
Submitted:
12 April 2023
Posted:
13 April 2023
You are already at the latest version
Abstract
Epithelial ovarian cancer (EOC) remains the most lethal gynecologic malignancy, largely due to metastasis and drug resistant recurrences. Fifteen percent of ovarian tumors carry mutations in BRCA1 or BRCA2, rendering them vulnerable to treatment with PARP inhibitors such as olaparib. Recent studies have shown that TGFβ can induce “BRCAness” in BRCA wild-type cancer cells. Given that TGFβ is a known driver of epithelial to mesenchymal transition (EMT), and the con-nection between EMT and metastatic spread in EOC and other cancers, we asked if TGFβ and EMT alter susceptibility of EOC to PARP inhibition. Epithelial EOC cells were transiently treated with soluble TGFβ and their clonogenic potential, expression and function of EMT and DNA repair genes, and response to PARP inhibitors compared with untreated controls. A second epithelial cell line was compared to its mesenchymal derivative for EMT and DNA repair gene expression and drug responses. We found that TGFβ and EMT resulted in downregulation of genes responsible for homologous recombination (HR) and sensitized cells to olaparib. HR efficiency was reduced in a dose-dependent manner. Furthermore, mesenchymal cells displayed sensitivity to olaparib, cis-platin, and the DNA-PK inhibitor Nu-7441. Therefore, treatment of disseminated, mesenchymal tumors may represent an opportunity to expand clinical utility of PARP inhibitors and similar agents.
Keywords:
Ovarian cancer
; PARP inhibitors
; epithelial to mesenchymal transition
; drug response
1. Introduction
Epithelial ovarian cancer (EOC) remains the leading cause of death among gynecologic malignancies, with approximately 20,000 new cases and over 13,000 deaths anticipated in 2023 [1]. Most EOC cases are diagnosed late, typically at stage III or IV, and despite generally favorable responses to chemotherapy, disease often recurs following treatment. Such recurrences are often widely metastatic and resistant to first line therapeutic agents. Five-year survival for stage III/IV disease hovers around 30% [2]. Therefore, new treatment paradigms are urgently needed.
Fifteen percent of EOC tumors carry mutations or deletions of BRCA1 or BRCA2 [3], a defect in homologous recombination (HR) that renders these tumor cells vulnerable to synthetic lethality induced by inhibitors of poly (ADP-ribose) polymerase (PARP). PARP is required for efficient repair of single stranded breaks (SSBs) by base excision repair (BER). SSBs left unchecked will become double strand breaks (DSBs) upon replication of DNA. Defective HR due to BRCA1/2 loss results in cells depending heavily on PARP-mediated repair such as BER, thus blockade of both HR and BER leads to synthetic lethality [4,5]. The PARP inhibitor (PARPi) olaparib has become standard therapy for ovarian tumors with BRCA mutations, in the settings of initial or recurrent maintenance therapy [6]. However, resistance can develop due to reversion of BRCA to wild-type (WT) [7,8]. Thus, whether reverted or simply never mutated, most EOC tumors are not currently considered candidates for olaparib therapy.
Recent work from our group has investigated the use of olaparib in combination with additional agents for treatment of BRCA-WT cancer. In particular, we have found that triapine, an inhibitor of ribonuclease reductase, can sensitize EOC cells to olaparib, topoisomerase inhibitors, or platinum chemotherapy to kill cancer cells in the presence of WT BRCA1/2 [9,10]. We also characterized an additional ribonucleotide reductase inhibitor, DB4, which showed a similar effect [11]. Further work found that combining olaparib with triapine and the vascular endothelial growth factor receptor (VEGFR) inhibitor cediranib was effective against BRCA-WT and PARPi-resistant EOC xenografts [12].
These studies demonstrate the potential of PARP inhibition in BRCA WT tumors, but in the present study we sought to identify intrinsic conditions that would sensitize BRCA WT cells to PARPi without the need for combination therapy. We therefore turned our attention to transforming growth factor beta (TGFβ), a well characterized cytokine and known oncogene that has been associated with tumor progression and epithelial to mesenchymal transition (EMT) [13]. TGFβ signals via serine/threonine kinase type receptors to activate a number of downstream signals, including SMAD-dependent and -independent pathways (Figure 1) [14,15,16]. Recent work has linked TGFβ to DNA damage response. TGFβ-deficient murine mammary cells display impaired response to DNA damage from ionizing radiation [17]. In a mouse model of prostate cancer, radiation increased TGFβ expression [18,19]. TGFβ also contributes to bone marrow failure in Fanconi anemia, while its inhibition promoted HR over the alternative pathways non-homologous end joining (NHEJ) [20]. In 2014, Liu et al. found that TGFβ could induce a state resembling BRCA1/2 loss, often termed “BRCAness” by downregulating DNA repair factors in breast cancer, thus sensitizing breast cancer cells to PARPi [21]. However, whether TGFβ plays a similar role in EOC is unknown.
In addition to TGFβ signaling, we investigated the impact of EMT as a whole on DNA repair and responses to PARPi. EMT involves the loss of cell adhesion molecules binding epithelial cells in a monolayer and upregulation of new cell surface proteins that favor motility and survival without attachment [22,23,24,25]. As such, EMT and its drivers such as TWIST1 have long been associated with metastatic spread of solid tumors, including ovarian[22,26,27]. The relationship of EMT to drug response, however, is complicated. In EOC, CD44+ cancer stem cells are inherently chemoresistant and are epithelial in character, while their CD44- mesenchymal progeny are fast-dividing and therefore chemosensitive [28]. However, across many other tumor types, EMT is correlated with stemness [29,30]. Among mesenchymal ovarian cancer cells, EMT correlates with drug resistance and reduction of EMT factors such as TWIST1 has been shown to sensitize such cells to chemotherapy [31,32]. Here we investigate the effect of EMT on HR and olaparib response.
We demonstrate that either transient TGFβ signaling or acquisition of mesenchymal phenotype via EMT can reduce HR factor expression in EOC cells, leading to sensitivity to olaparib. Given the prevalence of metastatic disease amongst recurrent EOC, the ability to efficiently eliminate mesenchymal cells shows great promise in combatting advanced disease. When combined with our prior studies on combination therapies, our findings support the continued investigation and clinical use of PARPi in BRCA WT EOC tumors.
2. Materials and Methods
2.1. Chemicals and Reagents
Cisplatin was obtained from Calbiochem/EMD Millipore (Billerica, MA, USA). Olaparib and cediranib were obtained from Selleck Chemicals (Houston, TX, USA). TGFβ and LY2109761 were obtained from Cayman Chemical (Ann Arbor, MI, USA). Triapine was synthesized in the Ratner laboratory at Yale University School of Medicine.
2.2. Cell Lines
PEO1 and PEO4 cells were a gift from Dr. Peter Glazer at Yale University. These lines were derived from a single patient at different points in the course of treatment for ovarian carcinoma [33]. PEO1 is a BRCA2-mutant, chemosensitive line with mesenchymal morphology. PEO4 was derived from recurrence following platinum-based chemotherapy. It carries a BRCA2 reversion to WT, is chemoresistant, and displays epithelial morphology. Both lines were maintained in DMEM with 10% FBS and 1% penicillin/streptomycin. R182 and TARA-R182 cells (also referred to as CD44+ OCSC-1 and CD44- OCC-1, respectively) were a gift from Dr. Gil Mor at Wayne State University and have been described previously [34,35]. Both lines were maintained in RPMI-1640 with 10% FBS, and 1% each of penicillin/streptomycin, non-essential amino acids, sodium pyruvate, and HEPES. SKOV-3 cells (HTB-77) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in McCoy’s 5A or RMPI-1640 with 10% FBS and 1% penicillin/streptomycin. All cells were grown in a humidified 37°C incubator at 5-6% CO2.
2.3. Clonogenic Assays
Cells were seeded in triplicate wells of six-well plates and allowed to adhere for 24h. Cells were then treated with cisplatin, olaparib, TGFβ, and/or LY2109761 and incubated undisturbed in standard cell culture conditions for 10-21 days as indicated. At the conclusion of this window, medium was removed, and cells were fixed and stained in 0.5% crystal violet in 50% methanol. Excess dye was washed out with water and plates were air dried. Images were obtained using a GelDoc imager, and colonies were counted using QuantityOne analysis software (both from Bio-Rad, Hercules, CA, USA). Where colony counting was impossible due to the growth pattern of cells, a standard digital camera image was obtained of each plate.
2.4. Western Blotting
Cell samples were lysed and protein concentrations determined by DC Protein Assay (Bio-Rad) or BCA Assay (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturers’ protocols. Equal masses of protein were run on Mini-PREOTEAN TGX gels (Bio-Rad) and transferred to nitrocellulose or PVDF membranes. Membranes were blocked in 5% milk in phosphate- or Tris-buffered saline with 0.5% Tween-20 (PBST or TBST) for >40 minutes at room temperature. Membranes were incubated with primary antibodies in 5% BSA in PBST/TBST overnight at 4°C. Membranes were then rinsed in PBST/TBST, incubated with HRP-conjugated secondary antibodies for 1-2h at room temperature, and rinsed again with PBST/TBST. Blot images were obtained using enhanced chemiluminescent substrate (Bio-Rad or Denville Scientific, South Plainfield, NJ, USA) and a ChemDoc imager from Syngene (Frederick, MD, USA). Beta actin and HSC-70 were used as loading controls. Anti-BRCA1 (D-9), anti-Rad51 (H-92), anti-β-Actin (C4), and anti-HSC70 (K-19) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-BRCA2 (A303-434A) was from Bethyl Laboratories (Fortis, Waltham, MA, USA). Anti-fibronectin (MS-1351) anti-snail (3879S), and anti-N-Cadherin (MA1-2002) were from ThermoFisher (Waltham, MA, USA). Anti-vimentin (3390S), anti-E-Cadherin (5296S), anti-AKT (4691S), and anti-pAKT (4058S) were from Cell Signaling Technology (Danvers, MA, USA).
