
Submitted:
14 April 2023
Posted:
17 April 2023
You are already at the latest version
Abstract
Differentiation of induced pluripotent stem cells to a range of useful, mature target cell types is ubiquitous in monolayer culture. To further improve the phenotype of the cells produced, 3D organoid culture is becoming increasingly prevalent. Mature organoids typically require the involvement of cells from multiple germ layers. The aim of this study was to produce pulmonary organoids from defined endodermal and mesodermal progenitors. Endodermal and mesodermal progenitors were differentiated from iPSCs then combined in 3D Matrigel hydrogels and differentiated for a further 14 days to produce pulmonary organoids. The organoids expressed a range of pulmonary cell markers such as SPA, SPB, SPC, AQP5 and T1α. Furthermore, organoids expressed ACE2 capable of binding SARS-Cov2 spike protein demonstrating the physiological relevance of the organoids produced. This study demonstrates the rapid production of pulmonary organoids using a multi-germ layer approach that could be used for studying novel respiratory disease interactions.
Keywords:
pulmonary organoids
; induced pluripotent stem cells (iPSCs)
; anterior foregut endoderm
; mesoderm
; alveoli epithelial cells
; SARS-Cov2
; iPSC disease modelling
1. Introduction
Induced pluripotent stem cells (iPSCs) are characterised by their capacity for indefinite self-renewal and their ability to differentiate into any of the mature cell types found in the adult human body. This includes differentiation to a pulmonary epithelial lineage such as alveolar cells [1]. Such differentiation schemes often mirror the embryogenesis of the cell or tissue type in question. In the case of alveolar cell development, this involves differentiation through definitive endoderm, anterior foregut endoderm (AFE), pulmonary progenitors and finally alveolar cells [2,3,4]. Often such differentiation schema were developed in simple 2D culture systems with growth factors and small molecules used to stimulate or inhibit specific cell signalling pathways. However, there is increasing appreciation that stem cell fate can be determined by a myriad of other parameters including the cellular microenvironment, extracellular matrix, 3D spatial arrangement, mechanical cues including fluid flow and the existence of multiple cell types [5]. This has led to the development of increasingly complex stem cell culture systems such as organoids or tissue matrix recellularisation [6,7].
In perhaps the first report on generating pulmonary organoids from iPSCs, Gotoh et al demonstrated the production of pulmonary organoids using NKX2.1+ (a marker of early pulmonary lineage commitment) ‘ventralised’ AFE cells cultured within Matrigel also containing human foetal lung fibroblasts [8]. The approach to 3D culture was based on previous studies using primary pulmonary cells to generate organoids in vitro [9] but the study resulted in two important findings, the need for a pure endodermal population to generate lung epithelial cells and the requirement for a mesodermal population of cells to support that differentiation. In a series of papers, Dye et al created ‘patterned lung organoids’ using anterior foregut endoderm spheroids [10,11]. They noted that whilst their AFE spheroids were 85-95% pure, a population of mesodermal cells persisted within and contributed to differentiation. Based on this, the authors were able to refine the growth factors necessary to generate mature pulmonary organoids. A more recent study also demonstrated that mesenchymal stem cells (MSC), when included in lung progenitor 3D cultures, improved alveolar differentiation of iPSC-derived cells, and MSC-conditioned medium alone was sufficient to promote alveolar organoid formation [12]. Taken together these studies underline the importance of multi-germ layer involvement in organoid development, however, the mesodermal component was poorly defined and the approaches themselves long and involved.
