Submitted:
04 April 2023
Posted:
05 April 2023
You are already at the latest version
Abstract
Macrophages are the key effector cells of innate immunity which show two polarized states: M1, classically activated and M2, alternatively activated. Tumor-associated macrophages (TAMs) which usually show M2 polarization, are immunosuppressive cells that enhance tumor metastasis and invasion. Also, enrichment of TAMs is known to be closely associated with poor prognosis of cancer patients. Therefore, TAMs are considered to be promising targets for immunotherapy. Importantly, tumor-derived exosomes emerged as a crucial player in immune regulation which can remodel the tumor microenvironment towards immunosuppressive state. Of note, few studies have shown that exosomes could induce polarization of macrophages towards M2 type in tumor condition, while others showed activation of M1 or mixed phenotype. Considering the role of TAMs in cancer, there is an urgent need to decipher the exosome-mediated cross-talk between tumor cells and macrophages.For this, we used murine model of Dalton’s lymphoma (DL), wherein, firstly, we evaluated increased exosome burden by assessing exosome level in serum as well as in various tissues of tumor-bearing host. Next, proteomic profiling of lymphoma-derived exosomes was done which revealed the presence of immunomodulatory proteins. Of note, these proteins were known to alter the macrophage polarization. In order to assess the effect of these exosomes on macrophages, we performed in vitro study using RAW264.7 cells. In vitro study revealed that frequent uptake of exosomes mediated a morphological change in macrophages which reduced their phagocytic activity. In parallel, exosomes increased reactive oxygen species (ROS) level and inhibited LPS-induced nitric oxide (NO) level in macrophages. Also, exosomes upregulated the expression of arginase-1 (an M2 marker) in macrophages but decreased the LPS-induced nitric oxide synthase 2 (NOS2) (an M1 marker) expression. Moreover, we observed a pro-tumoral cytokine profile in macrophages incubated with exosomes. Our study suggests that exosome load increases in lymphoma-bearing hosts systemically. Importantly, lymphoma-derived exosomes mediate the activation of macrophages towards pro-tumoral M2 type. Our results give an insight into the exosome-mediated tumor and immune cell cross-talk which could serve as an important prospect for targeting in cancer immunotherapy.
Keywords:
Exosomes
; lymphoma
; LC-MS
; pro-tumoral macrophages
; cytokines
1. Introduction
Exosomes, the nano-vesicles, are signalling entities which enclose molecular constituents of cell of its origin and deliver them to different target cells, at local and distant sites [1,2,3]. Accumulating evidence has established their crucial involvement in pathogenicity of various diseases, including cancer [1,2,3,4]. Cancer cells release an increased number of exosomes compared to non-transformed cells to promote its growth and metastasis [4]. Several reports have shown the role of tumor-derived exosomes in mediating pro-tumoral effects by reprogramming stromal cells to stimulate angiogenesis and activate immunosuppressive microenvironment [5,6,7]. Importantly, cancer development is critically governed by the immune response of the host. The crosstalk between tumor and immune cells has potential implications in cancer growth and metastasis. Indeed, these exosomes modulate the immune cell function at pre-metastatic sites and thus direct the organotropism of metastatic cancer cells [8,9,10]. In this line, recent studies have focused on the pathological relevance of tumor-derived exosomes in activation of immune cells towards immunosuppressive types [11,12,13,14,15].
Among various immunological effector cells present at primary and metastatic sites, macrophages are known to play a crucial role [8,9]. Macrophages broadly exhibit two polarized states, classically activated M1 type and alternatively activated M2 type [16]. Interestingly, they show plasticity i.e., they change their functional profile in response to different environmental cues. M1 macrophages are known to be pro-inflammatory and anti-tumorigenic while M2 macrophages show anti-inflammatory and pro-tumorigenic effect. M1/M2 macrophages are differentiated based on the expression of various markers such as nitric oxide synthase 2 (NOS2) and arginase-1, respectively [5]. The M2 macrophages are also known as tumor-associated macrophages (TAMs) and enrichment of TAMs is known to be closely associated with poor prognosis of cancer patients [5,17,18]. These TAMs receive signals from tumor cells within local tumor microenvironment, in turn, they promote cancer-related inflammation, matrix remodelling, immune escape which consequently lead to cancer metastases [8,19,20,21]. Of note, it has been revealed that tumor-derived exosomes are readily taken up by macrophages which alter their functional response, depending upon the exosomal cargoes [19,22,23,24]. For instance, tumor-derived exosomes activate the STAT3 signaling in macrophages via IL6 receptor gp130 which in turn establish a pro-tumoral microenvironment [24]. Although few studies have shown that exosomes could induce polarization of macrophages towards M2 type in tumor condition, others revealed activation of mixed phenotype [19,22,23,25,26]. Considering the critical role of pro-tumoral M2 macrophages in cancer, there is an urgent need to decipher the exosome-mediated cross-talk between tumor cells and macrophages.
In cancer research, Dalton’s lymphoma (DL) has been extensively used in pre-clinical system for screening drugs in cancer treatment [27,28]. DL is a kind of non-Hodgkin’s T cell lymphoma originating spontaneously in the thymus of murine host. It has been observed that spontaneously originated tumors resemble a condition more similar to human neoplasia than experimentally induced tumors. Importantly, DL growth is known to be associated with immunosuppression resulting from altered functions of B cells, T cells and macrophages [29]. However, whether this immune regulation is mediated via exosomes released by lymphoma cells or not, has not been revealed yet. In view of this, we aimed at investigating the role of lymphoma-derived exosomes in regulating immune cell, particularly the macrophage, function.
2. Materials and methods
2.1. Chemicals and reagents
Exosome isolation kit, Revert-Aid first strand cDNA synthesis kit, DNaseI and Power SYBR green master mix were purchased from ThermoFisher Scientific. PKH26 red fluorescent cell linker midi kit, Trizol, poly-L-lysine, H2O2 and citrate buffer were from Sigma-Aldrich. RNeasy Micro Kit and RNA protect tissue reagent were from Qiagen. Anti-CD63 was purchased from Cloud Clone Corp., anti-Alix and anti-flotillin-1, from Abcam. Alexa Fluor 568 goat anti-rabbit secondary antibody was from Invitrogen and HRP-labelled anti-mouse antibody was from Santa Cruz Biotechnology. Vectashield with DAPI was purchased from Vector laboratories. DHE (dihydroethidium) was from Invitrogen Molecular Probes. Xylene was purchased from Fisher Scientific and absolute alcohol was from Merck. Crystal violet, DPX, slides, cover slips and other reagents were of the highest analytical grade and were obtained from the common source.
2.2. Experimental animals
Inbred strains of healthy BALB/c mice (22-25g) of either sex, aged 3-4 months old, were obtained from the animal house facility of Department of Zoology, University of Delhi. Animals were housed in propylene cages, fed with standard pellet diet and water ad libitum at a constant environment (at 18-26°C with 12h light/dark cycles). The study was performed in accordance with the guidelines for the care and use of laboratory animals with approval of the Institutional Animal Ethics Committee, University of Delhi and protocols approved by the CPCSEA, India (Approval number: DU/ZOOL/IAECR/2019/16/E-1/2021).
2.3. Maintenance of Dalton’s lymphoma (DL) in vivo
The DL cells were obtained from the Department of Biotechnology, Banaras Hindu University and maintained in the peritoneum of BALB/c mice by i.p. transplantation as described earlier [30]. For experiments, growing DL cells were collected from the donor mice and immediately suspended in sterile phosphate buffer saline (PBS, 1X). The viability of cells was confirmed by the trypan blue staining and cell counting was done. The number of cells was adjusted to 1×106 cells/ml and i.p. injected in the healthy BALB/c mice.
2.4. Experimental groups
The mice, of either sex, were divided into two groups, with each group consisting of six mice. Group I represented the control group while group II animals were induced for DL development. The tumor was allowed to grow and mice were sacrificed through the cervical dislocation at day 18 post-tumor transplantation.
2.5. Cell culture and treatments
RAW264.7 cells (murine macrophage cell line) was obtained from the National cell repository NCCS, Pune, India. The cells were grown in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) in a humidified incubator at 370C with 5% CO2. Cells were sub-cultured upon they reached 80% confluence. All the experiments were performed in sterile and endotoxin free conditions. To investigate the polarization of macrophages by lymphoma-derived exosomes, we included four groups. Group I - control, group II- cells treated with lymphoma-derived exosomes (100µg/ml), group III- lipopolysaccharide (LPS, 2µg/ml)-stimulated cells and group IV- co-treatment of exosomes and LPS.
2.6. Immunofluorescence for CD63
The ascites was collected from the peritoneum of DL mice and centrifuged at 1500g for 10 min at 40C. The pellet of DL cells was washed with PBS three times and smeared on a clean glass slide, air dried and fixed in methanol. For T cells, thymus was dissected out from control mice and single cell suspension was made in PBS. The cells were pellet down and washed with PBS. The pellet was resuspended in PBS and smeared on a clean glass slide, air dried and fixed in methanol. EDTA anticoagulated peripheral blood samples were smeared on glass slides, air dried and fixed in methanol. Fixed slides were then permeabilized with 4% paraformaldehyde for 20 min at 40C followed by blocking with 5% normal goat serum for 1 h. Thereafter, the slides were incubated with anti-CD63 primary antibody (1, 100) at 40C overnight. Slides were washed three times with PBST (0.2% Tween-20) and incubated with Alexa Fluor 568 anti-rabbit secondary antibody for 2 h at room temperature in the dark. The slides were washed with PBST and mounted in Prolong Vectashield® mounting medium with DAPI. The slides were then observed under a fluorescence microscope (Nikon) and analyzed using NIS Elements software.
2.7. Immunohistochemistry for CD63
Immunohistochemistry was performed to check the expression of CD63 protein in peritoneum, liver, spleen and lungs. Tissue samples were fixed in 10% neutral buffer formalin (NBF), embedded in paraffin and were cut into 5 μm sections. Sections were deparaffinized in xylene, rehydrated with graded ethanol (100%, 95%, 80%, 70% and 50%) and then transferred to tap water. The endogenous peroxidase activity was blocked by incubating the sections with 3% H2O2 for 20 min. Thereafter, the slides were heated in sodium citrate buffer (pH 6.0) solution at 950C for 20 min for antigen retrieval. Non-specific reactivity was blocked by incubating the slides with 5% normal goat serum for 1 h. The slides were washed three times in PBST and incubated with anti-CD63 primary antibody (1, 100) at 40C overnight in a humidified chamber. Thereafter, slides were washed and incubated with Alexa Fluor anti-rabbit secondary antibody (1, 200) for 2h at room temperature in the dark. The slides were washed with PBST and mounted in Prolong Vectashield® mounting medium with DAPI. The slides were observed under a confocal microscope (Leica) and analyzed using Leica LASX software.
2.8. Isolation of exosomes
For isolation of exosomes from serum, blood was collected through retro-orbital method from control and DL mice. The blood was allowed to stand for an hour at room temperature followed by centrifugation at 1500g for 10 min at 40C. The supernatant was collected and passed through a 0.22µm syringe filter. Thereafter, exosomes were isolated using Total exosome isolation kit (from serum) as per the manufacturer’s instruction. Briefly, the required volume of serum was mixed with 0.2 volume of reagent and incubated for 30 min at 40C followed by centrifugation at 10,000g for 10 min. The supernatant was discarded and the exosome pellet was resuspended in PBS and used for further downstream analysis.
For isolation of lymphoma-derived exosomes, ascites was collected from the peritoneum of DL mice and centrifuged at 1500g for 10 min at 40C. The supernatant was collected and passed through a 0.22µm syringe filter and exosomes were isolated using Total exosome isolation kit (from other body fluids) as per the manufacturer’s instruction. Briefly, the required volume of ascites was mixed with 0.5 volume of reagent and incubated for 30 min at room temperature. Then the sample was centrifuged at 10,000g for 10 min at room temperature. The supernatant was discarded and the exosome pellet was washed with PBS. The exosome pellet was resuspended in PBS and used for further downstream analysis.
2.9. Electron microscopy
For morphometric analysis of isolated exosomes, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were performed. Briefly, the exosome pellet was resuspended in 100µl of 2% paraformaldehyde. 5µl of suspension was deposited on a grid and was allowed to dry, coated with gold palladium and microscopy was performed.
2.10. Dynamic light scattering (DLS)
The size and dispersity of the isolated exosomes was determined by DLS using Malvern Zetasizer Nano ZS analyzer. 50μL of exosome suspension isolated from serum and DL-ascites were diluted to 750μL in PBS, added to disposable micro brand UV cuvettes for measurement and three scattering measurements were recorded. The graph for size distribution by intensity and count rate of each sample was determined using the Zetasizer analyzer.
2.11. Western blotting
DL cells, ascites and lymphoma-derived exosomes were lysed in RIPA lysis buffer containing protease inhibitor cocktail and centrifuged at 10,000g at 40C to obtain clear supernatant. Protein concentrations were estimated by BCA assay. Protein samples (30μg) were mixed with protein loading dye followed by heating for 5 min at 950C and centrifugation at 10,000g. The protein samples were then electrophoresed on 10% sodium dodecyl sulphate (SDS) polyacrylamide gels followed by their transfer onto nitrocellulose membrane. The membranes were blocked with 5% bovine serum albumin (BSA) and incubated with primary antibodies (anti-Alix and anti-flotillin-1) overnight at 40C followed by incubation with their respective secondary antibodies. The specific antibody-bound protein bands were detected with ECL under Amersham Imager 600 (GE Healthcare).
2.12. Liquid chromatography-mass spectrometry (LC-MS) analysis
The exosome pellet was lysed in Urea lysis buffer (8 M urea buffer in 50 mM ammonium bicarbonate pH 8.5 with protease inhibitor cocktail) and protein concentration was determined by BCA assay. Exosomal protein (200µg) was reduced with dithiothreitol (DTT) with a final concentration of 20mM and alkylated with iodoacetamide (IAA) with a final concentration of 40mM. Alkylation reaction was quenched by adding DTT to a final concentration of 10mM followed by trypsin digestion with a final protease to protein ratio of 1, 20 to 1, 100 (w/w). The peptides were fractionated by reverse phase nano-Liquid Chromatography (Thermo Scientific Easy-nLC 1200) on PepMap RSLC C18 (2µm, 75µm x 50cm) column. After separation, the peptides were analyzed by Q Exactive Orbitrap (Thermo Scientific). The tandem mass spectra were queried against the Mus musculus on UniProt (https://www.uniprot.org/uniprotkb?facets=model_organism%3A10090&query=mus%20musculus) and analyzed using Proteome Discoverer 2.4. Thereafter, all the characterized proteins were analyzed for gene ontology using Gene Ontology Resource (http://geneontology.org/) wherein Fisher’s exact test with FDR correction was used to assess the enrichment levels of proteins. Further, the protein-protein interaction was determined using the STRING database (https://string-db.org/).
