
- Joseph, B.; et al. Cellulose nanocomposites: Fabrication and biomedical applications. J. Bioresour. Bioprod. 2020, 5, 223–237. [Google Scholar] [CrossRef]
- Kalka, S.; et al. Biodegradability of all-cellulose composite laminates. Compos. Part A Appl. Sci. Manuf. 2014, 59, 37–44. [Google Scholar] [CrossRef]
- Samir, A.; et al. Recent advances in biodegradable polymers for sustainable applications. NPJ Mater. Degrad. 2022, 6, 68. [Google Scholar] [CrossRef]
- Immonen, K.; et al. Thermoplastic cellulose-based compound for additive manufacturing. Molecules 2021, 26, 1701. [Google Scholar] [CrossRef] [PubMed]
- Norizan, M.N.; et al. Nanocellulose-based nanocomposites for sustainable applications: A Review. Nanomaterials 2022, 12, 3483. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Dutta, H. Characterization of nanocellulose extracted from short, medium and long grain rice husks. Ind. Crops Prod. 2020, 154, 112627. [Google Scholar] [CrossRef]
- Babu, N.K.; et al. Self-reinforced polymer composites: An opportunity to recycle plastic wastes and their future trends. J. Appl. Polym. Sci. 2022, 139, e53143. [Google Scholar] [CrossRef]
- Park, K.; et al. Recyclable and mendable cellulose-reinforced composites crosslinked with Diels–Alder adducts. Polymers 2019, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; et al. Effect of processing variables on the mechanical properties of sisal-fiber-reinforced polypropylene composites. Compos. Sci. Technol. 1999, 59, 1625–1640. [Google Scholar] [CrossRef]
- Rana, A.; et al. Short jute fiber reinforced polypropylene composites: Effect of compatibiliser, impact modifier and fiber loading. Compos. Sci. Technol. 2003, 63, 801–806. [Google Scholar] [CrossRef]
- Lu, J.Z.; et al. Chemical coupling in wood fiber and polymer composites: A review of coupling agents and treatments. Wood Fiber Sci. 2007, 32, 88–104. [Google Scholar]
- Amash, A.; Zugenmaier, P.J.P. Morphology and properties of isotropic and oriented samples of cellulose fibre–polypropylene composites. Polymer 2000, 41, 1589–1596. [Google Scholar] [CrossRef]
- Pothan, L.A.; et al. Dynamic mechanical analysis of banana fiber reinforced polyester composites. Compos. Sci. Technol. 2003, 63, 283–293. [Google Scholar] [CrossRef]
- George, J.; et al. A review on interface modification and characterization of natural fiber reinforced plastic composites. Polym. Eng. Sci. 2001, 41, 1471–1485. [Google Scholar] [CrossRef]
- Sue, H.-J.; et al. Epoxy nanocomposites based on the synthetic α-zirconium phosphate layer structure. Chem. Mater. 2004, 16, 242–249. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, S.; Qi, Z. Synthesis and characterization of elastomeric polyurethane/clay nanocomposites. J. Appl. Polym. Sci. 2001, 82, 1444–1448. [Google Scholar] [CrossRef]
- Lee, K.Y.; Goettler, L.A. Structure-property relationships in polymer blend nanocomposites. Polym. Eng. Sci. 2004, 44, 1103–1111. [Google Scholar] [CrossRef]
- Dan, C.H.; et al. Effect of clay modifiers on the morphology and physical properties of thermoplastic polyurethane/clay nanocomposites. Polymer 2006, 47, 6718–6730. [Google Scholar] [CrossRef]
- Morgan, A.B.; Gilman, J.W. Characterization of polymer-layered silicate (clay) nanocomposites by transmission electron microscopy and X-ray diffraction: A comparative study. J. Appl. Polym. Sci. 2003, 87, 1329–1338. [Google Scholar] [CrossRef]
- Chesworth, W.; et al. Clay-organic interactions, in Encyclopedia of Soil Science, W. Chesworth, Editor. 2008, Springer Netherlands: Dordrecht. p. 144-150.
- Vasudeo Rane, A.; et al. Chapter 4 - Clay–polymer composites: Design of clay polymer nanocomposite by mixing, in clay-polymer nanocomposites, K. Jlassi, M.M. Chehimi, and S. Thomas, Editors. 2017, Elsevier. p. 113-144.
