
Submitted:
30 March 2023
Posted:
03 April 2023
You are already at the latest version
Abstract
Glioblastoma (GBM) is the most common and aggressive adult brain cancer with an average survival rate of around 15 months in patients receiving standard treatment. Oncolytic adenovirus expressing therapeutic transgenes represent a promising alternative treatment for GBM. Of the many human adenoviral serotypes described to date, adenovirus 5 (Ad5) has been most utilized clinically and experimentally. However, the use of Ad5 as an anti-cancer agent may be hampered by naturally high seroprevalence rates to Ad5 coupled with infection of healthy cells via native receptors. To explore whether alternative natural adenoviral tropisms are better suited to GBM therapeutics, we pseudotyped an Ad5 based platform with the fiber knob protein from alternative serotypes. We demonstrate that the adenoviral entry receptors coxsackie and adenovirus receptor (CAR) and CD46 are highly expressed by both GBM and healthy brain tissue, whereas Desmoglein 2 (DSG2) is expressed at low level in GBM. We demonstrate that adenoviral pseudotypes, engaging CAR, CD46 and DSG2, effectively transduce GBM cells. However, the presence of these receptors on non-transformed cells presents the possibility of off-target effects and therapeutic transgene expression in healthy cells. To enhance specificity of transgene expression to GBM, we assessed the potential for tumour specific promoters hTERT and survivin to drive reporter gene expression selectively in GBM cell lines. We demonstrate tightly GBM specific transgene expression using these constructs, indicating that the combination of pseudotyping and tumour specific promoters approaches may enable the development of efficacious therapies better suited to GBM.
Keywords:
brain tumour
; oncolytic virus
; receptor
; therapy
1. Introduction
Glioblastoma (GBM), a tumour thought to arise from neuroglial stem or progenitor cells, is the most aggressive primary adult brain cancer. It has an average incidence of 3 cases per 100 000 people per year worldwide[1], making it the most common type of malignant adult brain neoplasm. The current standard treatment procedure for GBM involves the maximum surgical resection of the tumour (where possible), followed by radiation therapy and concomitant chemotherapy with the oral alkylating agent temozolomide (TMZ). Nevertheless, due to its heterogeneity, invasiveness, and rapid growth, the prognosis for GBM patients is very poor, where mean overall survival is only approximately 15 months [2].
Oncolytic viruses, which are viruses engineered to selectively infect and lyse tumour cells, constitute a promising alternative therapeutic agent for GBM (reviewed in [3]). Their ability to self-amplify within tumour cells not only expands the therapy at the point of need (i.e within the tumour microenvironment, TME), but also induces immunogenic cell death (ICD) through the lytic nature of cell death. The power of oncolytic viruses can be further augmented by engineering potent therapeutic transgenes into the viral genome that can further enhance immune activation and immune cell mediated tumour cell killing. Collectively, these traits have the potential to instigate an anti-glioma immune response against both the primary tumour and metastatic growth within the brain.
Whilst the array of viruses available for oncolytic applications are wide, adenoviruses (Ad) have proven most popular as oncolytic viruses against several types of cancer, as gauged by the volume of clinical trials conducted in the area to date [4]. They have a proven safety profile clinically, are relatively amenable to genetic manipulation, and are able to accommodate relatively large transgene inserts [5]. However, to date, the promising results demonstrated in vitro and in murine models of cancer have largely failed to translate into significant efficacy in clinical trials. Whilst these trials demonstrated feasibility and safety, improvements to cancer patient outcome have generally been modest . Limited efficacy may be due in part to the over reliance of many clinical studies focusing on adenovirus 5 (Ad5).
There have been over 100 human adenoviral serotypes described to date with differing entry receptors as well prevalence rates within the human population [8]. Ad5 has a particularly high prevalence in the human population, especially in Africa and Asia. Neutralising antibodies against Ad5, generated in response to a natural pathogenic infection, may hamper the efficacy of Ad5 based therapies when deployed clinically due to immune inactivation. Alternative serotypes including Ad10 have been explored to overcome the effects of anti-Ad5 neutralization [11]. In addition, Ad5 engages coxsackie and adenovirus receptor (CAR) [12] as a primary means of cell entry, a receptor that is expressed both on erythrocytes and the tight junctions of epithelial cells (reviewed in [13]), yet is commonly downregulated by some types of cancer . Furthermore, interactions between the major Ad5 capsid protein, hexon, and blood clotting factors, particularly factor (F) X, result in rapid and efficient cellular uptake of Ad5 virions via widely expressed heparan sulphate proteoglycans (HSPGs) [16,17,18]. This results in depletion of the therapeutic effects due to widespread off-target infection and sequestration with inefficient tumour cell infection by Ad5 [19]. Consequently, studies investigating Ad5 based therapies for recurrent GBM, have so far only been able to demonstrate modest success. Whilst demonstrating safety and oncolysis by the intratumoural delivery of the virus, overall median patient survival was not improved from standard of care (13 months) as the tumour invariably returned [20]. Local delivery is likely a necessity in GBM to overcome the physical obstacle of the blood brain barrier and to prevent local recurrence after surgical resection. We therefore investigated whether the development of Ad based therapeutics using alternative adenovirus entry receptors may represent a more potent virotherapy for GBM. We have focused on Ad5 vectors pseudotyped with fiber knob proteins derived from serotypes Ad26, Ad35 and Ad3 which utilise the cell entry receptors sialic acid (SA) [21], or CD46 [22] and Desmoglein 2 (DSG2) [23] respectively. We demonstrate that GBM cell lines and tissue express all three adenoviral entry receptors tested, namely CAR, CD46 and DSG2. In addition, sialic acid is known to be expressed at high levels in the brain[24]. We have also shown that GBM does not express αvβ6 integrin, which is upregulated in many other types of aggressive cancers (e.g., ovarian, lung, skin and cervical cancer [25], and is a promising target for novel adenoviral oncolytic therapies [16,17,18]. We also investigated the efficacy of various Ad5 pseudotypes to gauge which adenoviral serotype may be better suited for oncolytic therapy in GBM. Our experiments revealed that both GBM and GBM stem cells express recognized adenoviral receptors and can be transduced by Ad5 and Ad5 with pseudotyped fiber knob proteins binding CD46, sialic acid and DSG2. Since these entry receptors are not unique to transformed GBM cells, and are expressed on normal cells, we additionally investigated whether improving selectivity, through the incorporation of tumour specific promoters, could be employed to regulate the expression of potentially toxic or immunostimulatory transgenes to transformed GBM cells. Our findings indicate that a combination of alternative receptor usage, either through the use of pseudotyped or whole serotyped vectors, in combination with tumour specific promoters such as survivin, may offer a powerful and highly selective means to deliver cytotoxic or immunostimulatory payloads selectively to GBM cells.
