
Submitted:
29 March 2023
Posted:
30 March 2023
You are already at the latest version
Abstract
The development of oxidation processes with generating powerful radicals is the most interesting and thought-provoking dimension of peroxymonosulfate (PMS) activation. In this study, a magnetic spinel CuFe2O4 was successfully prepared by a facile, non-toxic, cost-efficient co-precipitation method with the synergetic effect of photocatalytic PMS oxidation against recalcitrant benzotriazole (BTA) pollutant. The high particle dispersion and large surface area (201.898 m2/g) provided a high photocatalytic activity with CuFe2O4. The obtained results from the central composite design (CCD) analysis confirmed the optimum degrading rate of BTA reached 81.4% at the optimum operational condition of CuFe2O4= 0.4 g/L, PMS= 2 mM, BTA= 20 mg/L after 70 min irradiation time. As such, the active species capture experiments revealed the presence of •OH, SO4•‒, O2•‒ and h+ species in CuFe2O4/UV/PMS system where SO4•‒ was dominated for BTA photodegradation. The combination of photocatalysis and PMS activation enhanced the consumption of metal ions in redox cycle reactions, thus preventing metal leaching and maintain the recyclability with reasonable mineralization efficiency which achieved more than 40% total organic carbon removal after four batch experiments. A retardant effect on BTA oxidation was observed with comparing the water’s common anion constituents as HCO3‒ > Cl‒ > NO3‒ > SO2‒. Results from intermediates identification showed that the transformation pathway of BTA developed as the deprotonation, hydrolysis and ring cleavage processes promoting the practical potential of photo-absorbing CuFe2O4 catalyzed PMS which can be a promising method due to convenient magnetic separation form the solution.
Keywords:
Peroxymonosulfate activation
; Copper ferrite
; Photocatalyst
; Benzotriazole
1. Introduction
Emerging Benzotriazole (BTA) contaminant has been widely use in industrial and domestic applications such as alloys protection against corrosion, automotive cooling and aircraft de-icing fluids, fastness to light in fabrics, antifreezes and in production of different detergents. BTA is a cyclic compound that has a benzene ring which is fused into a C-C bond with three nitrogen atoms [1,2]. The presence of BTA has been reported in the effluent-receiving water bodies (~ 7-18 μg/L) [3], sea water and urban runoff as the major sources into the natural environment [4]. The trait of the compound is low tendency for sorption to organic matters, extremely soluble in water and polar that causes its high environmental mobility [5]. Consequently, its persistence and toxicity in the aquatic environment makes it little affected by mechanical-biological treatment processes [4,5,6]. Moreover, studies argued that these features reflect the probable human carcinogen categorizing as a very toxic to aquatic organisms; hence, exploring the efficient water treatment technologies for BTA mineralization is vitally important.
Advanced oxidation processes (AOPs) have enormous potential for elimination of emerging and refractory pollutants using the reactive oxygen species (ROS) (such as singlet oxygen (1O2) molecules, sulfate (SO4•‒), hydroxyl (•OH) and superoxide (O2•‒) radicals). Several AOPs including photo-Fenton-like system [7], photocatalytic reactions [2], ozonation and ultrasound assisted oxidation [8] have been used to eliminate BTA. The sulfate based-AOPs such as peroxymonosulfate (HSO5‒, PMS) and peroxydisulfate (S2O82−, PDS), known as the emerging and promising AOPs, have expanded the treatment flexibility in different operational conditions, while Fenton process, in contrast, narrowed in pH adjustment with great difficulty [9]. PMS is environmentally benign because of its less harmful by-products (SO42‒) [10]. SO4•‒ with much higher oxidizing efficiency (E0 = 2.5–3.1 V) at neutral pH, better selectivity and higher persistency in aqueous solutions (30-40 µs) than •OH (E0 = 1.9–2.7 V) giving the opportunity of contacting with recalcitrant pollutants, and also with its remarkable durability for practical applications in terms of interfering water constituents, has produced promising results in water and wastewater treatment [11]. Therefore, SO4•‒ can overcome the unescapable deficiencies in a conventional AOPs [12]. In particular, the induction of the PMS or PDS precursors by UV or ultrasound irradiation, base and transition metals catalysts (Fe2+, Mn2+, Co2+, Cu2+ and so on), heat, and carbon-based materials, can produce SO4•‒ via electron transfer according to Eq. (1) [13,14]. In this regards, the homogeneous transition-metal catalyzed PMS has been preferred because it is easy to be initiated at room-temperature and pressure without low energy required through Eq. (2-3) forming SO4•‒ as the predominate oxidizing species [15]:
S2O82‒/HSO5‒ → SO4•‒ + (HSO5•, SO42‒, •OH)
HSO5‒ + e‒ → SO4•‒ + OH‒
HSO5‒ + e‒ → SO42‒ + •OH
However, the system suffers from releasing metal ions from solid phase during the reaction causing the secondary pollution by toxicity of transition metals and the difficulty of its recovery [16]. As a clean way to activate PMS, heterogeneous catalysts have been extended with a number of special features including hypotoxicity, low-cost and structural stability [17]. In view of difficulty associated with recovering these catalysts, Fe-based catalyst for easy separation under magnetic field is being studied to overcome this problem [18].
The heterogeneous catalysis of PMS closely depends on the interaction between PMS and catalyst. To fulfill this purpose, a large specific area, excellent catalytic activity, and high porous structure are needed so that the utilization rate of PMS can be increased [19]. Copper ferrites (CuFe2O4) with intrinsic surface hydroxyl sites and ferromagnetic property have been widely studied for efficaciously PMS oxidation in removal of organic pollutant that is refractory to the water treatment [20,21]. Although it is less catalytic effectiveness than CoFe2O4, safe carcinogenic potential has made it an applicable PMS activator. Furthermore, a significant linear correlation between the degradation rate and quantity of surface hydroxyl sites has been demonstrated [22]. More specifically, CuF2O4 as a Cu-Fe mixed-metal catalyst endows the surface with synergistic redox reactions for Cu3+/Cu2+, Cu+/Cu2+, and Fe2+/Fe3+ in a tetrahedral and/or octahedral structure to efficaciously generate SO4•‒ radical from PMS. The photocatalytic PMS activation by CuFe2O4 due to its suitable light absorption provides the photosynergistic effect which is favorable for organic pollutants degradation [21,23]. In this context, the photogenerated electrons are donated to the PMS electron acceptor (electron transfer), which cleaves O-O bond in PMS molecule into SO4•‒ and •OH radicals [24]. It can be also controlled the recombination phenomenon of photoinduced charges through the consumption of photoinduced electrons by PMS, thus amplifying the catalytic efficiency [13,25]. Therefore, this great ability to be synchronously interacted with both UV photon and PMS endow CuFe2O4 with the construction of CuFe2O4/UV/PMS reaction system in this study. As the authors know, no study has investigated BTA degradation by CuFe2O4/UV/PMS system. Up to now, several studies applied the CuFe2O4/PMS process and found the promising catalytic oxidation for different micropollutants elimination at neural pH [26,27,28,29]. In this work, we designed and synthesized the CuFe2O4 catalyst by a simple co-precipitation method obtaining high surface area and abundance active sites which is essential to activate PMS for BTA degradation. The structural, morphological, and physicochemical property of CuFe2O4 were investigated. At first, we studied the ability of different reaction systems for BTA degradation and found that UV-assisted CuFe2O4/ PMS couple transcended among other treatment methods. Then, the interactive effect of operational parameters (PMS dosage, catalyst dosage, different BTA concentrations, and reaction time) on catalytic performance was optimized using response surface methodology (RSM) via central composite design (CCD). The reusability and stability of CuFe2O4 was consecutively analyzed through four cycles and the treatment of BTA in presence of interfering water constituents was evaluated under the optimum conditions. In addition, the possible reaction mechanism of the CuFe2O4/UV/PMS system was proposed based on the identification of main activity radical species from quenching tests. Finally, the probable transformation pathways of BTA were explored. This study could provide a simple, easy, cheap, and trustworthy process with high catalytic performance for the elimination of organic pollutants.
2. Experimental Section
2.1. Chemicals
Benzotriazole (C6H5N3, 99%) in addition to cupper nitrate trihydrate (Cu(NO3)2·3H2O, 99%), ferric nitrate nonahydrate (Fe(NO3)3.9H2O, 99%) were provided from Merck Inc. Potassium peroxymonosulfate (Oxone, KHSO5.0.5KHSO4.0.5K2SO4), tert-butyl alcohol (TBA, (CH3)3COH), p-Benzoquinone (BQ, C6H4O2), methanol (MeOH, CH3OH) and potassium iodide (KI) were obtained from Sigma-Aldrich Inc., USA. All the aqueous solutions during experiments were prepared by deionized water (DI-water).
2.2. Preparation of Catalyst and Characterization Tests
Magnetic CuFe2O4 nanoparticles were prepared on the basis of co-precipitation method with the precursors Cu(NO3)2·3H2O and Fe(NO3)3·9H2O in molar ratio of one to two/1:2 referring to our previous study [30]. Powder X-ray diffraction (XRD) was performed utilizing an Quantachrome/NOVA 2000X-ray diffractometer with Cu Kα radiation (λ = 1.54 nm), operated at 30 mA and 40 kV in a 2θ range of 10 to 80°. Brunauer-Ennett-Teller (BET, ASAP 2010; Micromeritics, USA) was applied to identify the surface area, average pore size and volume . Magnetic feature of catalyst was conducted by vibrating sample magnetometer (VSM, Lakeshore 7400, USA). The structure and morphology of the catalyst was achieved by transmission electron microscopy (TEM, JEM-2100; Jeol; Japan) with high-resolution at 100 kV. Field emission scanning electron microscope (FESEM, Mira 3-XMU) coupled with an energy dispersive X-ray spectrometer (EDS) device and transmission electron microscopy (TEM, JEM-2100; Jeol; Japan) with high-resolution at 100 kV were served to explore the microstructure and morphology of the catalyst. The UV–Vis diffuse reflectance spectrum (DRS, Hitachi Ltd., Japan) was attained at the wavelength range of 190 to 800 nm.
2.3. Experimental Procedures and Analysis
A cylindrical quartz photoreactor with a reaction volume of 300 mL containing certain amount of BTA was used to perform photocatalytic experiments at 25 ± 5 °C. The initial pH in all experiment was considered to be unadjusted. For each experiment, at first, catalyst was put into the BPA solution and mechanically stirred (RW 20, IKA, Germany) at 250 r/min for 60 min to ensure the adsorption-desorption equilibrium among the BTA molecules and surface catalyst. A 6.0 W Hg UV-C lamp (Philips, India) coupled with a 254 nm filter and luminous intensity of 7800 cd provided the light source. It was immediately switched on as the addition of PMS and consequently the catalytic reaction was initiated. At the end of each course of experiment, 2 mL of BTA solution was withdrawn and then 1mL Na2S2O3.5H2O (20 mM) was added to be finally injected into the high-performance liquid chromatography (HPLC) device. HPLC (KNUER) was operated under the protocol of detection maximum wavelength of 254 nm with a C18 separation column (100–5; 4.6 mm × 250 mm, 5 µm) and a 2500 UV-visible detector. The mobile phase was a 50:50%(v/v) water-acetonitrile ratio with the injection flow rate of 1.0 mL min-1. The degradation efficiency (%) was used to describe the performance of the process by Eq. (4):
where C0 and Ct are the BTA concentration at the initial and certain treatment time, respectively. The photochemical stability of the catalyst was assessed for four repeated experiments. The concentrations of Fe and Cu filtrate were determined for each cycle by an atomic absorption spectrophotometer (AAS) (Analytikjena vario 6, Germany). Also, each cycle was completed with the total organic carbon (TOC) determination to monitor the mineralization efficiency by a Shimadzu VCHS/CSN, Japan. KI, BQ, TBA, and MeOH were applied to characterize the contribution of ROS. The effect of various anions exist in natural matrices was studied for BTA removal efficiency. Intermediates products were analyzed by liquid chromatography-mass spectrometer (LC-MS, Agilent 6500) to show the degradation routs for BTA.
