
Submitted:
10 April 2023
Posted:
10 April 2023
You are already at the latest version
Abstract
Glutathione (GSH) is an antioxidant that our cells utilize to prevent damage done by reactive oxygen species, free radicals, peroxides, lipid peroxides, and heavy metals [1]. Due to its immune role in tuberculosis (TB), GSH is hypothesized to play an important part in the immune response against M. tb. Furthermore, one of the hallmark structures of TB is granuloma formation, which involves many types of immune cells. T cells specifically are a major component and are involved in the release of cytokines and activation of macrophages. GSH acts as an important function in macrophages, natural killer cells, and T cells in modulating their activation, their metabolism, the proper cytokine release, the proper redox activity, and free radical levels. For more susceptible patients with HIV and Type 2 Diabetes, the demand for higher GSH levels is increased. This review compiles reports demonstrating the benefits of GSH in improving the immune cell responses against M. tb infection and the use of GSH as adjunctive therapy for TB.
Keywords:
glutathione
; tuberculosis
; mycobacteria
; interferons
; interleukins
; tumor necrosis factor
; trans-forming growth factor
1. Introduction
Glutathione (GSH) is a tripeptide and an antioxidant, which is mostly found in the cytosol, which is where it’s synthesized through two ATP-requiring enzymatic steps. The first involves the formation of γ-glutamyl cysteine from glutamate and cysteine, followed by the formation of γ-L-glutamyl-L-cysteinyl-glycine from γ-glutamylcysteine and glycine (Figure 1) [2]. The first step is the rate-limiting step and is catalyzed by glutamate–cysteine ligase (GCL) [3]. The second step is catalyzed by GSH synthase, which is subject to feedback inhibition by GSH itself [4]. In the presence of glutathione peroxidase, thiol-reduced glutathione (rGSH) reduces hydrogen peroxide formed from oxidative stress, resulting in the formation of oxidized glutathione (GSSG). Then, GSSG reductase (GSR) reduces GSSG back to rGSH using NADPH, forming a redox cycle [5]. Thus, accumulation of GSSG and depleted levels of rGSH can increase oxidative stress, which is sometimes measured in experiments using levels of reactive oxygen species. However, in the case of severe oxidative stress, the ability of the cell to reduce GSSG to rGSH is diminished and leads to an accumulation of GSSG and depleted levels of rGSH [6]. Glutathione (GSH) is synthesized at high levels by cells during reactive oxygen intermediate and nitrogen intermediate production. GSH scavenges peroxide species and has been recently shown to play an important role in apoptosis and to regulate antigen-presenting-cell functions [7]. Due to these immunomodulatory properties, GSH has been hypothesized to be an adjunctive therapy for Mycobacterium tuberculosis (M. tb) infection. In this review, we will analyze the role of GSH supplementation in forming proper immune response to M. tb and when used in adjunctive therapy for tuberculosis (TB) patients.
2. Granuloma and Cytokines
One of the hallmark structures of TB is granuloma formation. Granuloma is an aggregate of organized mature macrophages that wall off TB from the surroundings. Upon infection, M. tb contact induces phagocytosis by resident alveolar macrophages. These mature macrophages have increased cytoplasmic size and more organelles, and have ruffled cell membranes that makes them more phagocytic [8]. Other immune cells that make up granuloma include neutrophils, dendritic cells, B and T cells, and natural killer cells. T cells specifically are a major component of granulomas and are important in containing M. tb infection [9]. They release cytokines necessary for initiation, and for activation of macrophages. Granulomas are an important component of the primary immune response to TB and individuals who do not show signs of active TB could have had healed or sterile granulomas [10,11,12].
Granulomas play a complex role in M. tb infection. Granuloma forms within the lungs and keeps TB inactive, which is referred to as latent TB [13]. Mycobacteria can also exploit granuloma formation, proliferate inside the structure, and disseminate throughout the body [8]. But without granuloma formation, this dissemination would be greater. Thus, local control of the infection is crucial. There are also many cases where TB infiltrates the lungs and escapes trafficking and immune response by the host. This is especially evident in immunocompromised patients, who have deficiency of either tumor necrosis factor (TNF)-α, interferon (IFN)-γ, or interleukin (IL)-12 [8]. In immunocompromised individuals, GSH levels can also be significantly compromised [14]. Each granuloma is unique, even within the same host, and has a different proportion of different types of immune cells [9]. Furthermore, there are different proportions of cytokines in each granuloma. IL-2, IL-17, IFN-γ, and TNF-α all show inverse correlation to bacterial burden [9]. T cell secretion of IFN-γ, TNF-α and IL-17 is thought to be required for the activation of macrophages and inducing an immune response [9].
Cytokine balance is important for proper immune response against M. tb, and the proper ratio and proportion of the type of cytokines is crucial for the correct and efficient response against TB. In active TB, M. tb-specific T cells are suppressed, as is shown by suppression of proper IL-2 and IFN-γ production, and overproduction of immunosuppressive cytokines, IL-10 and transforming growth factor (TGF)-β [15]. TGF-β can itself diminish GSH levels by downregulating expression of enzyme subunits involved in GSH synthesis. Furthermore, in HIV positive patients, T-helper 1 (Th1) response is shifted to a T-helper 2 (Th2) response [16,17]. Thus, mechanisms utilized to reverse these changes in cytokine profile is important, since T cells and proper Th1 type cytokine responses are a hallmark for control of M. tb infection. Fortunately, in our previous studies, GSH supplementation resulted in an increase in Th1 cytokines, such as IL-2, IL-12, IFN-γ, and TNF-α [15,18].
In previous studies, we found that macrophages require both IFN-y and LPS, which is added to macrophages to result in TNF-α production, in order to have enhanced synthesis of inducible NO synthase (iNOS) and generation of nitric oxide (NO) [19]. NO is shown to have antimicrobial activity and inhibits growth of M. tb [20]. We have also observed decreased levels of IL-10, TGF-β, and IL-6 by GSH supplementation [18]. IL-6 is a proinflammatory cytokine that promotes Th2 differentiation, inhibits Th1 response, induces oxidative stress and causes systemic inflammation [21,22,23].