2.5. MTS Assay
1000-3000. cells per well were plated in 96-well plates and allowed to adhere for 24h. The following day, cells were treated with cisplatin, olaparib, or cediranib and incubated in the presence of drug and 100 μl medium per well for 72h. Wells containing media only and no cells were used to establish background. After 72h, 20 μl MTS (CellTiter 96 AQueous One Solution, Promega) was added to each well and cells were incubated for a further 2h. Absorbance at 490 nm was then read using a plate reader. Background was subtracted and absorbance was converted to percent viability compared with controls.
2.6. HRR Assay
SKOV-3-DR-GFP cells were pretreated with 5ng/ml or 20ng/ml TGFβ for 24 hours before being transiently transfected with the empty vector pcDNA/Neo (Thermo Fisher) or the I-SceI endonuclease expression vector pCBASceI using the TransFast reagent (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Five hours after transfection, cells were retreated with 5ng/ml TGFβ, 20ng/ml TGFβ, or 0.75μM triapine for 48 hours. Cells were trypsinized and analyzed for the percentage of GFP-positive cells by flow cytometry using a LSR II flow cytometer and FlowJo software (BD Biosciences, East Rutherford, NJ, USA).
2.7. Quantitative reverse transcription PCR
Total RNA was obtained from R182 and TARA-R182 cell pellets using the Total RNA Kit from IBI Scientific (Dubuque, IA, USA). RNA yield and quality was assayed using absorbance spectra on a NanoDrop 2000 (Thermo Fisher). cDNA was reverse transcribed using the qScript cDNA synthesis kit from Quantabio (Beverly, MA, USA). Quantitative PCR was run on an Applied Biosystems Quantstudio 5 qPCR machine (Thermo Fisher), using PerfeCTa SYBR Green FastMix, Low ROX (Quantabio) in 10 μl reactions. PCR was run for 40 cycles, followed by melt curve analysis. Beta actin was used as a housekeeping gene. Relative gene expression levels were calculated using the 2-ΔΔCt method. Primers used were: BRCA2-Fd, 5'-AAAACGTTGAGCTGTTGCCA-3'; BRCA2-Rv, 5'-TGTGTTTTGGTTGAATTGTACCTT-3'; RAD51 Fd, 5'-CCAGACCCAGCTCCTTTACC-3'; RAD51 Rv, 5'-CACTGCGACACCAAACTCATC-3'; BRCA1 Fd, 5′-GGCTATCCTCTCAGAGTGACATTT-3′; BRCA1 Rv, 5′-GCTTTATCAGGTTATGTTGCATGGT-3′; Actin Fd, 5'-TTCCTGGGCATGGAGTCC-3'; Actin Rv, 5'-CAGGTCTTTGCGGATGTCC-3'.
2.8. Wound healing assay
Cells were plated in 6-well plates and incubated 24 hours to allow formation of a complete monolayer. Where indicated, cells were treated with 5ng/ml TGFβ for one hour prior to scratching. Scratches were made using a sterile P200 pipet tip. Images of the wound were obtained immediately and after 24 hours.
2.9. Statistics
Data shown are mean ± S.E. qPCR data incorporate standard deviation in the calculation of fold change ranges. Data were compared with the respective control in each experiment using Student's t test in GraphPad Prism. A p value <0.05 was considered statistically significant. All experiments were repeated at least twice.
3. Results
3.1. TGFβ induces mesenchymal markers and morphology in ovarian cancer cells
We first chose to examine the effects of TGFβ on a pair of ovarian cancer cell lines derived from a single patient. PEO1 has mesenchymal-like morphology when grown as a monolayer (Figure 2A) and is BRCA2 mutant and therefore sensitive to olaparib. PEO4 was derived from a recurrence and has a reversion of BRCA2 to WT, rendering it resistant to olaparib. Interestingly, its morphology is more epithelial (Figure 2B) and its growth rate slower than PEO1, suggesting an epithelial-like subpopulation acquired a novel BRCA2 mutation that allowed it to outcompete a faster-growing but drug-responsive majority of tumor cells. The epithelial versus mesenchymal status of the two lines was verified by western blot, which showed elevated expression of Snail, Slug, and fibronectin in PEO1 compared to PEO4. Conversely, E-Cadherin was elevated in PEO4 compared to PEO1 (Figure 2C). When we treated PEO4 cells with the EMT inducing cytokine TGFβ, we observed that they adopt a more fibroblast-like morphology and ultimately resemble PEO1 (Figure 2D). To further examine mesenchymal character, we next assayed migratory potential of the two lines with and without TGFβ treatment. PEO1 exhibited higher migratory ability than PEO4, as shown by wound healing assays (Figure 2E). However, treatment of PEO4 with TGFβ was not sufficient to increase wound healing (Figure 2F).
3.2. TGFβ alters expression of EMT markers in PEO1 and PEO4
We next examined the expression of epithelial and mesenchymal markers in PEO1 and PEO4 cells, with and without TGFβ treatment. TGFβ resulted in increased expression of Snail in both lines, and upregulated fibronectin (FN) in PEO1. Co-treatment with the TGFβ receptor inhibitor LY2109761 abrogated these changes, showing that TGFβ signaling was responsible for the observed increase in marker expression (Figure 3A). TGFβ also upregulated phosphorylated Akt in PEO1, while LY2109761 reduced phospho-Akt in both lines. PEO4 did not express fibronectin, and TGFβ was not sufficient to induce fibronectin expression in this line (Figure 3A). Together, these data indicate that TGFβ induces a partial EMT in PEO4 while further upregulating mesenchymal genes and promoting growth and proliferation in the already mesenchymal PEO1.
3.3. TGFβ downregulates HR proteins in BRCA2 WT cells and sensitizes them to olaparib
Based on prior reports of TGFβ influencing the expression of DNA repair factors [19,20], we also asked whether HR pathway factors would be differentially expressed in our lines and following TGFβ treatment. BRCA1 and BRCA2 were downregulated in PEO1 compared to PEO4, while expression of RAD51 was similar between the two lines (Figure 3A). Treatment of PEO4 with TGFβ decreased expression of BRCA1, BRCA2, and to a lesser degree RAD51, but this effect was not seen in PEO1 (Fig. 3A). Treatment with LY2109761 rescues this effect on BRCA1, although BRCA2 expression following combination treatment remains low (Figure 3A). To determine if loss of HR protein expression translates to loss of HR efficiency, we utilized an HR reporter system built in the BRCA WT cell line SKOV-3 [9,10,11]. Briefly, SKOV-3-DR-GFP cells were treated with TGFβ, or the ribonuclease reductase inhibitor triapine as a positive control. Cells were then transfected with I-SceI nuclease, which cleaves the reporter cassette. Successful repair of the break via HR leads to expression of GFP, which we measured by flow cytometry. Pretreatment with 5 or 20 ng/ml TGFβ significantly reduced the number of GFP+ cells following I-SceI expression, indicating that TGFβ pretreatment impaired HR, though not to the extent seen with triapine (Figure 3B). Given the induction of “BRCAness” in PEO4 cells following TGFβ treatment, we next asked whether response to olaparib would be impacted. Clonogenic assays showed that as expected, PEO4 cells were substantially more resistant to olaparib compared to PEO1. Furthermore, while TGFβ treatment had no effect on the response of PEO1, TGFβ-treated PEO4 cells were sensitized to olaparib, and showed a dose response similar to that of BRCA2 mutant PEO1 (Figure 3C). LY2109761 completely blocked this effect, indicating that the change in response was due to TGFβ signaling (Figure 3D).
3.4. EMT also sensitizes ovarian cancer cells to PARP inhibition
As TGFβ treatment of PEO4 produced only a partial and transient EMT phenotype, we next sought to confirm our findings in EOC cells that had undergone EMT and retained their mesenchymal character indefinitely. To do this, we utilized the R182 and TARA-R182 (hereafter TARA) cell line models first developed and described by Mor and colleagues [28,34,35,36]. R182 cells exhibit classic epithelial “cobblestone” morphology (Figure 4A). TARA cells, which were derived from in vitro EMT of R182 cells, are smaller (Figure 4B), faster growing, and express mesenchymal markers, including the EMT driver TWIST1 (Figure 4C). We first performed clonogenic assays using R182 and TARA cells treated with varying doses of olaparib. The growth pattern of R182 was not conducive to accurate colony counts, however images of stained cells clearly show that for doses up to 5 μM, olaparib reduced the growth rate of R182 but had little effect on the number of colonies formed (Figure 4D). In contrast, treatment of TARA cells with as little as 0.625 μM olaparib produced a notable reduction in colony number, while a 5 μM dose completely abrogated TARA growth (Figure 4D). Cell proliferation assays comparing R182 and TARA cells also showed minimal inhibitory effects of olaparib in R182 cells but a clear dose response in TARA cells (Figure 4E). Thus, the pattern of olaparib sensitivity in mesenchymal cells holds for this model.
