
Submitted:
14 April 2023
Posted:
14 April 2023
You are already at the latest version
Abstract
The Americas, particularly Brazil, were greatly impacted by the widespread outbreak of Zika virus (ZIKV) in 2015 and 2016. Efforts were made to implement genomic surveillance of ZIKV as part of the public health responses. The accuracy of spatiotemporal reconstructions of the epidemic spread relies on the unbiased sampling of the transmission process. In the early stages of the outbreak, we recruited patients exhibiting clinical symptoms of arbovirus-like infection from Salvador and Campo Formoso, Bahia, in Northeast Brazil. Between May 2015 and June 2016, we identified 21 cases of acute ZIKV infection and subsequently recovered 14 near full-length sequences using the amplicon tiling multiplex approach with nanopore sequencing. We perform a time-calibrated discrete phylogeographic analysis to trace the spread and migration history of the ZIKV. Our phylogenetic analysis supports a consistent relationship between ZIKV migration from Northeast to Southeast Brazil and its subsequent dissemination beyond Brazil. Additionally, our analysis provides insights into the migration of ZIKV from Brazil to Haiti and the role Brazil played in the spread of ZIKV to other countries, such as Singapore, the USA and Dominican Republic. The data generated by this study enhances our understanding of ZIKV dynamics and supports the existing knowledge, which can aid in future surveillance efforts against the virus.
Keywords:
Zika
; arboviruses
; vector-borne infections
; genomic surveillance
; phylogenetics
1. Introduction
Zika virus (ZIKV) is an emerging arthropod-borne virus that causes severe neurotropic diseases, which include Guillain-Barré and Congenital Zika Syndrome [1]. The virus belongs to the Flavivirus genus of the Flaviviridae family. It has a positive-sense single-stranded RNA genome of approximately 11 kb [2,3] and a substitution rate of 7.55×10-4 to 1.66×10-3 substitutions per site per year [4]. ZIKV was first isolated in 1947 from a sentinel monkey in the Zika Forest, Uganda. ZIKV was described as causing sporadic infections for half a century. However, in 2007 and 2013, large outbreaks were reported in Micronesia [5] and French Polynesia [6]. In March 2015, ZIKV was identified in Brazil [7,8] and further spread to more than 40 countries in the Americas [9,10]. In February 2016, the World Health Organization (WHO) declared the ZIKV outbreak a Public Health Emergency of International Concern (PHEIC) due to its association with microcephaly and neurological diseases as Guillain-Barré syndrome [11,12]. This PHEIC declaration was lifted in November 2016 [13].
The revolution in molecular epidemiological surveillance with the advent of next-generation sequencing and advanced phylogenetic analyses allows to uncover unobserved transmission dynamics and helps to explain the introduction, spread, and evolution of infectious diseases [11,14]. Molecular dating analyses based on ZIKV sequences from patients identified during the Brazilian outbreak revealed that this virus possibly entered Brazil in the second half of 2013 [14]. Additionally, Lednicky et al. (2016) [15] and Campos et al. (2018) [16] described that ZIKV in Brazil possibly originated from Haiti. Both discoveries revealed, at the time of the event, a gap in our national surveillance system that hampered early detection and interventions to curb the spread of the virus in the country.
However, these findings need validation with new strategies of phylogenetic analyses and new sequences. This is because factors such as the limited number of ZIKV sequences compared to the proportion of cases reported during the outbreak in Brazil and the low sequence quality from the beginning of the epidemic (breadth of coverage < 70%) can directly affect the credibility of pathogen transmission dynamics reconstruction [17].
The low number of high-quality sequences available for ZIKV can be explained by several factors, including low viral load in ZIKV infection [18], low detection of ZIKV-infected patients due to a large number of asymptomatic cases (75% to 80%) or mild/unspecific symptoms [19], abrupt reduction in ZIKV cases [20] and the challenges faced by the Brazilian health system to perform real-time genomic surveillance. Retrospective sequencing of samples from the early stages of the outbreak in Brazil has the potential to increase the accuracy and precision of transmission dynamics reconstruction, which can help inform public health strategies for future outbreaks.