In the initial stages of pulmonary embryogenesis primitive lung buds form from a cluster of NKX2.1+ cells located in the anterior foregut of the epithelial tube that ultimately gives rise to the trachea and esophagus [2]. Surrounding these cells is a layer of mesoderm that expresses a range of growth factors, notably BMP4, FGF10 and WNT2, the absence of which inhibits lung bud maturation [13,14,15]. Consequently, many such growth factors can be found in culture medium designed to induce the differentiation of AFE to pulmonary cells. It has previously been shown that the transcription factor GATA4, localised to the developing pulmonary mesenchyme at the epithelial buds, is necessary for normal pulmonary development [16]. Furthermore, it has been shown that GATA4 can enhance the expression of FGF10 [17]. These findings suggest that an iPSC-derived GATA4+ mesoderm could be an appropriate cell population to support the differentiation of AFE to a pulmonary fate in vitro. Mesodermal cells are a common intermediary in the production of cardiomyocytes, indeed, cardiac mesoderm interacts with the pulmonary endoderm in utero. In an effort to increase the efficiency of iPSC differentiation to cardiomyocytes Gong et al refined the common BMP4 mediated differentiation of iPSCs to mesoderm to include the γ-secretase inhibitor DAPT [18]. Amongst their findings was that DAPT significantly increased the expression of GATA4, amongst other mesodermal markers, after 6 days of differentiation, a timing well aligned with the production of FOXA2+ AFE cells in vitro.
Based on our previous studies involving the differentiation of iPSCs to alveolar cells and adapting the work of Gong et al we sought to generate organoids via the co-culture of FOXA2+ iPSC-derived AFE and GATA4+ mesoderm encapsulated within Matrigel hydrogels. IPSC-derived organoid culture systems have become increasingly popular not just as means to study organogenesis but also to study and model disease and aid drug discovery. For example, Chen et al introduced HPS1 mutations into the iPSCs used to generate their pulmonary organoids as a way to recapitulate pulmonary fibrotic disease [19], and Suezawa et al created a model of pulmonary fibrosis using human PSC-derived alveolar organoids and identified inhibition of ALK5 or blocking integrin αVβ6 as potential therapeutic options [20]. Respiratory diseases, such as the recent COVID-19 pandemic caused by SARS-CoV2 infection, are highly prevalent and can cause extensive damage to the pulmonary epithelium. Several excellent studies have been published since the outbreak of the COVID-19 pandemic and some have identified candidate COVID-19 therapeutics [21,22,23,24]. The capacity to rapidly produce pulmonary organoids could be useful in understanding the pathogenicity of other respiratory viruses in the future. As such, we produced a method of generating pulmonary organoids by co-culture of defined endodermal and mesodermal progenitors, and also sought to demonstrate that the organoids were capable of interacting with SARS-CoV2 spike protein to highlight their physiological utility.
The introduction should briefly place the study in a broad context and highlight why it is important. It should define the purpose of the work and its significance. The current state of the research field should be carefully reviewed and key publications cited. Please highlight controversial and diverging hypotheses when necessary. Finally, briefly mention the main aim of the work and highlight the principal conclusions. As far as possible, please keep the introduction comprehensible to scientists outside your particular field of research. References should be numbered in order of appearance and indicated by a numeral or numerals in square brackets—e.g., [1] or [2,3], or [4,5,6]. See the end of the document for further details on references.
2. Materials and Methods
2.1. Cell culture and differentiation
Fully characterised iPSC lines (SERU7, SOJD3 and EIPL1) from HipSci stem cell bank, Public Health England via ECACC. were maintained in Essential 8 Medium on Matrigel coated plates. For AFE differentiation, iPSCs were dissociated with StemPro Accutase and then plated on Matrigel coated plates at a density of 30,000 cells/cm2 in Essential 8 with 10 µM Y27632. The following day the medium was changed to DMEM containing 100x Glutamax, 100x non-essential amino acids, 50x human serum replacement 3, 5 µM CHIR99021 and 100 ng/mL Activin A, termed day 0. On days 1-2, cells were cultured in the same medium minus CHIR99021. On day 3, cells were cultured in the same base medium with the addition of 1 µM Dorsomorphin and 2 µM SB43152. Finally, on day 4, cells were cultured in the same base medium with the addition of endo-IWR1 (200 nM) and SB43152 (2 µM). On day 5, AFE cells were characterised for phenotypic markers or used for pulmonary organoid construction. For mesoderm differentiation, iPSCs were plated on Matrigel coated plates at a density of 60,000 cells/cm2 in Essential 8 with 10 µM Y27632. The following day the medium was changed to DMEM containing 100x Glutamax, 100x non-essential amino acids, 50x human serum replacement 3 and BMP4, 0.5 ng/mL, termed day 0. On days 1-2, cells were cultured in the same base medium with the addition of Activin A, 3 ng/mL, BMP4, 10 ng/mL and FGF2, 5 ng/mL. Finally, on days 3-4, cells were cultured in the same medium but with the addition on 2 µM DAPT. On day 5, mesoderm cells were characterised for phenotypic markers or used for pulmonary organoid construction.