3.5. EMT alters HR gene expression
We next sought to determine if changes in olaparib response in TARA versus R182 cells were the result of changes in HR gene expression. Western blot showed that compared to R182, TARA cells expressed lower levels of BRCA2 and RAD51 protein (Figure 5A). However, qPCR demonstrated no significant change in mRNA expression between R182 and TARA for either of these genes, suggesting a post-transcriptional mechanism of BRCA2 and RAD51 loss (Figure 5B). Interestingly, BRCA1 mRNA was upregulated in TARA cells compared to R182, but this was not sufficient to give rise to olaparib resistance (Figure 4).
3.6. EMT induces an alternate DNA repair pathway and opens an additional therapeutic window
Given the fast growth rate of TARA cells despite lower expression of HR proteins, we hypothesized that alternate means of DNA repair may be upregulated. The most common alternate pathway for the repair of DSBs is canonical non-homologous end joining (c-NHEJ). In this process, Ku70 and Ku80 form a complex with the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) and DNA breaks, leading to recruitment of DNA ligase IV and XRCC4 and sealing of the break [37]. We found that in TARA cells, expression of Ku70 and Ku80 were upregulated compared to R182, as were levels of total and phosphorylated DNA-PKcs (Figure 5C). Since TARA cells are increasingly reliant on c-NHEJ for DSB repair, we next asked whether targeting of c-NHEJ could represent an additional therapeutic strategy against mesenchymal cells. We treated R182 and TARA cells with 10 μM Nu-7441, an inhibitor of DNA-PK. As expected, approximately 60% of TARA cells were killed by this dose, while less than 10% of R182 cells were (Figure 5D). However, at this dose, NU-7441 can also inhibit PI3K and mTOR, so we cannot be sure that inhibition of c-NHEJ alone is responsible for TARA cell growth inhibition, especially in the absence of exogenous DNA damage [38].
4. Discussion
Despite much research and the advent of PARPi-based therapy, ovarian cancer remains a highly deadly disease, with more than 13,000 deaths anticipated in the United States in 2023 [1]. Chief among the clinical challenges of EOC are recurrence and therapy resistance, which often coincide with widely metastatic disease. Only a minority of EOC tumors carry a BRCA mutation or other HR defect, meaning most patients are not normally candidates for PARPi-based therapy [3]. However, we have recently made the case for opening a therapeutic window for the use of olaparib in BRCA WT tumors by inducing “BRCAness”, for example using olaparib in combination with triapine [10,12]. In the present study, we sought additional cancer cell populations that would be vulnerable to PARPi. Recently, it was demonstrated that TGFβ sensitized breast cancer cells to PARPi by inducing “BRCAness” [39]. TGFβ has been well characterized as a driver of EMT, a developmental program that is reactivated in cancers in response to a stressful environment and gives rise to metastatic spread [22,40,41,42]. Given the association between metastatic progression and mortality in EOC, we asked whether TGFβ and EMT would impact DNA repair and PARPi response in EOC.
We first established the EMT status, DNA repair state, and drug response profile of PEO1 and PEO4 cells. PEO1 cells display mesenchymal-like character and sensitivity to olaparib on the basis of a BRCA2 mutation. Interestingly, PEO4 shows altered morphology with growth in islands, increased E-Cadherin expression, and loss of fibronectin expression, suggesting a more epithelial phenotype (Figure 2C). By restoring BRCA2 expression and function, PEO4 cells developed resistance to olaparib. However, treatment of PEO4 cells with the pro-EMT cytokine TGFβ was sufficient to reduce levels of BRCA2, RAD51, and BRCA1 protein and sensitize PEO4 cells to olaparib (Fig. 3). Notably, the dose response for these cells post-TGFβ was comparable to that of PEO1, a line that lacks functional BRCA2. Moreover, an inhibitor of TGFβ signaling was sufficient to abrogate changes in both gene expression and drug response, demonstrating that the effect is TGFβ-dependent. Furthermore, TGFβ upregulated phospho-Akt in PEO1 cells, while inhibition of TGFβ signaling reduced Akt activation. This is in alignment with prior work showing that EMT can impact expression and activation of Akt, and that Akt promotes survival and drug resistance [32,43].
As the PEO4 line was established at a later time than their PEO1 counterparts, it is possible that in addition to a reversion of BRCA2 to WT, PEO4 cells have undergone a mesenchymal to epithelial transition (MET) in vivo. EMT is often thought of as an irreversible process; it is more likely that tumor cells undergo a transient or incomplete EMT that allows them to retain some epithelial characteristics and seed secondary sites [44,45,46]. In the case of this model, PEO4 may have just shifted further back toward an epithelial state. Alternatively, the features of PEO4 may suggest that a subpopulation of drug-resistant epithelial cells were selected for by initial rounds of platinum-based chemotherapy. This interpretation is consistent with work that identified epithelial cancer stem cells that were resistant to therapy and could repopulate a tumor [28]. It is also consistent with the concept of tumor heterogeneity, which recognizes that tumors are mosaics of several lineages of cancer cells within a single patient [47]. Tumor heterogeneity represents a barrier to therapy due to the challenge of killing all lineages. The need to address multiple populations of cells may mean that combinations of therapeutics with different mechanisms of action (including targeting DNA repair) will be more successful than monotherapy or multiple lines of monotherapy.
Following our studies of PEO1 and PEO4, we went on to ask whether an additional model, representing EMT as opposed to transient TGFβ exposure, would show a similar trend. We therefore made use of the cell lines R182 and TARA-R182, also referred to as OCSC-1 and OCC-1, respectively [34,35]. These syngeneic cell lines have previously been shown to be epithelial-like and mesenchymal-like, respectively, and we reiterate this via analysis of morphology and expression of the EMT marker TWIST1 (Figure 4). As hypothesized, mesenchymal TARA cells were more sensitive to olaparib compared to their R182 forebears, as shown by both clonogenic and MTS cell quantitation assays (Figure 4). Moreover, TARA cells express lower levels of BRCA2 and RAD51 protein, despite little to no change in mRNA levels (Figure 5). Interestingly, BRCA1 mRNA is upregulated in TARA versus R182, though this is not sufficient to cause olaparib resistance. These findings are not consistent with the results of two studies in breast cancer. In the first, TGFβ treatment of breast cancer cells led to “BRCAness” through downregulation of a different set of HR genes, namely ATM, MSH2, and BRCA1 [39]. Wu et al. also showed that in triple negative breast cancer, EMT factors Slug and Snail led to repression of BRCA1 expression via recruitment of the demethylase LSD1 and direct binding of the BRCA1 promoter [48].
Finally, we hypothesized that due to loss of HR throughput, TARA cells would upregulate the c-NHEJ pathway to compensate. Indeed, we showed that TARA cells have an increased expression level of factors required for the c-NHEJ pathway, including Ku70, Ku80, and DNA-PKcs (Figure 5). Based on this observation, we assayed the response of TARA versus R182 cells to Nu-7441, an inhibitor of DNA-PK. As seen for olaparib, TARA cells were more sensitive to Nu-7441 than were R182. As noted earlier, the dose of Nu-7441 used is sufficient to impact additional targets, including PI3K and mTOR, and inhibition of these pathways may well contribute to TARA cell death. Furthermore, development of NU-7441 itself as a putative drug was halted due to concerns with solubility and thus bioavailability, meaning an alternative such as the DNA-PK and PI3K dual inhibitor KU-0060648 may be a better candidate for further development [49,50]. However, these results represent early data in support of broadening DNA repair targeted therapy in ovarian cancer, and in cancers at large. Walker et al. recently showed that the DNA repair landscape of ovarian tumors can predict survival, further supporting this assertion [51]. The same study found that HR and c-NHEJ defects were mutually exclusive, in agreement with our findings in TARA cells, and suggested that DNA repair status could be an important therapeutic consideration in non-ovarian tumors. Agents targeting DNA repair are a newer addition to our anti-cancer arsenal, and while they have faced challenges during development, they hold great promise for new avenues to treatment [49,52].
The role of EMT in determining drug responses is complicated, perhaps more so in EOC than in other tumor types. Nevertheless, we have developed a schematic to explain the relationship between EMT and drug responses in EOC over the course of the disease (Figure 6). Unlike many cancers, in which mesenchymal character correlates with stem-like phenotype [29,30], we and others have observed that CD44+ epithelial cells serve as cancer stem cells in EOC [27,28]. These stem cells, represented by the line R182, are inherently resistant to chemotherapy (Figure 6A), but give rise to fast-dividing mesenchymal-like cells that are chemosensitive (i.e., TARA; Figure 6B). Amongst mesenchymal cells, acquired resistance can manifest as EMT and tumor evolution progress (Figure 6C). Mesenchymal-like OVCAR8 cells displaying resistance to cisplatin were resensitized by the inhibition of TWIST1, leading to reduced Akt activation under cell culture conditions and reduced tumor growth in vivo [31,32]. Similarly, Craveiro et al showed that OCSC1-F2 cells, derived from R182 by in vivo rather than in vitro EMT, are initially sensitive to paclitaxel. However, despite mice carrying F2 tumors going into remission following paclitaxel treatment, tumors recur and are paclitaxel-resistant [34]. In the current study, we sought to determine whether the same pattern of drug responses as cells undergo EMT extends to PARP inhibitors, and more broadly, DNA repair-targeting therapies. In epithelial-like R182 and PEO4 cells, response to olaparib was low (Figure 6D) while in mesenchymal TARA and PEO1 cells, response was high (Figure 6E). PEO1 is expected to be sensitive to olaparib on the basis of its BRCA2 mutation, and TARA cells exhibit a similar trend due to their downregulated HR proteins. However, it remains unknown whether sensitivity to DNA repair-directed therapies, including olaparib or Nu-7441, either alone or in combination, will remain effective in advanced disease that has become resistant to platinum or taxane therapy (Figure 6F).
