
Submitted:
09 April 2023
Posted:
11 April 2023
You are already at the latest version
Abstract
Staphylococcus aureus (S. aureus), particularly Methicillin-resistant S. aureus (MRSA), is a life-threatening pathogen that causes a variety of infections in hospital and community settings. It poses a significant risk and challenge for global health that hinders our ability to control and treat bacterial infections. Recently, the emergence and spread of variant strains of bacteria, misuse of the limited available options of effective antibiotics, spread of fake drugs, and climate change that increased the contact between humans and animals’ populations carrying different bacterial has significantly increased the incidence of Multi-Drug Resistance strains (MDR). This in turn, has created a severe problem in the infection control treatment of S. aureus infected patients. Treatment options for MRSA infection are increasingly limited and complicated. Unfortunately, different strains of MRSA are showing tolerance and resistance toward vancomycin, which is the standard of care for complicated MRSA. Therefore, it is crucial now to invest developing new effective anti-biotics and synergies, develop and implement national action plans for the management of an-tibiotic resistance, as well as improving our understanding of the antimicrobial resistance mechanisms. Here, in this review we explain the main mechanisms of antibiotic resistance in MRSA and describes different approaches to manipulate it in MRSA, providing a basis for de-signing effective drugs, and shedding some light on the evolution of S. aureus.
Keywords:
Methicillin-resistant Staphylococcus aureus
; MRSA
; antibiotic resistance mechanism
; novel therapeutic approaches
1. Introduction
Methicillin-Resistant Staphylococcus aureus (MRSA) is a major emerging threat to the global health that causes Skin and Soft Tissue Infections (SSTIs), lethal pneumonia, septic arthritis, osteomyelitis, sepsis, and endocarditis [1]. MRSA infections are associated with significant morbidity, mortality, and severe socioeconomic impact due to the substantially increased treatment costs that burden patients and health systems [2]. MRSA was recently reported to be the leading drug-resistant cause of death pathogen in 27 European countries [3]. Recently, emergence of resistance to the currently available antibiotics has dramatically increased the incidence of Multi-Drug Resistance strains (MDR) and posed severe problems in the treatment and infection control of S. aureus [4]. Effective antibiotics for treating MRSA are exceedingly limited, costly, and complicated [4]. Besides resistance to β-lactam antibiotics, MRSA frequently exhibits resistance toward other antimicrobial agents such as macrolides, aminoglycosides, fluoroquinolones, and more recently vancomycin, which is the main current standard option of treatment for complicated infections caused by MRSA [5].
Moreover, MRSA strains exhibit resistance to topical drugs of choice, including mupirocin and fusidic acid, and many other new antibiotic compounds, including linezolid, daptomycin, and ceftaroline [5,6]. This has created a serious challenge for case management and infection control, particularly in limited resources settings. Therefore, there is an urgent need for novel effective antibiotics agents [7]. The primary strategies of pharmaceutical industries for developing new therapeutics is limited to pursue discovering new antibiotics or repurposing existing drugs, such as the groups of beta-lactams and aminoglycosides [8,9]. Other strategies that target intrinsic components in bacteria to mediate resistance to a particular antibiotic family, such as the use of Efflux Pump Inhibitors (EPI) and Methyltransferase Inhibitors to overcome the resistance mechanism of ciprofloxacin and Macrolide Lincosamide Streptogramin B (MLSB), respectively [10,11,12,13,14]. This review focuses on explaining the fundamental mechanisms of antibiotic resistance and novel therapeutic strategies to overcome the drug resistance mechanisms in MRSA.
2. Antimicrobial Resistance of MRSA
S. aureus has the ability to rapidly adapt, develop tolerance and resistance to new antibiotics [13]. Resistance to the most recently developed antibiotics such as linezolid, daptomycin, and fifth-generation cephalosporin (ceftaroline) have been reported among some strains of S. aureus. Several resistance mechanisms were described before among S. aureus including enzymatic modification of antibiotic targets, mutations that reduce antibiotic affinity for its target, and active efflux pumps that expel antibiotics outside the cell [15]. These mechanisms confer resistance to anti-MRSA drugs and are it is mediated by several factors (See Figure 1 and Table 1).
2.1. Penicillin & Methicillin Resistance
S. aureus resistance to penicillin is primarily conferred by the blaZ gene, which encodes a β-lactamase enzyme that inactivates penicillin by hydrolyzing the beta-lactam ring of penicillin. Methicillin, a semi-synthetic beta-lactam antibiotic, it was introduced in the late 1950s as a therapy for penicillin-resistant S. aureus (PRSA) infection; particularly for treating infections caused by beta-lactamase-producing staphylococci. However, interestingly within a year of its introduction, MRSA emerged [13]. MRSA strains were first reported in 1961 in the United Kingdom. The resistance was acquired via incorporating a mecA gene into the chromosome at a specific site [15]. mecA encodes an alternative penicillin-binding protein (PBP2a or PBP2`) that has a low affinity for most semi-synthetic penicillins, including methicillin, nafcillin, and oxacillin [11]. Briefly, PBPs are transpeptidase enzymes responsible for the final step of bacterial cell wall synthesis and are primary targets for all beta-lactam antibiotics [15]. Modified PBPs are the major cause of beta-lactam antibiotic resistance, especially among gram-positive cocci. In MRSA, PBP2a (PBP2’) is encoded by the mecA gene located on the mobile genetic element SCCmec, along with its regulators mecR1 and mecI [15]. The majority of healthcare-associated (HA)-MRSA infections are linked to SCCmec types II or III, whereas community-associated (CA)-MRSA is associated with SCCmec types IV [15,31]. Recently, mecC, which encodes a homolog of PBP2a (designated PBP2c by the European Committee on Antimicrobial Susceptibility Testing; EUCAST) from the S. aureus strain LGA251, has been described, which differs in beta-lactam binding characteristics [4]. Unlike penicillinase-mediated resistance, the resistance conferred by mecA is broad-spectrum to the entire class of lactam-related drugs, except for ceftaroline and ceftobiprole [15]. Therefore, MRSA is currently a major risk of hospital admission, particularly in immunocompromised people and a significant challenge for the infection control teams globally [32].
2.2. Vancomycin-resistant S. aureus (VRSA)
The major component of the S. aureus cell wall is peptidoglycan, a heavily cross-linked polymer consisting of N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) glycan chains, cross-linked by glycine bridges and pentapeptide stems (UDP-Mur-NAc-L-Ala-D-iso-Gln-L-Lys-D-Ala-D-Ala) [13]. Vancomycin disrupts downstream peptidoglycan assembled by binding non-covalently to the D-Ala-D-Ala residues of newly synthesized UDP-MurNAc-pentapeptides during the later stages of peptidoglycan synthesis [33]. The vanA operon confers resistance to vancomycin by hydrolyzing D-Ala-D-Ala peptidoglycan precursors and synthesizing D-Ala-D-lactate peptidoglycan precursors that are essential for vancomycin binding. The vanA operon consists of several genes including vanA, vanH, vanX, vanS, vanR, vanY, and vanZ [2]. The expression of the vanA operon is regulated by a two-component sensor-regulator system encoded by vanS and vanR, which activate the transcription of the operon [19]. VanA is a ligase that catalyzes the ester-bond formation of the D-Ala-D-Lac depsipeptide, and VanH is a dehydrogenase that forms D-Lac by reducing pyruvate [33]. VanA, VanH, and VanX are essential for producing vancomycin-resistant phenotypes. VanA and VanH synthesize the depsipeptide D-Ala-D-Lac, and VanX hydrolyzes the D-Ala-D-Ala ester bond, ensuring that the newly formed D-Ala-D-Lac depsipeptide has little competition to bind the UDP-linked tripeptide peptidoglycan precursor [15]. VanY is a D, D-carboxyl peptidase that facilitates the cleavage of D-Ala-D-Ala dipeptides already attached to the C-terminal end of stem pentapeptide structures [33]. On the other hand; the function of VanZ is not well understood, but it may be involved in the resistance mechanism to teicoplanin. Incorporating altered D-Ala-D-Lac into peptidoglycan results in a cell wall that is no longer susceptible to vancomycin [33].
2.3. Macrolide, Lincosamid and Streptogramine B (MLSB) Resistant S. aureus
MLSB antibiotics, including erythromycin, clindamycin, and streptogramin B, exert their antibacterial activity mainly by binding reversibly to the bacterial 50S ribosomal subunits, blocking the progression of nascent proteins through the exit tunnel in bacterial protein biosynthesis [34]. The resistance mechanisms to MLSB antibiotics primarily involve inhibiting protein synthesis via ribosomal binding site modification through methylation or creating mutations in the 23S rRNA gene. This process is encoded by the erythromycin ribosomal methylase (erm) genes; activating efflux mediated by msrA/B gene; or enzymatically modifying the antibiotics [16]. In S. aureus, the important mechanism of MLSB resistance is the N6 methylation of nucleotide A2058 in the macrolide binding site in 23S rRNA by the erm group of Methyltransferase (MTase) 5,6. This group of enzymes uses S-adenosyl methionine (SAM) to mono- or dimethylate a 50S ribosomal precursor substrate specifically [17]. The erm genes were initially identified in microorganisms that have natural macrolides as a mechanism of self-protection against their antibiotics. Furthermore, the dimethylation of adenine A2058 at the N6 position in the peptidyl transferase loop region in domain V of the 50S ribosomal subunit leads to cross-resistance between lincosamides, macrolides, and the streptogramin group B (MLSB) [18]. As the erythromycin binding site on the 50S ribosome subunit overlaps the binding site of the newer MLSB. The modification by methylase(s) reduces the binding of all three classes of antibiotics, resulting in the MLSB resistant phenotype [17].
2.4. Linezolid
Linezolid is a type of synthetic oxazolidinone antibiotic that can effectively inhibit enterococci, staphylococci, and most strains of Streptococcus [35]. The drug works by binding to the 23S site of ribosomal RNA on the 50S subunit of bacteria, it inhibits the 50S and 30S ribosomal subunits and ultimately prevents the formation of the 70S initiation complex, thus interfering with protein synthesis [35]. One of the most common mechanisms of resistance to linezolid is through drug-binding site mutations in the genes encoding the 23S rRNA [36]. Enzymatic modification of the 23S rRNA through methylation of adenine at position 2503 has also been observed in enterococci [37]. The gene responsible for this modification, Cfr, encodes for methylase and is a plasmid-borne determinant of resistance found in clinical isolates of E. faecalis and other gram-positive bacteria such as staphylococci [21,37]. The Cfr gene has been linked to the mobile transposable element IS256, which has a common sequence in multidrug-resistant staphylococci and enterococci. This sequence has been shown to mediate the transfer of antibiotic resistance genes, as well as alter the promoter sequence of regulatory proteins or activate the expression of existing resistance determinants [38]. This phenomenon could explain the ability of cfr to spread across species and the possibility of widespread dissemination in clinical settings. The Cfr protein is a methyltransferase that methylates C-8 of A2503 in 23S rRNA [39]. This base is located near the overlapping binding sites for these drugs, and its methylation prevents the molecules from binding [35].
2.5. Tigecycline
Tigecycline is a type of glycylcycline antibiotic that prevents amino acid incorporation into the bacterial peptide chains on the 30S ribosomal subunit. Structurally, tigecycline is similar to minocycline and tetracycline; however, it is still effective against most tetracycline-resistant isolates of S. aureus[24]. There have been reports of tigecycline resistance in S. aureus due to the serial passage of bacterial cultures in increasing drug concentrations. Exposed MRSA strains N315 and Mu3 resulted in mutants with 16- to 32-fold increased MICs. In both strains, mutations in a repressor gene resulted in increased gene transcription of a previously uncharacterized multidrug and toxin extrusion family transporter called MepA [24].
2.6. Ceftaroline and Ceftobiprole
Ceftaroline and Ceftobiprole are last-generation parenteral cephalosporins that have an extended spectrum of activity against multi-drug resistant bacteria including MRSA. They inhibit integral peptidoglycan transpeptidases, interfering with cell wall synthesis, and binding to abnormal PBPs like PBP2a in MRSA and PBP2b and PBP2x in β-lactam-resistant pneumococci, ultimately leading to rapid bacterial cell death [25]. X-ray crystallography has revealed that Ceftaroline can bind to two sites in PBP2a of MRSA, one inactivating the serine-active site, while the other attaches to an allosteric binding site, leading to a conformational change that increase the exposure to leathal dose of the drug [26]. However, the persistent usage of ceftaroline has placed a selection pressure that accelerated the development of resistant by mutations. These mutations are charcterized by amino acid substitutions close to the active site and the allosteric site of PBP2a [40]. Low-level resistance to Ceftaroline is associated with PBP2a mutations in the allosteric and transpeptidase domains, while high-level resistance is caused by mutations that modify the geometry of the active site [41]. While high-level Ceftaroline resistance was rendered by the transpeptidase domain mutations, which modifies the geometry of the active site. The low-level resistance was associated with modifications within the allosteric site and defined by a novel allosteric modulation of the active site [42].
