Submitted:
11 April 2023
Posted:
11 April 2023
You are already at the latest version
Abstract
Varunaivibrio sulfuroxidans type strain TC8T is a mesophilic, facultatively anaerobic, facultatively chemolithoautotrophic alphaproteobacterium, isolated from a sulfidic shallow-water marine gas vent located at Tor Caldara, Tyrrhenian Sea, Italy. V. sulfuroxidans belongs to the family Thalassospiraceae within the Alphaproteobacteria, with Magnetovibrio blakemorei as its closest relative. The genome of V. sulfuroxidans encodes the genes involved in sulfur, thiosulfate and sulfide oxidation, as well as nitrate and oxygen respiration. The genome encodes the genes involved in carbon fixation via Calvin-Benson-Bassham cycle, in addition to genes involved in glycolysis and TCA cycle, indicating a mixotrophic lifestyle. Genes involved in the detoxification of mercury and arsenate are also present. The genome also encodes a complete flagellar complex, one intact prophage and one CRISPR, as well as a putative DNA uptake mechanism mediated by the type IVc (aka Tad pilus) secretion system. Overall, the genome of Varunaivibrio sulfuroxidans indicates that the bacterium is metabolically versatile and well-adapted to the dynamic environmental conditions of sulfidic gas vents.
Keywords:
shallow-water vent
; Tyrrhenian Sea
; Tor Caldara
; chemolithotroph
; Alphaproteobacteria
; Thalasso-spiraceae
; genome
; geothermal
; sulfur oxidation
; DNA uptake
; nitrogen fixation
; carbon fixation
1. Introduction
Hydrothermal and gas vents are manifestations of volcanism on earth. Both environments are characterized by venting of gas and/or hydrothermal fluids, enriched in reduced, inorganic chemical species into the oxygenated water column. This creates a redox disequilibrium, which is then harnessed by lithotrophic microrganisms that convert chemical energy into ATP. Tor Caldara is a shallow-water gas vent located in the Mediterranean Sea, near the town of Anzio, Italy. The composition of gases released by the Tor Caldara vents is mostly CO2 (avg. 77 mol%) and H2S (avg. 23 mol%) [1]. The Tor Caldara gas vents are generated by degassing of CO2 of deep magmatic/mantle origin due to the alkali-potassic volcanoes present in the region, in particular the Colli Albani volcanic complex [2]. Since the gas venting at Tor Caldara is a peripheral manifestation of the quiescent Colli Albani volcano, whose activity began 600 ka ago [2], the system is thought to be very stable over time in comparison to the more dynamic venting associated with active hydrothermal vents.
The continuous venting of CO2 and H2S gas from the seafloor at Tor Caldara supports substrate-attached chemosynthetic microbial biofilms mostly composed of members of the Camplylobacterota (aka Epsilonproteobacteria) and Gammaproteobacteria [1,3], in line with previous studies of coastal and deep-sea marine hydrothermal environments e.g., [4,5,6,7,8,9] as well as more diverse microbial communities associated with the vent-associated sediments [10]. Alphaproteobacteria have also been observed to contribute abundantly to shallow-water vent microbial communities [11,12,13,14]. Consistent with these observations, Alphaproteobacteria were relatively abundant at Tor Caldara (4.5 to 15.1 % of the vent-associated sediment microbial community and 6 to 10 % of the substrate-attached biofilms) [1,10], and a novel genus of sulfur-oxidizing, chemolithoautotrophic alphaproteobacterium, Varunaivibrio sulfuroxidans strain TC8T, was isolated and characterized from the sediments associated with the gas vents of this site [15]. Currently, V. sulfuroxidans belongs to the family Thalassospiraceae (although a recent study proposed its reassignment to the new family Magnetovibrionaceae) [16], which includes four genera of marine bacteria: Magnetospira, Magnetovibrio, Thalassospira and Varunaivibrio [17]. In particular, Magnetospira thiophila, Magnetovibrio blakemorei and Varunaivibrio sulfuroxidans are facultative chemilthoautotrophs that conserve energy by anaerobic (nitrate) and/or microaerobic respiration, with reduced sulfur species as electron donors [15,18,19].
In this study we obtained the genome sequence of V. sulfuroxidans strain TC8 T and we analyzed it in light of its known physiological and metabolic traits, we reconstructed its central metabolism, its heavy metal detoxification and DNA uptake mechanism. This is one of the few chemolithoautotrophic alphaproteobacterial genomes that have been sequenced from a shallow-water hydrothermal vent, another being a Rhodovulum sp. from the coastal hydrothermal system of Kueishantao Island, Taiwan [20].
2. Materials and Methods
2.1. Genome Project History
The genome of V. sulfuroxidans strain TC8T was sequenced in August 2016 as part of a genome sequencing effort of newly characterized bacterial strains isolated at the Deep-Sea Microbiology Lab at Rutgers University (http://marine.rutgers.edu/deep-seamicrobiology/). The draft genome of V. sulfuroxidans was sequenced at the MicrobesNG facility (http://microbesng.uk/), and it was closed in house using the Oxford Nanopore MinION platform [21].
2.2. Growth conditions and genomic DNA preparation
Varunaivibrio sulfuroxidans strain TC8T was grown in modified medium 1011 [22] supplemented with with 0.2% (w/v) KNO3 under a gas phase of N2/CO2 (80:20 v/v; 200 kPA) according to Patwardhan and Vetriani, 2016 [15]. The genomic DNA was extracted from 0.5 – 1 g of cell biomass following a phenol:chloroform extraction protocol. In short, the bacterial pellet was added to 850 ul extraction buffer (50 mM Tris-HCl, 20mM EDTA, 100mM NaCl; pH 8.0) supplemented with100 ul of lysozyme (100 mg/ml) and incubated at 37°C for 30 min. This mix was then supplemented with 5 ul of proteinase K (20mg/ml) and incubated at 37°C for 30 min followed by the addition of 50 ul SDS (20%) and further incubated at 65°C for 1 hr. The genomic DNA was extracted by performing two consecutive phenol:chloroform:isoamylalcohol (25:24:1) extractions. The supernatant was precipitated in 3 M sodium-acetate and isopropanol, washed twice with 70% ice cold ethanol and re-suspended in ultra-pure water.
2.3. Genome sequencing and assembly
A hybrid approach involving short Illumina reads and long Nanopore MinIon reads was used to sequence the genome of V. sulfuroxidans. Genomic DNA was sequenced using a paired end 2x250 bp MiSeq library which generated a total of 1,004,710 quality checked short reads. A total of 40,150 long reads were also obtained on the Naonopre MinIon platform using the SQK-RAD004 library prep kit. Obtained short and long reads were assembled successively using Unicycler [23] and Geneious [24]. Assembly yielded 1 complete contig of a total length of 3,066,297 bp, with a 138x sequencing coverage. The genome sequencing project of V. sulfuroxidans strain TC8T is available under accession number: CP119676.
2.4. Genome annotation
The genome assembly was annotated using the Joint Genome Institute IMG system [25] and the RAST (Rapid Annotation using Subsystem Technology) server. The predicted proteins of Varunaivibrio sulfuroxidans TC8T involved in key metabolic pathways were searched using the KEGG database [26]. CRISPERs and tandem repeats were identified using CRISPRFinder [27]. Prophages in the genome were searched using Phaster [28]. Genomic islands were identified using IslandViewer [29].
2.5. Phylogenetic, average nucleotide identity and digital DNA-DNA hybridization analyses
Phylogenetic analyses were carried out as previously described [7,30]. In brief, 16S rRNA gene sequences were aligned with ClustalO [31] and the alignment was manually refined using SEAVIEW [32]. The maximum likelihood phylogeny tree was inferred from the alignment using PhyML [33], with the Jukes and Cantor substitution model [34] and 500 bootstrap resamplings. The aminoacid sequences of NifH, ComEC and CpaC from strain TC-8T and closest homologues were retrieved from GenBank, aligned in SEAVIEW [32] and the maximum likelihood phylogeny trees were inferred from the alignment using PhyML [33], with the Li and Gasquel (LG) aminoacid replacement matrix [35] and 500 bootstrap resamplings. The average nucleotide identity (ANI) between strain TC8T and its closest relative, Magnetovibrio blakemorei, was calculated using the EZBioCloud tool [36]. The digital DNA-DNA hybridization (dDDH) value between strain TC8T and M. blakemorei was obtained using the genome-to-genome distance calculator (GGDC) [37].
3. Results and Discussion
3.1. Overview of Varunaivibrio sulfuroxidans strain TC8T
V. sulfuroxidans strain TC8T is a mesophilic, facultatively anaerobic, facultatively chemolithoautotrophic bacterium isolated from a sulfidic shallow-water marine gas vent located at Tor Caldara, in the Tyrrhenian Sea, Italy [15]. Cells are Gram-stain-negative curved rods with one or more polar flagella. Cells were approximately 1–1.5 µm in length and 0.6 µm in width. Strain TC8T grows optimally at 30 °C, with 15-20 g NaCl l/1iter and optimum pH between 6.0 and 7.0. The generation time under optimal conditions is 8 h. Strain TC8T is a facultative chemolithoautotroph also capable of using organic substrates (tryptone, peptone, Casamino acids, pyruvate and glycerol). Chemolithoautotrophic growth occurs with sulfur and thiosulfate as the electron donors, CO2 as the carbon source, and nitrate or oxygen (5 %, v/v) as the electron acceptors. Strain TC8 T is able to grow in the absence of a source of fixed nitrogen (nitrate or ammonium) under a headspace of N2/CO2/O2 (53:45:2 v/v), which implies that it is able to fix gaseous nitrogen.
Phylogenetic analysis of the 16S rRNA gene sequence of strain TC8T placed this organism in a lineage within the family Thalassospiraceae, with Magnetovibrio blakemorei strain MV1T (91.25% sequence identity) as its closest cultured relative (Figure 1). When uncultured microorganisms known solely by their 16S rRNA gene are included in the analysis, the closest sequence to V. sulfuroxidans is clone 7M24_051 (98.89% sequence identity; Figure 1), obtained from a iron-rich, inactive sulfide associated with the deep-sea hydrothermal vent system of the East Pacific Rise at 9° North [38].