2. Materials and Methods
2.1. Cell lines and Adenoviral vectors
Human patient derived GBM stem cell lines, L0 and L1 [29] were cultured as described previously [16,17,18]. Briefly, cells were cultured in DMEM-F12 supplemented with N2 (Thermo Fisher, cat. no. 17502048) supplemented with 20 ng/ml EGF (R&D systems, cat. no. 236-EG-200), 20 ng/ml FGF (R&D systems, cat. no. 233-FB-010) and 100 μg/ml heparin (Sigma, cat. no. H4784). Cells were seeded at a density of 50,000 cells/ ml, cultured as neurospheres and passaged every 7 days using Accutase (Sigma, cat. no. A6964) to disassociate the cells. E51 and E55 were obtained from the Glioma Cellular Genetics Resource (GCGR) from Professor Steve Pollard (University of Edinburgh). E51 and E55 were derived from GBM patients as previously and cultured as previously described [32]. Cells were grown in DMEM-F12 (cat. No. D8437). Complete DMEM-F12 with 1.25% glucose (Sigma G8644), 2% l MEM-NEAA (Gibco, 11140-035) and Penicillin and Streptomycin, 0.16% BSA solution 7.5% (Gibco, 15260-037) 0.2% beta mercaptoethanol (Gibco 31350-010), 1% B27 Supplement (Gibco, 17504-044) and 0.5% N2 supplement (Gibco 17502-048). Complete media was supplemented with mouse EGF (10ng/ml, peprotech 315-09), human FGF (10ng/ml, peprotech 100-18b) and laminin (2µg/ml, Sigma L2020). HFCAR cells were generated in house and cultured in DMEM supplemented with 10% FBS, 2% Penicillin and Streptomycin and 1% L-Glutamine.
Adenoviral vectors were produced as described previously using recombineering techniques [33]. Replication deficient Ad5 expressing a luciferase reporter gene provided the backbone for replacement of fiber knob (K) protein to generate Ad5/26K, Ad5/35K and Ad5/49K [21]. Ad5/3K expressing GFP reporter gene was provided by Professor Andre Lieber (University of Washington). Ad5 vectors with luciferase expression under the control of the tumour specific promoters hTERT and Survivin and Survivin/hTERT fusion promoter (Survivin-luciferase-BHG poly A-hTERT) were generated in house using recombineering techniques. Promoter sequences have been included in Supplemental Table 1. Viral vectors were expanded in 293-TRex cells and purified using two step CsCl gradient ultracentrifugation.
2.2. Flow cytometry
Cells were incubated in FACS buffer (PBS (Thermo Fisher, cat. no. 10010023) with 5% fetal bovine serum (Sigma, cat. no. F9665)) and labelled with mouse primary antibodies: anti-αvβ6 10D5 (Millipore, cat no. mab20772), anti-CAR RmcB (Millipore, cat. no. 05-644), anti-CD46 258-MEM (Novus Biologicals, cat. no. LS-B5950-50) or anti-DSG2 CSTEM28 (Thermo Fisher, cat. no. 14-9159-82); all antibodies were diluted 1:500. Cells were subsequently stained with a secondary anti-mouse 647-conjugated antibody (Thermo Fisher) and fixed with 4% paraformaldehyde. Appropriate fluorescence gating parameters were established with unstained and matched isotype control IgG-stained cells. In all experiments, doublets were eliminated using pulse geometry gates (FSC-H versus FSC-A). Single-cell suspensions were analysed using the BD AccuriTM C6 flow cytometer; FlowJo software (FlowJo LLC) was used for subsequent analyses.
2.3. Viral infection and luciferase assays
Viral transduction assays were performed as described previously [34]. Briefly, cells were seeded at a density of 20,000 cells/ well in a 96-microwell plate (Nunc, Thermo Fisher) and infected with concentrations of 1000, 5000 or 10000 virus particles (vp)/ cell the following day. For each experimental condition triplicate wells were treated, and a no virus control was included. After 72 hrs, cells were lysed and the luciferase activity quantified using a BioTek microplate reader using the luciferase assay system (Promega, cat. no. E1500) according to the manufacturer’s instructions. In addition, total protein concentration per well was determined using a microplate absorbance plate reader (BioTek) using the Pierce Micro BCA Protein assay (Thermo Fisher, cat. no. 10249133) to enable correction to relative luminescence units (RLU) per mg total protein. Viral infections with GFP expressing vectors were carried out in the same manner however GFP levels were measured after 72 hours by fixing cells in 4% paraformaldehyde and measuring fluorescence using the BD AccuriTM C6 flow cytometer acquiring the data in the FL1 channel. Data was analysed using FlowJo software (FlowJo LLC).
2.4. mRNA expression analysis
The GBM multiforme RNA-seq data from the Cancer Genome Atlas (TCGA) (n= 153) and normal brain Genotype Tissue expression (GTEx) (n= 1141) RNA-seq datasets were downloaded from the UCSC Xena RNA-seq Compendium (https://xena.ucsc.edu/), where the samples had been normalised and re-analysed by the same RNA-seq pipeline to eliminate batch effects [35]. Expression of CAR, CD46, DSG2, Survivin and hTERT was compared between glioblastoma tumour and normal brain samples.
2.5. Statistical analysis
Statistical analyses were performed in Graphpad Prism 9 software. Normality of data sets was tested using the D’Agostino Pearson test and non-normally distributed data was compared using a Mann-Whitney U test. When comparing the means of ≥2 non-normally distributed groups involving one independent variable, the Kruskal-Wallis test with Dunn’s multiple comparisons was used. Statistically significant differences were marked as *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001
3. Results
3.1. Expression of the Adenoviral receptors CAR, CD46 and DSG2 in GBM.
The species C Ad5 is well described as engaging CAR as a primary cell entry receptor, which anatomically is localized at tight junctions. Species B adenovirus serotypes Ad35 and Ad3 use the ubiquitously expressed membrane protein, CD46 and the cell-cell adhesion protein DSG2, respectively. Using mRNA expression data taken from the TCGA dataset for GBM and GTex dataset for normal healthy brain tissue, the presence of CAR, CD46 and DSG2 was compared (Figure 1A). CAR and CD46 mRNA were abundant in both healthy brain and GBM. CAR mRNA levels in GBM were significantly higher than healthy brain. High levels of CD46 mRNA were observed in both healthy brain and GBM, although there were significantly higher mRNA levels in normal brain. DSG2 mRNA levels were substantially lower overall compared to CAR and CD46, however there was greater expression of DSG2 mRNA in GBM compared to normal brain tissue. To conclude, although there is differential mRNA expression of adenoviral receptors between healthy brain tissue and GBM, the presence of native adenoviral receptors on normal brain tissue represents an obstacle when considering the development of oncolytic adenoviruses using these receptors as potential GBM treatments.
Expression of adenoviral receptor on three GBM derived cell lines was analysed using flow cytometry. L1 is a suspension stem cell line derived from a high-grade GBM patient. E51 and E55 are adherent glioma stem cell lines derived from high-grade GBM patients. Figure 1B demonstrates receptor staining for CAR, CD46 and DSG2. L1 and E51 cells express high levels of all three adenoviral receptors. E55 cells were positive for CD46 but demonstrate low levels of both CAR (42.6%) and DSG2 (0.77%).
