
Submitted:
05 April 2023
Posted:
07 April 2023
You are already at the latest version
Abstract
With the growing hope that disease-modifying treatments could target the molecular basis of neurodegenerative diseases even before the onset of symptoms, there is mounting pressure to define disease entities based on pathophysiology rather than on clinical syndromes. The Alzheimer’s disease research community has recently transitioned from diagnostic criteria based on an amnestic syndrome to a purely biomarker-based disease definition, relying on the demonstration of amyloid-beta pathology, tau pathology, and neurodegeneration. In contrast, current diagnostic criteria for Parkinson’s disease still rely on the presence of the well-described clinical syndrome of parkinsonism, with the addition of characteristic motor- and non-motor signs and symptoms. However, there is now unequivocal evidence that Parkinson’s disease starts years before the onset of parkinsonism. Furthermore, neuropathologically defined Lewy body disease is clinically heterogeneous, combining a range of motor, non-motor, dopaminergic and non-dopaminergic features. Finally, clinically defined Parkinson’s disease has diverse underlying etiologies most, but not all, associated with α-synuclein positive Lewy pathology. In light of recent scientific advances, we propose a biologically based definition for the diagnosis of Parkinson's disease, initially to be used for research purposes. The criteria use a three-component ‘G-S-N’ system. The first is documentation of defined gene variants (‘G’), which cause or strongly predispose to PD as the most upstream component. The second is α-synuclein pathology (‘S’), currently defined as pathological α-synuclein deposition in tissue or positive α-synuclein seeding assays. The third is evidence of underlying neurodegeneration (‘N’), currently defined by specific neuroimaging procedures. The associated clinical syndrome (‘C’) is defined by a single high-specificity feature or multiple lower-specificity features. Initiating this transition will enable the field to fuel both basic and clinical research and move closer to the precision medicine required to develop clinically meaningful disease-modifying therapies. We acknowledge current limitations, ethical implications, and the need for prospective validation of this approach.
Keywords:
Parkinson’s disease
; genetics
; α-synuclein
; neurodegeneration
; clinical manifestation
; diagnosis
1. Introduction
A rapidly growing body of evidence is increasingly providing insights into the molecular etiology and pathogenesis of Parkinson's disease (PD), opening the realistic opportunity to develop clinically meaningful disease-modifying therapies.
Human genetics has taught us that PD, currently conceived as a clinicopathologic disease entity1, may have various genetic or environmental causes that initiate the disease along different, only partially overlapping pathways2-6.
Neuropathological findings have highlighted the pathogenic role of Lewy pathology (in Lewy bodies and Lewy neurites) and its major molecular component, the protein α-synuclein, which we refer to as “Parkinson’s type synucleinopathy”7,8. The neuropathological perspective has helped to clarify the difference between PD and other synucleinopathies that lack Lewy pathology, such as multiple system atrophy (MSA), but has also challenged us to question the traditional boundaries between PD and dementia with Lewy bodies9,10. Importantly, neuropathology has also taught us that Lewy pathology is neither sufficient nor necessary for clinically-defined PD and there are cases with clinical and genetic PD diagnoses without this neuropathological feature11.
Fluid, tissue, and imaging biomarkers are now sufficiently advanced to allow objective identification of genetic risk, pathological processes, and neurodegeneration, even before overt clinical symptoms have appeared.
Despite these advances, the currently accepted diagnostic criteria for PD are almost exclusively based on the identification of clinical features, and have been so for more than a century1,10,12. Notably, there is no single neurobiologically-based disease construct. Creating a biological diagnosis of PD as a disease entity with specific pathogenic mechanisms could serve as the basis for better preclinical and clinical objective diagnosis and staging, and for more accurate subdivision of PD according to active pathogenic mechanisms. In so doing, a biological diagnosis could advance research in multiple fields, including epidemiology, pathogenesis, biomarker discovery, and precision medicine development including disease-modifying therapies.
Here, we propose a biological definition of PD that includes genetic risk, the presence or absence of pathogenic α-synuclein in peripheral tissue or CSF, and the presence of characteristic features of neurodegeneration using selected imaging techniques. The establishment of a biological definition of PD recognizes that the biological processes that eventually lead to the development of the classic clinical features of PD are present long before the onset of these features, and that it is now possible to determine the presence of these changes much earlier than was previously feasible. We propose this definition and criteria initially for research purposes exclusively rather than as diagnostic criteria for clinical practice.
2. Development of the Disease Definition
Consensus Method
References for this position paper were identified as described in the Search Strategy section. GUH and AEL initiated and coordinated the process. After initial discussions on the overarching structure of the biological definition of PD, working groups were formed for PD-specific pathogenic gene variants (CK, GUH), PD synucleinopathy (CHA, TFO, AEL), PD-associated neurodegeneration (WP, AJS), and PD-associated clinical status (DB, RP). The working groups reviewed the available evidence underpinning the use of the different biological constructs and were asked to propose solutions for each component, which were presented to the group in a series of face-to-face virtual rounds (total N=16 between June 2022 and March 2023) for critical discussion combined with follow-up emails until a unanimous consensus was reached on each component and its integrated interpretation.
3. Biological Definition of Parkinson’s Disease
3.1. PD Disease Concept
Selected PD-specific pathogenic gene variants may serve as the earliest definable upstream cause or predisposition to a Parkinson’s type synucleinopathy, which is believed to result in PD-associated neurodegeneration, that eventually leads to corresponding clinical abnormalities in a wide range of domains. However, variations from this sequence are extremely common, such as the absence of PD-specific pathogenic gene variants in most cases of Parkinson’s type synucleinopathy, or pathological changes lacking Parkinson’s type synucleinopathy in a minority of cases with a definable genetic cause or predisposition. Moreover, the pattern of PD-associated neurodegeneration (and its subsequent clinical correlates) can vary considerably between individuals. Therefore, we have sought an overarching biological approach that encompasses this variability. We recognize that the clinical abnormalities associated with PD are part of a continuum that reflects disease stage and variability, but under a biologic definition, they will not be considered defining features of the disease. We propose that a biological disease definition provides an umbrella under which it is possible to harmonize different disease concepts that have been derived from a predominantly clinical perspective, including preclinical PD13, pre-motor PD13, prodromal PD12,14, defined non-motor syndromes (REM sleep behavior disorder (RBD)15, postganglionic pure autonomic failure (PAF)16) motor PD10,17,18, PD with dementia (PDD)19, and dementia with Lewy bodies (DLB)19-21.
3.2. Genetics
A monogenic pathogenic variant predisposing to PD can be detected in ~15% of all patients 22. Confirmed types of monogenic typical PD include four dominantly inherited forms (SNCA-PD, LRRK2-PD, VPS35-PD, CHCHD2-PD) and three recessively inherited forms (PRKN-PD, PINK1-PD, PARK7-PD)23. The likelihood of developing clinical PD depends strongly on the PD gene involved and, in the case of SNCA and GBA24, also on the type of the pathogenic variant. With respect to GBA-PD, for the biological definition of PD, we only include those pathogenic GBA variants that greatly increase the risk of manifesting PD and, thus, can be viewed as acting in a dominant fashion with highly reduced (age-dependent) penetrance25. Reduced penetrance, i.e. the conditional probability of being affected by a disease given a particular pathogenic genotype, is well-documented in inherited disorders.26 Detailed genotype and phenotype information on these conditions is available27,28, and www.mdsgene.org, except for GBA and CHCHD2, which are in progress).