2.4. Response Surface Experimental Design and Data Analysis
To find the synthetically effect of main factors for optimal degradation conditions of BTA, central composite design (CCD) was used under response surface methodology (RSM). In this experiment, the catalyst loading (x1), PMS dosage (x2), BTA concentration (x3), and irradiation time (x4) were varied at five various coded levels, as indicated in Table 1. The coded value of the ith variable (xi) was described by Eq. (5):
where Xi is the actual values of the variables, X0 is Xi at the center point, and ∆Xi is the step with maximum and minimum values of Xi. With the application of the CCD matrix, 30 tests (24 = 16 factorial points, 2 × 4 = 8 axial points, and 6 replications at the center points) were determined for four input variables (Table S1). The quadratic polynomial equation can estimate the effects of four independent variables on the efficiency of the photocatalytic PMS activation process. Here, y is a predicted response variable of BTA degradation efficiency, b0 is the intercept, bi, bij and bii are the linear, interaction, and quadratic regression coefficients, respectively, and is residual error which described according to the following equation:
The significance and adequacy of the quadratic regression model was estimated by analysis of variances (ANOVA). The R-squared (R2) and the adjusted R2 (R2adj) values were the criteria for verifying the statistical significance of the second order model.
3. Results and Discussion
3.1. The CuFe2O4 Spinel Catalyst Morphology and Characterization
To investigate the structures and morphologies of pure CuFe2O4, FESEM and TEM techniques were executed. Figure 1A showed an irregular polygon morphology and slightly agglomerated state coming from the nanoscaled size for CuFe2O4 with size of < 26 nm.Obviously, an uneven and rough surface was witnessed by TEM image of CuFe2O4 catalyst (Figure 1C,D), creating conditions favorable to the reactants’ diffusion which would boost the exposure of organic pollution and oxidant with active sites and consequently accelerate the redox reaction on the catalyst surface. In addition, EDS analysis linked to FESEM (Figure 1B) showed that the catalyst contained the elements including Cu, Fe, and O in molar ratio of nearly 1:1.82, which was almost equal to the molar ratio of the iron and copper precursors, proving the molecular formula of the CuFe2O4 catalyst.
The crystalline plane of CuFe2O4 were determined by XRD (Figure 2A). The main peaks at 2θ of 30.2, 35.85°, 43.32°, 57.39° and 62.87° ascribed to crystal planes of (2 2 0), (3 1 1), (4 0 0), (5 1 1), and (4 4 0) similarly described previously [20], which are typical of spinel ferrite structure of CuFe2O4 (JCPDS no. 25-0283). As demonstrated by XRD spectra, the narrow and strong diffraction intensities are indicative of the excellent crystallinity through which high catalyst activity can be expected [26]. Accordingly, with the Debye–Scherrer equation, D = Kλ/βcosθ; K is the Scherrer constant (value 0.94), λ is the X-ray wavelength (0.154 nm), β is the FWHM (full width at half maximum) of photocatalysts, D is calculated crystalline size and θ is the diffraction angle, the average size of CuFe2O4 nanoparticles was calculated as 16.7 nm.
N2 adsorption-desorption isotherm plot (Figure 2B) and the pore size distribution (Figure 2C) measured at -196.88 °C. The trend of Langmuir-isotherm related to type IV curve in the IUPAC classification with a hysteresis loop at the relative pressure (P/P0) range between 0.4 and 1, proving the mesoporous structure. Accordingly, the BET surface area of the pure CuFe2O4 nanoparticles (insert in Figure 2B) was calculated to be 201.898 m2/g, indicating a considerable surface area and significant adsorption capacity for the as-prepared catalyst. The total pore volume along with micropore volume were 0.191 and 0.0126 mL/g, respectively, and the average pore diameter was 3.7 nm. The pore size distribution in the range of 1-30 nm manifests mesoporous with size of about 2.6 nm (Figure 2C). Therefore, the large BET surface area with many micropores and mesopores could provide a rich source of surface reaction sites to activate PMS and also adsorb BTA [31].
VSM technique was applied in the presence of magnetic field ranging from - 40 and + 40 kOe to show the magnetic properties of CuFe2O4 (Figure 2D). The saturation magnetization value (Ms) of CuFe2O4 was equal to 35.2 emu g-1. Therefore, it is expected that CuFe2O4 as an ideal ferromagnetism can be separated from the aqueous in a few seconds, begetting high quality of recycling and utilization in the nano scale, thus avoiding the distribution of nanoparticles and secondary contamination into the environment and simplifying the practical application.
The light absorption capacity of the CuFe2O4 was exhibited by ultraviolet-visible-light (UV–Vis) diffuse reflection spectra (DRS) curve (Figure 2E). As observed, a wide range of light absorption from UV to visible region substantiates the formation of electron-hole pairs, which would recuperate photocatalysis, similar to the observation in former reports.
3.2. The Performance of Different Systems in Removal of BTA
Figure 3 describes the BTA removal profiles in various catalytic systems under controlled conditions. In condition of the addition of PMS or in the presence of UV light only 9.9 and 7.6% of the BTA was degraded within 60 min of reaction, respectively, showing a negligible contribution from direct oxidation in BTA degradation under ambient room temperature. This might be because PMS and UV were inert towards the recalcitrant nature of BTA and the contaminant couldn’t directly decomposed into striking oxidative radicals without activation. In contrast, the photolysis of PMS made the oxidation system stronger with approximately 29.4% of BTA being degraded. In fact, UV irradiation triggered the activated PMS to produce SO4•‒ and •OH radicals through breaking peroxide bond of PMS which increased the oxidative degradation in this condition. The presence of CuFe2O4 spinel (0.2 g L-1) promoted the BTA removal to 31%, indicating that the redox capacity and adsorption effect of CuFe2O4 nanoparticles towards BTA molecules was important providing more catalytic sites in its pores for contact between BTA and PMS molecules and absorbing light resulting in furtherance of the reaction rate constant [25]. Therefore, the adsorption equilibrium was established by a 60-min pre-adsorption time for all the tested samples. As expected, the sizeable degradation was observed for CuFe2O4/UV and CuFe2O4/PMS couples, producing BTA removal of 53.6 and 62.2%, respectively, demonstrating the catalytic ability of CuFe2O4 for the activation of PMS and promoted activity in coupling with UV light. It has been reported that the synergistic action of Cu and Fe sites on the CuFe2O4 surface induced the PMS decomposition (good interaction of catalyst surface with HSO5‒) to produce efficient SO4•‒ radicals in CuFe2O4/PMS oxidation system. It was observed from Figure 3 that the greatest synergystic effect was for PMS coupled with CuFe2O4 under UV light, e.g., CuFe2O4/UV/PMS system (up to 73.1% BTA degradation), thus confirming the photocatalytic PMS oxidation effectiveness towards BTA. It can be mentioned that performance of the CuFe2O4/UV/PMS system at its best results in a synergy depended on oxidation destruction interactions on the surface of the catalyst where the reactive species for BTA degradation adsorbed during UV irradiation [32]. Based on these results, it can be explained that: higher surface area (and hence the development of more accessible active areas) coincided with higher mass transfer of BTA molecules and consequently higher reaction rate constant [33], which in turn caused the enhancement of the catalytic reaction [9]; on the other hand, CuFe2O4 as a dispersive catalyst can make the material contact with BTA molecules and readily provide the photo-generated electrons to active PMS for consequently yielding SO4•‒ [17].
3.3. Influence of Operating Parameters on BTA Degradation Model
3.3.1. CCD Analysis
The CCD experiments and data of the experimental and predicted response values for BTA degradation as a function of catalyst dosage, PMS dosage, BTA initial concentration and irradiation time are depicted in Table S1. On the basis of multiple regression analysis, data showed a quadratic polynomial prediction equation agreement in coded factors terms (±α, ±1, 0) as follows:
BTA removal (%) = 58.04 + 8x1 + 3.01x2 – 4.38x3 + 3.92x4 + 0.51x1x2 + 2.23x1x3 + 1.83x1x4 + 2.76x2x3 + 0.42x2x4 – 1.34x3x4 + 2.37x12 + 0.95x22 + 0.09x32 + 1.61x42
Eq. (7) showed that the effects of catalyst loading (x1), PMS dosage (x2) and irradiation time (x4) on the predicted response were positive, whereas the effect of initial BTA concentration (x3) was negative. Also, the significant effect of x1, x2 and x4 on the BTA removal arranges in the order of x1 > x4 > x2. ANOVA analysis in Table. S2 showed that the quadratic regression model was highly significant (P-value > F-value: F-value = 24.18 and P-value = 0.0001) and reliable (Std. Dev. = 3.02). Furthermore, R2 of the quadratic model was 0.9576, providing a 0.957 confidence level for response variability (Figure S1). It was further observed by the normality stablishing between all residuals in the straight line (Figure S2) which can be randomly distributed in the range of ±3.00 (Figure S3), thus indicating an excellent approximation. In addition, R2adj of 0.918 was close to R2 verifying the goodness-of-fit of the model [34,35,36].
According to the prediction of CCD, the maximum removal of BTA was 81.46% (0.987 desirability) where the optimal values of influencing factors were catalyst loading (0.4 g L-1), PMS dosage (2 mM), BTA concentration (20 mg L-1) and irradiation time (70 min).
3.3.2. Interactions Analysis of Influencing Factors
The effect of PMS dosage and catalyst loading on BTA degradation efficiency are shown in Figure 4A. As can be seen, higher initial PMS and catalyst loads promoted the removal rate of BTA within the specified ranges, confirming the positive effect of both PMS and catalyst loading on the degradation efficiency. Such increasing the interaction between the catalyst and PMS have yielded greater reactive radicals by coupling more PMS to the further available active sites of the catalyst surface (according to Eq. (1)), especially the metal active species i.e., Cu and Fe sites would decompose PMS, which in turn, contribute to extensive oxidation of 30 mg L-1 of BTA within 50 min irradiation time.
At the constant level of PMS and BTA concentration (Figure 4B), the response surface plot depicted that the catalyst load was not a limiting factor; higher catalyst load under prolonged irradiation time caused the maximum removal of BTA. As measured, the BTA removal efficiency with CuFe2O4/UV/PMS process significantly improved from 50.2% to 81.9% with increasing the dosage of CuFe2O4 from 0.1 to 0.5 g L-1 after 50 min of reaction. It is because more SO4•‒ and •OH radicals were generated via the activation of PMS onto the CuFe2O4 surface (i.e. CuFe2O4 surf e- and CuFe2O4 surf h+) under the enhanced adsorption process of BTA molecules. These results suggested that the efficacy was affected by the simplification of availability of the total surface area as well as fiercely Cu/Fe active sites interactions in the catalyst for reaction with PMS during irradiation.
Figure 4C shows the effect of BTA initial concentration and irradiation time on the system performance. It is obvious that the degradation decreased sharply with an increase in BTA concentration from 10 to 50 mg L-1. The CuFe2O4/UV/PMS system governed by a certain catalyst level imposes limits on the amount of adsorption sites and subsequently catalytic sites to PMS molecules leading to a decline in the decomposition rate of PMS; therefore, fierce competition may arise between parent pollutant and intermediates for inadequate active sites and oxidative radicals on the catalyst surfaces with increasing the initial BTA concentration. More to the points, increasing concentration of BTA on the catalyst surface can decrease photocatalytic reactions that depend upon direct contact of photo-induced electron/hole pairs to generate active species, which in turn hamper the degradation of BTA. Despite of this, an increase in the degradation rate was observed by extension of irradiation time. For example, at initial BTA concentration of 30 mg L-1, the degradation rates increased from 50.9% to 75.1% when the reaction time was increased from 10 to 90 min.