2. Materials and Methods
During the ZIKV outbreak, our group actively recruited patients with arboviral-like symptoms in Salvador (n=948) [21] and Campo Formoso (n=230) [22], Bahia. A total of 21 cases of viremic ZIKV infection were identified between May 2015 to June 2016 in Bahia, Brazil. An amplicon tiling multiplex approach described by Quick et al. (2017) [23] was applied in conjunction with the MinION platform (Oxford Nanopore Technologies) for sequencing. For further details on diagnosis, sequencing and phylogenetic analysis, we refer to the Supplementary Appendix.
3. Results and Discussion
The sequencing efforts yielded near full-length sequences (>80% reference coverage, Genbank No. KJ776791.1) for 14 ZIKV-positive samples, with an average sequencing depth ranging from 377x to 1,438x (Table 1). The clinical and social-demographic characteristics are listed in Table S1.
A comprehensive dataset was compiled with 14 new genomes and 206 curated near full-length ZIKV genomes (Table S2). We performed a time-calibrated discrete phylogeographic reconstruction (details and references in Supplementary Appendix) to explore the geographic spread and transmission of ZIKV in Bahia and Brazil, as shown in Figure 1.
The newly sequenced ZIKV genomes analyzed in this study are distributed over four distinct clades, containing sequences from diverse regions of Brazil (North, Northeast, and Southeast) as well as other countries in the world, thus providing valuable insights into the migration patterns of ZIKV.
Ten of the newly sequenced isolates (TRDP238, TRDP256, TRDP257, TRDP274, TRDP282, TRDP300, TRDP309, TRDP317, TRDP333, and ZK0110) clustered in a clade together with genomes from Brazil (Southeast and Northeast), Singapore and Haiti (Clade I, Figure 1B). The estimated time to the most recent common ancestor (tMRCA) was August 2013, with 95% Bayesian high posterior density (HPD) between January 2013 and February 2014. The Singapore isolate was sequenced from a traveler from São Paulo (Brazil) to Singapore in May 2016 and was described as the first confirmed case of ZIKV infection in this country [24]. The isolates from Haiti have been previously associated to isolates from Brazil by Lednicky et al. (2016) [15] and Campos et al. (2018) [16], which also suggested that the 2014 Haitian ZIKV strain led to the spread of ZIKV in Brazil. However, our phylogeographic reconstruction with the new isolates, reveals a paraphyletic clustering of viruses from Haiti with respect to Brazilian lineages, suggesting that ZIKV spread from Brazil to Haiti. In addition, this reconstruction suggests that an outbreak in Salvador was responsible for seeding other locations in Northeast and Southeast Brazil and later spreading to Haiti. Noteworthy, this had already been suggested by Massad et al. (2017) [25], but based only on considering the limited numbers of ZIKV infections in Haiti compared to those obtained from the Brazilian epidemic.
Two of the newly sequenced isolates (TRDP173 and ZK0152) clustered together in a clade with genomes isolated from Brazil (Southeast and North) and USA (Clade II, Figure 1C), with a tMRCA estimated around February 2014 (95% HPD: July 2013, September 2014). The USA isolate was reported in a study by Grubaugh et al. (2019) [10], which clustered exclusively with Brazilian sequences from the Southeast [10]. However, in our analyses, the USA sequence formed a monophyletic clade with the TRDP173 isolated from Bahia.
The TRDP252 isolate clustered together in a clade with sequences from the Brazil (Southeast, North and Northeast), and Dominican Republic (Clade III, Figure 1D), with a tMRCA estimated around October 2013 (95% HPD: April 2013, March 2014). This Dominican Republic isolate was reported in a study by Metsky et al. (2017) [26], which clustered with Brazilian sequences, including from the Southeast, similar to our results.
The last isolate (TRDP433), sampled in July 2015, clustered together with sequences from Northeast Brazil, with a tMRCA estimated around March 2013 (95% HPD: September 2013, August 2014). All the isolates in this last clade were from Salvador with sampling dates between May 2015 and January 2016 (Clade IV, Figure 1E).