3.2. Genome Structure
The genome of V. sulfuroxidans strain TC8T contains 3,066,297 bp, with coding regions taking 88.1 % of the genome and a G+C content of 54.4 % (Table 1). The most abundant gene categories assigned to the clusters of orthologous genes (COGs) database code for amino acid transport and metabolism (10.3 %), energy conversion (8.9 %), translation and ribosomal structure (7.6 %) and inorganic ion transport and metabolism (7.5 %; Table 2). 27.1 % of the genes of strain TC8T were not in the COG database, while 4.7 % were classified as unknown function (Table 2).
The genome structure of V. sulfuroxidans strain TC8T, including the gene distribution, predicted genomic islands, prophage and genes involved in membrane transport, carbon and energy metabolism is summarized in Figure 2. Nine regions of the genome of strain TC8T were predicted as possible genomic islands (Figure 2A, line 2), while one region was found to be a complete bacteriophage (Figure 2B). The intact prophage found in the genome of TC8T has a size of 33.4Kb, with a total of 48 proteins and a GC content of 65.53% (Figure 2B). It consists of 22 different phage-like proteins, with the majority (31%) of proteins similar to prophage vB_RhkS_P1, which was induced in Rhodovulum sp. strain P5, an alphaproteobacterium isolated from a shallow-water hydrothermal vent off Kueishantao Islands [20,39].
The genome of strain TC8T encodes one short CRISPR array, which is 431 bp in length and has 6 spacers. The spacers did not show sufficient alignment to any sequences in the Genbank Phage or RefSeq plasmid databases.
The average nucleotide identity (ANI) between the genomes of V. sulfuroxidans and its closest cultured relative, M. blakemorei, is 68.86%, while the estimated digital DNA-DNA hybridization (dDDH) between the two genomes is 18.7%, confirming their assignment to different genera.
3.3. Carbon Metabolism and Microaerobic Respiration
The genome of strain TC8T encodes for all the enzymes necessary for carbon fixation using the Calvin-Benson-Bassham (CBB) cycle, including the key enzymes ribulose 1,5- bisphosphate carboxylase (RubisCO) and phosphoribulokinase (Figs. 2A and 3 and Table S1). The RubisCO enzyme catalyzes the carboxylation of ribulose 1,5- bisphosphate, which results in two molecules of 3-phosphoglycerate. Presently, there are three forms of RubisCO known that catalyze the CO2 fixation reaction, with form I being the most dominantly occurring [40]. From II, encoded by the cbbM gene, was first isolated from the alphaproteobacterium, Rhodospirillum rubrum. Similarly to other members of the family Thalassospiraceae, the genome of V. sulfuroxidans TC8T encodes a form II RubisCO closely related to the enzyme from M. blakemorei (sequence identity 88.45%). The carbon concentrating mechanisms of strain TC8T includes a type γ carbonic anhydrase, which catalyzes the interconversion between the dissociated ions of carbonic acid and carbon dioxide and water (Figure 3 and Table S1). The amino acid sequence of the carbonic anhydrase of V. sulfuroxidans is 69.4 % identical to that of M. blakemorei.
The genome of strain TC8T also encodes for all enzymes involved in the breakdown of organic carbon via the glycolysis pathway as well as for all the enzymes involved in the tricarboxylic acid (TCA) cycle, which provides reducing power for oxidative phosphorylation in heterotrophs (Figure 2 and Table S1). Thus, the genome of V. sulfuroxidans strain TC8T encodes for a heterotrophic as well as autotrophic metabolism, in line with its experimentally determined metabolic characteristics [15].
3.4. Sulfur Metabolism
V. sulfuroxidans TC8T was originally described as sulfur-oxidizing chemolithoautotroph [15]. Accordingly, the genome encodes the SoxABXYZ pathway that is responsible for thiosulfate oxidation (Figure 3 and Table S1). Strain TC8T lacks the SoxCD component which, in Paracoccus pantotrophus, is designated as a sulfur dehydrogenase [41]. Without SoxCD, thiosulfate is likely oxidized to elemental sulfur and not all the way to sulfate, as demonstrated experimentally in Allochromatium vinosum [42]. The aminoacid sequence of V. sulfuroxidans TC8T SoxA is 65.83% identical to that of the M. blakemorei enzyme. Besides the Sox gene cluster, the genome of strain TC8T encodes the sulfide:quinone oxidoreductases (SQR, 79.6 % identity to the M. blakemorei enzyme) and a putative flavocytochrome c-sulfide dehydrogenase (Fcc; Figure 3 and Table S1). The presence of these enzymes suggests that strain TC8T has the potential to oxidize sulfide to elemental sulfur [43]. Current knowledge indicates that, in chemolithotrophic sulfide-oxidizing bacteria, SQR-mediated sulfide oxidation is more energetically favorable than sulfide oxidation by Fcc, and that overall SQR is a more efficient enzyme [44]. However, due to its high affinity for sulfide, Fcc is hypothesized to be selectively expressed at low sulfide concentrations [3,45]. The dsr gene cluster, encoding several enzymes involved in oxidation of stored sulfur, is also present in the genome of strain TC8T (Figure 3 and Table S1). According to current understanding, sulfur is brought into the cytoplasm in a persulfidic form (e.g., glutathione persulfide) [43]. DsrL releases the sulfide from the persulfide, which then passes on to the reversely operating sulfite reductase DsrAB via DsrC and DsrEFH [46]. The DsrAB oxidizes sulfide to sulfite. Next, DsrAB interacts with the trans-membrane DsrMKJOP complex, which passes electrons in to the respiratory electron chain. Genes encoding for all the aforementioned proteins are present in the genome of strain TC8T. The sulfite formed is likely further oxidized to sulfate via AprAB and SAT enzymes, both encoded by the genome of strain TC8T. The presence of several sulfur oxidation pathways in the genome of strain TC8T indicates its metabolic flexibility and its ability to oxidize a variety of reduced sulfur compounds to conserve energy, which likely provide this bacterium with an advantage to thrive in its sulfidic habitat.
3.5. Nitrogen Metabolism
Dissimilatory nitrate reduction to ammonia (DNRA) and denitrification are two key pathways involved in nitrogen metabolism that occur commonly in vent organisms [47,48]. V. sulfuroxidans conserves energy via the oxidation of reduced sulfur species coupled to the reduction of nitrate to dinitrogen gas [15]. In line with this experimental evidence, the genome V. sulfuroxidans encodes all four proteins involved in the complete denitrification pathway (Figure 3 and Table S1). The first step of this pathway, i.e., the reduction of nitrate to nitrite, is catalyzed by a nitrate reductase, either the membrane bound NarG or the periplasmic NapAB. All four genes encoding the membrane bound nitrate reductase complex, namely narG, narH,narJ and narI are present in the genome of strain TC8T. The napA and napB genes encoding the periplasmic nitrate reductase NapAB are also present [47]. The aminoacid sequence of the periplasmic nitrate reductase (NapA) of V. sulfuroxidans is 80.26% identical to that of M. blakemorei. The genome has the entire nap gene cluster consisting of the quinol dehydrogenase NapC, the maturation chaperone NapD, the membrane bound dehydrogenases NapG and NapH and subunit NapF, whose role remains unclear (Figure 3). Several microorganisms, including Escherichia coli, encode both the membrane bound (Nar) and periplasmic (Nap) nitrate reductases. Potter et al. (1999) experimentally demonstrated that, in nitrate limited conditions, the presence of only the high-affinity Nap enzyme afforded E. coli a selective advantage over mutant strains that only encoded the Nar enzyme [49]. We hypothesize that, in the dynamic habitat of the Tor Caldara gas vents, the expression of the two nitrate reductases in V. sulfuroxidans is modulated by the availability of nitrate. The reduction of nitrite to nitric oxide (NO) is carried out by cytochrome cd1 protein NirS, encoded by the nirS gene. NorBC further reduces two molecules of NO to nitrous oxide (N2O). NosZ is involved in the reduction of N2O to N2, albeit is not present in all organisms [50]. However, NosZ is present in the genome of strain TC8T, which is consistent with its ability to generate dinitrogen gas during growth under nitrate reducing conditions [15]. Along with dissimilatory nitrate reduction, the genome also shows potential for assimilatory nitrate reduction, as the nasA and nasB genes that encode for cytoplasmic nitrate reductase NasAB are present (Figure 3). Genes for the assimilatory nitrite reductases NIT-6 and NirA were absent. However, the genome encoded for the NirBD protein which has also been shown to be involved in assimilatory nitrite reduction [51,52].
Nitrogen fixation, namely the reduction of N2 gas to two molecules of ammonia, is an important part of the marine nitrogen cycle, since it provides a source of newly fixed nitrogen to the ecosystem. The nitrogenase complex catalysing this reaction is made up of two proteins: dinitrogenase reductase and dinitrogenase encoded by three genes, nifH, nifD and nifK [53]. All three genes are found in the genome of V. sulfuroxidans, consistent with its ability to grow in the absence of fixed nitrogen (Figure 3 and Table S1) [15]. The genome of strain TC8T also encodes a nitrogenase-associated Fe-S ferredoxin (fdx gene; Table S1), which is 44.7 % identical to that of Rhodobacter capsulatus. We hypothesize that the fdx-encoded ferredoxin serves as electron donor to nitrogenase in V. sulfuroxidans, as experimentally demonstrated for R. capsulatus [54]. Phylogenetic analysis of the NifH placed the enzyme of V. sulfuroxidans in a cluster that includes several members of marine Alphaproteobacteria, including M. blakemorei, Cohaesibacter marisflavi, C. intestine and Rhodobium orientis, as well as Martelella endophytica, which was isolated from the rose rhizosphere (Figure 4). The NifH aminoacid sequences of V. sulfuroxidans and M. blakemorei are 88.8% identical. The genome of V. sulfuroxidans encodes several enzymatic complexes that catalyze the oxidoreduction of nitrogen (nitrate reduction, assimilation and nitrogen fixation) and sulfur species (thiosulfate, sulfur and possibly sulfide oxidation), effectively linking the nitrogen and sulfur cycles.