For pulmonary organoid construction, 12 mm cell culture inserts were first coated with a 60 µL Matrigel. AFE and mesoderm cells were dissociated in TrypLE and a cell suspension containing 500,000 cells each of AFE and mesoderm per organoid was mixed with an equal volume of Matrigel and 200 µL pipetted on to the first layer of Matrigel and set for 2 hours at 37°C, followed by adding a final 60 µL Matrigel and set for 2 hours at 37°C. Finally, DMEM containing 100x Glutamax, 100x non-essential amino acids, 50x human serum replacement 3, BMP4, 10 ng/mL, FGF2, 10 ng/mL, FGF7, 10 ng/mL and FGF10, 10 ng/mL was added to the cell culture wells and inserts and incubated at 37°C. All work was performed with cooled liquids and tips to prevent premature Matrigel polymerisation. Organoids were maintained in culture for 2-4 weeks and the medium changed every 2-3 days.
2.2. qRT-PCR
Total RNA from cells was extracted with an QIAGEN RNeasy Mini Kit and cDNA synthesised using an iScript cDNA Synthesis Kit. Five ng cDNA per sample was used for standard qPCR with SYBR Green chemistry and pre-designed primers (Table A1). Expression was normalised to GAPDH and statistical significance determined by Student’s T-test.
2.3. RNA sequencing
RNA sequencing (RNA-seq) was performed using total RNA derived from pulmonary organoids on an Illumina HiSeq 4000 sequencer. Detailed methods and data analysis are described in the supplemental information.
2.4. Flow cytometry
Cells were dissociated with Accutase fixed in 4% PFA and stained for cell surface proteins with antibodies found in Table A1. Samples were then permeablised with 0.1% Triton X-100 followed by staining for intracellular proteins using and antibodies found in Table A1. After filtering through a 40 µm cell strainer, flow cytometry was performed on an LSR II Flow Cytometer. Data was analysed with FACSDiva software.
2.5. Confocal imaging and histology
Pulmonary organoids were harvested for immunofluorescent staining and confocal imaging 9 days after the mixing of AFE and mesoderm. Sample preparation was performed as described by Dekkers et al [25]. After immunolabeling, the organoids were imaged on a Leica SP8 confocal microscope with 20× water immersion objective for multiphoton imaging. Details for the acquisition mode settings are listed in the supplemental information. Human lung sections were obtained following informed consent and ethical approval (National Research Ethics Service, REC 20/NW/0302). Tissues were deparaffinised and rehydrated with xylene and ethanol followed by antigen retrieval and sequential incubation with the primary and secondary as listed in supplemental table 1. After dehydration with ethanol gradient, the samples were mounted with mounting and imaged on an Olympus IX83 inverted microscope.
3. Results
3.1. Differentiation of iPSCs to endodermal and mesodermal progenitors
Prior to 3D pulmonary organoid, iPSCs were first differentiated to AFE and mesoderm (Figure 1A). After 5 days of endodermal differentiation the expression of the endodermal markers FOXA2 and SOX17 was significantly increased in AFE cells compared with iPSCs, p<0.001 and p<0.0001, respectively (Figure 1B). Across 3 different cell lines the mean number of FOXA2+ and SOX17+ cells following differentiation was 92% and 93%, respectively (Figure 1C). After 5 days of mesodermal differentiation the expression of the mesodermal markers GATA4 and PDGFRa was significantly increased in mesoderm cells compared with iPSCs, p<0.001 and p<0.0001, respectively (Figure 1D). The expression of TBX1 was detected in mesodermal cells whilst almost absent in iPSCs. Across 3 different iPSC lines the mean number of GATA4+, PDGFRa+ and TBX1+ cells following differentiation was 97%, 93% and 96%, respectively (Figure 1E).