3.2. Transduction of pseudotyped adenoviral vectors in GBM.
GBM derived cell lines L1, E51 and E55 were transduced with an Ad5 vector and Ad5 pseudotypes, Ad5/26K, Ad5/35K and Ad5/49K expressing the transgene under the control of a CMV IE promoter. L1 cells were transduced efficiently by all four viral vectors (Figure 2A). Interestingly, a dependent response was not observed in this cell line, suggesting that the threshold of infection at the lowest dose of 1000 vp/cell. E51 cells were transduced by all viral pseudotypes, where we did observe a dose dependent response with increasing transduction from 1000 vp/cell to 10,000 vp/cell (Figure 2B). Both L1 and E51 express high levels of CAR and CD46 therefore this transduction data is in line with the expected results. E55 cells express lower levels of CAR than L1 and E51 however it remained permissive to transduction by Ad5. All pseudotype vectors were able to transduce E55 cells (Figure 2C). Overall, a comparison of all four viral vectors tested, Ad5/49K, which uses an unknown cellular receptor[16,17,18], transduced all three cell lines most efficiently. This was also observed at a lower dose of 500 vp/cell used in L1 cells (Figure 2D). L1 cells were transduced with 500 vp/cell to determine whether this was under the threshold to observe a dose dependent response. Interestingly, at this dose it was apparent that Ad5, Ad5/26K and Ad5/35K were not transduced as efficiently as Ad5/49K.
Ad3 is recognized as engaging DSG2 for cell entry and therefore an Ad5/3K pseudotype vector expressing GFP as a reporter gene was used to investigate differential usage of DSG2 by adenoviral vectors in GBM. Transduction of Ad5/3K was quantified by expression of GFP reporter gene in L1 (DSG2 high) and E55 (DSG2 low) cells. L1 cells demonstrated a dose dependent transduction of Ad5/3K virus however Ad5 transduction remained consistent (approximately 60%) for all three doses (Figure 3A). Ad5/3K did not transduce L1 as efficiently as Ad5 despite the presence of DSG2 on this cell line (Figure 1B). E55 cells are negative for DSG2 (Figure 1B), and consequently Ad5/3K did not transduce E55 cells efficiently even at the highest dose of 10,000 vp/cell (Figure 3B). Representative microscopic images were taken (Figure 3C). This data suggests that although there is a degree of receptor specificity it cannot be relied upon for tumour specific uptake into GBM cells. Post entry selectivity must also be considered to improve safety of adenoviral vectors for the treatment of GBM.
3.3. Tumour specific promoter hTERT and survivin offer benefits in GBM cells over normal cells.
It is reported that 75% of GBM contain mutations in the human telomerase reverse transcriptase gene (hTERT) [38] Similarly, survivin, an inhibitor of apoptosis, is expressed in nearly 80% of GBMs [39]. Adenoviral vectors that contain tumour specific promoters exploiting the presence of hTERT and survivin may offer potential for treatment of GBM. The TCGA dataset was used to evaluate the expression levels of these mRNA expression for survivin and hTERT in GBM compared to normal brain (GTex). Figure 4A demonstrates that mRNA expression of survivin is significantly higher in GBM compared to normal brain tissue. hTERT expression levels are also significantly higher in tumour compared to healthy tissue (Figure 4B) supporting the reported literature. We therefore evaluated whether survivin or hTERT promoters demonstrated elevated selectivity of expression of a reporter gene in GBM derived cells compared to human derived fibroblasts expressing CAR (HF-CAR cells). Two GBM patient-derived cell lines L0 and L1, as well as HFCARs were transduced with Ad5 vectors containing luciferase under control of either survivin, hTERT promoter or a fusion promoter that requires both survivin and hTERT[40]. Cells were infected with 1000 vp/cell and luciferase activity was evaluated 72 hours post infection (Figure 4C). HFCARs demonstrated levels of transduction that were barely above background for all three adenoviral vectors. Conversely, when L0 and L1 were transduced efficiently by all three viral constructs containing tumour specific promoters, however Ad5.survivin was the most efficient for both GBM cell lines. Interestingly, Ad5.hTERT and Ad5.hTERT/survivin demonstrated similar levels of luciferase activity indicating that the combination of both hTERT and survivin did not improve the efficacy and may hamper the effects when compared to survivin alone.
4. Discussion
GBM is a common and aggressive brain cancer with poor prognosis and significant unmet clinical need. Oncolytic viruses have previously been evaluated for the treatment of GBM and a recombinant herpesvirus, Teserpaturev, has been licensed for treatment of GBM in Japan [41]. Adenoviral vectors have demonstrated safety in clinical trials however the presence of native adenoviral receptors has yet to be described in GBM [20]. We have evaluated the expression of three well described adenoviral receptors, CAR, CD46 and DSG2 in GBM compared to healthy brain tissue. Analysis of mRNA expression indicated that all three receptors are expressed in GBM and healthy brain however CAR and DSG2 are significantly upregulated in GBM. Adenoviral vectors such as Ad5 and Ad3 which use CAR and DSG2 respectively could be utilized to target GBM by local delivery into the tumour. It is important to consider whether the lack of specificity may impact healthy brain tissue surrounding the tumour.
We confirmed expression of CAR, CD46 and DSG2 on the surface of patient derived GBM stem cell lines, L1, E51 and E55. Both L1 and E51 were positive for CAR and DSG2 whereas E55 express lower levels of CAR and were negative for DSG2. All three cell lines express high levels of CD46. This data is consistent with the mRNA expression levels reported in GBM. Native adenoviral receptors must be considered when engineering oncolytic adenoviral therapies for GBM.
We assessed the transduction of GBM using Ad5 based pseudotype vectors. Ad5 binds CAR through engagement of the fiber knob protein. The fiber knob protein of Ad5 was replaced with the fiber knob protein from Ad26 (known to use sialic acid), Ad35 (binds CD46) and Ad49 (receptor usage unknown [16,17,18]) to generate Ad5 pseudotyped vectors. These vectors were then used to determine the transduction of these serotypes based on the fiber knob engagement. L1 cells were transduced consistently by all vectors at every concentration of virus. When the concentration of virus was reduced to 500vp/cell it was possible to determine that Ad5/49K demonstrated the highest transduction. This was also observed in E51 and E55 cells also. Interestingly, Ad5 pseudotyped vectors demonstrated improved transduction compared to Ad5 at all concentrations of virus. This may result from the availability of each receptor on the surface of the cells or additional factors such as engagement of the co-receptors αVβ3 and αvβ5 integrin that mediate internalization. E55 cells express low levels of CAR however demonstrate similar transduction of Ad5 when compared with E51, this suggests that there may be enough CAR present on the surface of these cells to mediate transduction. Differences between Ad5 transduction in E51 and E55 may be discerned if a lower viral concentration is used. L1 and E55 cells were transduced with Ad5 and Ad5/3K which engages DSG2 as a cell entry receptor [23]. Ad5/3K has previously been reported as demonstrating superior infectivity in glioma cells [43]. As previously demonstrated, Ad5 was able to transduce both cell lines efficiently. Ad5/3K demonstrated a dose dependent transduction in L1 cells however the transduction efficiency was lower than Ad5. Additional work is required to determine whether this is due to viral receptor usage or processing of the virus post entry. E55 cells were not efficiently transduced by Ad5/3K, consistent with the lack of DSG2 on this cell line. Considering the transduction data, Ad5/26K, Ad5/35K and Ad5/49K in particular appear to be superior candidates for pseudotype oncolytic virotherapy vector for GBM compared to Ad5 or Ad5/3K.
