
Submitted:
14 April 2023
Posted:
14 April 2023
You are already at the latest version
Abstract
In recent years, nonalcoholic fatty liver disease/steatohepatitis (NAFLD/NASH) has become a leading worldwide disease, and its therapies are facing many complexities. Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that belong to the nuclear receptor (NR) superfamily and have three subtypes: PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3). They have been identified to be essential in the regulation of lipid metabolism by controlling the transcription of genes related to fatty acids, bile acids, and cholesterol metabolism. Many PPAR agonists have been reported, such as natural agonists (fatty acids, eicosanoids, and phospholipids) and synthetic ligands (fibrates, thiazolidinediones, glitazars, and elafibranor). PPAR-based chemicals have a breakthrough in the innovation of therapy for fatty liver disease. This review will provide an overview of how PPARα and PPARγ play roles in lipid homeostasis, discussing the consideration of their agonists as potential therapeutic agents for NAFLD/NASH.
Keywords:
PPAR
; NAFLD
; NASH
1. Introduction
Hepatic steatosis (fatty liver) is one of the most prevalent chronic liver diseases worldwide, affecting approximately one quarter of the global population, and is predicted to become the leading indication for liver transplantation by 2030, posing a significant burden on global health [1,2,3,4,5]. According to the history of alcohol intake, fatty liver is artificially categorized into two common forms: alcoholic liver disease (ALD) and nonalcoholic fatty liver disease/steatohepatitis (NAFLD/NASH) [3,4,5,6,7]. While ALD is defined by the presence of hepatic steatosis associated with significant alcohol consumption, NAFLD is a generic term that includes a series of liver diseases with different injury severities and consequent fibrosis [3,5]. Among them, hepatic steatosis is referred to as NAFLD, which is defined as the composition of fat that takes up 5–10% of the liver weight. NASH is associated with inflammation and fibrosis, which may progress to cirrhosis and hepatocellular carcinoma (HCC) [8,9]. About 20% of patients with NAFLD develop NASH, and over 40% of patients with NASH progress to fibrosis [10]. It was proposed that the abnormality in lipid and lipoprotein metabolism accompanied by chronic inflammation and oxidative stress is the central pathway and/or the major risk factors involved in the pathogenesis of NAFLD/NASH. However, the pathogenesis of NAFLD/NASH is not fully understood. The "two-hit" hypothesis is now obsolete mainly because it is inadequate to explain some of the molecular and metabolic changes that occur in NAFLD/NASH. The "multiple hit" hypothesis is believed to describe more accurately the pathogenesis of NAFLD. The hits include insulin resistance (IR), hormones secreted by adipose tissue, nutritional factors, gut microbiota, and genetic and epigenetic factors. Thus, there are numerous factors involved in the pathogenesis of NAFLD/NASH [11].
Peroxisomes, originally called microbodies, were identified as intracellular organelles and named by de Dude et al. in 1960. Peroxisomes are present in most plant and animal cells and contain numerous enzymes, but they differ from other intracellular organelle membranes in their protein composition and are thin and fragile. The typical functions of peroxisomes are respiration through the H2O2 system and β-oxidation of fatty acids, and they are also involved in a wide range of other metabolic activities [12]. Proliferation of peroxisome is stimulated by compounds such as clofibrate and di[2'-ethylhexyl] phthalate. Peroxisome proliferator activated receptors (PPARs) have been identified as factors that mediate this proliferation, PPARs were first cloned in rodent hepatocytes in the 1990s and are ligand-activated transcription factors that belong to the nuclear receptor superfamily (NR). The PPARs are believed to control lipid metabolism via regulating the gene transcription involved in fatty acids (FAs), bile acid, and cholesterol metabolism. Up to now, three PPAR subtypes have been identified: PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3). PPARα is mainly expressed in tissues with high fatty acid metabolism, including the liver, kidney, and white and brown adipose tissue. PPARδ/β is ubiquitously and abundantly expressed in a broad range of tissues and regulates fatty acid oxidation [13]. PPARγ, consists of four isoforms in humans and rodents (PPARγ1-4), expressed in a variety of tissues such as liver, kidney heart. In contrast, PPARγ2 is restricted to adipose tissue. PPARγ3 is expressed in macrophage and colon and PPARγ4 is observed in endothelial cells [13,14]. PPARγ plays a variety of biological functions, including regulating fat and glucose metabolism, inducing tumor cell differentiation and apoptosis, promoting ovulation and anti-atherosclerotic activity, improving heart failure and ventricular remodeling, and inhibiting inflammatory reactions [13].
NAFLD/NASH progresses to lipid accumulation (hepatocyte), inflammation (inflammatory cells such as macrophage), and fibrosis (hepatic stellate cell). It is important for understanding pathogenesis of NAFLD/NASH to unveil molecular changes occurring in these liver cells. We will mainly outline the roles of PPARα and PPARγ in the NAFLD/NASH and discuss the potential of PPARs for the NAFLD/NASH therapy.
2. Structure of PPAR proteins
PPARs show four distinguishable functional domains [an N-terminal region (A/B domain), a central DNA-binding domain (DBD, C domain), a flexible hinge region (D domain), and a C-terminal ligand-binding domain (LBD, E domain)], as well as other NRs (Figure 1). The A/B domain, which is the least conserved domain among PPARs, harbors a ligand-independent activation function-1 (AF-1) region. The DBD, which is highly conserved and rich in cysteine and basic amino acids, consists of two zinc-finger binding motifs [15]. The domain is responsible for physical interaction with DNA. PPARs form a heterodimer with retinoid X receptors (RXRs). The PPAR-RXR heterodimer recognizes and binds to PPAR response elements (PPREs), which are localized in gene regulatory regions and organized as a direct repeat type 1 (DR-1). DR-1 consists of two copies of the hexameric nucleotide recognition motif 5′-AGGTCA-3′ separated by a single nucleotide [16]. The PPAR-RXR heterodimer can form independently of the PPAR ligand. The unliganded heterodimers recruit the corepressor protein complex and inhibit target gene transcription. Upon ligand binding, the corepressor complex is released from the heterodimer and then the coactivator complexes will be recruited to the promoter region of target genes, thereby initiating transcription [17]. The PPAR-LBD harbor a ligand-dependent transactivation function (AF-2) and has a structure consisting of an α-helical sandwich and a four-stranded β-sheet [15]. The LBDs differ among PPARs and the LBD of PPARα is more lipophilic than other PPARs, which potentially explains the greater affinity of PPARα to bind the more saturated FAs [18]. Following ligand binding, the AF-2 domain undergoes conformational changes, which allows for the interaction with various coactivators carrying LXXLL motifs (L, leucine; X, any amino acid) [19], such as cAMP response element binding protein (CBP)/p300 and steroid receptor coactivator-1 (SRC-1/NCoA-1) [20].