3.6. Heavy Metal Detoxification
Hydrothermal vent environments are known to have elevated concentrations of toxic heavy metals; hence, the resident microorganisms encode genes involved with heavy metal detoxification [55,56,57,58]. The genome of strain TC8T contains both the ars and the mer operons involved in detoxification of arsenate and mercury, respectively (Figure 3 and Table S1). Arsenic in the form of arsenite or arsenate is toxic for most microorganisms, and resistance is associated with a three gene or five gene ars operon. The five gene ars operon in strain TC8T consists of arsenate reductase ArsC, membrane efflux protein ArsB, two trans-acting repressors ArsR and ArsD, as well as another efflux protein ArsB that infers higher resistance to arsenic [59,60]. The mer operon of strain TC8T encodes for the periplasmic mercury-binding protein MerP, the trans-membrane transport protein MerT and the mercury reductase MerA [61,62]. V. sulfuroxidans did not grow in the presence of arsenate as the electron acceptor [15]. The genome does not encode for a selenate reductase; however, it has been shown that bacterial nitrate reductases, which the genome encodes for, are also capable of reducing selenate [63,64]. The genome also encodes for the DedA protein, which has shown to be involved selenite resistance [65]. The resistance of V. sulfuroxidans to mercury and selenite was not tested.
3.7. DNA uptake system
Knowledge of extracellular DNA uptake in Alphaproteobacteria is limited; however, recent studies suggest the involvement of a type IVc pilus known as the Tad pilus system [66]. In the naturally competent plant pathogen, Liberibacter crescens strain BT1, the tad pilus aids in survival through surface attachment and DNA uptake for nucleotide metabolism [67,68]. L. crescens, a member of the family Rhizobiaceae within the Alphaproteobacteria, was experimentally proven to be naturally competent, and the genes involved in this system were identified and found to be conserved among Liberibacter spp. [66,68].
Therefore, we used L. crescens strain BT1 as a reference organism to identify the putative DNA uptake system of V. sulfuroxidans. We discovered that the genome of V. sulfuroxidans encodes genes homologous to the tad genes organized in a cluster similar in structure to that of M. blakemorei and L. crescens (Figure 5). The tad pilus system consists of a biogenesis ATPase (tadZ/cpaE), motor ATPase (tadA/cpaF), prepilin peptidase (tadV/cpaA), inner membrane staging complex (tadB/cpaG and tadC/cpaH), secretin (rcpA/cpaC), periplasmatic subunits (tadG/cpaB and rcpB/cpaD), inner membrane anchor (rcpC/tadG), and pilotin (tadD/cpaO) [66]. V. sulfuroxidans have homologs to genes in this system except for pilotin (Figs. 2). The genome of strain TC8T also encodes the DNA translocation machinery homologous to L. crescens strain BT1, as well as core genes involved in DNA uptake homologous to those encoded by L. crescens. Phylogenetic tree reconstruction of CpaC, the enzyme secretin of the type IVc secretion system, revealed that the enzyme of V. sulfuroxidans was most closely related to that of M. blakemorei (44.79% aminoacid sequence identity; Figure 6). The CpaC from L. crescens was placed in a cluster with homologous enzymes from other members of the family Rhizobiaceae (33.48% aminoacid sequence identity with the CpaC from V. sulfuroxidans).
ComEC is also necessary for natural competence and DNA internalization in L. crescens, and a homolog was found in strain TC8T [68]. Phylogenetic analysis showed that the competence protein ComEC of V. sulfuroxidans was related to the homologous protein from M. blakemorei (45.47% aminoacid identity; Figure 7), while ComEC from L. crescens clustered with a group of proteins from various strains of Rhizobium and was 32.3% identical to that of V. sulfuroxidans.
Based on these results, we hypotesize that V. sulfuroxidians and M. blakemorei are naturally competent via the Tad pilus/Com system. However, the use of the Tad pilus system might differ between L. crescens and V. sulfuroxidans in the fact that former depends on this system for the essential uptake of nucleotides while strain TC8T can synthesize its own nucleotides.
4. Conclusions
In this study we sequenced and analyzed the genome of V. sulfuroxidans strain TC8T, whose physiology and metabolism were previously experimentally characterized. The genome of strain TC8T revealed that this facultative chemolithoautotroph fixes carbon via the CBB cycle, oxidize reduced sulfur species via the Sox, SQR and the oxidative Dsr pathways, encodes the nitrogenase gene cluster for nitrogen fixation and reduces nitrate via the periplasmic and/or cytoplasmic nitrate reductase complexes, Nap and Nar. Based on homology with the experimentally verified DNA uptake system of L. crescens, we further identified the Tad-mediated secretion system and competence gene cluster in the genome of V. sulfuroxidans and its closest relative, M. blakemorei. These findings suggest that both V. sulfuroxidans and M. blakemorei are naturally competent and can take up exogenous DNA.
Linking the genome sequence of V. sulfuroxidans with its observed metabolism establishes a direct association between genotype and phenotype, and enables genome-based metabolic inferences from environmental, sequence-based surveys, e.g., metagenome-assembled genomes (MAGs) of closely related strains (e.g., close relatives of V. sulfuroxidans identified by sequencing in hydrothermal and sedimentary habitats).
Overall, the genome of V. sulfuroxidans, along with its experimentally validated phenotype, reveals the metabolic versatility of this bacterium and corroborates its ability to thrive in the highly dynamic habitat of sulfidic gas vents.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
SP carried out the experiments, analyzed the data and wrote the manuscript, JP and CV analyzed the data and wrote the manuscript, FS carried out the experiments, analyzed the data and contributed to the writing of the manuscript.
Acknowledgments
This work was supported by NSF grants IOS 19-51690 and OCE 19-48623 and by NASA grant 20-EXO20-0084 to CV.
References
- Patwardhan, S.; Foustoukos, D.I.; Giovannelli, D.; Yücel, M.; Vetriani, C. Ecological Succession of Sulfur-Oxidizing Epsilon- and Gammaproteobacteria During Colonization of a Shallow-Water Gas Vent. Front. Microbiol. 2018, 9, 2970. [Google Scholar] [CrossRef]
- Carapezza, M.L.; Barberi, F.; Ranaldi, M.; Ricci, T.; Tarchini, L.; Barrancos, J.; Fischer, C.; Granieri, D.; Lucchetti, C.; Melian, G.; et al. Hazardous Gas Emissions from the Flanks of the Quiescent Colli Albani Volcano (Rome, Italy). Applied Geochemistry 2012, 27, 1767–1782. [Google Scholar] [CrossRef]
- Patwardhan, S.; Smedile, F.; Giovannelli, D.; Vetriani, C. Metaproteogenomic Profiling of Chemosynthetic Microbial Biofilms Reveals Metabolic Flexibility During Colonization of a Shallow-Water Gas Vent. Front. Microbiol. 2021, 12, 638300. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, P.; Duperron, S.; Philippot, P.; Foriel, J.; Susini, J.; Moreira, D. Bacterial Diversity in Hydrothermal Sediment and Epsilonproteobacterial Dominance in Experimental Microcolonizers at the Mid-Atlantic Ridge. Environ Microbiol 2003, 5, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, D.; d’Errico, G.; Manini, E.; Yakimov, M.M.; Vetriani, C. Diversity and Phylogenetic Analyses of Bacteria from a Shallow-Water Hydrothermal Vent in Milos Island (Greece). Front. Microbiol 2013, 4, 184. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.E.; Giovannelli, D.; Govenar, B.; Luther, G.W.; Lutz, R.A.; Shank, T.M.; Vetriani, C. Microbial Biofilms Associated with Fluid Chemistry and Megafaunal Colonization at Post-Eruptive Deep-Sea Hydrothermal Vents. Deep Sea Research Part II: Topical Studies in Oceanography 2015, 121, 31–40. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, I.; Bolognini, M.; Ricci, J.; Bini, E.; Vetriani, C. From Deep-Sea Volcanoes to Human Pathogens: A Conserved Quorum-Sensing Signal in Epsilonproteobacteria. ISME J 2015, 9, 1222–1234. [Google Scholar] [CrossRef]
- Huber, J.A.; Mark Welch, D.B.; Morrison, H.G.; Huse, S.M.; Neal, P.R.; Butterfield, D.A.; Sogin, M.L. Microbial Population Structures in the Deep Marine Biosphere. Science 2007, 318, 97–100. [Google Scholar] [CrossRef]
- Miranda, P.J.; McLain, N.K.; Hatzenpichler, R.; Orphan, V.J.; Dillon, J.G. Characterization of Chemosynthetic Microbial Mats Associated with Intertidal Hydrothermal Sulfur Vents in White Point, San Pedro, CA, USA. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Patwardhan, S.S. Prokaryotic Diversity, Physiology and Function at Tor Caldara, a Shallow-Water Gas Vent in the Tyrrhenian Sea. 2018. [CrossRef]
- Kerfahi, D.; Hall-Spencer, J.M.; Tripathi, B.M.; Milazzo, M.; Lee, J.; Adams, J.M. Shallow Water Marine Sediment Bacterial Community Shifts Along a Natural CO2 Gradient in the Mediterranean Sea Off Vulcano, Italy. Microb Ecol 2014, 1–10. [Google Scholar] [CrossRef]
- Gugliandolo, C.; Lentini, V.; Bunk, B.; Overmann, J.; Italiano, F.; Maugeri, T.L. Changes in Prokaryotic Community Composition Accompanying a Pronounced Temperature Shift of a Shallow Marine Thermal Brine Pool (Panarea Island, Italy). Extremophiles 2015, 19, 547–559. [Google Scholar] [CrossRef]
- Lentini, V.; Gugliandolo, C.; Bunk, B.; Overmann, J.; Maugeri, T.L. Diversity of Prokaryotic Community at a Shallow Marine Hydrothermal Site Elucidated by Illumina Sequencing Technology. Curr Microbiol 2014, 69, 457–466. [Google Scholar] [CrossRef]
- Tang, K. Microbial Communities in a Shallow-Sea Hydrothermal System. In Encyclopedia of Metagenomics; Nelson, K.E., Ed.; Springer New York, 2013; pp. 1–8 ISBN 978-1-4614-6418-1.
