
Submitted:
04 May 2023
Posted:
05 May 2023
Read the latest preprint version here
Abstract
In South Africa, drug-resistant tuberculosis (DR-TB) continues to be a serious public health issue. Gene mutations and the genotyping of DR-TB in a rural Eastern Cape Province have not previously been studied. We detected prevalent mutations of DR-TB, identified genetic diversity, and assigned lineages. Sputum specimens obtained from 1157 patients suspected with tuberculosis were assayed for rifam-pin-resistance using Xpert® MTB/RIF.and detection of mutations conferring resistance to anti-TB drugs was carried out using GenoType MTBDRplus VER 2.0. Thereafter, 441 isolates were spoligotyped. The most prevalent rifampin resistance-conferring mutations were in rpoB codon S531L. The INH resistant strains, 54.5% had mutations in katG gene at codon S315TB and 24.7% with mutations in inhA gene at codon C-15TB. Furthermore 69.9% strains displayed mutations involving both rpoB and katG genes, while 24.6% strains displayed mutations involving both rpoB and inhA genes. Overall prevalence of hetero-resistance was 17.9%. Beijing family was predominant from Spoligotyping analysis. The diversity of mutations in the study provides information for investigating the evolutional lineages of M. tuberculosis isolates. The recognition frequency of rpoB, katG and inhA mutations in different study areas may help to guide decision‑making about standardization of treatment regimens or individualized treatment in areas where these mutations have been found.
Keywords:
Drug-resistant TB
; heteroresistance
; mutations
; spoligotyping
1. Introduction
Global public health continues to be threatened by tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis (M. tuberculosis) and ranked among the top 10 causes of mortality globally [1]. In 2020, the estimated number of incident cases of TB stands at 9.9 million out of which Africa had a 25% share. South Africa is one of the 30 countries with a high TB burden and has the eighth highest TB incidence globally having more than 500 cases per 100,000 population, which towers above the global average incidence of 127 cases per 100,000 population [2].
In South Africa, TB continues to be a disease of major importance and remained the leading cause of death for three consecutive years that is from 2016 to 2018 [3]. In 2019 alone, an estimated 360,000 South Africans became ill with TB and 58,000 people were estimated to have died from the disease [2]. The COVID-19 pandemic's effects have undone years of progress made in reducing the number of TB deaths worldwide, with the predicted number of deaths in 2020 returning to the level of 2017 [4]. The eradication of TB by 2035, a strategic goal of World Health Organization (WHO) cannot be actualized unless the emergence of resistance in TB is seriously addressed and controlled [5].
Drug-resistant TB (DR-TB) has emerged as a major risk to global TB control. Different mutations in genes such as rpoB, katG, inhA, pncA, embB, rpsL, gyrA, ethA and rrs have been identified conferring resistance to TB first-line drugs, second-line drugs or injectables and fluoroquinolones [6,7]. Mutations in codon 507 to 533 regions of the rpoB gene, called the rifampicin-resistance determining region (RRDR) are majorly responsible for rifampicin resistance while mutations in the katG and inhA genes are associated with INH resistance. The katG and inhA mutations give rise to high-level and low-level INH resistance respectively. Although mutations in both gyrA and gyrB genes are responsible for fluoroquinolone resistance, gyrA accounts for 60%-70% of all mutations [8,9,10]. Recent research has revealed that different mutations in M. tuberculosis can confer varying levels of phenotypic resistance to anti-TB medications [6,7,11]. Consequently, the aggregation of mutations at several positions has a comprehensive effect on drug resistance [12].
In rural areas of the Eastern Cape, drug resistance and gene mutations remain major barriers to effective control and management of TB. However, there was no report on the frequency of gene mutations associated with resistance to rifampicin (RIF) and isoniazid (INH). Hence, in this study, we report the prevalence of mutations in drug resistance genes (rpoB, katG and inhA) and identified the strains and lineages of DR TB strains.
2. Materials and Methods
2.1. Study Site, Patient Population, and Specimen Collection
Sputum specimens were obtained from 1157 patients enrolled from different healthcare facilities and sent for testing and analysis to the National Health Laboratory Services (NHLS) at Mthatha over a 36-month duration (January 2018 to December 2020). These specimens were collected from patients showing clinical signs of TB. The patients were enrolled from one hundred and eighteen (118) healthcare facilities located in five districts including Oliver Reginald (OR) Tambo, Alfred Nzo, Amathole, Chris Hani, and Joe Gqabi.
2.2. Xpert® MTB/RIF Assay
This technique was used to find mutations in the RRDR of M. tuberculosis directly on TB sputum samples. The sputum samples were first decontaminated, then a 2:1 reagent buffer containing NaOH and isopropanol was added, followed by 15 minutes of incubation at room temperature [13]. The Xpert MTB/RIF cartridge was then put into the GeneXpert equipment, where the testing procedure is totally automated, with two milliliters of the final samples. The results of the experiment were categorised as RIF resistant or susceptible and M. tuberculosis negative or positive.
2.3. Phenotypic Drug Susceptibility Testing (DST)
According to the manufacturer's instructions, the test was performed on sputum samples using the automated BACTEC Mycobacterial Growth Indicator Tube (MGIT) 960 (Becton Dickinson, USA). Isoniazid (0.1 g/mL of medium), Rifampin (1.0 g/mL of medium), Ofloxacin (2.0 g/mL), Amikacin (1.0 g/mL), Kanamycin (2.5 g/mL), and Capreomycin (2.5 g/mL) were utilized as the critical concentrations for the first and second line anti-TB medications [14].
Briefly, reconstitution of PANTA powder with growth supplement (15mL) was done, followed by addition to MGIT tube and inoculation of each tube with 0.5ml of the processed specimen. The tubes were incubated at 37 0C in the BACTEC MGIT 960 instrument and monitored automatically every 60 min for increased fluorescence. Culture tubes were maintained until they became positive or for a maximum of 42 days to confirm if negative. Positive samples were removed from the instrument and recorded as positive along with the number of incubation days.
2.4. Genotypic DST
In this study, a Deoxyribonucleic acid (DNA) strip-based method called GenoType MTBDR plus version 2.0 (Hain Lifescience, Nehren, Germany) was used to simultaneously detect the most significant rpoB mutations, which confer RIF resistance, and katG and inhA mutations, which confer high- and low-level INH resistance, respectively. Following the manufacturer's instructions, three procedures—DNA extraction, multiplex amplification using biotinylated primers, and reverse hybridization—were carried out. [15].