It is also unknown what signals downstream from TGFβ mediate its effect on DNA repair. Effective leveraging of synthetic lethality in BRCA WT cells will depend on identifying novel targets within this pathway and designing drugs to inhibit them. Our future work will focus on these mechanistic and translational questions.
5. Conclusions
Our data clearly demonstrate that TGFβ treatment and acquisition of a mesenchymal phenotype in EOC cells results in downregulation of factors necessary for the HR pathway of DNA repair, and thus sensitization of these cells to PARP inhibition (Figure 7). Because mesenchymal, metastatic cells common to EOC recurrence can manifest resistance to first line chemotherapy, the addition of PARPi and other DNA repair targeting drugs to our arsenal, even for BRCA WT tumors, may represent a step forward in combating recurrent EOC and ultimately improving patient survival.
Supplementary Materials
TThe following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Full western images for Figure 2C; Figure S2: Full western images for Figure 3A. Figure S3: Flow cytometry data for Figure 3B. Figure S4: Full western images for Figure 4C and Figure 5A. Figure S5: Full western images for Figure 5C.
Author Contributions
Conceptualization: C.M.R., M.R.A., Z.P.L., and E.S.R. ; Methodology, Z.P.L.; Validation, C.M.R., M.R.A., and Z.P.L.; Formal Analysis, C.M.R. and M.R.A.; Investigation, C.M.R., M.R.A., R.E.H., Z.P.L.; Resources, E.S.R.; Data Curation, C.M.R. and M.R.A.; Writing – Original Draft Preparation, C.M.R. and M.R.A..; Writing – Review & Editing, R.E.H. and Z.P.L.; Visualization, C.M.R. and M.R.A; Supervision, Z.P.L.; Project Administration, C.M.R and E.S.R.; Funding Acquisition, C.M.R. Z.P.L. and E.S.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Discovery to Cure research grant at Yale University. C.M.R. was funded by American Cancer Society Postdoctoral Fellowship 131420-PF-17-236-01-DMC and by laboratory startup funds from Midwestern University. M.R.A. was supported by the William U. Gardner Memorial Student Research Fellowship and the James G. Hirsch, M.D. Endowed Medical Student Research Fellowship at Yale School of Medicine.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary material.
Acknowledgments
We thank Dr. Peter Glazer of Yale University School of Medicine for providing PEO1 and PEO4 cell lines and Dr. Gil Mor for providing R182 and TARA-R182. pCBASceI was a gift from Maria Jasin (Addgene plasmid # 26477 ; http://n2t.net/addgene:26477 ; RRID:Addgene_26477). We thank Yale Flow Cytometry for their assistance with and use of the LSR II flow cytometer. The Core is supported in part by an NCI Cancer Center Support Grant # NIH P30 CA016359. Finally, we wish to thank Dr. Ellen Kohlmeir and the Midwestern University Core Facility, Downers Grove, IL. MTS assays were performed using the PerkinElmer EnSpire Plate Reader, and cells were counted using the Denovix CellDrop automated cell counter located in the Core.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J Clin 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.-S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global Surveillance of Cancer Survival 1995–2009: Analysis of Individual Data for 25 676 887 Patients from 279 Population-Based Registries in 67 Countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian Cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.; Thomas, H.D.; Mitchell, J.; Curtin, N.J. Exploiting the Achilles Heel of Cancer: The Therapeutic Potential of Poly(ADP-Ribose) Polymerase Inhibitors in BRCA2-Defective Cancer. Br J Radiol 2008, 81 Spec No 1, S6–S11. [Google Scholar] [CrossRef]
- Ratner, E.S.; Sartorelli, A.C.; Lin, Z.P. Poly (ADP-Ribose) Polymerase Inhibitors: On the Horizon of Tailored and Personalized Therapies for Epithelial Ovarian Cancer. Curr Opin Oncol 2012, 24, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Mendez, I.; Bruijn, I. de; Tian, L.; Meisel, J.L.; Garcia-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin Cancer Res 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed]
- Francica, P.; Rottenberg, S. Mechanisms of PARP Inhibitor Resistance in Cancer and Insights into the DNA Damage Response. Genome Med 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Ratner, E.S.; Zhu, Y.L.; Penketh, P.G.; Berenblum, J.; Whicker, M.E.; Huang, P.H.; Lee, Y.; Ishiguro, K.; Zhu, R.; Sartorelli, A.C.; et al. Triapine Potentiates Platinum-Based Combination Therapy by Disruption of Homologous Recombination Repair. Br J Cancer 2016, 114, 777–786. [Google Scholar] [CrossRef]
- Lin, Z.P.; Ratner, E.S.; Whicker, M.E.; Lee, Y.; Sartorelli, A.C. Triapine Disrupts CtIP-Mediated Homologous Recombination Repair and Sensitizes Ovarian Cancer Cells to PARP and Topoisomerase Inhibitors. Mol Cancer Res 2014, 12, 381–393. [Google Scholar] [CrossRef]
- Lin, Z.P.; Zouabi, N.N.A.; Xu, M.L.; Bowen, N.E.; Wu, T.L.; Lavi, E.S.; Huang, P.H.; Zhu, Y.L.; Kim, B.; Ratner, E.S. In Silico Screening Identifies a Novel Small Molecule Inhibitor That Counteracts PARP Inhibitor Resistance in Ovarian Cancer. Sci Rep 2021, 11, 8042. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.P.; Zhu, Y.L.; Lo, Y.C.; Moscarelli, J.; Xiong, A.; Korayem, Y.; Huang, P.H.; Giri, S.; LoRusso, P.; Ratner, E.S. Combination of Triapine, Olaparib, and Cediranib Suppresses Progression of BRCA-Wild Type and PARP Inhibitor-Resistant Epithelial Ovarian Cancer. PLoS One 2018, 13, e0207399. [Google Scholar] [CrossRef] [PubMed]
- Bachman, K.E.; Park, B.H. Duel Nature of TGF-Beta Signaling: Tumor Suppressor vs. Tumor Promoter. Curr Opin Oncol 2005, 17, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-Beta Targeted Cancer Therapy. Int J Biol Sci 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Leivonen, S.K.; Kahari, V.M. Transforming Growth Factor-Beta Signaling in Cancer Invasion and Metastasis. Int J Cancer 2007, 121, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Syed, V. TGF-Beta Signaling in Cancer. J Cell Biochem 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Kirshner, J.; Jobling, M.F.; Pajares, M.J.; Ravani, S.A.; Glick, A.B.; Lavin, M.J.; Koslov, S.; Shiloh, Y.; Barcellos-Hoff, M.H. Inhibition of Transforming Growth Factor-Beta1 Signaling Attenuates Ataxia Telangiectasia Mutated Activity in Response to Genotoxic Stress. Cancer Res 2006, 66, 10861–10869. [Google Scholar] [CrossRef]
- Wu, C.T.; Chang, Y.H.; Lin, W.Y.; Chen, W.C.; Chen, M.F. TGF Beta1 Expression Correlates with Survival and Tumor Aggressiveness of Prostate Cancer. Ann Surg Oncol 2015, 22 (Suppl 3), S1587–93. [Google Scholar] [CrossRef]
- Wu, C.T.; Hsieh, C.C.; Yen, T.C.; Chen, W.C.; Chen, M.F. TGF-Beta1 Mediates the Radiation Response of Prostate Cancer. J Mol Med (Berl) 2015, 93, 73–82. [Google Scholar] [CrossRef]
- Tummala, H.; Dokal, I. TGF-Beta Pathway Inhibition Signals New Hope for Fanconi Anemia. Cell Stem Cell 2016, 18, 567–568. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, W.; Cheng, C.T.; Ren, X.; Somlo, G.; Fong, M.Y.; Chin, A.R.; Li, H.; Yu, Y.; Xu, Y.; et al. TGFbeta Induces “BRCAness” and Sensitivity to PARP Inhibition in Breast Cancer by Regulating DNA-Repair Genes. Mol Cancer Res 2014, 12, 1597–1609. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, Z.; Wu, X.; Zhou, Q.; Liao, C.; Liu, Y.; Li, D.; Shen, L.; Feng, D.; Yang, L. LMP1 Promotes Nasopharyngeal Carcinoma Metastasis through NTRK2-Mediated Anoikis Resistance. Am J Cancer Res 2020, 10, 2083–2099. [Google Scholar]
