
Submitted:
12 April 2023
Posted:
14 April 2023
You are already at the latest version
Abstract
The primary cilium plays critical roles in homeostasis and development of neurons. Recent studies demonstrate that cilia length is regulated by the metabolic state of cells, as dictated by processes such as glucose flux and O-GlcNAcylation (OGN). The study of cilia length regulation during neuron development, however, has been an area left largely unexplored. This project aims to elucidate the roles of O-GlcNAc in neuronal development through its regulation of the primary cilium. Here, we present findings suggesting that OGN levels negatively regulate cilia length on differentiated cortical neurons derived from human-induced pluripotent stem cells. In neurons, cilia length increased significantly during neurons maturation (after day 35), while OGN levels began to drop. Long-term perturbation of OGN via drugs, which inhibit or promote its cycling, during neuron development also have varying effects. Diminishing OGN levels increases cilia length until day 25, when neural stem cells expand and undergo early neurogenesis, before causing cell cycle exit defects and multinucleation. Elevating OGN levels induces greater primary cilia assembly but ultimately results in the development of premature neurons, which have higher insulin sensitivity. These results indicate that OGN levels and primary cilia length are jointly critical in proper neuron development and function. Understanding the interplays between these two nutrient sensors, O-GlcNAc and the primary cilium, during neuron development is important in paving connections between dysfunctional nutrient-sensing and early neurological disorders.
Keywords:
O-GlcNAc
; primary cilia
; neuronal development
; cortical neurons
; human induced-pluripotent stem cells
1. Introduction
The primary cilium, a solitary and non-mobile appendage, protrudes from the surface of nearly all mammalian cells [1]. Though overlooked as vestigial remnants of a long-lost, motile counterpart for decades following their discovery, phenotypes of cilia mutants have encouraged modern researchers to grasp a greater comprehension of ciliogenesis and the influence of developed primary cilia on physiological processes. Besides timing of cilia assembly and disassembly, which largely hinges on a bidirectional crosstalk with cell division, structural changes in cilia also bear tremendous implications in the overall function of the organelle [2,3,4,5,6,7]. To properly fulfill their various roles, primary cilia reach a specified range of sizes depending on the cell type from which they extend [8,9,10]. Disruptions in the form or function of these projections, commonly result in a myriad of debilitating disorders based on the affected organ, including cognitive deficiencies, blindness, deafness, and irregular breathing patterns, termed ciliopathies [11,12,13,14,15]. Beyond their transductory roles in special sensory cells, enabling the sensation of movement, light, odors, and various additional stimuli, primary cilia are instrumental in regulating several neurodevelopmental pathways [16,17,18,19,20,21,22]. Ablation and reduction of primary cilia through conditional knockout of certain genes such as Stumpy, which encodes a basal body protein, diminish hippocampal neurogenesis through an impairment of Hedgehog signaling [23,24]. In the nervous system, cilia are essential for proper patterning, maturation, general survival, and stem cell fate determination, as explained by their links with non-canonical Wnt, Hippo, Notch, mTOR, and TGF-β, among others [25,26,27,28]. Primary cilia are also highly dynamic and undergo alterations in shape, size, and composition constantly [29,30]. Such changes are brought about by enzymatic cascades, gene modifications, and even post-translational modifications on the cilium itself [31,32,33,34,35].
OGN, or the incorporation of O-linked-β-N-acetylglucosamine (O-GlcNAc) onto serine and threonine residues, is a post-translational modification which modulates protein function [36,37,38,39]. Despite being observed on over 9000 human proteins since its finding, the inclusion and cleavage of this moiety are driven by only a pair of enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) respectively [40]. OGT’s sensitivity to Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), its donor substrate and a central node in metabolism connecting glucose, fatty acid, amino acid, and nucleotide productions, makes OGN a ‘rheostat’ capable of adjusting cellular activity when faced with stress and/or changes in nutrient availability. In instances of glucose starvation, for example, O-GlcNAc modification of most proteins also tapers down, while a surplus of glucose results in enhanced OGN [41]. Prolonged dysregulation of OGN has been observed in diabetes, Alzheimer’s disease, cardiomyopathy, virtually all cancers, and other chronic illnesses of aging [42,43,44,45,46,47,48,49]. This single sugar addition regulates a diverse set of essential biological processes including transcription, mitochondrial functions, cell cycle progression, and cilia assembly [36,50,51,52,53,54]. Several studies have explored the separate effects of aberrant OGN on primary cilia and neuronal developments, but none have integrated the two. The following works attempt to record and understand how nutrient flux and inhibition of OGT/OGA impact the differentiation of pluripotent stem cells into mature neurons. Investigating how altered O-GlcNAc cycling affects primary cilia formation and subsequent neural development holds the potential in uncovering clues in how deficient metabolism gives rise to disease.
2. Materials and Methods
iPSC cell lines and maintenance
Human iPSCs were previously derived and characterized [69]. The cells were maintained as previously described [70]. Briefly, cells were cultured in feeder-free and chemically defined Essential 8 medium (Gibco, A15710-01) on 5 μg/ml vitronectin coated (Thermo Fisher/Life Technologies, A14700) cell culture plates, and were passaged with 0.5 mM EDTA (Corning, 46-034-Cl). The cells were maintained with 5% CO2 in a 37°C incubator.
Cortical neuron differentiation and treatments
Human iPSCs were induced into cortical neurons according to established protocols [71,72]. Briefly, when the cells reached 70-90% confluency, cells on two dishes were replated onto a single Geltrex (1:200, Gibco, A1413302)-coated culture plates. 100% confluency was evaluated after 24-hour replating. After washing the cells with DPBS twice, neural induction medium consisting of neural maintenance medium, 10 μM SB431542 (Tocris, 16-141-0), and 1 μM Dorsomorphin (Tocris, 30-931-0) was added to the cells. Neural maintenance medium contained 1:1 mixture of DMEM/F-12 GlutaMAX (Gibco, 10565018) and Neurobasal medium (Gibco, 12348-017), 0.5x B-27 (Gibco, 17504044), 0.5x N-2 (Gibco, 17502048), 0.5x Non-essential amino acids (Gibco, 11140-050), 0.5x GlutaMAX (Gibco, 35050-061), 500 mM sodium pyruvate (Sigma, S8636), 2.5 mg/ml Insulin (Sigma, I9278), 50 μM 2-mercaptoethanol, P/S (25 U/ml, Gibco, 15140122). On day 12, cells were detached using dispase (Stemcell, 7913) and replated with neural induction medium onto laminin-coated culture plates (10mg/ml, Sigma, L2020). 20 ng/ml of basic-FGF (R&D Systems, 4114-TC) was added to neural maintenance medium from day 13 to 16. On day 17, basic-FGF was withdrawn from the medium, and the cells were maintained in only neural maintenance medium. On day 25, cells were passaged as single cells with Accutase (Corning, 25-058-CI) onto laminin-coated culture plates. On day 35, the cells were replated at density of 100K cells/cm2 onto poly-L-ornithine (Sigma, P3655) and laminin-coated culture plates. For the following days until day 55 (the end of differentiation), cells were cultured in neural maintenance medium. For short-term OGN alteration, Ac-5SGlcNAc (Ac5S, a kind gift from Dr. Boons, 50μM) or Thiamet-G (TMG, Sigma, SML0244, 1μM) were used to treat neurons for 24 hours on day 55 after differentiation. For long-term perturbation of O-GlcNAc levels, Ac5S (25μM) or TMG (1μM) were added to the culture medium from day 0 throughout the differentiation process. For insulin treatment, cells were starved for 3 hours followed by insulin (Sigma, I9278) treatment at indicated concentration for 30 mins on day 55 of differentiation.