By analyzing sequences from other countries with the addition of new sequences from the Northeast region, we provide further evidence supporting the central role of ZIKV migration from the Northeast to the Southeast of Brazil and its subsequent spread outside the country. Unfortunately, the long time between the MRCA and the sequence dates does not allow more confident conclusions about the migration routes of ZIKV.
Although ZIKV was detected almost simultaneously in Brazil in the Salvador and Natal metropolitan regions during the period of 2015-2016, before the present study, only 5 sequences with >80% reference coverage (Genbank No. KJ776791.1) originating from this region were publicly available. The poor genomic sampling from this crucial moment and place of the Brazilian ZIKV outbreak limits the power of phylogenetic reconstructions and could bias the interpretation of disease transmission dynamics and may result in incorrect interpretations regarding the origin of ZIKV introduction and its routes of spread in Brazil.
Of note, since 2016, several independent efforts have been made in genomic surveillance of ZIKV to cover most of the Brazilian territories over different time periods, such as the prominent ZiBRA project that produced more than 140 sequences (Zika in Brazil Real Time Analysis) [23,27,28,29,30,31]. However, these great efforts were unable to rescue enough samples from this specific period due to logistic limitations in storing samples for long periods in Central Public Health Laboratories (LACEN).
5. Conclusions
Here we provide more credibility in the patterns of ZIKV spread from Northeast Brazil to the Americas based on the generation of 14 extra ZIKV sequences with good coverage (>80%), representing ~75% of the new publicly available dataset of this period, increasing our understanding of the dynamics of arbovirus infections worldwide and lending support to future public health responses against this virus.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Table S1: Clinical and social-demographic characteristics of the ZIKV positive cases in this study; Table S2: Dataset of ZIKV full or near full-length genomes with the date of sample collection and location; Table S3: Median and 95% HPD of estimated introductions of the clades I, II, III and IV; Supplementary Appendix: Extended text of study population, diagnosis, sequencing, collation of sequence dataset, phylogenetic and phylogeographic reconstructions.
Author Contributions
Conceptualization, G.S.R., L.A.S., P.L. and R.K.; Data curation, B.V., F.V.d.B., L.d.M. and L.A.S.; Formal analysis, B.V., F.V.d.B., L.d.M. and L.A.S.; Investigation, G.S.C., G.S.R., L.d.M., L.B.T., M.K., M.C., M.G.R., M.M.O.S., M.M.P. and V.S.B.; Methodology, G.S.R., P.L. and R.K.; Resources, A.-M.V., A.B., G.S.R., K.T., M.B.-N., P.L. and R.K.; Writing - original draft, L.d.M.; Writing - review & editing, A.-M.V., B.V., G.S.R., L.A.S., M.B.-N., M.M.P., P.L. and R.K. All the authors discussed the structure of the manuscript and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) [Finance Code 001 and 88881.130749/2016-01], Conselho Nacional de Desenvolvimento Científico e Tecnológico [439967/2016-3, 400830/2013-2, 440891/2016-7 and 311365/2021-3], Programa de Excelência em Pesquisa [420765/2017-4], Financiadora de Estudos e Projetos [MCTIC/FINEP/FNDCT 01/2016 ZIKA – 0416006000] and Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) [PET0026/2013, APP0044/2016, and PET0022/2016]. L.M. received a Ph.D. scholarship from CAPES. R.K., G.S.R., V.B., M.G.R., A.B. and M.B.-N. are CNPq fellows. The funding organizations had no role in the study design, data collection, data interpretation or writing of this report. B.V. was supported by a postdoctoral grant (12U7121N) of the Research Foundation – Flanders (Fonds voor Wetenschappelijk Onderzoek). P.L. acknowledges support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 725422-ReservoirDOCS), from the Wellcome Trust through project 206298/Z/17/Z and from the Research Foundation – Flanders (Fonds voor Wetenschappelijk Onderzoek – Vlaanderen, G0D5117N and G051322N).