3.4. Recyclability Performance of the CuFe2O4/UV/PMS System
Recycling experiments were carried out to determine the stability and multiple reusability potential of the catalyst in the CuFe2O4/UV/PMS system as one of the strengths of the spinel-structured heterogeneous catalytic system. After each cycle, the used catalyst was recovered magnetically followed by 3 times washing with deionized water and then heated at 80 °C for 1 hr. As shown in Figure 5A, the catalytic activity of CuFe2O4 towards BTA degradation could be maintained as high as 79% during four photocatalytic cycles (280 min), accompanied by more than 40% of TOC removal. Metal leaching detection indicated that CuFe2O4 also provided less Cu (0.33 mg L-1) and Fe (0.18 mg L-1) leaching (Figure 5B), corresponding to 0.10% and 0.06% of the total Cu and Fe contents in the catalyst, respectively. These findings exhibited that the coordination activities of Cu/Fe couples in spinel structure effectively minimized the leaching of metal ions which led to high stability and catalytic activity over several BTA degradation cycles (less than 4% loss of activity).
3.5. Feasibility of the Process (Effect of Inorganic Anions)
The role of inorganic anions in natural water and wastewater sources has been prominent on account of their interference with the efficient removal of target pollutants. Therefore, to investigate the practical applicability of CuFe2O4/UV/PMS system in the environmental systems, the BTA degradation in the presence of 10 mM of major anions i.e., bicarbonate (HCO3‒), chloride (Cl‒), sulfate (SO42‒), and nitrate (NO3‒), was studied. From Figure 6, it was found that presence of anions caused a disturbance in the BTA transformation reactions and thereby decreased the effective BTA degradation. Generally the effect of anions on the removal efficiency can be described as follow: (i) catalyzing PMS through electron exchange reaction to generate some radicals with lower redox potential, (ii) competing with pollutant molecules for reaction with free radicals, and (iii) covering the reactive sites and absorbing photons on the CuFe2O4 surface leading to deactivation of reactive sites [31,37]. As shown in Figure 6, the process performance was strongly deceased by 42.8% in the presence of HCO3‒ and 52.4% in the presence of Cl‒. However SO42‒ and NO3‒ anions slightly reduced the process efficiency to 76.8% and 73.3%, respectively which were almost close to the system efficiency without anions. As a result of low-speed reactions with SO4•‒/•OH, the demoting role of SO42‒ and NO3‒ as a radical scavenger in BTA degradation was lower than those of Cl‒ and HCO3‒, as shown by Eqs. (8) ‒ (14). Moreover, through Eq. (8) and Eq. (9) SO42‒ can be converted into the aqueous electron (e‒) and highly reactive SO4•‒, respectively. In this way, aqueous electron (E° = ‒2.9 V) could attack halogenated organic compounds [20].
SO4•‒ + SO42‒ → S2O82‒ + e‒ (4.4 × 108 M-1 s-1)
•OH + SO42‒ → SO4•‒ + OH‒ (3.5 × 105 M-1 s-1)
h+ + SO42‒ → SO4•‒
SO4•‒ + e‒ → SO42‒
SO4•‒ + NO3‒ → NO3•‒ + SO42‒ (5.5 × 105 M-1 s-1)
•OH + NO3‒ → NO3•‒ + OH‒ (< 5 × 105 M-1 s-1)
h+ + NO3‒ → NO3•
According to Eqs. (15) and (16), chloride ion can be thermodynamically oxidized by SO4•‒ to chlorine radicals and therefore chloride ion showed a radicals scavenging role in the reaction which decrease the process efficiency from 82.6 to 30.17% (Figure 6). Thus, the lower redox potential of Cl2•‒/2Cl‒ (E° = 2.09 V) and Cl•/Cl‒ (E° = 2.47 V) compared with that couple of SO4•‒/SO42‒ (E° = 2.5-3.1 V) may turn SO4•‒ to slower oxidants (i.e. Cl•, ClOH•‒, and Cl2•‒). PMS (E° = 1.75 V) could also be converted into Cl2 and HOCl (E° Cl2/2Cl‒ = 1.36 V and E° HOCl/Cl‒ = 1.48 V) in the presence of chloride ion according Eqs. (20) and (21) [38].
SO4•‒ + Cl‒ ↔ SO42- + Cl• (2.3 × 108 M-1 s-1)
•OH + Cl‒ ↔ ClOH•‒ (4.2 × 109 M-1 s-1)
ClOH•‒ + H+ → Cl• + H2O (6.1 × 109 M-1 s-1)
h+ + Cl‒ → Cl•
Cl• + Cl‒ ↔ Cl2•‒ (7.8 × 109 M-1 s-1)
HSO5‒ + Cl‒ ↔ SO42- + HOCl
HSO5‒ + 2Cl‒ + H+ ↔ SO42- + Cl2 + H2O
As shown in Figure 6, an obvious decrease (39.8%) in the degradation efficiency was observed in the presence of HCO3‒ which may be due to the fast scavenging of the photo-oxidizing species (SO4•‒, •OH, and h+) by HCO3‒ and production of CO3•‒ via Eqs. (22)–(24) thus, mitigating the consumption of BTA [18]. Despite high availability of HCO3•‒/CO3•‒, promoting effect on BTA removal did not occurred because of their low oxidation capability [39]. HCO3‒ was frequently found to inhibit the PMS degradation.
SO4•‒ + HCO3‒ → SO42‒ + CO3•‒ + H+ (1.6 × 106 M-1 s-1)
•OH + HCO3‒ → CO3•‒ + H2O (8.5 × 106 M-1 s-1)
h+ + HCO3‒ → HCO3•‒
3.6. Radical Identification Experiments
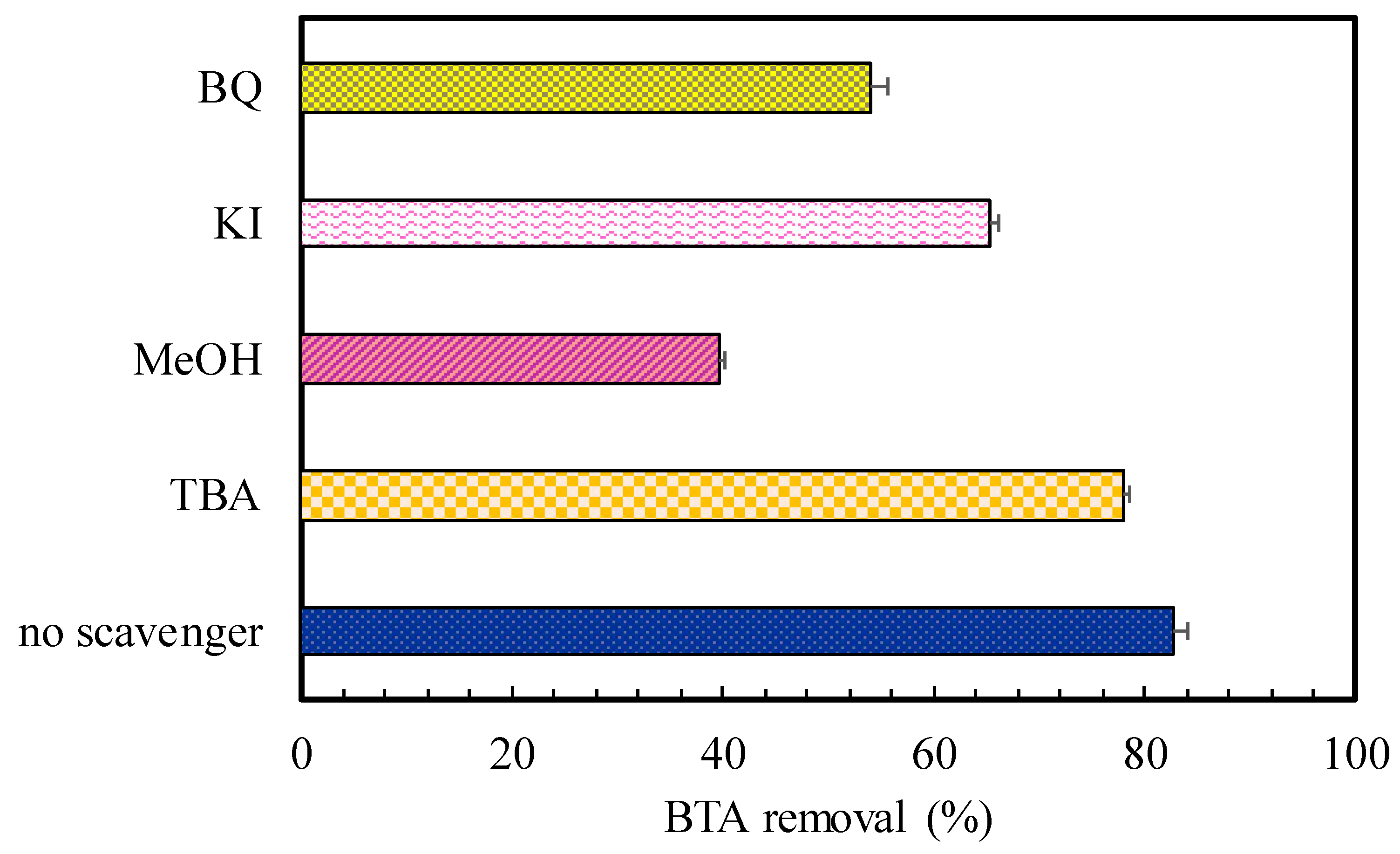
To investigate the active species which play a significant role in CuFe2O4/UV/PMS system as the most oxidizing agents in BTA degradation (Figure 3), quenching tests were performed. The quenching agents including TBA, MeOH, BQ, and KI were used in concentration of 10 mM to correspondingly trap •OH, SO4•‒, O2•‒ and h+, as the most probable ROS involved in the heterogeneous photocatalytic PMS oxidation process [9,16]. According to the different quenching reaction rates, MeOH can similarly quench both SO4•‒ and •OH (k MeOH/•OH = 9.7 × 108 M-1 s-1, k MeOH/SO4•‒ = 3.2 × 106 M-1 s-1) while TBA with 1000 times higher reactivity towards •OH compared to SO4•‒ (k TBA/•OH = 3.8-7.6 × 108 M-1 s-1, k TBA/SO4•‒ = 4.0-9.1 × 105 M-1 s-1), is only able to quickly scavenge •OH radicals [40]. Therefore, the difference in inhibiting abilities will clarify the dominant contribution of active spices in the reaction solutions. As the represented in Figure 7, the degradation efficiency was decreased from 82.6% (without any quenchers) to 78 and 39.6% in the system with TBA or MeOH, respectively. Additionally, it was found KI showed a 65.4% reduction in process efficiency, while BQ led to strong inhibitory effect (54.0% decrease) even more than TBA. These results confirmed the production of radicals which among different generated free radicals •OH, SO4•‒, O2•‒ and h+, SO4•‒ played a key role during the BTA decomposition process in the CuFe2O4/UV/PMS system.