In the current context, we propose different levels of pathogenic effect (Table 1;Supplement S1 for details):
- -
- The first level comprises the fully penetrant variants: SNCA triplications, SNCA missense variants, as well as bi-allelic PRKN, PINK1, and PARK7 missense, nonsense, small indels, and copy number variants.
- -
- The second level comprises variants that confer a strong predisposition to PD but not complete penetrance, including SNCA duplications and pathogenic variants in LRRK2, VPS35, and CHCHD2.
- -
- The third level, which results in an intermediate predisposition with highly reduced penetrance, consists of carriers of highly pathogenic GBA variants. GCH1 monoallelic pathogenic variants 29,30 and 22q11.2 deletion syndrome31 may also fall into this category, but are currently considered investigational due to limited knowledge.
A second aspect to consider with regard to the PD-specific pathogenic gene variants is the degree of predisposition for a Parkinson’s type synucleinopathy (Table 1):
- -
- Variants in SNCA32 and GBA25 unequivocally predispose to Parkinson’s type synucleinopathy.
- -
- LRRK2 monoallelic (or biallelic) pathogenic variants predispose to Parkinson’s type synucleinopathy in most cases, whereas neurodegeneration without synucleinopathy occurs in a minority30.
- -
- Biallelic variants in PRKN predispose to a Parkinson’s type synucleinopathy in approximately 20% of the cases33.
- -
- Only very few post-mortem reports are available for variants in PINK134,35, PARK736, and CHCHD237, and the 22q11.2 deletion syndrome31, some associated with Parkinson’s type synucleinopathy and others not, i.e. on a very low level of evidence.
- -
- For variants in VPS35 and GCH1, the predisposition for a Parkinson’s type synucleinopathy is currently unknown30.
We recommend reporting the PD-genetic status of a person as positive (GF+ or GP+, respectively), when a fully penetrant pathogenic variant or a pathogenic variant with strong or intermediate predisposition is confirmed (Table 1). All other conditions (pathogenic gene variants with low predisposition for PD or polygenic risk scores, or absent or unknown genetic contributions) are considered as G- “genetically indeterminate”.
3.3. Synucleinopathy
We recommend designating the Parkinson’s type synucleinopathy status of a person as positive (S+), when a pathological test specified in Table 2 is confirmed. All other conditions are considered as S.
The pathology of PD, with selected exceptions (see below), is defined by the presence of widespread aggregated α-synuclein as Lewy bodies and Lewy neurites in the central and peripheral nervous systems. It is widely believed that the deposition and spread of pathologic forms of misfolded α-synuclein are key aspects in the development and progression of the neurodegenerative process38. Therefore, we propose that pathological α-synuclein should be the defining attribute of the biochemical category of the disease definition (i.e., an α-synuclein positive or negative (S+/-) designation). All individuals classified in a new biological definition as having “sporadic” PD must be S+ while some genetic causes will be S- as they may lack α-synuclein aggregation (e.g., particularly most PRKN-PD (i.e. biallelic gene variant carriers)33 and a proportion of LRRK2-PD cases39. Thus, we propose to include α-synuclein-negative forms of PD (S-) in the biological definition of PD, if genetic markers with sufficient high penetrance are present.
Multiple methods have evaluated the presence of presumed pathogenic α-synuclein in vivo in biological fluids (CSF, saliva, blood, tears) and tissues (e.g., skin, salivary glands, gastrointestinal tract, olfactory mucosa) (Supplement S2). Currently, the evidence does not support using measurements of α-synuclein levels in biological fluids as a molecular marker of a biological diagnosis of PD40,41. Immunohistochemistry (IHC) and immunohistofluorescence (IHF) have been applied to multiple tissues. Currently, the data suggest that only skin biopsies, with specific methods, provide adequate sensitivity and specificity to be recommended for use as part of the biological definition of PD. The pattern and distribution of α-synuclein in the biopsy should be considered in differentiating PD from MSA.
The development of α-synuclein seeding amplification assays (SAAs) has revolutionized the potential for widespread application of a biological diagnosis of PD42,43 (Supplement S2). SAA α-synuclein positivity has been found in multiple biological samples, with skin and CSF having the highest sensitivities (0.92(0.87–0.95) and 0.90 (0.86–0.93) respectively)44. SAAs may be positive in patients at very early stages of the disease process (RBD and PAF)45,46. Many caveats must be considered with respect to different analysis methods, especially related to the differentiation of PD from MSA (potentially requiring additional exclusionary measures such as plasma neurofilament light chain levels or neuroimaging techniques) (Table 2 and Supplement S2). Rapid advances are expected, with an eventual evolution from the current binary (+/-) diagnostic test results to methods of monitoring disease status and progression.
Many biological pathways are postulated to be involved in PD. Numerous studies have evaluated other candidate biomarkers for PD, including markers of neurodegeneration, neuroinflammation, protein aggregation, or proteostasis network components (Supplement S2) but none reliably distinguish PD from controls, or PD from other neurodegenerative parkinsonian disorders. Given the biological heterogeneity, technological complexity, inter-laboratory variability, and the need for cross-validation by different laboratories, these approaches are not ready for use as diagnostic biomarkers47,48.
We recommend that the S+/- component of the biological definition of PD document the presence of pathological α-synuclein using validated IHC/IHF and/or SAA methods in skin biopsies or SAA assay in CSF while other tissues and fluids are still being investigated (Table 2).
3.4. Neurodegeneration
We recommend that evidence of any of the findings below is sufficient to define neurodegeneration in biologically suspected PD (Table 3, Supplement S3). However, the specificity of currently available methods to differentiate between PD and other neurodegenerative forms of parkinsonism is imperfect and restricted to a limited number of neuroanatomical systems.
A principal confirmation of PD-associated neurodegeneration is that of dopaminergic denervation. Reduced striatal uptake (typically asymmetric and with a caudal to rostral pattern of abnormality) can be detected using markers for the dopamine transporter (DAT), vesicular monoamine transporter 2 (VMAT2) or aromatic acid decarboxylase (F-dopa). While sensitivity is high, similar findings are seen in MSA, including the rostral-caudal gradient, and in PSP (which tends to affect caudate and putamen equally)49. Findings incompatible with PD, and more typical of these other neurodegenerative parkinsonisms, would include radioisotopic evidence of marked post-synaptic dopamine receptor loss (e.g. [11C]raclopride PET or [123I]iodobenzamide SPECT)50.
A second indication of PD-associated neurodegeneration is altered glucose metabolism, evidenced by FDG PET. While changes in glucose metabolic networks (Parkinson Disease Related Pattern; PDRP) are only indirectly related to loss of nigrostriatal DA neurons, the findings are so typical as to provide presumptive evidence of denervation to define PD. Similar changes are seen in prodromal disease (RBD)51, but have also been reported with neuroleptic use52. Specificity of this finding within degenerative forms of parkinsonism is high, as atypical parkinsonisms such as MSA, PSP or CBS are associated with different characteristic patterns53.