- Nonpanya, N.; Prakhongcheep, O.; Petsri, K.; Jitjaicham, C.; Tungsukruthai, S.; Sritularak, B.; Chanvorachote, P. Ephemeranthol A Suppresses Epithelial to Mesenchymal Transition and FAK-Akt Signaling in Lung Cancer Cells. Anticancer Res 2020, 40, 4989–4999. [Google Scholar] [CrossRef]
- Ko, H. Geraniin Inhibits TGF-Beta1-Induced Epithelial-Mesenchymal Transition and Suppresses A549 Lung Cancer Migration, Invasion and Anoikis Resistance. Bioorg Med Chem Lett 2015, 25, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Terauchi, M.; Kajiyama, H.; Yamashita, M.; Kato, M.; Tsukamoto, H.; Umezu, T.; Hosono, S.; Yamamoto, E.; Shibata, K.; Ino, K.; et al. Possible Involvement of TWIST in Enhanced Peritoneal Metastasis of Epithelial Ovarian Carcinoma. Clin Exp Metastasis 2007, 24, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Alvero, A.B.; Craveiro, V.; Holmberg, J.C.; Fu, H.H.; Montagna, M.K.; Yang, Y.; Chefetz-Menaker, I.; Nuti, S.; Rossi, M.; et al. Constitutive Proteasomal Degradation of TWIST-1 in Epithelial-Ovarian Cancer Stem Cells Impacts Differentiation and Metastatic Potential. Oncogene 2013, 32, 39–49. [Google Scholar] [CrossRef]
- Alvero, A.B.; Chen, R.; Fu, H.-H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.-A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular Phenotyping of Human Ovarian Cancer Stem Cells Unravels the Mechanisms for Repair and Chemoresistance. Cell cycle (Georgetown, Tex.) 2009, 8, 158–166. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist Modulates Breast Cancer Stem Cells by Transcriptional Regulation of CD24 Expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef]
- Kong, D.; Li, Y.; Wang, Z.; Sarkar, F.H. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins? Cancers (Basel) 2011, 3, 716–729. [Google Scholar] [CrossRef]
- Roberts, C.M.; Shahin, S.A.; Wen, W.; Finlay, J.B.; Dong, J.; Wang, R.; Dellinger, T.H.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Nanoparticle Delivery of SiRNA against TWIST to Reduce Drug Resistance and Tumor Growth in Ovarian Cancer Models. Nanomedicine 2017, 13, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 Drives Cisplatin Resistance and Cell Survival in an Ovarian Cancer Model, via Upregulation of GAS6, L1CAM, and Akt Signalling. Sci Rep 2016, 6, 37652. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and Properties of Nine Human Ovarian Adenocarcinoma Cell Lines. Cancer Res 1988, 48, 6166–6172. [Google Scholar] [PubMed]
- Craveiro, V.; Yang-Hartwich, Y.; Holmberg, J.C.; Joo, W.D.; Sumi, N.J.; Pizzonia, J.; Griffin, B.; Gill, S.K.; Silasi, D.A.; Azodi, M.; et al. Phenotypic Modifications in Ovarian Cancer Stem Cells Following Paclitaxel Treatment. Cancer Med 2013, 2, 751–762. [Google Scholar] [CrossRef]
- Steffensen, K.D.; Alvero, A.B.; Yang, Y.; Waldstrom, M.; Hui, P.; Holmberg, J.C.; Silasi, D.A.; Jakobsen, A.; Rutherford, T.; Mor, G. Prevalence of Epithelial Ovarian Cancer Stem Cells Correlates with Recurrence in Early-Stage Ovarian Cancer. J Oncol 2011, 2011, 620523. [Google Scholar] [CrossRef]
- Yang-Hartwich, Y.; Tedja, R.; Roberts, C.M.; Goodner-Bingham, J.; Cardenas, C.; Gurea, M.; Sumi, N.J.; Alvero, A.B.; Glackin, C.A.; Mor, G. P53-Pirh2 Complex Promotes Twist1 Degradation and Inhibits EMT. Mol Cancer Res 2019, 17, 153–164. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of Double-Strand Breaks by End Joining. Cold Spring Harb Perspect Biol 2013, 5, a012757. [Google Scholar] [CrossRef]
- Leahy, J.J.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a Highly Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor (NU7441) by Screening of Chromenone Libraries. Bioorg Med Chem Lett 2004, 14, 6083–6087. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, W.; Cheng, C.-T.; Ren, X.; Somlo, G.; Fong, M.Y.; Chin, A.R.; Li, H.; Yu, Y.; Xu, Y.; et al. TGFβ Induces “BRCAness” and Sensitivity to PARP Inhibition in Breast Cancer by Regulating DNA-Repair Genes. Mol Cancer Res 2014, 12, 1597–1609. [Google Scholar] [CrossRef]
- Savagner, P. The Epithelial-Mesenchymal Transition (EMT) Phenomenon. Ann Oncol 2010, 21 Suppl 7, vii89–92. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat Rev Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Lim, J.; Thiery, J.P. Epithelial-Mesenchymal Transitions: Insights from Development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Whicker, M.E.; Lin, Z.P.; Hanna, R.; Sartorelli, A.C.; Ratner, E.S. MK-2206 Sensitizes BRCA-Deficient Epithelial Ovarian Adenocarcinoma to Cisplatin and Olaparib. BMC Cancer 2016, 16, 550. [Google Scholar] [CrossRef] [PubMed]
- Portella, G.; Cumming, S.A.; Liddell, J.; Cui, W.; Ireland, H.; Akhurst, R.J.; Balmain, A. Transforming Growth Factor Beta Is Essential for Spindle Cell Conversion of Mouse Skin Carcinoma in Vivo: Implications for Tumor Invasion. Cell Growth Differ Mol Biology J Am Assoc Cancer Res 1998, 9, 393–404. [Google Scholar]
- Spaderna, S.; Schmalhofer, O.; Hlubek, F.; Berx, G.; Eger, A.; Merkel, S.; Jung, A.; Kirchner, T.; Brabletz, T. A Transient, EMT-Linked Loss of Basement Membranes Indicates Metastasis and Poor Survival in Colorectal Cancer. Gastroenterology 2006, 131, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Saunier, E.F.; Quigley, D.; Luu, M.T.; Sapio, A.D.; Hann, B.; Yingling, J.M.; Akhurst, R.J. Outgrowth of Drug-Resistant Carcinomas Expressing Markers of Tumor Aggression after Long-Term TβRI/II Kinase Inhibition with LY2109761. Cancer Res 2011, 71, 2339–2349. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-Q.; Li, X.-Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt Signaling Regulates Slug Activity and Links Epithelial–Mesenchymal Transition with Epigenetic Breast Cancer 1, Early Onset (BRCA1) Repression. Proc National Acad Sci 2012, 109, 16654–16659. [Google Scholar] [CrossRef]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA Repair Targeted Therapy: The Past or Future of Cancer Treatment? Pharmacol Therapeut 2016, 160, 65–83. [Google Scholar] [CrossRef]
- Munck, J.M.; Batey, M.A.; Zhao, Y.; Jenkins, H.; Richardson, C.J.; Cano, C.; Tavecchio, M.; Barbeau, J.; Bardos, J.; Cornell, L.; et al. Chemosensitization of Cancer Cells by KU-0060648, a Dual Inhibitor of DNA-PK and PI-3K. Mol Cancer Ther 2012, 11, 1789–1798. [Google Scholar] [CrossRef]
- Walker, T.D.J.; Faraahi, Z.F.; Price, M.J.; Hawarden, A.; Waddell, C.A.; Russell, B.; Jones, D.M.; McCormick, A.; Gavrielides, N.; Tyagi, S.; et al. The DNA Damage Response in Advanced Ovarian Cancer: Functional Analysis Combined with Machine Learning Identifies Signatures That Correlate with Chemotherapy Sensitivity and Patient Outcome. Brit J Cancer 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Tanikawa, M.; Sone, K.; Mori-Uchino, M.; Osuga, Y.; Fujii, T. Recent Advances in Targeting DNA Repair Pathways for the Treatment of Ovarian Cancer and Their Clinical Relevance. Int J Clin Oncol 2017, 22, 611–618. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
TGFβ signaling pathways. Canonical TGFβ binds to its receptor, leading to activation of Akt, SMAD proteins, and EMT.
Figure 1.
TGFβ signaling pathways. Canonical TGFβ binds to its receptor, leading to activation of Akt, SMAD proteins, and EMT.
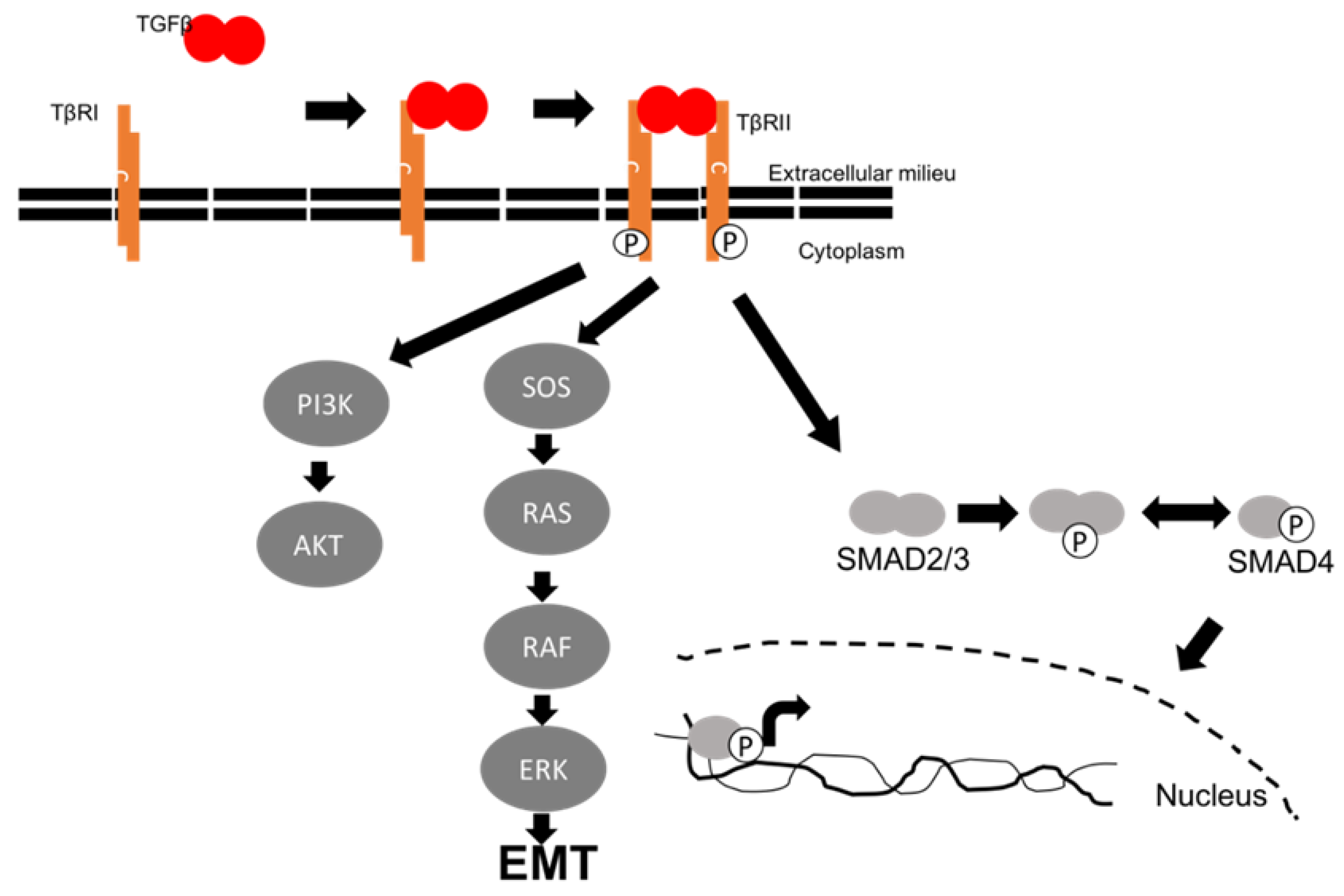
Figure 2.