At therapeutic concentrations, Ceftaroline activates the opening and acylation process of the active site by binding to the PBP2a allosteric site [43]. Mutations within the PBP2a allosteric domain can modulate the triggering mechanism, affecting protein-protein interactions needed for peptidoglycan biosynthesis [43]. However, additional factors contributing to Ceftaroline resistance remain to be analyzed. Reduced susceptibility to Ceftaroline has been described in S. aureus isolates predating its commercial introduction and in countries where it is not commercially available [22].
2.7. Daptomycin
Daptomycin is a lipopeptide antibiotic produced by Streptomyces roseosporus. It exhibits activity against a broad spectrum of gram-positive bacteria and is approved for the treatment of S. aureus bacteremia and endocarditis, making it a crucial antibiotic for MRSA treatment [44]. The antimicrobial activity of daptomycin is dependent on the presence of calcium, which forms a complex called Calcium-Daptomycin (Ca-Dap) [44]. The Ca-Dap complex binds to the negatively charged head groups of phosphatidylglycerol (PG) and penetrates the cell wall into the cytoplasmic membrane, leading to depolarization, permeabilization, and leakage of ions that ultimately results in cell death. Resistance to daptomycin arises due to mutations in genes that activate the bacterium's defenses towards cell envelope damage, including host cationic antimicrobial peptides [45]. The resistance usually occurs during prolonged therapy in infections involving high bacterial densities. Prior treatment of the patient with vancomycin is associated with a more rapid development of daptomycin resistance during therapy due to resistance mechanisms enhancement [23].
To resist daptomycin, S. aureus must prevent the drug from reaching the cytoplasmic membrane and interfere with its entry into the membrane. However, mutations in different genes can occur, resulting in the minimum inhibitory concentration (MIC) surpassing the susceptibility breakpoint [44]. The most common mutational changes conferring resistance to daptomycin are found in the multiple peptide resistance factors (mrpF) gene [39]. The mrpF protein plays a role in daptomycin resistance by adding a positively charged lysine residue to PG, forming lysylphosphatidyl glycerol (L-PG) [15]. L-PG is synthesized in the cytosol and is then translocated to the outer face of the membrane, resulting in increased levels of L-PG in the cell membrane, which increases the charge of the outer face of the membrane. This, in turn, repels the Ca-Dap complex, reducing the negatively charged PG needed for drug binding to initiate membrane damage [23].
2.8. Telavancin
Telavancin is a vancomycin derivative that contains a hydrophobic side chain on the vincamine sugar [46]. Like vancomycin, Telavancin inhibits bacterial cell wall synthesis by interacting with the polymerization and peptidoglycan biosynthesis[46]. In addition, Telavancin disrupts membrane barrier function by binding to the bacterial membrane, facilitated by targeted interaction with lipid II precursor in the cell wall [47]. Telavancin exerts concentration-dependent bactericidal activity against Gram-positive organisms in vitro, including a wide range of multi-drug resistant strains [46,47]. The FDA has approved Telavancin for treating cSSSI in adult patients caused by susceptible isolates of the following Gram-positive microorganisms: S. aureus (including methicillin-susceptible and -resistant isolates) [20].
2.9. Fusidic acid
Fusidic acid is commonly used topically to treat S. aureus skin infections and to reduce skin colonization in eczema patients. However, when the drug is used alone, resistance rapidly emerges by mutation due to the high selection pressure. A combination of rifampicin and fusidic acid effectively combats difficult-to-treat MRSA infections and is a useful alternative to linezolid. It is concerning that widespread topical usage could compromise the drug effectivity [27]. Fusidic acid targets elongation factor G, a cytoplasmic protein that binds to the ribosome after peptide bond formation and transformation of the growing peptide chain to the aa-tRNA at the A site [28]. Normally, the EF-G GTPase promotes translocation of the ribosome, such that the peptidyl-tRNA returns to the P site, and the deacylated tRNA is expelled from the E site. EF-G then detaches, allowing the next round of translation to begin with an aa-tRNA molecule entering the vacant A site. The presence of fusidic acid prevents the release of EF-G after the translocation event and blocks the next round of aa-tRNA binding [48]. There has been a recent increase in the reported resistance to fusidic acid, mainly involving plasmids expressing the FusB and FusC mechanisms. While the fusB gene is plasmid-encoded, fusC is located within SCC elements (SCCfus and SCCmec) and linked to the TirS gene, which encodes the TirS protein that inhibits Toll-like receptor signaling and likely contributes to virulence [27,48].
Furthermore, fusC and tirS seem are co-expressed because TirS expression is elevated in subinhibitory concentrations of fusidic acid. FusA resistance is due to spontaneous chromosomal mutants affecting EF-G that prevents the drug from binding to its target [48]. Flubs is a small ∼25 kDa protein that binds EF-G. It binds to the EF-G fusidic acid complex on the stalled ribosome in the post-translocation state. This triggers a conformational change in EF-G, promoting its release and allowing translation to proceed [48].
2.10. Mupirocin
Mupirocin is another topical antibiotic commonly used to reduce nasal carriage of MRSA among hospital patients and staff, as well as to treat skin infections. It targets the isoleucyl-tRNA synthetase, an enzyme involved in protein synthesis. Resistance to mupirocin can occur at both low and high levels [49]. Low-level resistance is usually caused by mutations (amino acid substitutions) in the target-site, thus reducing the drug's binding efficiency. On the other hand, high-level resistance is typically associated with plasmid-borne MupA determinants that specify an intrinsically insensitive IleRS enzyme, which allows the sensitive target to bypass the drug's inhibitory action [15].
2.11. Aminoglycosides
Neomycin, an aminoglycoside, is frequently used topically to prevent or treat skin infections and as a nasal ointment for decolonizing carriers. Also, Gentamicin was introduced in the 1970s to address severe nosocomial infections caused by S. aureus, but its efficacy was hindered by the emergence of high-level resistance caused by mobile genetic elements [50]. Aminoglycosides are the only ribosome-targeting antibiotics that have bactericidal properties due to their unique mode of action that causes misreading during translation. Clinical strains of S. aureus develop resistance to aminoglycosides through the acquisition of cytoplasmic aminoglycoside-modifying enzymes (AMEs), the most common mechanism of resistance encoded by mobile genetic elements [50]. AMEs are classified into three groups, which render aminoglycosides inactive via acetylation (acetyltransferases; AAC), adenylation (nucleotidyltransferases; ANT), or phosphorylation (phosphotransferases; APH). Genes encoding these enzymes often exist on mobile elements such as plasmids or transposons [15]. Drug modification prevents ribosome binding. Tn4001 encodes a bi-functional acetyltransferase-phosphotransferase (aacA-aphD), which confers gentamicin and neomycin resistance. Neomycin resistance is caused by Tn5405-encoded phosphotransferase (aphA) or plasmid pUB110-encoded adenyltransferase (aadD). In some MRSA strains, the plasmid is incorporated within the SCCmecII cassette [15].
2.12. Fluoroquinolone
Fluoroquinolones are a highly successful class of entirely synthetic antimicrobial drugs. The breakthrough in their development came with the discovery that fluorination at position C6 could enhance the spectrum and potency of the prototype molecule nalidixic acid [51]. Since then, several generations of fluoroquinolones have been introduced with additional substitutions, leading to improved potency and pharmacokinetics of the drug.
In S. aureus, the molecular targets for fluoroquinolones include DNA gyrase and topoisomerase IV. DNA gyrase introduces negative supercoils into chromosomal DNA, while topoisomerase IV promotes chromosome decatenation following replication. Both enzymes are heterotetramers of A and B subunits, with GyrA and GyrB in DNA gyrase and ParC and ParE in topoisomerase IV. These enzymes catalyze staggered double-stranded breaks, with the 5' end of each cleaved DNA strand forming a phosphotyrosine bond with the active site tyrosine in the two A subunits. A DNA strand is then passed through the break, the gap is resealed, and the phosphodiester bond between the backbone deoxyriboses is regenerated [29].
The fluoroquinolones form ternary complexes with Mg2+ and the A subunit of the topoisomerases at the intermediate covalent protein-DNA complex stage. This prevents resealing of the cleaved DNA strand, leading to double-stranded DNA breaks that cannot be repaired and result in a rapid bactericidal effect [30]. S. aureus resistance to fluoroquinolones involves mutational changes to the topoisomerases that reduce drug binding efficiency and elevate the expression of endogenous efflux pumps [30]. The most common type of mutational change that leads to resistance to fluoroquinolones is due to topoisomerase mutants. This results in amino acid substitutions in residues that constitute the drug-binding site, also known as the quinolone resistance-determining region. Additionally, resistance to fluoroquinolones is mediated by overexpressing chromosomally encoded efflux pumps [98]. Three pumps can efflux fluoroquinolones: NorA handles the hydrophilic molecules norfloxacin and ciprofloxacin, while NorB and NorC target hydrophobic drugs such as sparfloxacin and moxifloxacin [52].
Nor proteins belong to the major facilitator superfamily and they are related to the Tet efflux pump. However, the increase in minimum inhibitory concentration is usually determined by the transporter, which depends on the pump's affinity for each particular fluoroquinolone. The expression of NorA, NorB, and NorC is governed by complex interactions between regulatory proteins, with the small transcriptional regulator MgrA being centrally involved [30]. Nor proteins can bind directly to the promoters of the nor genes depending on the degree of phosphorylation catalyzed by kinase PknB. For example, phosphorylated MgrA reduced binding to the NorA promoter and increased binding to norB [52].
2.13. Extracellular vesicles and S. aureus antibiotics resistance
Extracellular vesicles (EVs) are naturally secreted from bacteria, carrying specific cargo molecules from the cell of origin. They play significant roles in protecting bacterial cells and contribute to their survival in the presence of antibiotics [53]. EV secretion from MRSA increases significantly under antibiotic-stressed conditions. EVs released under these conditions were found to contain abundant proteins related to the degradation of β-lactam antibiotics. Therefore, EVs from MRSA can function as a first-line defense against β-lactam antibiotics, playing a crucial role in the survival of β-lactam susceptible bacteria. As a result, antibiotic stress leads to the release of EVs with high defense activity [53].
3. The current situation of MRSA treatment
Infections caused by MRSA strains have become a major global health concern due to their high morbidity and mortality rates [6]. Therefore, there is an urgent need for developing novel effective drugs to treat multi-drug resistant MRSA infections [14]. This has led to global investment that in turn accelerated scientific research, resulting in the discovery of several potentially effective anti-MRSA drugs [37]. Vancomycin, a glycopeptide that inhibits peptidoglycan biosynthesis in gram-positive bacteria by forming a strong hydrogen bond interaction with D-alanyl-D-alanine precursors of peptidoglycan, became the primary antibiotic for treating MRSA infections in hospital settings in the late 1980s [54]. Another effective anti-MRSA drug is daptomycin, a cyclic lipopeptide that was approved in 2003 for use in skin and soft tissue infections. It works by binding to the cell membranes of gram-positive bacteria [55]. Additionally, Mupirocin is an effective topical antibiotic used to treat bacterial skin infections that inhibits bacterial protein synthesis by interfering with isoleucyl transfer RNA synthase in gram-positive bacteria [32]. Furthermore, Linezolid is a recent drug used to treat MRSA infections and other drug-resistant bacterial infections by inhibiting protein biosynthesis after binding to 50S ribosomal RNA [31].
Moreover, several semi-synthetic drugs, such as Tigecycline, Oritavancin, Dalbavancin, Iclaprim, Cethromycin, and Delafloxacin, have shown better activity against MRSA by inhibiting peptidoglycan biosynthesis or protein synthesis. Ceftaroline, a novel cephalosporin with a high affinity for the modified penicillin-binding protein (PBP 2A) and thus has in vitro activity against MRSA, is a recent anti-MRSA antibiotic [15]. Recently, several studies are investigating the safety and antibacterial effectiveness of using Snake Venom Phospholipases A2 as an innovative approach for treating drug resistant bacteria including S. aureus [56]. Nonetheless, the availability of a broad portfolio of antibiotics against MRSA is illustrated in Figure 2.
4. Novel Therapeutic Strategies for MRSA Treatment:
The emergence of MRSA strains with intrinsic resistance to the current used antimicrobials, particularly MDR strains, has reduced the effectivity of these drugs and made infection control and case management more challenging. Thus, it is crucial to develop efficient strategies to combat antimicrobials drug resistance and to develop innovative drugs for control of MRSA-associated infections (See Figure 3 and Table 2) [13].
4.1. Quorum Sensing Inhibition
In S. aureus, the accessory gene regulator (agr) locus is a crucial part of the global virulence response. The operon is composed of two transcripts, RNAII and RNAIII. The RNAII encodes the constituents of the agr system itself, namely agrB, agrD, agrC, and agrA [59]. The agr system is a two-component signal transduction system (TCS) that uses cyclic thiolactone peptides as signaling molecules. agrD produces auto-inducing peptides (AIPs) as pro-peptides, which are then processed into active AIPs by agrB, a transmembrane endopeptidase. The AIPs are secreted and accumulate in the extracellular environment. Upon reaching a certain concentration, they bind to agrC, the sensor kinase of the TCS. agrC, which is phosphorylated in an AIP-dependent manner, activates agrA, the response regulator. Activated agrA binds to RNAIII, a regulatory mRNA that acts as the effector of the agr QS system [57]. The agr system in S. aureus controls the expression of 70 to 150 genes and is mainly involved in the invasiveness of S. aureus. It upregulates toxins and enzymes (proteases, nucleases, lipases, etc.), and almost all known virulence factors are upregulated by agr [57].