- Patwardhan, S.; Vetriani, C. Varunaivibrio Sulfuroxidans Gen. Nov., Sp. Nov., a Facultatively Chemolithoautotrophic, Mesophilic Alphaproteobacterium from a Shallow-Water Gas Vent at Tor Caldara, Tyrrhenian Sea. International Journal of Systematic and Evolutionary Microbiology 2016, 66, 3579–3584. [Google Scholar] [CrossRef]
- Koziaeva, V.V.; Sorokin, D.Y.; Kolganova, T.V.; Grouzdev, D.S. Magnetospirillum Sulfuroxidans Sp. Nov., Capable of Sulfur-Dependent Lithoautotrophy and a Taxonomic Reevaluation of the Order Rhodospirillales. Systematic and Applied Microbiology 2023, 46, 126406. [Google Scholar] [CrossRef]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.-M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1,000+ Type-Strain Genomes Substantially Improves Taxonomic Classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Lefèvre, C.T.; Zhao, W.; Beveridge, T.J.; Bazylinski, D.A. Magnetospira Thiophila Gen. Nov., Sp. Nov., a Marine Magnetotactic Bacterium That Represents a Novel Lineage within the Rhodospirillaceae (Alphaproteobacteria). Int. J. Syst. Evol. Microbiol. 2012, 62, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Bazylinski, D.A.; Williams, T.J.; Lefevre, C.T.; Trubitsyn, D.; Fang, J.; Beveridge, T.J.; Moskowitz, B.M.; Ward, B.; Schubbe, S.; Dubbels, B.L.; et al. Magnetovibrio Blakemorei Gen. Nov., Sp. Nov., a Magnetotactic Bacterium (Alphaproteobacteria: Rhodospirillaceae) Isolated from a Salt Marsh. INTERNATIONAL JOURNAL OF SYSTEMATIC AND EVOLUTIONARY MICROBIOLOGY 2013, 63, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Zhang, Y.; Lin, D.; Han, Y.; Chen, C.-T.A.; Wang, D.; Lin, Y.-S.; Sun, J.; Zheng, Q.; Jiao, N. Cultivation-Independent and Cultivation-Dependent Analysis of Microbes in the Shallow-Sea Hydrothermal System Off Kueishantao Island, Taiwan: Unmasking Heterotrophic Bacterial Diversity and Functional Capacity. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol 2016, 17, 239. [Google Scholar] [CrossRef]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum Lithotrophicum Gen. Nov., Sp. Nov., a Novel Sulfur-Oxidizing Chemolithoautotroph within the ε-Proteobacteria Isolated from Okinawa Trough Hydrothermal Sediments. International Journal of Systematic and Evolutionary Microbiology 2004, 54, 1477–1482. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLOS Computational Biology 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Chen, I.-M.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated Genome and Metagenome Comparative Data Analysis System. Nucleic Acids Res 2017, 45, D507–D516. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. Journal of Molecular Biology 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Research 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B. Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded Prediction of Genomic Islands for Larger-Scale Datasets. Nucleic Acids Research 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Reed, A.J.; Dorn, R.; Van Dover, C.L.; Lutz, R.A.; Vetriani, C. Phylogenetic Diversity of Methanogenic, Sulfate-Reducing and Methanotrophic Prokaryotes from Deep-Sea Hydrothermal Vents and Cold Seeps. Deep Sea Research Part II: Topical Studies in Oceanography 2009, 56, 1665–1674. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol Syst Biol 2011, 7, 539. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Molecular Biology and Evolution 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Holmquist, R.; Cantor, C.; Jukes, T. Improved Procedures for Comparing Homologous Sequences in Molecules of Proteins and Nucleic Acids. Journal of Molecular Biology 1972, 64, 145–161. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Molecular Biology and Evolution 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.; Lim, J.; Kwon, S.; Chun, J. A Large-Scale Evaluation of Algorithms to Calculate Average Nucleotide Identity. Antonie van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Research 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Sylvan, J.B.; Toner, B.M.; Edwards, K.J. Life and Death of Deep-Sea Vents: Bacterial Diversity and Ecosystem Succession on Inactive Hydrothermal Sulfides. mBio 2012, 3, e00279-11. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Tang, K.; Han, Y.; Li, C.; Chen, X. Genome Sequence of an Inducible Phage in Rhodovulum Sp. P5 Isolated from the Shallow-Sea Hydrothermal System. Marine Genomics 2016, 30, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Tabita, F.R.; Hanson, T.E.; Li, H.; Satagopan, S.; Singh, J.; Chan, S. Function, Structure, and Evolution of the RubisCO-Like Proteins and Their RubisCO Homologs. Microbiol. Mol. Biol. Rev. 2007, 71, 576–599. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.G.; Rother, D.; Bardischewsky, F.; Quentmeier, A.; Fischer, J. Oxidation of Reduced Inorganic Sulfur Compounds by Bacteria: Emergence of a Common Mechanism? Appl. Environ. Microbiol. 2001, 67, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Hensen, D.; Sperling, D.; Trüper, H.G.; Brune, D.C.; Dahl, C. Thiosulphate Oxidation in the Phototrophic Sulphur Bacterium Allochromatium Vinosum. Molecular Microbiology 2006, 62, 794–810. [Google Scholar] [CrossRef]
- Dahl Christiane; Friedrich Cornelius; Kletzin Arnulf Sulfur Oxidation in Prokaryotes. eLS 2008. [CrossRef]
- Griesbeck, C.; Hauska, G.; Schütz, M. Biological Sulfide Oxidation: Sulfide-Quinone Reductase (SQR), the Primary Reaction. In Recent Research Developments in Microbiology; Trivadrum: Research Signpost; Vol. 4, pp. 179–203.
- Brune, D.C. Sulfur Compounds as Photosynthetic Electron Donors. In Anoxygenic Photosynthetic Bacteria; Advances in Photosynthesis and Respiration; Springer, Dordrecht, 1995; pp. 847–870 ISBN 978-0-7923-3681-5.
- Stockdreher, Y.; Venceslau, S.S.; Josten, M.; Sahl, H.-G.; Pereira, I.A.C.; Dahl, C. Cytoplasmic Sulfurtransferases in the Purple Sulfur Bacterium Allochromatium Vinosum: Evidence for Sulfur Transfer from DsrEFH to DsrC. PLOS ONE 2012, 7, e40785. [Google Scholar] [CrossRef] [PubMed]
- Vetriani, C.; Voordeckers, J.W.; Crespo-Medina, M.; O’Brien, C.E.; Giovannelli, D.; Lutz, R.A. Deep-Sea Hydrothermal Vent Epsilonproteobacteria Encode a Conserved and Widespread Nitrate Reduction Pathway (Nap). ISME J 2014, 8, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, I.; Bohnert, K.A.; Cuebas, M.; Keddis, R.; Vetriani, C. Detection and Phylogenetic Analysis of the Membrane-Bound Nitrate Reductase (Nar) in Pure Cultures and Microbial Communities from Deep-Sea Hydrothermal Vents. FEMS Microbiol Ecol 2013, 86, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.C.; Millington, P.; Griffiths, L.; Thomas, G.H.; Cole, J.A. Competition between Escherichia Coli Strains Expressing Either a Periplasmic or a Membrane-Bound Nitrate Reductase: Does Nap Confer a Selective Advantage during Nitrate-Limited Growth? Biochem J 1999, 344 Pt 1, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Heylen, K.; Keltjens, J. Redundancy and Modularity in Membrane-Associated Dissimilatory Nitrate Reduction in Bacillus. Front Microbiol 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vivian, C.; Cabello, P.; Martinez-Luque, M.; Blasco, R.; Castillo, F. Prokaryotic Nitrate Reduction: Molecular Properties and Functional Distinction among Bacterial Nitrate Reductases. J Bacteriol 1999, 181, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Malm, S.; Tiffert, Y.; Micklinghoff, J.; Schultze, S.; Joost, I.; Weber, I.; Horst, S.; Ackermann, B.; Schmidt, M.; Wohlleben, W.; et al. The Roles of the Nitrate Reductase NarGHJI, the Nitrite Reductase NirBD and the Response Regulator GlnR in Nitrate Assimilation of Mycobacterium Tuberculosis. Microbiology (Reading, Engl.) 2009, 155, 1332–1339. [Google Scholar] [CrossRef]
- Sohm, J.A.; Webb, E.A.; Capone, D.G. Emerging Patterns of Marine Nitrogen Fixation. Nat Rev Micro 2011, 9, 499–508. [Google Scholar] [CrossRef]
- Jouanneau, Y.; Meyer, C.; Naud, I.; Klipp, W. Characterization of an FdxN Mutant of Rhodobacter Capsulatus Indicates That Ferredoxin I Serves as Electron Donor to Nitrogenase. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1995, 1232, 33–42. [Google Scholar] [CrossRef]
- Price, R.E.; Lesniewski, R.; Nitzsche, K.; Meyerdierks, A.; Saltikov, C.; Pichler, T.; Amend, J. Archaeal and Bacterial Diversity in an Arsenic-Rich Shallow-Sea Hydrothermal System Undergoing Phase Separation. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- Rathgeber, C.; Yurkova, N.; Stackebrandt, E.; Beatty, J.T.; Yurkov, V. Isolation of Tellurite- and Selenite-Resistant Bacteria from Hydrothermal Vents of the Juan de Fuca Ridge in the Pacific Ocean. Appl. Environ. Microbiol. 2002, 68, 4613–4622. [Google Scholar] [CrossRef]
- Vetriani, C.; Chew, Y.S.; Miller, S.M.; Yagi, J.; Coombs, J.; Lutz, R.A.; Barkay, T. Mercury Adaptation among Bacteria from a Deep-Sea Hydrothermal Vent. Appl. Environ. Microbiol. 2005, 71, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Crepo-Medina, M.; Chatziefthimiou, A.D.; Bloom, N.S.; Luther, G.W.I.; Wright, D.D.; Reinfelder, J.R.; Vetriani, C.; Barkay, T. Adaptation of Chemosynthetic Microorganisms to Elevated Mercury Concentrations in Deep-Sea Hydrothermal Vents. Limnol. Oceanogr. 2009, 54, 41–49. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, T.; Kuroda, M.; Rosen, B.P. Metalloid Resistance Mechanisms in Prokaryotes. J Biochem 1998, 123, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Dopson, M.; Baker-Austin, C.; Koppineedi, P.R.; Bond, P.L. Growth in Sulfidic Mineral Environments: Metal Resistance Mechanisms in Acidophilic Micro-Organisms. Microbiology 2003, 149, 1959–1970. [Google Scholar] [CrossRef]
- Barkay, T.; Miller, S.M.; Summers, A.O. Bacterial Mercury Resistance from Atoms to Ecosystems. FEMS Microbiology Reviews 2003, 27, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Mathema, V.B.; Thakuri, B.C.; Sillanpää, M. Bacterial <Emphasis Type=“Italic”>mer</Emphasis> Operon-Mediated Detoxification of Mercurial Compounds: A Short Review. Arch Microbiol 2011, 193, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Sabaty, M.; Avazeri, C.; Pignol, D.; Vermeglio, A. Characterization of the Reduction of Selenate and Tellurite by Nitrate Reductases. Appl. Environ. Microbiol. 2001, 67, 5122–5126. [Google Scholar] [CrossRef]
- Oremland, R.S.; Blum, J.S.; Bindi, A.B.; Dowdle, P.R.; Herbel, M.; Stolz, J.F. Simultaneous Reduction of Nitrate and Selenate by Cell Suspensions of Selenium-Respiring Bacteria. Appl. Environ. Microbiol. 1999, 65, 4385–4392. [Google Scholar] [CrossRef]
- Ledgham, F.; Quest, B.; Vallaeys, T.; Mergeay, M.; Covès, J. A Probable Link between the DedA Protein and Resistance to Selenite. Res. Microbiol. 2005, 156, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Jain, M.; Sena-Vélez, M.; Jones, K.M.; Fleites, L.A.; Heck, M.; Gabriel, D.W. Tad Pilus-Mediated Twitching Motility Is Essential for DNA Uptake and Survival of Liberibacters. PLoS ONE 2021, 16, e0258583. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.; Wang, N. The Tad Pilus Apparatus of ‘ Candidatus Liberibacter Asiaticus’ and Its Regulation by VisNR. MPMI 2019, 32, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Cai, L.; Fleites, L.A.; Munoz-Bodnar, A.; Davis, M.J.; Gabriel, D.W. Liberibacter Crescens Is a Cultured Surrogate for Functional Genomics of Uncultured Pathogenic ‘ Candidatus Liberibacter’ Spp. and Is Naturally Competent for Transformation. Phytopathology® 2019, 109, 1811–1819. [Google Scholar] [CrossRef]
Figure 1.