Western blots
Cells were lysed in RIPA buffer with 1x protease inhibitor (Thermo, A32963), 1x phosphatase inhibitor (Thermo, A32957), and 10 μM PUGNAc (Sigma, A7229), then centrifuged at 13,200 rpm for 15 min at 4°C. The supernatant was collected, and total protein concentration was measured using the BCA Protein Assay kit (Thermo, 23227), according to the manufacturer’s protocols. For western blot experiments, 20 μg of total cell protein was combined with 4x Laemmli Sample Buffer (Bio-Rad, 1610747), diluted to 1x concentration, and heated at 95°C for 10 min. The samples were separated on 4-12% pre-cast Bis-Tris gels (Invitrogen, WG1402BOX) at 120V for ~90 min, transferred onto 0.2 mm PVDF membranes (GE healthcare, 10-6000-21) at 400 mA for 2 hours, and blocked with 5% BSA (Rockland, BSA-1000). The membranes were incubated with primary antibodies overnight at 4°C. They were washed three times with TBS-T (10 min each) and incubated with HRP secondary antibodies for 1 hour at room temperature. After three washes with TBS-T (10 min each), the samples were imaged. Antibodies used for western blotting analysis were specific to Nestin (Neuromics, MO22183, 1:1000), Tuj1 (Biolegend, 802001, 1:1000), CTIP2 (Biolegend, 650602, 1:1000), SatB2 (Santa Cruz, sc-81376, 1:500), Caspase3 (Cell signaling, 9662S, 1:1000), O-GlcNAc (CTD110.6, made in house, 1:1000), and β-actin (Cell Signaling, 3700S, 1:5000). Membrane imaging was completed using iBright FL1500 imaging system (Invitrogen), signal detection was developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo, 34578) and band intensity was quantified with NIH ImageJ (a public domain image analysis software) and normalized to β-actin.
Immunofluorescence staining
Immunofluorescence staining was carried out according to previous study [73]. Cell samples were fixed in Methanol (Sigma, 34860) for 10 minutes, permeabilized with 0.25% Triton X-100 (Sigma, T9284) in PBS, washed with PSB, and blocked for 30 minutes in blocking buffer containing 2% BSA, 0.2% gelatin (Sigma, G1890), 10 mM glycine (Sigma, G8898), and 50 mM NH4Cl (Acros, 393182500). The samples were incubated with primary antibodies in blocking buffer overnight at 4°C. After the samples were washed three times, they were incubated with fluorescence secondary antibodies and DAPI staining solution at room temperature. Antibodies used for immunofluorescence staining analysis were specific to NeuN (Abcam, ab104224, 1:500), Tuj1 (Biolegend, 802001, 1:500), Arl13b (Proteintech, 66739-1-Ig, 1:250), and IFT88 (Proteintech, 13967-1-AP, 1:200). Stained cells were post-fixed with 2% PFA in PBS for 10 mins, followed by imaging with an Echo Revolve microscope, and images were analyzed with ImageJ to measure cilia length and multinucleated cells percentage.
Quantification and statistical analysis
The GraphPad Prism software was used for statistical analysis. All statistical analyses were performed using t-test, one-way, or two-way ANOVA followed by multiple comparison test. Data in graphs are expressed as mean values ± standard deviation of the mean (mean ± SD). Error bars represented denote the SD.
3. Results
3.1. Results
3.1.1. Cilia length in cortical neurons is negatively correlated with O-GlcNAc levels
To study neuronal primary cilia, we differentiated human induced pluripotent stem cells (iPSC) into cortical neurons before culturing them in 30mM (high glucose level), 5mM (normal physiological glucose level in the brain), and 2mM (low glucose level) glucose medium for 24 h to induce ciliogenesis. The formation of cilia on neurons was analyzed by immunostaining with antibodies against IFT88 and Arl13b cilium markers as well as neuronal nuclear protein (NeuN), a neuronal marker (Figure 1A). We determined primary cilia length from NeuN-positive cells by measuring the distance from the base to the tip as indicated by IFT88. Cells cultured in 30mM glucose had a primary cilia length of 2.11 ± 0.82 μm, while cells cultured in 5mM and 2mM glucose had significantly longer cilia lengths of 2.31 ± 0.90 μm and 2.60 ± 0.95 μm, respectively. Since glucose deprivation decreases HBP flux, dampening the O-GlcNAc modification of proteins, we detected cellular OGN levels at different glucose concentrations via Western blotting. The immunoblots suggested that cellular OGN levels decreased significantly when cells were cultured in 5mM and 2mM glucose medium when compared to 30 mM glucose (Figure 1B). To verify that elongation of neuronal cilia length was indeed caused by changes in O-GlcNAc cycling, we treated cells with OGT and OGA inhibitors, Ac5S and TMG respectively, to decrease or increase OGN levels. After confirming the efficiency of chemical inhibitors by immunoblotting against O-GlcNAc, we analyzed the neuronal cilia length. The mean length of the 5mM control group was 2.26 ± 0.87 μm, the Ac5S treatment group had an elongated cilia length of 2.61 ± 0.85 μm, while the TMG treatment group had a shortened cilia length of 1.64 ± 0.77 μm; both differences are statistically significant. These results indicate that primary cilia length on cortical neuron cells is negatively regulated by cellular OGN levels.
3.1.2. Cilia formation is partially regulated by O-GlcNAc levels during cortical neuron differentiation
After demonstrating that short-term alterations in OGN levels negatively regulate cilia length in differentiated cortical neurons, we wondered if OGN levels also display other trends inversely correlated with cilia length during cortical neuron differentiation. To answer this question, we utilized human iPSC-derived cortical neuron as a platform to observe the interaction between cilia and OGN during cortical neuron differentiation. Throughout the differentiation process, cells underwent several stages, including neural stem cell, progenitor cells, early phase neurons, and functional neurons (Figure 2A). Cells were harvested at several key time points to determine the OGN levels via Western blotting (Figure 2B). The results showed that O-GlcNAc modification levels gradually increased after differentiation and reached their highest levels around day 35. After that, the O-GlcNAc levels started to descend as the neuron matured. Conversely, cilia length was elongated from 1.85± 0.61 on day 0 to 2.12 ± 0.54 µm on day 12 then gradually shortened to 1.85 ± 0.75 µm on day 35 at progenitor/early phase neuronal stages, while longer cilia were observed as the neurons matured over time (2.33 ± 0.90 µm on day 45 and 2.27 ± 0.79 µm on day 55) (Figure 2C). These results indicate that OGN levels might also be negatively associated with cilia length during neuron differentiation and thereby mediate multiple signaling pathways in the complex process of neuronal development.
3.1.3. Long-term perturbation of OGN causes ciliary defect during cortical neuron development
To further investigate the relationship between OGN and cilia of cortical neurons during development, we turned to pharmacological means of regulating OGN levels. The cortical neurons were generated from iPSCs using the same protocol with the addition of an OGT inhibitor, Ac5S, or OGA inhibitor, TMG, thorough out the differentiation process (Figure 3A). The efficacy of inhibitors was confirmed by Western blotting at multiple key points of differentiation (Figure 3B). OGN levels were effectively diminished by Ac5S treatment and elevated by TMG administration at all key time points. The lengths of cilia on cells, differentiated in the presence of Ac5S or TMG, were measured at several time points to reveal the effect of long-term OGN alteration (Figure 3C). On day 0 and day 12, Ac5S treatment groups displayed significantly elongated cilia length (2.11 ± 0.77 µm on day 0 and 2.27 ± 0.49 µm on day 12) when compared to control groups (1.89± 0.64 µm on day 0 and 2.05 ± 0.48 µm on day 12), while TMG treatments shortened cilia length (1.51 ± 0.49 µm and 1.51 ± 0.40 µm respectively). On day 25, mean cilia lengths of both Ac5S-treated and TMG-treated cells were remarkably longer (2.21 ± 0.60 µm for Ac5S and 2.19 ± 0.89 µm for TMG) than control cells (1.98 ± 0.61 µm). Interestingly, we observed a dramatically increased percentage of multinucleated cells at 69.27%, 74.47% and 80.83% by Ac5S treatment on days 35, 45 and 55, respectively. OGN and the cell cycle are inextricably linked, as a multitude of cell cycle-related proteins are dynamically modified by O-GlcNAc [55]. Hence, it is possible that abolishing O-GlcNAc directly affects cell division in early phases of differentiation. Since cilia were absent on the multinucleated cells of Ac5S-treated groups, we only compared cilia on control and TMG-treated cells after day 35. The TMG-treated cells displayed longer cilia, with mean lengths of 2.28 ± 0.92 µm on day 35, 2.79 ± 1.08 µm on day 45, and 3.37 ± 1.10 µm on day 55. Meanwhile, control cells had cilia measuring 1.85 ± 0.76 µm on day 35, 2.32 ± 0.90 µm on day 45, and 2.25 ± 0.79 µm on day 55. Altogether, decreased OGN caused aberrant cell cycle regulation and diminished cilia formation, while raised O-GlcNAc levels by TMG resulted in the formation of elongated cilia during cortical development.