3.7. Mechanistic Discussion
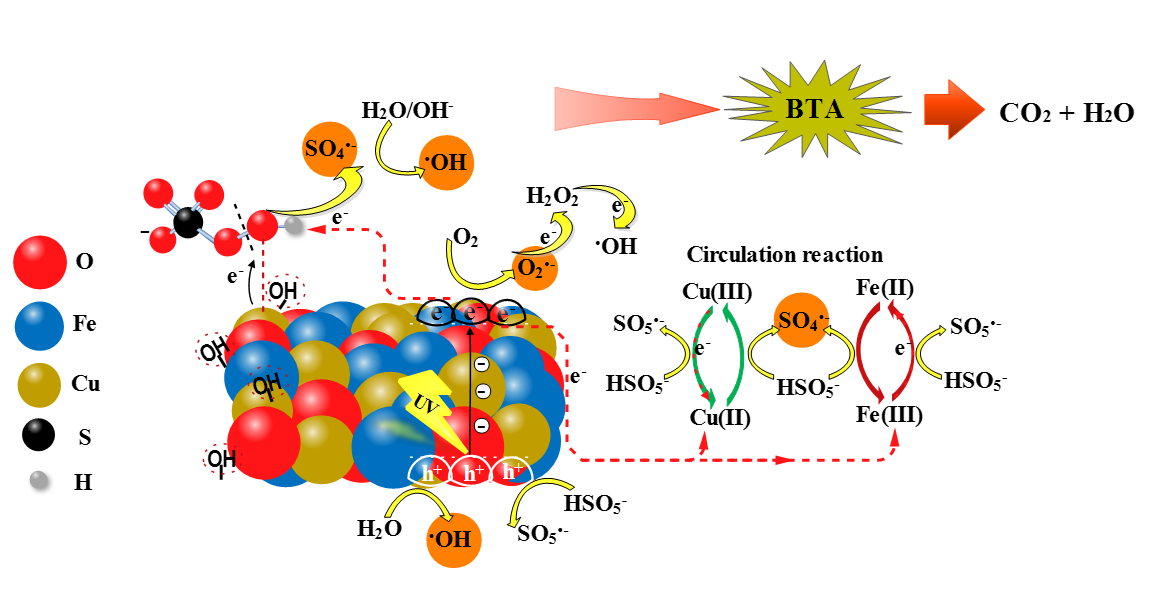
On the basis of the active species capture experiments and some previous studies [9,13,21], a possible mechanism for radicals transfer routes according to the simultaneous effect of photocatalysis and PMS activation in BTA degradation was evaluated (Scheme 1). The adsorbed light caused the excitation in the valence band (VB) of the catalyst, which could produce photo-induced electron and hole groups (e‒/h+) as Eq. (25) showed, with strong redox reaction ability. Conduction band electrons (eCB‒) of CuFe2O4 could quickly reduce dissolved O2 to O2•‒ radicals (Eq. (26)) based on the standard redox potential difference (E0eCB‒ = ˗0.48 eV vs. NHE compared to E0O2/O2•‒ = ˗0.46 eV vs. NHE) [21]. At the same time, h+ directly entered in the catalytic degradation when adsorbed H2O or OH‒ were thermodynamically able to be converted to •OH (Eq. 27), thereby establishing the photo-induced charges separation in CuFe2O4 [41]. O2•‒ could also trigger a series of redox reactions to produce •OH (Eq. (28) and (29)) [9].
CuFe2O4 + hv → hVB+ + eCB‒
CuFe2O4 (eCB‒) + O2 → O2•‒
CuFe2O4 (hVB+) + H2O → •OH + H+
O2•‒ + eCB‒ + 2H+ → H2O2
H2O2 + eCB‒ → •OH + OH‒
Additionally, it was observed that PMS further enhanced the catalytic reaction that can be attributed to the chemical stability arising from redox cycles of surface active centers. It was concluded from evidences that the process of CuFe2O4 catalyzed PMS for generating reactive species was initially related to the binding of hydroxyl groups (−OH) obtained from the dissociation of water on the surface metal (Fe or Cu) sites in CuFe2O4 catalyst [26]. In this regards, HSO5‒ could form bond with the surface Cu(II) of the catalyst via surface −OH displacement and generate Cu(II)-(OH)OSO3‒ intermediate by the inner-sphere complexation (Eq. (30)). Therefore, the favorability of the electron transfer from Cu(II), with the obtained high electron density, to OH of HSO5‒ could lead to the production of SO4•‒ radical directly and new surface −OH group which would bond to higher valence Cu(III) ion (Eq. (31)). Supposing that ≡Cu(III) −OH oxidized HSO5‒ to SO5•‒ bonded to copper initial valence (i.e., Cu(II)) (Eq. (32)), then the combination of surface SO5•‒ moieties would again generate SO4•‒ via Eq. (33). The efficient participation in redox process could result in the reductant character for Fe(II), reducing ≡Cu(III) to ≡Cu(II) (Eq. (34)), since E0≡Cu(III)/≡Cu(II) = 2.3 V is much higher than E0≡Fe(III)/≡Fe(II) = 0.77 V. The resulting ≡Fe(III) would be possibly turned to ≡Fe(II) during the process of HSO5‒ reduction (Eq. (35)), which would be afterward oxidized to ≡Fe(III) by the production of SO4•‒ radicals (Eq. (36)). Thus, redox mediators i.e., Cu(II-III-II) and Fe(III-II-III) by maintaining the reaction cycles not only enhance the activity of CuFe2O4 via Eq. (34), but also contribute to the further decomposition of PMS through Eqs. (35) and (36). From the hydrolization of SO4•‒ •OH can be released, which further yielded following the interaction of OH‒ (Eqs. (37) and (38)) [42]. Photocatalytic activation of PMS, under the action of photoinduced electrons, opened another route for direct generation of •OH and SO4•‒ radicals (Eq. (39)). In this way, the photoinduced holes could be captured by PMS to produce SO4•‒ via subsequent self-reaction of SO5•‒ radicals (Eqs. (40) and (41)). On the other hand, photo-induced electrons under the different valence states of Cu-Fe during the reaction, could provide a new equilibrium to get the cyclic Cu(III)/Cu(II) and Fe(III)/Fe(II) (Eq. (42)), leading to the fast photo charge transfer in the presence of PMS and it would be the reason for higher reactivity than the binary systems i.e., CuFe2O4/UV and CuFe2O4/PMS (Figure 3). Thus, PMS oxidation when combined with photocatalysis was synergistic with the production of more free active species. Negligible amount of SO4•‒, •OH, O2•‒, and h+ radicals, which might be leached from the surface bound layer to the solution bulk in the CuFe2O4/UV/PMS system, had key role in mineralization of BTA (Eq. (43)).
≡Cu(II)˗˗˗OH‒ + HSO5‒ → ≡Cu(II)˗˗˗(OH)˗OSO3‒ + OH‒
≡Cu(II)˗˗˗(OH)˗OSO3‒ → ≡Cu(III)˗˗˗OH‒ + SO4•‒
≡Cu(III)˗˗˗OH‒ + HSO5‒ → ≡Cu(II)˗˗˗SO5•‒ + H2O
2≡Cu(II)˗˗˗SO5•‒ + 2H2O → 2≡Cu(II)˗˗˗OH‒ + O2 + 2SO4•- + 2H+
≡Cu(III)˗˗˗OH‒ + ≡Fe(II) → ≡Cu(II)˗˗˗OH‒ + ≡Fe(III)
≡Fe(III) + HSO5‒ → ≡Fe(II) + SO5•‒ + H+
≡Fe(II) + HSO5‒ → ≡Fe(III) + SO4•‒ + OH‒
SO4•‒ + H2O → •OH + SO42‒ + H+
SO4•‒ + OH‒ → •OH + SO42‒
eCB‒ + HSO5‒ → SO4•‒ + OH‒
hVB+ + HSO5‒ → SO5•‒ + H+
2SO5•‒ → 2SO4•‒ + O2
Cu(III)/Fe(III) + eCB‒ → Cu(II)/Fe(II)
SO4•‒/•OH/O2•‒/h+ + BTA → products → CO2 + H2O + NH3
3.8. Reaction Pathway of BTA Degradation
The BTA degradation pathway in CuFe2O4/UV/PMS system was proposed and described in Figure 8 based on the identified intermediates by LC-MS analysis. As shown in Figure 8, radicals attack in active sites of the BTA molecule may first decompose the triazole ring of the molecule at site N14. In this way, the nitrogen double bond was destroyed and following the N–NH bond dissociation, it would be formed the yellow intermediate diazoimine. This primary diazo intermediate was a transient product, resulting in its stepwise oxidation to emit a nitrogen molecule and produce the colorless biradical intermediate under irradiation. On the basis of the recombination reaction, aniline would be yielded [5,43], which was in accordance with the identification of HPLC analysis with the help of the standard aniline (Figure S4). In pathway (I), the intermediates aniline radicals (P1) and benzoquinonimine (P2) appeared. In this step, aniline radicals either could be directly polymerized to dianiline (P3) and then the aromatic of aniline was opened into maleic acid (P7) [44], or give rise to produce the other intermediate 4-aminophenol (P4), that would be the main precursor for development of benzoquinone (P5) by the action of ROS. Besides, benzoquinone (P5) was made through the hydrolysis reaction by benzoquinonimine (P2) which was synchronized with the deamination. In addition, nitrobenzene (P6) was observed by the catalytic activity of ROS from the transformation of benzoquinonimine (P2) to lead the formation of maleic acid (P7) by the further deamination especially losing NO3‒ and finally converted into CO2 and H2O [45]. Under the attack of the ROS, the deprotonation process on the external carbons would also occur to produce the ring-cleavage intermediate 3-aminoprop-2-en-1-ol (P8) (pathway II) [2].
4. Conclusion
Herein, photosynergistic activation of PMS with a heterogenous CuFe2O4 catalyst was assessed to study the BTA degradation under the UV light in details . The novel designed system showed 1.17-fold and 1.3-fold increase in catalytic activity for BTA degradation (73%) than that of CuFe2O4/PMS (62.2%) and CuFe2O4/UV (53.6%) binary systems, respectively, which demonstrated synergy between UV, PMS, and catalyst. Radical quanching tests reveald the generation of multiple radicals including •OH, SO4•‒, O2•‒ and h+ through conversion of Cu(II)/Cu(III) and Fe(III)/Fe(II) on the surface of CuFe2O4 in which SO4•‒ worked dominantly. This resulted in neglegible leaching of metals and also the excellent recyclability of synthesized catalysts. As-prepared samples exhibited a good catalytic performance and low Fe and Cu leaching within four consecutive run. The possible transformation pathway of BTA was studied which started from triazol cleavge of the molecule. This study presented a feasible approach to improve the catalytic performance of wastewater treatment systems for environmental remediation.
Conflict of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledegment
This work is financed from Zabol university of medical sciences (IR.ZBMU.REC.1399.085).
References
- Bahnmueller, S.; Loi, C.H.; Linge, K.L.; Von Gunten, U.; Canonica, S. Degradation rates of benzotriazoles and benzothiazoles under UV-C irradiation and the advanced oxidation process UV/H 2 O 2. Water research 2015, 74, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, L.; Guo, C.; Zhang, Y.; Wang, S. Removal of benzotriazole from solution by BiOBr photocatalysis under simulated solar irradiation. Chemical engineering journal 2013, 221, 230–237. [Google Scholar] [CrossRef]
- Ye, J.; Zhou, P.; Chen, Y.; Ou, H.; Liu, J.; Li, C.; Li, Q. Degradation of 1H-benzotriazole using ultraviolet activating persulfate: Mechanisms, products and toxicological analysis. Chemical Engineering Journal 2018, 334, 1493–1501. [Google Scholar] [CrossRef]
- Felis, E.; Sochacki, A.; Magiera, S. Degradation of benzotriazole and benzothiazole in treatment wetlands and by artificial sunlight. Water research 2016, 104, 441–448. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, C.; Zhu, L.; Zhang, J. Photoelectrochemical activity of liquid phase deposited TiO2 film for degradation of benzotriazole. Journal of hazardous materials 2010, 175, 96–103. [Google Scholar] [CrossRef]
- Huang, H.; Wang, H.-L.; Shi, S.-B.; Jiang, W.-F. In-situ fabrication of AgI/AgnMoxO3x+ n/2/g-C3N4 ternary composite photocatalysts for benzotriazole degradation: Tuning the heterostructure, photocatalytic activity and photostability by the degree of molybdate polymerization. Separation and Purification Technology 2023, 307, 122874. [Google Scholar] [CrossRef]
- Ahmadi, M.; Kakavandi, B.; Jorfi, S.; Azizi, M. Oxidative degradation of aniline and benzotriazole over PAC@ Fe II Fe 2 III O 4: A recyclable catalyst in a heterogeneous photo-Fenton-like system. Journal of Photochemistry and Photobiology A: Chemistry, a: content-type="lang.