A third line of evidence indicating PD-associated neurodegeneration is cardiac sympathetic denervation. Reduced tracer uptake on delayed MIBG SPECT or F-dopamine PET provide sufficient evidence of peripheral cardiac sympathetic denervation to define the presence of neurodegeneration in PD, and may be seen in prodromal disease (RBD, PAF), but not in all cases of early PD54. Specificity is high, but imperfect, as abnormalities have been reported in PSP and especially MSA, and interpretation can be challenging.
Non-dopaminergic molecular imaging of other neurotransmitter systems using serotonin or noradrenaline transporter PET ligands such as 11C-DASB or 11C-MeNER respectively, or PET ligands of acetylcholine esterase to demonstrate peripheral cholinergic denervation are of interest but not sufficiently validated yet to serve as anchors for the definition of neurodegeneration55-57.
MR imaging of substantia nigra pathology using iron-sensitive MRI, free water or neuromelanin imaging are promising potential future imaging markers of neurodegeneration but currently still considered investigational49 (Table 3).
We recommend reporting the PD-associated neurodegeneration status of a person as positive (N+), when a pathological test specified in Table 3 is confirmed. All other conditions are considered as N.
3.5. Biological Designations
The biological designations resulting from different combinations of biomarkers are listed in Table 4. Any interpretation needs to consider the possibility of false negative findings in the categories G, S, and N due to current technical limitations.
Conscious of the fact that monogenic conditions may have a long preclinical period starting as early as birth or even conception, the field of hereditary neurodegenerative disease is beginning to adapt its classifications to consider this phase as the earliest disease stage58. In line with this reasoning, we recommend that carriers of fully penetrant pathogenic gene variants qualify for a diagnosis of genetic PD, based on the presence of this variant per se (Figure 1, Table 4). Pathogenic gene variants with reduced penetrance would qualify as genetic predisposition for PD, but require additional evidence of neurodegeneration for a diagnosis of genetic PD. Pathogenic gene variants with low predisposition for PD, polygenic risk scores, or absent or unknown genetic contributions are considered as genetically indeterminate in the current construct.
S+ qualifies for Parkinson’s type synucleinopathy, as long as N positivity is not yet present. S+ is an essential prerequisite for a biological PD diagnosis in G- individuals, but requires further evidence of N+ (since currently we have no other biological marker of neuronal dysfunction preceding neurodegeneration). As indicated above, genetic causes of PD are variably associated with Parkinson’s type synucleinopathy. In selected cases, including some designated as “genetic PD” (e.g., SNCA gene variants or triplications), G+ is expected to be associated with a S+ state as the biological process becomes established, while in others S positivity is not expected (e.g., most PRKN gene variants). Thus, in G+ individuals, the biological diagnosis of PD may also be established in S- conditions, if the specific gene does not unequivocally predispose to a Parkinson’s type synucleinopathy.
N+ generally indicates the transition from Parkinson’s type synucleinopathy to biologically defined PD (or to genetic synuclein-negative PD in rare instances).
Conditions not compatible with a diagnosis of PD are also reported in Table 4. A critical appraisal of these allocations is presented in Supplement S4.
3.6. Clinical Manifestations
G+ or S+ individuals must be further sub-classified by their clinical status, regardless of their N status, since signs and symptoms might arise due to neuronal dysfunction preceding neurodegeneration, or due to neurodegeneration that defies current methods of visualisation or measurement. This involves defining the presence of associated clinical symptoms or signs (C+), and determining whether any clinical signs or symptoms can be attributed to their biologically defined PD. We propose to apply these clinical criteria to any individual designated as G+, S+ or N+.
A. The concept of C+
Some important considerations should be noted in defining the concept of a C+ state (Supplement S5):
1) Early clinical symptoms of PD are diverse, may fluctuate in severity and are often predominantly non-motor, thereby reflecting pathology outside the area that currently defines clinical PD on standard diagnostic criteria (i.e., the substantia nigra). Although non-motor features frequently precede motor ones, generally there is no uniform order of appearance (precluding definition of a specific unitary ‘nonmotor-then-motor’ staging). Moreover, non-motor features and subtle motor features commonly co-exist. Therefore, a clinical status designation should be agnostic to the order and nature of appearance of clinical features.
2) Clinical symptoms differ in their specificity. In the context of defined biologic PD, some will be almost pathognomonic (e.g., core motor features, RBD, neurogenic orthostatic hypotension). However, many are common in the general population, and will remain non-specific even after diagnosing biologic PD.
3) Many clinical features are also early-phase markers of other synucleinopathies, including MSA and DLB without PD. Although clinical clues can help distinguish these conditions (e.g., normal olfaction suggesting possible MSA, MCI suggesting DLB) it is currently impossible to reliably determine which clinical condition will develop by using only clinical markers.
4) The definition of a C+ state should not be confused with a stage of disease. Markers of early PD include those that may have a very long average latency to full clinical PD (olfaction, autonomic dysfunction) and others that typically become abnormal only proximate to full clinical PD (cognitive changes, motor exam). The C+ state includes all stages of disease. Moreover, in this definition there is no distinction between ‘prodromal’ and later ‘defined’ disease stages. Rather, in parallel, clinical status could also be stratified according to its impact on activities of daily living (mild, moderate, severe functional impairment).
In summary, the core definition of a ‘C+’ state is that clinical symptoms and/or signs of PD have occurred as a consequence of the progressive biological process.
B. Methodology for diagnosing the C+ state.
We recommend that the clinical status should be reported in a three-component system, i.e. asymptomatic (C-), presence of clinical abnormalities possibly or probably related to PD (Cposs+, Cprob+).
The criteria for the C+ states are provided in Table 5. For each of these clinical features, it should be presumed that no other, more probable, explanation exists for the symptom (according to best clinical judgement). For example, if a subject has subthreshold parkinsonism while on medications capable of causing parkinsonism, or has urinary dysfunction likely explained by prostatism, the feature should not be scored as present. Moreover, the development and evolution of the feature should be consistent with early PD (e.g. a static symptom with onset before age 30 would generally be excluded). Also note that criteria are to be applied in persons with biologic evidence of PD (i.e. G+ or S+ or N+). If not, either the MDS prodromal PD criteria (which use a much lower pretest likelihood, and therefore a higher threshold for diagnosis)12,14 or MDS clinical PD criteria10 can be applied.
These clinical features are to be documented in individuals designated as G+ or S+ or N+ using the criteria outlined in the text. Unless otherwise noted, the definition of each feature from the MDS Prodromal Criteria12,14 applies. Note that for each feature, it should be verified that there is no other more likely explanation based on best clinical judgment and that the temporal evolution of the symptom is consistent with PD.
* N+ would only be considered when combined with G+ or S+.
See Supplement 5 for further details.