3.1.4. Altered OGN and subsequent cilia formation interferes with cortical neuronal differentiation
To observe the impact of OGN and cilia formation on cortical neurogenesis, we harvested cells differentiated with and without Ac5S or TMG treatments on day 55. We tracked neuronal generation by immunostaining against Tuj1, a neuronal marker. Both Tuj1 staining and phase images indicated that the generation of neurons in the control group was robust. Ac5S-treated cells barely differentiated into neurons, with most of the cells developing into multinucleated fibroblast-like cells instead (Figure 4A). Contrariwise, TMG-treated cells were able to generate Tuj1-positive neurons though a reduced cell number was noticed at later phases of differentiation. Upon further analysis by Western blotting, we found that neurons generated with TMG expressed neural progenitor maker, Nestin, and Tuj1 but lacked cortical neuronal markers CTIP2 and SatB2 (Figure 4B). Additionally, a higher level of cleaved-caspase3 was observed in TMG-treated neurons than control neurons (Figure 4C). These results suggest that longer primary cilia, as induced by TMG, might result in premature differentiation and a reduced pool of progenitors, before ultimately, triggering an abnormal fate specification and apoptosis. These results are congruent with findings in patients with Seckel syndrome, in which the mutation of a centrosomal-P4.1-associated protein (CPAP), hampers timely cilia disassembly [56]. In short, downregulating OGN during cortical neurogenesis may block cell cycle progression and result in multinucleated cells instead of neurons, while excessive OGN and, therefore, longer cilia lead to premature neurons with poor specification (Figure 4D). Our results emphasize the importance of finely coordinated OGN as well as cilia assembly and disassembly in the proper development of the human cortical neuron.
3.1.5. Increased primary cilia length leads to higher insulin sensitivity of TMG-treated neurons
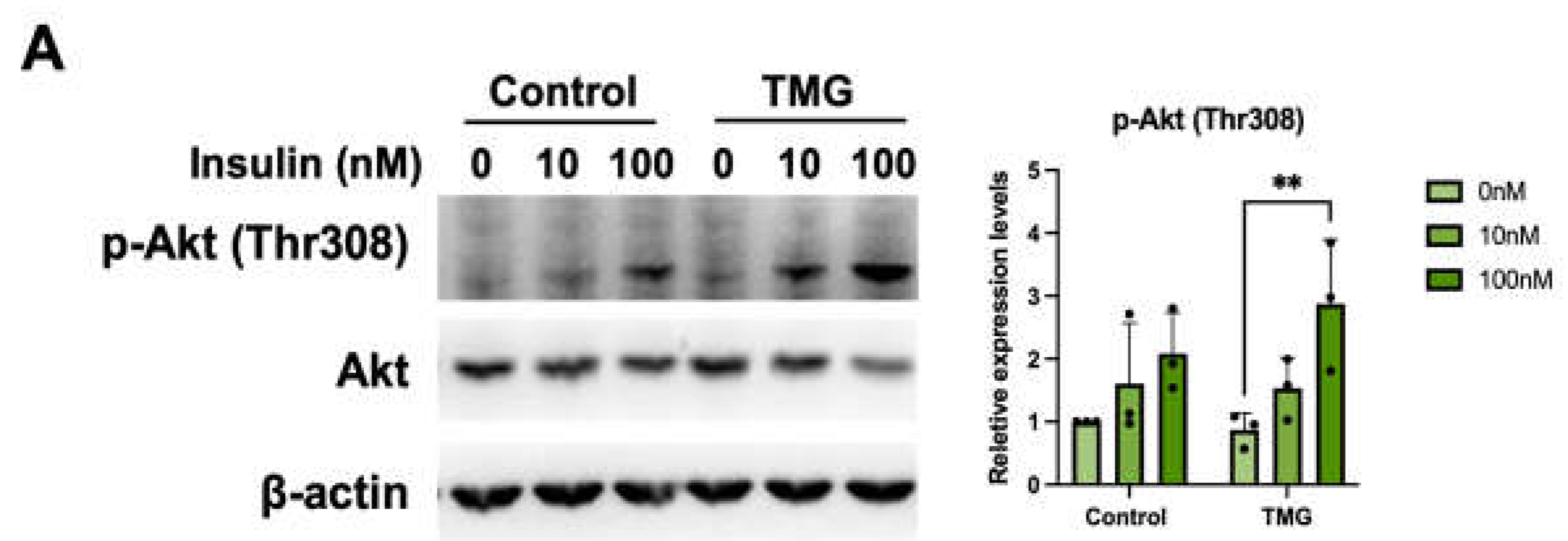
Cilia are known to harbor many signal cascade-initiating receptors such as Wnt, TGFB, IR, IGF-R1, and more [57]. Consequently, various signaling pathways have been shown to diminish when cilia are knocked out [58]. Understanding this, we wanted to determine if TMG-differentiated cells would show altered signaling due to their extended cilia. To do this, cells were serum starved and stimulated with insulin resulting in excitation of the PI3K/Akt pathway. Although both control and TMG-treated cells exhibited an increase in pT308 Akt phosphorylation upon insulin stimulation, the increase was 2.5-fold higher for TMG-treatment groups compared to a 1.4-fold increase for the control (Figure 5A). This result may suggest that the difference in Akt signaling was due to the extended cilia.
4. Discussion
In this study, we demonstrated that primary cilia assembly on differentiated cortical neurons was negatively regulated through either glucose or OGN levels and that the length was partially correlated with OGN levels during neuron differentiation. Furthermore, differentiating neurons when OGN levels are low caused cell cycle alterations and reduced cilia formation; higher OGN levels induced premature neurons and elongated cilia, which are more sensitive to insulin stimulation. Altogether, these findings suggest that regulations of cilia length have different mechanisms under short-term and long-term disruptions in OGN.
This series of experiments shows that glucose and OGN levels have negative impacts on cortical neuronal cilia length, which is consistent with previous studies stating that higher glucose and OGN levels significantly decrease primary ciliary length in human retinal pigment epithelia cells, diabetic mice, and diabetic patients [33,54]. Primary cilia of neurons are critical for regulating energy balance by sensing hormones and nutritional levels through various receptors, including insulin receptor (IR) and leptin receptor (LepR) [59,60]. Dysfunction in neuronal cilia result in metabolic disorder-related obesity and diabetes due to the mis-localization of hormone receptors and compromised satiety response [61]. Combined with the study of elevated OGN levels dampening the insulin signaling pathway, we can hypothesize that O-GlcNAc is a negative regulator of primary cilia length on neurons. It has been previously shown that chow-diet-fed lean mice have longer cilia length while obese mice, with a high fat and sucrose diet, have shorter cilia length on hypothalamus cells and are leptin resistant [62]. Future work focusing on studying the relationship between the conditional knockout of OGT and cilia length in neurons will aid us in better understanding the mechanisms of ciliopathy-associated obesity and diabetes.