Maximum likelihood phylogenetic tree derived from 16S rRNA gene sequences showing the position of Varunaivibrio sulfuroxidans strain TC8T (in bold) within the Alphaproteobacteria. Bootstrap values were based on 500 replicates and are shown at each node. Bar, 0.02% substitutions per position. Sequences belonging to the Rickettsia were used as the outgroup.
Figure 1.
Maximum likelihood phylogenetic tree derived from 16S rRNA gene sequences showing the position of Varunaivibrio sulfuroxidans strain TC8T (in bold) within the Alphaproteobacteria. Bootstrap values were based on 500 replicates and are shown at each node. Bar, 0.02% substitutions per position. Sequences belonging to the Rickettsia were used as the outgroup.
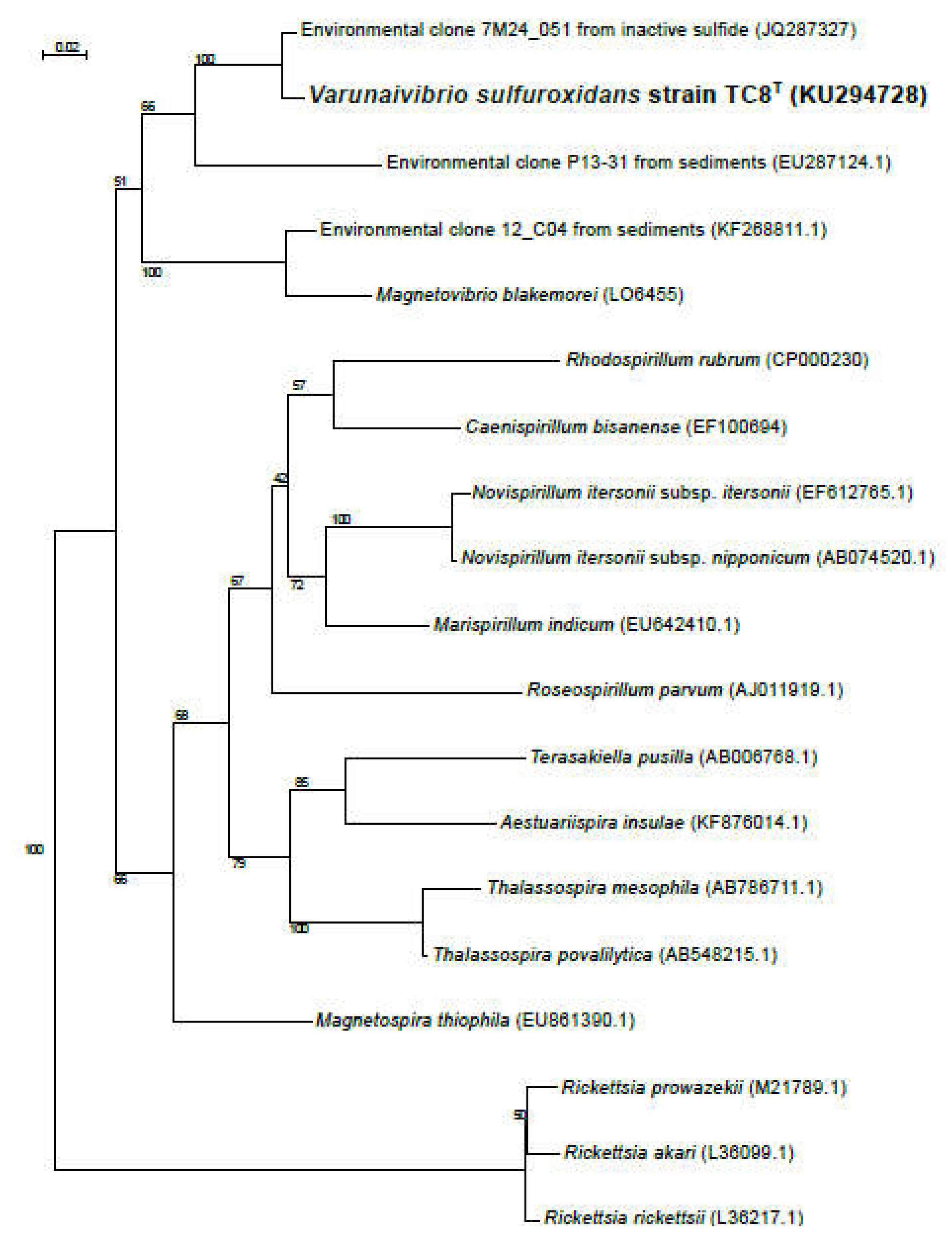
Figure 2.
A. Genome structure of Varunaivibrio sulfuroxidans strain TC8T. Features, starting with the outermost circle: 1. Gene distribution within strain TC8 T genome; 2. Predicted genomic islands in the genome of strain TC8 T by IslandPath-DIMOB (blue), SIGI-HMM (yellow); Predicted by all tools within IslandViewer (orange); 3. GC skew; 4. Genetic Information Processing (blue lines); 5. Membrane transport (orange lines); 6. Carbon metabolism: glycolysis/gluconeogenesis, TCA and CBB cycle (green lines); 7. nitrogen metabolism (purple lines) sulfur metabolism (yellow lines). B. Complete genome of the prophage of Varunaivibrio sulfuroxidans strain TC8T.
Figure 2.
A. Genome structure of Varunaivibrio sulfuroxidans strain TC8T. Features, starting with the outermost circle: 1. Gene distribution within strain TC8 T genome; 2. Predicted genomic islands in the genome of strain TC8 T by IslandPath-DIMOB (blue), SIGI-HMM (yellow); Predicted by all tools within IslandViewer (orange); 3. GC skew; 4. Genetic Information Processing (blue lines); 5. Membrane transport (orange lines); 6. Carbon metabolism: glycolysis/gluconeogenesis, TCA and CBB cycle (green lines); 7. nitrogen metabolism (purple lines) sulfur metabolism (yellow lines). B. Complete genome of the prophage of Varunaivibrio sulfuroxidans strain TC8T.
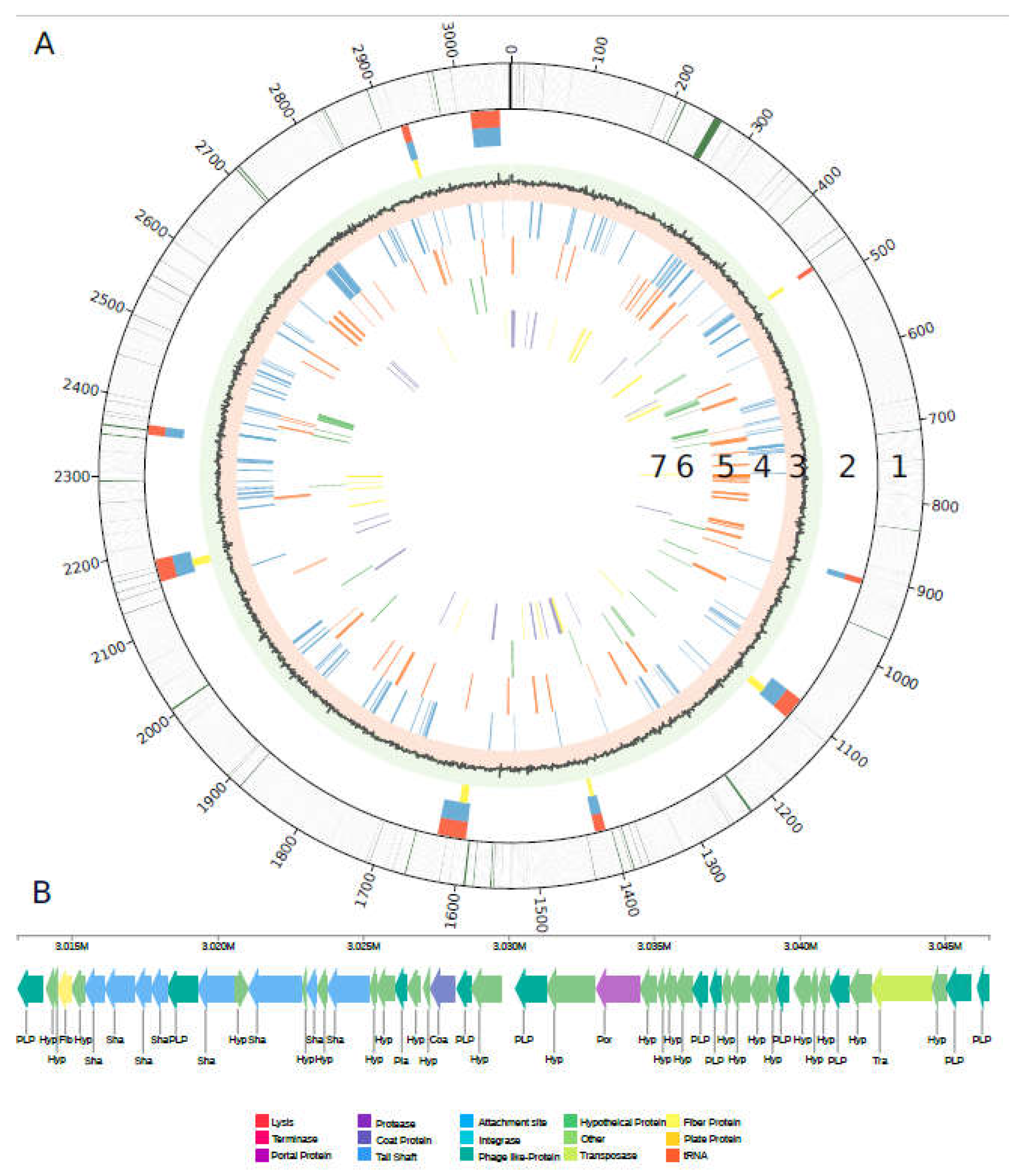
Figure 3.