Hence, inhibiting the agr quorum sensing can significantly attenuate bacterial virulence factors and result in the inhibition of biofilm formation, which plays a major role in MRSA drug resistance [58].
4.2. Iron Chelator
Iron (Fe) ions are essential nutrients for most organisms. Research has revealed that Fe ions play a significant role in essential biological enzymes such as oxidoreductase, and are involved in various life processes including electron transport, antioxidant reactions, and the nucleic acid synthesis [58]. Additionally, Fe carriers are essential for controlling pathogenic microorganisms. Chelation of Fe can prevent pathogenic bacteria from using Fe, thereby inhibiting their growth and metabolic activity. Since human cells do not possess a pathway for iron carrier synthesis, their biosynthesis and absorption pathways can be leveraged for antimicrobial treatment [62].
Targeting iron acquisition represents a potential strategy to overcome MRSA drug resistance by introducing new anti-MRSA agents that may act independently or synergistically support other antibiotics to enhance their efficacy. Thus, compounds that affect iron acquisition have been recognized as potential candidates [62]. S. aureus is dependent on iron and must acquire this essential nutrient from its host during infection via molecules such as transferrin, lactoferrin, and heme. Therefore, the dependence of S. aureus on Fe makes it an excellent target for Fe-chelating agents that could interfere with the bacterium's Fe acquisition mechanisms [49,63].
4.3. NorA efflux pumpsinhibitors:
To overcome drug resistance, another promising strategy is the inhibition of efflux pumps; compounds that prevent antibiotics from being pumped out. NorA, the most important efflux pump in S. aureus, is implicated in drug extrusion and subsequently decreased intracellular drug concentration [64]. Inhibiting the action of NorA efflux pumps with compounds represents a good opportunity to manipulate the antimicrobial resistance [69]. These compounds alone are not growth inhibitors but act synergistically with antibiotics, this makes the use of efflux pump inhibitors (EPIs) an effective approach to decreasing the MIC of drug agents. There are many anti-EPIs compounds, most of which are non-antibiotic drugs [70]. An early example of efflux pump inhibition in S. aureus that demonstrates the potential of the plant alkaloid, a known inhibitor of mammalian MDR efflux pumps, which reduced the ciprofloxacin MIC. Besides reserpine, various other natural products have been shown to inhibit bacterial efflux pumps. It appears that a variety of Berberis medicinal plants that produce berberine, a cationic antimicrobial, also synthesize the flavone 5 methoxyhydnocarpin (5′-MHC, 24), an inhibitor of the NorA efflux pump of S. aureus. Without 5-MHC, S. aureus readily extrudes the plant-derived antimicrobial alkaloid.
Efflux pump inhibitors are not restricted to natural products [71]. For instance, the nonsteroidal anti-inflammatory drug (NSAID) celecoxib has recently been demonstrated to re-sensitize S. aureus to multiple antibiotics. Sabatini and co-workers confirmed the efflux pump inhibitor activity of the NSAID and identified a new class of analogs acting as inhibitors of the S. aureus NorA efflux pump. The most active inhibitor showed only modest anti-staphylococcal activity and was able to restore the antibacterial activity of ciprofloxacin in norA-overexpressing S. aureus strains [69,71].
4.4. ErmC' methyltransferase (ermCMTases) Inhibitors:
S. aureus develops cross-resistance to MLSB antibiotics by modifying the drug target enzymatically [17]. This modification occurs through methylation of the 23S rRNA at A2058 that creates a macrolide binding site that is no longer accessible to the antibiotics. The erythromycin ribosomal methyltransferase encoded by erm genes (such as ermC, ermE, ermA) is responsible for this conversion, using S-adenosyl-L-Methionine (SAM) as a methyl donor for the process [72]. The crystal structure of the rRNA methyltransferase of ErmC and ErmE has provided a model for drug design aimed at overcoming and preventing MLSB cross-resistance [73]. Therefore, identifying novel compound combinations with antibiotics that act as potential inhibitors of resistance mechanisms could be useful in inhibiting the methylation of SAM substrates and thus restore the activity of antibiotics against bacteria [74].
Several strategies for developing inhibitors have been proposed and developed. Enzymatic activity was initially used to screen and identify combinations with potential inhibitory activity [74]. Followed by nuclear magnetic resonance-based screening and chemical synthesis based on the enzymatic activities. This approach led to the discovery of novel compounds that bound to the binding sites of ErmC and ErmB, such as SAM, and demonstrated the potential to overcome resistance [18].
Virtual screening of structural compounds and in vitro potency characterization of the Erm proteins helped design inhibitors based on the minimal substrate features [74]. Although, the SAM binding site is highly conserved across different methyltransferase (MTases) families, different conformational aspects of ErmC could be exploited to design new inhibitors. Additionally, the conserved water structures observed in the ErmC-SAM binding site could be substituted, and structural mimics of the transition state could be used to enhance the potency [65].
Similarly, the recent explication of the functions of the tentative carbon-oxygen (CH-O) hydrogen bond that was found between the methyl group of SAM and oxygen from particular active site residue(s) contributes to high-affinity SAM binding, limiting the motion of the SAM methyl group to maintain the alignment of the methyl group for optimal transfer geometry, and transition state stabilization. It could provide two distinct strategies for the specific inhibitor development of MTases, like designing ligands to act as CH- O hydrogen bond acceptors instead of SAM or mimicking SAM CH -O hydrogen-bond donors using cofactor or transition state analogs [65].
5. Synthetic Antimicrobial Peptides against multidrug resistant MRSA
Antimicrobial peptides (AMPs) and their synthetic analogs are from an extensive group of compounds of great structural diversity and multifunctionality, different modes of antimicrobial action, and considerable market potential. That proven antibacterial, antifungal, and antiviral activities [66]. Recently, the functions of the carbon-oxygen (CH-O) hydrogen bond that was found between the methyl group of SAM and oxygen from particular active site residue(s) have been elucidated. This bond contributes to high-affinity SAM binding, limiting the motion of the SAM methyl group to maintain the alignment of the methyl group for optimal transfer geometry, and stabilizing the transition state. This knowledge opened the door for two distinct strategies for the specific inhibitor development of MTases. One strategy is to design ligands that act as CH-O hydrogen bond acceptors instead of SAM, while the other strategy involves mimicking SAM CH-O hydrogen-bond donors using cofactor or transition state analogs.
Antimicrobial peptides (AMPs) and their synthetic analogs are a diverse group of compounds with multifunctionality, different modes of action, and significant market potential, with proven antibacterial, antifungal, and antiviral activity [67]. AMPs have emerged as promising therapeutic agents for treating infections caused by multidrug-resistant bacteria including MRSA. One of the unique characteristics of AMPs is their broad-spectrum activity against all pathogens, with minimal chance for developing resistance [67,75]. Therefore, this review explores the potential of synthetic AMPs, WR12 and D-IK8, for use as topical antimicrobials in the treatment of SSTIs caused by S. aureus [76].
5.1. Nanoparticles and Nanomedicine
Metal nanoparticles possess multiple antimicrobial properties that may be useful for combating intracellular infections as these properties reduce the selection pressure for developing antibiotic resistance. For instance, silver nanoparticles (AgNPs) release Ag+ ions that can disrupt electron transport and trigger ROS production, causing harm to bacteria [77]. Growing evidence has demonstrated that these AgNPs can kill 100% of extracellular bacteria and 76% of intracellular S. aureus within osteoblasts, with intracellular killing rates similar to those of rifampicin treatment, whereas gentamicin treatment alone did not show any intracellular killing effect [77]. In a recent study, S. aureus bacteria were isolated and tested on culture plates with AgNP-discs, which showed effective growth inhibition for both MRSA and VRSA variants. If AgNPs' efficacy is demonstrated in vivo, they could become valuable tools for combating drug-resistant S. aureus infections [78].
The physicochemical characteristics of nanocarriers signify their bactericidal effect, which differs for every material. The nanosize and higher surface area of NPs enable better interactions with bacterial cells compared to typical antibiotics [79,80]. Furthermore, these nanocarriers can effectively overcome the bacterial cell envelope, which limits the internalization of antibacterial drugs. For example, NPs such as metal NPs, dendrimers, carbon nanotubes, chitosan (CS), antimicrobial peptides (AMPs), and cyclodextrin (CD) can directly penetrate the bacterial envelope, causing envelope disruption. However, some delivery systems such as the fusogenic liposomes system do not penetrate bacteria but deliver the drug intracellularly to the bacteria [79].
When designing carriers for the specific and tolerable release of antibiotics in treating bacteria, several aspects must be considered. These aspects include minimizing antibiotic interactions with healthy tissue, measuring the accumulation of antibiotics at the infection site, improving drugs’ efficacy, and lowering the toxicity risks and vulnerability of commensal microflora to the antibiotic. One approach is to develop a stimuli-responsive system that can either react in a self-motivated way and recognize the bacterial microenvironment or is responsive to certain physical factors [81]. This approach is a promising strategy for improving antibiotic delivery systems, enhancing their efficiency, targeting properties, and effectiveness of antibiotics while minimizing the side effects [82]. Additionally, NPs can enter cells via endocytosis, a mechanism that can treat intracellular microbial infections. Several studies have shown that different types of NPs can overcome bacterial resistance to antibiotics by escaping from drug degradation enzymes such as β-lactamase, inhibiting efflux pumps, or penetrating thick cell walls. Several NP-based drug delivery systems designed for MRSA treatment are either candidates for clinical trials or have recently been approved [80]. In Figure 4, we demonstrate the potential usefulness of Synthetic Antimicrobial Peptides against multidrug resistant MRSA.
5.2. Inhibition of Teichoic acid biosynthesis
Most chronic MRSA infections are characterized by the formation of biofilms, in which group of bacterial pathogens including MRSA are attached to biological or non-biological surfaces such as medical devices. Biofilm bacteria are much more resistant to antibiotics than planktonic bacteria and more challenging to treat. Current treatments involve long-term antibiotic therapy, which can lead to increased tissue damage and persistence of the infection. Therefore, new agents that can eradicate or inhibit MRSA biofilm formation via novel mechanisms are needed [81].
Teichoic acid (TA) is a glycopolymer that represents a significant component of S. aureus cell walls. TAs consist of polyribitol-phosphate polymers cross-linked to MurNAc residues of peptidoglycan and covered with D-alanine and GlcNAc residues [77]. There are two types of TA: wall teichoic acid (WTA) and lipoteichoic acid (LTA) [77]. LTA is mainly anchored in the plasma membrane, while WTA is covalently linked to peptidoglycan. Although, WTA and LTA have similar structures, they are synthesized by different pathways, with WTA being a ribitol phosphate polymer and LTA being composed of glycerol phosphate [68]. Therefore, enzymes involved in biosynthesis and export could be effective drug targets. Several WTA biosynthesis inhibitors have been reported. For example, Targocil inhibits the membrane-bound ABC transporter, which exports WTA across the membrane. When used in combination with a β-lactam, a synergistic effect was observed, restoring the efficacy of the β-lactam towards MRSA [68,77].
Assessing potential antimicrobial strategies involves considering several factors, such as the ability to target specific bacterial infections, minimize harm to healthy tissue, improve drug efficacy, and reduce toxicity. One promising approach involves developing a stimuli-responsive drug delivery system that can react to the bacterial microenvironment or certain physical factors to enhance targeting properties and effectiveness while minimizing side effects [81].
5.3. Inhibition of S. aureus Aminoacyl-tRNA Synthetases (aaRSs)
Aminoacyl-tRNA synthetase (AaRS) enzymes have shown potential antimicrobial benefits because they play crucial roles in protein biosynthesis. Once these enzymes are inhibited, protein biosynthesis is halted, leading to inhibition of bacterial growth [77]. Recent studies suggest that AaRS could be used as therapeutic targets, and the loss of fitness associated with resistance could be compatible with antimicrobial restriction policies aimed at eliminating resistance [83].
AaRS enzymes meet several criteria defined by the pharmaceutical industry, making them ideal targets for antibacterial drug discovery. Although these enzymes are universally distributed in cell organisms, there has been evolutionary divergence between prokaryotic and eukaryotic enzymes, and these differences can be exploited to develop antibiotics that selectively inhibit bacterial enzymes. Moreover, AaRS enzymes are highly conserved across different bacterial pathogens, increasing the chances of making some discoveries of broad-spectrum antibiotics [83]. Their solubility, stability, and ease of purification in significant quantities by recombinant expression systems can be tested using one or more conventional methods amenable to high-throughput screening [84] (Figure 5).