3.2. Spontaneous pulmonary organoid formation by co-culture of endodermal and mesodermal progenitors
AFE and mesoderm progenitors were co-cultured in a 1:1 ratio for 9-14 days in Matrigel to induce pulmonary organoid formation. During the process of differentiation, spherical pulmonary organoids were gradually formed that possessed hollow internal lumen (Figure S1). AFE progenitors without co-culture with mesoderm cells could not form organoids (data not shown). The existence of a range of type I and II alveolar marker proteins in the organoids was analysed with immunofluorescent confocal microscopy (Figure 2A), and compared with normal human lung sections (Figure 2B). Robust levels of the alveolar type II markers surfactant proteins SPA, SPB and SPC was observed in pulmonary organoids (Figure 2A) and confirmed in normal human lung sections (Figure 2B). Similarly, the expression of the alveolar type I markers aquaporin 5 (AQP5) and podoplanin/T1α was also confirmed in the pulmonary organoids and in normal human lung (Figure 2A, 2B). The expression of NKX2.1 was weak in both the pulmonary organoids and normal human lung, suggesting relative maturation of the organoids. Secondary antibody staining controls were negative (Figure S2).
3.3. IPSC-derived pulmonary organoids express lung-specific marker genes
RNA sequencing (RNAseq) was then conducted on the pulmonary organoids differentiated from two different iPSC lines on day 14 after co-culture of AFE and mesoderm progenitors. The principle-component analysis (PCA) of the RNAseq data sets showed that the pulmonary organoids are clearly distinguished from the undifferentiated iPSCs (Figure 3A). The heatmap generated by DESeq analysis in Figure 3B shows that majority of lung-specific genes were enriched in the pulmonary organoids compared to the undifferentiated iPSCs. Among the lung-specific genes, subsets of genes on the Gene Ontology (GO) terms of lung development, lung epithelial development, and lung morphology were mostly upregulated, respectively, (Figures 3C, D & E, Appendix A1). The expression of key marker genes for type 1 (AQP5) and type II (SPA, SPC) alveolar cells as well as lung-specific genes in general (NKX2-1) were validated by RT-qPCR (Figure 3F-I).
3.4. Pulmonary organoids express ACE2 capable of binding SARS-CoV2 spike protein
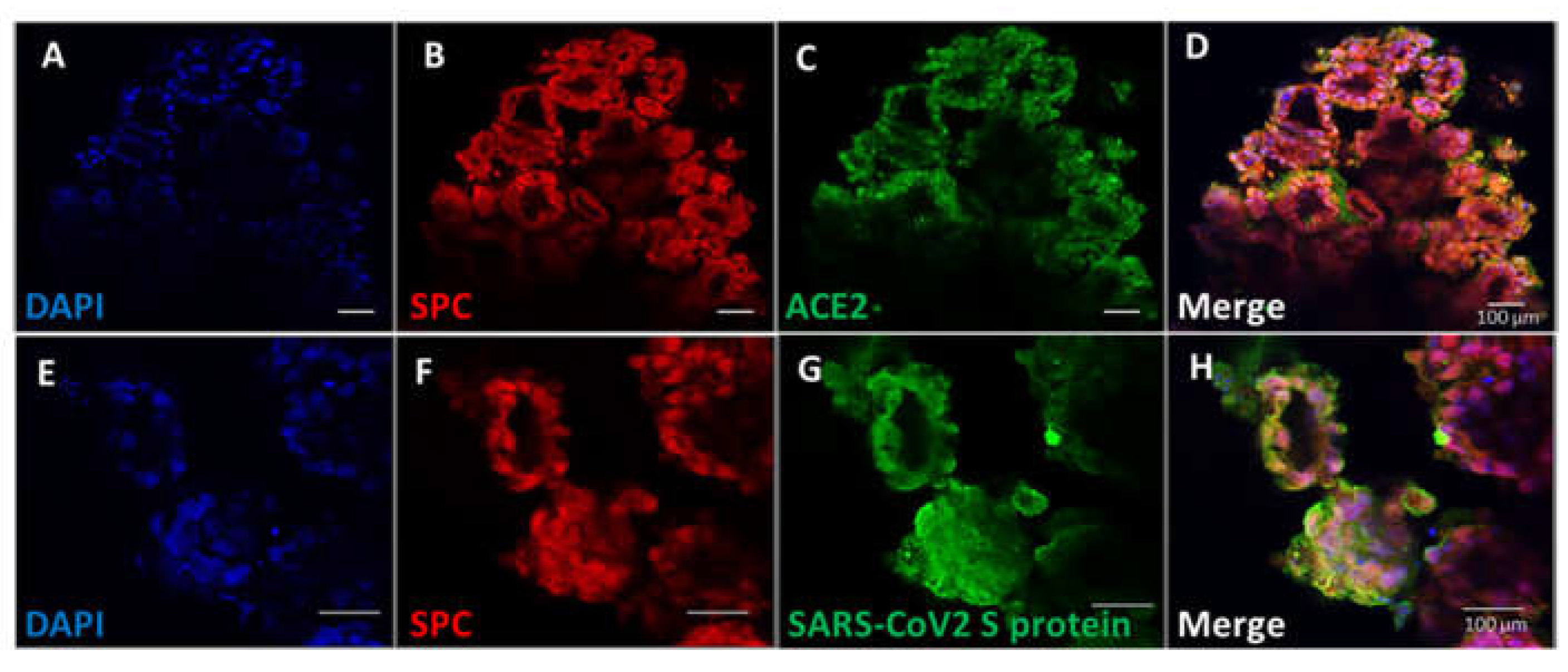
To demonstrate that the pulmonary organoids possessed physiological relevance, their interaction with SAR-CoV2 was investigated. The spike protein of SARS-CoV2 binds the host angiotensin-converting enzyme 2 (ACE2) receptor for viral entry [26]. We demonstrated that ACE2 proteins are co-express on cells that express SPC in pulmonary organoids (Figure 4A–D). To determine the virus binding capability, recombinant SARS-CoV2 spike proteins were incubated with the organoids and visualised by immunofluorescent microscopy. Results showed that there was clear detection of membrane bound SARS-CoV2 spike protein on alveolar cells of the organoids (Figure 4G), and the staining was also co-localised to cells expressing SPC (Figure 4E–H).