DNA extraction was performed using a Genolyse kit (Hain Lifescience, Nehren, Germany). Briefly, 1 ml of the decontaminated sputum was centrifuged for 15 min at 10000×g, and the supernatant was discarded. The sample was incubated at 950C for 5 minutes after the addition of 100 μl of lysis buffer in order to re-suspend the sediment. Subsequently, 100 μl of neutralization buffer was added, vortexed, and centrifuged for 5 minutes [16]. Multiplex amplification was carried out in different cycles at different temperatures as follows: 1 cycle of 15 min at 95°C, 20 cycles each of 30 s at 95°C and 2 min at 65°C, 30 cycles each of 25 s at 95°C, 40 s at 50°C, and 40 s at 70°C, and finally 1 cycle of 8 min at 70°C. Reverse hybridization was performed using an automated hybridization system, Auto-Lipa 48 system (Innogenetics) following the manufacturer’s instructions [15].
2.5. Evaluation and Interpretation of Results
The conjugate control and amplification control bands were lined up with the corresponding lines on the evaluation sheet before the generated strips were adhered to it in the prescribed fields. The mutation bands were deciphered using GenoLyse package inserts; the absence of a wild-type band and the presence of a mutant band for a particular gene on the strip denoted resistance.
2.6. Spoligotyping
441 randomly chosen M. tuberculosis isolates out of 1157 total isolates were genotyped using spoligotyping analysis. DNA was extracted after the samples had been heated to death. Following the manufacturer's instructions, the fluorescence intensity was measured with Luminex 200® (Austin, TX) and microbeads from the TB-SPOL Kit (Beamedex®, Orsay, France) to perform spoligotyping. Binary and octal formats were created from the hybridization patterns. The isolates' produced binary codes were added to the Pasteur Institute of Guadeloupe's SITVIT2 database and given particular spoligotype international types (SIT) [17]. As a positive control, M. tuberculosis H37Rv was used to confirm quality.
3. Results
3.1. Profile of the Isolates
A total of 1157 clinical isolates were analyzed over three years: 2018, 2019, and 2020 (Table 1), of which 950 (82.1%) were drug-resistant (DR-TB, multidrug-resistant TB (MDR-TB), extensively drug-resistant TB (XDR – TB)) and 207 (17.9%) were heteroresistant isolates. Table 1 displays the number of M. tuberculosis isolates from years 2018 to 2020. The heteroresistance rate increased over time as displayed in Table 1.
3.2. Mutations in the rpoB, katG, and inhA Genes
rpoB 531 and katG 315 mutations were the most prevalent variants linked to RIF and INH resistance, respectively, among 1157 M. tuberculosis clinical isolates. Among RIF- resistant strains, known mutations in rpoB were found in 761 (65.8%), wild type was found in 381 strains while missing mutation accounted for the remaining 15 strains. The most prevalent RIF resistance-conferring mutations were in rpoB codons S531L (70.2%). Others included D516V (18.3%), H526D (3.8%), and H526Y (3.4%) (Figure 1). Out of 761 isolates, 532 (69.9%) isolates displayed mutations involving both rpoB and katG genes while 187 (24.6%) isolates displayed mutations involving both rpoB and inhA genes.
The INH-resistant strains totaled 916 isolates with mutations in the katG and inhA genes accounting for 630 and 286 isolates respectively. Mutations in the katG gene which are associated with a high level of INH resistance occurred in codon S315TB of the 630 (68.8%) (Figure 2); mutations in inhA which are associated with a low level of INH resistance were found in 286 (31.2%) isolates and were detected in codons C-15TB (88.5%), T8A (8.4%), T-8C (2.1%), A-16G (0.7%) with double mutations in codons A-16G and T8A (0.3%) (Figure 3).
Out of 761 RIF-resistant strains, combined mutations involving the rpoB and katG or rpoB and inhA genes are displayed in Table 2 below.
3.3. Heteroresistance Mutations
Of 1157 DR-TB isolates, the overall prevalence of heteroresistance was 17.9% (n=207). This rate increased over time, ranging from 9.6% in 2018 to 32.2% in 2020. Heteroresistance to RIF was found in 58 (28%) cases whilst heteroresistance to INH was determined in 119 (57.5%) cases, of which 70 (58.8%) and 49 (41.2%) cases were katG and inhA-associated heteroresistance, respectively. In addition, combined heteroresistance to RIF and INH was found in 30 (14.5%) DR-TB isolates.
3.4. Spoligotyping
The 441 spoligotyped isolates produced 437 unique spoligotype patterns. A pre-existing SIT in the SITVIT2 database matched the patterns of 410 isolates (93.1%), but 27 unique patterns (6.1%) were not present in the database. With 185 (42%) isolates, the Beijing family was the most prevalent genotyping lineage, followed by the LAM family with 83 (18.8%), the X family with 48 (10.9%), and the T family with 34 (7.7%). In addition, the S family accounted for 31 (7.0%) isolates, the EAI family for 16 (3.6%), the H family for 6 (1.4%), and the CAS family for 5 (1.1%) isolates.
4. Discussion
Spontaneous chromosomal mutations in particular locations of the bacterial genome is generally thought to be the cause of resistance to RIF and INH which several studies have proven [9]. It is uncertain how M. tuberculosis strains are distributed, how much has been transmitted recently and in the past, and how DR strains transmit in rural Eastern Cape and according to our knowledge, this is the first study detailing the distribution of drug-resistant genes, mutation sites and genotypes.
Clinicians are concerned about MDR-TB produced by mutations in M. tuberculosis, which are mostly caused by the rpoB, katG, and inhA genes [18]. RIF resistance is typically thought to as a marker for MDR-TB. Hence, screening mutations in candidate genes constitute the foremost significant step to making a definite diagnosis in drug-resistant strains. The 81 bp core region of the rpoB gene's nucleotide sequences was examined for mutations. The prevalence of mutations in the rpoB gene in this study was higher than in the other genes (katG and inhA genes). Analysis of the RIF-associated mutations revealed a prevalence of 65.8% of the rpoB gene. This is comparable to the study done by Otchere et al., with a prevalence of 52% [19]. Contrarily, the prevalence of mutation reported in studies [9,20,21] was higher than that found in our investigation, at 93.5%, 94.9%, and 91.2%, respectively, in the katG gene. Our analysis found that the S531L codon was the site of the majority of rifampin resistance-causing mutations in the rpoB gene. This finding is consistent with earlier research [9,20,22,23] and may be related to the propagation of a common clone. This codon's mutation is known to be a hot area for rpoB gene mutations in M. tuberculosis, and it has also been observed in other South African provinces [24] showing that these mutations are prevalent in the country. This high frequency of occurrence may be due to the low fitness cost associated with rpoB S531L [24] and have been associated with major MDR-TB outbreaks. [23]. Low frequency of mutation was observed in codon 526 at 3.8% in this study but higher in Uganda at 12.5% [25]; China at 14.9% [20]; Brazil at 9.9%, [26] indicating that frequency varies with different geographic locations. Codons 526D and 531 co-occurred at a lesser rate in this study (2.8%) than they did in Iran (23.9%) [27].