- Ghanbari, F.; Khatebasreh, M.; Mahdavianpour, M.; Lin, K.-Y.A. Oxidative removal of benzotriazole using peroxymonosulfate/ozone/ultrasound: Synergy, optimization, degradation intermediates and utilizing for real wastewater. Chemosphere 2020, 244, 125326. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, X.; Liu, Y.; Xia, S.; Zhao, J. Activation of peroxymonosulfate by a floating oxygen vacancies-CuFe2O4 photocatalyst under visible light for efficient degradation of sulfamethazine. Science of The Total Environment 2022, 824, 153630. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, H.; Liu, C.; Luo, S.; Liu, Y.; Yu, X.; Ma, J.; Yin, K.; Feng, H. Fast and efficient removal of As (III) from water by CuFe2O4 with peroxymonosulfate: Effects of oxidation and adsorption. Water research 2019, 150, 182–190. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, H.; Zhang, S.; Wang, S.; Tang, H. Efficient degradation of carbamazepine by easily recyclable microscaled CuFeO2 mediated heterogeneous activation of peroxymonosulfate. Journal of Hazardous Materials 2016, 317, 686–694. [Google Scholar] [CrossRef]
- Lutze, H.V.; Kerlin, N.; Schmidt, T.C. Sulfate radical-based water treatment in presence of chloride: formation of chlorate, inter-conversion of sulfate radicals into hydroxyl radicals and influence of bicarbonate. Water research 2015, 72, 349–360. [Google Scholar] [CrossRef]
- Li, X.; Chen, T.; Qiu, Y.; Zhu, Z.; Zhang, H.; Yin, D. Magnetic dual Z-scheme g-C3N4/BiVO4/CuFe2O4 heterojunction as an efficient visible-light-driven peroxymonosulfate activator for levofloxacin degradation. Chemical Engineering Journal 2023, 452, 139659. [Google Scholar] [CrossRef]
- Wang, Z.; Bush, R.T.; Sullivan, L.A.; Chen, C.; Liu, J. Selective oxidation of arsenite by peroxymonosulfate with high utilization efficiency of oxidant. Environmental science & technology 2014, 48, 3978–3985. [Google Scholar]
- Anipsitakis, G.P.; Dionysiou, D.D. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt. Environmental science & technology 2003, 37, 4790–4797. [Google Scholar]
- Wu, X.; Sun, D.; Ma, H.; Ma, C.; Zhang, X.; Hao, J. Activation of peroxymonosulfate by magnetic CuFe2O4@ ZIF-67 composite catalyst for the study on the degradation of methylene blue. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2022, 637, 128278. [Google Scholar] [CrossRef]
- Dong, X.; Ren, B.; Sun, Z.; Li, C.; Zhang, X.; Kong, M.; Zheng, S.; Dionysiou, D.D. Monodispersed CuFe2O4 nanoparticles anchored on natural kaolinite as highly efficient peroxymonosulfate catalyst for bisphenol A degradation. Applied Catalysis B: Environmental 2019, 253, 206–217. [Google Scholar] [CrossRef]
- Fayyaz, A.; Saravanakumar, K.; Talukdar, K.; Kim, Y.; Yoon, Y.; Park, C.M. Catalytic oxidation of naproxen in cobalt spinel ferrite decorated Ti3C2Tx MXene activated persulfate system: Mechanisms and pathways. Chemical Engineering Journal 2021, 407, 127842. [Google Scholar] [CrossRef]
- Yang, S.; Qiu, X.; Jin, P.; Dzakpasu, M.; Wang, X.C.; Zhang, Q.; Yang, L.; Ding, D.; Wang, W.; Wu, K. MOF-templated synthesis of CoFe2O4 nanocrystals and its coupling with peroxymonosulfate for degradation of bisphenol A. Chemical Engineering Journal 2018, 353, 329–339. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Zhang, Y.; Lai, Y.; Fang, Q.; Duan, Y. Activation of peroxymonosulfate by CuFe2O4-CoFe2O4 composite catalyst for efficient bisphenol a degradation: Synthesis, catalytic mechanism and products toxicity assessment. Chemical Engineering Journal 2021, 423, 130093. [Google Scholar] [CrossRef]
- Xin, J. ; Liu, Y.; Niu, L.; Zhang, F.; Li, X.; Shao, C.; Li, X.; Liu, Y. Three-dimensional porous CuFe2O4 for visible-light-driven peroxymonosulfate activation with superior performance for the degradation of tetracycline hydrochloride. Chemical Engineering Journal 2022, 445, 136616. [Google Scholar] [CrossRef]
- Ren, Y.; Lin, L.; Ma, J.; Yang, J.; Feng, J.; Fan, Z. Sulfate radicals induced from peroxymonosulfate by magnetic ferrospinel MFe2O4 (M= Co, Cu, Mn, and Zn) as heterogeneous catalysts in the water. Applied Catalysis B: Environmental 2015, 165, 572–578. [Google Scholar] [CrossRef]
- He, S.; Chen, Y.; Li, X.; Zeng, L.; Zhu, M. Heterogeneous photocatalytic activation of persulfate for the removal of organic contaminants in water: a critical review. ACS ES&T Engineering 2022, 2, 527–546. [Google Scholar]
- Hasija, V.; Nguyen, V.-H.; Kumar, A.; Raizada, P.; Krishnan, V.; Khan, A.A.P.; Singh, P.; Lichtfouse, E.; Wang, C.; Huong, P.T. Advanced activation of persulfate by polymeric g-C3N4 based photocatalysts for environmental remediation: a review. Journal of hazardous materials 2021, 413, 125324. [Google Scholar] [CrossRef] [PubMed]
- Kakavandi, B.; Alavi, S.; Ghanbari, F.; Ahmadi, M. Bisphenol A degradation by peroxymonosulfate photo-activation coupled with carbon-based cobalt ferrite nanocomposite: Performance, upgrading synergy and mechanistic pathway. Chemosphere 2022, 287, 132024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhu, H.; Croué, J.-P. Production of sulfate radical from peroxymonosulfate induced by a magnetically separable CuFe2O4 spinel in water: efficiency, stability, and mechanism. Environmental science & technology 2013, 47, 2784–2791. [Google Scholar]
- Wang, Y.; Tian, D.; Chu, W.; Li, M.; Lu, X. Nanoscaled magnetic CuFe2O4 as an activator of peroxymonosulfate for the degradation of antibiotics norfloxacin. Separation and Purification Technology 2019, 212, 536–544. [Google Scholar] [CrossRef]
- Ding, Y.; Zhu, L.; Wang, N.; Tang, H. Sulfate radicals induced degradation of tetrabromobisphenol A with nanoscaled magnetic CuFe2O4 as a heterogeneous catalyst of peroxymonosulfate. Applied Catalysis B: Environmental 2013, 129, 153–162. [Google Scholar] [CrossRef]
- Guan, Y.-H.; Ma, J.; Ren, Y.-M.; Liu, Y.-L.; Xiao, J.-Y.; Lin, L.-q.; Zhang, C. Efficient degradation of atrazine by magnetic porous copper ferrite catalyzed peroxymonosulfate oxidation via the formation of hydroxyl and sulfate radicals. Water research 2013, 47, 5431–5438. [Google Scholar] [CrossRef]
- Golshan, M.; Kakavandi, B.; Ahmadi, M.; Azizi, M. Photocatalytic activation of peroxymonosulfate by TiO2 anchored on cupper ferrite (TiO2@ CuFe2O4) into 2, 4-D degradation: Process feasibility, mechanism and pathway. Journal of hazardous materials 2018, 359, 325–337. [Google Scholar] [CrossRef]
- Lei, X.; You, M.; Pan, F.; Liu, M.; Yang, P.; Xia, D.; Li, Q.; Wang, Y.; Fu, J. CuFe2O4@ GO nanocomposite as an effective and recoverable catalyst of peroxymonosulfate activation for degradation of aqueous dye pollutants. Chinese Chemical Letters 2019, 30, 2216–2220. [Google Scholar] [CrossRef]
- Xu, Y.; Ai, J.; Zhang, H. The mechanism of degradation of bisphenol A using the magnetically separable CuFe2O4/peroxymonosulfate heterogeneous oxidation process. Journal of Hazardous Materials 2016, 309, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H.; Ang, H.M.; Tadé, M.O.; Wang, S. Facile synthesis of hierarchically structured magnetic MnO2/ZnFe2O4 hybrid materials and their performance in heterogeneous activation of peroxymonosulfate. ACS Applied Materials & Interfaces 2014, 6, 19914–19923. [Google Scholar]
- Liu, F.; Wang, X.; Liu, Z.; Miao, F.; Xu, Y.; Zhang, H. Peroxymonosulfate enhanced photocatalytic degradation of Reactive Black 5 by ZnO-GAC: Key influencing factors, stability and response surface approach. Separation and Purification Technology 2021, 279, 119754. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, J.; Liu, D.; Li, L.; Liu, W.; Liu, S.; Feng, C. Construction of Z-scheme CuFe2O4/MnO2 photocatalyst and activating peroxymonosulfate for phenol degradation: Synergistic effect, degradation pathways, and mechanism. Environmental Research 2021, 200, 111736. [Google Scholar] [CrossRef] [PubMed]
- Kakavandi, B.; Ahmadi, M. Efficient treatment of saline recalcitrant petrochemical wastewater using heterogeneous UV-assisted sono-Fenton process. Ultrasonics Sonochemistry 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Li, D.; Yao, Z.; Lin, J.; Tian, W.; Zhang, H.; Duan, X.; Wang, S. Nano-sized FeVO4· 1.1 H2O and FeVO4 for peroxymonosulfate activation towards enhanced photocatalytic activity. Journal of Environmental Chemical Engineering 2022, 10, 107199. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D.; Gonzalez, M.A. Cobalt-mediated activation of peroxymonosulfate and sulfate radical attack on phenolic compounds. Implications of chloride ions. Environmental science & technology 2006, 40, 1000–1007. [Google Scholar]
- Nie, M.; Yang, Y.; Zhang, Z.; Yan, C.; Wang, X.; Li, H.; Dong, W. Degradation of chloramphenicol by thermally activated persulfate in aqueous solution. Chemical Engineering Journal 2014, 246, 373–382. [Google Scholar] [CrossRef]
- Li, Z.; Tang, X.; Huang, G.; Luo, X.; He, D.; Peng, Q.; Huang, J.; Ao, M.; Liu, K. Bismuth MOFs based hierarchical Co3O4-Bi2O3 composite: An efficient heterogeneous peroxymonosulfate activator for azo dyes degradation. Separation and Purification Technology 2020, 242, 116825. [Google Scholar] [CrossRef]
- Golshan, M.; Zare, M.; Goudarzi, G.; Abtahi, M.; Babaei, A.A. Fe3O4@ HAP-enhanced photocatalytic degradation of Acid Red73 in aqueous suspension: Optimization, kinetic, and mechanism studies. Materials Research Bulletin 2017, 91, 59–67. [Google Scholar] [CrossRef]
- Li, C.; Huang, Y.; Dong, X.; Sun, Z.; Duan, X.; Ren, B.; Zheng, S.; Dionysiou, D.D. Highly efficient activation of peroxymonosulfate by natural negatively-charged kaolinite with abundant hydroxyl groups for the degradation of atrazine. Applied Catalysis B: Environmental 2019, 247, 10–23. [Google Scholar] [CrossRef]
- Serdechnova, M.; Ivanov, V.L.; Domingues, M.R.M.; Evtuguin, D.V.; Ferreira, M.G.; Zheludkevich, M.L. Photodegradation of 2-mercaptobenzothiazole and 1, 2, 3-benzotriazole corrosion inhibitors in aqueous solutions and organic solvents. Physical Chemistry Chemical Physics 2014, 16, 25152–25160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, F.; Zhou, G.; Ni, Y. Aniline degradation by electrocatalytic oxidation. Chemosphere 2003, 53, 1229–1234. [Google Scholar] [CrossRef]
- Jorfi, S.; Kakavandi, B.; Motlagh, H.R.; Ahmadi, M.; Jaafarzadeh, N. A novel combination of oxidative degradation for benzotriazole removal using TiO2 loaded on FeIIFe2IIIO4@ C as an efficient activator of peroxymonosulfate. Applied Catalysis B: Environmental 2017, 219, 216–230. [Google Scholar] [CrossRef]
Figure 1.