6. Antibacterial Properties of Snake and Scorpion Venom: Potential for New Antimicrobial Agents
Snake and scorpion venoms are intricate mixtures of proteins, enzymes, and peptides that are primarily utilized for defense and capturing prey. Nonetheless, studies have indicated that some of the venom components possess antimicrobial activity. For instance, crotalicidin, a peptide extracted from the venom of South American rattlesnakes, has been demonstrated to be effective against a broad range of bacteria, including MRSA [85]. Similarly, cathelicidin-BF, a peptide present in the venom of Chinese cobras, has been found to have potent antibacterial activity against several bacteria, including multidrug-resistant strains [86].
Additionally, Pandinin 2, a peptide derived from the venom of the African scorpion Pandinus imperator, has demonstrated activity against both Gram-positive and Gram-negative bacteria [87]. Another peptide, IsCT, obtained from the venom of the North African scorpion Scorpio maurus, has also shown to have strong activity against various bacteria, including MRSA [88].
Although the antibacterial properties of snake and scorpion venom provide a promising avenue for the development of new antimicrobial agents, their toxicity should not be overlooked. Consequently, ongoing research is focused on identifying and isolating the most effective peptides for use in medical applications [56].
7. Antibacterial Properties of Snake and Scorpion Venom: Potential for New Antimicrobial Agents
In recent years, there has been a growing interest in developing vaccines for the prevention and control of MRSA infections [89]. The strategy of vaccination is to stimulate the body's immune system to recognize and destroy the pathogen, thus preventing the development of an infection [90]. Currently, there are no licensed vaccines available for MRSA, but many potential candidates are under development. One promising approach is to target the capsule polysaccharide of MRSA, which is a major virulence factor and plays a crucial role in the pathogenesis of the infection [89]. Several vaccine candidates have been developed to target this polysaccharide, such as conjugate vaccines and glycoconjugate vaccines, which link the polysaccharide to a carrier protein or a lipid molecule to enhance the immune response, respectively[89].
Another approach is to target specific surface proteins of MRSA, such as the clumping factor A (ClfA) and iron-regulated surface determinant B (IsdB), which are vital in adhesion and invasion of host cells. These proteins have shown promising results in preclinical studies [91,92].
The feasibility and potential use of an MRSA vaccine for the prevention and control of MRSA infections are significant. An effective vaccine would provide an essential tool in combating this highly resistant pathogen, potentially reducing the incidence of infections and the need for antibiotic treatment. Additionally, vaccination could assist in preventing the spread of MRSA in healthcare settings, where the risk of transmission is high. However, there are several challenges that need to be addressed in developing an MRSA vaccine, such as the high genetic variability of the organism and the requirement for effective adjuvants to enhance the immune response. Despite these challenges, the development of an effective MRSA vaccine remains a crucial goal in the fight against antibiotic-resistant bacteria.
8. Concluding Remarks
The emergence of multi-drug resistant S. aureus infections, particularly MRSA is serious threat to the global health that poses a significant challenge to the control of infections, particularly during the COVID-19 pandemic. It is crucial to make significant efforts to keep up with the development of antibiotic resistance and devise new strategies to manipulate the drug resistance mechanism and develop novel drugs to combat MRSA infections. Advances in microbial genomics, X-ray crystallography, and cryo-electron microscopy have provided promising avenues for the discovery of new antibacterial targets and the design and repurposing of antimicrobial compounds from existing antibiotics.
However, to effectively combat MRSA, it is essential to gain a deeper understanding of the molecular mechanisms of drug resistance. This understanding can be obtained by exploring the genetic and metabolic pathways involved in the development of drug resistance in MRSA. Moreover, investigating the bacterial cell's structural and functional features can provide crucial insights into the development of novel drugs and therapeutic strategies. In summary, the emergence of MRSA infections is a significant public health concern that demands urgent attention. It is essential to develop innovative strategies to combat this pathogen's drug resistance and discover new antibiotics to control MRSA infections including vaccination. Comprehensive molecular and structural investigations are critical to achieving these goals.
Author Contributions
N.M.H., A.E.A., A.A., and N.S.M.; conceptualization, N.M.H., S.I.M., A.E.A., A.M.B., A.E.A., M.S.E.A., A.A., and N.S.M.; writing—original draft preparation. A.A., R.A.O., E.E.S., A.A., writing—review and editing. N.M.H., A.A.; visualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schwendener, S.; Perreten, V. New MLSB Resistance Gene Erm (43) in Staphylococcus Lentus. Antimicrobial agents and chemotherapy 2012, 56, 4746–4752. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Srivastava, P.; Nirwan, P.S. Prevalence of MLSB Resistance and Observation of Erm A & Erm C Genes at A Tertiary Care Hospital. Journal of clinical and diagnostic research: JCDR 2015, 9, DC08. [Google Scholar] [PubMed]
- Mestrovic, T.; Aguilar, G.R.; Swetschinski, L.R.; Ikuta, K.S.; Gray, A.P.; Weaver, N.D.; Han, C.; Wool, E.E.; Hayoon, A.G.; Hay, S.I. The Burden of Bacterial Antimicrobial Resistance in the WHO European Region in 2019: A Cross-Country Systematic Analysis. The Lancet Public Health 2022, 7, e897–e913. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.; Yeh, A.J.; Cheung, G.Y.; Otto, M. Investigational Drugs to Treat Methicillin-Resistant Staphylococcus Aureus. Expert opinion on investigational drugs 2016, 25, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Davis, J.S. Eichenberger. E., Holland, TL, Fowler, VG Jr. Staphylococcus Aureus Infections: Epidemiology, Patho-Physiology, Clinical Manifestations, and Management. Clin Microbiol Rev 2015, 28, 603–661. [Google Scholar] [PubMed]
- Greer, N.D. Tigecycline (Tygacil): The First in the Glycylcycline Class of Antibiotics.; Taylor & Francis, 2006; Vol. 19, pp. 155–161.
- DiMondi, V.P.; Drew, R.H.; Chen, L.F. Ceftaroline Fosamil for Treatment of Communityacquired Pneumonia: Findings from FOCUS 1 and 2 and Potential Role in Therapy. Expert Review of Anti-infective Therapy 2011, 9, 567–572. [Google Scholar] [CrossRef]
- Leclercq, R.; Courvalin, P. Intrinsic and Unusual Resistance to Macrolide, Lincosamide, and Streptogramin Antibiotics in Bacteria. Antimicrobial agents and chemotherapy 1991, 35, 1273–1276. [Google Scholar] [CrossRef]
- Roberts, M.C.; Sutcliffe, J.; Courvalin, P.; Jensen, L.B.; Rood, J.; Seppala, H. Nomenclature for Macrolide and Macrolide-Lincosamide-Streptogramin B Resistance Determinants. Antimicrobial agents and chemotherapy 1999, 43, 2823–2830. [Google Scholar] [CrossRef]
- Li, L.; Feng, W.; Zhang, Z.; Xue, H.; Zhao, X. Macrolide-Lincosamide-Streptogramin Resistance Phenotypes and Genotypes of Coagulase-Positive Staphylococcus Aureus and Coagulase-Negative Staphylococcal Isolates from Bovine Mastitis. BMC Veterinary Research 2015, 11, 1–8. [Google Scholar] [CrossRef]
- Hall, B.G. Predicting the Evolution of Antibiotic Resistance Genes. Nature Reviews Microbiology 2004, 2, 430–435. [Google Scholar] [CrossRef]
- Jelić, D.; Antolović, R. From Erythromycin to Azithromycin and New Potential Ribosome-Binding Antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.; Ross, J.I.; Cove, J.H. Msr (A) and Related Macrolide/Streptogramin Resistance Determinants: Incomplete Transporters? International journal of antimicrobial agents 2003, 22, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Song, G.; Sun, M.; Wang, J.; Wang, Y. Prevalence and Therapies of Antibiotic-Resistance in Staphylococcus Aureus. Frontiers in cellular and infection microbiology 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Antibiotic Resistance in Staphylococcus Aureus. Current Status and Future Prospects. FEMS microbiology reviews 2017, 41, 430–449. [Google Scholar] [CrossRef]
- Champney, W.S.; Tober, C.L. Specific Inhibition of 50S Ribosomal Subunit Formation in Staphylococcus Aureus Cells by 16-Membered Macrolide, Lincosamide, and Streptogramin B Antibiotics. Current microbiology 2000, 41, 126–135. [Google Scholar] [CrossRef]
- Abdalla, A.E.; Kabashi, A.B.; Elobaid, M.E.; Hajhmed, N.M.H.; Modawyi, W.A.; Alameen, A.A.M.; Abosalif, K.O.A.; Ejaz, H. Methicillin and Inducible Clindamycin-Resistant Staphylococcus Aureus Isolated from Postoperative Wound Samples. Journal of Pure and Applied Microbiology 2019, 13, 1605–1609. [Google Scholar] [CrossRef]
- Sundlov, J.A.; Gulick, A.M. Insights into Resistance against Lincosamide Antibiotics. Structure 2009, 17, 1549–1550. [Google Scholar] [CrossRef]
- Duran, N.; Ozer, B.; Duran, G.G.; Onlen, Y.; Demir, C. Antibiotic Resistance Genes & Susceptibility Patterns in Staphylococci. The Indian journal of medical research 2012, 135, 389. [Google Scholar]
- Krause, K.M.; Renelli, M.; Difuntorum, S.; Wu, T.X.; Debabov, D.V.; Benton, B.M. In Vitro Activity of Telavancin against Resistant Gram-Positive Bacteria. Antimicrobial agents and chemotherapy 2008, 52, 2647–2652. [Google Scholar] [CrossRef]
- Locke, J.B.; Hilgers, M.; Shaw, K.J. Mutations in Ribosomal Protein L3 Are Associated with Oxazolidinone Resistance in Staphylococci of Clinical Origin. Antimicrobial agents and chemotherapy 2009, 53, 5275–5278. [Google Scholar] [CrossRef]
- Long, S.W.; Olsen, R.J.; Mehta, S.C.; Palzkill, T.; Cernoch, P.L.; Perez, K.K.; Musick, W.L.; Rosato, A.E.; Musser, J.M. PBP2a Mutations Causing High-Level Ceftaroline Resistance in Clinical Methicillin-Resistant Staphylococcus Aureus Isolates. Antimicrobial agents and chemotherapy 2014, 58, 6668–6674. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.M.; Peschel, A. Broad-spectrum Antimicrobial Peptide Resistance by MprF-mediated Aminoacylation and Flipping of Phospholipids. Molecular microbiology 2011, 80, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Beabout, K.; Hammerstrom, T.G.; Perez, A.M.; Magalhães, B.F.; Prater, A.G.; Clements, T.P.; Arias, C.A.; Saxer, G.; Shamoo, Y. The Ribosomal S10 Protein Is a General Target for Decreased Tigecycline Susceptibility. Antimicrobial agents and chemotherapy 2015, 59, 5561–5566. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.M.; Ba, X.; Blane, B.; Ellington, M.J.; Loeffler, A.; Hill, R.L.; Holmes, M.A.; Peacock, S.J. PBP2a Substitutions Linked to Ceftaroline Resistance in MRSA Isolates from the UK. Journal of Antimicrobial Chemotherapy 2016, 71, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.; Chiu, C.; Greninger, A.; Chatterjee, S.; Chan, L.; Hamilton, S.; Chambers, H.; Chiu, C. Whole-Genome Sequencing of Methicillin-Resistant Staphylococcus Aureus Resistant to Fifth-Generation Cephalosporins Reveals Potential Non-MecA Mechanisms of Resistance. 2016.