Due to its early significant bactericidal activity, INH is a first-line TB medication and a very important medicine for the treatment of TB. For doctors, finding mutations in the katG or inhA promoter region is crucial since it predicts the degree of INH resistance and helps them choose the best course of treatment [28]. Given the high degree of INH medication resistance, the catchment regions of the clinics in this study area need to be watched for any changes that may arise during patient TB treatment. In this investigation, both the katG S315T gene mutation and very resistant INH strains were seen. The majority of mutations occur at codon 315, which is present in 30% to 90% of INH-resistant bacteria [16]. This claim was corroborated by the study's findings, which showed a correlation between elevated levels of INH medication resistance and S315T mutations. Several nations, such as Zambia and Brazil, have observed a tendency of codon S315T mutation in the katG gene [9,29]. Mutations in katG occurred only in codon agc/acc S315Tb in this study but the study of Jagielski et al. [30] found that mutations occurred in eleven other codons. The prevalence of katG S315T varied according to the geographic region: Sub-Saharan Africa (94.9%) [9], West Africa (64%) [31], Southeast Asia (29.3%) [32], and the United States (38%) [33] while the global frequency of katG S315 is estimated at 64.2% [34]. In addition to the katG gene, the inhA, fabG1, and oxyR-ahpC genes are also associated with M. tuberculosis INH resistance. It has been discovered that 20–42% of INH-resistant bacteria carry mutations in the inhA promoter region [33,34]. Previous studies have shown that polymorphisms in the promoter region of the inhA gene cause low-level resistance to INH, which ranges from 8% to 43% [35]. In this study, low-level resistance ratio was 31.2%, close to the high limit of the reported range. Other percentages of inhA mutations have been found in various parts of South Africa, including Kwazulu Natal (27.5%) [36] and Free State (13.4%) [8]. On the other hand, INH-resistant strains were found in Zambia and Ethiopia at rates of 0.8% and 2.0%, respectively, of mutations in the inhA promoter region [9,37]. Contrarily, Lempens et al. [38], reported a lower percentage of occurrence of C-15T gene mutation with a high level of INH resistance. This suggests that mutation of the inhA gene does not always indicate a low resistance level. Mutations in the inhA gene also give cross-resistance to ethionamide (ETH), a second-line medication used in MDR therapy, and are therefore thought of as a surrogate marker for early diagnosis of ETH resistance [8,39,40]. This is because the two drugs share the same target of action. The ETH was once a component of the treatment plan for MDR-TB in South Africa, according to the National Department of Health (NDoH) in that country. In the presence of inhA mutations, employing ETH to treat MDR-TB would not have been successful due to this cross-resistance [8]. Consequently, in the clinical management of MDR-TB cases displaying inhA mutations, ETH must be excluded from the regimen. The majority of our C-15TB isolates (88.5%) included the most prevalent inhA gene mutation, supporting the findings of Seifert et al. [41], who stated that the most prevalent inhA gene mutation was frequently seen in C-15TB.
The clinical and molecular characteristics of the M. tuberculosis strains vary in different areas. This was observed in this study in consonance with the findings of Liu et al. [12]. Investigating the evolutionary lineages of M. tuberculosis can benefit from the range of mutations. The prevalence of rpoB, katG, and inhA mutations in various regions of Mthatha may aid in determining whether to standardize treatment plans or provide tailored care in each region where these mutations have been discovered. There is an indication of M. tuberculosis strains that are constantly mutating as we observed combined mutations (Table 2). This data can be used in the development of new anti-TB drugs.
Heteroresistance in the study area increased with time, by the third year of the study period, the heteroresistance rate was almost tripled that of the previous year (Table 1). The rpoB and katG combination had the highest number of heteroresistant isolates followed by rpoB and inhA combination. In rpoB and katG combination, the mutation regions rpoB S315L and katG 531ST had the highest number of isolates (Table 2). Under the selective pressure of inadequate anti-TB medication, separation into susceptible and resistant organisms most likely elucidates heteroresistance caused by infection with single strains. Several reports have documented the development of resistance as a result of insufficient treatment [42]. It has also been proven that mixed-strain infections caused by heteroresistant bacteria might have a negative effect on treatment outcomes. Treatment of such instances with first-line anti-tuberculosis medicine may select for and increase the drug-resistant strain in the host because heteroresistance makes it possible for the drug-resistant strain to go undetected [43]. The rate of 17.9% of heteroresistance in this study is similar to the finding by Rinder et al. [44], who reported a rate of 17%. Other studies have reported significantly lower rates [45]. In our study one strain of either the Beijing, LAM, or X genotype induced heteroresistance, indicating that the division of a single strain into susceptible and resistant organisms is most likely the main underlying mechanism.
The M. tuberculosis population in this study area was genetically diverse. From the 441 clinical isolates, 23 spoligotypes were observed and classified into major M. tuberculosis lineages; lineages 1, 2, 3, and 4 as shown in Table 3. Beijing and Euro-American (LAM, T, S, and X) strains dominate the population structure of rifampicin-resistant tuberculosis (RRTB) isolates in South Africa, which can be explained by the historical movement of strains as South Africa was located in a geographically central position on the historical trade route between East and West for hundreds of years [46].
One of the most widespread genotypes of M. tuberculosis found globally is the Beijing family, often known as lineage 2. It is usually linked to immune evasion and antibiotic resistance, which promotes rapid bacterial replication, spread, and transmission [47]. The Beijing family, which is more transmissible than other families [48] were prevalent in this study (42%). This lineage has been detected in studies reported from other parts of South Africa including Limpopo, Western Cape, and Mpumalanga [49,50]. According to Said et al. [49] Beijing family is predominant in the Eastern Cape followed by LAM. According to the study of Chihota et al. [51] based on the review of the repository and databases, South African M. tuberculosis strains revealed the greatest diversity and greatest abundance of Beijing families. Furthermore, the association between HLA-B27 and host-pathogen compatibility has accounted for the success of the Beijing lineage in South Africa [51]. Given the growing concern over the prevalence of Beijing strains and their success in evolving to fit into various human groups, suitable measures should be implemented for public health surveillance. The knowledge of the lineages circulating in the study area will help in understanding the drivers of drug resistance and their impact on treatment outcomes and management of TB transmission.
Beijing lineage was documented for the first time in East Asian nations, according to van Soolingen et al. [52], with a particular spoligotype pattern distinguished by the inclusion of the last 10 spacers (spacers 34– 43). The Beijing strains were historically introduced to South Africa, not from their primary origin (China), but from its secondary origin (Indonesia), according to historical evidence corroborated by genetic data [46]. Pokam et al. [53] argued that the dominance of the Beijing family is related to the current influx of Asian population into the African region, as well as increased trade relations of Africans returning from business trips to China, which led to the spread in the continent. Various theories have been proposed to support the introduction of the Beijing family into Africa. It is important to actively carry out surveillance of the Beijing family to verify its heightened transmission and understand its importance in the management plan of TB in this area.