Finally, given the lack of selectivity of CAR, CD46 and DSG2 expression in GBM, we considered the need for additional methods of intrinsic cellular selectivity when designing oncolytic virotherapies targeting GBM. We evaluated post entry selectivity of transgene expression mediated by tumour specific promoters utilizing the tumour markers, hTERT and survivin. mRNA analysis confirmed that both hTERT and survivin were significantly upregulated in GBM compared to normal brain tissue however the difference was more marked in the case of survivin. We designed three adenoviral vectors, based on Ad5, expressing luciferase under an hTERT promoter, survivin promoter and a fusion hTERT and survivin promoter [40]. These vectors were used to transduce GBM L0 and L1 cells as well as non-transformed, human fibroblasts, HF-CARs. As expected, adenoviral vectors were not able express luciferase in the HF-CAR cells due to the absence of hTERT and survivin. Both L0 and L1 cells were efficiently transduced by all three vectors however Ad5-survivin was able to transduce these cells more efficiently than the Ad5-hTERT and Ad5-hTERT/survivin fusion, suggesting that survivin may be the most promising tumour specific promoter for future therapeutic applications in GBM both in terms of the quantity and selectivity of transgene expression in GBM cell lines. The levels of survivin and hTERT was not evaluated in these cells, and the levels could vary between patients however we have demonstrated that these vectors can selectively express transgenes in GBM cells. Future work will include developing therapeutics under control of the survivin promoter for treatment of GBM, potentially coupled as part of an Ad5/K49 pseudotyped vector to maximise uptake and activity in GBM cells.
5. Conclusions
To conclude, we have evaluated the expression of well described adenoviral vectors in GBM. We identified that CAR, CD46 and DSG2 expression in GBM and that patient derived GBM cells and GBM stem cells can be transduced by Ad5 based adenoviral serotypes, in particular Ad5/49K. Given the lack of natural GBM selectivity of adenoviral receptors, we highlighted the need for additional selectivity and demonstrated that tumour specific promoters can enhance the specificity of GBM cells, where the survivin promoter appears particularly well suited. This data informs the design of future oncolytic adenoviral therapies expressing therapeutic transgenes for the treatment of GBM.
Author Contributions
Conceptualization, EAB, FAS, ALP; methodology, EAB, CL, ARP, JAD; formal analysis, EAB, CL, ARP; investigation, EAB, CL, ARP.; resources, FAS; writing—original draft preparation, EAB, ALP.; writing—review and editing, ALP, CL, ARP, JAD, FAS, ALP; supervision, ALP; funding acquisition, ALP. All authors have read and agreed to the published version of the manuscript.
Funding
EAB and ARP were funded by the Experimental Cancer Medicine Centre award to Cardiff University (reference C7838/A25173). EAB was additionally funded by Cancer Research Wales (reference 516619). CL was funded by a grant from the Department of Health and Social Care (DHSC) and supported by the National Institute for Health Research (NIHR; NIHR135073). JAD was funded by Cancer Research UK/Stand up to Cancer Biotherapeutics Programme grant award to ALP (reference C52915/A29104). FAS and ALP are funded by the Higher Education Funding Council for Wales (HEFCW).
Data Availability Statement
The data presented in this study are available on request from the corresponding author. Publicly available datasets were also analyzed in this study. This data can be found here https://xena.ucsc.edu/.
Acknowledgments
We thank Professor Steve Pollard and Dr Gillian Morrison of Edinburgh University and the Glioma Cellular Genetics Resource (GCGR) for the E51 and E55 cell lines.
Conflicts of Interest
ALP is founder and chief scientific officer of Trocept Therapeutics. All other authors declare no conflict of interest.
References
- Q. T. Ostrom et al., ‘CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011’, Neuro-Oncol., vol. 16 Suppl 4, no. Suppl 4, pp. iv1-63, Oct. 2014. [CrossRef]
- D. T. Di Carlo, F. Cagnazzo, N. Benedetto, R. Morganti, and P. Perrini, ‘Multiple high-grade gliomas: epidemiology, management, and outcome. A systematic review and meta-analysis’, Neurosurg. Rev., vol. 42, no. 2, pp. 263–275, Jun. 2019. [CrossRef]
- M. Lim, Y. Xia, C. Bettegowda, and M. Weller, ‘Current state of immunotherapy for glioblastoma’, Nat. Rev. Clin. Oncol., vol. 15, no. 7, pp. 422–442, Jul. 2018. [CrossRef]
- N. Macedo, D. M. Miller, R. Haq, and H. L. Kaufman, ‘Clinical landscape of oncolytic virus research in 2020’, J. Immunother. Cancer, vol. 8, no. 2, p. e001486, Oct. 2020. [CrossRef]
- A. J. Bett, L. Prevec, and F. L. Graham, ‘Packaging capacity and stability of human adenovirus type 5 vectors’, J. Virol., vol. 67, no. 10, pp. 5911–5921, Oct. 1993. [CrossRef]
- D. H. Sterman et al., ‘Adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir gene therapy in patients with localized malignancy: results of a phase I clinical trial in malignant mesothelioma’, Hum. Gene Ther., vol. 9, no. 7, pp. 1083–1092, May 1998. [CrossRef]
- Z. Ram et al., ‘Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells’, Nat. Med., vol. 3, no. 12, pp. 1354–1361, Dec. 1997. [CrossRef]
- ‘HAdV Working Group’. http://hadvwg.gmu.edu/ (accessed Mar. 29, 2023).