Genomic reconstruction of the central metabolism and DNA uptake system of Varunaivibrio sulfuroxidans strain TC8T. Enzymes are reported as in Table S1. Abbreviations: DENITRIFICATION: NapABCFGHL: periplasmic nitrate reductase complex; NarGHJI: cytoplasmic nitrate reductase complex; NirS: nitrite reductase; NorBC: nitric oxide reductase; NosZ: nitrous oxide reductase. ENERGY CONSERVATION: ATP synthetase complex; THIOSULFATE/SULFUR OXIDATION: Sqr: sulfide:quinone oxidoreductase; Fcc: putative cytochrome C sulfide dehydrogenase; SoxYZABX: thiosulfate oxidation; Dsr complex: sulfite reductase; AprAB: adenylylsulfate reductase; Sat: sulfate adenylyltransferase. MICROAEROBIC RESPIRATION: Cyt bd2 oxidase: cytochrome bd2; Cyt cbb3: cytochrome c oxidase; Cyt bc1: ubiquinol cytochrome c reductase. NITRATE ASSIMILATION: Nas. NITROGEN FIXATION: NifHDK: nitrogenase complex. FLAGELLAR COMPLEX: for simplicity single unit names are not reported. CITRIC ACID CYCLE: citrate synthase (672); aconitate hydratase (2402); isocitrate dehydrogenase (1364); 2-oxoglutarate dehydrogenase complex (2460/2463/2464); succinyl-CoA ligase (2458/2459); succinate dehydrogenase (2454/2455); fumarate hydratase (946); malate dehydrogenase (2457). GLYCOLYSIS: Eno: enolase; Pgm: phosphoglycerate mutase; Pgk: phosphoglycerate kinase; Gapdh: glyceraldehyde 3-phosphate dehydrogenase; Fba: fructose-bisphosphate aldolase; Tim: triosephosphate isomerase; Fbp: fructose-1,6-bisphosphatase; Pgi: phosphoglucose isomerase; Pgm: Phosphoglucomutase. CALVIN-BENSON-BASSHAM CYCLE: Prk: phosphoribulokinase. CARBON CONCENTRATING MECHANISM (CCM): CA: carbonic anhydrase. SECRETION SYSTEM and COMPETENCE: TadBCDG and CpaABCDEF: Type IV secretion system complex and pilus assembly; ComEC: DNA internalization competence protein. MERCURY REDUCTION: MerA: Mercuric ion reductase. ARSENATE REDUCTION: ArsC: arsenate reductase.
Figure 3.
Genomic reconstruction of the central metabolism and DNA uptake system of Varunaivibrio sulfuroxidans strain TC8T. Enzymes are reported as in Table S1. Abbreviations: DENITRIFICATION: NapABCFGHL: periplasmic nitrate reductase complex; NarGHJI: cytoplasmic nitrate reductase complex; NirS: nitrite reductase; NorBC: nitric oxide reductase; NosZ: nitrous oxide reductase. ENERGY CONSERVATION: ATP synthetase complex; THIOSULFATE/SULFUR OXIDATION: Sqr: sulfide:quinone oxidoreductase; Fcc: putative cytochrome C sulfide dehydrogenase; SoxYZABX: thiosulfate oxidation; Dsr complex: sulfite reductase; AprAB: adenylylsulfate reductase; Sat: sulfate adenylyltransferase. MICROAEROBIC RESPIRATION: Cyt bd2 oxidase: cytochrome bd2; Cyt cbb3: cytochrome c oxidase; Cyt bc1: ubiquinol cytochrome c reductase. NITRATE ASSIMILATION: Nas. NITROGEN FIXATION: NifHDK: nitrogenase complex. FLAGELLAR COMPLEX: for simplicity single unit names are not reported. CITRIC ACID CYCLE: citrate synthase (672); aconitate hydratase (2402); isocitrate dehydrogenase (1364); 2-oxoglutarate dehydrogenase complex (2460/2463/2464); succinyl-CoA ligase (2458/2459); succinate dehydrogenase (2454/2455); fumarate hydratase (946); malate dehydrogenase (2457). GLYCOLYSIS: Eno: enolase; Pgm: phosphoglycerate mutase; Pgk: phosphoglycerate kinase; Gapdh: glyceraldehyde 3-phosphate dehydrogenase; Fba: fructose-bisphosphate aldolase; Tim: triosephosphate isomerase; Fbp: fructose-1,6-bisphosphatase; Pgi: phosphoglucose isomerase; Pgm: Phosphoglucomutase. CALVIN-BENSON-BASSHAM CYCLE: Prk: phosphoribulokinase. CARBON CONCENTRATING MECHANISM (CCM): CA: carbonic anhydrase. SECRETION SYSTEM and COMPETENCE: TadBCDG and CpaABCDEF: Type IV secretion system complex and pilus assembly; ComEC: DNA internalization competence protein. MERCURY REDUCTION: MerA: Mercuric ion reductase. ARSENATE REDUCTION: ArsC: arsenate reductase.
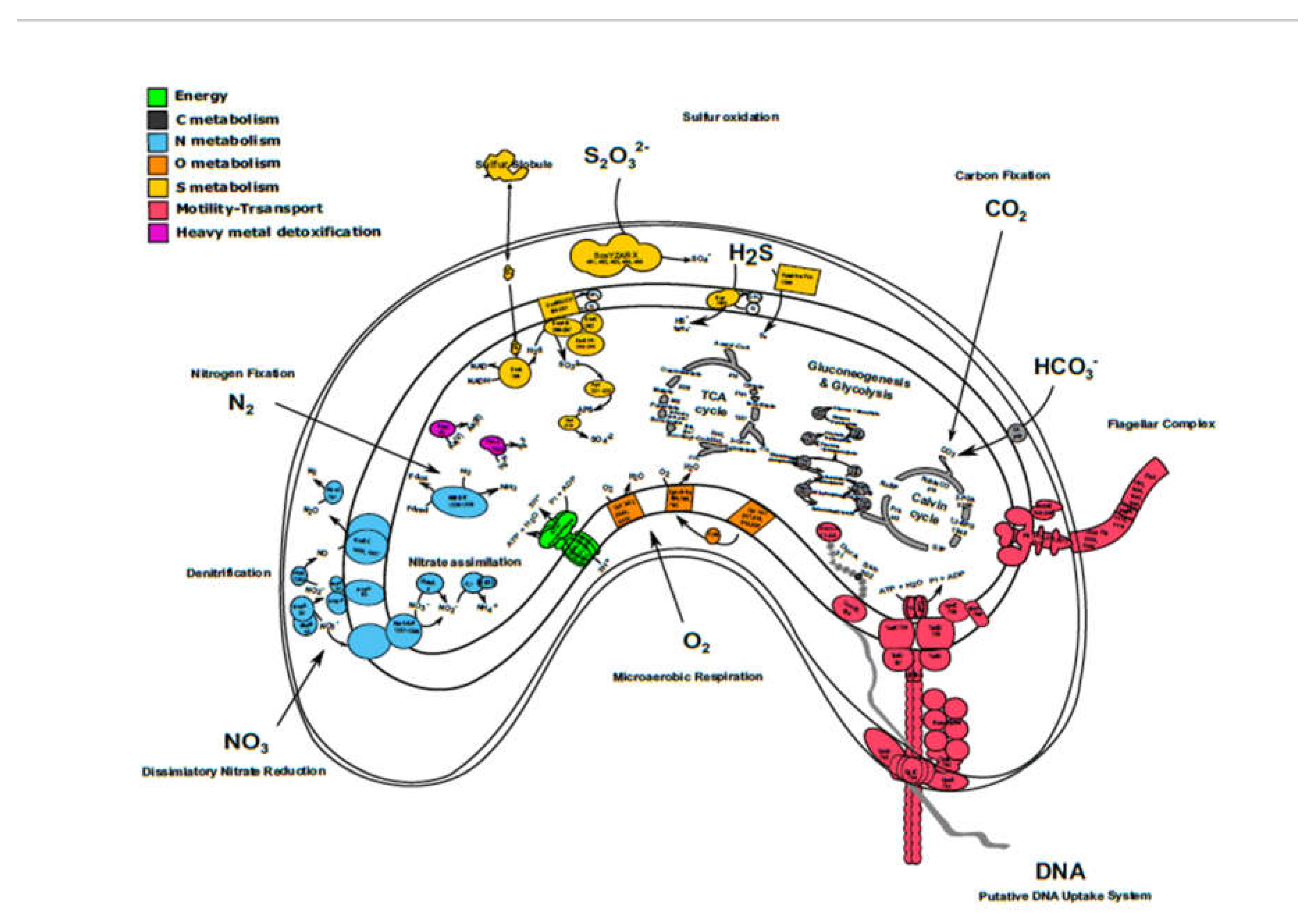
Figure 4.