In our investigation, the LAM lineage was the next most common (18.8%). This is not unexpected given that this genotype has been found to be widespread in the provinces of the Eastern Cape and the Free State [54,55]. This suggests there is a continuous TB transmission strain still ongoing throughout the province. Nonetheless, the LAM genotype predominates in Gauteng, Northern Cape, and KwaZulu Natal [49] and some other neighboring countries in Southern Africa, including Zambia and Zimbabwe [56,57]; but with least prevalence in Western Cape [49] The LAM strain is predominant in KwaZulu Natal, which is a neighboring Province to Eastern Cape; this might eventually bring it to par with the predominant Beijing strain thereby causing more havoc. Hence, continuous surveillance of the genetic diversity must be carried out to profile these strains. Concerning delineation of the LAM sub-lineages, five were found in our study namely LAM 3, 4, 5, 9 and LAM II-ZWE with LAM 3 being predominant at 74.7%. On the other hand, another study in South Africa reported that out of the 12 sub-lineages delineated globally, six sub-lineages were found of which LAM4 was the most predominant [54]. LAM11-ZWE has been reported to be a dominant subfamily in Zambia and Zimbabwe [56] whose origin has been traced to Portugal [51]. Two strains from the LAM 3 sub-lineage were labeled as orphans because they had no matches in the SITVIT2 database. Compared to the results obtained by Maguga-Phasha et al., LAM 3 in our investigation, which corresponds to SIT33 and SIT 719, represented 8.2% and 3.9%, respectively [50] in Limpopo had LAM 3 (7.0%) corresponding to SIT 33 only. LAM 1, LAM 2 and LAM 6 were not reported in this study but reported elsewhere [29].
5. Conclusions
The identification of areas where DR-TB is concentrated in rural areas where TB is a burden could assist policy makers to implement targeted interventions aimed at the pre-vention and management of TB transmission. This is crucial in environments with little resources and in places with a high prevalence of DR-TB, such as the rural districts around Mthatha. Targeted interventions to rural populations are essential since it is impossible to deliver DR-TB treatments to all groups in these locations because DR-TB diagnosis and treatment are complicated by a number of factors, including co-infection with HIV and poor treatment adherence.
The distribution of gene mutations and spoligotypes among treating physicians, as well as the importance of laboratories reporting these mutations, are of utmost importance because little is known about the effects of antibiotic-resistance conferring mutations on the bacterial fitness and spoligotypes of M. tuberculosis. Continuous surveillance is advised in this study region since genetic alterations are to blame for the high rate of resistance shown in tuberculosis treatment. Early drug-resistant tuberculosis identification is crucial for the prevention and management of the spread of these drug-resistant variants.
Author Contributions
Conceptualization, L.M.F.; methodology, L.M.F.; formal analysis, L.M.F., A.D., S.O. and R.W.; investigation, L.M.F.; resources, L.M.F. and R.W.; data curation, A.D. and S.O.; writing—original draft preparation, L.M.F., N.S. and M.C.H.; writing—review and editing, L.M.F., N.S. and M.C.H.; visualization, L.M.F. and M.C.H.; supervision, R.M.W., S.V. and T.A.; project administration, L.M.F.; funding acquisition, L.M.F. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for this study was obtained from the South African Medical Research Council (SAMRC) Research development grant (Pilot grant).
Institutional Review Board Statement
This study was conducted in accordance with the Decla-ration of Helsinki and approved by the Research Ethics and Biosafety Committee of the Faculty of Health Sciences of Walter Sisulu University (Ref. No. 026/2019) and Eastern Cape Department of Health (Ref No EC_201904_011).
Informed Consent Statement
Not applicable. This study used routine samples received in National Health Laboratory Services (NHLS) TB laboratory.
Data Availability Statement
Data will be made available upon request from the corresponding author.
Acknowledgments
The authors are grateful to the NHLS TB laboratory staff and participating clinics for their support during sample analysis and data collection.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bwalya, P.; Yamaguchi, T.; Solo, E.S.; Chizimu, J.Y.; Mbulo, G.; Nakajima, C.; Suzuki, Y. Characterization of Mutations Associated with Streptomycin Resistance in Multidrug-Resistant Mycobacterium tuberculosis in Zambia. Antibiotics 2021, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global tuberculosis report, 2020 World Health Organization. Geneva, Switzerland, available at https://apps.who.int/ iris/handle/10665/336069 [accessed August 6, 2022].
- Statistics South Africa. Mortality and causes of death in South Africa. Findings from death notification. Pretoria, South Africa. Stats SA’s June 2021 report [accessed 21 January 2022].
- World Health Organization. Global tuberculosis report 2021: Supplementary material [accessed August 8, 2022].
- Kabir, S.; Junaid, K.; Rehman, A. Variations in rifampicin and isoniazid resistance associated genetic mutations among drug naïve and recurrence cases of pulmonary tuberculosis. Int. J. Infect. Dis. 2021, 103, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Libiseller-Egger, J.; Phelan, J.; Campino, S.; Mohareb, F.; Clark, T.G. Robust detection of point mutations involved in multidrug-resistant Mycobacterium tuberculosis in the presence of co-occurrent resistance markers. PLoS Comput. Biol. 2020, 16, e1008518. [Google Scholar] [CrossRef]
- Wan, L.; Liu, H.; Li, M.; Jiang, Y.; Zhao, X.; Liu, Z.; Wan, K.; Li, G.; Guan, C-x. Genomic Analysis Identifies Mutations Concerning Drug-Resistance and Beijing Genotype in Multidrug-Resistant Mycobacterium tuberculosis Isolated From China. Front. Microbiol. 2020, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Pitso, L.; Potgieter, S.; Van der Spoel van Dijk, A. Prevalence of isoniazid resistance-conferring mutations associated with multidrug-resistant tuberculosis in Free State Province, South Africa. S Afr. Med. J. 2019, 109, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Solo, E.S.; Nakajima, C.; Kaile, T.; Bwalya, P.; Mbulo, G.; Fukushima, Y.; Chila, S.; Kapata, N.; Shah, Y.; Suzuki, Y. Mutations in rpoB and katG genes and the inhA operon in multidrug-resistant Mycobacterium tuberculosis isolates from Zambia. J. Glob. Antimicrob. Res. 2020, 22, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Diriba, G.; Kebede, A.; Tola, H.H.; Alemu, A.; Yenew, B.; Moga, S.; Addise, D.; Mohammed, Z.; Getahun, M.; Fantahun, M.; Tadesse, M. Utility of line probe assay in detecting drug resistance and the associated mutations in patients with extra pulmonary tuberculosis in Addis Ababa, Ethiopia. SAGE Open Med. 2022, 10, 20503121221098241. [Google Scholar] [CrossRef]
- Valafar, S.J. Systematic review of mutations associated with isoniazid resistance points to continuing evolution and subsequent evasion of molecular detection, and potential for emergence of multidrug resistance in clinical strains of Mycobacterium tuberculosis. Antimicrob. Agents. Chem. 2021, 65, e02091–e20. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, X.; Wu, X.; Li, S.; Liu, B.; Rajaofera, M.J.; Zeng, Y.; Dong, S.; Bei, Z.; Pei, H.; Xia, Q. Prevalence and molecular characteristics of drug-resistant Mycobacterium tuberculosis in Hainan, China: From 2014 to 2019. BMC. Microbiol. 2021, 21, 185. [Google Scholar] [CrossRef]
- Bodmer, T.; Ströhle, A. Diagnosing pulmonary tuberculosis with the Xpert MTB/RIF test. J.Vis. Exp. 2012, 62, e3547. [Google Scholar]
- World Health Organization. The impact of the roll-out of rapid molecular diagnostic testing for tuberculosis on empirical treatment in Cape Town, South Africa, Bulletin. WHO 2017, 95, 545–608. [Google Scholar]
- Hain Lifescience. Company history and product releases. 2016, http://www.hain-lifescience.de/en/company/history.html. (Accessed 11 February 2021).