- Fernandes, P. Fusidic Acid: A Bacterial Elongation Factor Inhibitor for the Oral Treatment of Acute and Chronic Staphylococcal Infections. Cold Spring Harbor perspectives in medicine 2016, 6, a025437. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.J.; Chopra, I. Molecular Basis of FusB-mediated Resistance to Fusidic Acid in Staphylococcus Aureus. Molecular microbiology 2006, 59, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of Drug Resistance: Quinolone Resistance. Annals of the New York academy of sciences 2015, 1354, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Lekshmi, M.; Ammini, P.; Adjei, J.; Sanford, L.M.; Shrestha, U.; Kumar, S.; Varela, M.F. Modulation of Antimicrobial Efflux Pumps of the Major Facilitator Superfamily in Staphylococcus Aureus. AIMS microbiology 2018, 4, 1. [Google Scholar] [CrossRef]
- Qiu, P.-L.; Liu, S.-Y.; Bradshaw, M.; Rooney-Latham, S.; Takamatsu, S.; Bulgakov, T.S.; Tang, S.-R.; Feng, J.; Jin, D.-N.; Aroge, T. Multi-Locus Phylogeny and Taxonomy of an Unresolved, Heterogeneous Species Complex within the Genus Golovinomyces (Ascomycota, Erysiphales), Including G. Ambrosiae, G. Circumfusus and G. Spadiceus. BMC microbiology 2020, 20, 1–16. [Google Scholar] [CrossRef]
- Mendes, R.E.; Deshpande, L.M.; Castanheira, M.; DiPersio, J.; Saubolle, M.A.; Jones, R.N. First Report of Cfr-Mediated Resistance to Linezolid in Human Staphylococcal Clinical Isolates Recovered in the United States. Antimicrobial agents and chemotherapy 2008, 52, 2244–2246. [Google Scholar] [CrossRef]
- Gutmann, L.; Billot-Klein, D.; Al-Obeid, S.; Klare, I.; Francoual, S.; Collatz, E.; Van Heijenoort, J. Inducible Carboxypeptidase Activity in Vancomycin-Resistant Enterococci. Antimicrobial agents and chemotherapy 1992, 36, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Saderi, H.; Emadi, B.; Owlia, P. Phenotypic and Genotypic Study of Macrolide, Lincosamide and Streptogramin B (MLSB) Resistance in Clinical Isolates of Staphylococcus Aureus in Tehran, Iran. Medical science monitor: international medical journal of experimental and clinical research 2011, 17, BR48. [Google Scholar] [CrossRef]
- Sazdanovic, P.; Jankovic, S.M.; Kostic, M.; Dimitrijevic, A.; Stefanovic, S. Pharmacokinetics of Linezolid in Critically Ill Patients. Expert Opinion on Drug Metabolism & Toxicology 2016, 12, 595–600. [Google Scholar]
- Long, K.S.; Vester, B. Resistance to Linezolid Caused by Modifications at Its Binding Site on the Ribosome. Antimicrobial agents and chemotherapy 2012, 56, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.B.; Zurenko, G.E.; Shaw, K.J.; Bartizal, K. Tedizolid for the Management of Human Infections: In Vitro Characteristics. Clinical infectious diseases 2014, 58, S35–S42. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.; Kiratisin, P.; Mendes, R.E.; Panesso, D.; Singh, K.V.; Arias, C.A. Transferable Plasmid-Mediated Resistance to Linezolid Due to Cfr in a Human Clinical Isolate of Enterococcus Faecalis. Antimicrobial agents and chemotherapy 2012, 56, 3917–3922. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, Y.; Cai, J.; Schwarz, S.; Cui, L.; Hu, Z.; Zhang, R.; Li, J.; Zhao, Q.; He, T. A Novel Gene, OptrA, That Confers Transferable Resistance to Oxazolidinones and Phenicols and Its Presence in Enterococcus Faecalis and Enterococcus Faecium of Human and Animal Origin. Journal of Antimicrobial Chemotherapy 2015, 70, 2182–2190. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus Aureus. Annual review of biochemistry 2015, 84, 577–601. [Google Scholar] [CrossRef]
- Strommenger, B.; Layer, F.; Klare, I.; Werner, G. Pre-Use Susceptibility to Ceftaroline in Clinical Staphylococcus Aureus Isolates from Germany: Is There a Non-Susceptible Pool to Be Selected? PLoS One 2015, 10, e0125864. [Google Scholar] [CrossRef]
- Zhong, N.S.; Sun, T.; Zhuo, C.; D’Souza, G.; Lee, S.H.; Lan, N.H.; Chiang, C.-H.; Wilson, D.; Sun, F.; Iaconis, J. Ceftaroline Fosamil versus Ceftriaxone for the Treatment of Asian Patients with Community-Acquired Pneumonia: A Randomised, Controlled, Double-Blind, Phase 3, Non-Inferiority with Nested Superiority Trial. The Lancet Infectious Diseases 2015, 15, 161–171. [Google Scholar] [CrossRef]
- Kelley, W.L.; Jousselin, A.; Barras, C.; Lelong, E.; Renzoni, A. Missense Mutations in PBP2A Affecting Ceftaroline Susceptibility Detected in Epidemic Hospital-Acquired Methicillin-Resistant Staphylococcus Aureus Clonotypes ST228 and ST247 in Western Switzerland Archived since 1998. Antimicrobial Agents and Chemotherapy 2015, 59, 1922–1930. [Google Scholar] [CrossRef]
- Arbeit, R.D.; Maki, D.; Tally, F.P.; Campanaro, E.; Eisenstein, B.I. ; Daptomycin 98-01 and 99-01 Investigators The Safety and Efficacy of Daptomycin for the Treatment of Complicated Skin and Skin-Structure Infections. Clinical Infectious Diseases 2004, 38, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.-J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The Bacterial Defensin Resistance Protein MprF Consists of Separable Domains for Lipid Lysinylation and Antimicrobial Peptide Repulsion. PLoS pathogens 2009, 5, e1000660. [Google Scholar] [CrossRef]
- Higgins, D.L.; Chang, R.; Debabov, D.V.; Leung, J.; Wu, T.; Krause, K.M.; Sandvik, E.; Hubbard, J.M.; Kaniga, K.; Schmidt Jr, D.E. Telavancin, a Multifunctional Lipoglycopeptide, Disrupts Both Cell Wall Synthesis and Cell Membrane Integrity in Methicillin-Resistant Staphylococcus Aureus. Antimicrobial agents and chemotherapy 2005, 49, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Lunde, C.S.; Hartouni, S.R.; Janc, J.W.; Mammen, M.; Humphrey, P.P.; Benton, B.M. Telavancin Disrupts the Functional Integrity of the Bacterial Membrane through Targeted Interaction with the Cell Wall Precursor Lipid II. Antimicrobial agents and chemotherapy 2009, 53, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Thompson, G.S.; Jenkins, H.T.; Peske, F.; Savelsbergh, A.; Rodnina, M.V.; Wintermeyer, W.; Homans, S.W.; Edwards, T.A.; O’Neill, A.J. Ribosome Clearance by FusB-Type Proteins Mediates Resistance to the Antibiotic Fusidic Acid. Proceedings of the National Academy of Sciences 2012, 109, 2102–2107. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Hothersall, J.; Willis, C.L.; Simpson, T.J. Resistance to and Synthesis of the Antibiotic Mupirocin. Nature Reviews Microbiology 2010, 8, 281–289. [Google Scholar] [CrossRef]
- Serio, A.W.; Keepers, T.; Andrews, L.; Krause, K.M. Aminoglycoside Revival: Review of a Historically Important Class of Antimicrobials Undergoing Rejuvenation. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Truong-Bolduc, Q.; Dunman, P.; Strahilevitz, J.; Projan, S.; Hooper, D. MgrA Is a Multiple Regulator of Two New Efflux Pumps in Staphylococcus Aureus. Journal of bacteriology 2005, 187, 2395–2405. [Google Scholar] [CrossRef]
- Zárate, S.G.; Morales, P.; Świderek, K.; Bolanos-Garcia, V.M.; Bastida, A. A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters. Antibiotics 2019, 8, 25. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, J.S.; Park, S.B.; Lee, A.R.; Jung, J.W.; Chun, J.H.; Lazarte, J.M.S.; Kim, J.; Seo, J.-S.; Kim, J.-H. The Importance of Porins and β-Lactamase in Outer Membrane Vesicles on the Hydrolysis of β-Lactam Antibiotics. International journal of molecular sciences 2020, 21, 2822. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, W.; Ni, M.; Liu, Y.; Zhang, J.; Xia, F.; He, W.; Wang, Q.; Wang, Z.; Cao, B. Linezolid-Resistant Clinical Isolates of Enterococci and Staphylococcus Cohnii from a Multicentre Study in China: Molecular Epidemiology and Resistance Mechanisms. International journal of antimicrobial agents 2013, 42, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.; Xiong, L.; Arias, C.A.; Villegas, M.V.; Lolans, K.; Quinn, J.; Mankin, A.S. Acquisition of a Natural Resistance Gene Renders a Clinical Strain of Methicillin-resistant Staphylococcus Aureus Resistant to the Synthetic Antibiotic Linezolid. Molecular microbiology 2007, 64, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, Z.U.; Musa, S.S.; Abu-Odah, H.; Ahmed, A.; Lawan, A.A.; Bello, U.M. Bactericidal Effects of Snake Venom Phospholipases A2: A Systematic Review and Analysis of Minimum Inhibitory Concentration. Physiologia 2023, 3, 30–42. [Google Scholar] [CrossRef]
- Pérez-Pérez, M.; Jorge, P.; Pérez Rodríguez, G.; Pereira, M.O.; Lourenço, A. Quorum Sensing Inhibition in Pseudomonas Aeruginosa Biofilms: New Insights through Network Mining. Biofouling 2017, 33, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.P.; Williams, P.; Chan, W.C. Attenuating Staphylococcus Aureus Virulence Gene Regulation: A Medicinal Chemistry Perspective. Journal of medicinal chemistry 2013, 56, 1389–1404. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Y.; Ge, Y.; Zhu, X.; Pan, J. Regulatory Mechanisms and Promising Applications of Quorum Sensing-Inhibiting Agents in Control of Bacterial Biofilm Formation. Frontiers in microbiology 2020, 11, 589640. [Google Scholar] [CrossRef]
- Yin, S.-L.; Chang, Y.-J.; Deng, S.-P.; Wang, Q.-C.; Yu, W.-G.; Gong, Q.-H. Research Progress of New Antibacterial Drugs That Target Bacterial Quorum Sensing Systems. Yao xue xue bao= Acta Pharmaceutica Sinica 2011, 46, 613–621. [Google Scholar]
- Kalia, V.C.; Purohit, H.J. Quenching the Quorum Sensing System: Potential Antibacterial Drug Targets. Critical reviews in microbiology 2011, 37, 121–140. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends in biochemical sciences 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Parquet, M. del C.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B.E. Novel Iron-Chelator DIBI Inhibits Staphylococcus Aureus Growth, Suppresses Experimental MRSA Infection in Mice and Enhances the Activities of Diverse Antibiotics in Vitro. Frontiers in Microbiology 2018, 9, 1811. [Google Scholar] [CrossRef] [PubMed]
- Markham, P.N.; Westhaus, E.; Klyachko, K.; Johnson, M.E.; Neyfakh, A.A. Multiple Novel Inhibitors of the NorA Multidrug Transporter of Staphylococcus Aureus. Antimicrobial agents and chemotherapy 1999, 43, 2404–2408. [Google Scholar] [CrossRef] [PubMed]