3. Lipid homeostasis in the liver
Hepatic lipid homeostasis is regulated mainly by uptake, synthesis, degradation, and excretion of fatty acid (FAs) in hepatocytes [21]. Upon eating, glucose and insulin promote FA synthesis (lipogenesis) through synergistic effects between sterol regulatory element binding protein 1C (SREBP1c) and carbohydrate response element binding protein (ChREBP) [22,23]. Insulin induces SREBP1c gene expression via AKT signaling and enhances SREBP1c activity. Hepatic SREBP-1c activation leads to the regulation of lipogenic genes such as fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC) in the diet-related condition [24,25]. Elevated glucose levels induce phosphorylation-dependent nuclear relocalization of ChREBP and the ChREBP induces FAS and ACC gene expression [26]. In addition to the de novo synthesis, hepatic lipid stores consist of FAs derived from breakdown of triglycerides (lipolysis) in adipose tissue and dietary intake [27]. FAs are transported in the hepatocytes through a fatty acid transport protein (FATP) and a fatty acid translocase (FAT), CD36, which facilitate the uptake of long-chain fatty acids. Once inside the hepatocytes, FAs bind to fatty acid binding protein-1 (FABP1) and change their fate depending on the condition of hepatocytes. Generally, FAs undergo the peroxisomal β-oxidation by acyl-CoA oxidase 1 (ACOX1) or are transported into mitochondria by the shuttle of carnitine-palmitoyl transferase/carnitine-acyl carnitine carrier (CPT/CAC) going through β-oxidation. In mitochondria, FA β-oxidation is mediated mainly by medium-chain acyl-CoA dehydrogenase (MCAD) or long-chain acyl-CoA dehydrogenase (LCAD) [28]. When these β-oxidations can no longer process FAs, FAs are esterified to cholesteryl esters by acyl CoA cholesterol acyltransferases (ACAT1 and 2) [29], and to triglycerides (TGs) by diacylglycerol-O-acyltransferases (DGAT1 and 2) in the endoplasmic reticulum [30,31] and become contents of VLDL (secreted from hepatocytes via the Golgi apparatus) and lipid droplet (accumulated in hepatocytes). DGAT1 mainly localizes to the endoplasmic reticulum and catalyzes TG formation using FAs supplied from foods [30,31]. On the other hand, DGAT2 can translocate from the endoplasmic reticulum to lipid droplets [30,31] and promote TG formation using de novo synthesized FAs [31]. Hepatocytes perform lipolysis and autophagy to recruit FAs to mitochondrial/peroxisomal β-oxidation or the VLDL secretion. FAs for VLDL assembly derive from re-esterification of triglycerides stored in lipid droplets [32]. In many kinds of cells, adipose triglyceride lipase (ATGL) and its cofactor CGI-58 work with hormone sensitive lipase (HSL) and monoacylglycerol lipase (MGL) to cleave fatty acids from the TG [33,34]. However, in hepatocyte, contribution of these lipases to lipolysis remains unclear. HSL is very low expressed in human liver. Moreover, overexpression or knockdown of ATGL, HSL and MGL did not substantially alter hepatic VLDL secretion [35]. Thus, other lipases such as TG hydrolase existing endoplasmic reticulum are thought to contribute to hepatic VLDL formation. On the other hand, Liver-specific deletion of either ATGL or CGI-58 results in steatosis and fibrosis in mice following enhanced hepatic β-oxidation [33,35]. The ATGL and CGI-58 may mobilize lipids for β-oxidation.
4. The pathogenesis of NAFLD/NASH
It is generally believed that the occurrence of fatty liver is not only related to insulin resistance and disorder of fat metabolism, but also related to biological processes, such as glucometabolic disorder, oxidative stress, and intracellular inflammatory response. Moreover, these processes are correlated and/or coordinated with each other and accelerate the progress of NAFLD [36].
Increased fatty acid levels can trigger enhancement of lipid peroxidation in the liver. The enhanced lipid peroxidation results in persistent reactive oxygen species (ROS) production [37]. In addition to the pre-existing factors related to the elevated ROS levels, other new or additional factors can enhance lipid peroxidation in the liver, such as inflammatory cytokines, adipokines, endotoxins, and mitochondrial inactivation [11]. The oxidative stress following accumulation of fatty acids will eventually lead to NAFLD progression that promotes cell death and inflammation. Especially, the inflammation is positively correlated with liver injury, negatively correlated with hepatic lipolysis, and induces further lipid peroxidation aggravating the pathogenesis of NAFLD/NASH [38]. Additionally, endoplasmic reticulum (ER) stress is another important factor in the pathogenesis of NAFLD/NASH [39,40]. Metabolic disorders, such as obesity and diabetes, can cause ER stress, leading to the accumulation of unfolded protein response and affecting the normal physiological functions of hepatocytes [41]. The ER stress can activate sterol-regulatory element binding protein (SREBP), promoting the transcription of FAS and ACC, resulting in enhancement of fatty acid synthesis and lipid droplet formation in the liver [42]. Considering that the lipid peroxidation can also induce the ER stress, the fatty acid accumulation and lipid peroxidation in hepatocytes is an important factor for aggravating the NAFLD/NASH [43]. We summarized the relationships between fatty acid homeostasis in the liver and PPARα/ γ in (Figure 2). The homeostasis of fatty acids in the liver and the functions of PPARα/γ are described below.
5. PPARα and PPARγ functions in hepatocytes
PPARα serves as an intracellular lipid sensor and is activated by a wide range of endogenous or naturally occurring biological molecules encompassing a variety of FAs and FA derivatives, including acyl-CoAs, oxidized FAs, eicosanoids, endocannabinoids, phytanic acids [19]. PPARα promotes the transport and oxidation of fatty acid and ketogenesis, pointing out the role of PPARα as a master regulator of the hepatic lipid metabolism in fasting conditions. PPARα positively regulates CD36 (uptake into hepatocyte) and FABP1 (transfer to mitochondria) gene expressions [44]. In addition, PPARα induces expressions of mitochondrial FA β-oxidation-related genes, such as MCAD and LCAD [19]. Moreover, PPARα upregulates the gene expressions related with biosynthesis of ketone bodies, such as mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase (a key ketogenic enzyme which catalyzes the condensation of acetyl-CoA and acetoacetyl-CoA to generate 3-hydroxy-3-methylglutaryl-CoA, the first ketone body) [44]. Therefore, PPARα contributes to decrease in hepatic FA levels in the liver and reduces lipid peroxidation and ROS production via enhancement of mitochondrial β-oxidation and ketone body generation, resulting in protection from hepatic lipotoxicity. Notably, PPARα are activated under condition of a carbohydrate-rich diet and can stimulate lipogenesis via promoting SREBP1C gene transcription [28]. The SREBP1C induces lipogenic genes such as FAS and ACC [42]. In addition, PPARα can indirectly control the SREBP1C activity by inducing expression of the liver X receptor that regulates the SREBP1C gene transcription [45]. PPARα induces expression of lipoprotein lipase (LPL) that catalyzes the hydrolysis of plasma triglycerides contained in lipoproteins, while inducing gene expression of apolipoproteins A-I (APOAI) and A-II (APOAII), the major proteins of HDL [44,46]. Thus, PPARα also influences lipoprotein metabolism by reduction in hepatic VLDL synthesis, leading to increase of high-density lipoproteins (HDL). In hepatocytes, PPARα works as a metabolic switch between fasting and postprandial states and as a part of the triglyceride/fatty acid cycle [44].