- Ogari, C.O.; Nyamache, A.K.; Nonoh, J.; Amukoye, E. Prevalence and detection of drug resistant mutations in Mycobacterium tuberculosis among drug naïve patients in Nairobi, Kenya. BMC Infect. Dis. 2019, 19, 279. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; David, A.; Zozio, T.; Rastogi, N. Macro-geographical specificities of the prevailing tuberculosis epidemic as seen through SITVIT2, an updated version of the Mycobacterium tuberculosis genotyping database. Infect. Genet.Evol. 2019, 72, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Bhembe, N.L.; Green, E. Characterization of mutations in the rpoB gene conferring rifampicin resistance in Mycobacterium tuberculosis complex isolated from lymph nodes of slaughtered cattle from South Africa. Braz. J. Microbiol. 2020, 51, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Otchere, I.D.; Asante-Poku, A.; Osei-Wusu, S.; Baddoo, A.; Sarpong, E.; Ganiyu, A.H.; Aboagye, S.Y.; Forson, A.; Bonsu, F.; Yahayah, A.I.; Koram, K. Detection and characterization of drug-resistant conferring genes in Mycobacterium tuberculosis complex strains: A prospective study in two distant regions of Ghana. Tuberculosis (Edinb). 2016, 99, 147–154. [Google Scholar] [CrossRef]
- Jia, H.; Xu, Y.; Sun, Z. Analysis on Drug-Resistance-Associated Mutations among Multidrug-Resistant Mycobacterium tuberculosis Isolates in China. Antibiotics. 2021, 10, 1367. [Google Scholar] [CrossRef]
- Isakova, J.; Sovkhozova, N.; Vinnikov, D.; Goncharova, Z.; Talaibekova, E.; Aldasheva, N.; Aldashev, A. Mutations of rpoB, katG, inhA and ahp genes in rifampicin and isoniazid-resistant Mycobacterium tuberculosis in Kyrgyz Republic. BMC Microbiol. 2018, 18, 22. [Google Scholar] [CrossRef]
- Uddin, M.K.; Rahman, A.; Ather, M.F.; Ahmed, T.; Rahman, S.M.; Ahmed, S.; Banu, S. Distribution and frequency of rpoB mutations detected by Xpert MTB/RIF assay among Beijing and non-Beijing rifampicin resistant Mycobacterium tuberculosis isolates in Bangladesh. Infect. Drug Resist. 2020, 13, 789. [Google Scholar] [CrossRef]
- Meftahi, N.; Namouchi, A.; Mhenni, B.; Brandis, G.; Hughes, D.; Mardassi, H. Evidence for the critical role of a secondary site rpoB mutation in the compensatory evolution and successful transmission of an MDR tuberculosis outbreak strain. J. Antimicrob. Chemother. 2016, 71, 324–332. [Google Scholar] [CrossRef]
- Evans, J.; Stead, M.C.; Nicol, M.P.; Segal, H. Rapid genotypic assays to identify drug-resistant Mycobacterium tuberculosis in South Africa. J. Antimicrob. Chemother. 2009, 63, 11–16. [Google Scholar] [CrossRef]
- Ssengooba, W.; Meehan, C.J.; Lukoye, D.; Kasule, G.W.; Musisi, K.; Joloba, M.L.; Cobelens, F.G.; de Jong, B.C. Whole genome sequencing to complement tuberculosis drug resistance surveys in Uganda. Infect. Genet. Evol. 2016, 40, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Salvato, R.S.; Schiefelbein, S.; Barcellos, R.B.; Praetzel, B.M.; Anusca, I.S.; Esteves, L.S.; Halon, M.L.; Unis, G.; Dias, C.F.; Miranda, S.S.; de Almeida, I.N. Molecular characterisation of multidrug-resistant Mycobacterium tuberculosis isolates from a high-burden tuberculosis state in Brazil. Epidemiol. Infec. 2019, 147. [Google Scholar]
- Tajbakhsh, A.; Ghasemi, F.; Mirbagheri, S.Z.; Heravi, M.M.; Rezaee, M.; Meshkat, Z. Investigation of the rpoB mutations causing rifampin resistance by rapid screening in Mycobacterium tuberculosis in North-East of Iran. Iran. J. Pathol. 2018, 13, 429. [Google Scholar] [PubMed]
- Bollela, V.R. , Namburete, E.I.; Feliciano, C.S.; Macheque, D.; Harrison, L.H.; Caminero, J.A. Detection of katG and inhA mutations to guide isoniazid and ethionamide use for drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2016, 20, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Verza, M.; Scheffer, M.C.; Salvato, R.S.; Schorner, M.A.; Barazzetti, F.H.; Machado, H.D.; Medeiros, T.F.; Rovaris, D.B.; Portugal, I.; Viveiros, M.; Perdigão, J. Genomic epidemiology of Mycobacterium tuberculosis in Santa Catarina, Southern Brazil. Sci. Rep. 2020, 10, 12891. [Google Scholar] [CrossRef]