FESEM image (A) and EDS spectrum (B) of CuFe2O4; TEM microgrphs of CuFe2O4 (C, D) with different scale bar.
Figure 1.
FESEM image (A) and EDS spectrum (B) of CuFe2O4; TEM microgrphs of CuFe2O4 (C, D) with different scale bar.

Figure 2.
(A) XRD patterns (A), (B) N2 adsorption-desorption isotherms (insert: pore size distribution), (C) Magnetic hysteresis loops, and (D) and UV–Vis absorption spectra of CuFe2O4 (E).
Figure 2.
(A) XRD patterns (A), (B) N2 adsorption-desorption isotherms (insert: pore size distribution), (C) Magnetic hysteresis loops, and (D) and UV–Vis absorption spectra of CuFe2O4 (E).
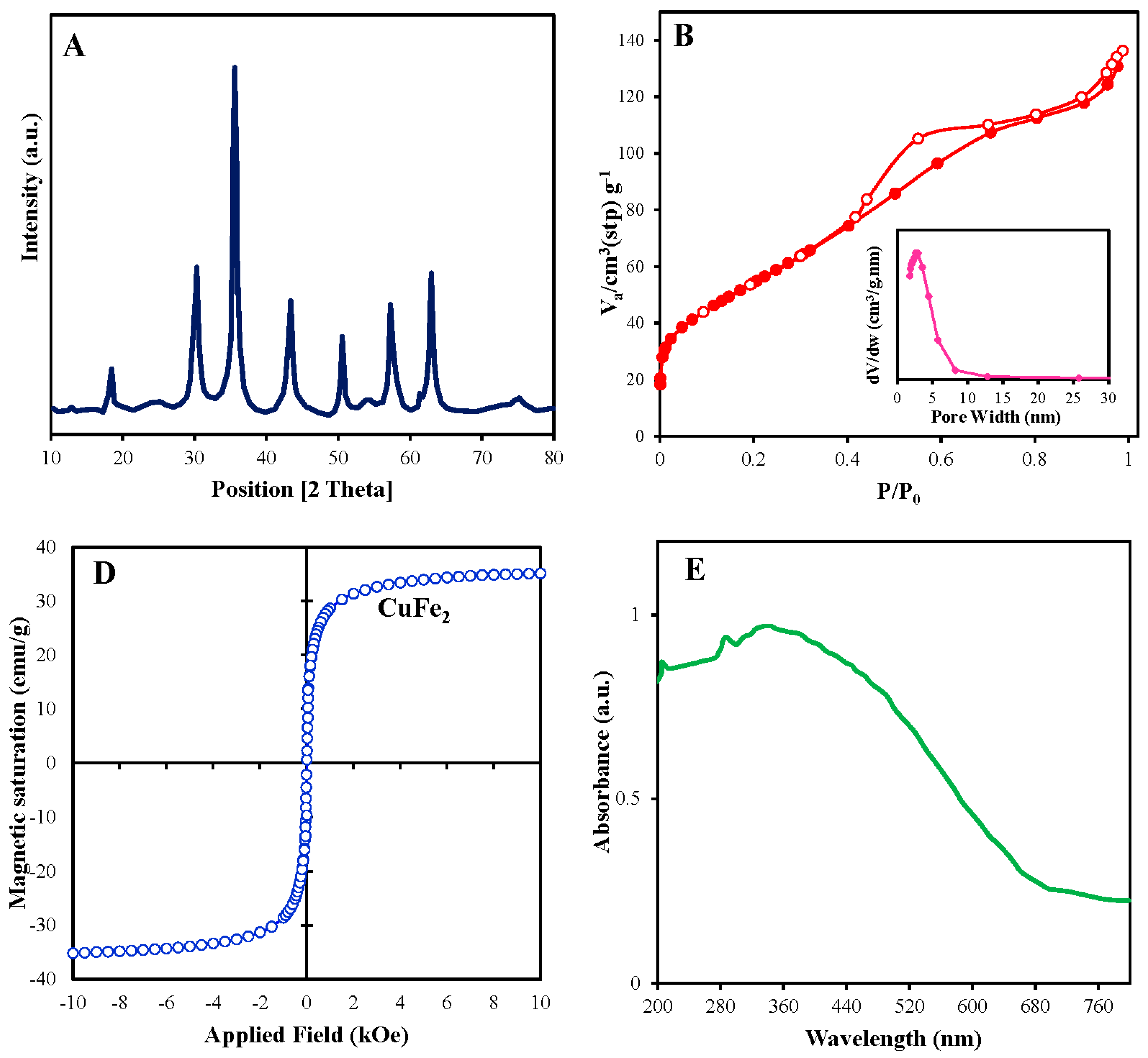
Figure 3.
Removal efficiencies of BTA in different reaction systems. Conditions: BTA = 40 mg L-1, catalyst (CuFe2O4) = 0.2 g L-1 and PMS = 1.5 mM during 60 min reaction time.
Figure 3.
Removal efficiencies of BTA in different reaction systems. Conditions: BTA = 40 mg L-1, catalyst (CuFe2O4) = 0.2 g L-1 and PMS = 1.5 mM during 60 min reaction time.
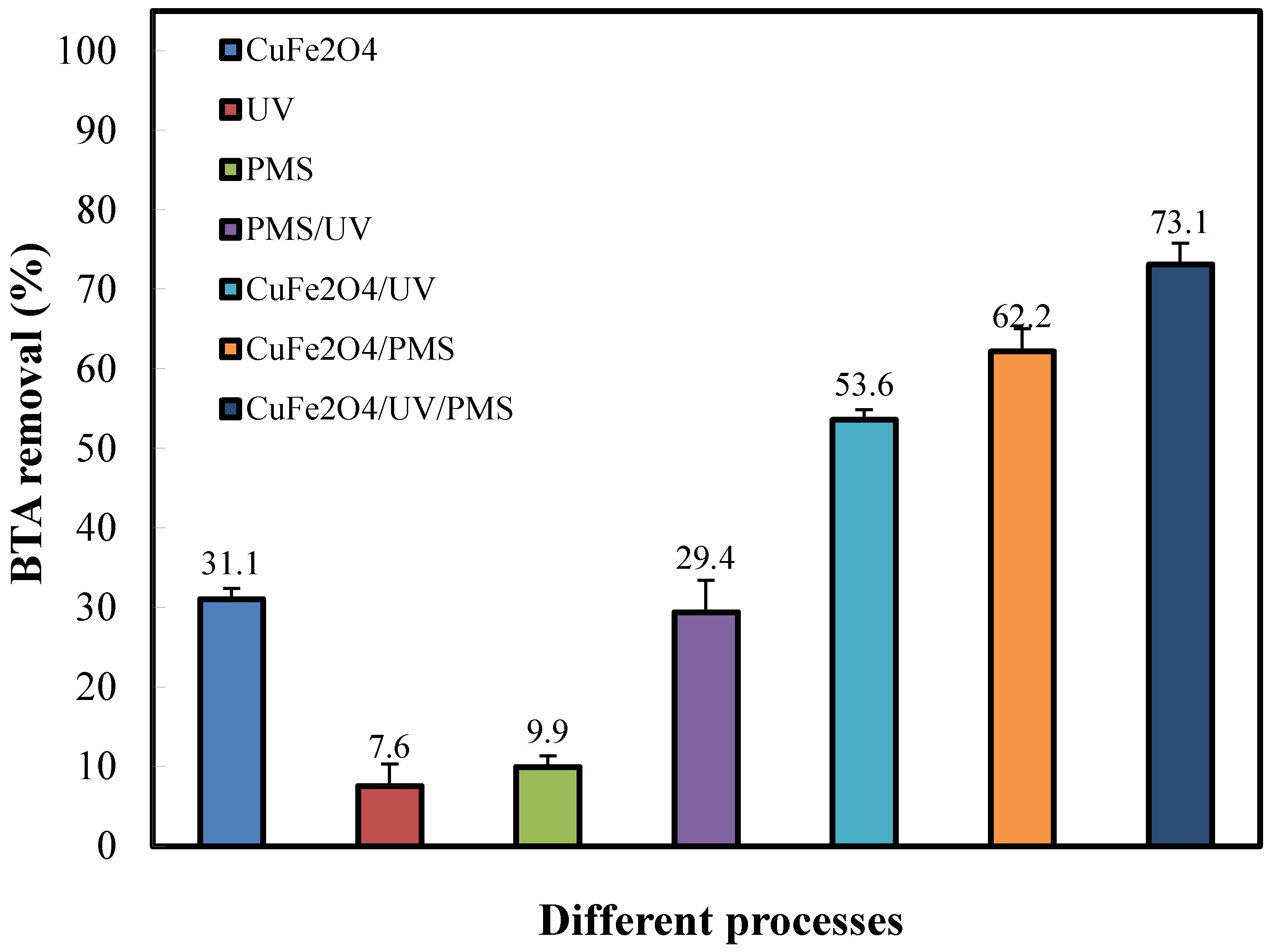
Figure 4.
Effect of operating factors on the BTA removal rate over CuFe2O4/UV/PMS system: Catalyst dosage vs PMS concentration (A), catalyst dosage vs reaction time (B), BTA concentration vs reaction time (C). Other variables were kept at zero level.
Figure 4.
Effect of operating factors on the BTA removal rate over CuFe2O4/UV/PMS system: Catalyst dosage vs PMS concentration (A), catalyst dosage vs reaction time (B), BTA concentration vs reaction time (C). Other variables were kept at zero level.