3. Role of GSH during Ferroptosis and in Macrophages
Cell death during M. tb infection is considered detrimental as it allows the mycobacteria to spread. Ferroptosis is one type of cell death in which accumulation of iron and toxic lipid peroxides leads to the necrosis of the cell [24]. It was observed that macrophage necrosis was stimulated by reduced levels of GSH and glutathione peroxidase-4 (Gpx4), and under conditions of iron overload lipid peroxidation is triggered and if Gpx4 is suppressed, cell death presumes [24,25,26,27]. During usual conditions, lipid peroxides are reduced by Gpx4 through GSH oxidation, but when Gpx4 expression or activity is inhibited during iron overload, lipid peroxide levels lead to cell death [24,28].
GSH also plays other crucial roles in macrophages. Mycobacteria defective in the transport of peptides such as GSH, are resistant to antimicrobial effects of GSH. In a study that examined the role of GSH in macrophages, and the intracellular growth of mycobacteria, mutant mice defective in the transport of GSH were found to be resistant to toxicity caused by GSH [19]. Furthermore, another study found that a virulent strain of M. tb, H37Rv, was sensitive to GSH and nitrosoglutathione (GSNO), in vitro [7]. To test the role of GSH in antimycobacterial activity, macrophages infected with H37Rv were treated with IFN-γ, LPS, and buthionine sulfoximine (BSO), and growth of H37Rv inside macrophages was measured. It should be noted that BSO inhibits activity of the first step in the synthesis of GSH and can be used to differentiate antimycobacterial activity due to GSH levels. Venketaraman et al. demonstrated that BSO treated macrophages had a significant decrease in intracellular GSH levels, and observed a significant increase in the intracellular growth of H37Rv [7]. However, the mechanism of action of GSH remains unclear, and it is hypothesized that high concentrations of GSH could result in an imbalance in redox activity within a bacterium containing alternative thiol responsible for the redox activity [7].
4. Natural Killer Cells
Natural killer (NK) cells are cytotoxic lymphocytes part of the innate immune system, which controls spread of multiple types of microbial infections and tumors. They are also in the same family as T and B cells and similar to cytotoxic T cells, and release granules that contain enzymes, such as granulysin, that can kill infected cells by lysis [29]. They respond to virus-infected cells, cells infected with intracellular pathogens, and to tumor cells. Research has highlighted the importance of NK cells as regulatory cells that interact with dendritic cells, macrophages, T cells, and endothelial cells [30].
NK cell activation and lytic capacity is controlled by a balance between cell surface activating and inhibitory receptors. Activating receptors detect the presence of ligands, such as the stress-induced self-ligands that are recognized by NKG2D activating receptors [31]. The ideal cytokine environment provides the right interactions between NK cells and other immune cells, and cytotoxic and cytokine response of NK cells depends on this interaction. Type I IFN cytokines, such as IL-2, IL-12, IL-15, are potent activators of NK cells [32]. Specifically, IL-2 can influence cytotoxic, proliferative, and cytokine producing activities of NK cells, and T cells are important for the production of this cytokine [33]. One study found that GSH in combination with IL-2 and IL-12 augments NK cell functions and leads to control of M. tb infection [29]. Liposomal GSH supplementation restores redox homeostasis, and improves the immune response against M. tb infection, by further shifting cytokine profile to a Th1 response [18]. Considering the antimycobacterial and immune-modulating properties of GSH through its action on NK cell activity and restoration of Th1 cytokine response, GSH in combination with Th1 cytokines, such as IL-2 and IL-12, enhances the host response against M. tb infection [18,34,35].
NK cells inhibit intracellular H37Rv growth in monocytes in vitro [29]. Furthermore, GSH supplementation along with cytokines augments NK cells’ function to completely inhibit H37Rv growth inside human monocytes [35]. In the same study, NK cells treated with N-acetyl cysteine (NAC), a GSH prodrug, in conjunction with cytokines such as IL-2 and IL-12, had increased expression of cytotoxic ligands, FasL and CD40L. NK cell cytotoxic receptors, NK activation receptors, and NK cytotoxic ligands on the cell surface of NK cells are correlated with inhibition of M. tb growth [35]. In the end, NK cells require adequate levels of GSH and particular cytokines to function properly and to play their intended role in the innate immune response against tumor cells and parasitic infections, especially M. tb infection.
5. T cells
Th1 cells are important for the control of M. tb infection. Their production of IL-2, IFN-γ, and TNF-α is necessary for the activation and cell recruitment of macrophages [9]. T cells are also a major component of granulomas and play a critical role for the immune response against M. tb infection [9]. Active TB is characterized by the suppression of this type of T cell response, as can be seen with the decreased levels of Il-2 and IFN-γ [36,37,38,39,40]. Furthermore, there is overproduction of immunosuppressive cytokines IL-10 and TGF-β when T cell function is decreased during TB. Treatment of T cells derived from HIV patients with NAC results in reduction in the growth of M. tb inside monocytes [15]. GSH levels are lower in T cells isolated from HIV patients. However, NAC-treatment of T cells also resulted in an increase in the levels of GSH, which leads to enhanced functions of T lymphocytes in controlling M. tb infection [15].
Activated T cells produce reactive oxygen species (ROS), triggering antioxidative GSH responses [41]. Thus, GSH is important for T cell effector functions. In a knock-out mice experiment, GSH lacking T cells undergo normal activation but cannot meet energy and biosynthetic requirements [41]. Activated T cells undergo clonal expansion, which raises the need for glucose and glutamine utilization, and increases the production of ROS [42,43,44,45,46]. T cell receptor activation activates mitochondrial ROS production, and prevention of lipid oxidation by glutathione peroxidase 4 (Gpx4) is important for survival and further activation [41]. Additionally, GSH-deficient T cells can undergo activation but cannot reprogram metabolism to meet their energy needs. GSH deficiency compromised activation of mTOR and expression of Myc transcription factor [41]. Myc is linked to glutaminolysis and glycolysis types of metabolism, and T cells with reduced Myc, switch to fatty-acid beta oxidation utilization [47,48]. In early proliferation, T cells require fatty acids for lipid and membrane synthesis. These components are provided by processes that increase acetyl-CoA synthesis from glucose and glutamine and fuels energy [41]. Without proper GSH levels, this process is disturbed, and if rising demand for acetyl-CoA levels are not met by the T cells, they eventually undergo necrosis.