A. Micrograph of PEO1 cells, showing monolayer growth. B. Micrograph of PEO4 cells, showing growth in islands. C. Representative western blot of epithelial and mesenchymal markers in PEO1 and PEO4. HSC-70: loading control. D. Following TGFβ treatment, PEO4 cells adopt PEO1-like morphology. E. Wound healing assays show that PEO1 rapidly migrates to close a wound, while PEO4 has a slower rate of healing. F. Treatment with TGFβ does not appreciably change migratory potential in either line.
Figure 2.
A. Micrograph of PEO1 cells, showing monolayer growth. B. Micrograph of PEO4 cells, showing growth in islands. C. Representative western blot of epithelial and mesenchymal markers in PEO1 and PEO4. HSC-70: loading control. D. Following TGFβ treatment, PEO4 cells adopt PEO1-like morphology. E. Wound healing assays show that PEO1 rapidly migrates to close a wound, while PEO4 has a slower rate of healing. F. Treatment with TGFβ does not appreciably change migratory potential in either line.
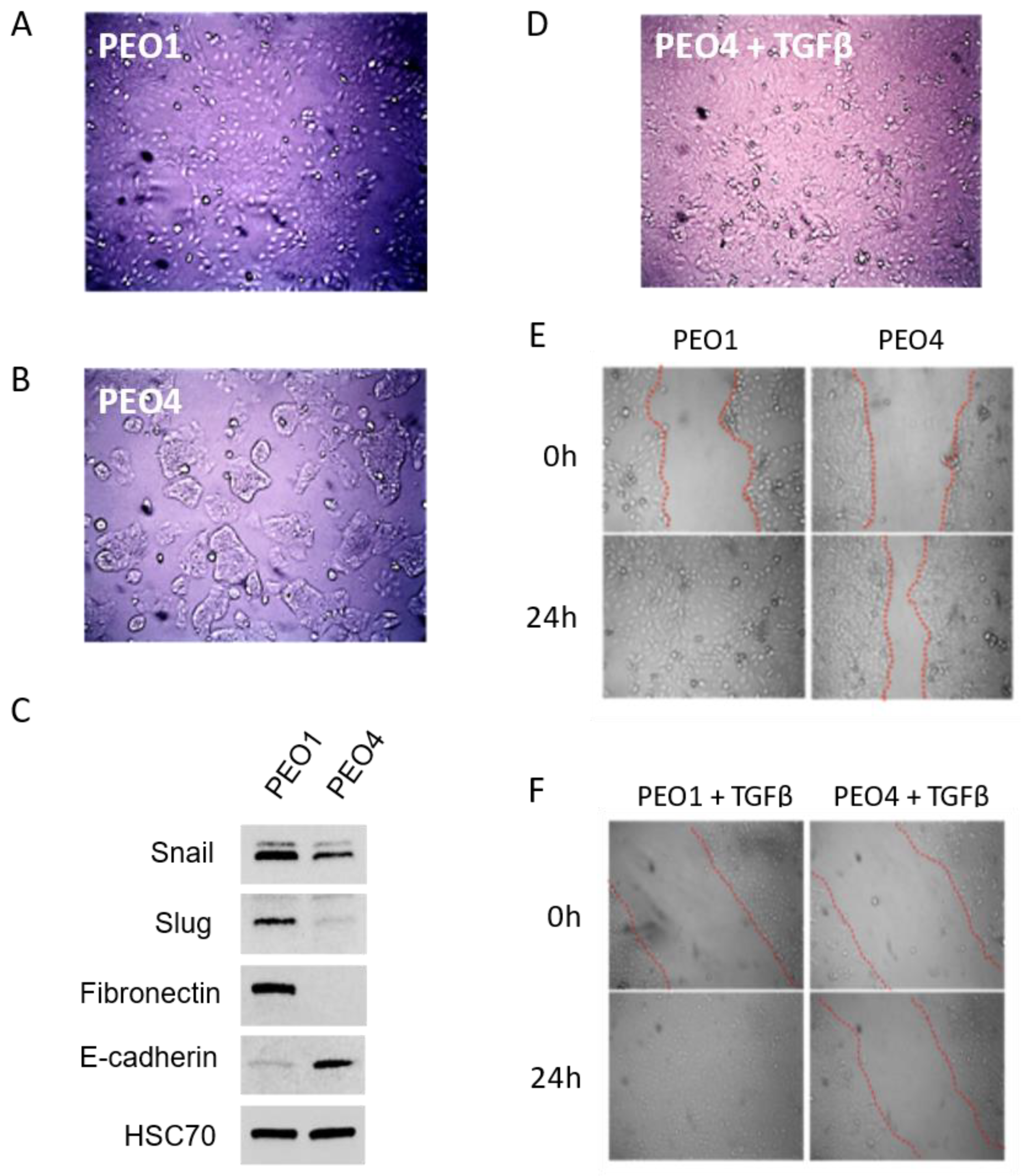
Figure 3.
A. Top: TGFβ upregulates Snail in both PEO1 and PEO4 cells, and fibronectin (FN) and phosphor-Akt in PEO1. TGFβ receptor inhibitor LY2109761 (LY) abrogates these changes and lowers pAkt in both lines. Bottom: TGFβ decreases BRCA2, BRCA1, and RAD51 expression in PEO4. HSC-70: loading control. Representative blot image shown. B. Percent GFP positive SKOV-3 cells following the treatments shown in HR reporter assay. C. Clonogenic assay demonstrates that PEO4 is olaparib resistant, while PEO4 + TGFβ is sensitive and shows similar response to PEO1 with or without TGFβ. D. Clonogenic assay shows LY2109761 reverses the effect of TGFβ on olaparib response in PEO4.
Figure 3.
A. Top: TGFβ upregulates Snail in both PEO1 and PEO4 cells, and fibronectin (FN) and phosphor-Akt in PEO1. TGFβ receptor inhibitor LY2109761 (LY) abrogates these changes and lowers pAkt in both lines. Bottom: TGFβ decreases BRCA2, BRCA1, and RAD51 expression in PEO4. HSC-70: loading control. Representative blot image shown. B. Percent GFP positive SKOV-3 cells following the treatments shown in HR reporter assay. C. Clonogenic assay demonstrates that PEO4 is olaparib resistant, while PEO4 + TGFβ is sensitive and shows similar response to PEO1 with or without TGFβ. D. Clonogenic assay shows LY2109761 reverses the effect of TGFβ on olaparib response in PEO4.