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Research Ethics Committee of the Faculdade de Medicina/UFBA (CEP/CAAE: 56910516.3.0000.5577; approval number: 1.657.324) and by the Research Ethics Committee of the Instituto Gonçalo Moniz/FIOCRUZ (CEP/CAAE: 55904616.4.0000.0040, approval number 3.363.703; CEP/CAAE: 55904616.4.0000.0040, approval number: 1.642.535).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The new sequences have been deposited in NCBI GenBank under accession numbers OQ727565-OQ727578; and the XML files and datasets analyzed in this study are available in the GitHub repository (https://github.com/khourious/Early-ZIKV-genomes-NE-BR-to-the-Americas).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Broutet, N.; Krauer, F.; Riesen, M.; Khalakdina, A.; Almiron, M.; Aldighieri, S.; Espinal, M.; Low, N.; Dye, C. Zika Virus as a Cause of Neurologic Disorders. N. Engl. J. Med. 2016, 374, 1506–1509. [Google Scholar] [CrossRef]
- Kuno, G.; Chang, G.-J.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the Genus Flavivirus. J Virol. 1998, 72, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G.-J.J. Full-Length Sequencing and Genomic Characterization of Bagaza, Kedougou, and Zika Viruses. Arch Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; Fajardo, A.; Moreno, P.; Moratorio, G.; Cristina, J. An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region. Viruses. 2018, 10, 698. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and Serologic Properties of Zika Virus Associated with an Epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Aubry, M.; Teissier, A.; Huart, M.; Merceron, S.; Vanhomwegen, J.; Roche, C.; Vial, A.-L.; Teururai, S.; Sicard, S.; Paulous, S.; et al. Zika Virus Seroprevalence, French Polynesia, 2014–2015. Emerg. Infect. Dis. 2017, 23, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; Melo, V.C.A. de; Mosimann, A.L.P.; Santos, G.I.V. dos; Santos, C.N.D. dos; Luz, K. First Report of Autochthonous Transmission of Zika Virus in Brazil. Mem. Inst. Oswaldo Cruz. 2015, 110, 569–572. [Google Scholar] [CrossRef]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika Virus Outbreak, Bahia, Brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Kraemer, M.U.G.; Dudas, G.; Tan, A.L.; Gangavarapu, K.; Wiley, M.R.; White, S.; Thézé, J.; Magnani, D.M.; et al. Genomic Epidemiology Reveals Multiple Introductions of Zika Virus into the United States. Nature. 2017, 546, 401–405. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Saraf, S.; Gangavarapu, K.; Watts, A.; Tan, A.L.; Oidtman, R.J.; Ladner, J.T.; Oliveira, G.; Matteson, N.L.; Kraemer, M.U.G.; et al. Travel Surveillance and Genomics Uncover a Hidden Zika Outbreak during the Waning Epidemic. Cell. 2019, 178, 1057–1071.e11. [Google Scholar] [CrossRef]
- Chitti, S. V.; Prasad, A.K.; Saxena, S.K. Emerging Zika Virus Disease: A Public Health Emergency of Global Concern. VirusDisease. 2016, 27, 211–214. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Director-General Summarizes the Outcome of the Emergency Committee Regarding Clusters of Microcephaly and Guillain-Barré Syndrome. Available online: https://www.who.int/en/news-room/detail/01-02-2016-who-director-general-summarizes-the-outcome-of-the-emergency-committee-regarding-clusters-of-microcephaly-and-guillain-barré-syndrome (accessed on 01 April 2023).
- WHO. Fifth Meeting of the Emergency Committee under the International Health Regulations (2005) Regarding Microcephaly, Other Neurological Disorders and Zika Virus. Available online: https://www.who.int/en/news-room/detail/18-11-2016-fifth-meeting-of-the-emergency-committee-under-the-international-health-regulations-(2005)-regarding-microcephaly-other-neurological-disorders-and-zika-virus (accessed on 01 April 2023).