Hepatic PPARγ expression is low under normal physiological conditions but induced during the development of steatosis. The PPARγ promotes accumulation of lipid droplets in hepatocyte via inducing hepatic fat-specific protein 27 (Fsp27) expression that is involved in formation of lipid droplet [47]. In addition, Hepatocyte-specific PPARγ deletion improves hepatic steatosis via attenuating expressions of de novo lipogenesis genes (FAS, ACC, SCD1). PPARα activation contributes to decrease in hepatic lipid accumulation under the condition of NAFLD/NASH, although PPARα has the ability to induce the de novo lipogenesis genes. Thus, hepatocyte PPARγ is thought to play an important factor in the development of hepatic steatosis, rather than PPARα [48]. On the other hand, PPARγ is a ligand-activated nuclear receptor with potent anti-inflammatory properties [49]. A PPARγ agonist exerted an anti-inflammatory effect by inhibiting NLR family pyrin domain containing 3 (NLRP3) inflammasome activation in lipid-laden hepatocytes [50]. Furthermore, PPARγ deficiency enhanced ROS generation, NLRP3 inflammasome activation, and IL1β secretion in hepatocytes after treatment with FA in in vitro study, and modulation of PPARγ activity attenuated high fat diet-induced NAFLD by regulating lipid metabolism and oxidative stress in hepatocytes via nuclear factor erythroid 2-related factor 2 activation [51]. Interestingly, enhancement of PPARγ activity in lipid-laden hepatocytes reversed macrophage M1 polarization and reduced the toll-like receptor 4/nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) pathway activation, and the hepatocyte-specific PPARγ deletion accelerated the macrophage M1 polarization and toll-like receptor 4/NF-κB pathway activation. Hepatocyte-specific PPARγ deletion highly has been to augment TNFα-induced production of C-X-C Motif Chemokine Ligand 1 (CXCL1) [52]. Thus, hepatic PPARγ can work as a potent regulator for hepatic inflammation.
6. PPARγ function in liver macrophages and hepatic stellate cells
PPARγ mainly works as suppressor of inflammation in livers of NASH. Macrophage-specific PPARγ deficient (PPARγΔLyz2) showed that upregulated differentially expressed genes were enriched in the NOD-like receptor signaling pathway, NF-κB signaling pathway, chemokine-signaling pathway and cytokine–cytokine receptor interaction, suggesting an enhancement of NLRP3 and NF-κB-driven inflammatory cytokine and chemokine production [53]. While downregulated differentially expressed genes were enriched in the PPAR signaling pathway and insulin resistance, suggesting severe metabolic disorder. In addition, Macrophage-specific PPARγ deficient showed significantly increased expression of liver fibrosis-related genes, such as TGFβ1, ACTA2, COL1A1 and TIMP metallopeptidase inhibitor 1, in the NASH livers. The PPARγ deletion in macrophages can affect macrophage phenotypes and hence influence hepatic stellate cell (HSC) activation and NASH development in mice [52,13]. Activation of HSCs results in excess accumulation of extracellular matrix such as type I collagen (COL1A) in the liver (called liver fibrosis).
7. PPARα and PPARγ functions in adipose tissue
The role of PPARα is not fully understood in adipose tissue because its role overlaps with that of PPARγ, which has potent activity, although some studies indicated that the role of PPARα in brown adipose tissue thermogenesis and white adipose tissue browning [54]. The lipolysis of white adipose tissue results in increased FFA levels and enhanced TAG synthesis in the liver. Among the several factors involved in adipose tissue regulation, PPARγ is considered the “master regulator” of adipogenesis [55,56]. The PPARγ is a well-characterized regulator of energy metabolism mainly by improving uptake and expenditure of fatty acid in adipocyte and is clearly involved in the pathophysiology of obesity and related complications. Adipocyte PPARγ deletion has been linked to insulin resistance in mice [54]. Furthermore, PPARγ activation has been demonstrated to enhance expression of adiponectin in adipocytes. In addition, PPARγ is known to induce adipocyte expression of Fsp27 which is a regulator of lipid droplet formation. Adipocyte-specific PPARγ deletion leads to severe lipoatrophy, highlighting the role of PPARγ in mouse adipocyte development. Adipocyte-specific Fsp27 deletion also inhibits lipid accumulation in mouse adipose tissue as well as the PPARγ deletion. Furthermore, the adipocyte-specific Fsp27 deletion exacerbates hepatic steatosis after a challenge of high-fat diet. The adipocyte PPARγ-FSP27 cascade may be a key regulator for hepatic fatty homeostasis and suppress the procession of NAFLD/NASH [57,58,59].
8. Clinical drugs and trial for NASH (The potency of PPAR ligands as clinic therapeutic agents.)
PPARs can be activated by various natural agonists, such as fatty acids (FAs), eicosanoids, and phospholipids derived from cellular FA metabolism or dietary lipids. Synthetic ligands, including fibrates, thiazolidinediones (TZDs), glitazars, elafibranor, and others, can also activate PPARs (Table 1). These PPAR agonists have been widely used in basic research and are progressing to clinical development as therapeutic agents for NAFLD/NASH [60].
8.1. PPARα agonists
Studies using mouse models have shown that PPARα agonists, such as Wy-14643, can prevent hepatic triglyceride accumulation induced by a methionine and choline-deficient (MCD) diet in wild-type mice but have no effect on the PPARα deficient mice [61,62]. The potential use of PPARα agonists, particularly fibrates, in the treatment of NASH has been of interest for over two decades [63,64]. Gemfibrozil can reduce levels of liver enzymes (aspartate aminotransferase, alanine aminotransferase, and gamma-glutamyl transferase) and very low-density lipoprotein in NASH patients. Fenofibrate has been shown to decrease serum levels of the inflammatory marker RANTES in type 2 diabetes patients with hypertriglyceridemia, indicating potential anti-inflammatory properties related to NAFLD. However, a 48-week trial of fenofibrate in NASH patients with biopsy-proven NAFLD did not show any improvement in liver histology despite improving metabolic syndrome-related parameters. Clofibrate was also assessed for its potential anti-NASH properties but did not show histological improvements in the liver enzyme levels. Pemafibrate, by enhancing lipid metabolism and reducing inflammation, has shown potential to ameliorate liver dysfunction in patients with type 2 diabetes and to ameliorate NASH pathology in mouse and rat models [46,64,65,66].
8.2. PPARγ agonists
Function of PPARγ agonists have been most investigated with TZDs. TZDs (pioglitazone and rosiglitazone) are a class of insulin-sensitizing drugs and currently used in clinical therapy [67]. These drugs enhance the uptake and storage of FAs in adipose tissue, thereby protecting the skeletal muscle and the liver. Rosiglitazone shown to prevent form development of NASH induced by MCD diet [64]. Pioglitazone exhibits anti-inflammatory and antifibrotic properties by suppressing the expression of platelet-derived growth factor and tissue inhibitor of metalloproteinase-2 in mouse model. [62]. However, rosiglitazone was withdrawn from the European market due to its association with an increased risk of myocardial infarction and heart failure. The PIVENS (Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis) trial evaluated the potential efficacy of the pioglitazone/vitamin E for NASH therapy. In the PIVENS trial, long-term pioglitazone treatment resulted in the resolution of NASH in more than half of the studied patients with prediabetes or type 2 diabetes [64]. However, we need to keep it in mind that pioglitazone is associated with an increased risk of bladder cancer. Consequently, the AASLD and EASL now recommend the use of pioglitazone/vitamin E for treating the biopsy-proven NASH but mentioned that pioglitazone is off-label in patients without type 2 diabetes and may cause weight gain. Lobeglitazone, another PPARγ agonist, has also been evaluated in type 2 diabetes patients with NAFLD, resulting in moderate weight gain and attenuated hepatic steatosis with improvement of glucose and lipid homeostasis. Further randomized controlled trials are needed to assess the effect of lobeglitazone [64].