In addition to their importance on differentiated neurons, primary cilia also play crucial roles during brain development through the transduction of important signaling pathways, such as Wnt and Shh signaling [63]. Primary cilia are required for the differentiation of neural progenitors and neural stems cells into multiple types of brain cells [64,65]. Dysfunction of primary cilia in early brain development will induce a plethora of severe abnormalities, such as neuron tube defects, corpus callosum agenesis, cerebellar hypoplasia, and hydrocephalus [66]. Perhaps due to the diverse ways in which ciliary defects present themselves, very little is known about how cilia length is regulated during neuron differentiation. Here, we used a human iPSC model and showed that cilia length was indeed oscillating during the neuron differentiation process, rather than following a continuous trend. Cilia elongated significantly on days 45 and 55 in differentiated neurons, while OGN levels hindered significantly compared to during differentiation, at day 35. These results suggest that O-GlcNAc is one of the factors involved in regulating cilia length during neuron differentiation. Future studies could investigate the molecular mechanisms behind how OGN levels regulate cilia length, and whether primary cilia transduce different signals at different stages of differentiation. Interestingly, the primary cilium may also facilitate cell-cell communication functions in addition to its role as signal hub to transduce signals [63,67]. Our immunostaining results showed that neuronal cilia form clusters (Figure 2C, Day 55), which possibly allows for cell-cell communication.
Differentiating neurons with abnormal OGN levels for extended periods of time showed that cilia length and cell morphology were drastically different from short-term conditions. Differentiating neurons in conditions limiting OGN showed longer cilia length until day 25, however, cells gradually died and surviving cells were mostly de-ciliated and multinucleated. It has been previously known that attenuated cellular OGN levels results in multinucleation by blocking cytokinesis [68]. This could be explained by the prolonged depletion of OGN arresting the cell cycle by inhibiting cytokinesis and, thus, prohibiting neuron differentiation. On the other hand, when differentiating neurons underwent prolonged elevations in OGN levels, we observed that cells had significantly longer cilia length compared to the control group since day 25. Cells also developed into neuron progenitors earlier but were unable to develop into cortical neurons. This is consistent with previous findings that long-cilia-iPSCs derived from Seckel syndrome patients showed signs of early differentiation, as indicated by neuronal maker Tuj1, decreased proliferative capacity, and delayed cell cycle re-entry [56]. These results suggest that long-term TMG treatment has different effects on primary cilia length regulation compared to short-term treatment. Future research on how fluctuations in OGN levels regulate cilia length through cell cycle progression holds potential in greatly expanding our knowledge of neuron differentiation and its relationship with disease.
Author Contributions
Conceptualization, J.L.T. and C.W.H.; Methodology, J.L.T and C.W.H.; Formal Analysis, J.L.T. and C.W.H.; Investigation, J.L.T., C.W.H., F.E., M.P.M., and R.L.M.; Visualization, J.L.T., C.W.H., and F.E.; Supervision, G.W.H.; Writing – Original Draft, J.L.T., C.W.H., F.E., M.P.M., and R.L.M.; Writing – Review & Editing, all authors.
Funding
The authors are supported by the Georgia Research Alliance (GRA), the University of Georgia and NIH R01 DK124366.
Acknowledgments
We would like to thank Dr. Michael Tiemeyer and Dr. Geert-Jan Boons at the University of Georgia for providing us with the necessary cell line and chemicals.
Conflicts of Interest
The authors declare no conflict of interest. GWH receives a share of royalty received on sales of the CTD 110.6 antibody, managed by JHU.
References
- Pazour, G.J. and G.B. Witman, The vertebrate primary cilium is a sensory organelle. Current Opinion in Cell Biology, 2003. 15(1): p. 105-110. [CrossRef]
- Goto, H., A. Inoko, and M. Inagaki, Cell cycle progression by the repression of primary cilia formation in proliferating cells. Cell Mol Life Sci, 2013. 70(20): p. 3893-905. [CrossRef]
- Mirvis, M., T. Stearns, and W. James Nelson, Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem J, 2018. 475(14): p. 2329-2353. [CrossRef]
- Plotnikova, O.V., E.A. Golemis, and E.N. Pugacheva, Cell cycle-dependent ciliogenesis and cancer. Cancer Res, 2008. 68(7): p. 2058-61. [CrossRef]
- Inoko, A., et al., Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J Cell Biol, 2012. 197(3): p. 391-405. [CrossRef]
- Besschetnova, T.Y., et al., Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr Biol, 2010. 20(2): p. 182-7. [CrossRef]
- Resnick, A. and U. Hopfer, Force-response considerations in ciliary mechanosensation. Biophys J, 2007. 93(4): p. 1380-90. [CrossRef]
- Sun, S., et al., Three-dimensional architecture of epithelial primary cilia. Proc Natl Acad Sci U S A, 2019. 116(19): p. 9370-9379. [CrossRef]
- Wang, L., et al., Alterations in renal cilium length during transient complete ureteral obstruction in the mouse. J Anat, 2008. 213(2): p. 79-85. [CrossRef]
- Verghese, E., et al., Renal primary cilia lengthen after acute tubular necrosis. J Am Soc Nephrol, 2009. 20(10): p. 2147-53. [CrossRef]
- Collin, G.B., et al., Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet, 2002. 31(1): p. 74-8.
- Romani, M., A. Micalizzi, and E.M. Valente, Joubert syndrome: congenital cerebellar ataxia with the molar tooth. The Lancet Neurology, 2013. 12(9): p. 894-905. [CrossRef]
- Hernandez-Hernandez, V., et al., Bardet-Biedl syndrome proteins control the cilia length through regulation of actin polymerization. Hum Mol Genet, 2013. 22(19): p. 3858-68. [CrossRef]
- May-Simera, H.L., et al., Primary Cilium-Mediated Retinal Pigment Epithelium Maturation Is Disrupted in Ciliopathy Patient Cells. Cell Rep, 2018. 22(1): p. 189-205. [CrossRef]
- Shi, H., et al., Mutations in OSBPL2 cause hearing loss associated with primary cilia defects via sonic hedgehog signaling. JCI Insight, 2022. 7(4). [CrossRef]
- Luo, N., et al., Primary cilia signaling mediates intraocular pressure sensation. Proc Natl Acad Sci U S A, 2014. 111(35): p. 12871-6. [CrossRef]
- McEwen, D.P., et al., Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc Natl Acad Sci U S A, 2007. 104(40): p. 15917-22.
- Butler, M.T. and J.B. Wallingford, Planar cell polarity in development and disease. Nat Rev Mol Cell Biol, 2017. 18(6): p. 375-388. [CrossRef]
- Ehnert, S., et al., TGF-beta1 impairs mechanosensation of human osteoblasts via HDAC6-mediated shortening and distortion of primary cilia. J Mol Med (Berl), 2017. 95(6): p. 653-663.
- Zullo, A., et al., Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum Mol Genet, 2010. 19(14): p. 2792-803. [CrossRef]
- Grisanti, L., et al., Primary cilia maintain corneal epithelial homeostasis by regulation of the Notch signaling pathway. Development, 2016. 143(12): p. 2160-71. [CrossRef]
- Kim, M., et al., The MST1/2-SAV1 complex of the Hippo pathway promotes ciliogenesis. Nat Commun, 2014. 5: p. 5370.