- Foik, I.P.; Tuszynska, I.; Feder, M.; Purta, E.; Stefaniak, F.; Bujnicki, J.M. Novel Inhibitors of the RRNA ErmC’methyltransferase to Block Resistance to Macrolides, Lincosamides, Streptogramine B Antibiotics. European Journal of Medicinal Chemistry 2018, 146, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of Short Synthetic Antimicrobial Peptides for Treatment of Drug-Resistant and Intracellular Staphylococcus Aureus. Scientific reports 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Aristizábal, I.; Ocampo-Ibáñez, I.D. Antimicrobial Peptides with Antibacterial Activity against Vancomycin-Resistant Staphylococcus Aureus Strains: Classification, Structures, and Mechanisms of Action. International journal of molecular sciences 2021, 22, 7927. [Google Scholar] [CrossRef]
- Naclerio, G.A.; Onyedibe, K.I.; Sintim, H.O. Lipoteichoic Acid Biosynthesis Inhibitors as Potent Inhibitors of S. Aureus and E. Faecalis Growth and Biofilm Formation. Molecules 2020, 25, 2277. [Google Scholar] [CrossRef]
- Kaatz, G.W.; Seo, S.M.; O’Brien, L.; Wahiduzzaman, M.; Foster, T.J. Evidence for the Existence of a Multidrug Efflux Transporter Distinct from NorA in Staphylococcus Aureus. Antimicrobial agents and chemotherapy 2000, 44, 1404–1406. [Google Scholar] [CrossRef]
- Schaenzer, A.J.; Wright, G.D. Antibiotic Resistance by Enzymatic Modification of Antibiotic Targets. Trends in Molecular Medicine 2020, 26, 768–782. [Google Scholar] [CrossRef]
- Astolfi, A.; Felicetti, T.; Iraci, N.; Manfroni, G.; Massari, S.; Pietrella, D.; Tabarrini, O.; Kaatz, G.W.; Barreca, M.L.; Sabatini, S. Pharmacophore-Based Repositioning of Approved Drugs as Novel Staphylococcus Aureus NorA Efflux Pump Inhibitors. Journal of medicinal chemistry 2017, 60, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jhang, S.T.; Jin, H.J. Potential Target Site for Inhibitors in MLSB Antibiotic Resistance. Antibiotics 2021, 10, 264. [Google Scholar] [CrossRef]
- Stsiapanava, A.; Selmer, M. Crystal Structure of ErmE-23S RRNA Methyltransferase in Macrolide Resistance. Scientific reports 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schluckebier, G.; Zhong, P.; Stewart, K.D.; Kavanaugh, T.J.; Abad-Zapatero, C. The 2.2 Å Structure of the RRNA Methyltransferase ErmC′ and Its Complexes with Cofactor and Cofactor Analogs: Implications for the Reaction Mechanism. Journal of molecular biology 1999, 289, 277–291. [Google Scholar] [CrossRef]
- Lesiuk, M.; Paduszyńska, M.; Greber, K.E. Synthetic Antimicrobial Immunomodulatory Peptides: Ongoing Studies and Clinical Trials. Antibiotics 2022, 11, 1062. [Google Scholar] [CrossRef]
- Nuti, R.; Goud, N.S.; Saraswati, A.P.; Alvala, R.; Alvala, M. Antimicrobial Peptides: A Promising Therapeutic Strategy in Tackling Antimicrobial Resistance. Current medicinal chemistry 2017, 24, 4303–4314. [Google Scholar] [CrossRef] [PubMed]
- Lade, H.; Kim, J.-S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus Aureus. Antibiotics 2021, 10, 398. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ye, W.; Qi, Y.; Ying, Y.; Xia, Z. Overcoming Multidrug Resistance in Bacteria through Antibiotics Delivery in Surface-Engineered Nano-Cargos: Recent Developments for Future Nano-Antibiotics. Frontiers in Bioengineering and Biotechnology 2021, 569. [Google Scholar] [CrossRef] [PubMed]
- Pelgrift, R.Y.; Friedman, A.J. Nanotechnology as a Therapeutic Tool to Combat Microbial Resistance. Advanced drug delivery reviews 2013, 65, 1803–1815. [Google Scholar] [CrossRef]
- Vermote, A.; Van Calenbergh, S. Small-Molecule Potentiators for Conventional Antibiotics against Staphylococcus Aureus. ACS infectious diseases 2017, 3, 780–796. [Google Scholar] [CrossRef]
- Vanamala, K.; Tatiparti, K.; Bhise, K.; Sau, S.; Scheetz, M.H.; Rybak, M.J.; Andes, D.; Iyer, A.K. Novel Approaches for the Treatment of Methicillin-Resistant Staphylococcus Aureus: Using Nanoparticles to Overcome Multidrug Resistance. Drug discovery today 2021, 26, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Masimen, M.A.A.; Harun, N.A.; Maulidiani, M.; Ismail, W.I.W. Overcoming Methicillin-Resistance Staphylococcus Aureus (MRSA) Using Antimicrobial Peptides-Silver Nanoparticles. Antibiotics 2022, 11, 951. [Google Scholar] [CrossRef]
- Elbaramawi, S.S.; Ibrahim, S.M.; Lashine, E.-S.M.; El-Sadek, M.E.; Mantzourani, E.; Simons, C. Exploring the Binding Sites of Staphylococcus Aureus Phenylalanine TRNA Synthetase: A Homology Model Approach. Journal of Molecular Graphics and Modelling 2017, 73, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Weeks, S.D.; Van Aerschot, A. Aminoacyl-TRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. International Journal of Molecular Sciences 2021, 22, 1750. [Google Scholar] [CrossRef]
- Falcao, C.B.; Radis-Baptista, G. Crotamine and Crotalicidin, Membrane Active Peptides from Crotalus Durissus Terrificus Rattlesnake Venom, and Their Structurally-Minimized Fragments for Applications in Medicine and Biotechnology. Peptides 2020, 126, 170234. [Google Scholar] [CrossRef] [PubMed]
- Rádis-Baptista, G.; Gopalakrishnakoneal, P.; Inagaki, H.; Mukherjee, A.; Rahmy Carl-Wilhelm Vogel, T. Vipericidins, Snake Venom Cathelicidin-Related Peptides, in the Milieu of Reptilian Antimicrobial Polypeptides. Snake Venoms 2015, 1–25. [Google Scholar]
- CORZO, G.; ESCOUBAS, P.; VILLEGAS, E.; BARNHAM, K.J.; HE, W.; NORTON, R.S.; NAKAJIMA, T. Characterization of Unique Amphipathic Antimicrobial Peptides from Venom of the Scorpion Pandinus Imperator. Biochemical journal 2001, 359, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.L.; Abdel-Rahman, M.A.; Strong, P.N.; Tawfik, M.M.; Miller, K. Characterisation of Three Alpha-Helical Antimicrobial Peptides from the Venom of Scorpio Maurus Palmatus. Toxicon 2016, 117, 30–36. [Google Scholar] [CrossRef]
- Giersing, B.K.; Dastgheyb, S.S.; Modjarrad, K.; Moorthy, V. Status of Vaccine Research and Development of Vaccines for Staphylococcus Aureus. Vaccine 2016, 34, 2962–2966. [Google Scholar] [CrossRef]
- Byrne, F.; Wilcox, M. MRSA Prevention Strategies and Current Guidelines. Injury 2011, 42, S3–S6. [Google Scholar] [CrossRef]
- Hu, D.-L.; Narita, K.; Hyodo, M.; Hayakawa, Y.; Nakane, A.; Karaolis, D.K. C-Di-GMP as a Vaccine Adjuvant Enhances Protection against Systemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Infection. Vaccine 2009, 27, 4867–4873. [Google Scholar] [CrossRef]
- Yu, L.; Wang, N.; Ma, J.; Tong, C.; Song, B.; Chi, J.; Ma, G.; Zhu, Z.; Cui, Y. Improved Protective Efficacy of a Chimeric Staphylococcus Aureus Vaccine Candidate Iron-regulated Surface Determinant B (N 126–P 361)-target of RNAIII Activating Protein in Mice. Microbiology and immunology 2013, 57, 857–864. [Google Scholar] [CrossRef]
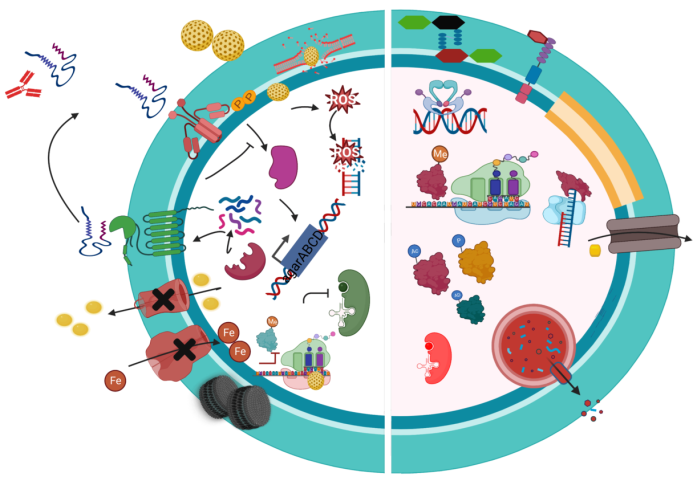
Figure 1.
Mechanism of resistance of anti-MRSA. 1. Vancomycin: Alteration of the antibiotic target from D-ala D-ala to D-ala D-lac. 2. Daptomycin: Modification of phosphatidylglycerol (PG) by MrpF protein, which adds a positively charged lysine residue to PG, forming lysylphosphatidyl glycerol (L-PG). 3. Fusidic acid: Mutation in Elongation Factor-G (EF-G). 4. Mupirocin: Mutation in Isoleucyl-tRNA synthetase (IleRS). 5. Fluoroquinolones: Mutation in DNA gyrase/Topoisomerase IV. 6. Degradation of antibiotics and release by extracellular vesicles. 7. Aminoglycosides: Enzymatic modification of antibiotics, such as Acetyltransferase. 8. MLSB and Linezolid: Mutation/Methylation of 23s rRNA, MLSB (erm MTase), Linezolid (cfr MTase). 9. Ceftaroline and Ceftobiprole: Mutation/Alteration of PBP2a. 10. Ciprofloxacin and Tigecycline: Active efflux pump (EP): NorA for ciprofloxacin, TetM, and MepA EP for tigecycline. Created with BioRender.com.
Figure 1.
Mechanism of resistance of anti-MRSA. 1. Vancomycin: Alteration of the antibiotic target from D-ala D-ala to D-ala D-lac. 2. Daptomycin: Modification of phosphatidylglycerol (PG) by MrpF protein, which adds a positively charged lysine residue to PG, forming lysylphosphatidyl glycerol (L-PG). 3. Fusidic acid: Mutation in Elongation Factor-G (EF-G). 4. Mupirocin: Mutation in Isoleucyl-tRNA synthetase (IleRS). 5. Fluoroquinolones: Mutation in DNA gyrase/Topoisomerase IV. 6. Degradation of antibiotics and release by extracellular vesicles. 7. Aminoglycosides: Enzymatic modification of antibiotics, such as Acetyltransferase. 8. MLSB and Linezolid: Mutation/Methylation of 23s rRNA, MLSB (erm MTase), Linezolid (cfr MTase). 9. Ceftaroline and Ceftobiprole: Mutation/Alteration of PBP2a. 10. Ciprofloxacin and Tigecycline: Active efflux pump (EP): NorA for ciprofloxacin, TetM, and MepA EP for tigecycline. Created with BioRender.com.
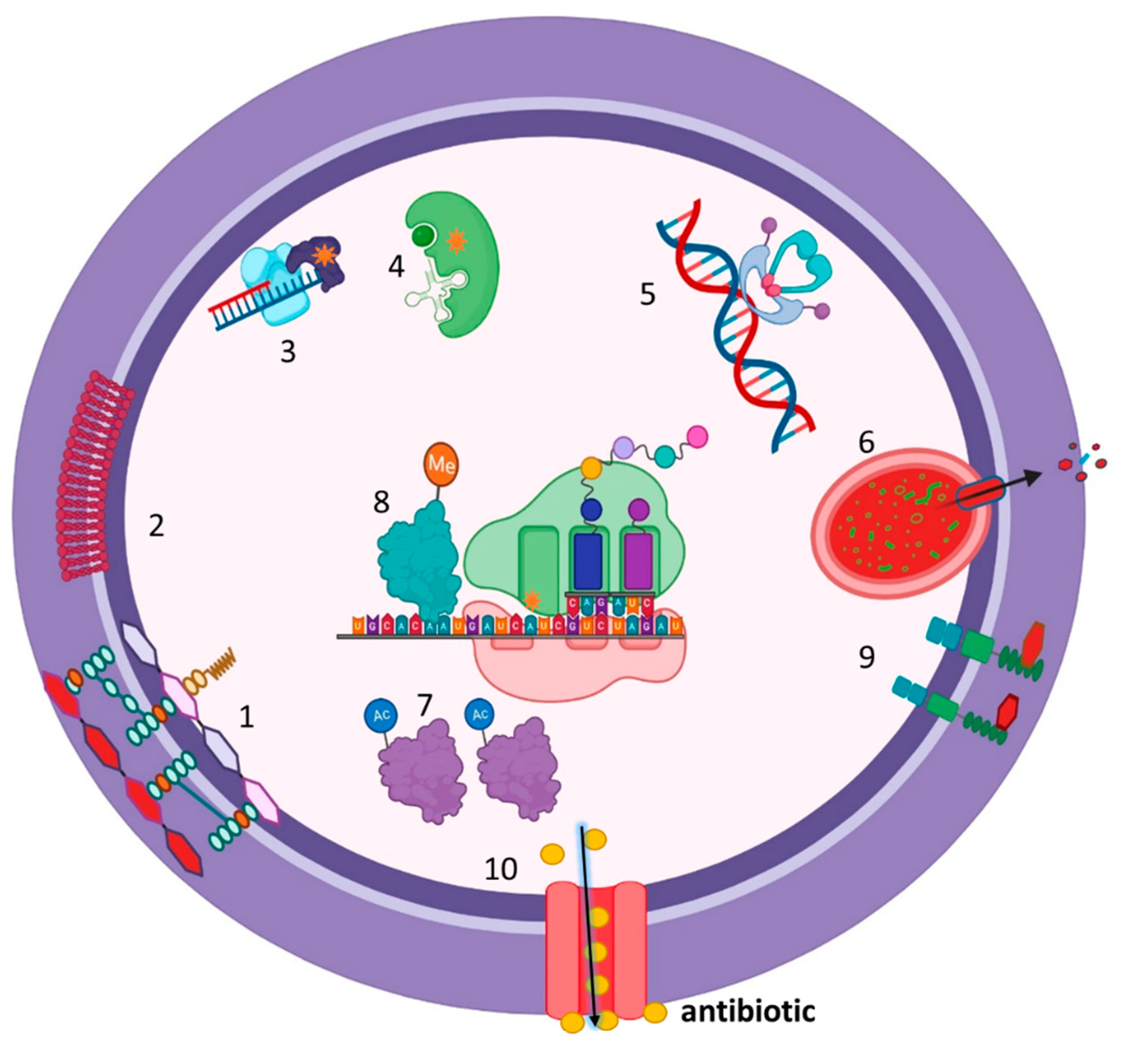
Figure 2.
Mechanisms of action of anti-MRSA. 1. Vancomycin and Telavancin: interfere with late-stage peptidoglycan synthesis by forming non-covalent hydrogen bonds with the D-Ala-D-Ala residues of newly synthesized UDP-MurNAc-pentapeptides, thereby disrupting downstream peptidoglycan assembly. 2. Daptomycin: inserts into the cytoplasmic membrane by binding to negatively charged phosphatidylglycerol (PG) head groups, inducing strain into the lipid bilayer, which results in depolarization, permeabilization, and leakage of ions, notably K+, and ultimately cell death. 3. Fusidic acid: prevents the release of EF-G after the translocation event and blocks the next round of aa-tRNA binding. 4. Mupirocin: binds to isoleucyl-tRNA synthetase, which recognizes amino acids, ATP, and cognate tRNA. 5. Fluoroquinolones: inhibit DNA gyrase and topoisomerase IV. 6. Ceftaroline: inhibits the action of peptidoglycan transpeptidases, which interfere with cell wall synthesis through binding to abnormal PBPs like PBP2a. 7. Macrolides, lincosamides, and linezolid: bind to the bacterial 23S rRNA on 50S ribosomal subunits and block nascent protein synthesis. Created with BioRender.com.