- A. L. Parker et al., ‘Effect of neutralizing sera on factor x-mediated adenovirus serotype 5 gene transfer’, J. Virol., vol. 83, no. 1, pp. 479–483, Jan. 2009. [CrossRef]
- T. C. Mast et al., ‘International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: correlates of high Ad5 titers and implications for potential HIV vaccine trials’, Vaccine, vol. 28, no. 4, pp. 950–957, Jan. 2010. [CrossRef]
- E. A. Bates et al., ‘Development of a low-seroprevalence, αvβ6 integrin-selective virotherapy based on human adenovirus type 10’, Mol. Ther. Oncolytics, vol. 25, pp. 43–56, Jun. 2022. [CrossRef]
- J. M. Bergelson et al., ‘Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5′, Science, vol. 275, no. 5304, pp. 1320–1323, Feb. 1997. [CrossRef]
- C. B. Coyne and J. M. Bergelson, ‘CAR: a virus receptor within the tight junction’, Adv. Drug Deliv. Rev., vol. 57, no. 6, pp. 869–882, Apr. 2005. [CrossRef]
- M. Anders et al., ‘Loss of the coxsackie and adenovirus receptor contributes to gastric cancer progression’, Br. J. Cancer, vol. 100, no. 2, pp. 352–359, Jan. 2009. [CrossRef]
- K. Matsumoto, S. F. Shariat, G. E. Ayala, K. A. Rauen, and S. P. Lerner, ‘Loss of coxsackie and adenovirus receptor expression is associated with features of aggressive bladder cancer’, Urology, vol. 66, no. 2, pp. 441–446, Aug. 2005. [CrossRef]
- D. M. Shayakhmetov, A. Gaggar, S. Ni, Z.-Y. Li, and A. Lieber, ‘Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity’, J. Virol., vol. 79, no. 12, pp. 7478–7491, Jun. 2005. [CrossRef]
- S. N. Waddington et al., ‘Adenovirus serotype 5 hexon mediates liver gene transfer’, Cell, vol. 132, no. 3, pp. 397–409, Feb. 2008. [CrossRef]
- A. L. Parker et al., ‘Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes’, Blood, vol. 108, no. 8, pp. 2554–2561, Oct. 2006. [CrossRef]
- R. C. Carlisle et al., ‘Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1’, Blood, vol. 113, no. 9, pp. 1909–1918, Feb. 2009. [CrossRef]
- F. F. Lang et al., ‘Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma’, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol., vol. 36, no. 14, pp. 1419–1427, May 2018. [CrossRef]
- A. T. Baker, R. M. Mundy, J. A. Davies, P. J. Rizkallah, and A. L. Parker, ‘Human adenovirus type 26 uses sialic acid-bearing glycans as a primary cell entry receptor’, Sci. Adv., vol. 5, no. 9, p. eaax3567, Sep. 2019. [CrossRef]
- A. Gaggar, D. M. Shayakhmetov, and A. Lieber, ‘CD46 is a cellular receptor for group B adenoviruses’, Nat. Med., vol. 9, no. 11, pp. 1408–1412, Nov. 2003. [CrossRef]
- H. Wang et al., ‘Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14′, Nat. Med., vol. 17, no. 1, pp. 96–104, Jan. 2011. [CrossRef]
- P. Rawal and L. Zhao, ‘Sialometabolism in Brain Health and Alzheimer’s Disease’, Front. Neurosci., vol. 15, 2021, Accessed: Mar. 27, 2023. [Online]. Available: https://www.frontiersin.org/articles/10.3389/fnins.2021.648617. [CrossRef]
- L. A. K. Van Aarsen et al., ‘Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism’, Cancer Res., vol. 68, no. 2, pp. 561–570, Jan. 2008. [CrossRef]
- H. Uusi-Kerttula et al., ‘Ad5NULL-A20: A Tropism-Modified, αvβ6 Integrin-Selective Oncolytic Adenovirus for Epithelial Ovarian Cancer Therapies’, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res., vol. 24, no. 17, pp. 4215–4224, Sep. 2018. [CrossRef]
- J. A. Davies et al., ‘Efficient Intravenous Tumor Targeting Using the αvβ6 Integrin-Selective Precision Virotherapy Ad5NULL-A20’, Viruses, vol. 13, no. 5, p. 864, May 2021. [CrossRef]
- H. Uusi-Kerttula et al., ‘Pseudotyped αvβ6 integrin-targeted adenovirus vectors for ovarian cancer therapies’, Oncotarget, vol. 7, no. 19, pp. 27926–27937, May 2016. [CrossRef]
- S. G. M. Piccirillo et al., ‘Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells’, Nature, vol. 444, no. 7120, pp. 761–765, Dec. 2006. [CrossRef]
- L. B. Hoang-Minh et al., ‘Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma’, EMBO J., vol. 37, no. 23, p. e98772, Dec. 2018. [CrossRef]
- F. A. Siebzehnrubl et al., ‘The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance’, EMBO Mol. Med., vol. 5, no. 8, pp. 1196–1212, Aug. 2013. [CrossRef]
- S. M. Pollard et al., ‘Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens’, Cell Stem Cell, vol. 4, no. 6, pp. 568–580, Jun. 2009. [CrossRef]
- ‘The AdZ adenovirus cloning system | AdZ.cf.ac.uk’. https://adz.cf.ac.uk/ (accessed Mar. 27, 2023).
- H. Uusi-Kerttula et al., ‘Incorporation of Peptides Targeting EGFR and FGFR1 into the Adenoviral Fiber Knob Domain and Their Evaluation as Targeted Cancer Therapies’, Hum. Gene Ther., vol. 26, no. 5, pp. 320–329, May 2015. [CrossRef]
- M. J. Goldman et al., ‘Visualizing and interpreting cancer genomics data via the Xena platform’, Nat. Biotechnol., vol. 38, no. 6, pp. 675–678, Jun. 2020. [CrossRef]
- A. T. Baker et al., ‘The Fiber Knob Protein of Human Adenovirus Type 49 Mediates Highly Efficient and Promiscuous Infection of Cancer Cell Lines Using a Novel Cell Entry Mechanism’, J. Virol., vol. 95, no. 4, pp. e01849-20, Jan. 2021. [CrossRef]
- R. S. Dakin, A. L. Parker, C. Delles, S. A. Nicklin, and A. H. Baker, ‘Efficient transduction of primary vascular cells by the rare adenovirus serotype 49 vector’, Hum. Gene Ther., vol. 26, no. 5, pp. 312–319, May 2015. [CrossRef]
- H. N. Nguyen et al., ‘Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy’, Neuro-Oncol., vol. 19, no. 3, pp. 394–404, Mar. 2017. [CrossRef]
- A. Das, W.-L. Tan, J. Teo, and D. R. Smith, ‘Expression of survivin in primary glioblastomas’, J. Cancer Res. Clin. Oncol., vol. 128, no. 6, pp. 302–306, Jun. 2002. [CrossRef]
- I. V. Alekseenko et al., ‘Activity of the Upstream Component of Tandem TERT/Survivin Promoters Depends on Features of the Downstream Component’, PLOS ONE, vol. 7, no. 10, p. e46474, Oct. 2012. [CrossRef]
- H. Fukuhara, Y. Ino, and T. Todo, ‘Oncolytic virus therapy: A new era of cancer treatment at dawn’, Cancer Sci., vol. 107, no. 10, pp. 1373–1379, Oct. 2016. [CrossRef]
- E. A. Bates et al., ‘In Vitro and In Vivo Evaluation of Human Adenovirus Type 49 as a Vector for Therapeutic Applications’, Viruses, vol. 13, no. 8, p. 1483, Jul. 2021. [CrossRef]
- A. A. Stepanenko et al., ‘Superior infectivity of the fiber chimeric oncolytic adenoviruses Ad5/35 and Ad5/3 over Ad5-delta-24-RGD in primary glioma cultures’, Mol. Ther. - Oncolytics, vol. 24, pp. 230–248, Mar. 2022. [CrossRef]
Figure 1.
Expression of recognized adenoviral receptors on GBM, L1 cells, and glioma stem cells (E51 and E55). (A) mRNA expression analysis of adenoviral receptors CAR, CD46 and DSG2 in GBM vs normal brain. The data was collated from TCGA (tumour) and GTEx (normal) and represents n=153 and n=1141, respectively. Statistical significance was determined using Mann-Whitney U test *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001. (B) Flow cytometry to determine receptor staining for CAR, CD46 and DSG2 on the surface of L1 (GBM) and glioma stem cells (E51 and E55). Histograms are representative example from an individual experiment repeated in triplicate.