- Guo, F.; et al. A review of the synthesis and applications of polymer–nanoclay composites. Appl. Sci. 2018, 8, 1696. [Google Scholar] [CrossRef]
- Poletto, M.; Ornaghi, H.L.; Zattera, A.J.J.M. Native cellulose: Structure, characterization and thermal properties. Materials 2014, 7, 6105–6119. [Google Scholar] [CrossRef]
- Fan, M.; Dai, D.; Huang, B. Fourier transform infrared spectroscopy for natural fibres. Fourier Transform.-Mater. Anal. 2012, 3, 45–68. [Google Scholar]
- Ellis, T.S.; D’Angelo, J.S. Thermal and mechanical properties of a polypropylene nanocomposite. Mater. Des. 2003, 90, 1639–1647. [Google Scholar] [CrossRef]
- Botev, M.; et al. Mechanical properties and viscoelastic behavior of basalt fiber-reinforced polypropylene. J. Appl. Polym. Sci. 1999, 74, 523–531. [Google Scholar] [CrossRef]
- Bondeson, D.; et al. All cellulose nanocomposites produced by extrusion. J. Biobased Mater. Bioenergy 2007, 1, 367–371. [Google Scholar] [CrossRef]
- Jonoobi, M.; et al. Mechanical properties of cellulose nanofiber (CNF) reinforced polylactic acid (PLA) prepared by twin screw extrusion. Compos. Sci. Technol. 2010, 70, 1742–1747. [Google Scholar] [CrossRef]