- Town, T., et al., The stumpy gene is required for mammalian ciliogenesis. Proc Natl Acad Sci U S A, 2008. 105(8): p. 2853-8. [CrossRef]
- Breunig, J.J., et al., Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A, 2008. 105(35): p. 13127-32. [CrossRef]
- Baudoin, J.P., et al., Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron, 2012. 76(6): p. 1108-22. [CrossRef]
- Bae, J.E., et al., Primary cilia mediate mitochondrial stress responses to promote dopamine neuron survival in a Parkinson's disease model. Cell Death Dis, 2019. 10(12): p. 952. [CrossRef]
- Li, A., et al., Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol, 2011. 13(4): p. 402-11. [CrossRef]
- Lee, C.H., et al., Primary cilia mediate early life programming of adiposity through lysosomal regulation in the developing mouse hypothalamus. Nat Commun, 2020. 11(1): p. 5772. [CrossRef]
- Choi, H., et al., Nucleus pulposus primary cilia alter their length in response to changes in extracellular osmolarity but do not control TonEBP-mediated osmoregulation. Sci Rep, 2019. 9(1): p. 15469.
- Matsumoto, M., et al., Dynamic Changes in Ultrastructure of the Primary Cilium in Migrating Neuroblasts in the Postnatal Brain. J Neurosci, 2019. 39(50): p. 9967-9988. [CrossRef]
- Stilling, S., et al., PIP2 determines length and stability of primary cilia by balancing membrane turnovers. Commun Biol, 2022. 5(1): p. 93. [CrossRef]
- Sachamitr, P., et al., PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat Commun, 2021. 12(1): p. 979. [CrossRef]
- Tian, J.L. and H. Qin, OGN Regulates Primary Ciliary Length by Promoting Microtubule Disassembly. iScience, 2019. 12: p. 379-391.
- Yang, Y., et al., Mixed-lineage leukemia protein 2 suppresses ciliary assembly by the modulation of actin dynamics and vesicle transport. Cell Discov, 2019. 5: p. 33. [CrossRef]
- Yang, Y., et al., CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res, 2014. 24(11): p. 1342-53. [CrossRef]
- Ozcan, S., S.S. Andrali, and J.E. Cantrell, Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta, 2010. 1799(5-6): p. 353-64.
- Li, C., et al., Hypertonic stress modulates eNOS function through O-GlcNAc modification at Thr-866. Sci Rep, 2021. 11(1): p. 11272.
- Wang, S., et al., Extensive crosstalk between OGN and phosphorylation regulates Akt signaling. PLoS One, 2012. 7(5): p. e37427.
- Yi, W., et al., Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science, 2012. 337(6097): p. 975-80. [CrossRef]
- Hart, G.W., Three Decades of Research on OGN - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front Endocrinol (Lausanne), 2014. 5: p. 183.
- Swamy, M., et al., Glucose and glutamine fuel protein OGN to control T cell self-renewal and malignancy. Nat Immunol, 2016. 17(6): p. 712-20.
- Guo, H. Guo, H., et al., O-Linked N-Acetylglucosamine (O-GlcNAc) Expression Levels Epigenetically Regulate Colon Cancer Tumorigenesis by Affecting the Cancer Stem Cell Compartment via Modulating Expression of Transcriptional Factor MYBL1. J Biol Chem, 2017. 292(10): p. 4123-4137.
- Gelinas, R., et al., AMPK activation counteracts cardiac hypertrophy by reducing OGN. Nat Commun, 2018. 9(1): p. 374.
- Umapathi, P., et al., Excessive OGN Causes Heart Failure and Sudden Death. Circulation, 2021. 143(17): p. 1687-1703.
- Ferrer, C.M., et al., OGN regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell, 2014. 54(5): p. 820-31.
- de Queiroz, R.M., et al., Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the p53 Pathway in Ovarian Cancer Cells. J Biol Chem, 2016. 291(36): p. 18897-914.
- Peterson, S.B. and G.W. Hart, New insights: A role for OGN in diabetic complications. Crit Rev Biochem Mol Biol, 2016. 51(3): p. 150-61.
- Park, J., et al., OGN ameliorates the pathological manifestations of Alzheimer's disease by inhibiting necroptosis. Sci Adv, 2021. 7(3).
- Liu, F., et al., OGN regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A, 2004. 101(29): p. 10804-9.
- Constable, S., et al., O-GlcNAc transferase regulates transcriptional activity of human Oct4. Glycobiology, 2017. 27(10): p. 927-937.
- Akinbiyi, E.O., et al., Blocked O-GlcNAc cycling alters mitochondrial morphology, function, and mass. Sci Rep, 2021. 11(1): p. 22106.
- Park, S.J., et al., Increased OGN of Drp1 by amyloid-beta promotes mitochondrial fission and dysfunction in neuronal cells. Mol Brain, 2021. 14(1): p. 6.
- Shin, H., et al., OGN of the Tumor Suppressor FOXO3 Triggers Aberrant Cancer Cell Growth. Cancer Res, 2018. 78(5): p. 1214-1224.
- Yu, F., et al., Ciliary defects caused by dysregulation of O-GlcNAc modification are associated with diabetic complications. Cell Res, 2019. 29(2): p. 171-173.
- Zachara, N.E. and G.W. Hart, Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta, 2006. 1761(5-6): p. 599-617.
- Gabriel, E., et al., CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J, 2016. 35(8): p. 803-19. [CrossRef]
- Ma, R., et al., Primary cilia and ciliary signaling pathways in aging and age-related brain disorders. Neurobiol Dis, 2022. 163: p. 105607. [CrossRef]
- Ishii, S., et al., Primary cilia safeguard cortical neurons in neonatal mouse forebrain from environmental stress-induced dendritic degeneration. Proc Natl Acad Sci U S A, 2021. 118(1). [CrossRef]
- Guemez-Gamboa, A., N.G. Coufal, and J.G. Gleeson, Primary cilia in the developing and mature brain. Neuron, 2014. 82(3): p. 511-21. [CrossRef]
- Tian, J.L. and F.I. Gomeshtapeh, Potential Roles of OGN in Primary Cilia- Mediated Energy Metabolism. Biomolecules, 2020. 10(11).
- Han, Y.M., et al., Leptin-promoted cilia assembly is critical for normal energy balance. J Clin Invest, 2014. 124(5): p. 2193-7. [CrossRef]
- Kirk, S.L., et al., Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One, 2009. 4(6): p. e5870. [CrossRef]
- Rocha, C. and P. Prinos, Post-transcriptional and Post-translational Modifications of Primary Cilia: How to Fine Tune Your Neuronal Antenna. Front Cell Neurosci, 2022. 16: p. 809917. [CrossRef]
- Wang, G., et al., Regulation of neural progenitor cell motility by ceramide and potential implications for mouse brain development. J Neurochem, 2008. 106(2): p. 718-33. [CrossRef]
- He, Q., et al., Primary cilia in stem cells and neural progenitors are regulated by neutral sphingomyelinase 2 and ceramide. Mol Biol Cell, 2014. 25(11): p. 1715-29. [CrossRef]
- Thomas, S., et al., Cilia in hereditary cerebral anomalies. Biol Cell, 2019. 111(9): p. 217-231. [CrossRef]
- Gerdes, J.M., E.E. Davis, and N. Katsanis, The vertebrate primary cilium in development, homeostasis, and disease. Cell, 2009. 137(1): p. 32-45. [CrossRef]
- Li, Z., et al., Checkpoint kinase 1-induced phosphorylation of O-linked beta-N-acetylglucosamine transferase regulates the intermediate filament network during cytokinesis. J Biol Chem, 2017. 292(48): p. 19548-19555.
- Si-Tayeb, K., et al., Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol, 2010. 10: p. 81. [CrossRef]
- Wu, H.F. and N. Zeltner, Efficient Differentiation of Postganglionic Sympathetic Neurons using Human Pluripotent Stem Cells under Feeder-free and Chemically Defined Culture Conditions. J Vis Exp, 2020(159).