Figure 2.
Mechanisms of action of anti-MRSA. 1. Vancomycin and Telavancin: interfere with late-stage peptidoglycan synthesis by forming non-covalent hydrogen bonds with the D-Ala-D-Ala residues of newly synthesized UDP-MurNAc-pentapeptides, thereby disrupting downstream peptidoglycan assembly. 2. Daptomycin: inserts into the cytoplasmic membrane by binding to negatively charged phosphatidylglycerol (PG) head groups, inducing strain into the lipid bilayer, which results in depolarization, permeabilization, and leakage of ions, notably K+, and ultimately cell death. 3. Fusidic acid: prevents the release of EF-G after the translocation event and blocks the next round of aa-tRNA binding. 4. Mupirocin: binds to isoleucyl-tRNA synthetase, which recognizes amino acids, ATP, and cognate tRNA. 5. Fluoroquinolones: inhibit DNA gyrase and topoisomerase IV. 6. Ceftaroline: inhibits the action of peptidoglycan transpeptidases, which interfere with cell wall synthesis through binding to abnormal PBPs like PBP2a. 7. Macrolides, lincosamides, and linezolid: bind to the bacterial 23S rRNA on 50S ribosomal subunits and block nascent protein synthesis. Created with BioRender.com.
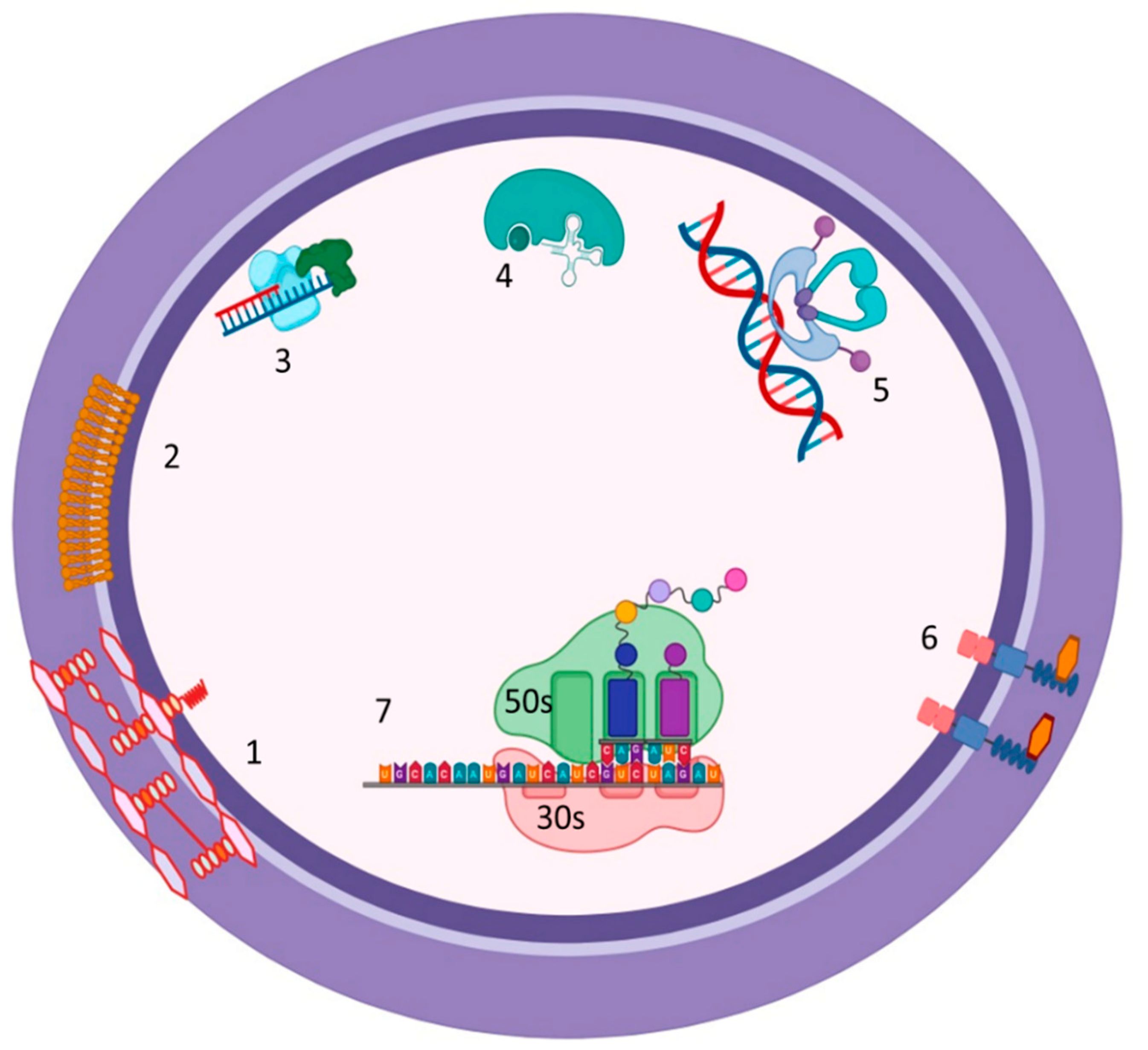
Figure 3.
Novel approaches for MRSA treatment. 1. Inhibition of NorA efflux pump. 2. Blocking of Ironsiderophore by Iron chelators. 3. Inhibition of ermC methyltransferases. 4. Synthetic Anti-microbial Peptides to disrupt the cell wall. 5. Inhibition of the accessory gene regulator (agr) locus. 6. Autoinducing peptides (AIPs) synthase. 7. Pro-AIP. 8. Inhibition of the type I signal peptidase (SpsB). 9. AgrB: a transmembrane endopeptidase. 10. AgrC: Histidine Kinase, the sensor kinase of the TCS. 11. AIPs. 12. AgrA: which binds to RNAIII. 13. Inhibition of quorum sensing by inhibiting AIP synthase, which inhibits the synthesis of pro-AIP. 14. Inhibition of quorum sensing by inhibiting the regulatory factor agrA. 15. Inhibition of quorum sensing by using monoclonal antibodies against AIP to inhibit AIP binding to AgrC Histidine Kinase. Created with BioRender.com.
Figure 3.
Novel approaches for MRSA treatment. 1. Inhibition of NorA efflux pump. 2. Blocking of Ironsiderophore by Iron chelators. 3. Inhibition of ermC methyltransferases. 4. Synthetic Anti-microbial Peptides to disrupt the cell wall. 5. Inhibition of the accessory gene regulator (agr) locus. 6. Autoinducing peptides (AIPs) synthase. 7. Pro-AIP. 8. Inhibition of the type I signal peptidase (SpsB). 9. AgrB: a transmembrane endopeptidase. 10. AgrC: Histidine Kinase, the sensor kinase of the TCS. 11. AIPs. 12. AgrA: which binds to RNAIII. 13. Inhibition of quorum sensing by inhibiting AIP synthase, which inhibits the synthesis of pro-AIP. 14. Inhibition of quorum sensing by inhibiting the regulatory factor agrA. 15. Inhibition of quorum sensing by using monoclonal antibodies against AIP to inhibit AIP binding to AgrC Histidine Kinase. Created with BioRender.com.
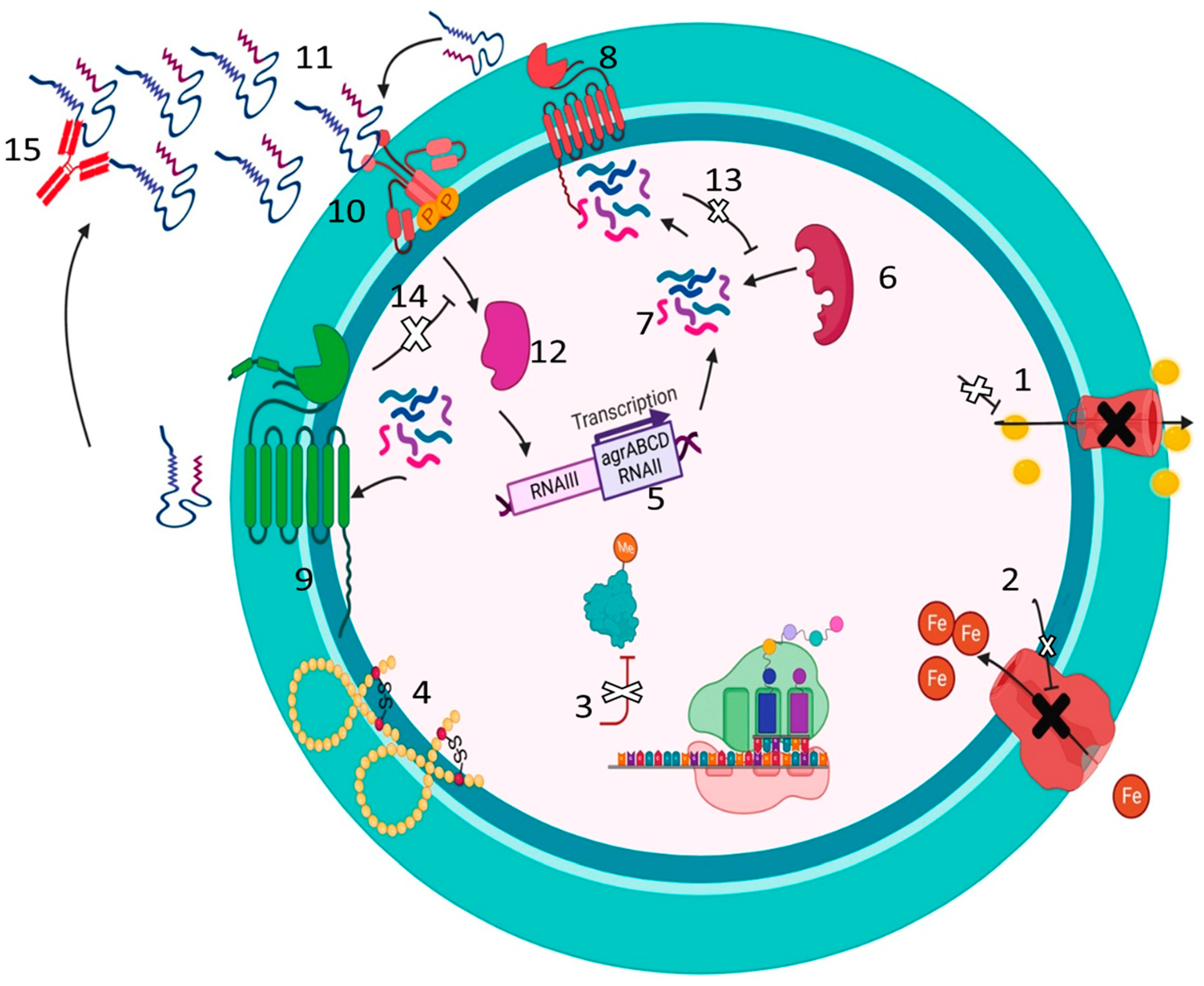
Figure 4.
Synthetic Antimicrobial Peptides against multidrug resistant MRSA 1. Nanoparticles ex: silver nanoparticles (AgNPs). 2. cellular toxicity by the generation of ROS. 3. ROS oxidize and damage the DNA. 4. ROS oxidize the proteins. 5. Nanoparticles can directly be destabilized and denature the proteins. 6. Nanoparticles interact and act with mitochondria leading to mitochondrial dysfunction. 7. Nanoparticles interact and destabilize ribosomes. 8. Nanoparticle adhesion to the cell membrane alters membrane structure and permeability, resulting in leakage of electrolyte and cellular content. Created with BioRender.com.
Figure 4.
Synthetic Antimicrobial Peptides against multidrug resistant MRSA 1. Nanoparticles ex: silver nanoparticles (AgNPs). 2. cellular toxicity by the generation of ROS. 3. ROS oxidize and damage the DNA. 4. ROS oxidize the proteins. 5. Nanoparticles can directly be destabilized and denature the proteins. 6. Nanoparticles interact and act with mitochondria leading to mitochondrial dysfunction. 7. Nanoparticles interact and destabilize ribosomes. 8. Nanoparticle adhesion to the cell membrane alters membrane structure and permeability, resulting in leakage of electrolyte and cellular content. Created with BioRender.com.