- Lee, S.-Y.; et al. Thermal behavior of hwangto and wood flour reinforced high density polyethylene (HDPE) composites. J. Korean Wood Sci. Technol. 2006, 34, 59–66. [Google Scholar]
- Lee, S.; et al. Thermal, mechanical and morphological properties of polypropylene/clay/wood flour nanocomposites. eXPRESS Polym. Lett. 2008, 2, 78–87. [Google Scholar] [CrossRef]
- Wang, K.H.; et al. Morphology and physical properties of polyethylene/silicate nanocomposite prepared by melt intercalation. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 1454–1463. [Google Scholar] [CrossRef]
Figure 1.
Cellulose fiber’s chemical structure.
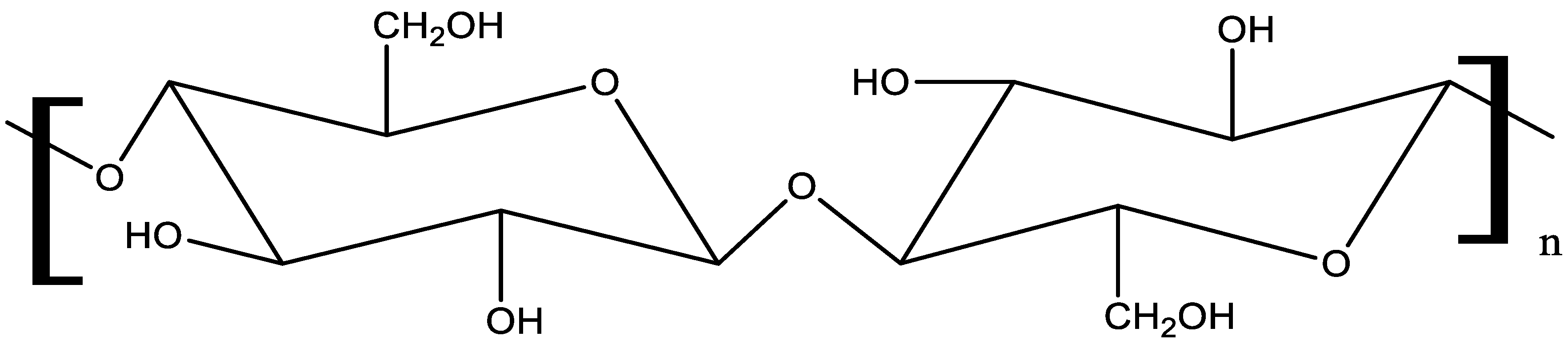
Figure 2.
FTIR spectra of the prepared samples.
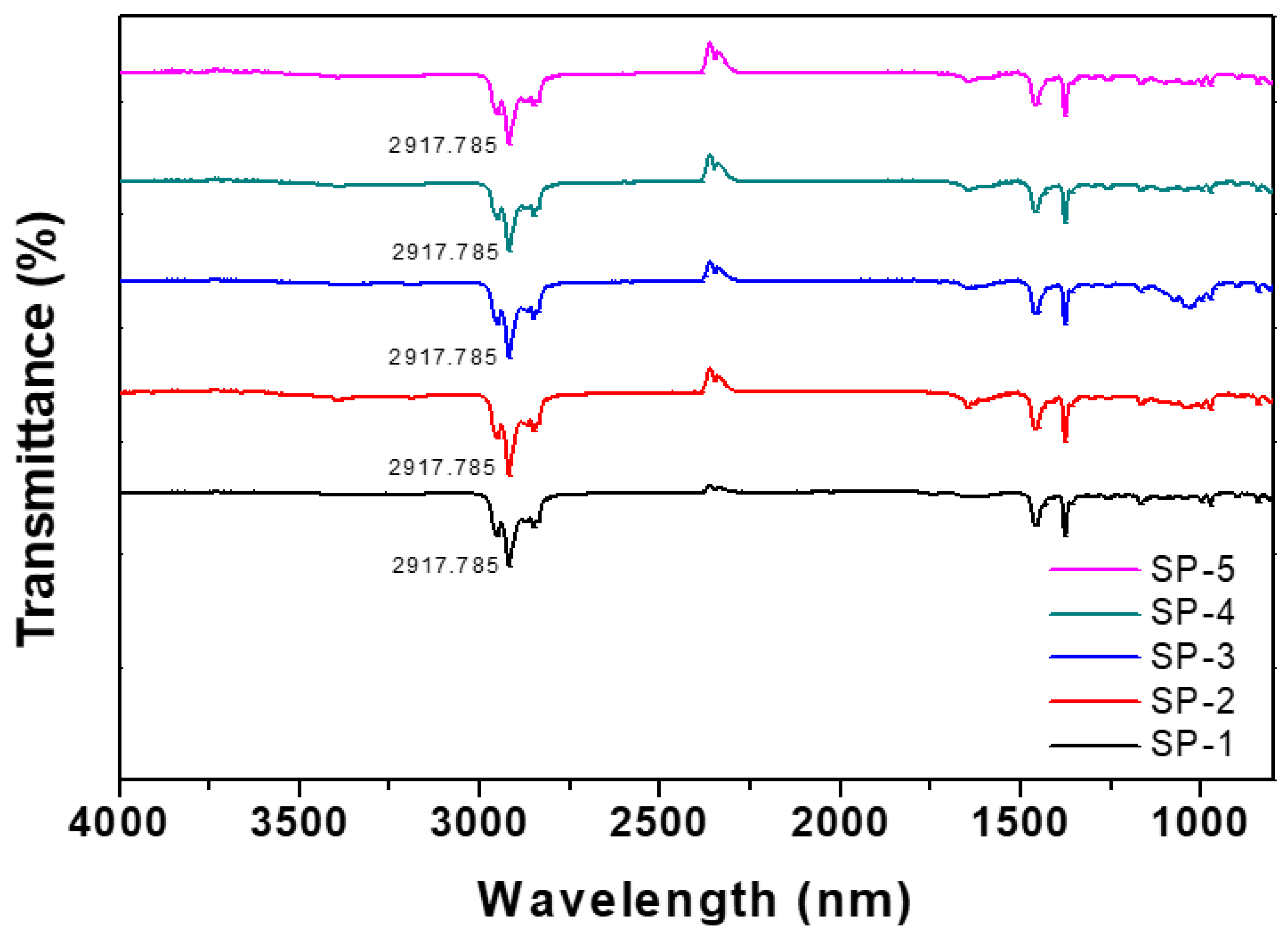
Figure 3.
(a, b) SEM images of Cellulose powder (low and high magnification) (c) Cellulose composite.
Figure 3.
(a, b) SEM images of Cellulose powder (low and high magnification) (c) Cellulose composite.

Figure 4.
Stress-Strain curve for the prepared samples.
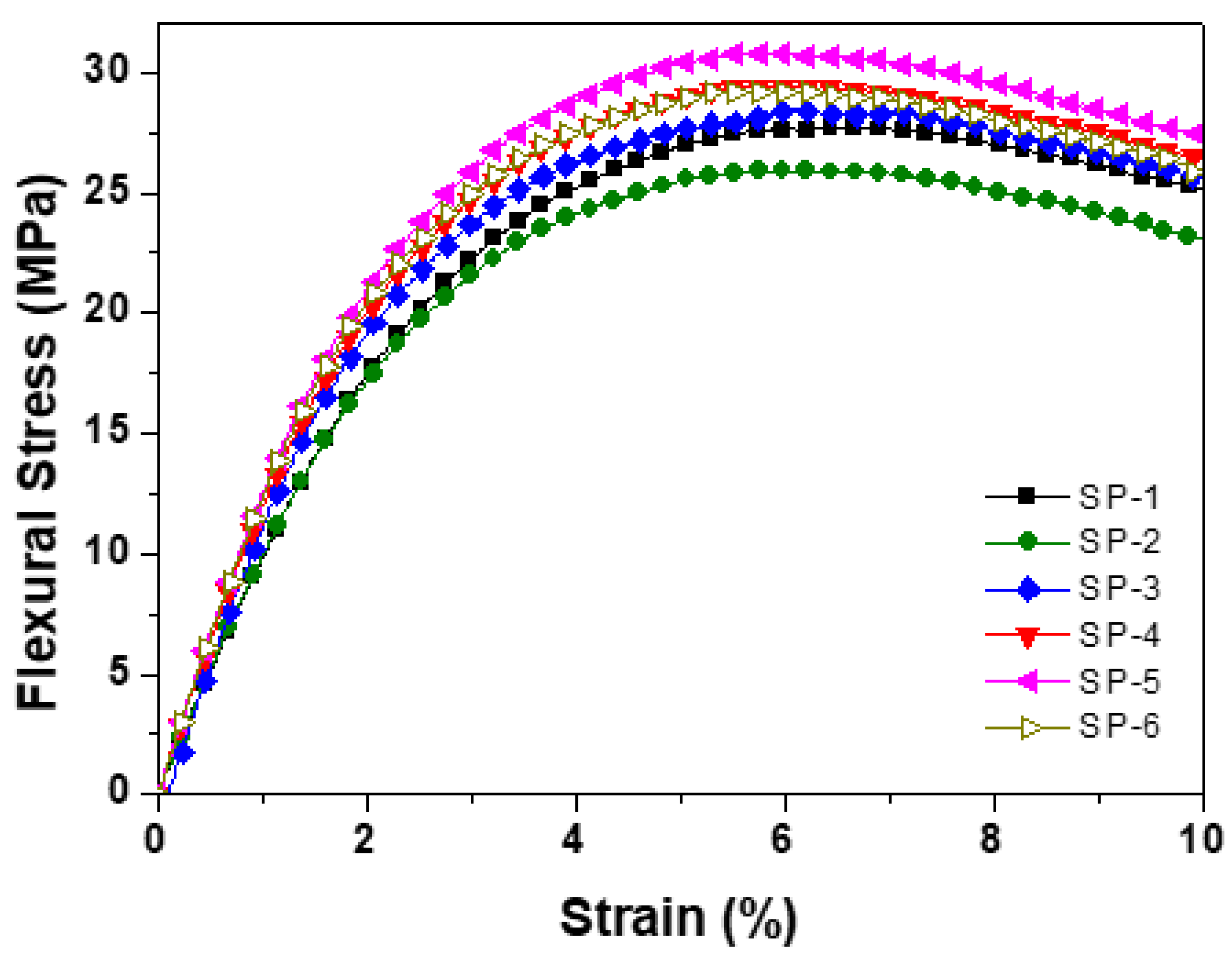
Figure 5.