4. Discussion
The aim of this study was to demonstrate a rapid production of pulmonary organoids via the co-culture of iPSC-derived endodermal and mesodermal progenitors and demonstrate that the organoids would be responsive to the novel respiratory virus SAR-CoV2. Using a well-established approach of encapsulating cells in 3D hydrogels based on Matrigel it was shown that pulmonary organoids spontaneously formed within 14 days of co-culturing endodermal and mesodermal progenitors. The resulting organoids expressed a range of proteins associated with alveolar type I and II cells. The pulmonary organoids also expressed ACE2 that was capable of binding SARS-CoV2 spike protein.
Previous studies demonstrated a clear need for a mesodermal component in establishing iPSC-derived pulmonary organoids [11,19], which is unsurprising given the role of mesoderm in driving pulmonary organogenesis. However, these studies utilised a mesoderm that was an unintentional by-product of endodermal differentiation. Based on the phenotype of pulmonary mesoderm in utero we produced a GATA4+ mesoderm to support the differentiation of endodermal progenitors in vitro. Most of current protocols in literature use defined factors including CHIR and BMP4 to provide mesodermal cues for lung organoid differentiation [27,28]. Albeit successful, a few defined factors unlikely recapitulate the entire role that mesoderm plays during lung organogenesis. We have used a highly controlled approach to co-culture AFE and mesoderm progenitors. We found that the mesoderm progenitors are absolutely necessary for pulmonary organoid generation. AFE progenitors alone were not able to form organoids. Although we introduced an additional step for pulmonary organoid generation in the initial AFE and mesoderm differentiation, the protocols produce AFE and mesoderm progenitors in 5-6 days, which can be co-cultured directly for pulmonary organoid construction on Matrigel, without the need for selection or purification of the progenitors. The organoids start to form in 4 days after co-culture and robustly express lung-specific markers as earlier as day 9. Thus, the total protocol takes less than 20 days to produce lung organoids. This is much quicker than the existing methods published, which usually take 30 – 60 days [23,28], and similar to the protocol by Chen et al which was 25 days [27].