6. GSH in HIV Patients
Studies on HIV-positive individuals have shown that GSH increases lymphocyte activation, which is crucial to the pathophysiology of HIV infection [49]. These studies have demonstrated that these individuals have low GSH levels. Decreased function of the immune system is correlated with HIV progression. Free radical overload of monocytes and granulocytes is one way immunological function declines. This results in a lack of antioxidant defenses, which could cause the CD4 cell loss that is frequently observed when HIV progresses [50]. According to further studies, when GSH levels are elevated, nuclear factor kB (NFkB), which is required for the HIV provirus’s active transcription, is activated [51,52,53]. Replenishing GSH is a potential therapeutic treatment in individuals with HIV because the primary pathophysiology of the HIV virus is the loss of CD4+ T cells.
In a previous study, our lab showed that treatment with 10 mM NAC, a GSH precursor, leads to reduction of growth of M. tb inside monocytes from HIV patients compared to other treatment conditions but did not show complete stasis of M. tb growth as was seen in healthy individuals [15]. T cells from healthy participants treated with NAC were shown to have higher levels of IL-12, IL-2 and IFN-γ. These cytokines are essential in controlling intracellular infections like M. tb as they induce the Th1 response. Cell-mediated immunity is greatly diminished as a result of HIV’s high affinity for infecting and killing CD4+ T cells. This lowered immunity makes opportunistic infections, including M. tb, more frequent. GSH at low levels is important for cell-mediated immunity since it has been related to CD4+ T cell death, the main pathology of HIV infection [54,55,56,57]. Consequently, GSH may be an effective adjuvant treatment for those patients with HIV and M. tb infection [15].
GSH is produced by almost all cell types and as mentioned earlier, it comes in two forms: reduced (rGSH) and oxidized (GSSG). The synthesis of rGSH has two distinct pathways ( Figure 1). The two-step de novo production of rGSH requires the separate enzymes glutathione synthetase (GSS) and glutamate-cysteine ligase (GCL) (Figure 1). Additionally, GSSG is reduced by glutathione reductase (GSR) to create rGSH [52]. We have further conducted experiments to identify the reasons for decreased levels of GSH in HIV-infected people by comparing the extent to which the levels of GCLC, GSS, and GSR are lower in RBCs separated from HIV-infected patients compared to healthy participants. Levels of GSS and GCLC in the RBCs of HIV-infected individuals were found to be significantly lower than that of healthy individuals [15,58,59]. These studies have demonstrated that GSR expression is dramatically reduced in RBCs taken from HIV-positive subjects. This supports the cause of low GSH levels and the effects of GSH shortage, such as the decline in immunological function seen in HIV patients. Important research revealing lower levels of GSS, GCLC, and GSR in HIV-infected subjects confirms the notion and earlier findings that those with HIV-infections had lower amounts of GSH than healthy individuals [60].
The increased production of proinflammatory cytokines (Table 1), such as IL-1, IL-17, and TNF-α, has been hypothesized to be linked to chronic HIV infection. This rise in proinflammatory cytokine levels in the HIV-infected correlated with an increase in free radical production. Free GSH is able to scavenge free radicals produced as a result of the proinflammatory cytokines’ chronic overproduction. The increased generation of free radicals in HIV-positive individuals will lead to the depletion of GSH [58]. Additionally, it was found that HIV-infected patients had significantly higher levels of TGF-β in their plasma and macrophage supernatants. This increase correlated with a decrease in the expression of the GCL gene in macrophages [58]. Moreover, increased TGF-β inhibits the formation of GCLC, which lowers the generation of new GSH molecules.
7. GSH in Type 2 Diabetes Patients
According to recent data, individuals with diabetes are two to three times more likely to develop TB than the general public. Diabetes is related to 10% of TB cases, and diabetic patients are more likely to die during TB therapy and experience relapse following treatment [61,62]. Another risk for diabetic patients is an increase in pro-inflammatory cytokines, such as IL-6 and IL-17, which cause chronic inflammation [63]. Furthermore, reduced expression of GSH synthesizing enzymes such GCLC, GSS, and GGT is associated with lower GSH levels in people with Type 2 diabetes mellitus (T2DM) [64].
There is evidence that individuals with T2DM have altered GSH synthesis and metabolism, as well as higher amounts of ROS and pro-inflammatory cytokines. Studies have shown that GSH concentrations in erythrocytes and plasma are lower in patients with T2DM, associated with reduced levels of the enzymes necessary for GSH production [65,66]. Depletion of GSH and excessive production of ROS cause an increase in the prevalence of TB-T2DM co-infection, which alters the immunological response and leads to worse outcomes in these individuals [67].
GSH has been studied as a possible TB adjunctive therapy in patients with T2DM. Our lab has demonstrated that three months of liposomal GSH (lGSH) supplementation in T2DM individuals can lower overall oxidative stress, lower levels of oxidized GSH (GSSG), and maintain levels of reduced GSH in different blood components [68]. Additionally, lGSH supplementation helps patients with T2DM and was shown to increase control of in vitro mycobacterial infections by reducing redox imbalance. lGSH combination therapy with everolimus, a mammalian target of rapamycin (mTOR) inhibitor, has also been studied as an adjunctive treatment for TB in T2DM patients. Everolimus is a small molecule that could modulate autophagy to enhance the killing of mycobacteria via inhibition of the mTOR pathway. In human studies, mTOR inhibition by everolimus has been shown to promote autophagy and improve the cellular innate immune response [69]. Everolimus has been shown to be effective in controlling M. tb infection in the granulomas and has demonstrated additive effects when combined with isoniazid and pyrazinamide [70].
Another treatment option, a first-line antibiotic for M. tb infection, is rifampin (RIF). By interacting with and inhibiting mycobacterial DNA-dependent RNA polymerase, RIF prevents M. tb from being able to proliferate but causes oxidative stress [71,72,73]. When RIF and lGSH are provided together in diabetic mouse models, malondialdehyde (MDA) measurements of oxidative stress are dramatically reduced, indicating a possibility for lGSH to counteract the negative oxidative effects of RIF while boosting its bactericidal benefits [72,73,74,75]. IL-6, a proinflammatory cytokine, is also thought to contribute to the development of oxidative stress. The levels of IL-6 in the lungs of diabetic mice treated with RIF and GSH and infected with M. tb was significantly decreased [76]. By reducing oxidative stress and boosting RIF bactericidal action, these findings highlight the value of lGSH as an adjuvant therapy for M. tb infection. Levels of IL-12 and IFN-γ, cytokines important in controlling M. tb infections, were markedly elevated in the lungs of diabetic mice infected with M. tb across all treatment groups and were noticeably elevated further in animals treated with a combination of RIF and lGSH. lGSH can be added to RIF therapy to help control the immune system, which will improve the ability to manage M. tb infection and lessen the risk of serious complications in diabetics [76]. The findings from lGSH trials on mice should encourage additional research into the potential of lGSH as an adjuvant therapy for M. tb infection in humans.