4. Discussion
Here, we propose a biological definition of PD that comprises PD-specific pathogenic gene variants, Parkinson’s type synucleinopathy, and PD-associated neurodegeneration. Recent advances, most notably the establishment of sensitive and specific in vivo biomarkers for the presence of underlying α-synuclein pathology42-44, have placed the field in the critical position of evolving from largely clinically based diagnostic approaches to an emphasis on the biological underpinnings of a disease that affects the central and peripheral nervous systems decades before our clinical approaches permit diagnostic consideration. A biological definition of PD is mandatory for the next stage of a broad range of basic and clinical research studies as well as serving as a framework for future biomarker-based subclassification and staging that may be essential pre-requisites for successful implementation of precision medicine approaches to disease modification.
New criteria that incorporate biological components have been proposed for other neurodegenerative diseases, most notably Huntington disease (HD)58 and Alzheimer’s disease (AD)59, and these are contributing to ongoing clinical research advances in their respective fields.
Acknowledging that 15% of PD is monogenetic22 we distinguish between genetic and non-genetic cases and acknowledge a category of genetic PD lacking other biological criteria in individuals who have high penetrance gene variants. In analogy to the recommendations in HD58, this might be defined as a ‘Stage 0’ PD. One could consider PD patients with incompletely penetrant genetic predisposition (i.e. LRRK2 and GBA) as comparable to HD at-risk individuals with 36-39 CAG repeats. Beyond this, our approach is distinctly different from the Integrated Staging System proposed for HD58. In contrast to staging of the disease, we are proposing a Clinical State component with the understanding that other efforts are ongoing to establish a staging system for PD. It is not currently possible in PD to determine many of the background data used to develop the HD staging system (e.g. progressive neuroimaging changes such as caudate atrophy) prior to the development of overt clinical symptoms. However, the wide application of a biological definition of PD as we are proposing would enhance and permit such research to be conducted.
Our biological definition of PD using three binary classes (G/S/N) is comparable but not identical to the Amyloid/Tau/Neurodegeneration (A/T/N) approach proposed for AD59. Like the ATN system, which was particularly triggered by the development of tau biomarkers, the PD biological definition has been made possible by the development of tools to detect α-synuclein pathology in vivo.
There are similarities and clear differences between these two approaches. For example, ATN is agnostic to temporal ordering while GSN implies an order to the three components. ATN does not specify clinical disease status; our proposed approach includes a clinical component layered onto the binary GSN components.
The incorporation of a S- designation is critical to our definition. This acknowledges that α-synuclein is not necessary for the development of clinical PD and is notably absent in a proportion of patients with selected genetic forms of PD, including an important minority of LRRK2 cases30 and the majority of patients with biallelic PRKN pathogenic variant33s. However, the majority of patients with LRRK2 gene variants are S+, and some patients with biallelic pathogenic PRKN gene variants do demonstrate classical Lewy body pathology, as do most patients with clinical parkinsonism carrying PRKN heterozygous pathogenic variants. There is insufficient information about what to expect with biallelic pathogenic PINK1 and PARK7 gene variants, although both have been reported to be associated with α-synuclein-positive Lewy body pathology34-36. Given this knowledge, we believe that defining PD exclusively as a “synucleinopathy" misrepresents our current understanding of the pathology and pathogenesis of PD and the inclusion and formal acknowledgment of S- cases will advance our understanding of PD.
Of note, this biologic definition makes no distinctions between prodromal and clinical stages, and makes no distinctions between clinical PD and DLB. This should not be interpreted as an invalidation of diagnostic criteria for these states. Rather, the biologic and clinical definitions overlap, and serve different purposes. The criteria for prodromal PD12 and DLB21 useful for identifying patients in whom the biological criteria are not met, for situations in which there is no opportunity to identify markers for G/S and N, and for identifying patients with MCI or neuropsychiatric symptoms who have early-stage DLB. The criteria for clinical PD10 remain important in making accurate clinical diagnoses of PD as the cause of parkinsonism, while the criteria for DLB21 remain important for identifying DLB as the underlying cause of dementia. The criteria for the biological definition of PD, in contrast, recognize the unifying biologic factors underlying these conditions.
5. Limitations/Implementation/Future Directions
We believe that establishing a biological definition of PD will advance research on a number of fronts including epidemiology, natural history, neuroimaging, clinical trials, the development of newer biomarkers, to name just a few. However, we acknowledge important limitations and concerns raised by this approach. Although we propose the biological definition of PD for the exclusive purposes of advancing research, we recognize that some may obtain testing and apply these criteria to asymptomatic individuals in a non-research setting. This has important ethical implications, particularly given our limited understanding of the natural history of individuals in the various biological categories we propose and our current inability to prevent progression of PD from its early stages. Prospective studies must validate the evolution of these biological categories and provide more reliable methods predicting the underlying biological processes in asymptomatic individuals. We expect continuous advances in our understanding of genetic (including polygenic risk scores) and of environmental risk factors, and, therefore, these may be incorporated into future iterations or revisions of this system. We also expect further optimization of S and N biomarkers that will improve the sensitivity and specificity of the currently recommended testing. For example, we expect the development of successful S imaging ligands that can easily be incorporated once available. We expect that newer tools will successfully identify relevant disease mechanisms in subgroups of patients, allowing their incorporation into the biological scheme (e.g., a marker of inflammation or mitochondrial dysfunction). Finally, other classification components could be combined with GSN designations where appropriate (e.g., an additional ATN designation59 in patients with cognitive dysfunction). We emphasize that the current proposal is a first step in the critical process of moving the field from a purely clinical to a biological approach to the disease.
Contributors
GUH, CHA, DB, CK, TFO, WP, RP, AJS, and AEL contributed to the study design, literature search, data collection, data analysis, data interpretation, and writing of this article. GUH designed and drafted the figures.