- Jagielski, T.; Grzeszczuk, M.; Kamiński, M.; Roeske, K.; Napiórkowska, A.; Stachowiak, R.; Augustynowicz-Kopeć, E.; Zwolska, Z.; Bielecki, J. Identification and analysis of mutations in the katG gene in multidrug-resistant Mycobacterium tuberculosis clinical isolates. Adv. Resp. Med. 2013, 81, 298–307. [Google Scholar] [CrossRef]
- Abanda, N.N.; Djieugoué, J.Y.; Lim, E.; Pefura-Yone, E.W.; Mbacham, W.; Vernet, G.; Penlap, V.M.; Eyangoh, S.I.; Taylor, D.W.; Leke, R.G. Diagnostic accuracy and usefulness of the Genotype MTBDRplus assay in diagnosing multidrug-resistant tuberculosis in Cameroon: A cross-sectional study. BMC infect. Dis. 2017, 17, 379. [Google Scholar] [CrossRef]
- Tseng, S.T.; Tai, C.H.; Li, C.R.; Lin, C.F.; Shi, Z.Y. The mutations of katG and inhA genes of isoniazid-resistant Mycobacterium tuberculosis isolates in Taiwan. J. Microbiol. Immunol. Infect. 2015, 48, 249–255. [Google Scholar] [CrossRef]
- Charoenpak, R.; Santimaleeworagun, W.; Suwanpimolkul, G.; Manosuthi, W.; Kongsanan, P.; Petsong, S.; Puttilerpong, C. Association between the phenotype and genotype of isoniazid resistance among Mycobacterium tuberculosis isolates in Thailand. Infect. Drug Resist. 2020, 13, 627. [Google Scholar] [CrossRef]
- Norouzi, F.; Moghim, S.; Farzaneh, S.; Fazeli, H.; Salehi, M.; Esfahani, B.N. Significance of the coexistence of non-codon 315 katG, inhA, and oxyR-ahpC intergenic gene mutations among isoniazid-resistant and multidrug-resistant isolates of Mycobacterium tuberculosis: A report of novel mutations. Pathog. Glob. Health. 2022, 116, 22–29. [Google Scholar] [CrossRef]
- Tessema, B.; Beer, J.; Emmrich, F.; Sack, U.; Rodloff, A.C. Analysis of gene mutations associated with isoniazid, rifampicin and ethambutol resistance among Mycobacterium tuberculosis isolates from Ethiopia. BMC Infect. Dis. 2012, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Gliddon, H.D.; Frampton, D.; Munsamy, V.; Heaney, J.; Pataillot-Meakin, T.; Nastouli, E.; Pym, A.S.; Steyn, A.J.; Pillay, D.; McKendry, R.A. A Rapid Drug Resistance Genotyping Workflow for Mycobacterium tuberculosis, Using Targeted Isothermal Amplification and Nanopore Sequencing. Microbiol. Spectr. 2021, 9, e00610. [Google Scholar] [CrossRef] [PubMed]
- Abate, D.; Tedla, Y.; Meressa, D.; Ameni, G. Isoniazid and rifampicin resistance mutations and their effect on second-line anti-tuberculosis treatment. Int. J. Tuberc. Lung Dis. 2014, 18, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Lempens, P.; Meehan, P.J.; Vandelannoote, K.; Fissette, K.; de Rijk, P.; Van Deun, A.; Rigouts, L.; de Jong, B.C. Isoniazid resistance levels of Mycobacterium tuberculosis can largely be predicted by high confidence resistance-conferring mutations. Sci. Rep. 2018, 8, 3246. [Google Scholar] [CrossRef] [PubMed]
- Sarin, R.; Bhalla, M.; Kumar, G.; Singh, A.; Myneedu, V.P.; Singhal, R. Correlation of inhA mutations and ethionamide susceptibility: Experience from national reference center for tuberculosis. Lung India. 2021, 38, 520–523. [Google Scholar]
- Maitre, T.; Morel, F.; Brossier, F.; Sougakoff, W.; Jaffre, J.; Cheng, S.; Veziris, N.; Aubry, A.; NRC-MyRMA. How a PCR Sequencing Strategy Can Bring New Data to Improve the Diagnosis of Ethionamide Resistance. Microorganisms 2022, 10, 1436. [Google Scholar] [CrossRef]
- Seifert, M.; Catanzaro, D.; Catanzaro, A.; Rodwell, T.C. Genetic Mutations Associated with Isoniazid Resistance in Mycobacterium tuberculosis: A Systematic Review. PLoS ONE, 2015, 10, e0119628. [Google Scholar] [CrossRef]
- Hofmann-Thiel, S.; van Ingen, J.; Feldmann, K.; Turaev, L.; Uzakova, G.T.; Murmusaeva, G.; van Soolingen, D.; Hoffmann, H. Mechanisms of heteroresistance to isoniazid and rifampin of Mycobacterium tuberculosis in Tashkent, Uzbekistan. Eur. Respir. J. 2009, 33, 368–374. [Google Scholar] [CrossRef]
- Shin, S.S.; Modongo, C.; Baik, Y.; Allender, C.; Lemmer, D.; Colman, R.E.; Engelthaler, D.M.; Warren, R.M.; Zetola, N.M. Mixed Mycobacterium tuberculosis–strain infections are associated with poor treatment outcomes among patients with newly diagnosed tuberculosis, independent of pretreatment heteroresistance. J. Infect. Dis. 2018, 218, 1974–1982. [Google Scholar] [CrossRef]