Tensile Stress-Strain curves of prepared samples.
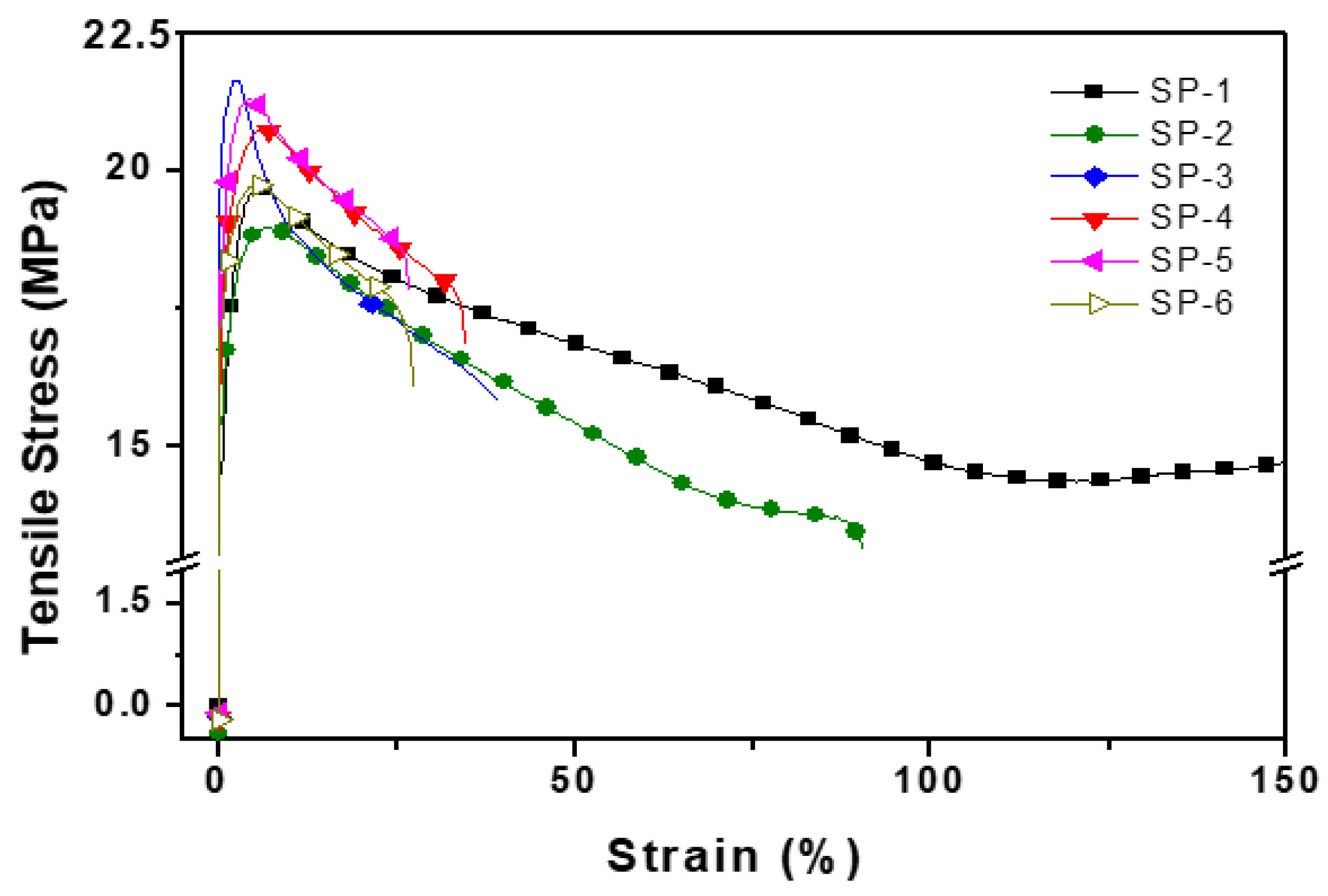
Figure 6.