Figure 1.
Expression of recognized adenoviral receptors on GBM, L1 cells, and glioma stem cells (E51 and E55). (A) mRNA expression analysis of adenoviral receptors CAR, CD46 and DSG2 in GBM vs normal brain. The data was collated from TCGA (tumour) and GTEx (normal) and represents n=153 and n=1141, respectively. Statistical significance was determined using Mann-Whitney U test *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001. (B) Flow cytometry to determine receptor staining for CAR, CD46 and DSG2 on the surface of L1 (GBM) and glioma stem cells (E51 and E55). Histograms are representative example from an individual experiment repeated in triplicate.
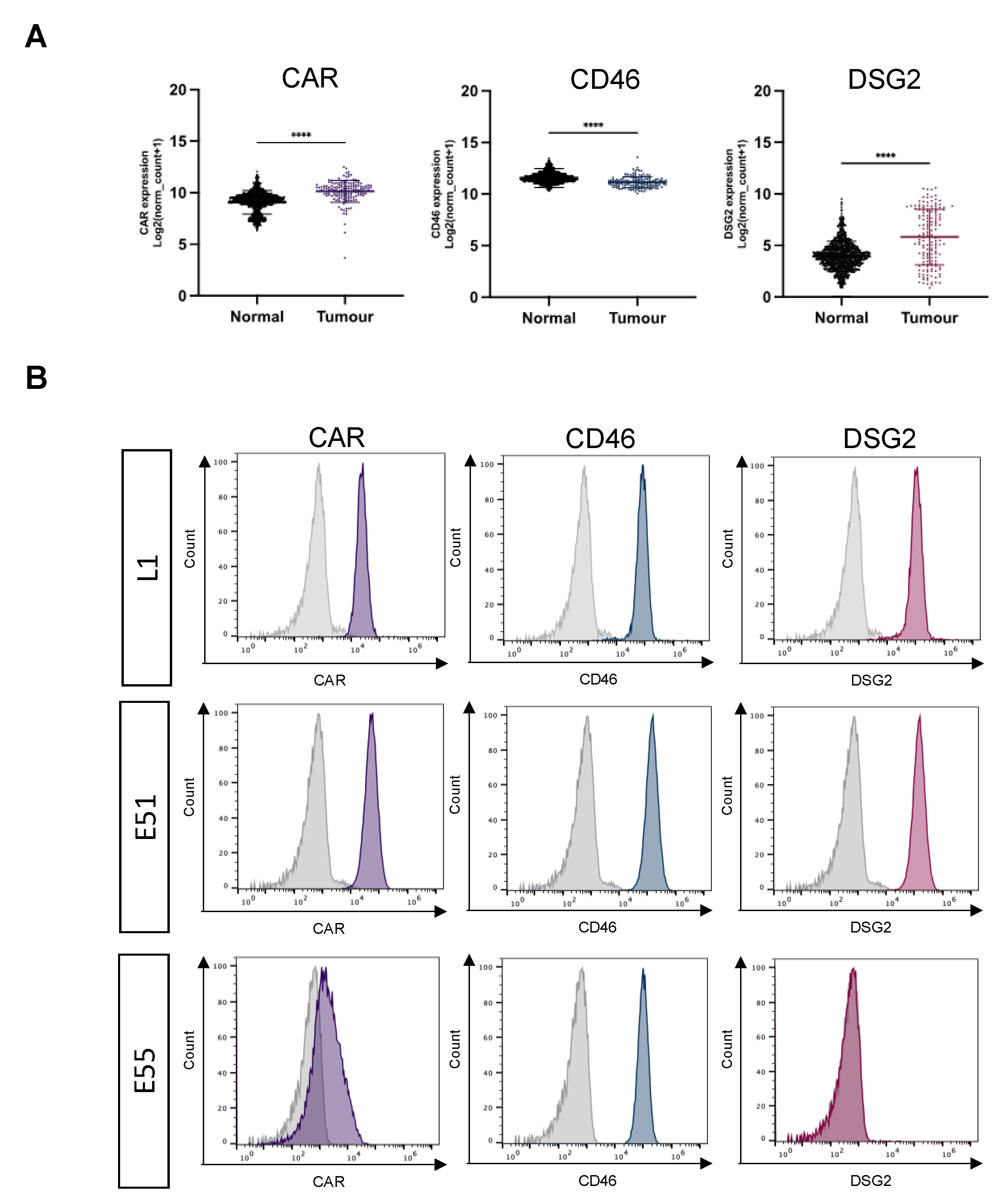
Figure 2.
Transduction of L1, E51 and E55 cells by adenoviral pseudotype vectors. (A) L1 cells (B) E51 cells and (C) E55 cells were transduced with 1000, 5000 and 10,000 vp/cell of adenoviral pseudotype vectors, Ad5, Ad5/26K, Ad5/35K and Ad5/49K. (D) L1 cells were transduced with 500 vp/cell to investigate the threshold for infection. Luciferase activity was measured 72 hours post infection. Data represents mean of triplicates with individual data points shown. Error bars represent standard deviation and data has been presented on a log scale.
Figure 2.
Transduction of L1, E51 and E55 cells by adenoviral pseudotype vectors. (A) L1 cells (B) E51 cells and (C) E55 cells were transduced with 1000, 5000 and 10,000 vp/cell of adenoviral pseudotype vectors, Ad5, Ad5/26K, Ad5/35K and Ad5/49K. (D) L1 cells were transduced with 500 vp/cell to investigate the threshold for infection. Luciferase activity was measured 72 hours post infection. Data represents mean of triplicates with individual data points shown. Error bars represent standard deviation and data has been presented on a log scale.