Maximum likelihood phylogenetic tree showing the position of the nitrogenase (NifH) of Varunaivibrio sulfuroxidans strain TC8T (in bold). Bootstrap values based on 500 replications are shown at branch nodes. Bar, 0.05% substitutions per position.
Figure 4.
Maximum likelihood phylogenetic tree showing the position of the nitrogenase (NifH) of Varunaivibrio sulfuroxidans strain TC8T (in bold). Bootstrap values based on 500 replications are shown at branch nodes. Bar, 0.05% substitutions per position.
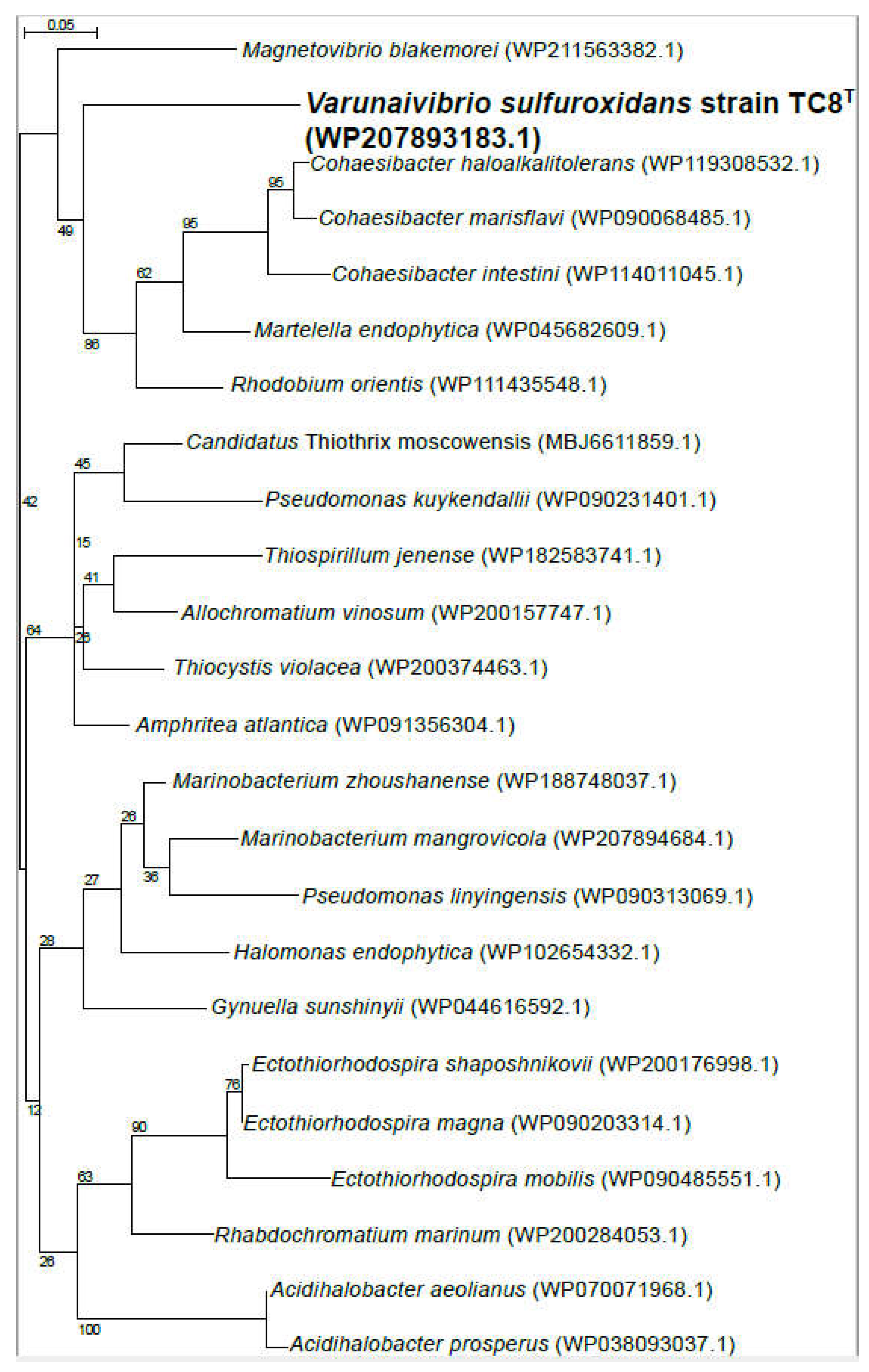
Figure 5.
Organization of the tad/cpa gene cluster in Magnetovibrio blakemorei strain MV-1, Varunaivibrio sulfuroxidans strain TC8T and Liberibacter crescens strain BT-1. Individual genes are drawn to scale.
Figure 5.
Organization of the tad/cpa gene cluster in Magnetovibrio blakemorei strain MV-1, Varunaivibrio sulfuroxidans strain TC8T and Liberibacter crescens strain BT-1. Individual genes are drawn to scale.
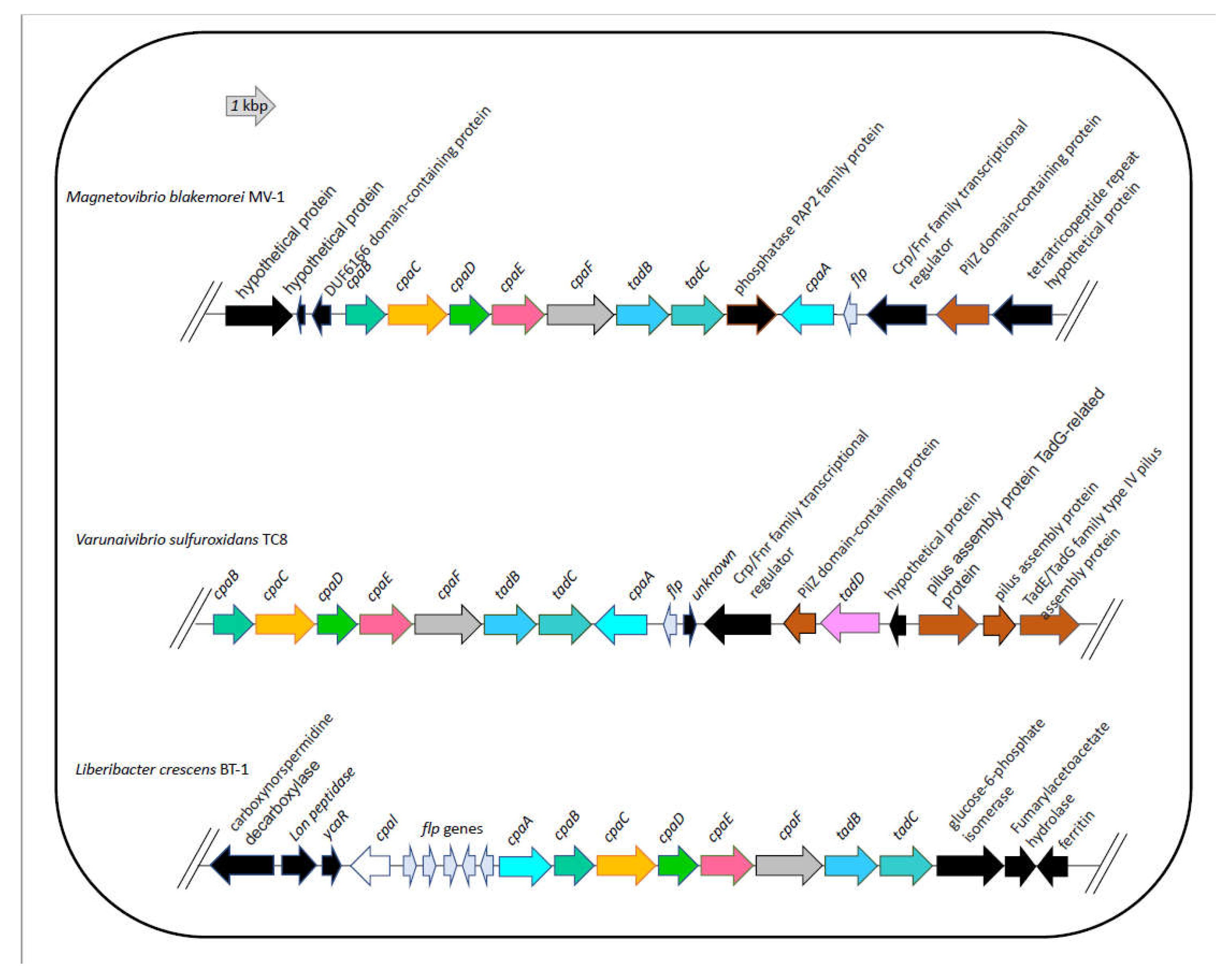
Figure 6.
Maximum likelihood phylogenetic tree showing the position of the CpaC component of the secretion system of Varunaivibrio sulfuroxidans strain TC8T (in bold). Bootstrap values based on 500 replications are shown at branch nodes. Bar, 0.2% substitutions per position.
Figure 6.
Maximum likelihood phylogenetic tree showing the position of the CpaC component of the secretion system of Varunaivibrio sulfuroxidans strain TC8T (in bold). Bootstrap values based on 500 replications are shown at branch nodes. Bar, 0.2% substitutions per position.