2.13. Exosome uptake assay
Exosomes were isolated from ascites and labelled with PKH26 dye according to the manufacturer’s protocol. Briefly, the exosome pellet was resuspended in 1ml of diluent C. Separately, 1ml of diluent C was mixed with 4μl of PKH26 dye. Immediately, the exosome suspension was mixed with the staining solution and incubated for 5 min. Subsequently, the reaction was stopped by adding an equal volume of DMEM supplemented with 2% exosome depleted-FBS or d-FBS. Labelled exosomes were further re-isolated using Exosome isolation kit (from cell culture media) as per manufacturer’s instructions. Briefly, 0.5volume of reagent was mixed with the sample and incubated at 40C overnight. Thereafter, the samples were centrifuged at 10,000g for 1hr at 40C. Supernatant was discarded and labelled exosome pellet was washed twice with PBS to remove unbound dye and resuspended in PBS. PKH labelled exosomes were quantified using BCA protein estimation kit and a total of 100μg of exosomes were incubated with RAW264.7 cells for different time points (30 min, 2hrs and 4hrs) and untreated cells were considered as control (0 min). After incubation time, the cells were washed with PBS thrice and fixed with methanol followed by rehydration with PBS for 5 min. The cells were mounted in Prolong Vectashield® mounting medium with DAPI and observed under a confocal microscope (Nikon).
2.14. Morphological analysis
To study the effect of lymphoma-derived exosomes on morphology of macrophages, crystal violet staining was performed. RAW 264.7 cells (5x103) were seeded in a 24 well plate and treated with exosomes and LPS, alone and combined. After 24h, the culture medium was removed and cells were washed with PBS. Cells were fixed with methanol followed by crystal violet staining. The morphology of cells was analysed under Nikon inverted microscope.
2.15. Phagocytic assay
To assess the phagocytic function of macrophages, 1x105 RAW264.7 cells per well were seeded in 6 well plate and treatment was given. After 24 hrs, the media was removed, cells were washed with PBS and heat killed yeast (0.5mg/ml) were flooded over macrophages in each well and incubated for 15 min to allow phagocytosis. Then cells were washed with PBS thrice and fixed in methanol and stained with crystal violet. Percent phagocytosis was calculated by counting the number of cells showing engulfment of yeast under Nikon inverted microscope.
2.16. Reactive oxygen species (ROS) detection
RAW264.7 cells were seeded on glass coverslips placed in a 24 well plate and given treatment of exosomes and LPS, alone and combined. After 24 hrs, the media was removed, cells were washed with PBS and incubated with dihydroethidium (DHE). DHE, a reduced form of ethidium bromide, can passively diffuse into the cells and reacts with the superoxide anion to form a red fluorescent product, 2-hydroxyethidium. It is commonly used as the most specific fluorescent probe for superoxide detection. DHE was suspended in DMSO at a stock concentration of 10mM and diluted for a final working concentration of 10μM in PBS. After incubation, the cells were washed with PBS and coverslips were mounted in Prolong Vectashield® mounting medium with DAPI and observed under inverted fluorescence microscope (Nikon).
2.17. Nitrite estimation
Nitric oxide (NO) level in the culture supernatant was estimated by measuring nitrite concentration, the stable end product of NO, using Griess reagents. Briefly, 100µl culture conditioned medium and Griess reagent (1, 1 of 0.1% naphthylethylenediamine dihydrochloride and 1% of sulfanilamide in 2.5% phosphoric acid) were mixed and incubated for 10 min at room temperature. Thereafter, absorbance was taken at 540 nm using microplate reader (EPOCH2, BioTek). Sodium nitrite was taken as standard and unknown concentrations in the samples were extrapolated from the standard curve.
2.18. RNA extraction and real-time quantitative PCR
To assess the expression of genes at transcriptional level, total RNA was isolated from cells in different treatment groups using RNeasy Mini Kit as per the manufacturer’s protocol. The quality of RNA was checked by analyzing the absorbance ratio at 260/280 nm using Nanodrop. The RNA was treated with DNaseI and then reverse transcribed using cDNA synthesis kit. The cDNA was used to analyze the expression of NOS2, arginase-1, IL6, IL10, IL12, TGFβ, TNFα and IL1β by performing real time PCR (qPCR) using QuantStudio™6 Flex System Thermal cycler. All sets of genes were run in duplicate with a reaction volume of 6µl using Power SYBR green master mix. The qPCR was run for 3min at 950C followed by 40 cycles, each consisting of 15s at 950C and 45s at 600C. The fold changes were calculated by 2-(ΔΔCt) value and data were normalized by β-actin. The primers were designed using NCBI primer BLAST. Sequence of the primers used in the study are listed in Table 1,
2.19. Statistical analysis
The statistical analysis was performed using GraphPad Prism 8 software by Student t-test for comparison between two groups and by one-way ANOVA followed by Tukey post hoc test for comparison between multiple groups. P≤0.05 was considered a statistically significant difference.
3. Results
3.1. Experimental design
In the present work, we utilized murine tumor model of Dalton’s lymphoma (DL), a kind of non-Hodgkin’s T cell lymphoma. DL represents an excellent model for assessment of various parameters of tumor growth and progression and also for screening therapeutic efficacy of various drugs [27,28]. DL was induced by i.p. injection of 1x106 cells/ml of tumor cell suspension. The tumor was allowed to grow wherein growth was assessed by their body weight and girth size. Mice were sacrificed at day 18 and the samples, including blood, ascites and tissues, such as peritoneum, liver, spleen and lungs, were collected for experiments (Figure 1).
3.2. Exosome level increases in tumor cells and in peripheral blood of tumor-bearing mice
Accumulating evidence has established the role of tumor-derived exosomes in cancer-induced pathogenicity [5,6,7]. Importantly, there is increased biogenesis of exosomes within the tumor cells which are consequently release out into the circulation. Notably, there are reports which suggest elevated exosome level in systemic circulation of patients with breast, pancreatic and ovarian cancer [31,32]. In view of this, we also investigated the exosome level in tumor-bearing hosts. In the present study, we used murine tumor model of Dalton’s lymphoma (DL) wherein firstly we evaluated increased exosome biogenesis within the tumor cells. We performed immunofluorescence staining of DL cells using an exosome-specific marker, CD63. CD63 is a tetraspanin family member, which is a principal component of exosomes. We observed a significantly high level of CD63 in DL cells compared to the T cells that revealed increased exosome biogenesis (Figure 2A). Next, we looked for the level of exosomes in circulation of the tumor-bearing host. For this, we collected blood from control and DL mice, isolated exosomes from their serum. As shown in Figure 2B, SEM images revealed the morphometric analysis of exosomes wherein we observed vesicle-like structure. Further, the DLS results evaluated the size distribution of exosomes in our sample (Figure 2C). The exosome size approximately ranges from 30-150 nm and we observed the average exosome size of 100 nm in our sample. Moreover, DLS data revealed increased count rate of nanoparticles (exosomes) in serum from tumor-bearing mice compared to the control group (Figure 2C(iii)). In DL mice, the count rate was found to be approximately 1.8 fold high (p<0.0001) compared to the control group. The count rate represents the concentration of exosomes in each sample. So, our results suggested a high level of exosomes in serum of DL mice compared to the control mice.
Considering increased exosome load in peripheral blood of tumor-bearing hosts, we were interested to know whether these exosomes are taken up by the blood cells or not. For this, we performed immunofluorescence staining using anti-CD63 antibody on blood smear. Interestingly, we observed high levels of CD63 within the cells that revealed high exosome uptake in peripheral blood of tumor-bearing hosts compared to the normal groups (Figure 2D). Also, we observed that the number of cells showing exosome uptake were more in DL-bearing groups compared to the control groups.
3.3. Exosome level increases systemically in tumor-bearing hosts
Several lines of evidence have shown that exosomes act as key drivers for pre-metastatic niche formation in various cancers. Importantly, tumor-derived exosomes, once they enter the circulation, reach at different sites throughout the body and establish an immunosuppressive pre-metastatic niche, where the tumor cells actually metastasize [7,33,34]. In this line, after evaluating increased exosome levels in serum, we were interested to investigate the status of exosomes at systemic level in tumor-bearing mice. Previously, we have shown that tumor burden in DL mice is associated with disturbed histoarchitecture of various organs, such as peritoneum, liver, spleen and lungs [30]. So, we considered these organs to investigate whether there is any change in exosome level or not. For this, we performed immunohistochemistry using anti-CD63 antibody and analysed the expression by confocal microscopy. Interestingly, we observed an increased expression of CD63 which revealed high exosome level in all the examined organs of tumor-bearing hosts (Figure 3).
3.4. LC-MS analysis of lymphoma-derived exosomes reveals presence of immunomodulatory proteins
After evaluating increased systemic exosome load in DL mice, we were interested to know the cargoes of lymphoma-derived exosomes. In DL mice, with progressive stages of tumor growth, ascites accumulates in the peritoneum and represent the rich source of tumor-derived factors. So, we isolated exosomes from fully grown ascites of DL mice and confirmed the presence of exosomes in our sample by electron microscopy, DLS and immunoblotting. SEM and TEM images showed heterogenous population of vesicle-like structures in our sample (Figure 4A). DLS results further revealed the size distribution of exosomes, wherein average exosome size ranged from 30-150 nm in diameter (Figure 4B). Additionally, immunoblotting with pan-exosomal markers, Alix, a cytoplasmic protein, and flotillin-1, a lipid-raft protein, which are involved in exosome biogenesis, confirmed the presence of exosomes in our sample (Figure 4C).
Thereafter, we performed proteomic profiling of these exosomes wherein LC-MS analysis revealed the presence of 537 proteins in the exosome sample. Firstly, these proteins were hit with the proteins present in ExoCarta (database of exosomal proteins) which showed the presence of 206 common proteins (Figure 4D(i)). Also, we screened these proteins with EMTome, a web-based portal which include epithelial to mesenchymal (EMT) related genes and signatures across various cancer types. With EMTome database, we observed presence of 55 common proteins (Figure 4D(ii)) which suggest EMT potential of lymphoma-derived exosomes (Table 2).
Next, we performed the GO enrichment analysis of exosomal proteins using Gene ontology resource. GO enrichment analysis show the biological domain of proteins with respect to: cellular components, molecular functions and biological processes. Figure 4E show the enrichment of exosomal proteins in top 20 biological domains, sorted based on their p-values. Interestingly, we observed involvement of exosomal proteins in immunomodulatory pathways such as in regulating immune response, immune system process, complement activation, humoral immune response, defense response (Figure 4E(iii)). Subsequently, we selected 200 immunomodulatory proteins and studied their interaction using STRING database. In the protein-protein interaction (PPI) network (Figure 5), we observed 131 nodes and 362 edges (PPI enrichment value, p<0.05). These immunomodulatory proteins formed a strong interactome network (hub) which was majorly constituted by complement proteins, such as C1q(a,b,c), C3, C4 and C9 (Figure 5).
Presence of immunomodulatory proteins in lymphoma-derived exosomes led us to look into their potential in altering activation state of macrophages. The macrophages are the key immune infiltrate within tumor microenvironment which are also known to be associated with poor prognosis of cancer patients [17,18]. Interestingly, we observed that lymphoma-derived exosomes have various proteins which could either promote macrophage recruitment within tumor microenvironment or impact their polarization towards M1 or M2 type. Moreover, we observed proteins which are overexpressed by M2 macrophages and thus suggest that these exosomes might mediate M2 polarization of macrophages (Table 3).
3.5. Alteration in macrophage morphology and their reduced phagocytic activity upon uptake of lymphoma-derived exosomes
From LC-MS analysis, it was clear that lymphoma-derived exosomes could modulate the activation state of macrophages. To elucidate their effect on macrophages, we performed in vitro study using murine macrophage cell line, RAW264.7. Firstly, we performed exosome uptake assay to check whether these exosomes are taken up by macrophages or not. For this, we labelled the isolated exosomes with PKH26 dye. PKH26 is a non-toxic, red fluorescent dye which stains membranes by intercalating their aliphatic portion into the lipid bilayer [72]. Then, we incubated the macrophages with these labelled exosomes for different time points and internalization of labelled exosomes was analysed by confocal fluorescence microscopy. We observed time-dependent uptake of exosomes by macrophages which was shown by fluorescence signal (Figure 6A). Interestingly, we observed the exosome uptake within 30 min. of incubation time which thus revealed fast uptake of exosomes by macrophages.
Macrophages are highly plastic cells which show distinct phenotypic and functional profiles based on their activation states. In order to decipher the polarized state of macrophages induced by lymphoma-derived exosomes, we included an LPS-stimulated group in our in vitro study which represent positive control for M1 macrophages. Firstly, we looked for the morphological changes in different groups. Alterations in morphology is the key characteristic of macrophages which changes in accordance with their polarization. Inactivated macrophages show round, more or less spherical morphology, while inflammatory conditions induce them to become more flattened with expanded cytoplasm, characteristics of M1 macrophages [73]. Similarly, we observed round cells in the control group while macrophages looked more flattened with more phagocytic extensions in the LPS-stimulated group.