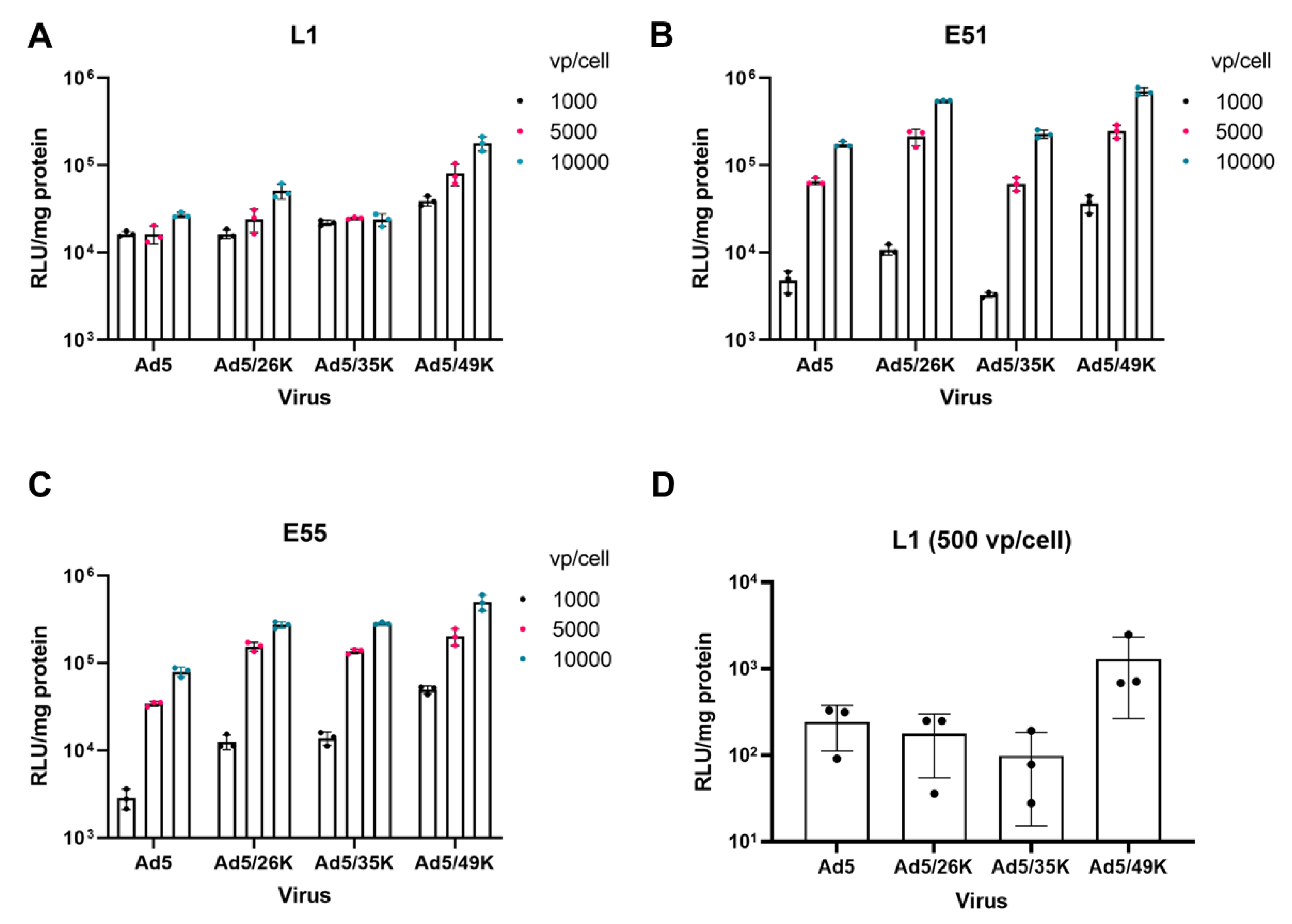
Figure 3.
L1 and E55 cells were transduced with Ad5/3K vector expressing GFP. GFP levels were measured by flow cytometry and data was analysed in FlowJo. (A) L1 cells and (B) E55 were transduced with Ad5.GFP and Ad5/3K.GFP at concentrations of 1000, 5000 and 10, 000 vp/cell. Data is presented as mean of triplicate values of an independent experiment and individual data points have been shown. (C) Representative images of E55 cells taken at 10X magnification bars using the EVOS cell imaging system (ThermoFisher). Images were taken both in TRANS and GFP channels and merged. Scale bar represents 300 µm.
Figure 3.
L1 and E55 cells were transduced with Ad5/3K vector expressing GFP. GFP levels were measured by flow cytometry and data was analysed in FlowJo. (A) L1 cells and (B) E55 were transduced with Ad5.GFP and Ad5/3K.GFP at concentrations of 1000, 5000 and 10, 000 vp/cell. Data is presented as mean of triplicate values of an independent experiment and individual data points have been shown. (C) Representative images of E55 cells taken at 10X magnification bars using the EVOS cell imaging system (ThermoFisher). Images were taken both in TRANS and GFP channels and merged. Scale bar represents 300 µm.
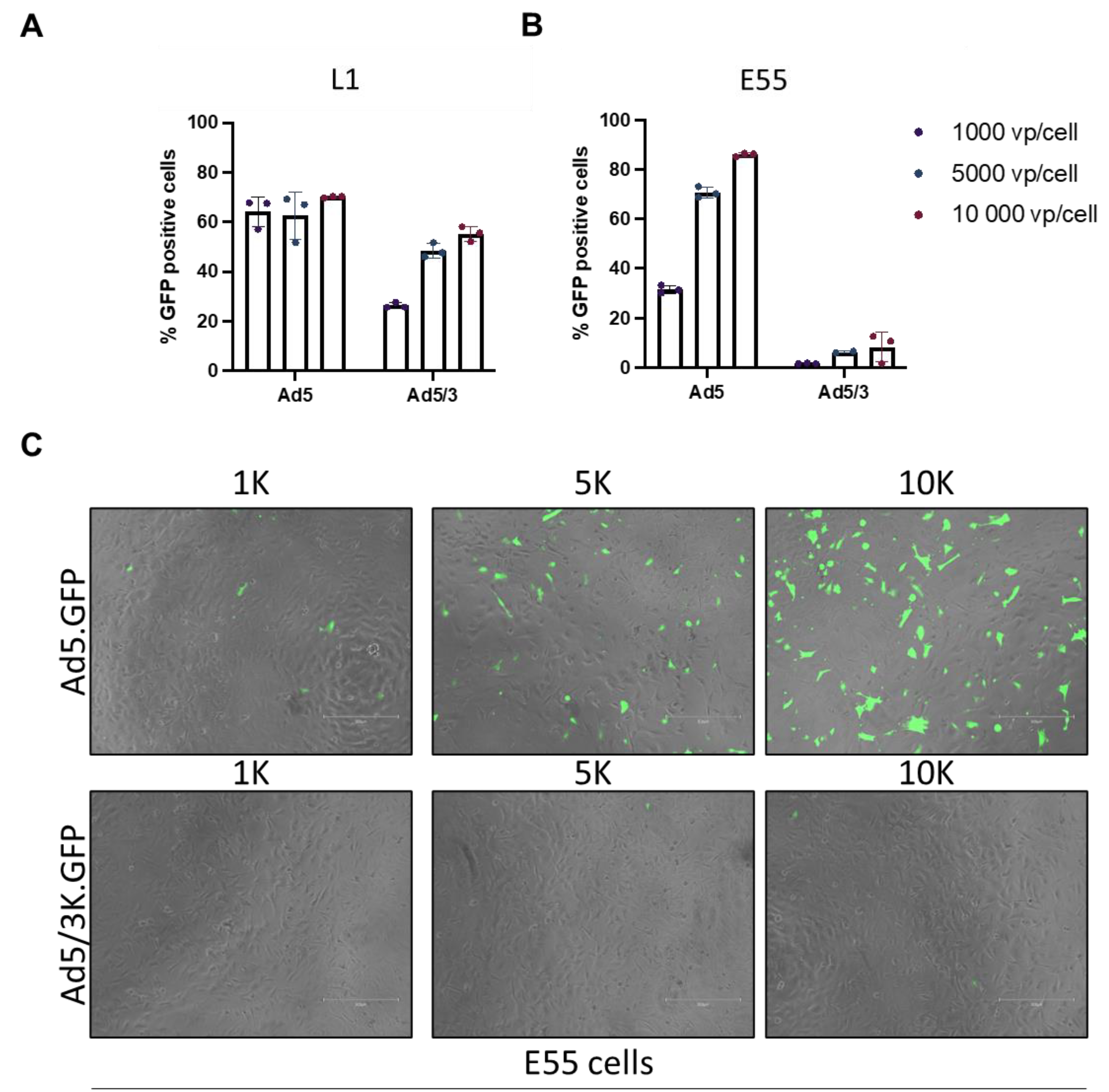
Figure 4.