- Rinder, H.; Mieskes, K.T.; Löscher, T. Heteroresistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2001, 5, 339–345. [Google Scholar]
- de Assis Figueredo, L.J.; de Almeida, I.N.; Augusto, C.J.; Soares, V.M.; Suffys, P.N.; da Silva Carvalho, W.; de Miranda, S.S. Characterization of Mycobacterium tuberculosis heteroresistance by genotyping. Int. J. Mycobacteriol. 2020, 9, 368–372. [Google Scholar] [PubMed]
- Said, H.; Ratabane, J.; Erasmus, L.; Gardee, Y.; Omar, S.; Dreyer, A.; Ismail, F.; Bhyat, Z.; Lebaka, T.; van der Meulen, M.; Gwala, T.; Adelekan, A.; Diallo, K.; Ismail, N. Distribution and Clonality of drug-resistant tuberculosis in South Africa. BMC Microbiol. 2021, 21, 157. [Google Scholar] [CrossRef] [PubMed]
- Mokrousov, I.; Ly, H.M.; Otten, T.; Lan, N.N.; Vyshnevskyi, B.; Hoffner, S.; Narvskaya, O. Origin and primary dispersal of the Mycobacterium tuberculosis Beijing genotype: Clues from human phylogeography. Genome Res. 2005, 15, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- María Irene, C.C.; Juan Germán, R.C.; Gamaliel, L.L.; Dulce Adriana, M.E.; Estela Isabel, B.; Brenda Nohemí, M.C.; Payan Jorge, B.; Zyanya Lucía, Z.B.; Myriam, B.D.; Adrian, O.L.; Martha Isabel, M. Profiling the immune response to Mycobacterium tuberculosis Beijing family infection: A perspective from the transcriptome. Virulence 2021, 12, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Trauer, J.M.; Ascher, D.B.; Denholm, J.T. ; Hyper transmission of Beijing lineage Mycobacterium tuberculosis: Systematic review and meta-analysis. J. Infect. 2019, 79, 572–581. [Google Scholar] [CrossRef]
- Maguga-Phasha, N.T.; Munyai, N.S.; Mashinya, F.; Makgatho, M.E.; Mbajiorgu, E.F. Genetic diversity and distribution of Mycobacterium tuberculosis genotypes in Limpopo, South Africa. BMC Infect. Dis. 2017, 17, 764. [Google Scholar] [CrossRef]
- Chihota, V.N.; Niehaus, A.; Streicher, E.M.; Wang, X.; Sampson, S.L.; Mason, P.; Källenius, G.; Mfinanga, S.G.; Pillay, M.; Klopper, M.; Kasongo, W. Geospatial distribution of Mycobacterium tuberculosis genotypes in Africa. PLoS ONE 2018, 13, e0200632. [Google Scholar] [CrossRef] [PubMed]
- van Soolingen, D.; Qian, L.; De Haas, P.E.; Douglas, J.T.; Traore, H.; Portaels, F.; Qing, H.Z.; Enkhsaikan, D.; Nymadawa, P.; van Embden, J.D. Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia. J. Clin. Microbiol. 1995, 33, 3234–3238. [Google Scholar] [CrossRef]
- Pokam, B.D.; Yeboah-Manu, D.; Amiteye, D.; Asare, P.; Guemdjom, P.W.; Yhiler, N.Y.; Morton, S.N.; Ofori-Yirenkyi, S.; Laryea, R.; Tagoe, R.; Asuquo, A.E. Molecular epidemiology and multidrug resistance of Mycobacterium tuberculosis complex from pulmonary tuberculosis patients in the Eastern region of Ghana. Heliyon 2021, 7, e08152. [Google Scholar] [CrossRef]
- Bhembe, N.L.; Nwodo, U.U.; Okoh, A.I.; Obi, C.L.; Mabinya, L.V.; Green, E. Clonality and genetic profiles of drug-resistant Mycobacterium tuberculosis in the Eastern Cape Province, South Africa. Microbiology Open 2019, 8, e00449. [Google Scholar] [CrossRef]
- Van der Spoel van Dijk, A.; Makhoahle, P.M.; Rigouts, L.; Baba, K. Diverse molecular genotypes of Mycobacterium tuberculosis complex isolates circulating in the Free State, South Africa. Int. J. Microbiol. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Solo, E.S.; Suzuki, Y.; Kaile, T.; Bwalya, P.; Lungu, P.; Chizimu, J.Y.; Shah, Y.; Nakajima, C. Characterization of Mycobacterium tuberculosis genotypes and their correlation to multidrug resistance in Lusaka, Zambia. Int. J. Infect. Dis. 2021, 102, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Sagonda, T.; Mupfumi, L.; Manzou, R.; Makamure, B.; Tshabalala, M.; Gwanzura, L.; Mason, P.; Mutetwa, R. Prevalence of extensively drug resistant tuberculosis among archived multidrug resistant tuberculosis isolates in Zimbabwe. Tuberc. Res. Treat. 2014, 349141. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of rpoB gene mutations.
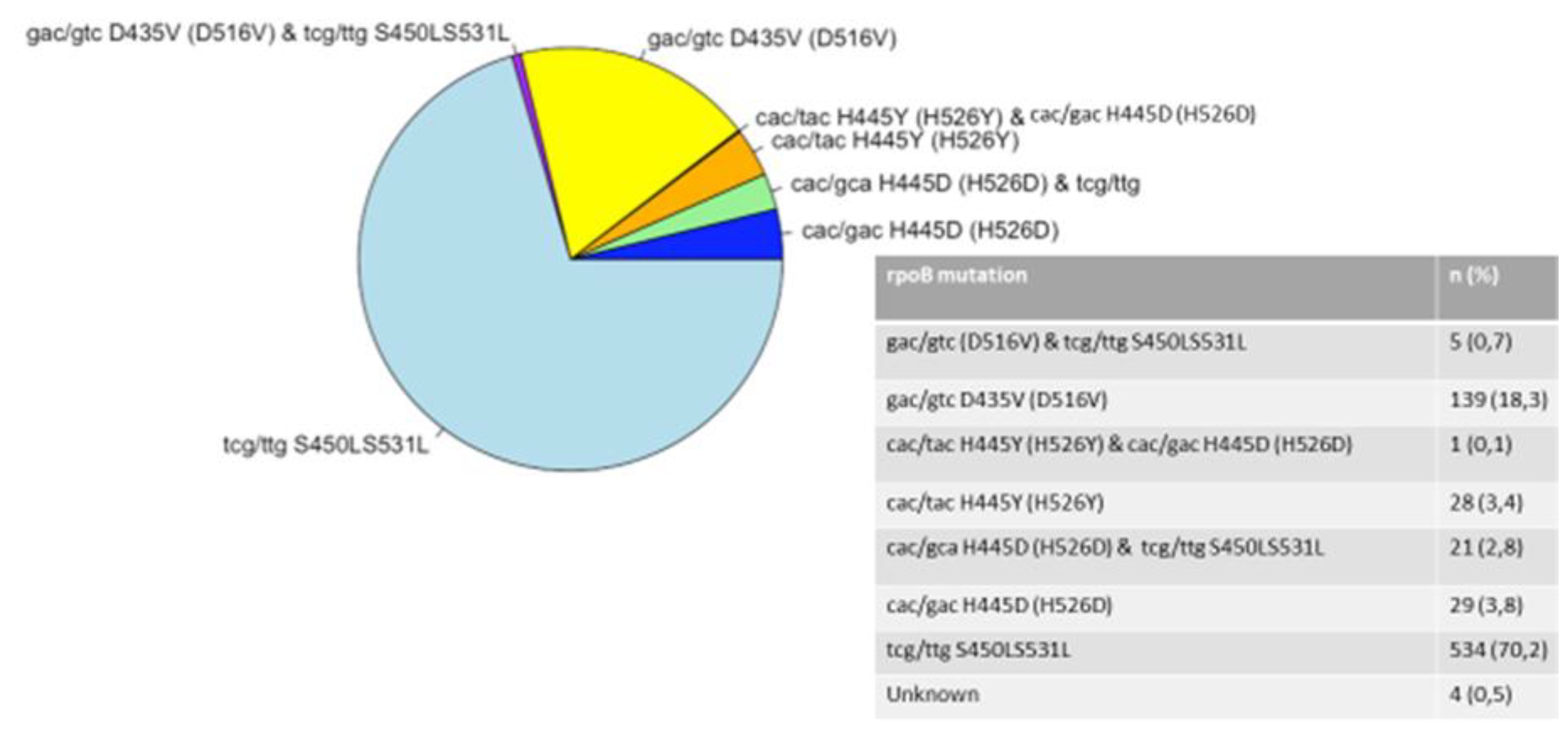
Figure 2.
Distribution of katG gene mutations.