8. Clinical Trials using GSH as Adjunct Therapy
TGF-β, a cytokine produced by regulatory T cells (T-Regs) and macrophages, is well known for its function in controlling immune responses by limiting the proliferation and expansion of T cells. The levels of GSH in the peripheral blood mononuclear cells (PBMCs) has been shown to be significantly decreased in HIV-infected individuals, which was linked to elevated TGF-β levels. At 13 weeks post treatment, studies show a decline in TGF-β, taking into consideration how supplementing with GSH lowers levels of immunosuppressive cytokines [18]. Interestingly, liposomal glutathione (lGSH) supplementation for 13 weeks resulted in a significant decrease in the levels of IL-6 in HIV-positive individuals. In line with the diminished levels of GSH and increased levels of TGF-β, IL-6, and free radicals, we found decreased levels of Th1-specific cytokines such as IL-2, IL-12, and IFN-γ in HIV-positive individuals (Table 1). Interestingly, 13 weeks of lGSH supplementation led to a significant drop in IL-6 levels in HIV-positive individuals [18].
Lower levels of Th1-specific cytokines such IL-2, IL-12, and IFN-γ were detected in HIV-positive people, which is consistent with the decreased levels of GSH and higher levels of TGF-β, IL-6, and free radicals. These findings show a considerable rise in IFN-γ levels in HIV-infected individuals 13 weeks after lGSH administration. Further findings have demonstrated a significant rise in IL-1 and TNF-α levels in HIV-infected persons 13 weeks after lGSH intake. According to current data, there is no considerable change in the levels of IL-2 or IL-17 following lGSH administration. At 13 weeks, lGSH supplementation was successful in lowering IL-10 levels. Further results show that HIV disrupts the body’s physiological processes, causing an imbalance in the cytokine profiles. Supplementing with lGSH can restore immunological responses, which could be beneficial for HIV patients in treating opportunistic infections [18].
In a double-blinded study, this lab gave either an empty liposomal supplement or an lGSH supplement to a group of HIV-infected people with CD4+ T cell counts < 350 cells/mm3 to take over a 3-month period. Measurements showed that IL-2, IL-12, and IFN-γ levels were significantly lower at baseline in HIV-positive patients compared to healthy individuals, but levels of IL-6, IL-10, TGF-β, and free radicals were significantly higher [77]. When HIV-positive participants were given lGSH for three months, their levels of IL-12, IL-2, and IFN-γ increased significantly in comparison to those of the placebo group, while their levels of IL-6, IL-10, and free radicals decreased, and their levels of TGF-β, IL-1, and IL-17 remained stable. While GSH levels rose in the treatment group, free radical levels in CD4+ T cells stabilized. For the course of the study, there was no discernible difference in the placebo group. In summary, supplementing with lGSH can assist with restoring redox homeostasis and cytokine balance in HIV-infected patients with CD4+ T cell counts below 350 cells/mm3, supporting the immune system’s ability to fight opportunistic infections, such as M. tb [77].
One of the features of TB is oxidative stress, which is evident by elevated lipid peroxidation products such as malondialdehyde (MDA) as well as decreased antioxidant capacity [78,79]. TB patients are low in vitamins A, C, E, and also Selenium, and GSH [80,81]. NAC, a GSH precursor, was shown to limit M. tb infection by suppressing the oxidative stress induced by the infection and also having direct antimicrobial activity [82,83]. The direct effect of GSH involves enhancing nitric oxide (NO). When combined with GSH, NO forms S-nitrosoglutathione (GSNO) [79]. NAC is a synthetic form of cysteine that has been shown to lower ROS levels and improve control of M. tb infection [19]. As a precursor to GSH, NAC can also prevent ferroptosis given that it can restore proper GSH levels to prevent oxidative stress. Notably, patients taking NAC showed significant increase in GSH levels and improvement in antioxidative activity compared to the control group [82].
9. Conclusions
Adequate GSH levels and balanced levels of free radicals are important for an effective immune response against a vast number of infections. Specifically, M. tb is endemic and one of the leading causes of death in every part of the world. Through research and experiments, our growing understanding of TB and patients’ responses to treatments has improved and increased the number of available therapy options for TB patients. In this review article, we have demonstrated the cytokine imbalance and changes in the levels of free radicals that follow M. tb infection. One molecule that has shown promising results is GSH. GSH in addition to other treatment options, such as Th1 cytokines, everolimus, and first-line antibiotics like rifampin, show significant improvements in the levels of GSH, free radicals, and cytokine profile. We have also highlighted the importance of GSH for patients with HIV or Type 2 Diabetes, who are more susceptible to TB. GSH is a naturally synthesized molecule by our body’s cells that shifts the immune response towards a beneficial state and should be a part of an adjunctive therapy against TB.