In contrast, the macrophages in the exosome incubated group, attained more elongated, spindle-shaped morphology (Figure 6B). Importantly, with combined treatment of exosomes and LPS, we observed both kinds of cellular morphologies, remarkably, the majority of cells resembled spindle-shaped (Figure 6B). Besides morphological alterations, macrophages are known to show differential phagocytic activity with respect to their altered activation state. Phagocytosis plays crucial role in host defense by eliminating pathogens, such as microbes, cellular debris as well as the cancer cells. However, this function is known to get impaired in tumor condition leading to escape of tumor cells from killing and thus support its survival [74,75]. In view of this, we were also interested to investigate the phagocytic function of macrophages activated by lymphoma-derived exosomes. We assayed percent phagocytosis by counting the number of macrophages showing engulfment of yeast. Interestingly, we observed remarkably reduced percent phagocytosis of macrophages in presence of lymphoma-derived exosomes compared to the control group. Contrastingly, LPS-stimulated macrophages showed significantly increased phagocytic activity, however it was significantly inhibited in presence of exosomes and showed only basal level of phagocytic activity (Figure 6C).
3.6. Lymphoma-derived exosomes increase ROS levels and arginase-1 expression (M2 marker) in macrophages
Reactive oxygen species (ROS) level within tumor-microenvironment has been emerged as a crucial factor that promote tumor growth and progression. Importantly, infiltrating immune cells, such as myeloid-derived suppressor cells (MDSCs) and TAMs, are known to be one of the key producers of ROS [76,77]. So, we investigated whether lymphoma-derived exosomes could induce the ROS production in macrophages or not. Interestingly, our results showed significantly high ROS levels in macrophages when incubated with lymphoma-derived exosomes compared to the control group and was comparable to the LPS-stimulated group (Figure 7A). Additionally, combined treatment of exosomes and LPS induced highest levels of ROS in macrophages.
High ROS levels also govern the differentiated state of macrophages towards M2 type. As we observed high ROS production by macrophages in presence of exosomes, we further looked for their differentiated state. Macrophage polarization is governed by two opposing metabolic pathways for a single amino acid, arginine. Arginine can be metabolized either to nitric oxide (NO) and citrulline by nitric oxide synthase (NOS2) of M1 macrophages or to ornithine and urea by arginase-1 of M2 macrophages [78].
Firstly, we estimated the nitrite level, stable metabolite of NO, in the culture supernatant of different groups by Griess reaction. NO assay is a rapid and efficient method to study the impact of any stimulus in inducing M1 polarized state of macrophages. In our study, we found a basal level of NO in macrophages activated by exosomes which was similar to the control group. In contrast, we observed a significantly high level of NO in the LPS-stimulated group compared to the control group. Importantly, with the simultaneous treatment of exosomes and LPS, although we observed significantly high NO level but the level was remarkably low when compared to the LPS-stimulated group (Figure 7B). We further corroborated the NO levels by assessing the expression of NOS2 and arginase-1. We observed that exosomes did not induce NOS2 expression in macrophages and also inhibited the heightened expression of LPS-induced NOS2 which was observed with LPS-stimulation alone (Figure 7C). Further, we found that exosomes significantly upregulated the arginase-1 expression in presence and absence of LPS (Figure 7D).
3.6. Lymphoma-derived exosomes induce an M2-specific cytokine profile in macrophages
M1/M2 macrophage polarization is governed by their functional response determined by the set of cytokines which they release. Therefore, next, we investigated the effect of lymphoma-derived exosomes on functional response of macrophages by assessing the expression profile of important cytokines at transcriptional level by qRT-PCR. Key cytokine markers, indicative of macrophage polarization, include IL10, TGFβ, IL6, IL1β, IL12 and TNFα.
As shown in Figure 8, lymphoma-derived exosomes induced the expression of IL10, TGFβ, IL6 and IL1β while expression of IL12 and TNFα was comparable to the control group. Further, IL10 expression was significantly enhanced in presence of LPS. Intriguingly, LPS induced significantly high levels of IL6 and IL1β compared to the exosome group, however in presence of exosomes their expression level was remarkably inhibited. Also, exosomes inhibited the expression of LPS-induced IL12 and TNFα which was comparable to the control group (Figure 8).
4. Discussion
Exponentially emerging reports have established the role of exosomes in cancer growth and its metastatic progression. Importantly, tumor-derived exosomes play a crucial role in mediating intercellular communication networks with stromal cells, including fibroblast cells and immune cells, at local and distant sites [10,79,80,81]. Indeed, immunosuppressive tumor microenvironment is known to be promoted by tumor-derived exosomes [11,12,13,14,15]. Among various immunosuppressive cells, macrophages are the key immune cells whose enrichment within tumor microenvironment is associated with poor prognosis of cancer patients [5,17,18]. Therefore, macrophages or tumor-associated macrophages are considered to be the key target in cancer immunotherapy. In recent years, although researchers have shown the impact of tumor-derived exosomes on M1/M2 activation state of macrophages, there are contrasting reports [19,22,23,25,26]. Considering the pivotal role played by tumor-derived exosomes in mediating immunosuppressive environment and macrophages being the key players in governing prognosis of cancer in the host, there is an urgent need to decipher the cross-talk between tumor cells and macrophages via exosomes. In this line, we were interested in deciphering the effect of lymphoma-derived exosomes on macrophage activation. We observed increased exosome level in peripheral blood as well as in all the examined organs of lymphoma-bearing hosts. Further, we found that lymphoma-derived exosomes have immunomodulatory proteins which mediated a pro-tumoral change in macrophages by altering morphology, reducing phagocytic ability, increasing the expression of arginase-1 (M2-marker) and also by inducing the release of pro-tumoral cytokine profile.
Cancer cells are the transformed cells which show increased exosome biogenesis that carry oncogenic cargoes and alter the target cell behaviour towards pro-tumoral type [7]. Indeed, tumor-derived exosomes play a crucial role in governing aggressiveness and metastatic potential of cancer cells [5,6,7]. There are various molecular mechanisms which upregulate the exosome biogenesis machinery in cancer cells. Also, it has been revealed that inhibiting exosome machinery could reduce the pathogenicity of cancer [7]. In the present study, we evaluated the exosome machinery in tumor cells wherein we observed increased level of exosomes in DL cells compared to the normal cells (Figure 2A). Our result is in line with the existing reports which show increased production of exosomes by tumor cells [7]. Once formed, the exosomes are released out of the cells and account for their increased level in peripheral blood. Importantly, various reports suggest a significantly high exosome level in systemic circulation of cancer patients than healthy individuals [31]. Likewise, we observed increased count rate of exosomes in serum isolated from the DL-bearing group compared to the normal group (Figure 2C(iii)) reflecting increased exosome level in the circulation of tumor-bearing hosts. Of note, we also observed increased uptake of exosomes by peripheral blood leukocytes in the DL-bearing group compared to the normal group (Figure 2D). Although it has been revealed that increased exosome level in blood is not only contributed by tumor-derived exosomes, but, upon uptake, the immune cells further release exosomes either to suppress or promote the tumor growth, depending upon the molecular constituents of tumor-derived exosomes [6]. However, in our study, whether the increased exosome level in blood is also contributed by immune cells or not needs to be further investigated.
Several lines of evidence have shown that exosomes act as key drivers for pre-metastatic niche formation in various cancers [33]. Importantly, tumor-derived exosomes, once they enter the circulation, reach at different sites throughout the body and establish an immunosuppressive pre-metastatic niche, where the tumor cells actually metastasize [7]. For instance, Peinado et al 2012, observed organotropism of melanoma cell-derived exosomes in the interstitium of lungs, liver, spleen and bone marrow [82]. In contrast, another report suggests the presence of tumor-derived exosomes in metastatic as well as non-metastatic organs of tumor-bearing hosts [34]. In our study, we also observed increased exosome level in various tissues (peritoneum, liver, spleen and lungs) of DL-bearing group (Figure 3). Likewise, several line of evidence showed that systemically administered exosomes accumulate mainly in the vital organs, such as in liver, spleen, lungs and gastrointestinal tract [83]. Previously, we have shown that DL represents a systemic disease wherein we observed disturbed histoarchitecture and infiltration of leukocytes in most of the organs, including peritoneum, liver, spleen and lungs [30]. Presence of exosomes in these organs suggest that the exosomes might be mediating a change in functioning of stromal cells as well as infiltrating immune cells to make the sites favorable for metastasis. There are reports that show tumor microenvironment-associated cells also release more exosomes than in normal environments to promote cell to cell communication [84]. So, we speculate that increased systemic exosome load might be majorly contributed by tumor-derived exosomes while stromal cells and infiltrating immune cells may also be releasing some exosomes in response to tumor burden. We recently reviewed various mechanisms which affect exosome biogenesis machinery in cancer cells, among them, hypoxia is known to be the key trigger [7]. Conversely, exosome release by cancer cells has been found to be reduced under hyperoxic conditions [85]. Moreover, accumulating evidence suggests that exosome biogenesis under hypoxic conditions is considered to be universal because in such conditions non-transformed cells also release a high number of exosomes [86,87]. In view of this, we correlated our previous study, wherein we found high systemic ROS levels in DL mice [30], and speculate that increased systemic exosome load could be due to high oxidative stress which might be causing immune cells as well as other stromal cells to release more exosomes.
Several lines of evidence have also shown the role of key exosomal cargoes in promoting various aspects of tumor growth and progression, including oncogenic transformation, stromal remodelling, angiogenesis, immune evasion and metastasis. For instance, in multiple myeloma, tumor-derived exosomes have been characterized to have VEGF, bFGF, HGF, MMP-9 and Serpin E1 [10] while glioblastoma-derived exosomes carry VEGF, TGFβ, gelatinases and plasminogen activators to promote angiogenesis [88]. Moreover, there are reports which show the presence of immunomodulatory proteins within tumor-derived exosomes, such as presence of TGFβ1, IL-10 and Fas Ligand in ovarian cancer cell-derived exosomes and presence of NKG2D ligands and TGFβ1 in prostate cancer, mesothelioma and B-lymphoblastoid cell-derived exosomes, which could mediate immune evasion [15,89]. In this line, we investigated the proteomic constituents of lymphoma-derived exosomes, which is not yet revealed, by LC-MS analysis. Although most of the existing studies have used culture conditioned media of cancer cell lines for isolation of their exosomes, we isolated lymphoma-derived exosomes from the ascites of growing DL cells in the in vivo system. This negotiated the differences caused by in vitro culture of tumor cells and thus provided a more rational approach to investigate the molecular constituents of these exosomes. Interestingly, we found various oncogenic proteins in proteome profile of lymphoma-derived exosomes (data not shown). Also, we observed that many of these proteins are known to be EMT-related markers in several cancer types (Table 2). This suggests invasive and metastatic potential of lymphoma-derived exosomes. Moreover, we observed enrichment of these proteins in immunomodulatory pathways (Figure 4E(iii)). These immunomodulatory proteins formed a strong interactome network principally constituted by complement proteins, such as C1q (a,b,c), C3, C4 and C9 (Figure 5). The immunomodulatory potential of complement proteins has been very well-established (25, 26). Importantly, within the tumor microenvironment, different complement proteins are known to alter the activation state of various immune cells (25). Also, evidence suggests the presence of complement proteins with extracellular vesicles which may modulate the innate and adaptive immune response (27, 28). Moreover, we observed presence of proteasomal proteins, including PSMA1, PSME1, PSMB8, PSMB9 and PSMB10 (Figure 5). Besides their role in proteasome-mediated catabolic processes, the functional enrichment analysis revealed their involvement in immune system processes. Previous studies also reported presence of proteasomal subunits within exosomes [90]. For instance, Ding et al, 2020, revealed the presence of various proteasomal proteins, including PSMA1, in exosomes isolated from serum of gastric cancer patients [90]. Altogether, our results revealed that lymphoma-derived exosomal proteins could modulate the immune response of the host. As we were focused on investigating the impact of these exosomes on macrophage activation, we further screened these immunomodulatory proteins for their impact on macrophage function. From literature, it was observed that the exosomal cargoes such as Hsp90α, Myh9, CFHR4 and galectin3 are known to promote the recruitment of macrophages at tumor site in different cancer types (Table 3). For instance, Hsp90α stabilizes macrophage migration inhibitory factor (MIF) which consequently promote cancer growth via recruitment of macrophages. These recruited macrophages release high level of VEGF and thus show pro-tumoral behavior [35]. Moreover, we observed proteins which could induce M1 polarization such as STAT1, vitronectin, clusterin, or M2 polarization, such as S100A8/A9, annexin, nidogen 1, MMP-8, cathepsin S, cathepsin L, either in tumor conditions or in other pathological conditions (Table 3). Depending upon the abundance value of these proteins, exosomes would switch the polarized state of macrophages either towards M1 or M2 type. This suggests that macrophages might be recruited at the tumor site or pre-metastatic site by the lymphoma-derived exosomes wherein their polarization would have been altered in the tumor-bearing host. Of note, we also observed proteins which are known to be overexpressed by M2 macrophages, such as arginase-1, macrophage mannose receptor 1, TGFβi, Tgm2, Vps35, which implied that lymphoma-derived exosomes could induce M2 macrophage polarization (Table 3).
To investigate the impact of lymphoma-derived exosomes on macrophage polarization, we did in vitro study using RAW264.7 cell line. Importantly, we observed frequent uptake of lymphoma-derived exosomes, i.e., within 30min., by macrophages which further increased in a time-dependent manner (Figure 6A). Our observation was in line with other studies, wherein macrophages have been shown to be the key player in the rapid uptake of exosomes [22]. Also, it is evident that monocytes show frequent uptake of exosomes compared to other PBMCs [11] and thus suggest that macrophages could readily accept the biological signals via exosomes. Further, we observed that exosomes induced a morphological change in macrophages wherein they become elongated and resembled spindle-shaped (Figure 6B). Our results were consistent with previous reports which showed a phenotypic change in macrophages when incubated with tumor-derived exosomes [5,24]. For instance, Piao et al, 2018, observed a similar elongated morphology in pro-tumoral macrophages upon uptake of breast cancer cell-derived exosomes [5]. Also, it is suggested that M2 macrophages exhibit more elongated morphology compared to M1 type [91]. In a study, monocytes when treated with cord blood-stem cell-derived exosomes become M2 macrophages and displayed an elongated, spindle-shaped morphology [92]. These studies suggest that lymphoma-derived exosomes mediated a pro-tumoral M2 like phenotypic change in macrophages. However, in another study, it has been revealed that different sources of tumor-derived exosomes induce different morphologies in monocytes due to cytoskeletal rearrangements [11]. Besides morphological alterations, macrophages are known to show differential phagocytic activity with respect to their different polarized state. For instance, activation of immunosuppressive M2 macrophages in tumor conditions are known to show reduced phagocytosis [74,75]. Likewise, we observed reduced phagocytic activity of macrophages when treated with lymphoma-derived exosomes. Importantly, these exosomes also inhibited the increased phagocytic response induced by LPS (Figure 6C). This suggests the activation of pro-tumoral M2 macrophages by lymphoma-derived exosomes.