Storage modulus variation with temperature.
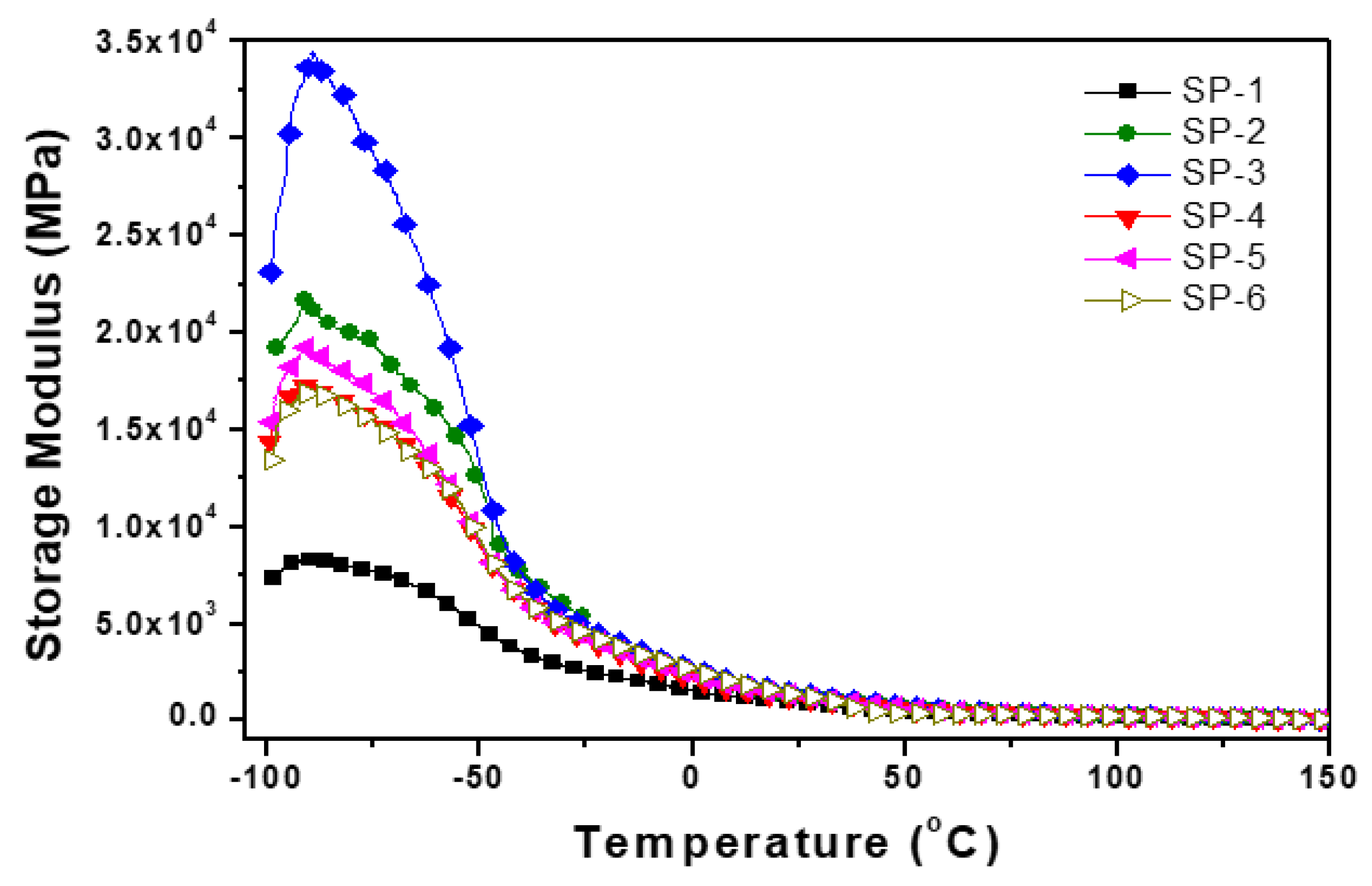
Figure 7.
Tan Delta variation with temperature.