Author Contributions
Conceptualization, V.V., A.A., J.V., K.S. and M.K.; writing—original draft preparation, A.A., J.V., K.S. and M.K.; writing—review and editing, A.A., J.V., K.S. and M.K.; supervision, V.V. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A. F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, L.; Wellner, V.P.; Griffith, O.W.; Meister, A. Glutathione synthetase. Purification from rat kidney and mapping of the substrate binding sites. J. Biol. Chem. 1979, 254, 5184–5190. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Checa, J. C.; Kaplowitz, N.; García-Ruiz, C.; Colell, A.; Miranda, M.; Marí, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. C. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Venketaraman, V.; Dayaram, Y. K.; Talaue, M. T.; Connell, N.D. Glutathione and nitrosoglutathione in macrophage defense against Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 2012, 12, 352–366. [Google Scholar] [CrossRef]
- Gideon, H. P.; Phuah, J.; Myers, A. J.; Bryson, B. D.; Rodgers, M. A.; Coleman, M. T.; Maiello, P.; Rutledge, T.; Marino, S.; Fortune, S. M.; et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS pathogens 2015, 11, e1004603. [Google Scholar] [CrossRef]
- Cosma, C.L.; Sherman, D.R.; Ramakrishnan, L. The secret lives of the pathogenic mycobacteria. Annu. Rev. Microbiol. 2003, 57, 641–676. [Google Scholar] [CrossRef]
- Feldman, W. H.; Baggenstoss, A. H. The residual infectivity of the primary complex of tuberculosis. Am. J. Pathol. 1938, 14, 473–490. [Google Scholar] [PubMed]
- Dutta, N. K.; Karakousis, P. C. Latent tuberculosis infection: myths, models, and molecular mechanisms. Microbiol. Mol. Biol. Rev. 2014, 78, 343–371. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Ippolito, G.; Mfinanga, S.; Ntoumi, F.; Yeboah-Manu, D.; Vilaplana, C.; Zumla, A.; Maeurer, M. Latent TB Infection (LTBI) - Mycobacterium tuberculosis pathogenesis and the dynamics of the granuloma battleground. Int. J. Infect. Dis. 2019, 80S, S58–S61. [Google Scholar] [CrossRef] [PubMed]
- Venketaraman, V.; Millman, A.; Salman, M.; Swaminathan, S.; Goetz, M.; Lardizabal, A.; Hom, D.; Connell, N. D. Glutathione levels and immune responses in tuberculosis patients. Microb. Pathog. 2008, 44, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Morris, D.; Sipin, A.; Kung, S.; Franklin, M.; Gray, D.; Tanzil, M.; Guilford, F.; Khasawneh, F. T.; Venketaraman, V. Glutathione and adaptive immune responses against Mycobacterium tuberculosis infection in healthy and HIV infected individuals. PLoS One 2011, 6, e28378. [Google Scholar] [CrossRef] [PubMed]
- Osakwe, C. E.; Bleotu, C.; Chifiriuc, M. C.; Grancea, C.; Oţelea, D.; Paraschiv, S.; Petrea, S.; Dinu, M.; Băicuş, C.; Streinu-Cercel, A.; Lazăr, V. TH1/TH2 cytokine levels as an indicator for disease progression in human immunodeficiency virus type 1 infection and response to antiretroviral therapy. Roum. Arch. Microbiol. Immunol. 2010, 69, 24–34. [Google Scholar] [PubMed]
- Klein, S. A.; Dobmeyer, J. M.; Dobmeyer, T. S.; Pape, M.; Ottmann, O. G.; Helm, E. B.; Hoelzer, D.; Rossol, R. Demonstration of the Th1 to Th2 cytokine shift during the course of HIV-1 infection using cytoplasmic cytokine detection on single cell level by flow cytometry. AIDS 1997, 11, 1111–1118. [Google Scholar] [CrossRef]
- Ly, J.; Lagman, M.; Saing, T.; Singh, M. K.; Tudela, E. V.; Morris, D.; Anderson, J.; Daliva, J.; Ochoa, C.; Patel, N.; Pearce, D.; Venketaraman, V. Liposomal Glutathione Supplementation Restores TH1 Cytokine Response to Mycobacterium tuberculosis Infection in HIV-Infected Individuals. J. Interferon Cytokine Res. 2015, 35, 875–887. [Google Scholar] [CrossRef]
- Venketaraman, V.; Dayaram, Y. K.; Amin, A. G.; Ngo, R.; Green, R. M.; Talaue, M. T.; Mann, J.; Connell, N. D. Role of glutathione in macrophage control of mycobacteria. Infect. Immun. 2003, 71, 1864–1871. [Google Scholar] [CrossRef]
- Chan, J.; Xing, Y.; Magliozzo, R. S.; Bloom, B. R. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 1992, 175, 1111–1122. [Google Scholar] [CrossRef]
- Maeda, K.; Mehta, H.; Drevets, D. A.; Coggeshall, K. M. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood 2010, 115, 4699–4706. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Rincón, M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E. P.; Costa, D. L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K. D.; Andrade, B. B.; Sher, A. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef]
- Dixon, S. J.; Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B. R.; Friedmann Angeli, J. P.; Bayir, H.; Bush, A. I.; Conrad, M.; Dixon, S. J.; Fulda, S.; Gascón, S.; Hatzios, S. K.; Kagan, V. E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V. E.; Bayir, H.; Pagnussat, G. C.; Head, B.; Traber, M. G.; Stockwell, B. R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef]
- Amaral, E. P.; Foreman, T. W.; Namasivayam, S.; Hilligan, K. L.; Kauffman, K. D.; Barbosa Bomfim, C. C.; Costa, D. L.; Barreto-Duarte, B.; Gurgel-Rocha, C.; et al. GPX4 regulates cellular necrosis and host resistance in Mycobacterium tuberculosis infection. J. Exp. Med. 2022, 219, e20220504. [Google Scholar] [CrossRef]
- Millman, A. C.; Salman, M.; Dayaram, Y. K.; Connell, N. D.; Venketaraman, V. Natural killer cells, glutathione, cytokines, and innate immunity against Mycobacterium tuberculosis. J. Interferon Cytokine Res. 2008, 28, 153–165. [Google Scholar] [CrossRef]
- Long, E. O. Ready for prime time: NK cell priming by dendritic cells. Immunity 2007, 26, 385–387. [Google Scholar] [CrossRef]