Declaration of Interests
Günter U. Höglinger participated in industry-sponsored research projects from Abbvie, Bial, Biogen, Biohaven, Novartis, Sanofi, Takeda, UCB; served as a consultant for Abbvie, Alzprotect, Aprineua, Asceneuron, Bial, Biogen, Biohaven, Kyowa Kirin, Lundbeck, Novartis, Retrotope, Roche, Sanofi, UCB; received honoraria for scientific presentations from Abbvie, Bayer Vital, Bial, Biogen, Bristol Myers Squibb, Kyowa Kirin, Roche, Teva, UCB, Zambon; received publication royalties from Academic Press, Kohlhammer, and Thieme; holds a patent on the Treatment of Synucleinopathies. United States Patent No.: US 10,918,628 B2 / European Patent Patent No.: EP 17 787 904.6-1109 / 3 525 788. Charles H. Adler received consultant fees from Cionic, CND Life Science, Jazz, Neurocrine, Precon Health, XW Pharma. Daniela Berg served on Advisory boards of UCB Pharma GmbH and ACImmune SA and received honoraria from Biogen, UCB Pharma GmbH, Novartis. Her research was supported by UCB Pharma GmbH, EU, Novartis Pharma GmbH, Lundbeck. Christine Klein is deputy editor of Movement Disorders and associate editor of Annals of Neurology; she serves as a medical adviser to Centogene for genetic testing reports in the fields of movement disorders and dementia, excluding Parkinson’s disease and to Tetromer Therapeuticsand does not hold any stocks or stock options with any companies that are connected to Parkinson’s disease or to any of the topics in this paper. She received honoraria for scientific presentations from Bial and Desitin. Tiago Outeiro reports no conflicts of interests. Werner Poewe reports personal fees from AbbVie, AFFiRiS, AstraZeneca, Bial, Boston Scientific, Britannia, Intec, Ipsen, Lundbeck, NeuroDerm, Neurocrine, Denali Pharmaceuticals, Novartis, Orion Pharma, Prexton, Teva, UCB, and Zambon; royalties from Thieme, Wiley Blackwell, Oxford University Press, and Cambridge University Press. Ronald Postuma received personal fees as an advisor from Takeda, Roche, Biogen, Abbvie, Curasen, Lilly, Novartis, Eisai, Merck, Vaxxinity, and a stipend from the International Parkinson and Movement Disorders Society A Jon Stoessl chairs a DSMB for Neurocrine, serves on a DSMB for AskBio and serves as an advisor to Capsidia. He receives a stipend from the International Parkinson & Movement Disorders Society as Editor-in-Chief of Movement Disorders. Anthony E. Lang has served as an advisor for AbbVie, AFFiRis, Alector, Amylyx, Aprinoia, Biogen, BioAdvance, BlueRock, Biovie, BMS, CoA Therapeutics, Denali, Janssen, Jazz, Lilly, Novartis, Paladin, Pharma 2B, PsychoGenetics, Retrophin, Roche, Sun Pharma, and UCB; received honoraria from Sun Pharma, AbbVie and Sunovion; is serving as an expert witness in litigation related to paraquat and Parkinson’s disease, received publishing royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press.
Acknowledgments
Günter U. Höglinger was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198), within the Hannover Cluster RESIST (EXC 2155 – - project number 39087428) and the E-Rare Project MSAomics (HO2402/18-1); the EU/EFPIA/Innovative Medicines Initiative [2] Joint Undertaking (IMPRIND grant n° 116060); the German Federal Ministry of Education and Research (BMBF, 01KU1403A EpiPD; 01EK1605A HitTau; 01DH18025 TauTherapy); European Joint Programme on Rare Diseases (Improve-PSP); the Niedersächsisches Ministerium für Wissenschaft und Kunst (MWK, ZN3440.TP); the Volkswagen Foundation (Niedersächsisches Vorab); the Petermax-Müller Foundation (Etiology and Therapy of Synucleinopathies and Tauopathies); the German Parkinson Society (DPG, ProAPS project). Charles H. Adler received research funding from: NIH, Michael J. Fox Foundation, State of Arizona, Mayo Clinic, Banner Health. Daniela Berg received research funding from Deutsche Forschungsgemeinschaft (DFG), German Parkinson’s Disease Association (dPV), BMBF, Parkinson Fonds Deutschland gGmbH, Damp foundation, Michael J Fox Foundation. Christine Klein received a grant (FOR2488) from the German Research Foundation to support a part of this work and is the recipient of research funding from the BMBF and the Michael J. Fox Foundation (including the ASAP program for GP2). Tiago Outeiro was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2067/1- 390729940, and by SFB1286 (B8). Werner Poewe received grant support from The Michael J. Fox Foundation, EU FP7, and Horizon 2020. Ronald Postuma was supported by the Fonds de Recherche du Quebec – Sante ́, the Canadian Institutes of Health Research, the Parkinson Society of Canada, the Weston-Garfield Foundation, the Michael J. Fox Foundation, the Webster Foundation and the National Institute of health. A Jon Stoessl was supported by Brain Canada, CIHR, Canada Foundation for Innovation, Weston Brain Institute and Michael J. Fox Foundation. Anthony E. Lang received grants from Brain Canada, Canadian Institutes of Health Research, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, Parkinson Foundation, Parkinson Canada, and W. Garfield Weston Foundation
References
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, P.; Bonnet, A.M.; Debarges, B.; et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson's disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Toledo, J.B.; Walker, L.; et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: A multi-centre study. Acta Neuropathol. 2021, 141, 159–172. [Google Scholar] [CrossRef]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Berg, D.; Postuma, R.B.; Bloem, B.; et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson's disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Postuma, R.B.; Adler, C.H.; et al. MDS research criteria for prodromal Parkinson's disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D. Advances in markers of prodromal Parkinson disease. Nat. Rev. Neurol. 2016, 12, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Berg, D.; Gasser, T.; et al. Update of the MDS research criteria for prodromal Parkinson's disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- REM sleep behaviour disorder. Nat. Rev. Dis. Primers 2018, 4, 20. [CrossRef] [PubMed]
- Coon, E.A.; Singer, W.; Low, P.A. Pure Autonomic Failure. Mayo Clin. Proc. 2019, 94, 2087–2098. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The significance of the Lewy body in the diagnosis of idiopathic Parkinson's disease. Neuropathol. Appl. Neurobiol. 1989, 15, 27–44. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson's disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Walker, Z.; Possin, K.L.; Boeve, B.F.; Aarsland, D. Lewy body dementias. Lancet 2015, 386, 1683–1697. [Google Scholar] [CrossRef]
- McKeith, I.G.; Ferman, T.J.; Thomas, A.J.; et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 2020, 94, 743–755. [Google Scholar] [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Skrahina, V.; Gaber, H.; Vollstedt, E.J.; et al. The Rostock International Parkinson's Disease (ROPAD) Study: Protocol and Initial Findings. Mov. Disord. 2021, 36, 1005–1010. [Google Scholar] [CrossRef]
- Lange, L.M.; Gonzalez-Latapi, P.; Rajalingam, R.; et al. Nomenclature of Genetic Movement Disorders: Recommendations of the International Parkinson and Movement Disorder Society Task Force - An Update. Mov. Disord. 2022, 37, 905–935. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, G.; Schulte, C.; Jost, W.H.; et al. GBA-associated PD: Chances and obstacles for targeted treatment strategies. J. Neural. Transm (Vienna) 2022, 129, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Zeldenrust, F.M.J.; Huang, J.; et al. Genotype-phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 2018, 33, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Wittke, C.; Petkovic, S.; Dobricic, V.; et al. Genotype-Phenotype Relations for the Atypical Parkinsonism Genes: MDSGene Systematic Review. Mov. Disord. 2021, 36, 1499–1510. [Google Scholar] [CrossRef]