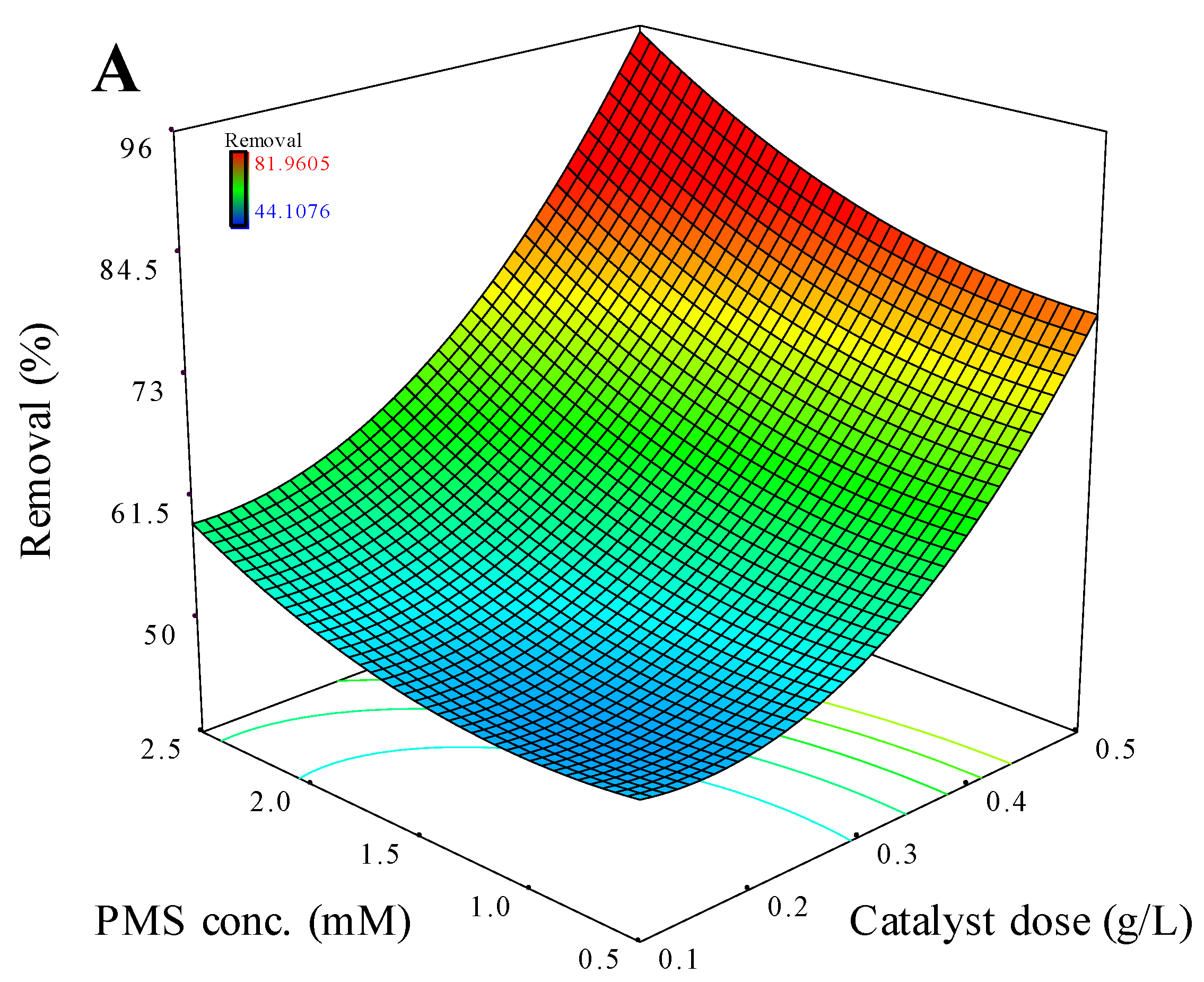
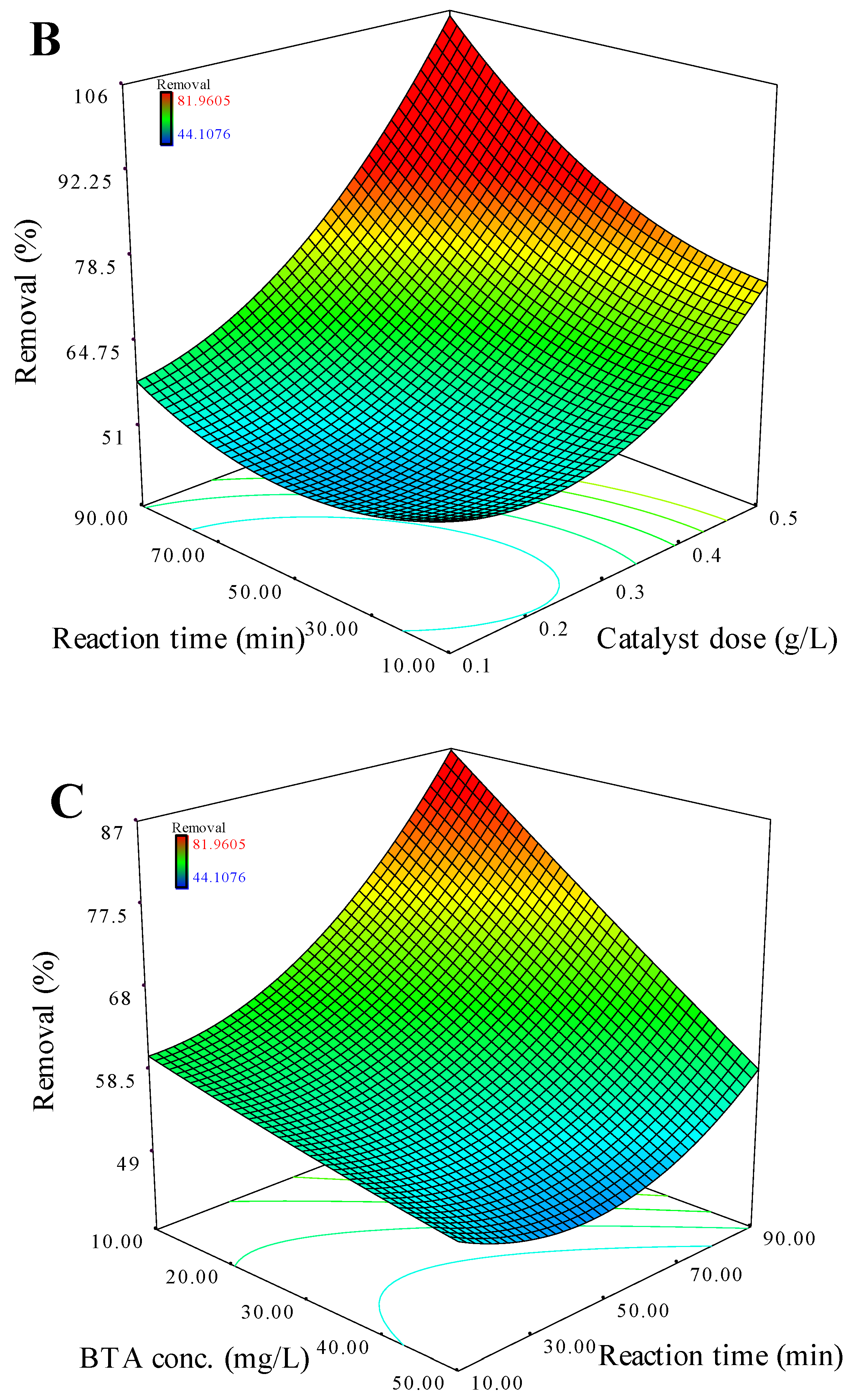
Figure 5.
Performance of the recycled catalysts in the CuFe2O4/UV/PMS system on BTA mineralization (A) and Cu and Fe leaching from CuFe2O4 after each cycle (B). Conditions: C0 = 20 mg L-1, CuFe2O4 = 0.4 g L-1, PMS = 2 mM, time = 70 min.
Figure 5.
Performance of the recycled catalysts in the CuFe2O4/UV/PMS system on BTA mineralization (A) and Cu and Fe leaching from CuFe2O4 after each cycle (B). Conditions: C0 = 20 mg L-1, CuFe2O4 = 0.4 g L-1, PMS = 2 mM, time = 70 min.
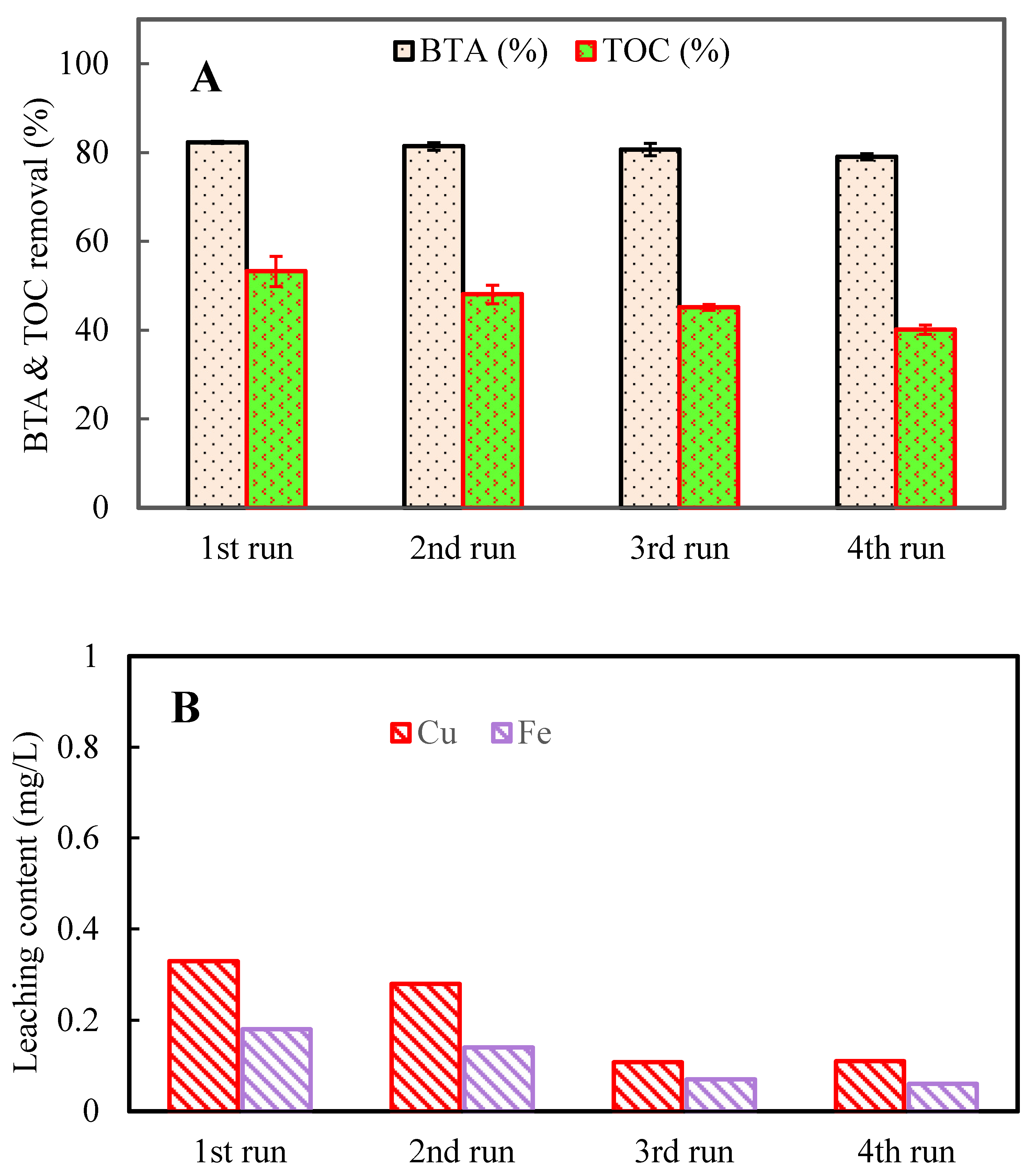
Figure 6.
Percentage of BTA removal in presence of different anions. Conditions: C0 = 20 mg L-1, CuFe2O4 = 0.4 g L-1, PMS = 2 mM, time = 70 min.
Figure 6.
Percentage of BTA removal in presence of different anions. Conditions: C0 = 20 mg L-1, CuFe2O4 = 0.4 g L-1, PMS = 2 mM, time = 70 min.