- Lanier, L. L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Walzer, T.; Dalod, M.; Robbins, S. H.; Zitvogel, L.; Vivier, E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood 2005, 106, 2252–2258. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Bailey, C.; Cahatol, I.; Dodge, L.; Yim, J.; Kassissa, C.; Luong, J.; Kasko, S.; Pandya, S.; Venketaraman, V. Mechanisms of Control of Mycobacterium tuberculosis by NK Cells: Role of Glutathione. Front. Immunol. 2015, 6, 508. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Johal, K.; Morris, D.; Moreno, S.; Alvarado, O.; Gray, D.; Tanzil, M.; Pearce, D.; Venketaraman, V. Control of Mycobacterium tuberculosis growth by activated natural killer cells. Clin. Exp. Immunol. 2012, 168, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Gong, J. H.; Zhang, M.; Modlin, R. L.; Linsley, P. S.; Iyer, D.; Lin, Y.; Barnes, P. F. Interleukin-10 downregulates Mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infect. Immun. 1996, 64, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Toossi, Z.; Kleinhenz, M. E.; Ellner, J. J. Defective interleukin 2 production and responsiveness in human pulmonary tuberculosis. J. Exp. Med. 1986, 163, 1162–1172. [Google Scholar] [CrossRef]
- Huygen, K.; Van Vooren, J. P.; Turneer, M.; Bosmans, R.; Dierckx, P.; De Bruyn, J. Specific lymphoproliferation, gamma interferon production, and serum immunoglobulin G directed against a purified 32 kDa mycobacterial protein antigen (P32) in patients with active tuberculosis. Scand. J. Immunol. 1988, 27, 187–194. [Google Scholar] [CrossRef]
- Torres, M.; Mendez-Sampeiro, P.; Jimenez-Zamudio, L.; Teran, L.; Camarena, A.; Quezada, R.; Ramos, E.; Sada, E. Comparison of the immune response against Mycobacterium tuberculosis antigens between a group of patients with active pulmonary tuberculosis and healthy household contacts. Clin. Exp. Immunol. 1994, 96, 75–78. [Google Scholar] [CrossRef]
- Mehra, S.; Pahar, B.; Dutta, N. K.; Conerly, C. N.; Philippi-Falkenstein, K.; Alvarez, X.; Kaushal, D. Transcriptional reprogramming in nonhuman primate (rhesus macaque) tuberculosis granulomas. PLoS One 2010, 5, e12266. [Google Scholar] [CrossRef]
- Mak, T. W.; Grusdat, M.; Duncan, G. S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brüstle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Carr, E. L.; Kelman, A.; Wu, G. S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A. M.; Frauwirth, K. A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K. A.; Riley, J. L.; Harris, M. H.; Parry, R. V.; Rathmell, J. C.; Plas, D. R.; Elstrom, R. L.; June, C. H.; Thompson, C. B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K. N.; Powell, J. D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014, 14, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Gülow, K.; Kaminski, M.; Darvas, K.; Süss, D.; Li-Weber, M.; Krammer, P. H. HIV-1 trans-activator of transcription substitutes for oxidative signaling in activation-induced T cell death. J. Immunol. 2005, 174, 5249–5260. [Google Scholar] [CrossRef]
- Sena, L. A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D. A.; Wang, C. R.; Schumacker, P. T.; Licht, J. D.; Perlman, H.; Bryce, P. J.; Chandel, N. S. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Verbist, K. C.; Guy, C. S.; Milasta, S.; Liedmann, S.; Kamiński, M. M.; Wang, R.; Green, D. R. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 2016, 532, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C. P.; Shi, L. Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L. L.; Fitzgerald, P.; Chi, H.; Munger, J.; Green, D. R. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Buhl, R.; Jaffe, H. A.; Holroyd, K. J.; Wells, F. B.; Mastrangeli, A.; Saltini, C.; Cantin, A. M.; Crystal, R. G. Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet 1989, 2, 1294–1298. [Google Scholar] [CrossRef]
- Dobmeyer, T. S.; Findhammer, S.; Dobmeyer, J. M.; Klein, S. A.; Raffel, B.; Hoelzer, D.; Helm, E. B.; Kabelitz, D.; Rossol, R. Ex vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: role for lymphocyte loss in HIV infection. Free Radic. Biol. Med. 1997, 22, 775–785. [Google Scholar] [CrossRef]
- Staal, F. J.; Roederer, M.; Herzenberg, L. A.; Herzenberg, L. A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. U S A 1990, 87, 9943–9947. [Google Scholar] [CrossRef] [PubMed]
- Staal, F. J. Glutathione and HIV infection: reduced reduced, or increased oxidized? Eur. J. Clin. Invest. 1998, 28, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Duh, E. J.; Maury, W. J.; Folks, T. M.; Fauci, A. S.; Rabson, A. B. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. U S A 1989, 86, 5974–5978. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Holm, E. Role of cysteine and glutathione in HIV infection and other diseases associated with muscle wasting and immunological dysfunction. FASEB J. 1997, 11, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L. A.; De Rosa, S. C.; Dubs, J. G.; Roederer, M.; Anderson, M. T.; Ela, S. W.; Deresinski, S. C.; Herzenberg, L. A. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. U S A 1997, 94, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Levy, J. A. HIV pathogenesis and long-term survival. AIDS 1993, 7, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, G.; Graziosi, C.; Fauci, A. S. The immunopathogenesis of human immunodeficiency virus infection. N. Engl. J. Med. 1993, 328, 327–335. [Google Scholar] [CrossRef]
- Morris, D.; Guerra, C.; Donohue, C.; Oh, H.; Khurasany, M.; Venketaraman, V. Unveiling the mechanisms for decreased glutathione in individuals with HIV infection. Clin. Dev. Immunol. 2012, 2012, 734125. [Google Scholar] [CrossRef]
- Venketaraman, V.; Rodgers, T.; Linares, R.; Reilly, N.; Swaminathan, S.; Hom, D.; Millman, A. C.; Wallis, R.; Connell, N. D. Glutathione and growth inhibition of Mycobacterium tuberculosis in healthy and HIV infected subjects. AIDS Res. Ther. 2006, 3, 5. [Google Scholar] [CrossRef]
- Morris, D.; Ly, J.; Chi, P. T.; Daliva, J.; Nguyen, T.; Soofer, C.; Chen, Y. C.; Lagman, M.; Venketaraman, V. Glutathione synthesis is compromised in erythrocytes from individuals with HIV. Front. Pharmacol. 2014, 5, 73. [Google Scholar] [CrossRef]
- Restrepo, B. I.; Camerlin, A. J.; Rahbar, M. H.; Wang, W.; Restrepo, M. A.; Zarate, I.; Mora-Guzmán, F.; Crespo-Solis, J. G.; Briggs, J.; McCormick, J. B.; Fisher-Hoch, S. P. Cross-sectional assessment reveals high diabetes prevalence among newly-diagnosed tuberculosis cases. Bull. World Health Organ. 2011, 89, 352–359. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://apps.who.int/iris/handle/10665/44698 (accessed on 20 February 2023).