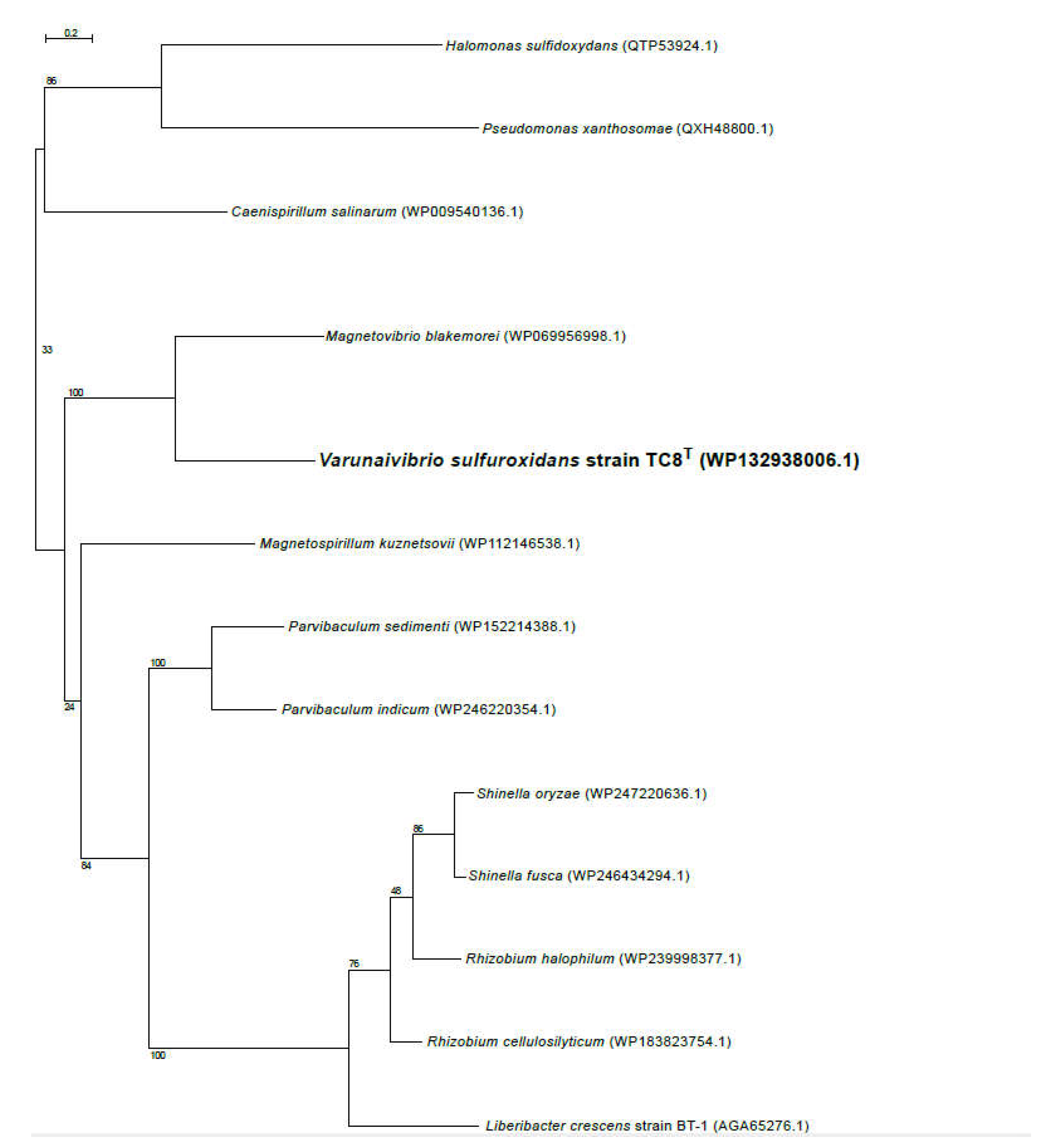
Figure 7.
Maximum likelihood phylogvvvvvvvenetic tree showing the position of the ComEC component of the competence apparatus of Varunaivibrio sulfuroxidans strain TC8T (in bold). Bootstrap values based on 500 replications are shown at branch nodes. Bar, 0.5% substitutions per position.
Figure 7.
Maximum likelihood phylogvvvvvvvenetic tree showing the position of the ComEC component of the competence apparatus of Varunaivibrio sulfuroxidans strain TC8T (in bold). Bootstrap values based on 500 replications are shown at branch nodes. Bar, 0.5% substitutions per position.
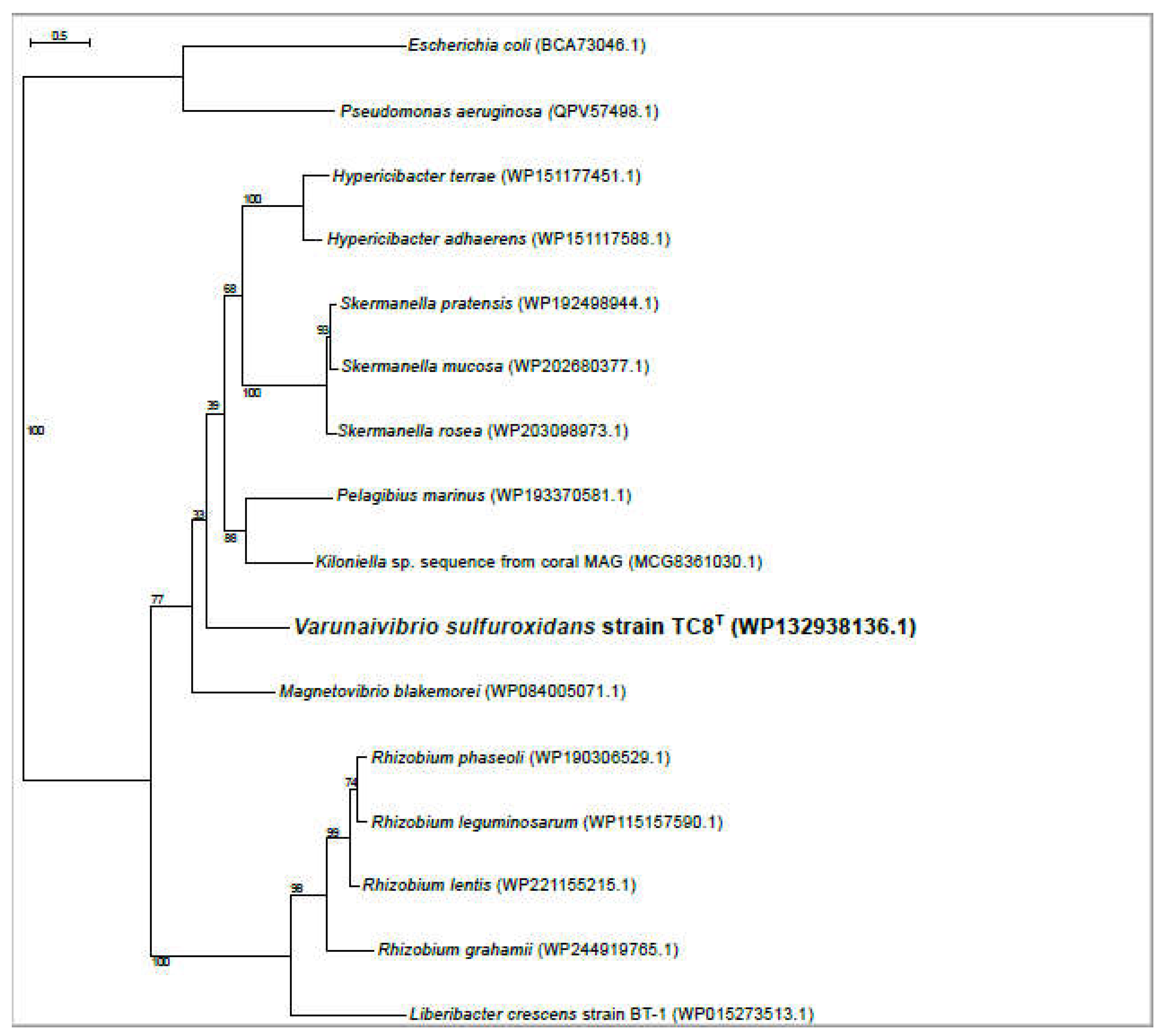

Table 1.
Genome statistics.
| Attribute | Genome (Total) | |
|---|---|---|
| Value | % Total1 | |
| Size (bp) | 3066297 | 100.00% |
| G+C content (bp) | 1821569 | 59.41% |
| Coding region (bp) | 2702087 | 88.12% |
| Total genes | 2890 | 100.00% |
| RNA genes | 61 | 2.11% |
| Protein-coding genes | 2829 | 97.89% |
| Genes assigned to Pfam3 | 2450 | 84.78% |
| Genes assigned to COGs | 2105 | 72.84% |
| Genes assigned in KEGG Orthology (KO) | 1641 | 56.78% |
| Genes assigned to Subsystems | 1568 | 54.25% |
| Genes coding signal peptides | 160 | 5.54% |
| Genes coding transmembrane proteins | 745 | 25.78% |
| CRISPRs | 1 | |
| Intact prophage | 1 | |
1 The % total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
Table 2.
Number of genes associated with the 25 general COG functional categories.
| Code | Value | % Total1 | Description |
|---|---|---|---|
| E | 10.32% | 239 | Amino acid transport and metabolism |
| C | 8.93% | 207 | Energy production and conversion |
| J | 7.60% | 176 | Translation, ribosomal structure and biogenesis |
| P | 7.51% | 174 | Inorganic ion transport and metabolism |
| R | 6.78% | 157 | General function prediction only |
| H | 5.91% | 137 | Coenzyme transport and metabolism |
| T | 5.91% | 137 | Signal transduction mechanisms |
| O | 5.74% | 133 | Posttranslational modification, protein turnover, chaperones |
| M | 5.44% | 126 | Cell wall/membrane/envelope biogenesis |
| K | 4.96% | 115 | Transcription |
| G | 4.92% | 114 | Carbohydrate transport and metabolism |
| S | 4.75% | 110 | Function unknown |
| I | 3.84% | 89 | Lipid transport and metabolism |
| L | 3.63% | 84 | Replication, recombination and repair |
| F | 2.81% | 65 | Nucleotide transport and metabolism |
| N | 2.68% | 62 | Cell motility |
| Q | 1.90% | 44 | Secondary metabolites biosynthesis, transport and catabolism |
| V | 1.77% | 41 | Defense mechanisms |
| U | 1.42% | 33 | Intracellular trafficking, secretion, and vesicular transport |
| X | 1.38% | 32 | Mobilome: prophages, transposons |
| D | 1.25% | 29 | Cell cycle control, cell division, chromosome partitioning |
| W | 0.47% | 11 | Extracellular structures |
| B | 0.04% | 1 | Chromatin structure and dynamics |
| - | 27.16% | 785 | Not in COG |
1 The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