- Mencacci, N.E.; Isaias, I.U.; Reich, M.M.; et al. Parkinson's disease in GTP cyclohydrolase 1 mutation carriers. Brain 2014, 137 Pt 9, 2480–2492. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Butcher, N.J.; Kiehl, T.R.; Hazrati, L.N.; et al. Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: Identification of a novel genetic form of Parkinson disease and its clinical implications. JAMA Neurol. 2013, 70, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.A.; Schmidt, S.I.; Blaabjerg, M.; Meyer, M. Interaction between Parkin and alpha-Synuclein in PARK2-Mediated Parkinson's Disease. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Samaranch, L.; Lorenzo-Betancor, O.; Arbelo, J.M.; et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 2010, 133 Pt 4, 1128–1142. [Google Scholar] [CrossRef]
- Takanashi, M.; Li, Y.; Hattori, N. Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology 2016, 86, 2212–2213. [Google Scholar] [CrossRef]
- Taipa, R.; Pereira, C.; Reis, I.; et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139 Pt 6, 1680–1687. [Google Scholar] [CrossRef]
- Ikeda, A.; Nishioka, K.; Meng, H.; et al. Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum. Mol. Genet. 2019, 28, 3895–3911. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; et al. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.N.; et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef]
- Magalhaes, P.; Lashuel, H.A. Opportunities and challenges of alpha-synuclein as a potential biomarker for Parkinson's disease and other synucleinopathies. NPJ Parkinsons Dis. 2022, 8, 93. [Google Scholar] [CrossRef]
- Zubelzu, M.; Morera-Herreras, T.; Irastorza, G.; Gomez-Esteban, J.C.; Murueta-Goyena, A. Plasma and serum alpha-synuclein as a biomarker in Parkinson's disease: A meta-analysis. Parkinsonism Relat. Disord. 2022, 99, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, G.; De Luca, C.M.G.; Paoletti, F.P.; Gaetani, L.; Moda, F.; Parnetti, L. alpha-Synuclein Seed Amplification Assays for Diagnosing Synucleinopathies: The Way Forward. Neurology 2022, 99, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Concha-Marambio, L.; Pritzkow, S.; Shahnawaz, M.; Farris, C.M.; Soto, C. Seed amplification assay for the detection of pathologic alpha-synuclein aggregates in cerebrospinal fluid. Nat. Protoc. 2023. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Bang, J.I.; Ahn, C.; et al. Diagnostic value of alpha-synuclein seeding amplification assays in alpha-synucleinopathies: A systematic review and meta-analysis. Parkinsonism Relat. Disord. 2022, 104, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, A.; Fairfoul, G.; Ayudhaya, A.C.N.; et al. Detection of alpha-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: A longitudinal observational study. Lancet Neurol. 2021, 20, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Singer, W.; Schmeichel, A.M.; Shahnawaz, M.; et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain Predict Phenoconversion of Pure Autonomic Failure. Ann. Neurol. 2021, 89, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Bartl, M.; Dakna, M.; Galasko, D.; et al. Biomarkers of neurodegeneration and glial activation validated in Alzheimer's disease assessed in longitudinal cerebrospinal fluid samples of Parkinson's disease. PLoS ONE 2021, 16, e0257372. [Google Scholar] [CrossRef] [PubMed]
- Schulz, I.; Kruse, N.; Gera, R.G.; et al. Systematic Assessment of 10 Biomarker Candidates Focusing on alpha-Synuclein-Related Disorders. Mov. Disord. 2021, 36, 2874–2887. [Google Scholar] [CrossRef]
- Mitchell, T.; Lehericy, S.; Chiu, S.Y.; Strafella, A.P.; Stoessl, A.J.; Vaillancourt, D.E. Emerging Neuroimaging Biomarkers Across Disease Stage in Parkinson Disease: A Review. JAMA Neurol. 2021, 78, 1262–1272. [Google Scholar] [CrossRef]
- Antonini, A.; Leenders, K.L.; Vontobel, P.; et al. Complementary PET studies of striatal neuronal function in the differential diagnosis between multiple system atrophy and Parkinson's disease. Brain 1997, 120 Pt 12, 2187–2195. [Google Scholar] [CrossRef]
- Holtbernd, F.; Gagnon, J.F.; Postuma, R.B.; et al. Abnormal metabolic network activity in REM sleep behavior disorder. Neurology 2014, 82, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Kotomin, I.; Korotkov, A.; Solnyshkina, I.; Didur, M.; Cherednichenko, D.; Kireev, M. Parkinson's Disease-Related Brain Metabolic Pattern Is Expressed in Schizophrenia Patients during Neuroleptic Drug-Induced Parkinsonism. Diagnostics (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.C.; Poston, K.L.; Eckert, T.; et al. Differential diagnosis of parkinsonism: A metabolic imaging study using pattern analysis. Lancet Neurol. 2010, 9, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, K.; Imamura, T.; Shinya, T. Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson's disease. Parkinsonism Relat. Disord. 2010, 16, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Yarnall, A.J.; Weil, R.S.; et al. Cholinergic system changes in Parkinson's disease: Emerging therapeutic approaches. Lancet Neurol. 2022, 21, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Nahimi, A.; Kinnerup, M.B.; Sommerauer, M.; Gjedde, A.; Borghammer, P. Molecular Imaging of the Noradrenergic System in Idiopathic Parkinson's Disease. Int. Rev. Neurobiol. 2018, 141, 251–274. [Google Scholar] [PubMed]
- de Natale, E.R.; Wilson, H.; Politis, M. Serotonergic imaging in Parkinson's disease. Prog. Brain Res. 2021, 261, 303–338. [Google Scholar] [PubMed]
- Tabrizi, S.J.; Schobel, S.; Gantman, E.C.; et al. A biological classification of Huntington's disease: The Integrated Staging System. Lancet Neurol. 2022, 21, 632–644. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Norcliffe-Kaufmann, L.; Kaufmann, H.; Palma, J.A.; et al. Orthostatic heart rate changes in patients with autonomic failure caused by neurodegenerative synucleinopathies. Ann. Neurol. 2018, 83, 522–531. [Google Scholar] [CrossRef]
Figure 1.
Research framework of PD biological definitions. Temporal sequence and variability of the components contributing to the biological definition of PD. Green bars indicate a physiological condition, other colours indicate pathological conditions. 1 Fully penetrant pathogenic gene variants (GF+) qualify for diagnosis of „genetic PD“, based on G positivity per se. 2 Pathogenic gene variants with strong or intermediate predisposition (GP+) for PD qualify as „genetic predisposition“; they require additional N positivity for a diagnosis of PD. 3 Pathogenic gene variants with low predisposition for PD or polygenic risk scores, or absent or unknown genetic contributions are considered as “genetically indeterminate” (G-); they require additional S and N positivity for a diagnosis of PD. 4 Synuclein-negative PD may be diagnosed in absence of S positivity in cases with pathogenic gene variants that do not consistently predispose for a Parkinson’s type synucleinopathy (e.g. GLRRK2+ S- N+). G+ or S+ or N+ individuals must be further sub-classified by their clinical status, regardless of their N status, since signs and symptoms might arise already due to neuronal dysfunction, preceding overt neurodegeneration. Note that the graph does not imply any temporal alignment of the occurrence of the different C states in relation to the G, S, or N states.
Figure 1.