- Faria, N.R.; Azevedo, R. do S. da S.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Thézé, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika Virus in the Americas: Early Epidemiological and Genetic Findings. Science. 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.; Beau De Rochars, V.M.; El Badry, M.; Loeb, J.; Telisma, T.; Chavannes, S.; Anilis, G.; Cella, E.; Ciccozzi, M.; Rashid, M.; et al. Zika Virus Outbreak in Haiti in 2014: Molecular and Clinical Data. PLoS Negl. Trop. Dis. 2016, 10, e0004687. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.D.L.; Durães-Carvalho, R.; Rezende, A.M.; de Carvalho, O.V.; Kohl, A.; Wallau, G.L.; Pena, L.J. Revisiting Key Entry Routes of Human Epidemic Arboviruses into the Mainland Americas through Large-Scale Phylogenomics. Int. J. Genomics. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Villabona-Arenas, C.J.; Hanage, W.P.; Tully, D.C. Phylogenetic Interpretation during Outbreaks Requires Caution. Nat. Microbiol. 2020, 5, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Rasche, A.; Baronti, C.; Aldabbagh, S.; Cadar, D.; Reusken, C.B.; Pas, S.D.; Goorhuis, A.; Schinkel, J.; Molenkamp, R.; et al. Assay Optimization for Molecular Detection of Zika Virus. Bull. World Health Organ. 2016, 94, 880–892. [Google Scholar] [CrossRef]
- Rathore, A.P.S.; St. John, A.L. Cross-Reactive Immunity Among Flaviviruses. Front. Immunol. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Cardoso, C.W.; Paploski, I.A.D.; Kikuti, M.; Rodrigues, M.S.; Silva, M.M.O.; Campos, G.S.; Sardi, S.I.; Kitron, U.; Reis, M.G.; Ribeiro, G.S. Outbreak of Exanthematous Illness Associated with Zika, Chikungunya, and Dengue Viruses, Salvador, Brazil. Emerg. Infect. Dis. 2015, 21, 2274–2276. [Google Scholar] [CrossRef]
- Silva, M.M.O.; Tauro, L.B.; Kikuti, M.; Anjos, R.O.; Santos, V.C.; Gonçalves, T.S.F.; Paploski, I.A.D.; Moreira, P.S.S.; Nascimento, L.C.J.; Campos, G.S.; et al. Concomitant Transmission of Dengue, Chikungunya, and Zika Viruses in Brazil: Clinical and Epidemiological Findings From Surveillance for Acute Febrile Illness. Clin. Infect. Dis. 2019, 69, 1353–1359. [Google Scholar] [CrossRef]
- de Moraes, L.; Cerqueira-Silva, T.; Nobrega, V.; Akrami, K.; Santos, L.A.; Orge, C.; Casais, P.; Cambui, L.; Rampazzo, R. de C.P.; Trinta, K.S.; et al. A Clinical Scoring System to Predict Long-Term Arthralgia in Chikungunya Disease: A Cohort Study. PLoS Negl. Trop. Dis. 2020, 14, e0008467. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Ho, Z.J.M.; Hapuarachchi, H.C.; Barkham, T.; Chow, A.; Ng, L.C.; Lee, J.M.V.; Leo, Y.S.; Prem, K.; Lim, Y.H.G.; de Sessions, P.F.; et al. Outbreak of Zika Virus Infection in Singapore: An Epidemiological, Entomological, Virological, and Clinical Analysis. Lancet Infect. Dis. 2017, 17, 813–821. [Google Scholar] [CrossRef] [PubMed]
- MASSAD, E.; BURATTINI, M.N.; KHAN, K.; STRUCHINER, C.J.; COUTINHO, F.A.B.; WILDER-SMITH, A. On the Origin and Timing of Zika Virus Introduction in Brazil. Epidemiol. Infect. 2017, 145, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika Virus Evolution and Spread in the Americas. Nature. 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Sabino, E.C.; Nunes, M.R.T.; Alcantara, L.C.J.; Loman, N.J.; Pybus, O.G. Mobile Real-Time Surveillance of Zika Virus in Brazil. Genome Med. 2016, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Quick, J.; Claro, I.M.; Thézé, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and Cryptic Transmission of Zika Virus in Brazil and the Americas. Nature. 