8.3. PPAR-α/γ Agonists
In randomized trials for NAFLD or NASH treatment, pioglitazone (PPARγ ligand) and elafibranor (dual PPARα and β/δ ligand) were tested. These drug candidates have strong side effects [68,69]. Thus, it is required to minimize undesirable side effects. Saroglitazar (PPARα/γ-dual agonist) was designed to have both PPARα and PPARγ agonisms. The PPARα agonism enhances fatty acid oxidation in the liver, reduces the synthesis and secretion of triglycerides, and improves circulating lipoprotein profiles. The PPAR-γ agonism regulates insulin-responsive genes, increases insulin sensitivity, and reduces blood glucose and glycosylated hemoglobin A1c levels. A phase 3 clinical trial with saroglitazar treatment is currently underway in NAFLD/NASH patients. Lanifibranor (IVA-337) is a pan agonist that has shown promising results in improving NASH histology, reducing weight gain, and normalizing plasma glucose and insulin levels in mouse model. Additionally, IVA337 has been shown to be more effective at preventing and reversing liver fibrosis than any of the single agonists [70]. Phase 2 trials have been conducted to assess the efficacy of IVA337 in humans with intrahepatic triglycerides and histologically proven NASH (now phase 3 is going) [71]. These results may suggest that dual and pan agonists are available for the NAFLD/NASH treatment.
9. Conclusion
PPAR is activated by ligand and has metabolic actions that regulate energy, glucose, and lipid homeostasis. PPARα enhances FA oxidation in the liver and brown adipose tissue, while PPARγ promotes lipid storage and adipogenesis in the liver and white adipose tissue. In the liver, these functions contribute to reduced free FA level and results in suppressing lipid peroxidation and ROS generation in the hepatocytes. The PPARα and PPARγ agonists show promise for the treatment of NAFLD/NASH, but will be required for a contrivance, such as liver-specific delivery system, to avoid the side effects.

Table 1.
The potency of PPAR ligands as clinic therapeutic agents.
| PPAR Ligands | Isotype | Status | Source | Reference |
|---|---|---|---|---|
| Arachidonic acid | α | Nature | [72] | |
| Leukotriene B4 | α | Nature | [73,74] | |
| Phosphatidylcholine | α | Nature | [75,76] | |
| Resveratrol | α | Nature | [77,78] | |
| Linoleic Acid | γ | Nature | [79] | |
| Prostaglandin D2 | γ | Nature | [80] | |
| 15-deoxy-delta 12,14-prostaglandin J2 | γ | Nature | [81] | |
| Lysophosphatidic acid | γ | Nature | [82] | |
| Clofibrate | α | Clinical | Synthesis | [83] |
| Fenofibrate | α | Clinical | Synthesis | [84,85] |
| Bezafibrate | α | Clinical | Synthesis | [86] |
| Gemfibrozil | α | Clinical | Synthesis | [87] |
| Pemafibrate | α | Clinical | Synthesis | NCT03350165 [65,88,89,90,91,92,93] |
| WY14643 | α | Basic | Synthesis | [94] |
| GW9578 | α | Basic | Synthesis | [95] |
| GW7647 | α | Basic | Synthesis | [96,97] |
| Pioglitazone | γ | Clinical | Synthesis | NCT00063622 NCT00062764 NCT00013598 NCT03646292 NCT04976283NCT04501406 |
| Ciglitazone | γ | Clinical | Synthesis | [98] |
| Troglitazone | γ | Clinical | Synthesis | [99,100] |
| Rosiglitazone | γ | Clinical | Synthesis | [101,102,103] |
| S26948 | γ | Clinical | Synthesis | [104] |
| INT131 | γ | Clinical | Synthesis | [105,106] |
| Saroglitazar | α/γ | Clinical | Synthesis | NCT05011305 NCT03639623 NCT03061721 NCT02265276 NCT03863574 NCT03617263 NCT03112681 NCT04193982 |
| Lobeglitazone | α/γ | Clinical | Synthesis | [107,108] |
| Lanifibranor (IVA-337) | pan | Clinical | Synthesis | NCT03008070 NCT02503644 NCT05232071 NCT04849728 |
Abbreviations
ACAD, Acyl-CoA dehydrogenase; ACC, Acetyl-CoA carboxylase; ACOX, Acyl-CoA oxidase; ADIPOQ, Adiponectin; AKT, Alpha serine/threonine-protein kinase; APOC2, Apolipoprotein C2; APOC3 Apolipoprotein C3; APOE; Apolipoprotein E, ATA, Ataxia-telangiectasia group A; CD36, Cluster of differentiation 36; ChREBP, Carbohydrate response element binding protein; CPT1/2, Carnitine palmitoyltransferase 1/2; DGAT1/2 Diacylglycerol O-acyltransferase 1/2; EHHADH, Enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase; FABP4, Fatty acid binding protein 4; FAS, Fatty acid synthase; FSP27, Fat-specific protein 27; G6P, Glucose-6-phosphate; HMGCS2, 3-Hydroxy-3-methylglutaryl-CoA synthase 2; LPL; Lipoprotein lipase; MCP1, Monocyte chemoattractant protein 1; MOGAT1, Monoacylglycerol O-acyltransferase 1; MTTP, Microsomal triglyceride transfer protein; PI3K, Phosphoinositide 3-kinase; SLC27A1, Solute carrier family 27 member 1; SREBP1C, Sterol regulatory element-binding protein; TFEB, Transcription factor EB; TGFβ, Transforming growth factor beta; TNFα, Tumor necrosis factor alpha.
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.; Masson, S.; Anstee, Q.M. The bidirectional impacts of alcohol consumption and the metabolic syndrome: Cofactors for progressive fatty liver disease. J. Hepatol. 2018, 68, 251–267. [Google Scholar] [CrossRef]
- Inan-Eroglu, E.; Huang, B.-H.; Ahmadi, M.N.; Johnson, N.; El-Omar, E.M.; Stamatakis, E. Joint associations of adiposity and alcohol consumption with liver disease-related morbidity and mortality risk: findings from the UK Biobank. Eur. J. Clin. Nutr. 2022, 76, 74–83. [Google Scholar] [CrossRef]
- Kupčová, V.; Fedelešová, M.; Bulas, J.; Kozmonová, P.; Turecký, L. Overview of the Pathogenesis, Genetic, and Non-Invasive Clinical, Biochemical, and Scoring Methods in the Assessment of NAFLD. Int. J. Environ. Res. Public Heal. 2019, 16, 3570. [Google Scholar] [CrossRef]
- Geier, A.; Tiniakos, D.; Denk, H.; Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 2021, 70, 1570–1579. [Google Scholar] [CrossRef]
- Sheka, A.C.; et al. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Shi, K.; Li, R.; Xu, Z.; Zhang, Q. Identification of Crucial Genetic Factors, Such as PPARγ, that Regulate the Pathogenesis of Fatty Liver Disease in Dairy Cows Is Imperative for the Sustainable Development of Dairy Industry. Animals 2020, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Imanaka, T. Biogenesis and Function of Peroxisomes in Human Disease with a Focus on the ABC Transporter. Biol. Pharm. Bull. 2019, 42, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism-Clinical and Experimental 2020, 114, 154338. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-Y.; Sun, P.-M. Estrogen receptor-associated receptor α and peroxisome proliferator-activated receptor γ in metabolism and disease (Review). Mol. Med. Rep. 2020, 23, 156. [Google Scholar] [CrossRef] [PubMed]
- Miyamae, Y. Insights into Dynamic Mechanism of Ligand Binding to Peroxisome Proliferator-Activated Receptor γ toward Potential Pharmacological Applications. Biol. Pharm. Bull. 2021, 44, 1185–1195. [Google Scholar] [CrossRef]
- Hassan, F.-U.; Nadeem, A.; Li, Z.; Javed, M.; Liu, Q.; Azhar, J.; Rehman, M.S.-U.; Cui, K.; Rehman, S.U. Role of Peroxisome Proliferator-Activated Receptors (PPARs) in Energy Homeostasis of Dairy Animals: Exploiting Their Modulation through Nutrigenomic Interventions. Int. J. Mol. Sci. 2021, 22, 12463. [Google Scholar] [CrossRef]