- Shi, Y., et al., Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci, 2012. 15(3): p. 477-86, S1. [CrossRef]
- Shi, Y., P. Kirwan, and F.J. Livesey, Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc, 2012. 7(10): p. 1836-46. [CrossRef]
- King, C.R., et al., Fbxo41 Promotes Disassembly of Neuronal Primary Cilia. Sci Rep, 2019. 9(1): p. 8179. [CrossRef]
Figure 1.
Cilia length in cortical neurons is negatively correlated with O-GlcNAc levels. (A) Immunofluorescent images of primary cilia on cortical neuron at different glucose concentrations; cells were co-immunostained with antibodies against NeuN, Arl13, and IFT88. Scatter plots represent the quantification of mean cilia lengths. (B) Glucose deprivation decreased cellular OGN levels. Cortical neurons were cultured in the medium with a glucose concentration of 30mM, 5mM, or 2mM for 24 h. Cell lysates were analyzed by Western blotting with O-GlcNAc antibody; β-actin was used as loading control. Bar graph showed that the statistical analysis of O-GlcNAc level differences is significant.(C) Effects of OGT and OGA chemical inhibitors on cellular OGN levels. Cortical neurons were treated with and without inhibitors Ac5S or TMG for 24 h. Cell lysates were analyzed by Western blotting with O-GlcNAc antibody; β-actin was used as loading control. Bar graph showed that the statistical analysis of O-GlcNAc level differences is significant.(D) OGN levels negatively regulate neuronal cilia length. Immunofluorescent image of cortical neuron primary cilia at different OGN levels. Scatter plots represent the quantification of mean cilia lengths. Scale bars, 40 µm. All data are mean SD from three independent experiments (> 80 cells per experiment). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; n.s., not significant.
Figure 1.
Cilia length in cortical neurons is negatively correlated with O-GlcNAc levels. (A) Immunofluorescent images of primary cilia on cortical neuron at different glucose concentrations; cells were co-immunostained with antibodies against NeuN, Arl13, and IFT88. Scatter plots represent the quantification of mean cilia lengths. (B) Glucose deprivation decreased cellular OGN levels. Cortical neurons were cultured in the medium with a glucose concentration of 30mM, 5mM, or 2mM for 24 h. Cell lysates were analyzed by Western blotting with O-GlcNAc antibody; β-actin was used as loading control. Bar graph showed that the statistical analysis of O-GlcNAc level differences is significant.(C) Effects of OGT and OGA chemical inhibitors on cellular OGN levels. Cortical neurons were treated with and without inhibitors Ac5S or TMG for 24 h. Cell lysates were analyzed by Western blotting with O-GlcNAc antibody; β-actin was used as loading control. Bar graph showed that the statistical analysis of O-GlcNAc level differences is significant.(D) OGN levels negatively regulate neuronal cilia length. Immunofluorescent image of cortical neuron primary cilia at different OGN levels. Scatter plots represent the quantification of mean cilia lengths. Scale bars, 40 µm. All data are mean SD from three independent experiments (> 80 cells per experiment). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; n.s., not significant.
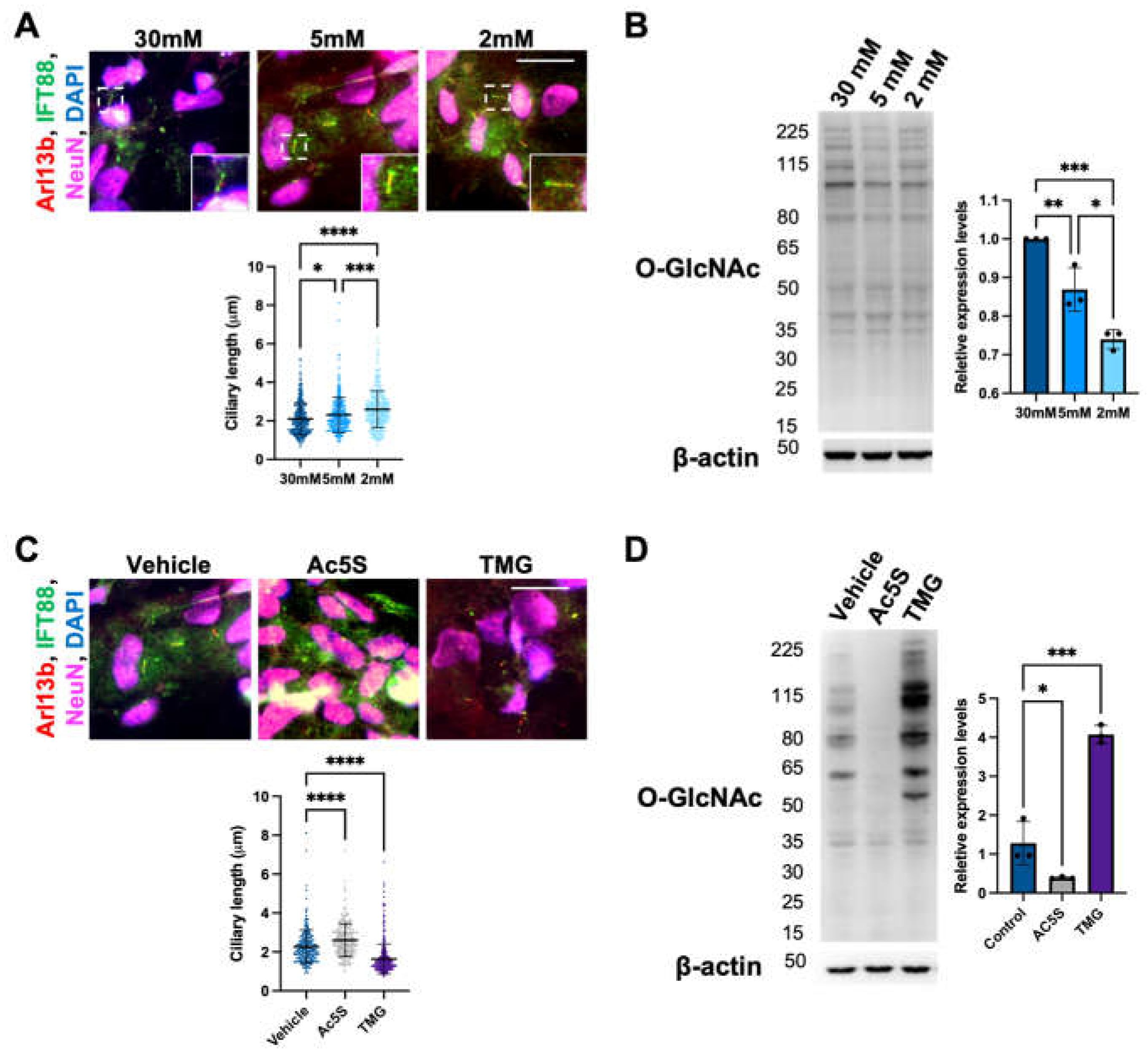
Figure 2.
Cilia formation is partially regulated by O-GlcNAc levels during cortical neuron differentiation.(A) Schematics and phase images of iPSC-derived cortical neurons at key time points. (B) Western blotting result and quantification. Cells were harvested at indicated time points during differentiation, and O-GlcNAc levels were analyzed via Western blotting. (C) Representative images of immunofluorescence staining and the quantification of cilia length. Cells were harvested on days 0, 12, 25, 34, 45, and 55 and stained for IFT88, Arl13b, and NeuN (after day 25). Cilia lengths were measured on NeuN-positive cells after day 25. Scale bars, 40 µm. All data are mean SD from three independent experiments (> 80 cells per experiment). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; n.s., not significant.
Figure 2.