To further confirm the polarized state of macrophages in presence of lymphoma-derived exosomes, we investigated various other parameters. Particularly, we assessed hypoxic condition, a predominant factor known to play a crucial role in mediating immunosuppressive pro-tumoral microenvironment. Indeed, high ROS level is a key determinant of hypoxic conditions. Within intra-tumoral hypoxic regions, infiltrating immunosuppressive leukocytes, such as myeloid-derived suppressor cells (MDSCs) and TAMs, are known to regulate T cell function to inhibit tumor cell killing via ROS production [76,77]. Likewise, we observed a significantly high level of ROS in macrophages when activated by lymphoma-derived exosomes (Figure 7A). Importantly, high ROS levels not only mediate T cell deregulation but also govern the differentiated state of macrophages towards M2 type within tumor-microenvironment [76]. In view of this, we hypothesized that lymphoma-derived exosomes might be polarizing macrophages towards pro-tumoral M2 type. To confirm the polarized state of macrophages induced by lymphoma-derived exosomes, we assessed their arginine metabolism. Macrophage polarization is governed by two opposing metabolic pathways for a single amino acid, arginine. Arginine can be metabolized either to nitric oxide (NO) and citrulline by nitric oxide synthase (NOS2) of M1 macrophages or to ornithine and urea by arginase-1 of M2 macrophages [78]. We observed that macrophages in presence of exosomes produced only basal levels of NO. Importantly, presence of exosomes inhibited the effect of LPS for production of heightened levels of NO in macrophages (Figure 7B). This result was further corroborated with increased expression of arginase-1 while NOS2 expression was unaltered in macrophages activated by exosomes (Figure 7C & 7D). Importantly, LPS-induced NOS2 expression was found to be significantly inhibited in presence of exosomes (Figure 7C) and thus explained the reduction in LPS-induced NO level in presence of exosomes compared LPS-stimulated group. Several lines of evidence have evaluated the M1 or M2 polarized state of macrophages based on the expression level of arginase-1 and NOS2 [5,19,22,26]. For instance, Bardi et al, 2018, demonstrated that melanoma-derived exosomes promoted mixed polarized state of macrophages (M1 and M2) as they expressed NOS2 as well as arginase-1 [26]. Piao et al, 2017, evaluated that M2 polarized macrophages express arginase-1 when activated by tumor-derived exosomes and they further associated their polarization with lymph node metastasis [5]. In view of the existing studies, we suggest that lymphoma-derived exosomes induced M2 polarized state in macrophages. Although, we also observed arginase-1 expression in LPS-stimulated macrophages (Figure 7D) which is suggested to be a counter regulatory response wherein LPS induces NOS2 expression earlier than arginase-1 expression [93]. Of note, enhanced level of arginase-1 in macrophages is not only important for immunosuppressive M2 polarization but it also weakens the NO-mediated cytotoxic response against the tumor cells. Arginase-1 not only enables the production of polyamines important for tumor cell proliferation but it also limits the substrate availability for the activity of NOS2 enzyme, consequently the NO level and thus prevents tumor cell death [78].
M1/M2 activation of macrophages represent two extreme polarized states. While M1 macrophages are known to inhibit tumor growth, M2 macrophages, or TAMs, are known to promote tumor growth and metastasis via releasing pro-tumoral factors and immunosuppressive cytokines [23]. Cytokines are low molecular weight soluble proteins that act in autocrine or paracrine manner to mediate intercellular communication. Principally, they are synthesized by immune cells which then regulate cell survival, differentiation as well as the overall immune response in the host [94]. In particular, M2 macrophages are immunosuppressive cells which release anti-inflammatory cytokines, such as IL10 and TGFβ, and are known to promote the stemness and migratory properties of cancer cells [8,95,96]. Also, it has been revealed that tumor-derived exosomes induce high level of IL10 and TGFβ in macrophages and thus mediate their polarization towards M2 type [8,19,96,97]. Similarly, we observed increased expression of these immunosuppressive cytokines (Figure 8) which thus suggest activation of M2 macrophages. Besides these anti-inflammatory cytokines, it is evident that TAMs also release certain pro-inflammatory cytokines, such as IL6, IL1β and TNFα, with different sources of exosomes and show the pro-tumoral behavior [22,25,26]. For instance, Linton et al, 2018 observed that pancreatic cancer cell-derived exosomes induced release of IL6, IL1β and TNFα in M2 polarized macrophages [25]. In contrast, few reports also evaluated increased expression of these cytokines by M1 macrophages. However, these M1 macrophages showed the pro-tumoral role and promoted malignant properties of cancer cells [22,26]. Intriguingly, we observed increased expression of IL6 and IL1β while no significant change in the TNFα expression was found in macrophages activated by lymphoma-derived exosomes (Figure 8). Importantly, in our study, the LPS-stimulated group showed the highest expression of IL6, IL1β and TNFα, however in presence of exosomes, the heightened expression of these cytokines was found to be significantly reduced (Figure 8). It could be explained by the fact that the mechanism for the production of these inflammatory cytokines by M1 and M2 macrophages is different. For instance, LPS-induced IL6 production is principally mediated by NFĸB activation [98]; on the other hand, in TAMs, IL6 production is known to be induced via p38-MAPK pathway [99]. So, we speculate that lymphoma-derived exosomes might have inhibited the LPS-induced pathway for pro-inflammatory cytokine production. Additionally, we observed remarkably no difference in the expression profile of the key pro-inflammatory cytokine, IL12, in presence of exosomes. Importantly, we found significantly reduced levels of LPS-induced IL12 in presence of lymphoma-derived exosomes compared to LPS treatment alone (Figure 8). This showed the effectiveness of lymphoma-derived exosomes to inhibit the LPS-induced M1 polarization of macrophages. Taken together, our results substantiated the pro-tumoral M2-specific cytokine profile in macrophages activated by lymphoma-derived exosomes.
5. Conclusion
Accumulating evidence has established the role of tumor-derived exosomes in mediating immunosuppressive microenvironment at local and distant sites. Macrophages are considered to be the key immune infiltrate within tumor microenvironment and their enrichment at tumor site is associated with poor prognosis. Our study demonstrated systemic increase in exosome load in tumor-bearing hosts. Importantly, these exosomes were equipped with key immunomodulatory proteins which mediate the activation of macrophages towards pro-tumoral M2 type. However, in-depth mechanistic studies need to be conducted to better understand the impact of tumor-derived exosomes. Altogether, our study gives beneficial insights into exosome-mediated tumor and immune cell cross-talk which could provide novel avenues for designing efficient immunotherapeutic agents against cancer.
Funding
The work was supported by Faculty Research Programme Grant-IoE (Institute of Eminence), University of Delhi. The author, Saima Syeda received the research fellowship in the form of SRF from ICMR and Kavita Rawat received research fellowship in the form of JRF/SRF from UGC, Government of India.
Author’s Contributions
SS and AS conceptualized and designed the experiments. SS performed the experiments and wrote the original manuscript text. All the authors did the formal analysis and reviewed and approved the final manuscript.
Acknowledgments
The authors would like to thank Prof. Yogendra Singh from Department of Zoology, University of Delhi, for providing funding for LC-MS analysis and Prof. Sunil K. Sharma from Department of Chemistry, University of Delhi, for providing DLS facility. We are grateful to the Central Instrumentation Facility (CIF), Department of Zoology. Also, we would like to acknowledge University Science Instrumentation Centre (USIC), University of Delhi for SEM and confocal facility, IIT-Delhi for TEM facility and CIF, South campus, University of Delhi, for LC-MS analysis and confocal microscopy.
Conflicts of Interest
The authors declare no competing interests
References
- Li, X.; Corbett, A.L.; Taatizadeh, E.; Tasnim, N.; Little, J.P.; Garnis, C.; et al. Challenges and opportunities in exosome research—Perspectives from biology, engineering, and cancer therapy. APL bioengineering. 2019, 3, 011503. [Google Scholar] [CrossRef] [PubMed]
- Atay, S.; Godwin, A.K. Tumor-derived exosomes: A message delivery system for tumor progression. Communicative & integrative biology. 2014, 7, e28231. [Google Scholar] [CrossRef]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Scientific reports. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Nguyen, T.; Cao, H.; Lee, J.; Chung, J. Emerging roles of exosomes in cancer invasion and metastasis. BMB reports. 2016, 49, 18. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget. 2018, 9, 7398. [Google Scholar] [CrossRef]
- Chen, Q.; Li, Y.; Liu, Y.; Xu, W.; Zhu, X. Exosomal non-coding RNAs-mediated crosstalk in the tumor microenvironment. Frontiers in cell and developmental biology. 2021, 9, 646864. [Google Scholar] [CrossRef]
- Syeda, S.; Rawat, K.; Shrivastava, A. Pharmacological Inhibition of Exosome Machinery: An Emerging Prospect in Cancer Therapeutics. Current Cancer Drug Targets. 2022, 22, 560–76. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, NM.; Ocean, AJ.; Singh, S.; Zhang, H.; Thakur, BK.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nature cell biology. 2015, 17, 816–26. [Google Scholar] [CrossRef]
- Chow, A.; Zhou, W.; Liu, L.; Fong, MY.; Champer, J.; Van Haute, D.; et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Scientific reports. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, MA.; Ribatti, D.; Vacca, A.; et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. The Journal of pathology. 2016, 239, 162–73. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, AL.; Ott, M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-β1. Haematologica. 2011, 96, 1302–9. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Molecular cancer. 2018, 17, 1–16. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J-P.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. The Journal of clinical investigation. 2010, 120, 457–71. [Google Scholar] [CrossRef]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PloS one. 2010, 5, e11469. [Google Scholar] [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Frontiers in immunology. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast cancer research. 2015, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-associated macrophages: recent insights and therapies. Frontiers in oncology. 2020, 10, 188. [Google Scholar] [CrossRef]
- Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; et al. Lung tumor cell-derived exosomes promote M2 macrophage polarization. Cells. 2020, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Xie, L.; Lu, C.; Gu, C.; Xia, Y.; Lv, J.; et al. Gastric cancer-derived exosomal miR-519a-3p promotes liver metastasis by inducing intrahepatic M2-like macrophage-mediated angiogenesis. Journal of Experimental & Clinical Cancer Research. 2022, 41, 1–20. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Frontiers in physiology. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Zhang, J.; Chen, W.; Chen, W. M1-like tumor-associated macrophages activated by exosome-transferred THBS1 promote malignant migration in oral squamous cell carcinoma. Journal of Experimental & Clinical Cancer Research. 2018, 37, 1–15. [Google Scholar] [CrossRef]
- Chen, Q.; Li, Y.; Gao, W.; Chen, L.; Xu, W.; Zhu, X. Exosome-Mediated Crosstalk Between Tumor and Tumor-Associated Macrophages. Frontiers in Molecular Biosciences. 2021, 977. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Lima, LG.; Chai, E.P.Z.; Muller, A.; Lobb, RJ.; Krumeich, S.; et al. Breast cancer-derived exosomes alter macrophage polarization via gp130/STAT3 signaling. Frontiers in immunology. 2018, 9, 871. [Google Scholar] [CrossRef] [PubMed]
- Linton, SS.; Abraham, T.; Liao, J.; Clawson, GA.; Butler, PJ.; Fox, T.; et al. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PLoS One. 2018, 13, e0206759. [Google Scholar] [CrossRef]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine. 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Rawat, K.; Syeda, S.; Shrivastava, A. A novel role of Tinospora cordifolia in amelioration of cancer-induced systemic deterioration by taming neutrophil infiltration and hyperactivation. Phytomedicine. 2023, 108, 154488. [Google Scholar] [CrossRef]
- Kumari, R.; Rawat, K.; Kumari, A.; Shrivastava, A. Amelioration of Dalton’s lymphoma–induced angiogenesis by melatonin. Tumor Biology. 2017, 39, 1010428317705758. [Google Scholar] [CrossRef] [PubMed]
- Koiri, R.; Mehrotra, A.; Trigun, S. Dalton’s lymphoma as a murine model for understanding the progression and development of t-cell lymphoma and its role in drug discovery. Int j immunother cancer res. 2017, 3, 1–6. [Google Scholar]
- Rawat, K.; Syeda, S.; Shrivastava, A. Hyperactive neutrophils infiltrate vital organs of tumor bearing host and contribute to gradual systemic deterioration via upregulated NE, MPO and MMP-9 activity. Immunology Letters. 2022, 241, 35–48. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. The Journal of clinical investigation. 2016, 126, 1208–15. [Google Scholar] [CrossRef] [PubMed]
- Konoshenko, M.; Sagaradze, G.; Orlova, E.; Shtam, T.; Proskura, K.; Kamyshinsky, R.; et al. Total blood exosomes in breast cancer: Potential role in crucial steps of tumorigenesis. International Journal of Molecular Sciences. 2020, 21, 7341. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, Z.; Fu, W.; Lu, K.; Gu, X.; Xu, F.; et al. Exosome-derived ADAM17 promotes liver metastasis in colorectal Cancer. Frontiers in pharmacology. 2021, 12. [Google Scholar] [CrossRef]
- Satake, T.; Suetsugu, A.; Nakamura, M.; Kunisada, T.; Saji, S.; Moriwaki, H.; et al. Color-coded imaging of the fate of cancer-cell-derived exosomes during pancreatic cancer metastases in a nude-mouse model. Anticancer Research. 2019, 39, 4055–60. [Google Scholar] [CrossRef]
- Klemke, L.; De Oliveira, T.; Witt, D.; Winkler, N.; Bohnenberger, H.; Bucala, R.; et al. Hsp90-stabilized MIF supports tumor progression via macrophage recruitment and angiogenesis in colorectal cancer. Cell death & disease. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Feng, L.; Weng, J.; Yao, C.; Wang, R.; Wang, N.; Zhang, Y.; et al. Extracellular Vesicles Derived from SIPA1high Breast Cancer Cells Enhance Macrophage Infiltration and Cancer Metastasis through Myosin-9. Biology. 2022, 11, 543. [Google Scholar] [CrossRef]
- Qinglin, D.; Zhigao, X.; Hanluo, L.; Kanghong, H.; Qifa, Y. Identification of CFHR4 Associated With Poor Prognosis of Hepatocellular Carcinoma. 2021.