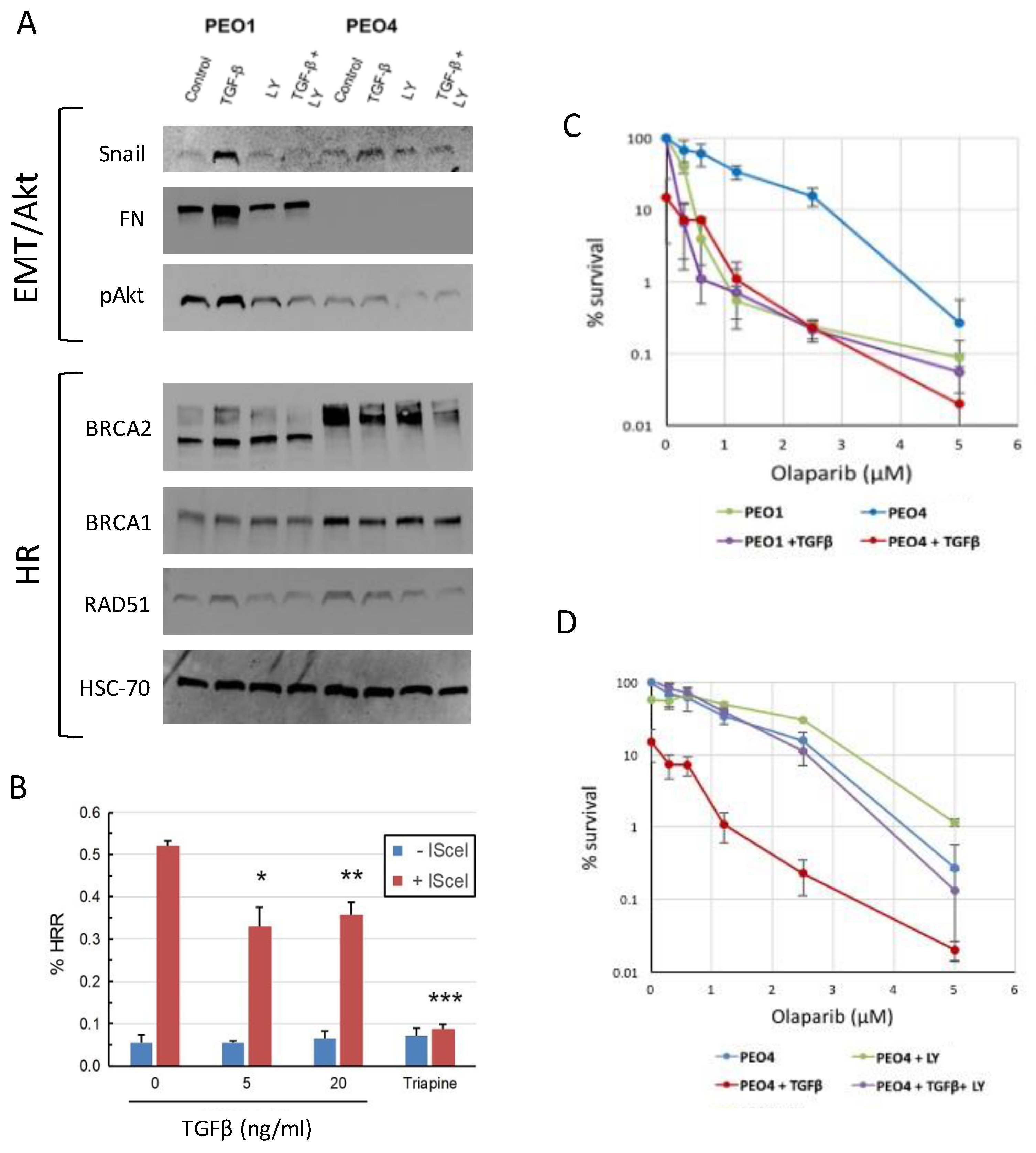
Figure 4.
A. R182 cells show cobblestone-like epithelial morphology. B. TARA-R182 cells show slightly angular morphology reminiscent of PEO1. C. TARA-R182 cells are mesenchymal as shown by expression of the EMT marker TWIST1, as compared to the epithelial, TWIST1 negative R182. Representative blot shown. D. Top: Clonogenic assay of R182 cells treated with the indicated doses of olaparib. Increasing dose yields smaller colonies, but little change in colony number. Bottom: The same assay in TARA cells shows TARA cells are more sensitive to doses ≥ 1.25μM. E. MTS assay measuring cell proliferation shows robust dose response in TARA cells treated with olaparib, while R182 is unaffected. Error bars, S.E. of technical triplicates; representative graph of three independent experiments shown.
Figure 4.
A. R182 cells show cobblestone-like epithelial morphology. B. TARA-R182 cells show slightly angular morphology reminiscent of PEO1. C. TARA-R182 cells are mesenchymal as shown by expression of the EMT marker TWIST1, as compared to the epithelial, TWIST1 negative R182. Representative blot shown. D. Top: Clonogenic assay of R182 cells treated with the indicated doses of olaparib. Increasing dose yields smaller colonies, but little change in colony number. Bottom: The same assay in TARA cells shows TARA cells are more sensitive to doses ≥ 1.25μM. E. MTS assay measuring cell proliferation shows robust dose response in TARA cells treated with olaparib, while R182 is unaffected. Error bars, S.E. of technical triplicates; representative graph of three independent experiments shown.
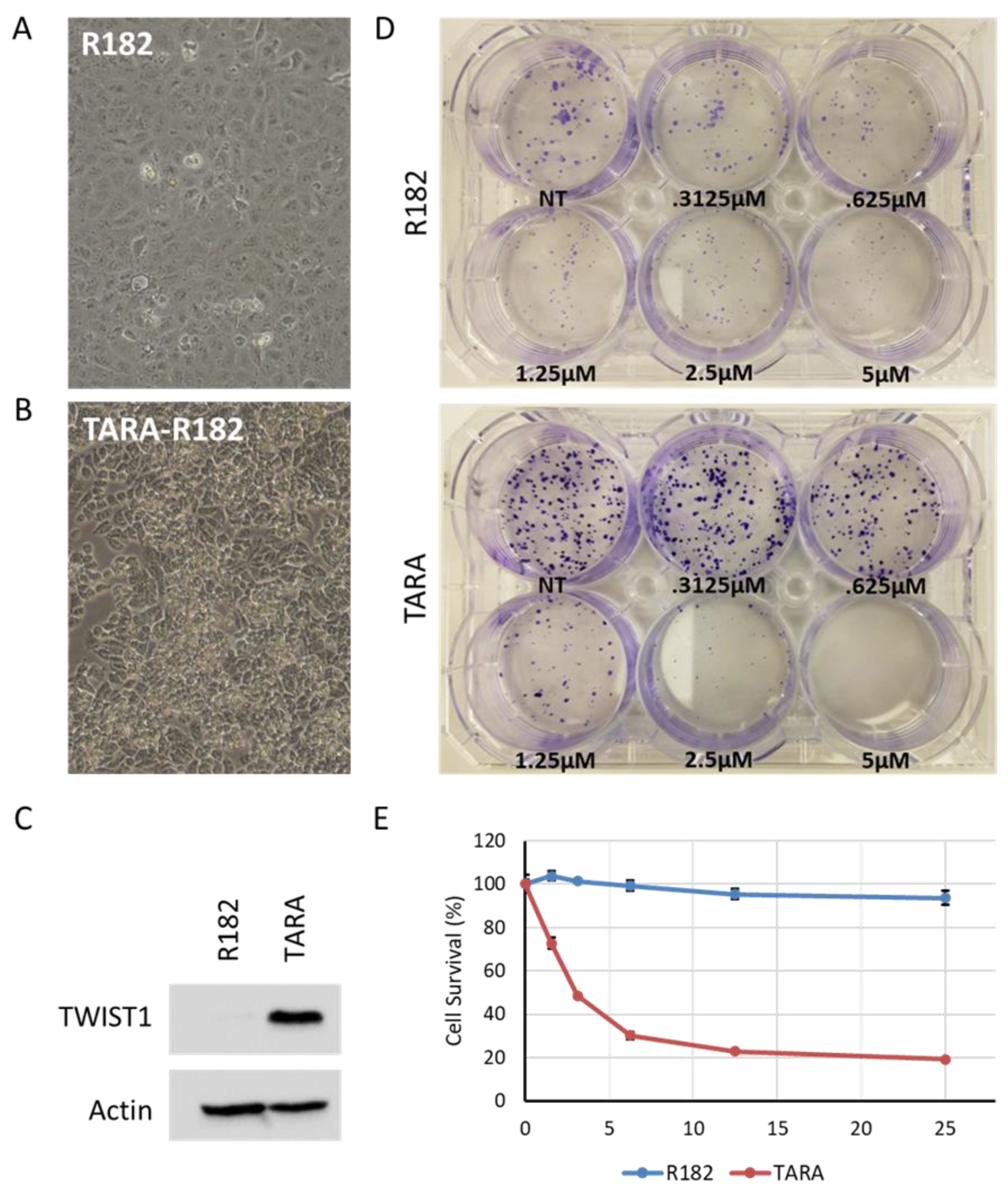
Figure 5.
A. Western blot shows that expression of BRCA2 and RAD51 are reduced in TARA vs R182. HSC-70, loading control. Representative blot shown. B. qPCR shows that BRCA2 and RAD51 mRNA levels are not substantially changed between R182 and TARA cells, while BRCA1 mRNA expression trends higher in TARA. Error bars, range of fold changes; representative graph of three independent experiments shown. C. Western blot shows levels of Ku70, Ku80 and DNA-PKcs proteins are elevated in TARA vs R182. Actin, loading control. Representative blots shown. D. MTS assay of R182 and TARA cells treated with indicated drugs. Cisplatin, 10 μM; olaparib, 25 μM; Nu-7441, 10 μM. Error bars: S.E. of technical triplicates. Representative graph of three independent experiments shown. **** p<.0001.
Figure 5.
A. Western blot shows that expression of BRCA2 and RAD51 are reduced in TARA vs R182. HSC-70, loading control. Representative blot shown. B. qPCR shows that BRCA2 and RAD51 mRNA levels are not substantially changed between R182 and TARA cells, while BRCA1 mRNA expression trends higher in TARA. Error bars, range of fold changes; representative graph of three independent experiments shown. C. Western blot shows levels of Ku70, Ku80 and DNA-PKcs proteins are elevated in TARA vs R182. Actin, loading control. Representative blots shown. D. MTS assay of R182 and TARA cells treated with indicated drugs. Cisplatin, 10 μM; olaparib, 25 μM; Nu-7441, 10 μM. Error bars: S.E. of technical triplicates. Representative graph of three independent experiments shown. **** p<.0001.