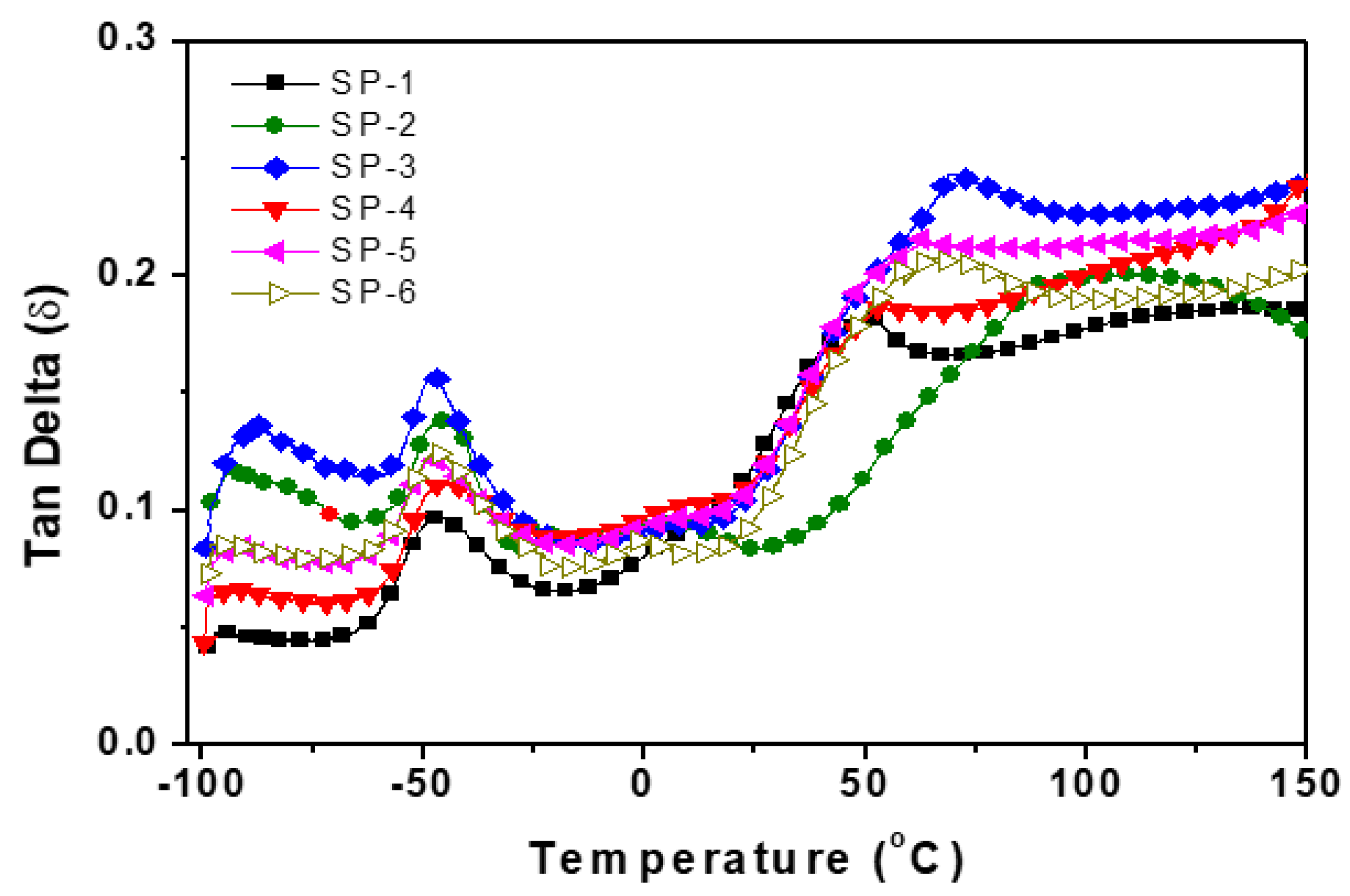
Figure 8.
Overlay of the crystallographic analysis of the SP samples.