- Jia, W.; Kidoya, H.; Yamakawa, D.; Naito, H.; Takakura, N. Galectin-3 accelerates M2 macrophage infiltration and angiogenesis in tumors. The American journal of pathology. 2013, 182, 1821–31. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, C.; Chen, X.; Wu, J.; Guan, G.; Zou, C.; et al. Dual role of ARPC1B in regulating the network between tumor-associated macrophages and tumor cells in glioblastoma. Oncoimmunology. 2022, 11, 2031499. [Google Scholar] [CrossRef]
- Martinez-Marin, D.; Jarvis, C.; Nelius, T.; De Riese, W.; Volpert, OV.; Filleur, S. PEDF increases the tumoricidal activity of macrophages towards prostate cancer cells in vitro. PLoS One. 2017, 12, e0174968. [Google Scholar] [CrossRef]
- Zhu, Y.; Wan, N.; Shan, X.; Deng, G.; Xu, Q.; Ye, H.; et al. Celastrol targets adenylyl cyclase-associated protein 1 to reduce macrophages-mediated inflammation and ameliorates high fat diet-induced metabolic syndrome in mice. Acta Pharmaceutica Sinica, B. 2021, 11, 1200–12. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Wang, Y.; Bao, W.; Zhou, Y.; Dang, W.; et al. BIG1 controls macrophage pro-inflammatory responses through ARF3-mediated PI (4, 5) P2 synthesis. Cell death & disease. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Ho, T.-C.; Yeh, S.-I.; Chen, S.-L.; Tsao, Y.-P. Integrin αv and Vitronectin Prime Macrophage-Related Inflammation and Contribute the Development of Dry Eye Disease. International Journal of Molecular Sciences. 2021, 22, 8410. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Zhao, H.; Guan, Q.; Shi, G.; Feng, S.; Gleave, ME.; et al. Clusterin regulates macrophage expansion, polarization and phagocytic activity in response to inflammation in the kidneys. Immunology and Cell Biology. 2021, 99, 274–87. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, F.; Xu, X.; Zhang, J.; Xu, G. The dual role of STAT1 in ovarian cancer: insight into molecular mechanisms and application potentials. Frontiers in Cell and Developmental Biology. 2021, 9, 636595. [Google Scholar] [CrossRef]
- Dominguez, M.; Brüne, B.; Namgaladze, D. Exploring the role of ATP-citrate lyase in the immune system. Frontiers in Immunology. 2021, 12, 632526. [Google Scholar] [CrossRef]
- Atay, S.; Roberson, C.D.; Gercel-Taylor, C.; Taylor, D.D. Ovarian cancer-derived exosomal fibronectin induces pro-inflammatory IL-1β. exosomes and microvesicles. 2013, 1, 2. [Google Scholar] [CrossRef]
- Dan, H.; Liu, S.; Liu, J.; Liu, D.; Yin, F.; Wei, Z.; et al. RACK1 promotes cancer progression by increasing the M2/M1 macrophage ratio via the NF-κB pathway in oral squamous cell carcinoma. Molecular Oncology. 2020, 14, 795–807. [Google Scholar] [CrossRef]
- Wang, S.; Sun, Z.; Zhao, W.; Wang, Z.; Wu, M.; Pan, Y.; et al. CD97/ADGRE5 Inhibits LPS Induced NF-κB Activation through PPAR-γ Upregulation in Macrophages. Mediators of Inflammation. 2016, 2016. [Google Scholar] [CrossRef]
- Pireaux, V.; Sauvage, A.; Bihin, B.; Van Steenbrugge, M.; Rousseau, A.; Van Antwerpen, P.; et al. Myeloperoxidase-Oxidized LDLs Enhance an Anti-Inflammatory M2 and Antioxidant Phenotype in Murine Macrophages. Mediators of inflammation. 2016, 2016. [Google Scholar] [CrossRef]
- Hong, J.; Qu, P.; Wuest, T.; Lin, P.C. Neutrophilic granule protein is a novel murine LPS antagonist. The Journal of Immunology 2020, 204 (1_Supplement), 60.21. [Google Scholar] [CrossRef]
- Zbinden, A.; Layland, SL.; Urbanczyk, M.; Carvajal Berrio, DA.; Marzi, J.; Zauner, M.; et al. Nidogen-1 mitigates ischemia and promotes tissue survival and regeneration. Advanced Science. 2021, 8, 2002500. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood, The Journal of the American Society of Hematology. 2015, 125, 111–23. [Google Scholar] [CrossRef]
- Tang, Y.; Lin, X.; Chen, C.; Tong, Z.; Sun, H.; Li, Y.; et al. Nucleolin Improves Heart Function During Recovery From Myocardial Infarction by Modulating Macrophage Polarization. Journal of Cardiovascular Pharmacology and Therapeutics. 2021, 26, 386–95. [Google Scholar] [CrossRef]
- Oelschlaegel, D.; Weiss Sadan, T.; Salpeter, S.; Krug, S.; Blum, G.; Schmitz, W.; et al. Cathepsin inhibition modulates metabolism and polarization of tumor-associated macrophages. Cancers. 2020, 12, 2579. [Google Scholar] [CrossRef]
- Kim, J.H.; Sun-Hee, O.; Kim, E-J.; Park, SJ.; Hong, SP.; Cheon., JH.; et al. The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochemical and biophysical research communications. 2012, 423, 60–6. [Google Scholar] [CrossRef]
- Ni, Y.; Wu, G-H.; Cai, J-J.; Zhang, R.; Zheng, Y.; Liu, J-Q.; et al. Tubule-mitophagic secretion of SerpinG1 reprograms macrophages to instruct anti-septic acute kidney injury efficacy of high-dose ascorbate mediated by NRF2 transactivation. International journal of biological sciences. 2022, 18, 5168. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, K.; Fujiwara, Y.; Pan, C.; Esumi, S.; Saito, Y.; Bi, J.; et al. α1-Acid Glycoprotein Enhances the Immunosuppressive and Protumor Functions of Tumor-Associated MacrophagesAGP Enhances Protumor Function of TAM in Tumor Development. Cancer Research. 2021, 81, 4545–59. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, AR.; Zulcic, M.; Bao, L.; Messer, K.; Ideker, T.; et al. Rac2 controls tumor growth, metastasis and M1-M2 macrophage differentiation in vivo. PloS one. 2014, 9, e95893. [Google Scholar] [CrossRef]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI insight 2020, 5. [Google Scholar] [CrossRef]
- Wen, G.; Zhang, C.; Chen, Q.; Mustafa, A.; Ye, S.; Xiao, Q. A novel role of matrix metalloproteinase-8 in macrophage differentiation and polarization. Journal of Biological Chemistry. 2015, 290, 19158–72. [Google Scholar] [CrossRef]
- Moraes, LA.; Kar, S.; Foo, SL.; Gu, T.; Toh, YQ.; Ampomah, PB.; et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Scientific reports. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Ochieng, J.; Nangami, G.; Sakwe, A.; Moye, C.; Alvarez, J.; Whalen, D.; et al. Impact of Fetuin-A (AHSG) on tumor progression and type 2 diabetes. International Journal of Molecular Sciences. 2018, 19, 2211. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Caskey, L.; Holtzhausen, A.; Hunter, D.; Story, C.; Earp, H.S. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. The Journal of clinical investigation. 2018, 128, 2356–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mi, J.; Liu, S.; Chen, H.; Jiang, L. PSMB5 overexpression is correlated with tumor proliferation and poor prognosis in hepatocellular carcinoma. FEBS open bio. 2022, 12, 2025–41. [Google Scholar] [CrossRef]
- Fu, W.; Hu, W.; Yi, Y-S.; Hettinghouse, A.; Sun, G.; Bi, Y.; et al. TNFR2/14-3-3ε signaling complex instructs macrophage plasticity in inflammation and autoimmunity. The Journal of clinical investigation 2021, 131. [Google Scholar] [CrossRef]
- Martinez, FO.; Helming, L.; Milde, R.; Varin, A.; Melgert, BN.; Draijer, C.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood, The Journal of the American Society of Hematology. 2013, 121, e57–e69. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nature communications. 2019, 10, 4353. [Google Scholar] [CrossRef]
- Yang, X.; Pang, Y.; Zhang, J.; Shi, J.; Zhang, X.; Zhang, G.; et al. High expression levels of ACTN1 and ACTN3 indicate unfavorable prognosis in acute myeloid leukemia. Journal of Cancer. 2019, 10, 4286. [Google Scholar] [CrossRef]
- Zheng, P.; Luo, Q.; Wang, W.; Li, J.; Wang, T.; Wang, P.; et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein, E. Cell death & disease. 2018, 9, 1–14. [Google Scholar]
- Steitz, AM.; Steffes, A.; Finkernagel, F.; Unger, A.; Sommerfeld, L.; Jansen, JM.; et al. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell death & disease. 2020, 11, 249. [Google Scholar] [CrossRef]
- Dominkuš, PP.; Stenovec, M.; Sitar, S.; Lasič, E.; Zorec, R.; Plemenitaš, A.; et al. PKH26 labeling of extracellular vesicles: Characterization and cellular internalization of contaminating PKH26 nanoparticles. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2018, 1860, 1350–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lee, M.Y.; Jeon, Y.J. Silymarin inhibits morphological changes in LPS-stimulated macrophages by blocking NF-κB pathway. Korean J Physiol Pharmacol. 2015, 19, 211–8. [Google Scholar] [CrossRef]
- Wang, X.; Luo, X.; Chen, C.; Tang, Y.; Li, L.; Mo, B.; et al. The Ap-2α/Elk-1 axis regulates Sirpα-dependent tumor phagocytosis by tumor-associated macrophages in colorectal cancer. Signal Transduction and Targeted Therapy. 2020, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Gordon, SR.; Maute, RL.; Dulken, BW.; Hutter, G.; George, BM.; McCracken, MN.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017, 545, 495–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell research. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive oxygen species in the tumor microenvironment: an overview. Cancers. 2019, 11, 1191. [Google Scholar] [CrossRef]
- Chang, C.-I.; Liao, J.C.; Kuo, L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer research. 2001, 61, 1100–6. [Google Scholar]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, PM.; Hare, RJ.; Gryczynski, I.; et al. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer MetastasisExosomal Anx II in Angiogenesis and Metastasis. Molecular Cancer Research. 2017, 15, 93–105. [Google Scholar] [CrossRef]
- Beckham, CJ.; Olsen, J.; Yin, P-N.; Wu, C-H.; Ting, H-J.; Hagen, FK.; et al. Bladder cancer exosomes contain EDIL-3/Del1 and facilitate cancer progression. The Journal of urology. 2014, 192, 583–92. [Google Scholar] [CrossRef]
- Rahman, M.A.; Barger, J.F.; Lovat, F.; Gao, M.; Otterson, G.A.; Nana-Sinkam, P. Lung cancer exosomes as drivers of epithelial mesenchymal transition. Oncotarget. 2016, 7, 54852. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nature medicine. 2012, 18, 883–91. [Google Scholar] [CrossRef] [PubMed]
- Parada, N.; Romero-Trujillo, A.; Georges, N.; Alcayaga-Miranda, F. Camouflage strategies for therapeutic exosomes evasion from phagocytosis. Journal of Advanced Research. 2021, 31, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Möller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nature Reviews Cancer. 2020, 20, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, K.; Tian, J.; Xia, X.; Ma, J.; Tang, X.; et al. Granulocytic Myeloid-Derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Advanced Science. 2019, 6, 1901278. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, L.; Rong, Y.; Qian, D.; Chen, J.; Zhou, Z.; et al. Hypoxic mesenchymal stem cell-derived exosomes promote bone fracture healing by the transfer of miR-126. Acta Biomaterialia. 2020, 103, 196–212. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. American Journal of Physiology-Renal Physiology. 2017, 313, F906–F13. [Google Scholar] [CrossRef]
- Giusti, I.; Delle Monache, S.; Di Francesco, M.; Sanità, P.; D’Ascenzo, S.; Gravina, GL.; et al. From glioblastoma to endothelial cells through extracellular vesicles: messages for angiogenesis. Tumor Biology. 2016, 37, 12743–53. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. The Journal of Immunology. 2008, 180, 7249–58. [Google Scholar] [CrossRef]
- Ding, X.-Q.; Wang, Z.-Y.; Xia, D.; Wang, R.-X.; Pan, X.-R.; Tong, J.-H. Proteomic profiling of serum exosomes from patients with metastatic gastric cancer. Frontiers in oncology. 2020, 10, 1113. [Google Scholar] [CrossRef]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proceedings of the National Academy of Sciences. 2013, 110, 17253–8. [Google Scholar] [CrossRef]
- Hu, W.; Song, X.; Yu, H.; Sun, J.; Zhao, Y. Released exosomes contribute to the immune modulation of cord blood-derived stem cells. Frontiers in Immunology. 2020, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Klasen, S.; Hammermann, R.; Fuhrmann, M.; Lindemann, D.; Beck, KF.; Pfeilschifter, J.; et al. Glucocorticoids inhibit lipopolysaccharide-induced up-regulation of arginase in rat alveolar macrophages. British journal of pharmacology. 2001, 132, 1349–57. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. Journal of immunology research. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, Y.; Li, Y.; Wang, D.; Liu, S.; Wang, D.; et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. International Journal of Cancer. 2019, 145, 1099–110. [Google Scholar] [CrossRef]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology. 2020, 9, 1747677. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, Y.; Zhang, Y.; Jia, Z.; Zhang, Z.; Yang, J. Cancer derived exosomes induce macrophages immunosuppressive polarization to promote bladder cancer progression. Cell Communication and Signaling. 2021, 19, 1–13. [Google Scholar] [CrossRef]
- Lee, A.-J.; Cho, K.-J.; Kim, J.-H. MyD88–BLT2-dependent cascade contributes to LPS-induced interleukin-6 production in mouse macrophage. Experimental & molecular medicine. 2015, 47, e156. [Google Scholar] [CrossRef]
- Radharani, N.; Yadav, AS.; Nimma, R.; Kumar, T.; Bulbule, A.; Chanukuppa, V.; et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via Stat-3 pathway. Cancer cell international. 2022, 22, 1–19. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of experimental design.