Evaluation of tumour specific promoters hTERT and survivin. mRNA expression of (A) survivin and (B) hTERT were compared for GBM vs normal brain. The data was collated from TCGA (tumour) and GTEx (normal) and represents n=153 and n=1141, respectively. Statistical significance was determined using Mann-Whitney U test *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001. (C) Adenoviral vectors expressing luciferase under control of three different tumour specific promoters; survivin, hTERT and a combination of survivin and hTERT. Virus was added to human derived fibroblasts (HFCARs) and GBM derived, L0 and L1, cells at a concentration of 1000 vp/cell and luciferase activity was measured 72 hours after infection. The data has been plotted on a log scale and represents mean of triplicate values. Statistical significance was determined by Kruskal-Wallis test with Dunn’s multiple comparisons was used. Only significant values have been shown *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001.
Figure 4.
Evaluation of tumour specific promoters hTERT and survivin. mRNA expression of (A) survivin and (B) hTERT were compared for GBM vs normal brain. The data was collated from TCGA (tumour) and GTEx (normal) and represents n=153 and n=1141, respectively. Statistical significance was determined using Mann-Whitney U test *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001. (C) Adenoviral vectors expressing luciferase under control of three different tumour specific promoters; survivin, hTERT and a combination of survivin and hTERT. Virus was added to human derived fibroblasts (HFCARs) and GBM derived, L0 and L1, cells at a concentration of 1000 vp/cell and luciferase activity was measured 72 hours after infection. The data has been plotted on a log scale and represents mean of triplicate values. Statistical significance was determined by Kruskal-Wallis test with Dunn’s multiple comparisons was used. Only significant values have been shown *= p ≤ 0.05; **= p ≤ 0.01; ***=p ≤ 0.001 and ****= p ≤ 0.0001.
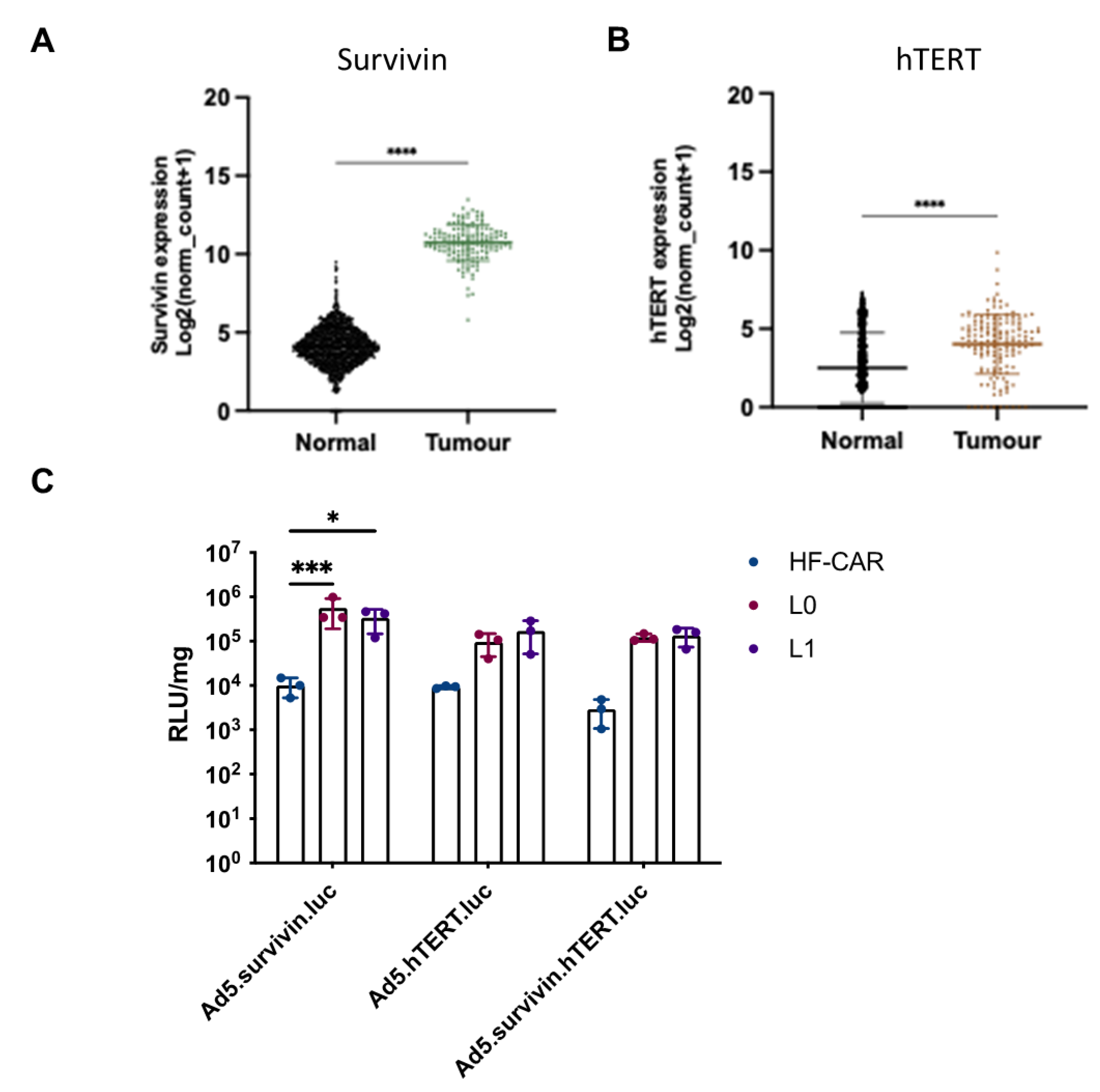

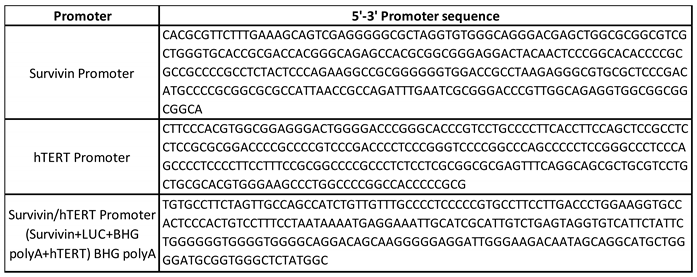
Table 1.
Promoter sequences inserted into Ad5 vector expressing luciferase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