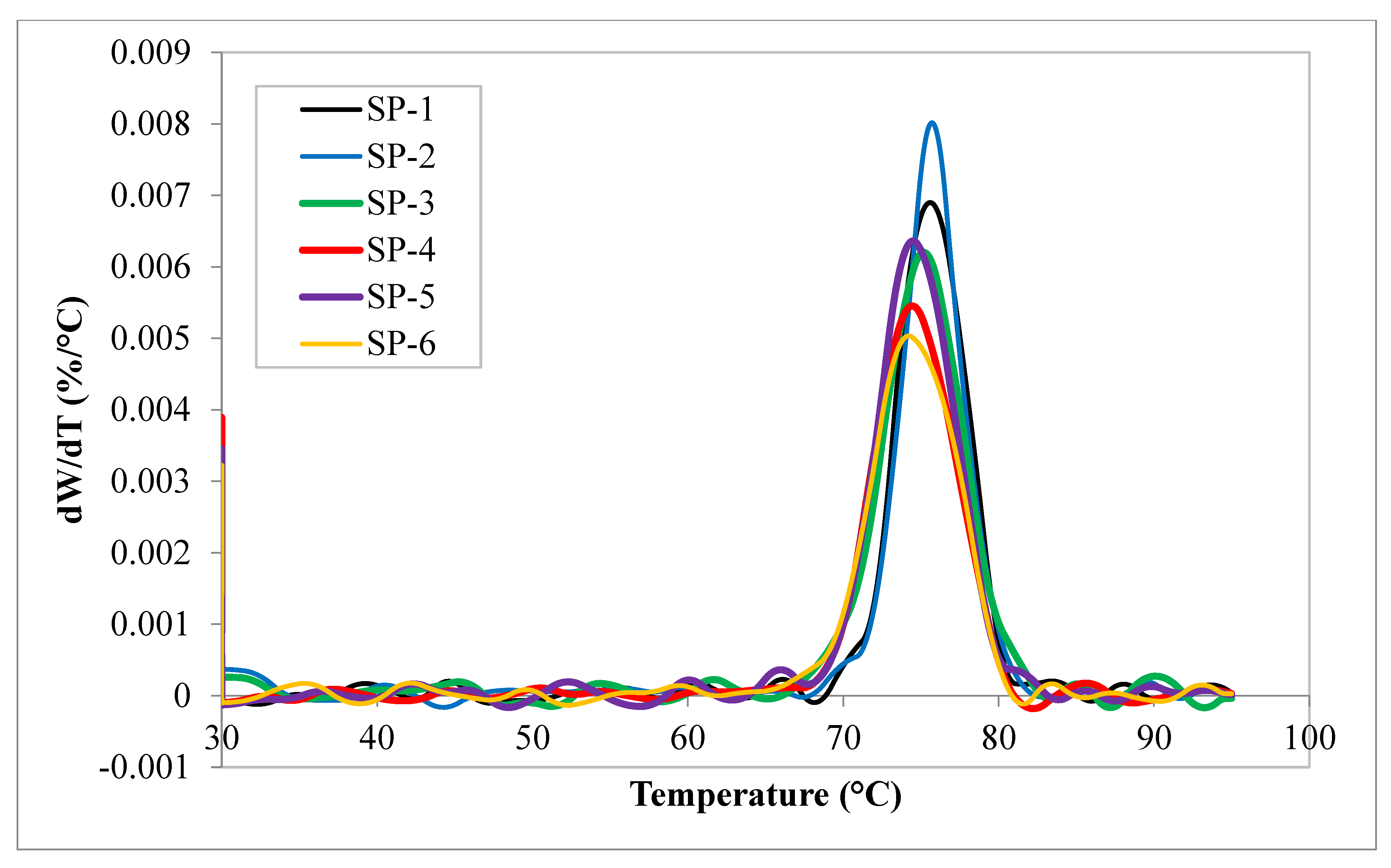
Figure 9.
TGA curves of prepared samples.
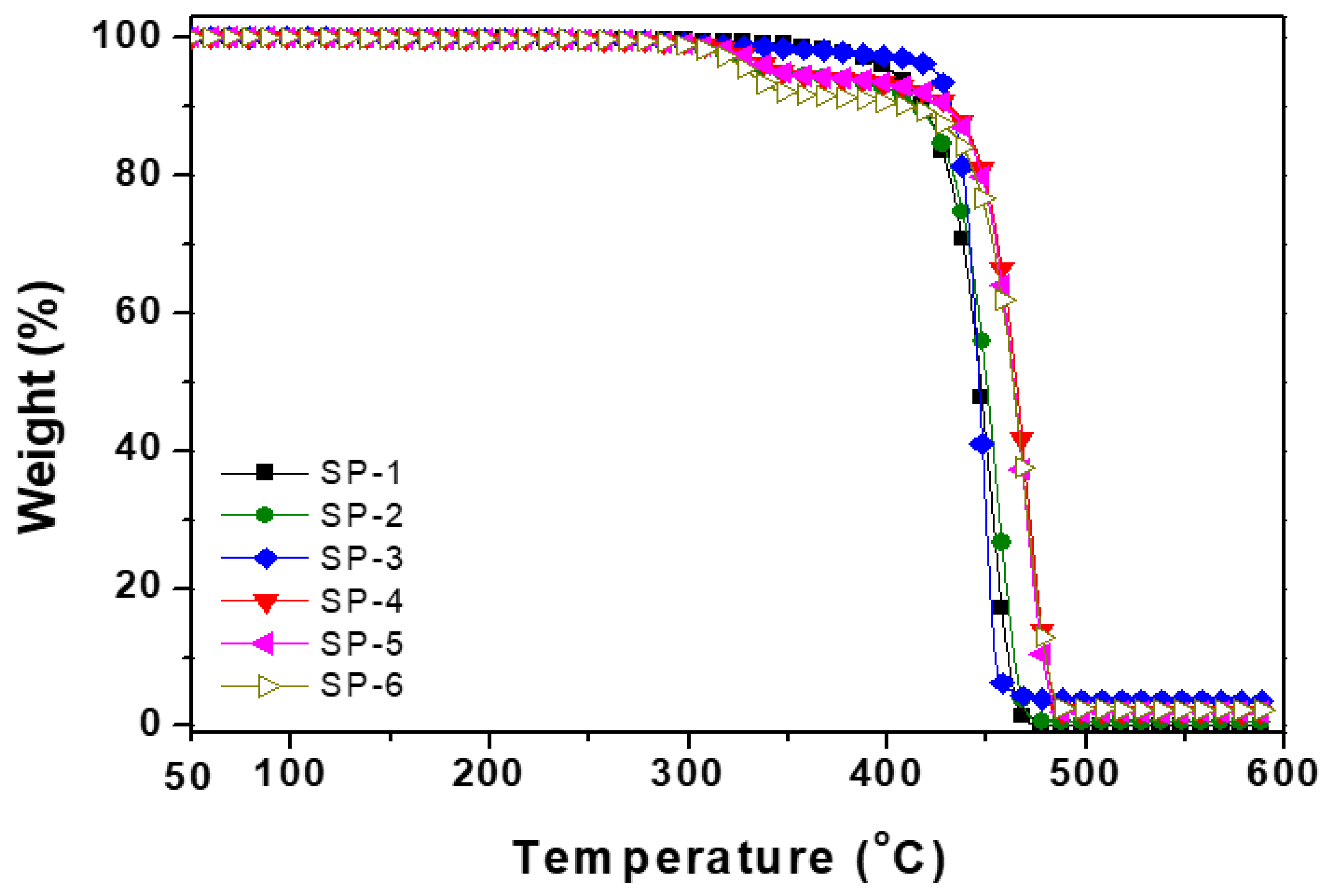

Table 1.