Cilia formation is partially regulated by O-GlcNAc levels during cortical neuron differentiation.(A) Schematics and phase images of iPSC-derived cortical neurons at key time points. (B) Western blotting result and quantification. Cells were harvested at indicated time points during differentiation, and O-GlcNAc levels were analyzed via Western blotting. (C) Representative images of immunofluorescence staining and the quantification of cilia length. Cells were harvested on days 0, 12, 25, 34, 45, and 55 and stained for IFT88, Arl13b, and NeuN (after day 25). Cilia lengths were measured on NeuN-positive cells after day 25. Scale bars, 40 µm. All data are mean SD from three independent experiments (> 80 cells per experiment). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; n.s., not significant.
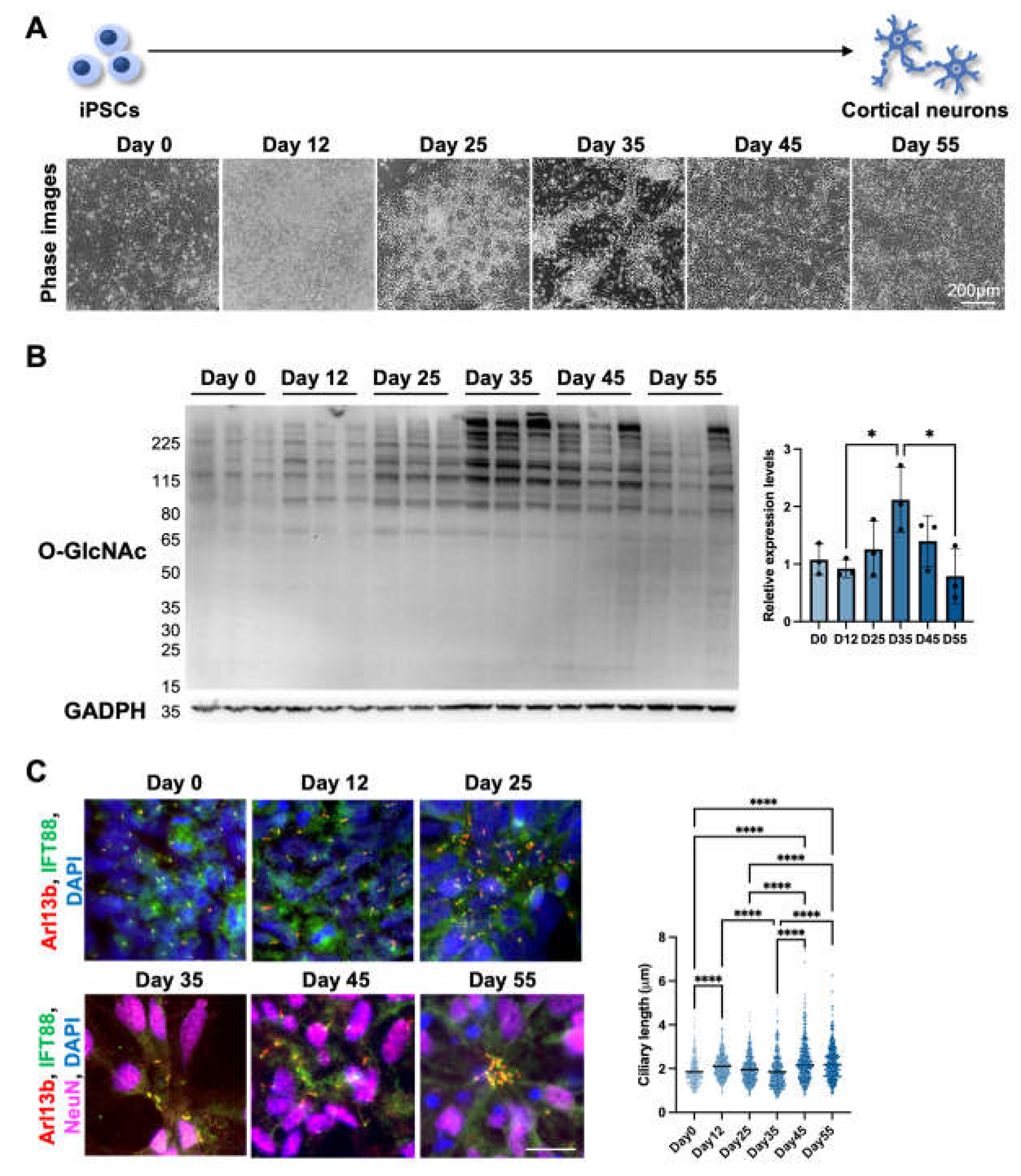
Figure 3.
Long-term perturbation of OGN causes ciliary defect during cortical neuron development. (A) Schematic illustrating Ac5S and TMG treatment during cortical neuron differentiation. (B) Cells differentiated with or without inhibitors treatments were harvested at indicated time points. Western blotting results show that O-GlcNAc levels in cells were efficiently suppressed or elevated by Ac5S (A) or TMG (T) as compared to control (Ct), respectively. (C) Representative images and quantification of multinucleated cells percentage and cilia length on cells differentiated with or without inhibitors treatments at indicated time points. Cells were harvested on days 0, 12, 25, 35, 45, and 55 and stained for IFT88, Arl13b, and NeuN (after day 25). Cilia lengths were measured on NeuN-positive cells after day 25. Scale bars, 40 µm. All data are mean SD from three independent experiments (> 80 cells per experiment). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; n.s., not significant.
Figure 3.
Long-term perturbation of OGN causes ciliary defect during cortical neuron development. (A) Schematic illustrating Ac5S and TMG treatment during cortical neuron differentiation. (B) Cells differentiated with or without inhibitors treatments were harvested at indicated time points. Western blotting results show that O-GlcNAc levels in cells were efficiently suppressed or elevated by Ac5S (A) or TMG (T) as compared to control (Ct), respectively. (C) Representative images and quantification of multinucleated cells percentage and cilia length on cells differentiated with or without inhibitors treatments at indicated time points. Cells were harvested on days 0, 12, 25, 35, 45, and 55 and stained for IFT88, Arl13b, and NeuN (after day 25). Cilia lengths were measured on NeuN-positive cells after day 25. Scale bars, 40 µm. All data are mean SD from three independent experiments (> 80 cells per experiment). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; n.s., not significant.