Figure 2.
Exosome level increases in tumor cells and in peripheral blood of DL-bearing mice. A) Representative immunofluorescence images of T cells and DL cells stained with CD63 antibody (magnification, X 200) and A(i) represents the mean fluorescence intensity analysed by ImageJ software. SEM imaging of exosomes isolated from serum of B(i) control and B(ii) DL mice. C(i) and (ii) represent the size distribution of exosomes in control and DL mice, respectively, by dynamic light scattering (DLS). C(iii) Bar graph represents the count rate of exosomes in the sample determined by DLS. D) Immunofluorescence staining of blood smear of control and DL mice with antibody directed against CD63 (magnification, X 400) and A(i) represents the mean fluorescence intensity analysed by ImageJ software. Data are shown from one of the three independent experiments with a similar pattern of results and are expressed as mean ±SD. The statistical significance between control and tumor group was determined by Student t-test where *p<0.05 and ***p<0.001). .
Figure 2.
Exosome level increases in tumor cells and in peripheral blood of DL-bearing mice. A) Representative immunofluorescence images of T cells and DL cells stained with CD63 antibody (magnification, X 200) and A(i) represents the mean fluorescence intensity analysed by ImageJ software. SEM imaging of exosomes isolated from serum of B(i) control and B(ii) DL mice. C(i) and (ii) represent the size distribution of exosomes in control and DL mice, respectively, by dynamic light scattering (DLS). C(iii) Bar graph represents the count rate of exosomes in the sample determined by DLS. D) Immunofluorescence staining of blood smear of control and DL mice with antibody directed against CD63 (magnification, X 400) and A(i) represents the mean fluorescence intensity analysed by ImageJ software. Data are shown from one of the three independent experiments with a similar pattern of results and are expressed as mean ±SD. The statistical significance between control and tumor group was determined by Student t-test where *p<0.05 and ***p<0.001). .
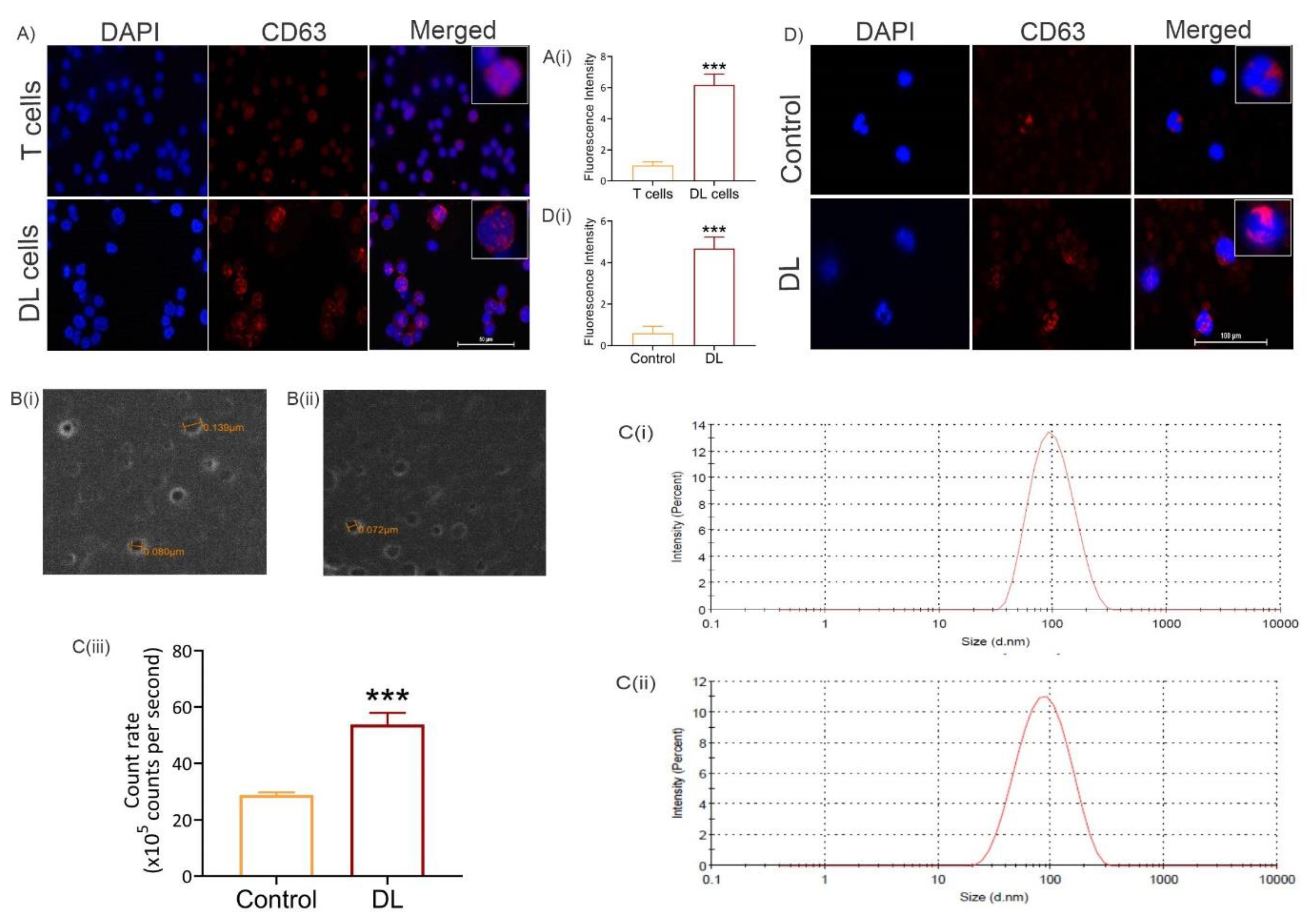
Figure 3.
Exosome level increases systemically in tumor-bearing mice. Representative immunofluorescence images of peritoneum, liver, spleen and lungs stained with anti-CD63 antibody (magnification, X 600, scale bar, 50µm). Bar graphs show the quantitative analysis of immunofluorescence data analysed by ImageJ software. Data are shown from one of the three independent experiments with a similar pattern of results and are expressed as mean ±SD. The statistical significance between the control and tumor group was determined by Student t-test where ***p≤0.001.
Figure 3.
Exosome level increases systemically in tumor-bearing mice. Representative immunofluorescence images of peritoneum, liver, spleen and lungs stained with anti-CD63 antibody (magnification, X 600, scale bar, 50µm). Bar graphs show the quantitative analysis of immunofluorescence data analysed by ImageJ software. Data are shown from one of the three independent experiments with a similar pattern of results and are expressed as mean ±SD. The statistical significance between the control and tumor group was determined by Student t-test where ***p≤0.001.
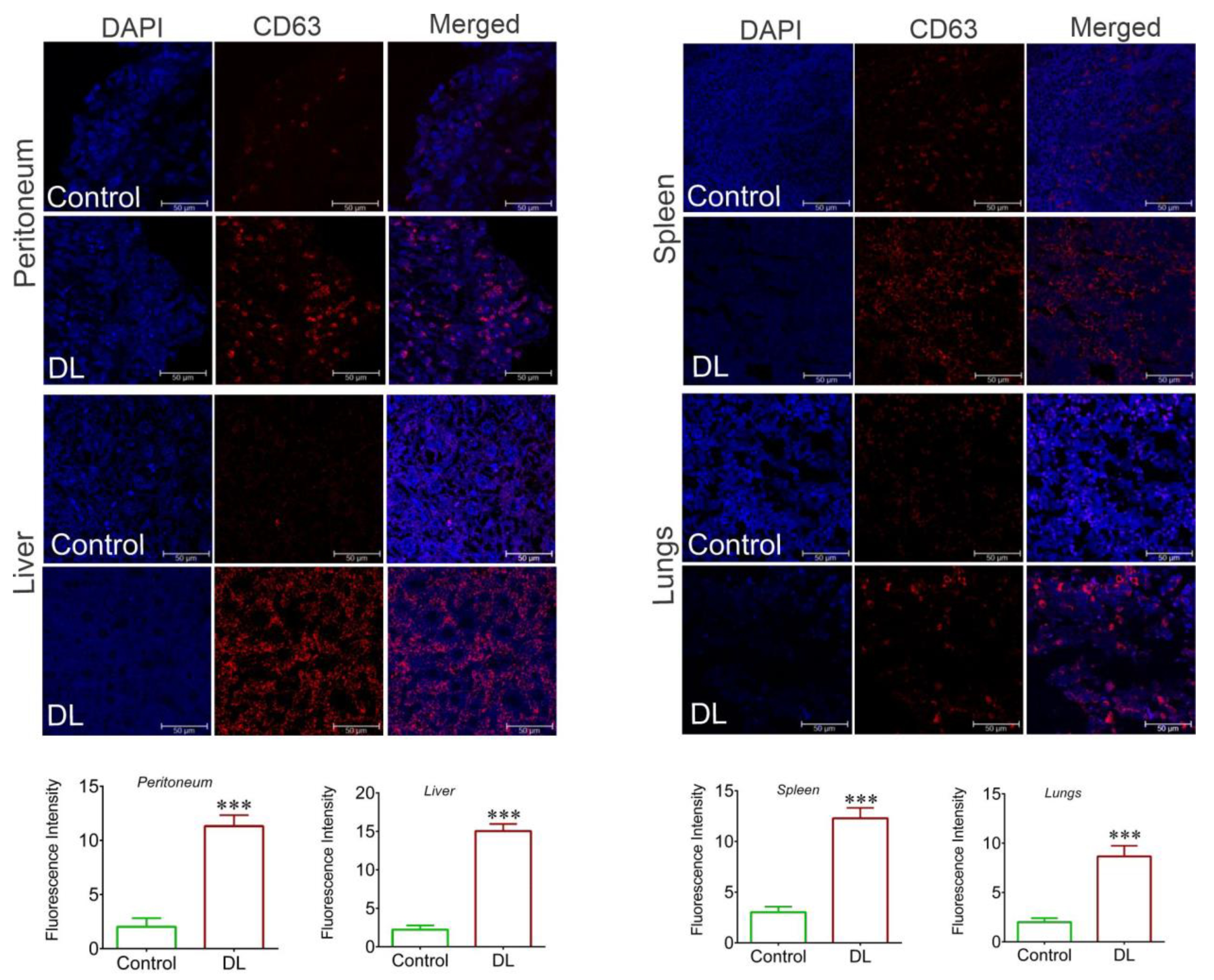
Figure 4.
GO enrichment analysis of lymphoma-derived exosomal proteins characterized by LC-MS analysis. Lymphoma-derived exosomes were assessed for morphology using electron microscopy, Ai) scanning electron microscopy (SEM) and Aii) transmission electron microscopy (TEM), scale bar, 200nm. The size distribution of exosomes was determined by B) dynamic light scattering (DLS) and presence of exosomes was confirmed by C) immunoblotting using exosome-specific markers, Alix and flotillin-1. D(i) and D(ii) Venn diagrams depict the presence of overlap proteins in lymphoma-derived exosomes with the proteins present in ExoCarta and EMTome database, respectively. Bar graphs represent GO enrichment analysis of the identified proteins in lymphoma-derived exosomes in the E(i) cellular component, E(ii) molecular function and E(iii) biological process using Gene ontology resource. Top 20 significantly enriched GO terms (p<0.05) are displayed.
Figure 4.
GO enrichment analysis of lymphoma-derived exosomal proteins characterized by LC-MS analysis. Lymphoma-derived exosomes were assessed for morphology using electron microscopy, Ai) scanning electron microscopy (SEM) and Aii) transmission electron microscopy (TEM), scale bar, 200nm. The size distribution of exosomes was determined by B) dynamic light scattering (DLS) and presence of exosomes was confirmed by C) immunoblotting using exosome-specific markers, Alix and flotillin-1. D(i) and D(ii) Venn diagrams depict the presence of overlap proteins in lymphoma-derived exosomes with the proteins present in ExoCarta and EMTome database, respectively. Bar graphs represent GO enrichment analysis of the identified proteins in lymphoma-derived exosomes in the E(i) cellular component, E(ii) molecular function and E(iii) biological process using Gene ontology resource. Top 20 significantly enriched GO terms (p<0.05) are displayed.