Nanocomposites fabrication formulation.
| Sample name | PP% |
TPO EG8407 |
Ethanox 310 | Ethanox-368 | EV-91 | EV-12 | Clay% | Cellulose powder% |
| SP-1 | 79.96 | 20 | 0.01 | 0.01 | 0.01 | 0.01 | 0 | 0 |
| SP-2 | 79.46 | 20 | 0.01 | 0.01 | 0.01 | 0.01 | 0 | 5.5 |
| SP-3 | 74.46 | 20 | 0.01 | 0.01 | 0.01 | 0.01 | 5.5 | 0 |
| SP-4 | 72.46 | 20 | 0.01 | 0.01 | 0.01 | 0.01 | 2.0 | 5.5 |
| SP-5 | 68.46 | 20 | 0.01 | 0.01 | 0.01 | 0.01 | 2.0 | 9.5 |
| SP-6 | 67.4.6 | 20 | 0.01 | 0.01 | 0.01 | 0.01 | 3.0 | 9.5 |
Table 2.
Mechanical properties of the composites.
| Sample name |
Flexural Strength MPa |
Flexural Modulus MPa |
Tensile strength MPa |
Tensile Modulus MPa |
Yield strain | Elongation % | Notched Izod strength kg. cm/cm2 |
| SP-1 | 27.5 | 134.8 | 18.5 | 110 | 0.168182 | 82 | 42.5 |
| SP-2 | 26.0 | 112.8 | 18.7 | 115 | 0.162609 | 102 | 30 |
| SP-3 | 28.0 | 130 | 21.75 | 120 | 0.18125 | 93 | 38 |
| SP-4 | 29.0 | 145 | 21.0 | 125 | 0.168 | 86.2 | 27 |
| SP-5 | 31.0 | 155 | 21.85 | 122 | 0.179098 | 79 | 25 |
| SP-6 | 28.8 | 135 | 19.5 | 120 | 0.1625 | 89 | 22 |
Table 3.
Dynamic mechanical properties of the composites.
| Sample name |
Storage modulus E’(MPa) −100°C |
Storage modulus E’(MPa) −80°C |
Tg (°C) |
tanδ1 | Loss modulus E’’ T°C | tanδ2 |
Loss modulus E’’(MPa) |
| SP-1 | 7500 | 8000 | 52 | 0.08 | 55 | 0.19 | 1425 |
| SP-2 | 18000 | 22000 | 57 | 0.14 | 100 | 0.22 | 3960 |
| SP-3 | 26000 | 33000 | 55 | 0.16 | 75 | 0.24 | 6240 |
| SP-4 | 14000 | 17500 | 52 | 0.12 | 57 | 0.2 | 2800 |
| SP-5 | 16000 | 19000 | 52 | 0.13 | 65 | 0.18 | 2880 |
Table 4.
DSC analysis results of the composites.
| Composites |
Melting point °C |
Enthalpy of melting (J/g) ΔH | Crystallization exotherm maximum(°C) |
Enthalpy of crystallization (J/g) ΔH |
Crystallinity % |
| SP-1 | 157.35 | 62.37 | 111.34 | 68.92 | 36 |
| SP-2 | 161.15 | 60.58 | 121.25 | 69.51 | 36.6 |
| SP-3 | 163.15 | 63.48 | 113.25 | 72.86 | 38.4 |
| SP-4 | 163.85 | 65.58 | 113.00 | 76.45 | 40.2 |
| SP-5 | 163.89 | 55.08 | 113.55 | 66.68 | 35.1 |
Table 5.
TGA analysis results of the composites.
| Composites | Onset 1 temperature °C | Onset 2 temperature °C | Residue wt.% |
| SP-1 | 400 | 425.25 | 0.76 |
| SP-2 | 410 | 435.00 | 3.782 |
| SP-3 | 290.0 | 434.65 | 0.580 |
| SP-4 | 313.6 | 448.00 | 1.926 |
| SP-5 | 309.0 | 446.00 | 2.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