- Chung, S. S.; Ho, E. C.; Lam, K. S.; Chung, S. K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236. [Google Scholar] [CrossRef] [PubMed]
- Oni, T.; Stoever, K.; Wilkinson, R. J. Tuberculosis, HIV, and type 2 diabetes mellitus: a neglected priority. Lancet Respir. Med. 2013, 1, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Lagman, M.; Ly, J.; Saing, T.; Kaur Singh, M.; Vera Tudela, E.; Morris, D.; Chi, P. T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS One 2015, 10, e0118436. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Hirokawa, J.; Tagami, S.; Kawakami, Y.; Urata, Y.; Kondo, T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: regulation of glutathione synthesis and efflux. Diabetologia 1995, 38, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B. I. Diabetes and Tuberculosis. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- To, K.; Cao, R.; Yegiazaryan, A.; Owens, J.; Nguyen, T.; Sasaninia, K.; Vaughn, C.; Singh, M.; Truong, E.; Medina, A.; et al. Effects of Oral Liposomal Glutathione in Altering the Immune Responses Against Mycobacterium tuberculosis and the Mycobacterium bovis BCG Strain in Individuals With Type 2 Diabetes. Front. Cell. Infect. Microbiol. 2021, 11, 657775. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J. B.; Del Giudice, G.; Lattanzi, M.; Valiante, N. M.; Praestgaard, J.; Huang, B.; Lonetto, M. A.; Maecker, H. T.; Kovarik, J.; Carson, S.; et al. mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 2014, 6, 268ra179. [Google Scholar] [CrossRef]
- Ashley, D.; Hernandez, J.; Cao, R.; To, K.; Yegiazaryan, A.; Abrahem, R.; Nguyen, T.; Owens, J.; Lambros, M.; Subbian, S.; Venketaraman, V. Antimycobacterial Effects of Everolimus in a Human Granuloma Model. J. Clin. Med. 2020, 9, 2043. [Google Scholar] [CrossRef]
- Campbell, E. A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S. A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Sharma, S. K.; Sharma, A.; Kadhiravan, T.; Tharyan, P. Rifamycins (rifampicin, rifabutin and rifapentine) compared to isoniazid for preventing tuberculosis in HIV-negative people at risk of active TB. Cochrane Database Syst. Rev. 2013, 2013, CD007545. [Google Scholar] [CrossRef] [PubMed]
- Wehrli, W. Rifampin: mechanisms of action and resistance. Rev. Infect. Dis. 1983, 5, S407–S411. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A. M.; Berning, S. E.; Iseman, M. D.; Peloquin, C. A. Failure of drug penetration and acquisition of drug resistance in chronic tuberculous empyema. Tuber. Lung Dis. 1995, 76, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Maug, A. K. J.; Hossain, M. A.; Gumusboga, M.; Decroo, T.; Mulders, W.; Braet, S.; Buyze, J.; Arango, D.; Schurmans, C.; Herssens, N.; et al. First-line tuberculosis treatment with double-dose rifampicin is well tolerated. Int. J. Tuberc. Lung Dis. 2020, 24, 499–505. [Google Scholar] [CrossRef]
- Beever, A.; Kachour, N.; Owens, J.; Sasaninia, K.; Kolloli, A.; Kumar, R.; Ramasamy, S.; Sisliyan, C.; Khamas, W.; Subbian, S.; Venketaraman, V. L-GSH Supplementation in Conjunction With Rifampicin Augments the Treatment Response to Mycobacterium tuberculosis in a Diabetic Mouse Model. Front. Pharmacol. 2022, 13, 879729. [Google Scholar] [CrossRef]
- Valdivia, A.; Ly, J.; Gonzalez, L.; Hussain, P.; Saing, T.; Islamoglu, H.; Pearce, D.; Ochoa, C.; Venketaraman, V. Restoring Cytokine Balance in HIV-Positive Individuals with Low CD4 T Cell Counts. AIDS Res. Hum. Retroviruses 2017, 33, 905–918. [Google Scholar] [CrossRef]
- Vidhya, R.; Rathnakumar, K.; Balu, V.; Pugalendi, K. V. Oxidative stress, antioxidant status and lipid profile in pulmonary tuberculosis patients before and after anti-tubercular therapy. Indian J. Tuberc. 2019, 66, 375–381. [Google Scholar] [CrossRef]
- Ejigu, D. A.; Abay, S. M. N-Acetyl Cysteine as an Adjunct in the Treatment of Tuberculosis. Tuberc. Res. Treat. 2020, 2020, 5907839. [Google Scholar] [CrossRef]
- Madebo, T.; Lindtjørn, B.; Aukrust, P.; Berge, R. K. Circulating antioxidants and lipid peroxidation products in untreated tuberculosis patients in Ethiopia. Am. J. Clin. Nutr. 2003, 78, 117–122. [Google Scholar] [CrossRef]
- Muzembo, B. A.; Mbendi, N. C.; Ngatu, N. R.; Suzuki, T.; Wada, K.; Ikeda, S. Serum selenium levels in tuberculosis patients: A systematic review and meta-analysis. J. Trace Elem. Med. Biol. 2018, 50, 257–262. [Google Scholar] [CrossRef]
- Safe, I. P.; Amaral, E. P.; Araújo-Pereira, M.; Lacerda, M. V. G.; Printes, V. S.; Souza, A. B.; Beraldi-Magalhães, F.; Monteiro, W. M.; Sampaio, V. S.; Barreto-Duarte, B.; et al. Adjunct N-Acetylcysteine Treatment in Hospitalized Patients With HIV-Associated Tuberculosis Dampens the Oxidative Stress in Peripheral Blood: Results From the RIPENACTB Study Trial. Front. Immunol. 2021, 11, 602589. [Google Scholar] [CrossRef] [PubMed]
- Mapamba, D. A.; Sauli, E.; Mrema, L.; Lalashowi, J.; Magombola, D.; Buza, J.; Olomi, W.; Wallis, R. S.; Ntinginya, N. E. Impact of N-Acetyl Cysteine (NAC) on Tuberculosis (TB) Patients-A Systematic Review. Antioxidants 2022, 11, 2298. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Thiol-reduced form of Glutathione (rGSH) is synthesized through two different pathways. The first and rate-limiting step involves the formation of γ-glutamyl cysteine from the joining of glutamate and cysteine using glutamine-cysteine ligase (GCL) as the enzyme. The second step uses glutathione synthase (GSS) to join glycine to γ-glutamyl cysteine, to form rGSH. An alternative pathway involves reduction of oxidized GSH (GSSG), which is catalyzed by glutathione reductase (GSR) using NADPH as a cofactor, to reduce GSSG to two rGSH molecules.