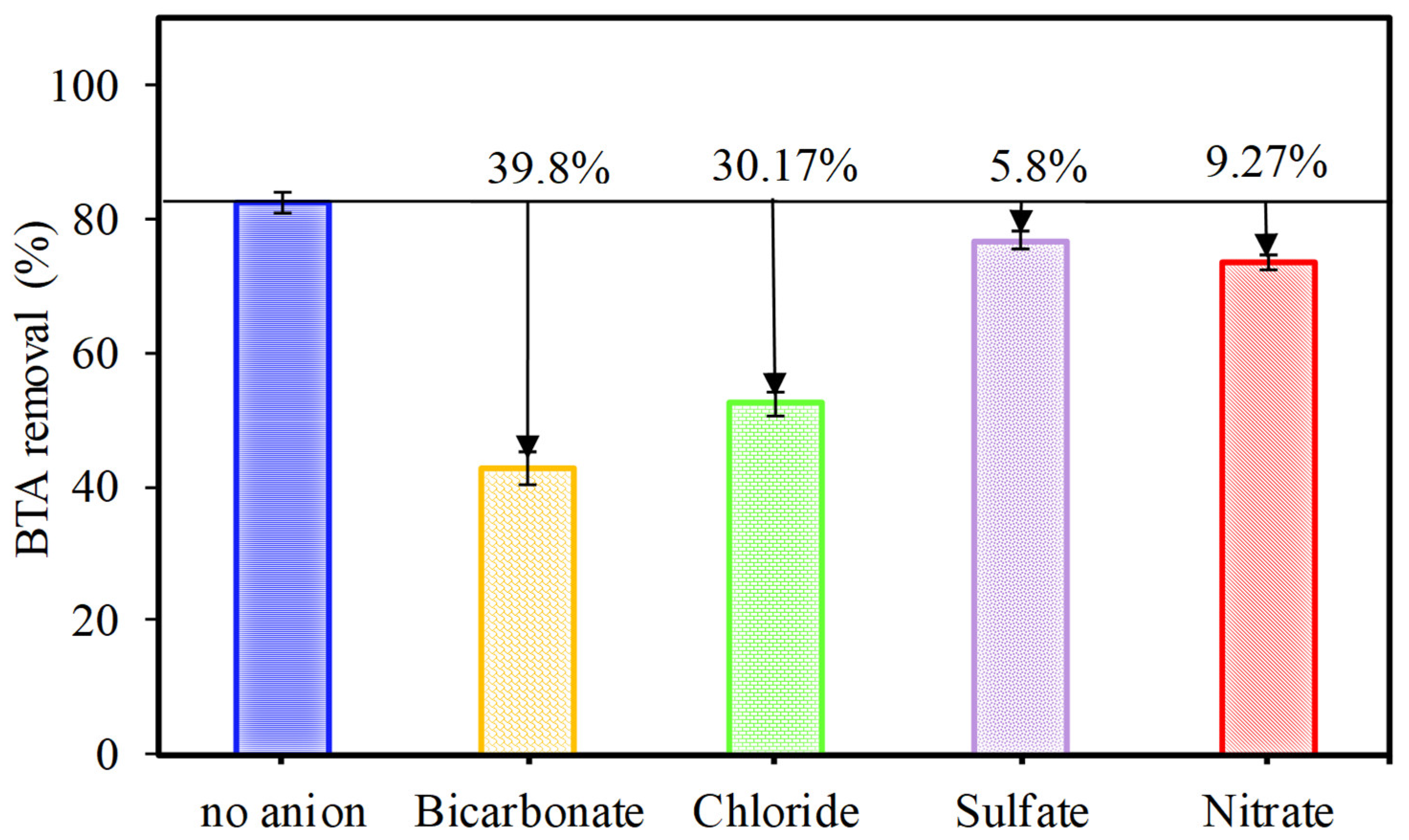
Figure 7.
The effects of different radical scavengers on BTA degradation. Conditions: C0 = 20 mg L-1, CuFe2O4 = 0.4 g L-1, PMS = 2 mM, time = 70 min.
Figure 7.
The effects of different radical scavengers on BTA degradation. Conditions: C0 = 20 mg L-1, CuFe2O4 = 0.4 g L-1, PMS = 2 mM, time = 70 min.
Scheme 1.
Schematic diagram of the reaction mechanism for degradation of BTA by CuF2O4/UV/PMS process.
Scheme 1.
Schematic diagram of the reaction mechanism for degradation of BTA by CuF2O4/UV/PMS process.
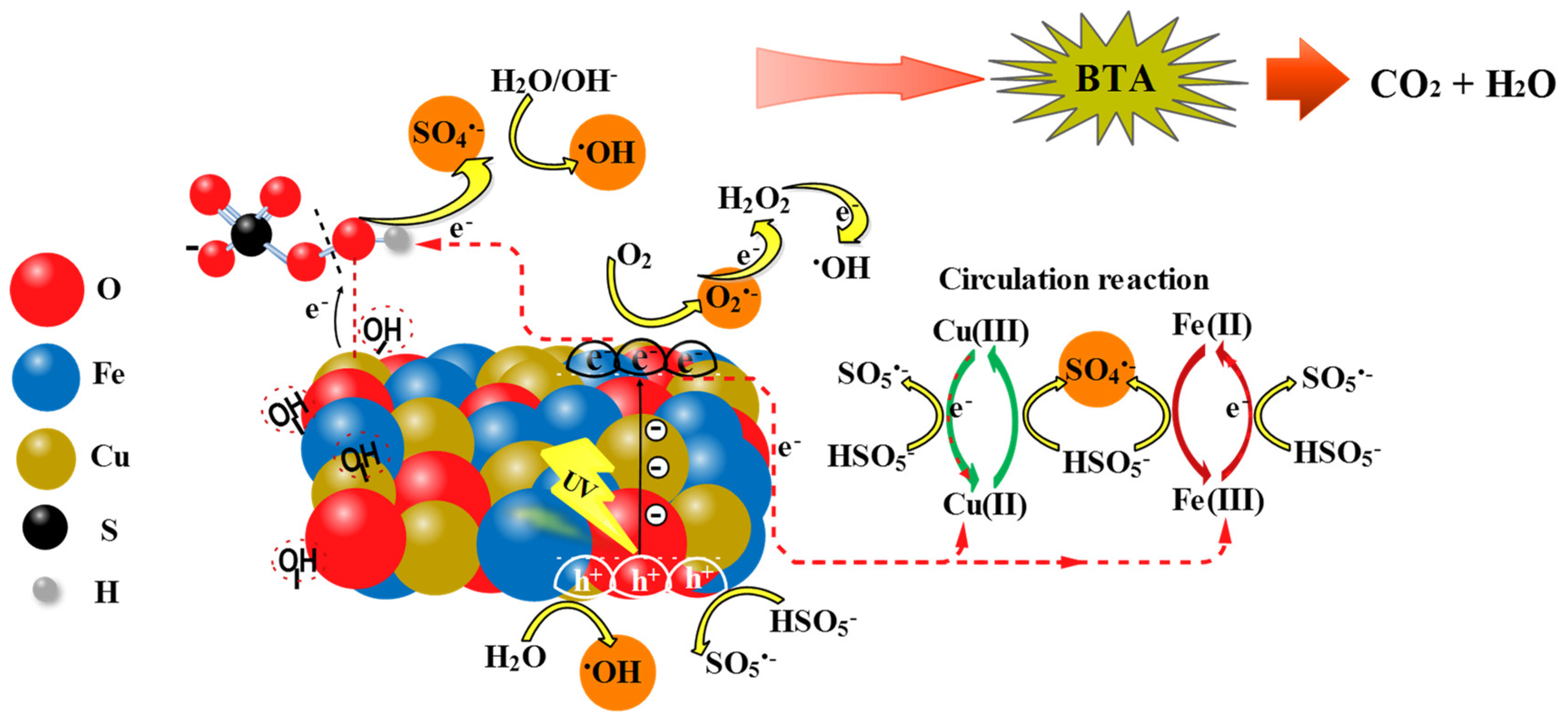
Figure 8.
Shematic degradation pathways of BTA during the CuFe2O4/UV/PMS process based on the potential consequences.
Figure 8.
Shematic degradation pathways of BTA during the CuFe2O4/UV/PMS process based on the potential consequences.
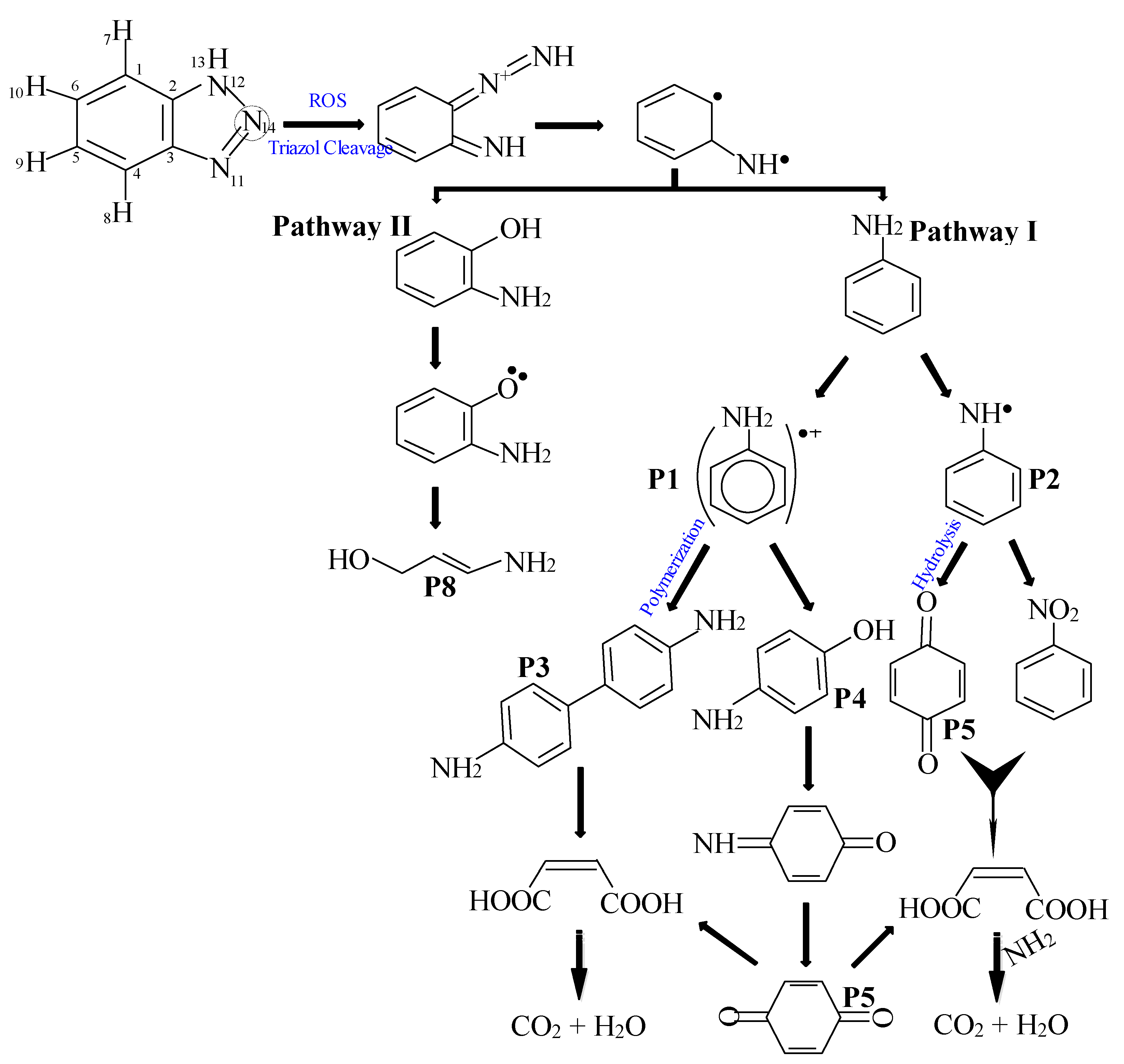

Table 1.
Four independent factors and their ranges at various levels.
| Independent factors | Unit | Symbols | Ranges and levels | ||||
| -α | Low(-1) | Middle(0) | High(+1) | +α | |||
| Catalyst loading | g L-1 | x1 | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 |
| PMS dosage | mM | x2 | 0.5 | 1 | 1.5 | 2 | 2.5 |
| Initial BTA concentration | mg L-1 | x3 | 10 | 20 | 30 | 40 | 50 |
| Irradiation time | min | x4 | 10 | 30 | 50 | 70 | 90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