- Zhao, B.; Xin, Z.; Ren, P.; Wu, H. The Role of PPARs in Breast Cancer. Cells 2023, 12, 130. [Google Scholar] [CrossRef]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Matsuoka, H.; Michihara, A. Identification of the RORα Transcriptional Network Contributes to the Search for Therapeutic Targets in Atherosclerosis. Biol. Pharm. Bull. 2021, 44, 1607–1616. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Linden, A.G.; et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice [S]. Journal of Lipid Research 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. The Roles of Carbohydrate Response Element Binding Protein in the Relationship between Carbohydrate Intake and Diseases. Int. J. Mol. Sci. 2021, 22, 12058. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lin, X.; Wang, G. Targeting SREBP-1-Mediated Lipogenesis as Potential Strategies for Cancer. Front. Oncol. 2022, 12, 952371. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.V.; Mondal, P. Causative and Sanative dynamicity of ChREBP in Hepato-Metabolic disorders. Eur. J. Cell Biol. 2020, 99, 151128. [Google Scholar] [CrossRef]
- Geng, Y.; et al. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatology International 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Judyta, Z.; Tomasz, S.; Ewa, S. Acyl-Coenzyme A: Cholesterol Acyltransferase Inhibition in Cancer Treatment. Anticancer Research 2019, 39, 3385. [Google Scholar]
- Li, S.; Wu, T.; Lu, Y.-X.; Wang, J.-X.; Yu, F.-H.; Yang, M.-Z.; Huang, Y.-J.; Li, Z.-J.; Wang, S.-L.; Huang, L.; et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020, 36, 101596. [Google Scholar] [CrossRef]
- Løvsletten, N.G.; Vu, H.; Skagen, C.; Lund, J.; Kase, E.T.; Thoresen, G.H.; Zammit, V.A.; Rustan, A.C. Treatment of human skeletal muscle cells with inhibitors of diacylglycerol acyltransferases 1 and 2 to explore isozyme-specific roles on lipid metabolism. Sci. Rep. 2020, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Q.; Pan, Y.; Chen, S.; Zhao, Y.; Hu, Y. New insights into the role of dietary triglyceride absorption in obesity and metabolic diseases. Front. Pharmacol. 2023, 14, 1097835. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.Y.; Yu, J.; Gupta, A.; Guo, T.; Iyengar, P.; Infante, R.E. Cytokine-Mediated STAT3 Transcription Supports ATGL/CGI-58-Dependent Adipocyte Lipolysis in Cancer Cachexia. Front. Oncol. 2022, 12, 841758. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A. Genetically modified mouse models to study hepatic neutral lipid mobilization. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Tsamos, G.; Vasdeki, D.; Koufakis, T.; Michou, V.; Makedou, K.; Tzimagiorgis, G. Therapeutic Potentials of Reducing Liver Fat in Non-Alcoholic Fatty Liver Disease: Close Association with Type 2 Diabetes. Metabolites 2023, 13, 517. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.-Y.; Roh, Y.-S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef]
- Koo, J.H.; Han, C.Y. Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules 2021, 11, 242. [Google Scholar] [CrossRef]
- Zhou, L.; Shen, H.; Li, X.; Wang, H. Endoplasmic reticulum stress in innate immune cells - a significant contribution to non-alcoholic fatty liver disease. Front. Immunol. 2022, 13, 951406. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Batchuluun, B.; Pinkosky, S.L.; Steinberg, G.R. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 2022, 21, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- orotea, D.; Koya, D.; Ha, H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPARα Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef]
- Decara, J.; Rivera, P.; López-Gambero, A.J.; Serrano, A.; Pavón, F.J.; Baixeras, E.; De Fonseca, F.R.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef]
- Meng, Q.-Q.; et al. PPAR-γ Activation Exerts an Anti-inflammatory Effect by Suppressing the NLRP3 Inflammasome in Spinal Cord-Derived Neurons. Mediators of Inflammation 2019, 2019, 6386729. [Google Scholar] [CrossRef]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARγ expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; et al. Regulation of PPAR-gamma activity in lipid-laden hepatocytes affects macrophage polarization and inflammation in nonalcoholic fatty liver disease. World J. Hepatol. 2022, 14, 1365–1381. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Gao, M.; Yang, P.; Liu, D.; Wang, D.; Song, F.; Zhang, X.; Liu, Y. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR-γ signaling during diabetic wound healing. J. Cell. Physiol. 2019, 234, 4217–4231. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mao, S.; Chen, S.; Zhang, W.; Liu, C. PPARs-Orchestrated Metabolic Homeostasis in the Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 8974. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhou, W.; Xu, R.; Xing, L.; Ji, G.; Dang, Y. Natural PPARs agonists for the treatment of nonalcoholic fatty liver disease. Biomed. Pharmacother. 2022, 151, 113127. [Google Scholar] [CrossRef] [PubMed]
- Faghfouri, A.H.; Khajebishak, Y.; Payahoo, L.; Faghfuri, E.; Alivand, M. PPAR-gamma agonists: Potential modulators of autophagy in obesity. Eur. J. Pharmacol. 2021, 912, 174562. [Google Scholar] [CrossRef] [PubMed]
- Aibara, D.; Matsuo, K.; Yamano, S.; Matsusue, K. Fat-specific protein 27b is regulated by hepatic peroxisome proliferator-activated receptor γ in hepatic steatosis. Endocr. J. 2020, 67, 37–44. [Google Scholar] [CrossRef]
- Sharma, R.; Luong, Q.; Sharma, V.M.; Harberson, M.; Harper, B.; Colborn, A.; E Berryman, D.; Jessen, N.; Jørgensen, J.O.L.; Kopchick, J.J.; et al. Growth hormone controls lipolysis by regulation of FSP27 expression. J. Endocrinol. 2018, 239, 289–301. [Google Scholar] [CrossRef]
- Sharma, V.M.; Vestergaard, E.T.; Jessen, N.; Kolind-Thomsen, P.; Nellemann, B.; Nielsen, T.S.; Vendelbo, M.H.; Møller, N.; Sharma, R.; Lee, K.Y.; et al. Growth hormone acts along the PPARγ-FSP27 axis to stimulate lipolysis in human adipocytes. Am. J. Physiol. Metab. 2018, 316, E34–E42. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.-X.; Pan, W.-S.; Khan, F.U.; Qian, C.; Qi-Li, F.-R.; Xu, X. Administration of methyl palmitate prevents non-alcoholic steatohepatitis (NASH) by induction of PPAR-α. Biomed. Pharmacother. 2019, 111, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef]
- Liss, K.H.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; et al. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. Journal of Clinical Lipidology 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. Thiazolidinediones: the Forgotten Diabetes Medications. Curr. Diabetes Rep. 2019, 19, 151. [Google Scholar] [CrossRef]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C.; Koduru, S.V.; Philip, B.; Jain, M.R.; Giri, S.R.; et al. The PPAR α/γ Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 9330. [Google Scholar] [CrossRef]
- Gawrieh, S.; et al. Saroglitazar, a PPAR-alpha/gamma Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1829–1832. [Google Scholar] [CrossRef]
- Wettstein, G.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatology Communications 2017, 16. [Google Scholar] [CrossRef]
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.-F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef] [PubMed]
- Sztolsztener, K.; Chabowski, A.; Harasim-Symbor, E.; Bielawiec, P.; Konstantynowicz-Nowicka, K. Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development. Biomolecules 2020, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Narala, V.R.; Adapala, R.K.; Suresh, M.V.; Brock, T.G.; Peters-Golden, M.; Reddy, R.C. Leukotriene B4 Is a Physiologically Relevant Endogenous Peroxisome Proliferator-activated Receptor-α Agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, L.; Roinestad, K.; Van, T.; Springman, E. Recent advances in clinical development of leukotriene B4 pathway drugs. Semin. Immunol. 2017, 33, 65–73. [Google Scholar] [CrossRef]