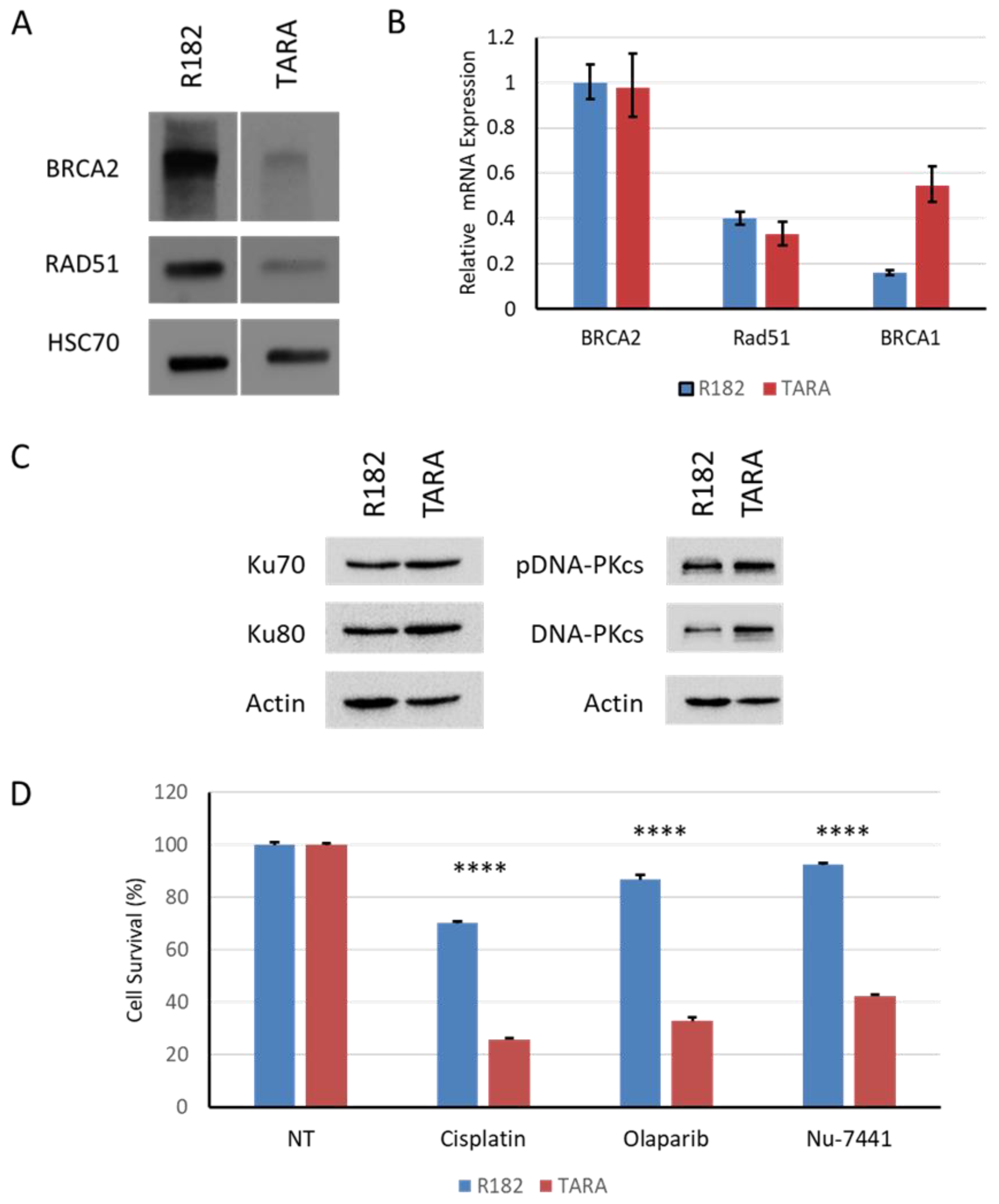
Figure 6.
A-C. As shown here and in other studies, EMT gives rise to fast-dividing, chemosensitive cells that go on to develop secondary resistance to cytotoxic chemotherapy (blue curve). D-E. We show here that EMT also leads to increased efficacy of DNA repair-targeted drugs, such as olaparib and Nu-7441 (orange curve). F. Further work is needed to determine if chemoresistant cells retain sensitivity to DNA repair-targeted agents, either alone or in combination (orange dashes).
Figure 6.
A-C. As shown here and in other studies, EMT gives rise to fast-dividing, chemosensitive cells that go on to develop secondary resistance to cytotoxic chemotherapy (blue curve). D-E. We show here that EMT also leads to increased efficacy of DNA repair-targeted drugs, such as olaparib and Nu-7441 (orange curve). F. Further work is needed to determine if chemoresistant cells retain sensitivity to DNA repair-targeted agents, either alone or in combination (orange dashes).
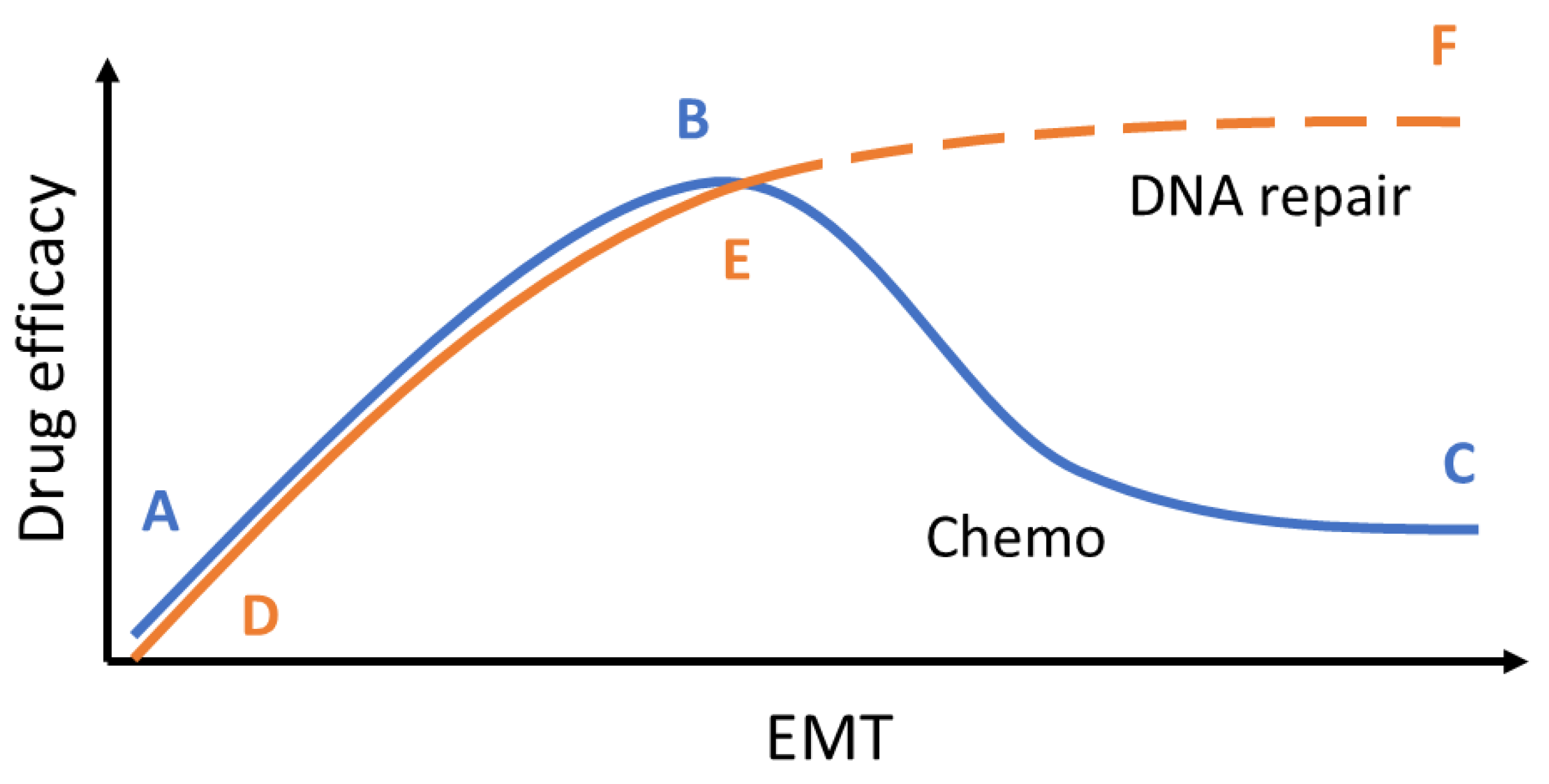
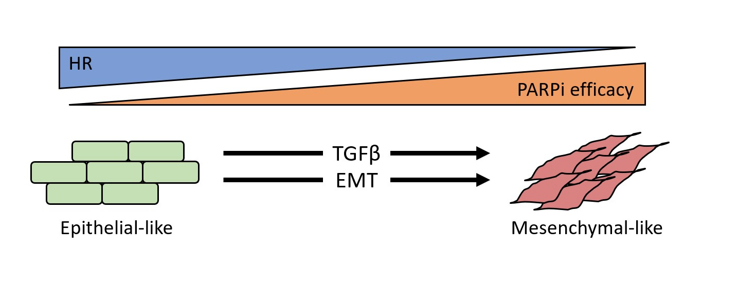
Figure 7.
Schematic of overall findings. TGFβ signaling or EMT can give rise to olaparib lethality even in the presence of WT HR proteins.
Figure 7.
Schematic of overall findings. TGFβ signaling or EMT can give rise to olaparib lethality even in the presence of WT HR proteins.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