Research framework of PD biological definitions. Temporal sequence and variability of the components contributing to the biological definition of PD. Green bars indicate a physiological condition, other colours indicate pathological conditions. 1 Fully penetrant pathogenic gene variants (GF+) qualify for diagnosis of „genetic PD“, based on G positivity per se. 2 Pathogenic gene variants with strong or intermediate predisposition (GP+) for PD qualify as „genetic predisposition“; they require additional N positivity for a diagnosis of PD. 3 Pathogenic gene variants with low predisposition for PD or polygenic risk scores, or absent or unknown genetic contributions are considered as “genetically indeterminate” (G-); they require additional S and N positivity for a diagnosis of PD. 4 Synuclein-negative PD may be diagnosed in absence of S positivity in cases with pathogenic gene variants that do not consistently predispose for a Parkinson’s type synucleinopathy (e.g. GLRRK2+ S- N+). G+ or S+ or N+ individuals must be further sub-classified by their clinical status, regardless of their N status, since signs and symptoms might arise already due to neuronal dysfunction, preceding overt neurodegeneration. Note that the graph does not imply any temporal alignment of the occurrence of the different C states in relation to the G, S, or N states.
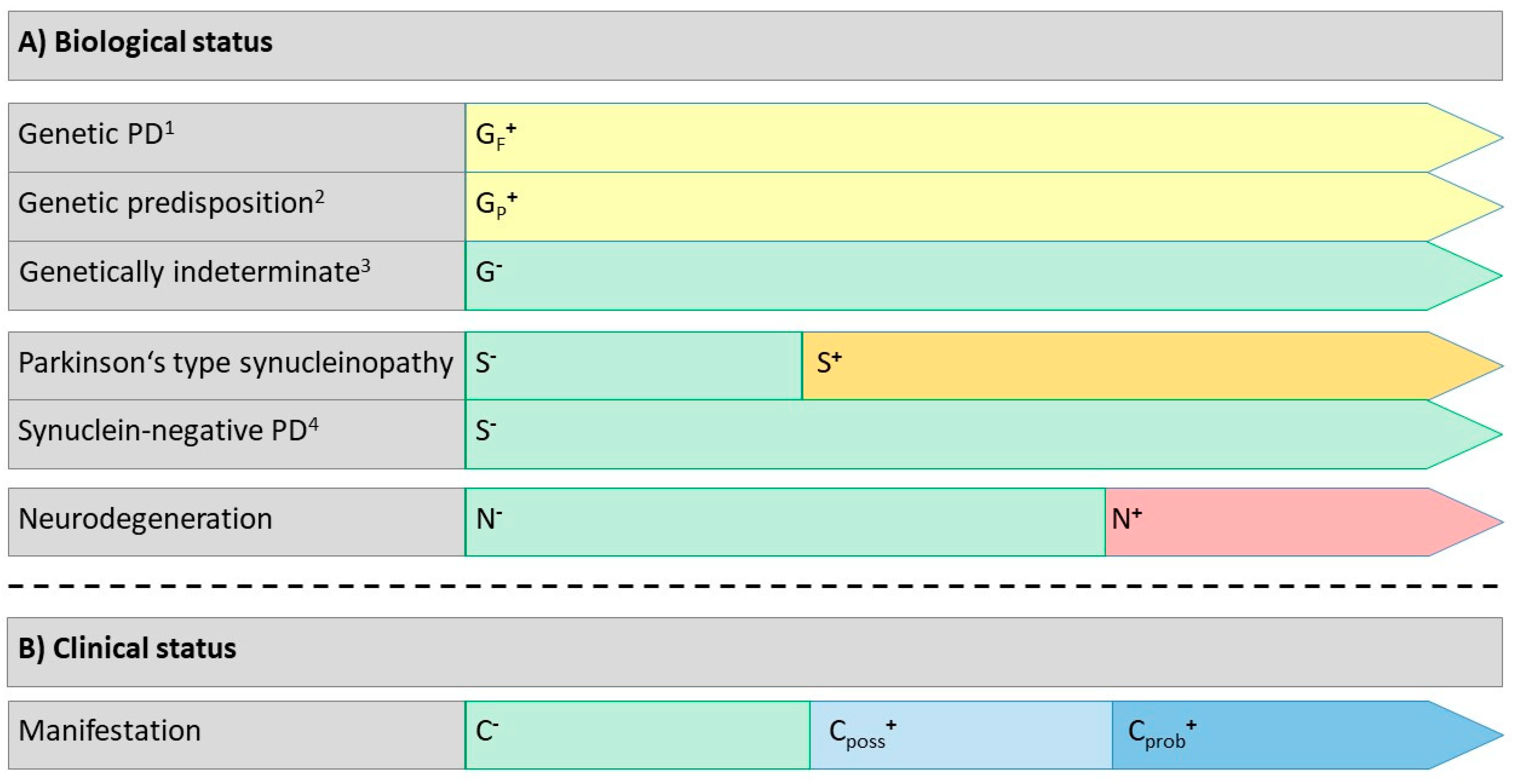

Table 1.
PD-specific pathogenic gene variants.
| Designation | Biomarker Status | Abnormality | Pathogenic effect | Presdisposing for |
|---|---|---|---|---|
| GF+ | Recommended | SNCA monoallelic triplication30,32 | fully penetrant | Parkinson’s type synucleinopathy |
| GF+ | Recommended | SNCA monoallelic pathogenic single nucleotide variants30,32 | fully penetrant | Parkinson’s type synucleinopathy |
| GF+ | Recommended | PRKN biallelic pathogenic variants30,33 | fully penetrant | Neurodegeneration without synucleinopathy in ~80% and Parkinson’s type synucleinopathy in ~20% of the cases |
| GF+ | Recommended | PINK1 biallelic pathogenic variants30,34,35 | fully penetrant | Parkinson’s type synucleinopathy in a single case and neurodegeneration without synucleinopathy in another |
| GF+ | Recommended | PARK7 biallelic pathogenic variants30,36 | fully penetrant | Parkinson’s type synucleinopathy in the single case published |
| GP+ | Recommended | SNCA monoallelic duplication30,32 | strong predisposition | Parkinson’s type synucleinopathy |
| GP+ | Recommended | LRRK2 monoallelic (or biallelic) pathogenic variants30 | strong predisposition | Parkinson’s type synucleinopathy in most cases; neurodegeneration without synucleinopathy in a minority |
| GP+ | Recommended | VPS35 monoallelic pathogenic variants30 | strong predisposition | unknown |
| GP+ | Recommended | CHCHD2 monoallelic pathogenic variants30,37 | strong predisposition | unknown |
| GP+ | Recommended | GBA monoallelic severely pathogenic variants25,30 | medium predisposition | Parkinson’s type synucleinopathy |
| G- | Investigational | GCH1 monoallelic pathogenic variants30 | medium predisposition | Parkinson’s type synucleinopathy in the single case published |
| G- | Investigational | 22q11.2 deletion syndrome30,31 | medium predisposition | Neurodegeneration without synucleinopathy in 1/3 and Parkinson’s type synucleinopathy in 2/3 published cases |
GF+: fully penetrant pathogenic gene variants, GP+: pathogenic gene variants with strong or intermediate predisposition. See for further details.
Table 2.