- Carlsson, E.R.; Allin, K.H.; Madsbad, S.; Fenger, M. Phosphatidylcholine and its relation to apolipoproteins A-1 and B changes after Roux-en-Y gastric bypass: a cohort study. Lipids Heal. Dis. 2019, 18, 169. [Google Scholar] [CrossRef]
- Mao, L.; Wang, M.; Li, Y.; Liu, Y.; Wang, J.; Xue, C. Eicosapentaenoic acid-containing phosphatidylcholine promotes osteogenesis:mechanism of up-regulating Runx2 and ERK-mediated phosphorylation of PPARγ at serine 112. J. Funct. Foods 2019, 52, 73–80. [Google Scholar] [CrossRef]
- Toupchian, O.; et al. The effects of resveratrol supplementation on PPARα, p16, p53, p21 gene expressions, and sCD163/sTWEAK ratio in patients with type 2 diabetes mellitus: A double-blind controlled randomized trial. Phytotherapy Research 2021, 35, 3205–3213. [Google Scholar] [CrossRef]
- Okudaira, N.; Ishizaka, Y.; Tamamori-Adachi, M. Resveratrol blocks retrotransposition of LINE-1 through PPAR α and sirtuin-6. Sci. Rep. 2022, 12, 7772. [Google Scholar] [CrossRef]
- Shrestha, N.; Cuffe, J.S.; Holland, O.J.; Perkins, A.V.; McAinch, A.J.; Hryciw, D.H. Linoleic Acid Increases Prostaglandin E2 Release and Reduces Mitochondrial Respiration and Cell Viability in Human Trophoblast-Like Cells. Cell. Physiol. Biochem. 2019, 52, 94–108. [Google Scholar]
- Harris, S.G.; Phipps, R.P. Prostaglandin D2, its metabolite 15-d-PGJ2, and peroxisome proliferator activated receptor-γ agonists induce apoptosis in transformed, but not normal, human T lineage cells. Immunology 2002, 105, 23–34. [Google Scholar] [CrossRef]
- Raman, P.; et al. 15-Deoxy-Δ12,14-prostaglandin J2-Glycerol Ester, a Putative Metabolite of 2-Arachidonyl Glycerol, Activates Peroxisome Proliferator Activated Receptor γ. Molecular Pharmacology 2011, 80, 201–209. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; Hilaire, A.S.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Laurin, J.; et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of Fenofibrate and Niacin on Intrahepatic Triglyceride Content, Very Low-Density Lipoprotein Kinetics, and Insulin Action in Obese Subjects with Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Shimada, T.; Iizuka, S.; Suzuki, W.; Makihara, H.; Teraoka, R.; Tsuneyama, K.; Hokao, R.; Aburada, M. Effects of bezafibrate in nonalcoholic steatohepatitis model mice with monosodium glutamate-induced metabolic syndrome. Eur. J. Pharmacol. 2011, 662, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: a systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 38. [Google Scholar] [CrossRef]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef]
- Hatanaka, T.; Kakizaki, S.; Saito, N.; Nakano, Y.; Nakano, S.; Hazama, Y.; Yoshida, S.; Hachisu, Y.; Tanaka, Y.; Kashiwabara, K.; et al. Impact of Pemafibrate in Patients with Hypertriglyceridemia and Metabolic Dysfunction-associated Fatty Liver Disease Pathologically Diagnosed with Non-alcoholic Steatohepatitis: A Retrospective, Single-arm Study. Intern. Med. 2021, 60, 2167–2174. [Google Scholar] [CrossRef]
- Shinozaki, S.; Tahara, T.; Lefor, A.K.; Ogura, M. Pemafibrate decreases markers of hepatic inflammation in patients with non-alcoholic fatty liver disease. Clin. Exp. Hepatol. 2020, 6, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Tahara, T.; Lefor, A.K.; Ogura, M. Pemafibrate improves hepatic inflammation, function and fibrosis in patients with non-alcoholic fatty liver disease: a one-year observational study. Clin. Exp. Hepatol. 2021, 7, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Alimentary Pharmacology & Therapeutics 2021, 54, 1263–1277. [Google Scholar]
- Veiga, F.M.S.; Graus-Nunes, F.; Rachid, T.L.; Barreto, A.B.; Mandarim-De-Lacerda, C.A.; Souza-Mello, V. Anti-obesogenic effects of WY14643 (PPAR-alpha agonist): Hepatic mitochondrial enhancement and suppressed lipogenic pathway in diet-induced obese mice. Biochimie 2017, 140, 106–116. [Google Scholar] [CrossRef]
- Brown, P.J.; Winegar, D.A.; Plunket, K.D.; Moore, L.B.; Lewis, M.C.; Wilson, J.G.; Sundseth, S.S.; Koble, C.S.; Wu, Z.; Chapman, J.M.; et al. A Ureido-Thioisobutyric Acid (GW9578) Is a Subtype-Selective PPARα Agonist with Potent Lipid-Lowering Activity. J. Med. Chem. 1999, 42, 3785–3788. [Google Scholar] [CrossRef]
- Okishio, S.; et al. PPARα agonist and metformin co-treatment ameliorates NASH in mice induced by a choline-deficient, amino acid-defined diet with 45% fat. Scientific Reports 2020, 10, 19578. [Google Scholar] [CrossRef]
- Brown, P.J.; Stuart, L.; Hurley, K.P.; Lewis, M.C.; Winegar, D.A.; Wilson, J.G.; Wilkison, W.O.; Ittoop, O.R.; Willson, T.M. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorganic Med. Chem. Lett. 2001, 11, 1225–1227. [Google Scholar] [CrossRef]
- Aleshin, S.; Grabeklis, S.; Hanck, T.; Sergeeva, M.; Reiser, G. Peroxisome Proliferator-Activated Receptor (PPAR)-γ Positively Controls and PPARα Negatively Controls Cyclooxygenase-2 Expression in Rat Brain Astrocytes through a Convergence on PPARβ/δ via Mutual Control of PPAR Expression Levels. Mol. Pharmacol. 2009, 76, 414–424. [Google Scholar] [CrossRef]
- Masubuchi, Y. Metabolic and Non-Metabolic Factors Determining Troglitazone Hepatotoxicity: A Review. Drug Metab. Pharmacokinet. 2006, 21, 347–356. [Google Scholar] [CrossRef]
- Caldwell, S.H.; et al. A Pilot Study of A Thiazolidinedione, Troglitazone, in Nonalcoholic Steatohepatitis. Official journal of the American College of Gastroenterology|ACG 2001, 96. [Google Scholar] [CrossRef]
- Idilman, R.; Mizrak, D.; Corapcioglu, D.; Bektas, M.; Doganay, B.; Sayki, M.; Coban, S.; Erden, E.; Soykan, I.; Emral, R.; et al. Clinical trial: insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2008, 28, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Akyüz, F.; Demir, K.; Özdil, S.; Aksoy, N.; Poturoğlu, S.; İbrişim, D.; Kaymakoğlu, S.; Beşışık, F.; Boztaş, G.; Çakaloğlu, Y.; et al. The Effects of Rosiglitazone, Metformin, and Diet with Exercise in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2007, 52, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; LeNaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; LIDO Study Group. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.-A.K.; Cruciani-Guglielmacci, C.; Kassis, N.; Clément, L.; Ouali, F.; Caüzac, M.; Lebègue, N.; Berthelot, P.; Caignard, D.-H.; Pégorier, J.-P.; et al. S26948, a new specific peroxisome proliferator activated receptor gamma modulator improved in vivo hepatic insulin sensitivity in 48 h lipid infused rats. Eur. J. Pharmacol. 2009, 608, 104–111. [Google Scholar] [CrossRef]
- Dunn, F.L.; Higgins, L.S.; Fredrickson, J.; DePaoli, A.M. Selective modulation of PPARγ activity can lower plasma glucose without typical thiazolidinedione side-effects in patients with Type 2 diabetes. J. Diabetes its Complicat. 2011, 25, 151–158. [Google Scholar] [CrossRef]
- DePaoli, A.M.; et al. Can a Selective PPARγ Modulator Improve Glycemic Control in Patients With Type 2 Diabetes With Fewer Side Effects Compared With Pioglitazone? Diabetes Care 2014, 37, 1918–1923. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.-J.; Cho, Y.M.; Lee, B.-W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef]
Figure 1.