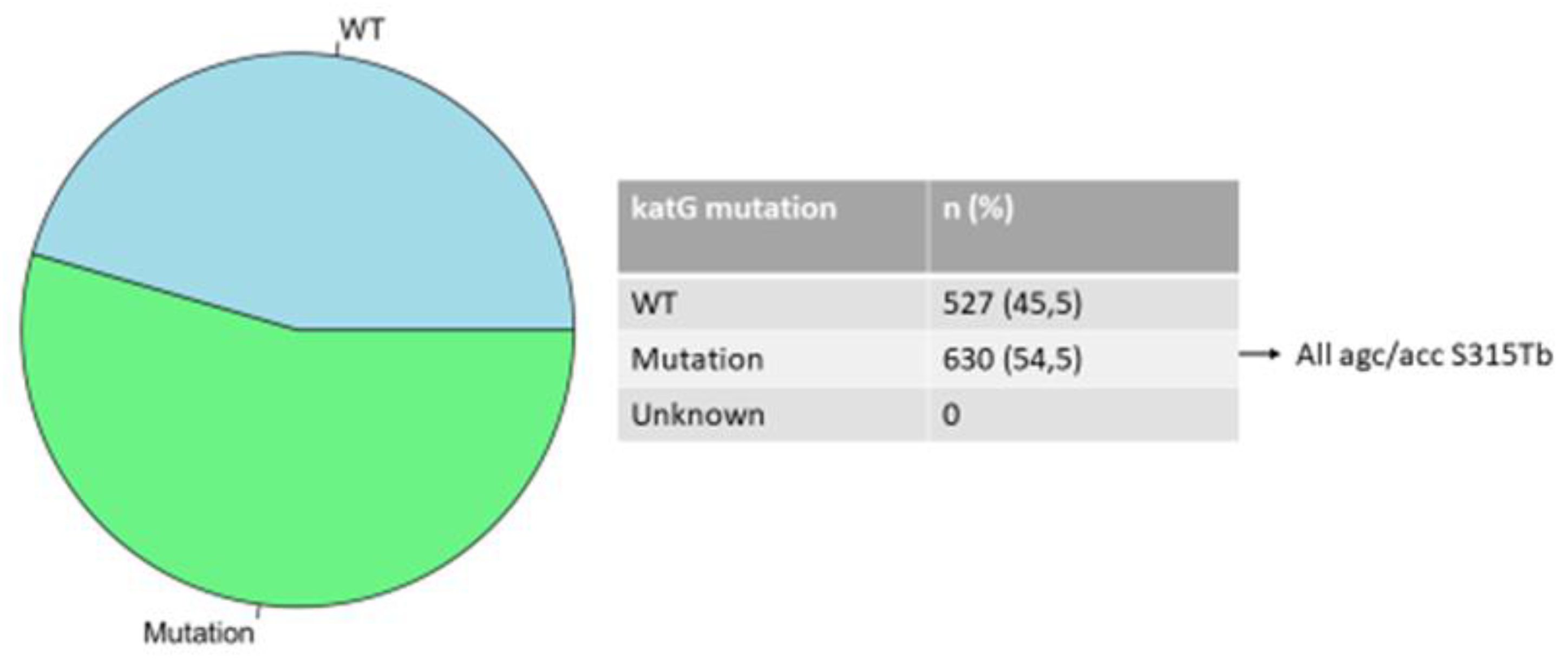
Figure 3.
Distribution of inhA gene mutations.
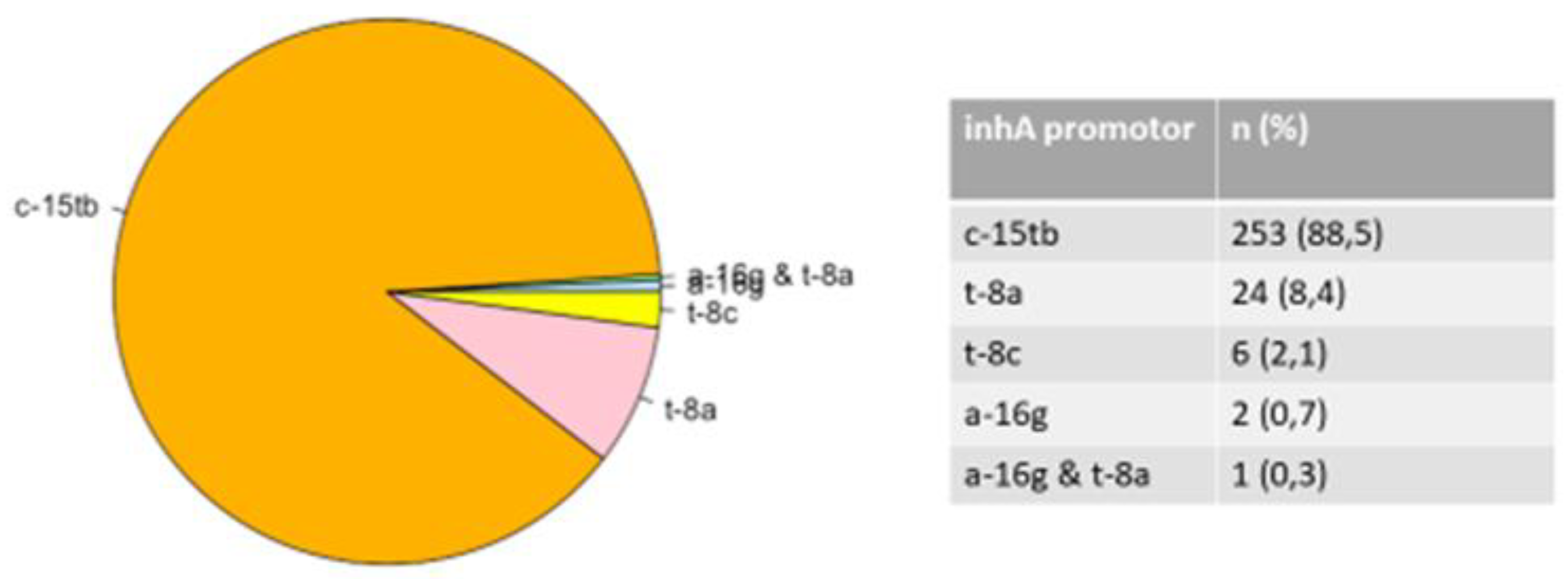

Table 1.
Number of DR-TB isolates over a 3-year period.
| Year of Diagnosis | DR-TB Case n (%) | Annual Heteroresistance Rate n (%) |
|---|---|---|
| 2018 | 385 (33.3) | 37 (9.6) |
| 2019 | 376 (32.5) | 43 (11.4) |
| 2020 | 396 (34.2) | 127 (32.2) |
Table 2.
Isolates with predominant gene mutations and mutation regions.
| Combined Mutation | Number of Isolates (%) | Mutation Regions | Number of Isolates (%) |
|---|---|---|---|
| rpoB and katG | 532 (69.9) | rpoB S315L and katG 531ST | 366 (68,8%) |
| rpoB and inhA | 187 (24.6) | rpoB S315L and inhA c-15tb | 171 (91,4%) |
| rpoB S315L and inhA a-16g | 1 (0,5%) |
rpoB S315L and inhA t-8c 2 (1,1%) rpoB S315L and inhA t-8a 13 (6,9%)
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