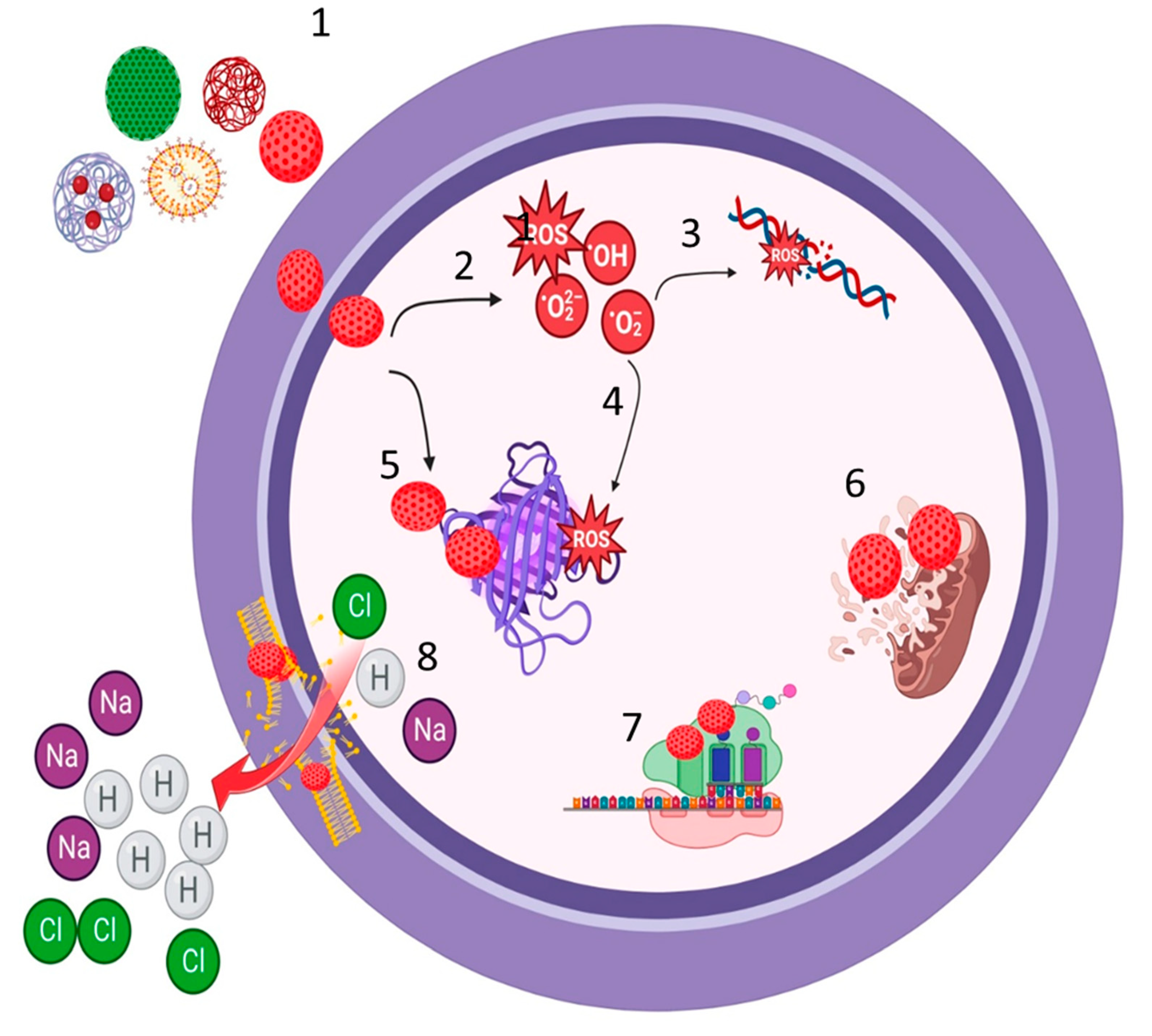
Figure 5.
Inhibition of S. aureus Aminoacyl-tRNA Synthetases (aaRSs) 1. Aa-tRNA synthetases recognize specific amino acids. 2. Aa-tRNA synthetases bind to specific amino acids and recognize specific cognate uncharged tRNA. 3. Aa-tRNA synthetases bind to uncharged tRNA. 4. Aa-tRNA synthetases transfer amino acids to uncharged tRNA and become charged tRNA with specific amino acid. 5. Charged or acylated tRNA goes to the ribosome, and the peptide grows. 6. Inhibition of aa-tRNA synthetases: Inhibition of these stages leads to the accumulation of uncharged tRNA, which attaches to ribosomes, resulting in an interruption in the elongation of the peptide chain and inhibition of protein synthesis. Created with BioRender.com.
Figure 5.
Inhibition of S. aureus Aminoacyl-tRNA Synthetases (aaRSs) 1. Aa-tRNA synthetases recognize specific amino acids. 2. Aa-tRNA synthetases bind to specific amino acids and recognize specific cognate uncharged tRNA. 3. Aa-tRNA synthetases bind to uncharged tRNA. 4. Aa-tRNA synthetases transfer amino acids to uncharged tRNA and become charged tRNA with specific amino acid. 5. Charged or acylated tRNA goes to the ribosome, and the peptide grows. 6. Inhibition of aa-tRNA synthetases: Inhibition of these stages leads to the accumulation of uncharged tRNA, which attaches to ribosomes, resulting in an interruption in the elongation of the peptide chain and inhibition of protein synthesis. Created with BioRender.com.
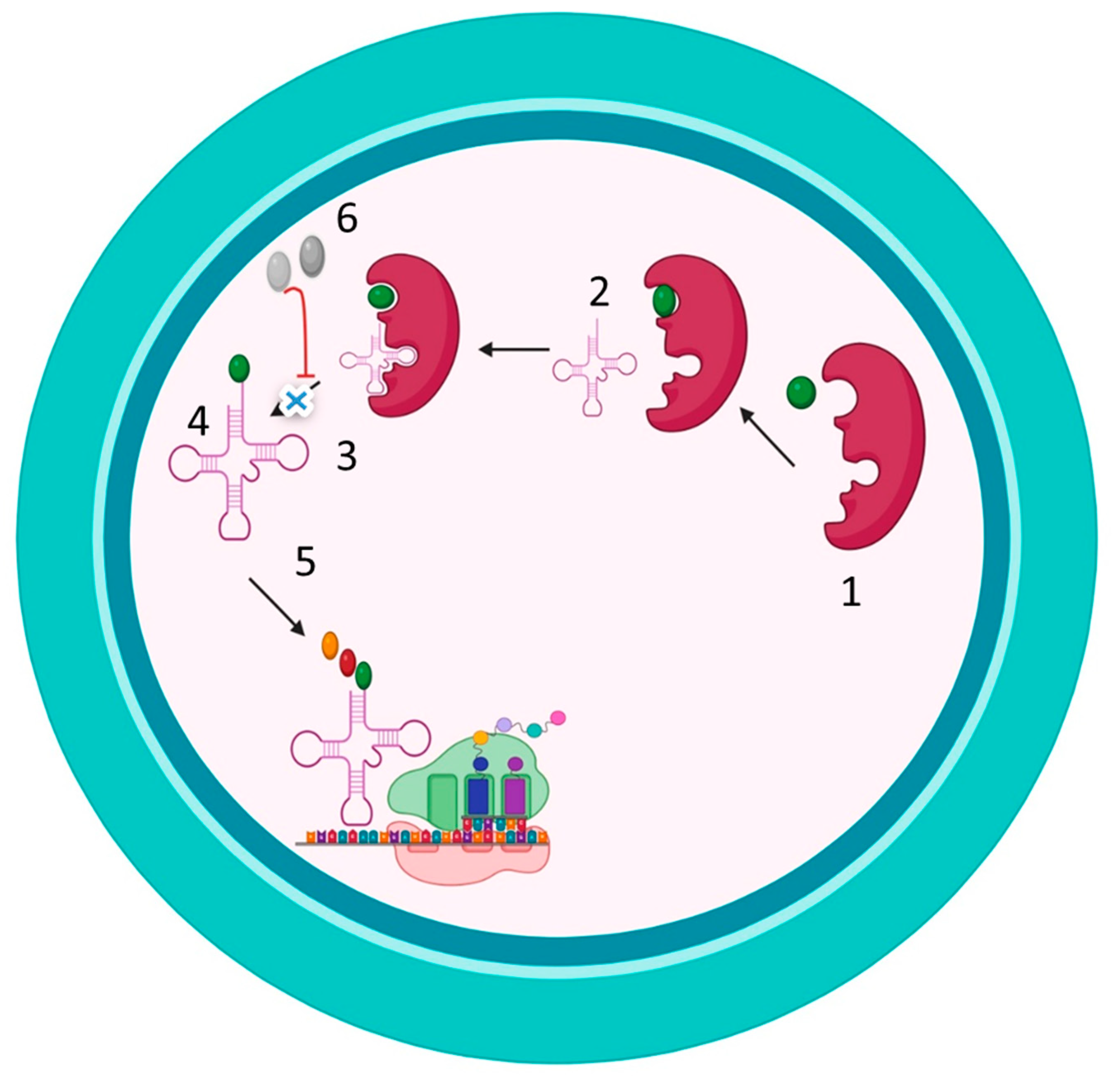

Table 1.
Mechanism of resistance of MRSA to current antibiotics.
| Drugs | Mechanism of resistance |
| Macrolide, Licosamid, and streptogramin B (MLSB) | MLSB resistance involves two possible mechanisms: (a) modification of ribosomal binding site by methylation in the A 2058 in 23S rRNA, mediated by methyltransferase encoded by erm genes, or (b) expression of active efflux mediated by msrA/B gene [16,17,18]. |
| Vancomycinand Telavavin | Resistance to vancomycin is mainly by modification of peptidoglycan precursors to D-alanine-D-lactate instead of D-alanine-Alanine, mediated by vanA operon (vanH and vanA) and regulated by two genes (vanS and vanR) [15,19,20]. |
| Linezolid | Linezolid resistance is mediated by mutation or changes in ribosomal proteins on the drug binding site and methylation of the drug target on 23S rRNA, encoded by the cfr gene [21,22]. |
| Daptomycin | Daptomycin resistance occurs due to modification of the drug target by an increased positive charge of the bacterial cell surface that is mediated by Multiple Peptide Resistance Factor encoded by mrpf gene [23]. |
| Tigecycline | Resistance to tigecycline is conferred by alterations of rpsJ protein or by expression of an active efflux pump called mepA [24]. |
| Ceftaroline | Mutations within the PBP2a allosteric domain alter the triggering mechanism and possibly affect protein-protein interactions needed for peptidoglycan biosynthesis [25,26]. |
| Fusidic acid | A mutation in the chromosomal fusA gene encoding Factor-G (EF-G) confers resistance to fusidic acid [27,28]. |
| Mupirocin | A point mutation in Isoleucyl-tRNA synthetase (IIeRT), which is the drug target [15]. |
| Fluoroquinolone | Fluoroquinolone resistance is conferred by a mutation in the quinolone determining region on DNA gyrase and topoisomerases IV that reduce drug binding efficiency and/or by overexpression of endogenous efflux pumps such as NorA efflux pump [29,30]. |
| Aminoglycoside | Aminoglycoside resistance is conferred by modification of antibiotics to prevent binding to the ribosomal target, which is mediated by bi-functional acetyltransferase-phosphotransferase enzymes [15]. |
Table 2.
Novel Therapeutic Strategies for treating MRSA infection.
| New approaches | Mechanism of action / Uses | Examples |
| Quorum Sensing Inhibitor | Inhibits the switch of agr quorum sensing, preventing expression of virulence factors and subsequent inhibition of biofilm formation [57,58]. | Alanine-5 (D5A), tr-AIP-1, triAIP2, solonamide A and B, Savirin, Hamamelitannin, anti-autoinducer monoclonal antibody AP4-24H11 [59,60,61]. |
| Iron Chelators | Inhibit S. aureus iron acquisition mechanisms, suppressing MRSA wound infection and nares MRSA carriage, and potentially enhancing antibiotic activities [62]. | Deferiprone, Deferasirox, Hydroxypyridinone (DIBI) [63]. |
| NorA efflux pump inhibitors | Inhibit the NorA efflux pump [64]. | Reserpine, Thioridazine, Orizabin XV, Verapamil, Omeprazole, Paroxetine, Chlorpromazine [30,52]. |
| ErmC' methyl transferase inhibitor | Inhibit bacterial methyl transferase [65]. | Sinefungin, UK–10573, ZINC database (ZINC code 327479060 compound) [65]. |
| Antimicrobial peptide (AMP) | Display a direct activity by disrupting the plasma membrane and/or acting on specific intracellular targets to inhibit DNA, RNA, or protein synthesis processes, or inactivate essential intracellular enzymes [66]. | CAMA, LL-37, Magainin-II, Nisin, WR12, D-IK8 [66,67]. |
| Teichoic acid and lipoteichoic acid Inhibition | Inhibition Inhibit teichoic acid and lipoteichoic acid biosynthesis, resulting in the inhibition of biofilm formation and disruption of bacterial growth, cell wall physiology, and membrane homeostasis [68]. | Tunicamycin, Targocil, HSGN-189, and HGSN-94 [68]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