Figure 1.
Thiol-reduced form of Glutathione (rGSH) is synthesized through two different pathways. The first and rate-limiting step involves the formation of γ-glutamyl cysteine from the joining of glutamate and cysteine using glutamine-cysteine ligase (GCL) as the enzyme. The second step uses glutathione synthase (GSS) to join glycine to γ-glutamyl cysteine, to form rGSH. An alternative pathway involves reduction of oxidized GSH (GSSG), which is catalyzed by glutathione reductase (GSR) using NADPH as a cofactor, to reduce GSSG to two rGSH molecules.
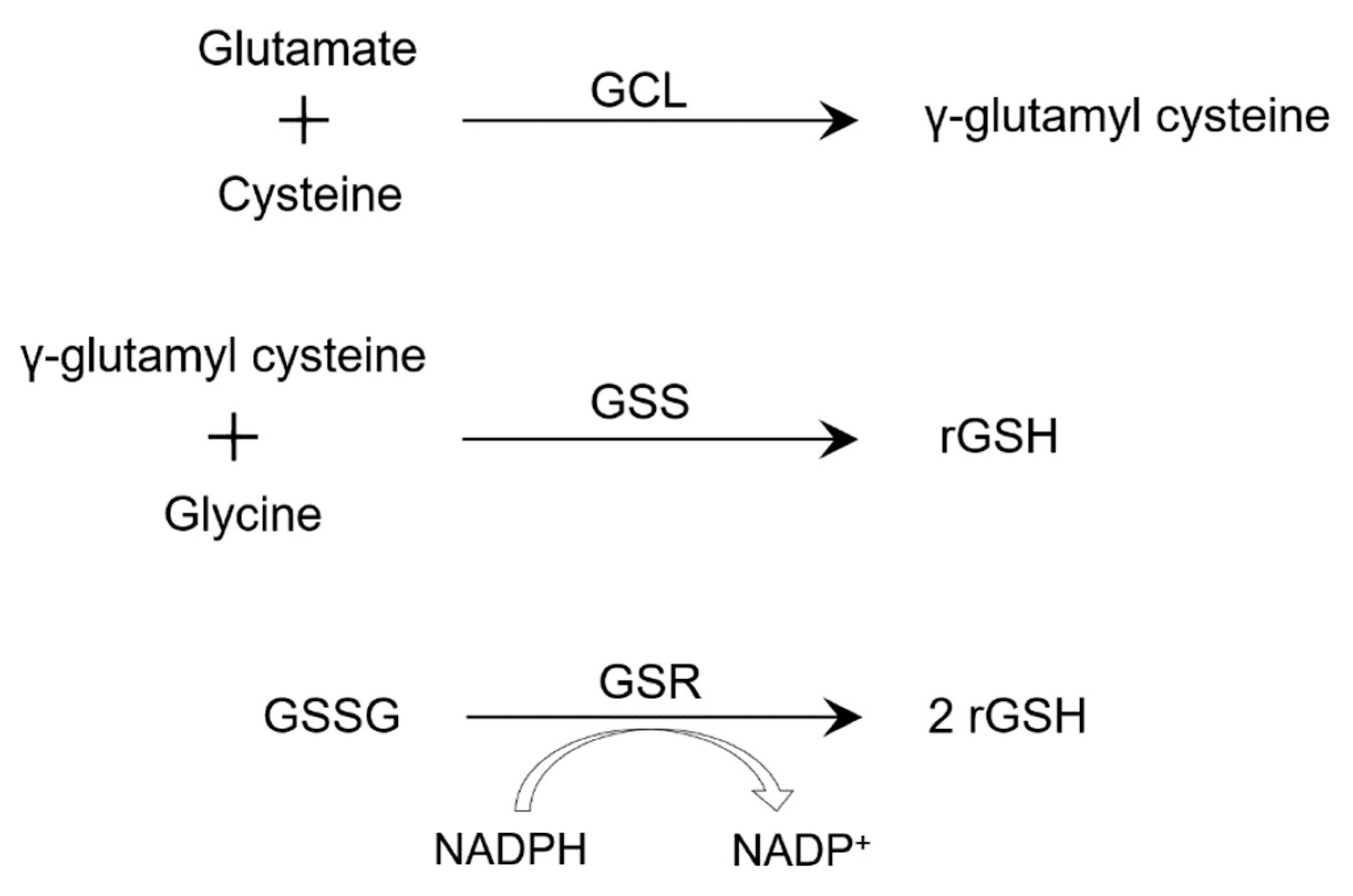

Table 1.
Types and functions of pro- and anti-inflammatory cytokines. Grouped into three categories: T helper 1, pro-inflammatory, and anti-inflammatory cytokines.
Table 1.
Types and functions of pro- and anti-inflammatory cytokines. Grouped into three categories: T helper 1, pro-inflammatory, and anti-inflammatory cytokines.
| Type | Cytokine | Functions |
|---|---|---|
| T helper 1 | IFN-γ | Activates macrophages and induces differentiation; stimulates NK cells and neutrophils |
| TNF-α | Produced predominately by macrophages, induces necrosis or apoptosis | |
| IL-12 | Induces production of IFN-γ, Th1 T-cell response and forms link between innate and adaptive immune responses; activates NK cells | |
| IL-2 | Enhances T-cell viability and proliferation; enhances the generation of effector and memory cells; activates NK cells | |
| Pro-inflammatory | IL-17 | Cytokine that links T-cell activation to neutrophil mobilization and activation |
| IL-6 | Elevated in chronic inflammation and high oxidative stress, recruitment of neutrophils and macrophages | |
| Anti-inflammatory | IL-10 | Immunosuppressive cytokine. Inhibits IFN-γ and IL-12 production |
| TGF-β | Immunosuppressive cytokine. Inhibits T-cell proliferation and function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