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Iani, F.C.M.; Giovanetti, M.; Fonseca, V.; Souza, W.M.; Adelino, T.E.R.; Xavier, J.; Jesus, J.G.; Pereira, M.A.; Silva, M.V.F.; Costa, A.V.B.; et al. Epidemiology and Evolution of Zika Virus in Minas Gerais, Southeast Brazil. Infect. Genet. Evol. 2021, 91, 104785. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Pereira, L.A.; Adelino, T.É.R.; Fonseca, V.; Xavier, J.; de Araújo Fabri, A.; Slavov, S.N.; da Silva Lemos, P.; de Almeida Marques, W.; Kashima, S.; et al. A Retrospective Overview of Zika Virus Evolution in the Midwest of Brazil. Microbiol. Spectr. 2022, 10. [Google Scholar] [CrossRef]
- Giovanetti, M.; Faria, N.R.; Lourenço, J.; Goes de Jesus, J.; Xavier, J.; Claro, I.M.; Kraemer, M.U.G.; Fonseca, V.; Dellicour, S.; Thézé, J.; et al. Genomic and Epidemiological Surveillance of Zika Virus in the Amazon Region. Cell Rep. 2020, 30, 2275–2283.e7. [Google Scholar] [CrossRef]
- Balm, M.N.D.; Lee, C.K.; Lee, H.K.; Chiu, L.; Koay, E.S.C.; Tang, J.W. A Diagnostic Polymerase Chain Reaction Assay for Zika Virus. J. Med. Virol. 2012, 84, 1501–1505. [Google Scholar] [CrossRef]
- Black, A.; Moncla, L.H.; Laiton-Donato, K.; Potter, B.; Pardo, L.; Rico, A.; Tovar, C.; Rojas, D.P.; Longini, I.M.; Halloran, M.E.; et al. Genomic Epidemiology Supports Multiple Introductions and Cryptic Transmission of Zika Virus in Colombia. BMC Infect. Dis. 2019, 19, 963. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics. 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics. 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience. 2021, 10, 1–4. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An Automated System for Virus Identification from High-Throughput Sequencing Data. Bioinformatics. 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
Figure 1.
Bayesian phylogeographic reconstruction of ZIKV in Brazil. (A) Maximum clade credibility (MCC) phylogeny estimated from full or near full-length ZIKV sequences from Brazil. For visual clarity, clades highlighted in gray represent the sequences reported in this study. Branch colors indicate the most probable ancestral lineage location from America countries and Brazilian states. (B–E) Highlighted clades of the inferred geographic migration history of ZIKV, showing the movement from Northeast into Southeast Brazil, and subsequently to other countries such as (B) Haiti and Singapore, (C) USA, (D) Dominican Republic and (E) a single introduction of ZIKV outbreak in Salvador, Northeast Brazil. Circles are sequences from Genbank; Squares and triangles are sequences from this study. The numbers show the posterior probabilities > 0.80.
Figure 1.
Bayesian phylogeographic reconstruction of ZIKV in Brazil. (A) Maximum clade credibility (MCC) phylogeny estimated from full or near full-length ZIKV sequences from Brazil. For visual clarity, clades highlighted in gray represent the sequences reported in this study. Branch colors indicate the most probable ancestral lineage location from America countries and Brazilian states. (B–E) Highlighted clades of the inferred geographic migration history of ZIKV, showing the movement from Northeast into Southeast Brazil, and subsequently to other countries such as (B) Haiti and Singapore, (C) USA, (D) Dominican Republic and (E) a single introduction of ZIKV outbreak in Salvador, Northeast Brazil. Circles are sequences from Genbank; Squares and triangles are sequences from this study. The numbers show the posterior probabilities > 0.80.