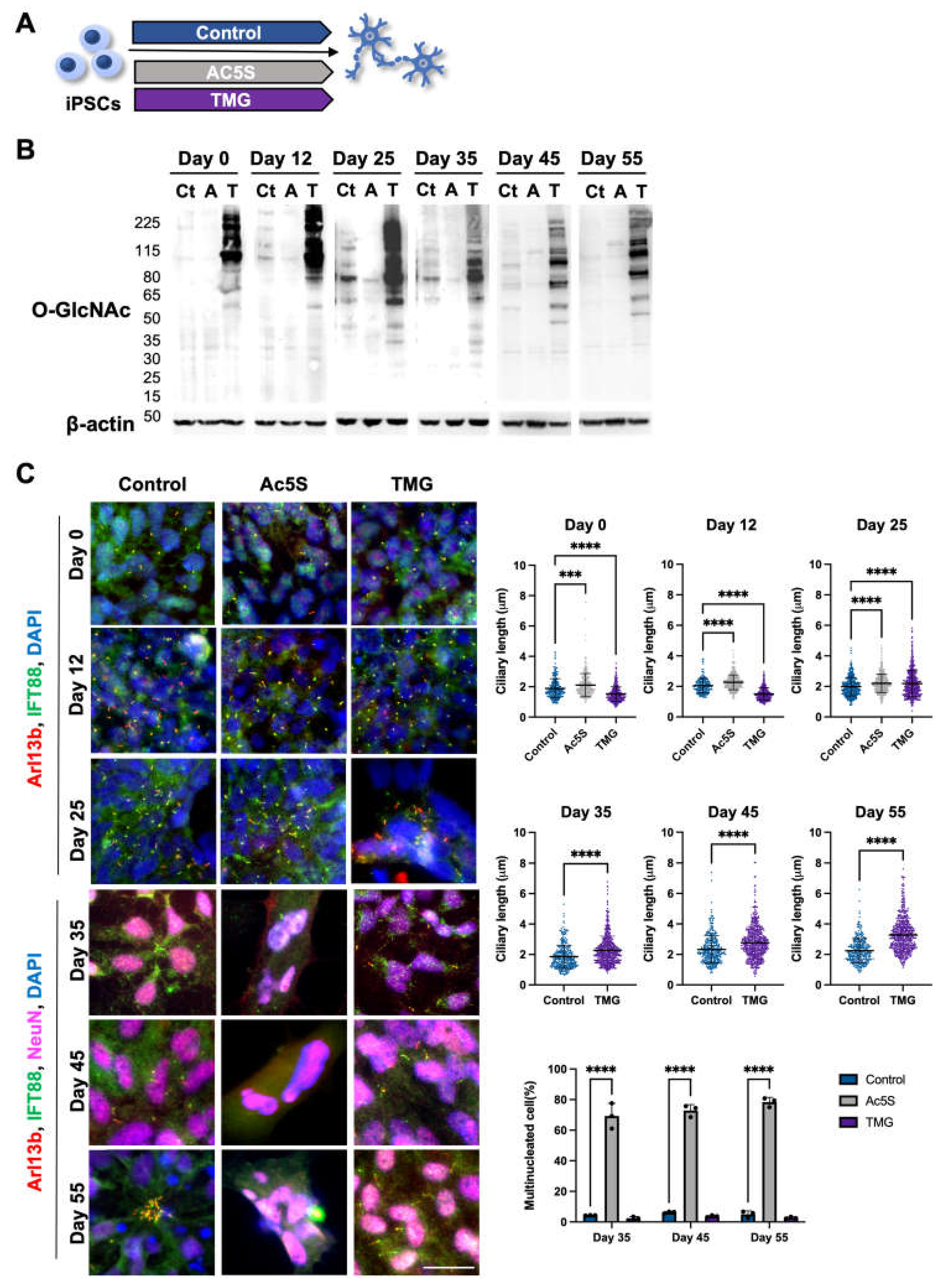
Figure 4.
Altered OGN and subsequent cilia formation interferes with cortical neuronal differentiation. (A) Phase immunofluorescence images of Tuj1 staining. Cells differentiated with or without inhibitors treatment were observed on day 55. (B and C) Neuronal markers (B) and apoptosis marker (C) were detected by Western blotting on day 55. Ct: control, A: Ac5S, and T: TMG. (D) Schematics illustrating cells generated with or without inhibitors treatment possessed different cilia length, morphology, and identity.
Figure 4.
Altered OGN and subsequent cilia formation interferes with cortical neuronal differentiation. (A) Phase immunofluorescence images of Tuj1 staining. Cells differentiated with or without inhibitors treatment were observed on day 55. (B and C) Neuronal markers (B) and apoptosis marker (C) were detected by Western blotting on day 55. Ct: control, A: Ac5S, and T: TMG. (D) Schematics illustrating cells generated with or without inhibitors treatment possessed different cilia length, morphology, and identity.
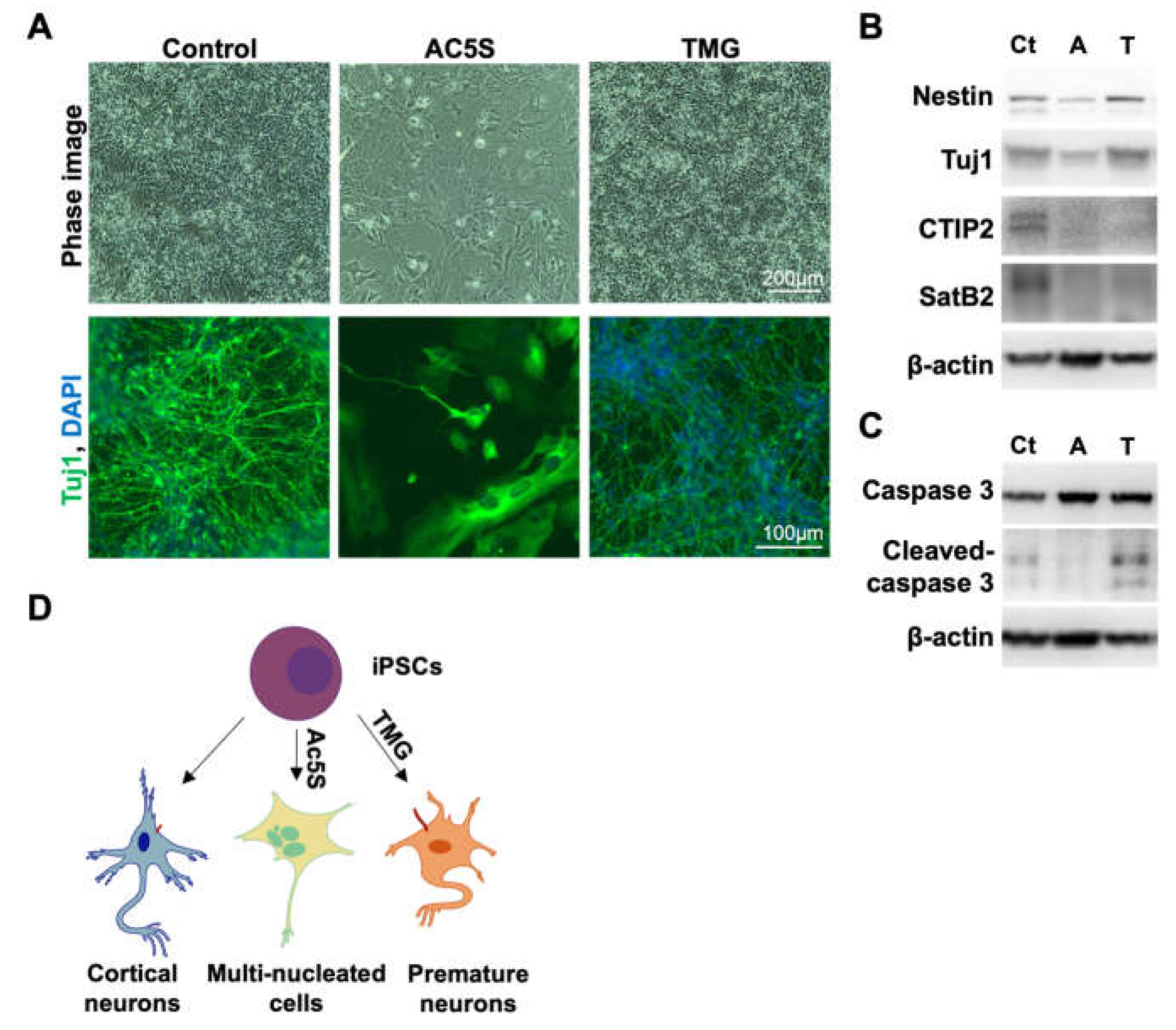
Figure 5.
Increased primary cilia length leads to higher insulin sensitivity of TMG-treated neurons. (A) Neurons differentiated under normal (control) and TMG-treatment conditions were serum-starved for 3 hours prior to the addition of 10 or 100 nM insulin for 30 minutes. Lysates were analyzed by Western blotting and probed for pAkt (pT308), Akt and β-actin act as loading controls. (n = 3, **P<0.01.)
Figure 5.
Increased primary cilia length leads to higher insulin sensitivity of TMG-treated neurons. (A) Neurons differentiated under normal (control) and TMG-treatment conditions were serum-starved for 3 hours prior to the addition of 10 or 100 nM insulin for 30 minutes. Lysates were analyzed by Western blotting and probed for pAkt (pT308), Akt and β-actin act as loading controls. (n = 3, **P<0.01.)
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