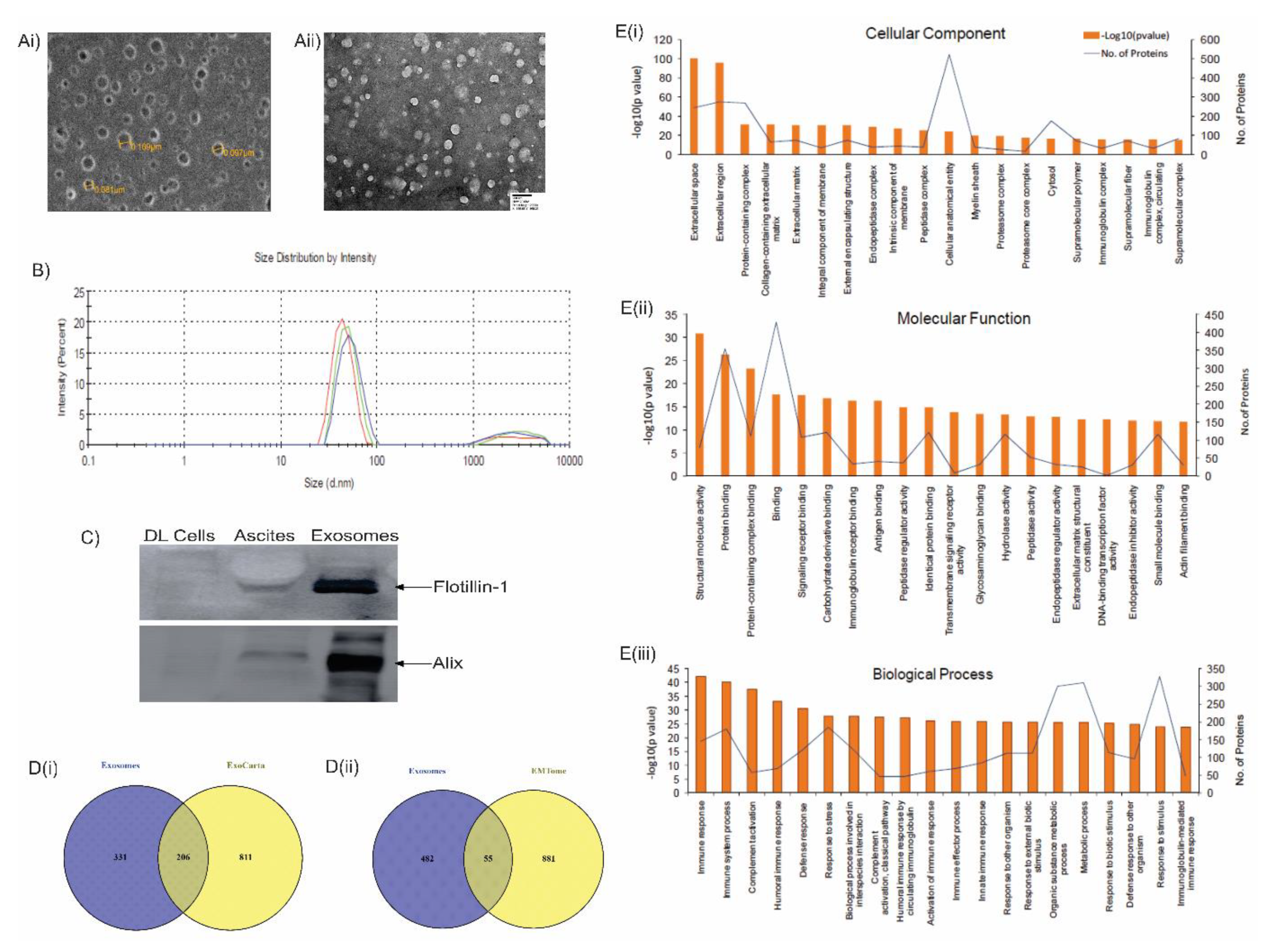
Figure 5.
Interactome network of the immunomodulatory proteins identified in lymphoma-derived exosomes by LC-MS analysis. The proteins enriched in immunomodulatory processes were screened from the top 20 GO enriched biological processes.
Figure 5.
Interactome network of the immunomodulatory proteins identified in lymphoma-derived exosomes by LC-MS analysis. The proteins enriched in immunomodulatory processes were screened from the top 20 GO enriched biological processes.
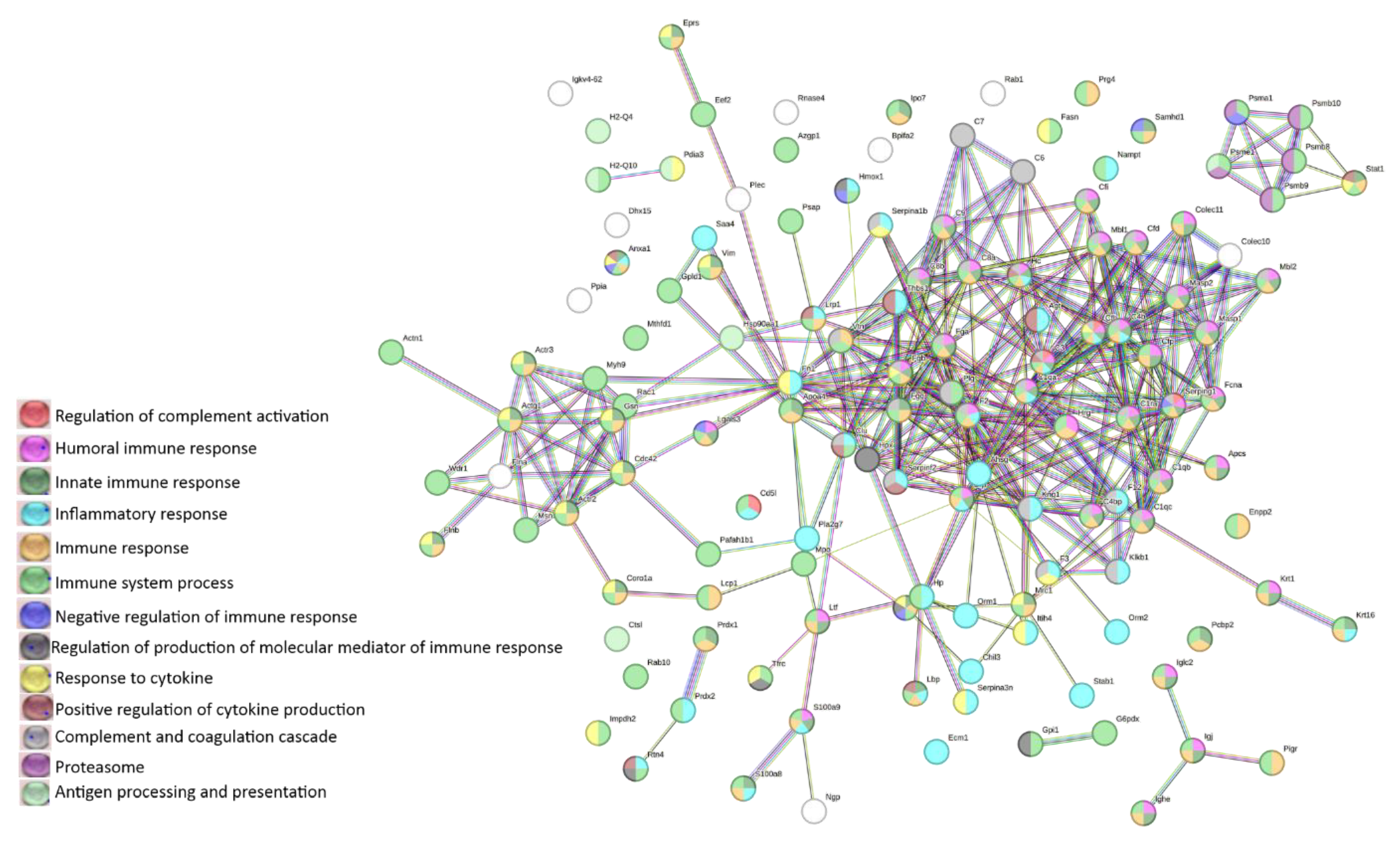
Figure 6.
Uptake of lymphoma-derived exosomes mediate a morphological change in macrophages and reduces their phagocytic activity in the in vitro system. A) Fluorescence images showing the time kinetics for the uptake of lymphoma-derived exosomes by macrophages. The exosomes were pre-labelled with PKH dye and incubated with macrophages for different time points. The red fluorescence represents the uptake of pre-labelled exosomes by macrophages, (magnification, x 400, scale bar, 20µm). B) Representative images showing the morphological change in macrophages in different treatment groups by crystal violet staining (magnification, x 200, scale bar, 50µm). C) Bar graph represents the percent phagocytosis shown by macrophages in different treatment groups. The results are expressed as the mean ± SD from three independent experiments. Statistical significance between the groups was determined by one-way ANOVA followed by Tukey post hoc test where *p<0.05, ***p<0.001 vs control group and ###p<0.001 vs LPS-stimulated group.
Figure 6.
Uptake of lymphoma-derived exosomes mediate a morphological change in macrophages and reduces their phagocytic activity in the in vitro system. A) Fluorescence images showing the time kinetics for the uptake of lymphoma-derived exosomes by macrophages. The exosomes were pre-labelled with PKH dye and incubated with macrophages for different time points. The red fluorescence represents the uptake of pre-labelled exosomes by macrophages, (magnification, x 400, scale bar, 20µm). B) Representative images showing the morphological change in macrophages in different treatment groups by crystal violet staining (magnification, x 200, scale bar, 50µm). C) Bar graph represents the percent phagocytosis shown by macrophages in different treatment groups. The results are expressed as the mean ± SD from three independent experiments. Statistical significance between the groups was determined by one-way ANOVA followed by Tukey post hoc test where *p<0.05, ***p<0.001 vs control group and ###p<0.001 vs LPS-stimulated group.
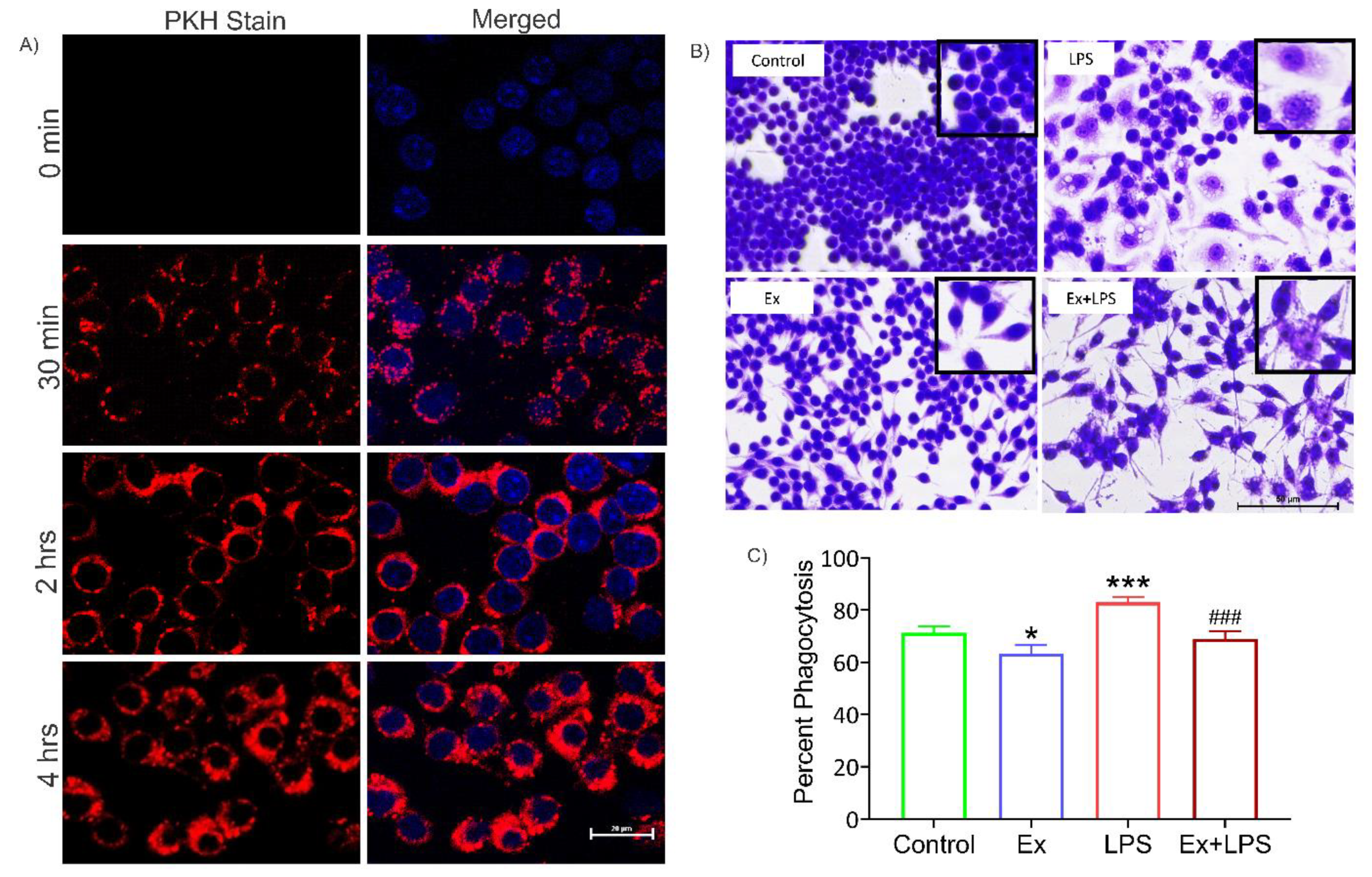
Figure 7.
Lymphoma-derived exosomes increase the ROS levels and upregulate arginase-1 expression in macrophages. A) Representative images showing ROS level assessed by DHE staining in different treatment groups (magnification, x 200, scale bar, 50µm). A(i) Bar graph represents the mean fluorescence intensity of ROS analysed by ImageJ software. B) Bar graph represents the nitrite level assessed by Griess reaction in different treatment groups. C) and D) Representative mRNA expression of NOS2 and arginase-1, respectively, assessed by quantitative RT-PCR in different treatment groups. β-actin was used as an internal control. The results are expressed as the mean ± SD from three independent experiments. Statistical significance between the groups was determined by one-way ANOVA followed by Tukey post hoc test where *p<0.05, ***p<0.001 vs control group and ##p<0.01, ###p<0.001 vs LPS-stimulated group.
Figure 7.
Lymphoma-derived exosomes increase the ROS levels and upregulate arginase-1 expression in macrophages. A) Representative images showing ROS level assessed by DHE staining in different treatment groups (magnification, x 200, scale bar, 50µm). A(i) Bar graph represents the mean fluorescence intensity of ROS analysed by ImageJ software. B) Bar graph represents the nitrite level assessed by Griess reaction in different treatment groups. C) and D) Representative mRNA expression of NOS2 and arginase-1, respectively, assessed by quantitative RT-PCR in different treatment groups. β-actin was used as an internal control. The results are expressed as the mean ± SD from three independent experiments. Statistical significance between the groups was determined by one-way ANOVA followed by Tukey post hoc test where *p<0.05, ***p<0.001 vs control group and ##p<0.01, ###p<0.001 vs LPS-stimulated group.