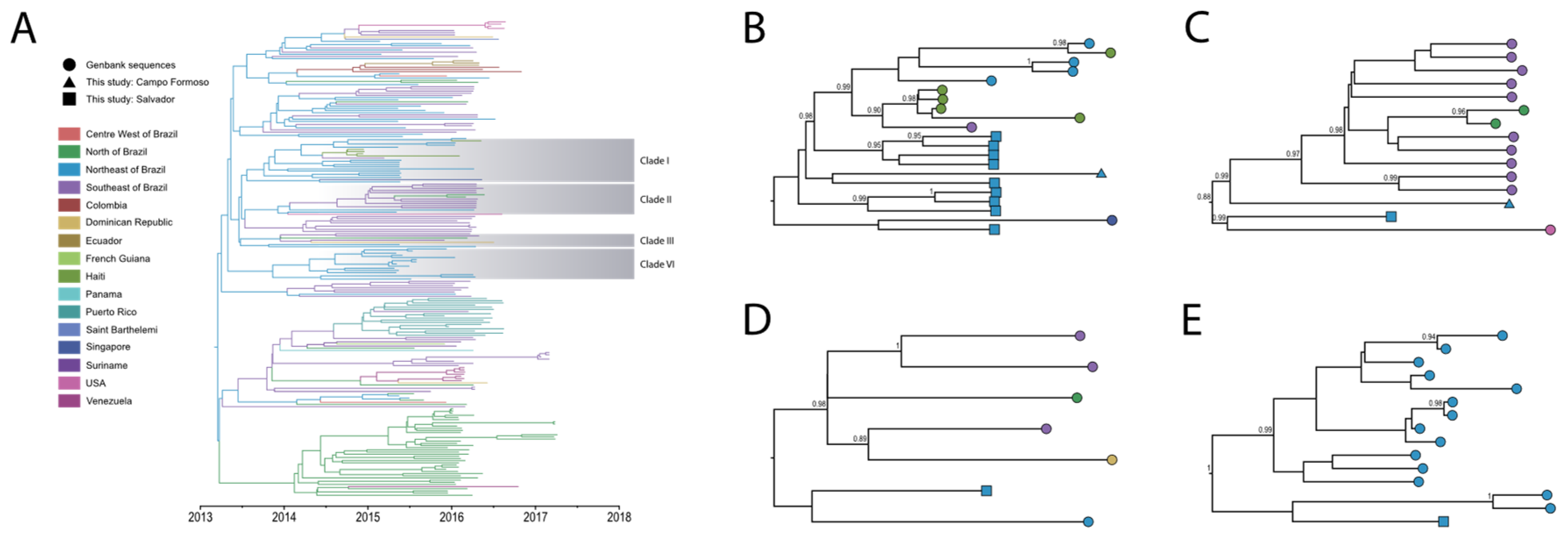

Table 1.
Summary of sample collection, location, Cq value and sequencing metrics for the 14 ZIKV new genome sequences.
Table 1.
Summary of sample collection, location, Cq value and sequencing metrics for the 14 ZIKV new genome sequences.
| ID | Municipality | Collection Date | Cq Value | No. of Mapped Reads |
Avg. Depth Coverage |
Reference Covered |
|---|---|---|---|---|---|---|
| TRDP173 | Salvador | 2015-05-06 | 30.96 | 39,617 | 1,207.66 | 94.03 |
| TRDP238 | Salvador | 2015-05-18 | 31.16 | 44,842 | 1258.69 | 90.22 |
| TRDP252 | Salvador | 2015-05-19 | 28.64 | 38,452 | 1,197.17 | 88.76 |
| TRDP256 | Salvador | 2015-05-19 | 29.95 | 39,530 | 1,170.28 | 89.10 |
| TRDP257 | Salvador | 2015-05-19 | 37.96 | 58,214 | 1,372.15 | 83.82 |
| TRDP274 | Salvador | 2015-05-20 | 37.84 | 35,944 | 1,116.41 | 84.62 |
| TRDP282 | Salvador | 2015-05-21 | 35.49 | 21,026 | 727.62 | 88.89 |
| TRDP300 | Salvador | 2015-05-22 | 30.03 | 19,670 | 678.25 | 81.89 |
| TRDP309 | Salvador | 2015-05-26 | 33.87 | 20,459 | 705.72 | 87.94 |
| TRDP317 | Salvador | 2015-05-26 | 33.60 | 20,414 | 704.03 | 87.99 |
| TRDP333 | Salvador | 2015-05-27 | 39.07 | 11,078 | 377.35 | 80.62 |
| TRDP433 | Salvador | 2015-07-09 | 27.63 | 60,626 | 1,438.09 | 96.64 |
| ZK0110 | Campo Formoso | 2016-04-08 | 21.91 | 40,452 | 1,301.48 | 97.18 |
| ZK0152 | Campo Formoso | 2016-04-09 | 34.94 | 17,820 | 562.33 | 84.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