PPARs structure and functions. PPARs has four functional domains: a ligand-independent activation domain, a DNA binding domain, a hinge domain, and a ligand-binding domain.
Figure 1.
PPARs structure and functions. PPARs has four functional domains: a ligand-independent activation domain, a DNA binding domain, a hinge domain, and a ligand-binding domain.
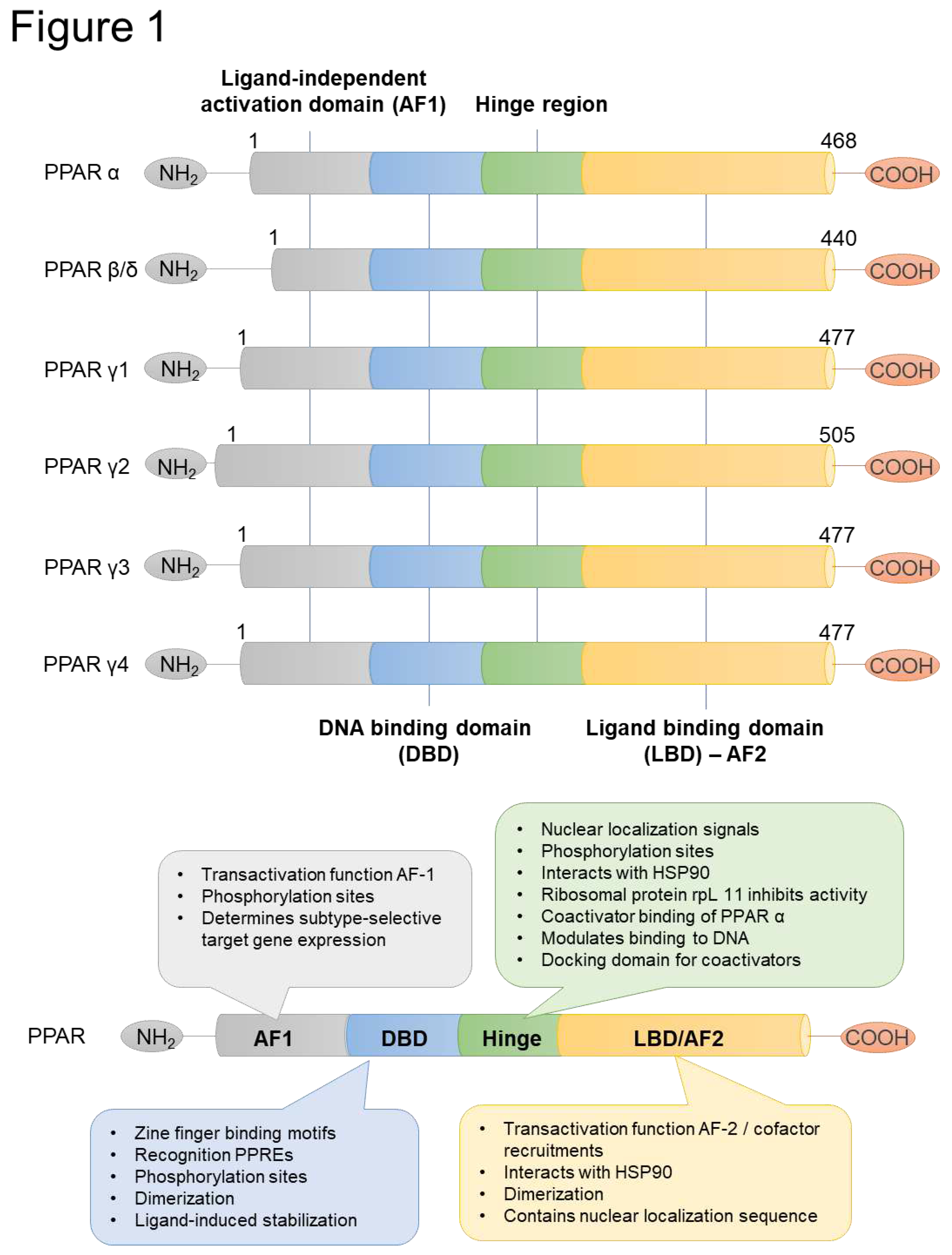
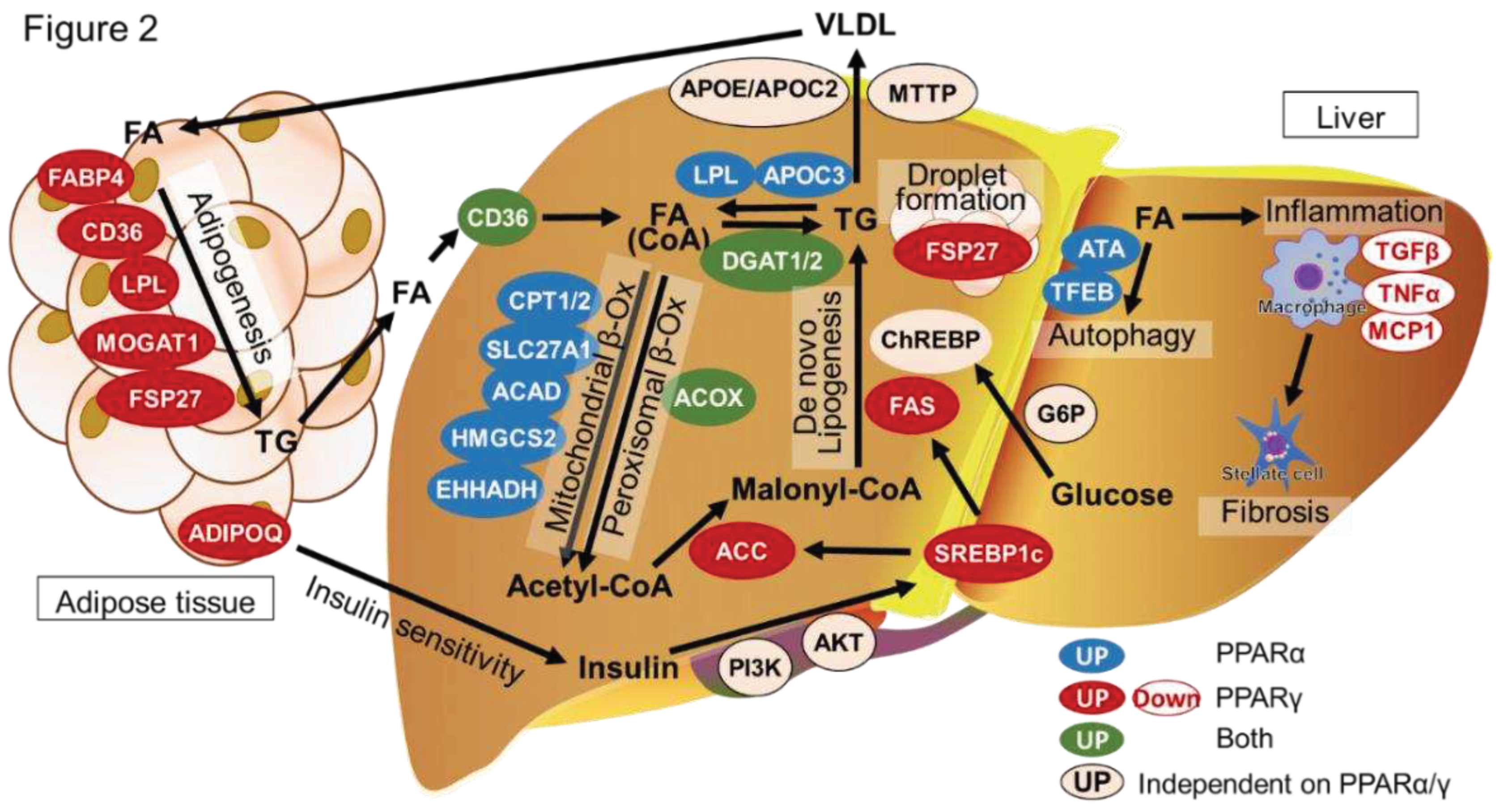
Figure 2.