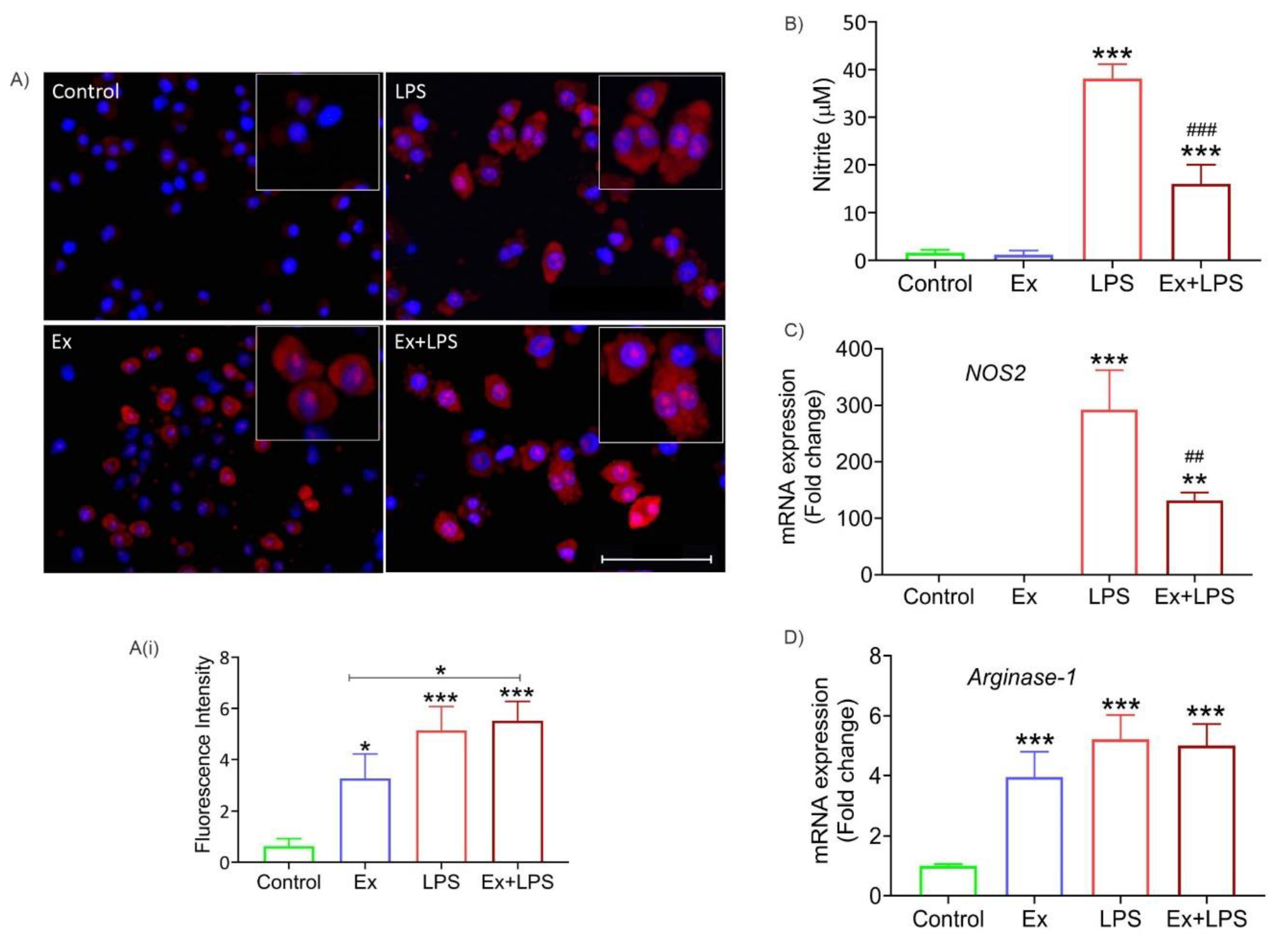
Figure 8.
Lymphoma-derived exosomes induce an M2-specific cytokine profile in macrophages in vitro. Representative mRNA expression of IL10, IL6, TNFα, TGFβ, IL1β and IL12 assessed by quantitative RT-PCR in different treatment groups. β-actin was used as an internal control. The results are expressed as the mean ± SD from three independent experiments. Statistical significance between the groups was determined by one-way ANOVA followed by Tukey post hoc test where *p<0.05, **p<0.01, ***p<0.001 vs control group and #p<0.05, ##p<0.01, ###p<0.001 vs LPS-stimulated group.
Figure 8.
Lymphoma-derived exosomes induce an M2-specific cytokine profile in macrophages in vitro. Representative mRNA expression of IL10, IL6, TNFα, TGFβ, IL1β and IL12 assessed by quantitative RT-PCR in different treatment groups. β-actin was used as an internal control. The results are expressed as the mean ± SD from three independent experiments. Statistical significance between the groups was determined by one-way ANOVA followed by Tukey post hoc test where *p<0.05, **p<0.01, ***p<0.001 vs control group and #p<0.05, ##p<0.01, ###p<0.001 vs LPS-stimulated group.
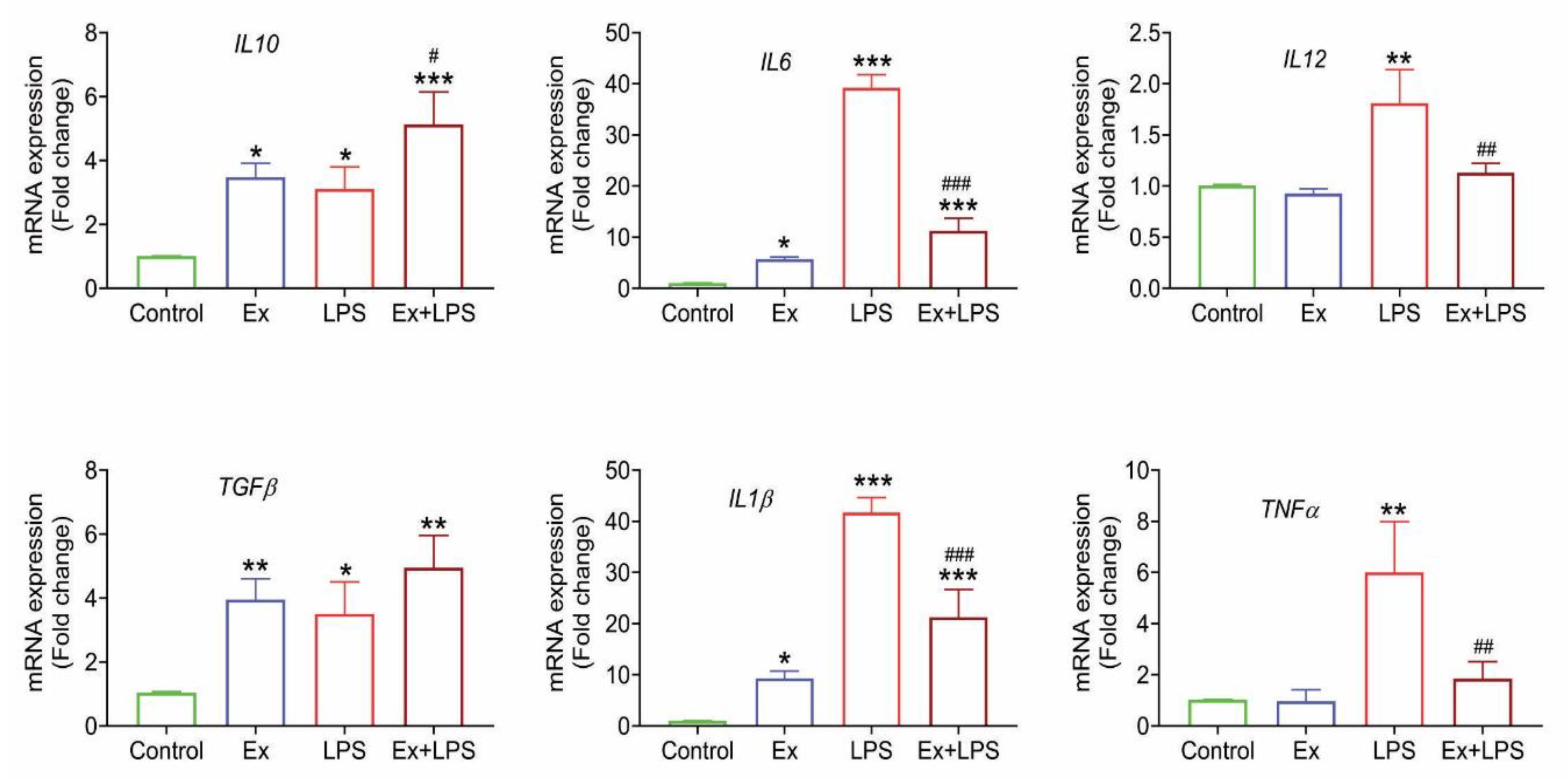

Table 1.
List of primer sequence used for qPCR.
| Gene | Forward | Reverse |
|---|---|---|
| Arginase-1 | CTTAGAGATTATCGGAGCGCCT | AAGTTTTTCCAGCAGACCAGC |
| NOS2 | ACAACAGGAACCTACCAGCTC | TACAGTTCCGAGCGTCAAAGA |
| IL6 | CTTCTTGGGACTGATGCTGGT | GCCATTGCACAACTCTTTTCTCA |
| IL10 | TGAGGCGCTGTCATCGATTT | TGGCCTTGTAGACACCTTGG |
| IL1β | GCCACCTTTTGACAGTGATGAG | ATGTGCTGCTGCGAGATTTG |
| IL12 | TGTGGAATGGCGTCTCTGTC | AGTTCAATGGGCAGGGTCTC |
| TGFβ | ATGCTAAAGAGGTCACCCGC | ACTGCTTCCCGAATGTCTGA |
| TNFα | ACGCTGATTTGGTGACCAGG | CCCGTAGGGCGATTACAGTC |
| β-actin | CTTCTTGGGTATGGAATCCTG | GTAATCTCCTTCTGCATCCTG |
Table 2.
Common proteins found in lymphoma-derived exosomes and EMTome database (http://www.emtome.org./).
Table 2.
Common proteins found in lymphoma-derived exosomes and EMTome database (http://www.emtome.org./).
| Fibronectin |
|---|
| Lymphocyte cytosolic protein 1 |
| Complement factor H |
| Sulfhydryl oxidase 1 |
| Filamin α |
| Vimentin |
| Moesin |
| Alpha actinin 1α |
| Keratin 5 |
| Keratin 15 |
| Xanthine dehydrogenase/oxidase |
| Epidermal growth factor receptor |
| Serine protease HTRA1 |
| Nidogen-1 |
| Keratin 8 |
| Protein S100-A9 |
| Platelet-activating factor acetylhydrolase |
| Fibrillar collagen NC1 domain-containing protein |
| Collagen α-1(I) chain |
| Keratin 19 |
| Keratin 14 |
| Junction plakoglobin |
| Keratin 16 |
| Transforming growth factor-β-induced (TGFβi) |
| Fibulin-1 |
| Laminin subunit Ƴ-2 |
| Fibrillar collagen NC1 domain-containing protein |
| Protein S100-A8 |
| Extracellular matrix protein 1 |
| Desmoplakin |
| Ectonucleotide pyrophosphatase 2 |
| Laminin B1 |
| Filamin-β |
| Coronin |
| D-3-phosphoglycerate dehydrogenase |
| ZnMc domain-containing protein (MMP3) |
| Fibulin-2 |
| Periostin isoform M2 |
| Procollagen-lysine 5-dioxygenase |
| Kallikrein related-peptidase 10 |
| Basement membrane-specific heparan sulfate proteoglycan core protein |
| Procollagen C-endopeptidase enhancer 1 |
| Decorin |
| Low density lipoprotein receptor-related protein 1 |
| Guanine nucleotide-binding protein subunit β-4 |
| Thrombospondin 1 |
| Prolyl 3-hydroxylase 2 |
| Collagen, type IV, α2 |
| Platelet-activating factor acetylhydrolase IB subunit α |
| Heme oxygenase (biliverdin-producing) (HMOX1) |
| Neuropilin 1 |
| Doublecortin like kinase 1 |
| Protein-glutamine Ƴ-glutamyltransferase 2 |
| Collagen, type VI, α3 |
| Signal transducer and activator of transcription (STAT1) |
Table 3.
List of exosomal proteins that affect either macrophage recruitment or macrophage polarization.
Table 3.
List of exosomal proteins that affect either macrophage recruitment or macrophage polarization.
| Proteins affecting recruitment of macrophages | References |
| Heat shock protein 90α (Hsp90α) | [35] |
| Myosin-9 (Myh9) | [36] |
| Complement factor H-related 4 (CFHR4) | [37] |
| Galectin 3 | [38] |
| Actin-related protein 2/3 complex subunit | [39] |
| Proteins associated with M1 macrophage polarization | References |
| Pigment epithelium-derived factor | [40] |
| Adenylyl cyclase-associated protein | [41] |
| ADP-ribosylation factor 3 | [42] |
| Vitronectin | [43] |
| Clusterin | [44] |
| Signal transducer and activator of transcription 1 (STAT1) | [45] |
| ATP-citrate synthase | [46] |
| Proteins associated with M2 macrophage polarization | References |
| Fibronectin | [47] |
| Receptor of activated protein C kinase 1 | [48] |
| Adhesion G protein-coupled receptor E5 | [49] |
| Myeloperoxidase | [50] |
| Neutrophilic granule protein | [51] |
| Nidogen 1 | [52] |
| Nicotinamide phosphoribosyl transferase | [53] |
| Nucleolin | [54] |
| Cathepsin B | [55] |
| Protein S100A8 | [56] |
| Plasma protease C1 inhibitor | [57] |
| α-1-acid glycoprotein | [58] |
| Protein S100A9 | [56] |
| Ras-related C3 botulinum toxin substrate 2 | [59] |
| Heme oxygenase | [60] |
| ZnMc domain-containing protein | [61] |
| Annexin | [62] |
| α-2-HS-glycoprotein | [63] |
| Vitamin K-dependent protein S | [64] |
| Procathepsin L | [55] |
| Nidogen | [52] |
| Proteasome subunit β5 | [65] |
| 14-3-3 protein € | [66] |
| Arginase 1 | [67] |
| Macrophage mannose receptor 1 (Mrc1) | [67] |
| Chitinase-like protein 3 | [68] |
| Protein-glutamine Ƴ-glutamyltransferase 2 (Tgm2) | [67] |
| α actinin 1 A | [69] |
| Vacuolar protein sorting-associated protein 35 (Vps35) | [67] |
| Apolipoprotein E (ApoE) | [70] |
| Cytoplasmic FMR1-interacting protein (Cyfip1) | [71] |
| Transferrin receptor protein 1 | [71] |
| Transforming growth factor-beta-induced protein (TGFβi) | [71] |
| Lipoprotein lipase | [71] |
| Lipopolysaccharide binding protein | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