Parkinson’s type synucleinopathy.
| Designation | Biomarker Status | Abnormality | Sensitivity* | Specificity* |
|---|---|---|---|---|
| S+ | Recommended | α-syn SAA in CSF | High | High |
| S+ | Recommended | α-syn SAA in skin | High | High |
| S+ | Recommended | α-syn ICH/ICF in skin | Moderate | High |
| S+ | Investigational | α-syn SAA in neuronal exosomes from plasma | Insufficient evidence | Insufficient evidence |
| S+ | Investigational | α-syn SAA in plasma | Insufficient evidence | Insufficient evidence |
| S+ | Investigational | α-syn SAA in submandibular gland | Insufficient evidence | Insufficient evidence |
| Exclusion criterion ruling out S+ | For S+ testing unable to differentiate PD from MSA (e.g., selected SAA methodologies in CSF and SAA in skin to date) | Elevated Neurofilament Light chain (NfL) | High for atypical parkinsonism (e.g., MSA) | High for MSA but low for specific diagnosis (e.g., also elevated in PSP but these cases would be S- in the absence of copathology) |
| Exclusion criterion ruling out S+ | For S+ testing unable to differentiate PD from MSA* | Neuroimaging features of MSA (e.g., characteristic changes in the putamen, cerebellum and pons) | Moderate | High |
*high> 80%; moderate >70 < 80%; low < 70%. α-syn: α-synuclein, CSF: cerebrospinal fluid, IHC: immunohistochemistry, IHF: immunohistofluorescence; MSA: multiple system atrophy, PD: Parkinson’s disease, PSP: progressive supranuclear palsy, SAA: seeding amplification assay. See Supplement S2 for further details.
Table 3.
PD-associated neurodegeneration.
| Designation | Biomarker Status | Examination | Interpretation | Sensitivity* | Specificity* |
|---|---|---|---|---|---|
| N+ | Recommended | DAergic PET/SPECT | Striatal dopaminergic deficit | High | Low |
| N+ | Recommended | Metabolic FDG PET | PD related brain metabolic pattern | High | High |
| N+ | Recommended | Cardiac MIBG SPECT | Sympathetic cardiac denervation | Moderate to high | Moderate |
| N+ | Investigational | Neuromelanin MRI | Limited test-retest stability | Moderate to high | Low |
| N+ | Investigational | Iron-sensitive MRI | Sophisticated method restricted to specialized centers, may not directly prove neurodegeneration | Moderate to high | Low |
| N+ | Investigational | Substantia nigra free water MRI | Sophisticated method restricted to specialized centers, may not directly prove neurodegeneration | High | Moderate to high if applied to extra-nigral sites |
| N+ | Investigational | Structural MRI (T1) | Sophisticated method restricted to specialized centers | Low | Moderate to high |
| N+ | Investigational | Diffusion tensor imaging | Sophisticated method restricted to specialized centers | Low to moderate | High |
| N+ | Investigational | Multimodal MRI | Sophisticated method restricted to specialized centers | Moderate to high | High |
| Exclusion criterion ruling out N+ | Recommended | Structural MRI | Findings characteristic of atypical parkinsonism: e.g. PSP (midbrain/superior cerebellar atrophy); e.g. MSA (pontine atrophy, hot-cross bun sign, cerebellar atrophy, increased basal ganglia iron with putaminal rim), e.g. CBS (parietal atrophy) | Moderate, stage dependent | High |
| Exclusion criterion ruling out N+ | Recommended | FDG PET | PSP and MSA each have characteristic patterns that are distinct from PD | High | High |
| Exclusion criterion ruling out N+ | Investigational | 11C-raclopride PET or 123I-iodobenzamide SPECT | Major reductions in dopamine receptor binding in early disease are more suggestive of PSP or MSA (modest reductions can be seen in treated PD) | Moderate | High |
*high> 80%; moderate >70 < 80%; low < 70%. See Supplement 3 for further details. CBS: corticobasal syndrome, FDG: fluoro-deoxy-glucose, MIBG: metaiodbenzylguanidin, MRI: magnetic resonance imaging, MSA: multiple system atrophy, PD: Parkinson’s disease, PET: positron emission tomography, PSP: progressive supranuclear palsy, SPECT: single-photon emission computerized tomography.
Table 4.
Biological designations in biomarker-positive individuals.
| Gene pathogenic variant | Synucleinopathy | Neurodegeneration | Biological designation |
|---|---|---|---|
| G- | S+ | N+ | Sporadic PD |
| N- | Sporadic Parkinson’s type synucleinopathy | ||
| S- | N+ | Non-PD neurodegeneration (or false negative S test) |
|
| N- | No evidence for PD | ||
| GF+ | S+ or S- | N+ or N- | Genetic PD (e.g. SNCA-PD) |
| GP+ | S+ | N+ | Genetic PD (e.g. GBA-PD) |
| N- | Genetic Parkinson’s type synucleinopathy (e.g. GBA-Parkinson’s type synucleinopathy) |
||
| S- | N+ (gene predisposing for either Parkinson’s type synucleinopathy or non-synucleinopathy) | Genetic synuclein-negative PD (e.g. LRRK2-PD, PRKN-PD) |
|
| N+ (gene consistently predisposing for Parkinson’s type synucleinopathy) | Non-PD neurodegeneration (or false neg. S-test) |
||
| N- | Genetic predisposition for PD (e.g. GBA-predisposition for PD) |
GF+: fully penetrant pathogenic gene variants, GP+: pathogenic gene variants with strong or intermediate predisposition, G-: pathogenic gene variants with low predisposition for PD or polygenic risk scores, or absent or unknown genetic contributions are considered as “genetically indeterminate”. See Supplement S4 for critical appraisal.
Table 5.
Clinical manifestations in patients with biologic evidence of PD.
| A. Clinical abnormalities possibly related to PD (Cposs+) |
|---|
| - Option 1: If S+ or N+ *: at least 1 feature from 1 of the following categories - Option 2: If isolated G+ (S- and N-): at least 1 feature from 2 of the following categories |
| 1. Motor |
| a. A single cardinal manifestation of parkinsonism (i.e. expert-examined bradykinesia, or rest tremor) b. Abnormal quantitative motor testing (>1 standard deviation below age-adjusted normal motor speed) |
| 2. Sensory |
| a. Olfactory loss |
| 3. Autonomic |
| a. Chronic constipation |
| b. Urinary dysfunction |
| c. Severe erectile dysfunction (onset at an age <60) |
| d. Likely neurogenic orthostatic hypotension (i.e. heart rate increase <0.5 bpm/mmHg systolic BP drop)60 |
| 4. Sleep |
| History of REM sleep behavior disorder (without polysomnographic confirmation) |
| b. Excessive daytime somnolence |
| 5. Affective / Cognitive |
| a. Depression |
| b. Mild cognitive impairment |
| B. Clinical abnormalities probably related to PD (Cprob+) |
| - Option 1: If S+ or N+ *: at least 1 feature from at least 2 of the categories of A, or - Option 2: If isolated G+ (S- and N-): at least 1 feature from at least 3 of the categories of A, or - Option 3: If G+, S+, or N+: at least 1 of the following features: |
| a. Parkinsonism10 (bradykinesia plus one of rigidity or rest tremor) |
| b. Dementia |
| c. REM sleep behavior disorder (polysomnography-confirmed) |
| d. Neurogenic orthostatic hypotension (≥20/10 mmHg blood pressure drop within 3 minutes of standing or head-up tilt test)60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