PPAR-targets and contributing factors in NAFLD/NASH. Lipid homeostasis is an essential process in the liver by keeping the balance between lipid acquisition and disposal. In lipid metabolism, insulin resistance plays a crucial role in the pathology of liver disease. Insulin level is regulated by ADIPOQ which is secreted by PPARγ from the adipose tissue. In the liver, insulin via PI3K and AKT signaling pathway regulates sterol regulatory element-binding protein 1c (SREBP-1c) by activation of PPARα. SREBP-1c regulates the essential glycolytic enzymes, acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) via PPARα, which play a crucial role in the synthesis of triglycerides (TG) from carbohydrates. This process is known as de novo lipogenesis. Carbohydrate response element binding protein (ChREBP), which expression level is controlled by glucose from the bloodstream via PPARα activation through glucose-6-phosphate (G6P) signaling, is also involved in triglyceride synthesis. After, lipoprotein lipase (LPL) and apolipoprotein C3 (APOC3), secreted by PPARα, promote TG hydrolyzation into fatty acids for further homeostasis processes, called mitochondrial β-oxidation of fatty acids to acetyl-CoA. In mitochondrial β-oxidation CPT1/2, SLC27A1, ACAD, HMGCS2, and EHHADH are involved via regulating of PPARα. In further, with the presence of ACC that is regulated by SREBP-1c via PPARα activation, acetyl-CoA is converted to malonyl-CoA which will be involved in the de novo lipogenesis to TG, completing a circle of lipid homeostasis. When fatty acids are over-elevated, mitochondrial β-oxidation can be overburdened leading to hepatic accumulation and steatosis. As a compensatory effect, the peroxisomal β-oxidation is activated via ACOX signaling that causes oxidative stress, and a part of the fatty acid is converted back into TG via DGAT1/2, and both processes are controlled through PPARγ activation. Excess TG further will be transported to adipose tissue by very-low-density lipoprotein (VLDL) synthesis, via expression of APOE/APOC2 and MTTP, and transport by apolipoprotein. In adipose tissue fatty acids are converted into TGs in storage form by the adipogenesis process via the regulation of FABP4, CD36, LPL, MOGAT1 and FSP27 through PPARγ activation. Impaired VLDL synthesis and transport by apolipoprotein expression in the liver, which is essential for TG conversion into VLDL, may also contribute to hepatic fat accumulation by droplet formation via FSP27 which is also regulated by PPARγ. Overburden of fatty acids in the liver causes hepatic cell autophagy by ATA and TFEB regulation via PPARα activation. The progression of NAFLD to non-alcoholic steatohepatitis (NASH) is mainly caused by hepatic inflammation, which is triggered by downregulation of TGFβ, TNFα and MCP1 through PPARγ activation of macrophage that further activate the hepatic stellate cell, leading to fibrosis.
Figure 2.
PPAR-targets and contributing factors in NAFLD/NASH. Lipid homeostasis is an essential process in the liver by keeping the balance between lipid acquisition and disposal. In lipid metabolism, insulin resistance plays a crucial role in the pathology of liver disease. Insulin level is regulated by ADIPOQ which is secreted by PPARγ from the adipose tissue. In the liver, insulin via PI3K and AKT signaling pathway regulates sterol regulatory element-binding protein 1c (SREBP-1c) by activation of PPARα. SREBP-1c regulates the essential glycolytic enzymes, acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) via PPARα, which play a crucial role in the synthesis of triglycerides (TG) from carbohydrates. This process is known as de novo lipogenesis. Carbohydrate response element binding protein (ChREBP), which expression level is controlled by glucose from the bloodstream via PPARα activation through glucose-6-phosphate (G6P) signaling, is also involved in triglyceride synthesis. After, lipoprotein lipase (LPL) and apolipoprotein C3 (APOC3), secreted by PPARα, promote TG hydrolyzation into fatty acids for further homeostasis processes, called mitochondrial β-oxidation of fatty acids to acetyl-CoA. In mitochondrial β-oxidation CPT1/2, SLC27A1, ACAD, HMGCS2, and EHHADH are involved via regulating of PPARα. In further, with the presence of ACC that is regulated by SREBP-1c via PPARα activation, acetyl-CoA is converted to malonyl-CoA which will be involved in the de novo lipogenesis to TG, completing a circle of lipid homeostasis. When fatty acids are over-elevated, mitochondrial β-oxidation can be overburdened leading to hepatic accumulation and steatosis. As a compensatory effect, the peroxisomal β-oxidation is activated via ACOX signaling that causes oxidative stress, and a part of the fatty acid is converted back into TG via DGAT1/2, and both processes are controlled through PPARγ activation. Excess TG further will be transported to adipose tissue by very-low-density lipoprotein (VLDL) synthesis, via expression of APOE/APOC2 and MTTP, and transport by apolipoprotein. In adipose tissue fatty acids are converted into TGs in storage form by the adipogenesis process via the regulation of FABP4, CD36, LPL, MOGAT1 and FSP27 through PPARγ activation. Impaired VLDL synthesis and transport by apolipoprotein expression in the liver, which is essential for TG conversion into VLDL, may also contribute to hepatic fat accumulation by droplet formation via FSP27 which is also regulated by PPARγ. Overburden of fatty acids in the liver causes hepatic cell autophagy by ATA and TFEB regulation via PPARα activation. The progression of NAFLD to non-alcoholic steatohepatitis (NASH) is mainly caused by hepatic inflammation, which is triggered by downregulation of TGFβ, TNFα and MCP1 through PPARγ activation of macrophage that further activate the hepatic stellate cell, leading to fibrosis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