The mesoderm population is also desirable for the future production of more complicated lung organoids. By adding minimum further factors such as VEGF, this protocol has the potential to generate vascularised lung organoids. The vascular components would derive from the mesoderm population, as the growth factors employed in the protocol are very similar to the endothelial cell differentiation from iPSCs in our previous work [29]. In fact, we have already observed some expression of endothelial marker CD31 in the organoids surrounding the AQP5+ alveolar cells (Figure S3), suggesting the possible emergence of an endothelial population in the matrix of the organoids. Further work will be required to understand the fate and contribution of the mesodermal progenitors to the organoid formation, and to optimise a protocol for the vascularisation of lung organoids. Such effort would potentially open the prospect for the study of lung barrier function and modelling diseases under a more physiological context.
The expression of ACE2 receptor and subsequent interaction with SARS-CoV2 spike protein suggest that pulmonary organoids could in the future be used to study the interaction of respiratory viruses with the pulmonary epithelium in vitro. A recent study by Porotto et al demonstrated the infection of pulmonary organoids with parainfluenza virus localised to alveolar epithelial cells as would occur in vivo [30]. The more recent publication by Han et al [21] attempted drug screening using pulmonary organoids to identify blockers of SARS-Cov2 infection for the treatment of COVID-19. However, severe respiratory viral infections often cause extensive damage mediated by an inflammatory cascade involving pro-inflammatory cytokines and alveolar macrophages and neutrophils. In order for pulmonary organoids to fully recapitulate the pathogenesis of respiratory virus infection, future work could incorporate iPSC-derived alveolar macrophages [31] and neutrophils [32], although considerable work will be needed to understand the timing of introducing immune cells and how to maintain their phenotype over prolonged culture.
In summary we have developed a novel multi-germ layer approach for rapid production of pulmonary organoids that exhibited the capacity for infection by SARS-CoV2 and upon further development may be useful in understanding the pathogenesis of future novel respiratory viruses and pulmonary disease modelling.
Limitations of the study
One limitation of this study is that the organoids we differentiated were an alveolar model rather than an airway model. Although the alveolar-focused organoids derived from iPSCs have good values for the future study of a range of human conditions, it is worthwhile to further develop the protocol to build a full lung model. Additionally, we have aimed to develop a rapid protocol to produce the organoids, but did not culture the organoids longer beyond the time point when specific alveolar cell markers were detected. Depending on the desired applications, a prolonged culture may benefit a further maturation of the organoids.
Author Contributions
Conceptualization, Adam Mitchell, Timothy Felton and Tao Wang; Formal analysis, Adam Mitchell and Xiangjun Zhao; Funding acquisition, Timothy Felton and Tao Wang; Investigation, Adam Mitchell, Chaowen Yu and Xiangjun Zhao; Methodology, Adam Mitchell, Chaowen Yu, Xiangjun Zhao and Laurence Pearmain; Resources, Laurence Pearmain, Rajesh Shah, Karen Hanley and Tao Wang; Supervision, Timothy Felton and Tao Wang; Writing – original draft, Adam Mitchell; Writing – review & editing, Chaowen Yu, Karen Hanley, Timothy Felton and Tao Wang.
Funding
This report is independent research supported by the North West Lung Centre Charity at Manchester University NHS Foundation Trust (Ref: LT038). CY is supported by a Post-Doctoral Fellowship from the China Scholarship Council (ref No.201908500055). KPH and LP are supported by The Medical Research Council (KPH, MR/P023541/1 and LP, MR/R00191X/1). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the North West Lung Centre Charity or the Department of Health.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
The authors would like to acknowledge the Manchester Allergy, Respiratory and Thoracic Surgery Biobank and the North West Lung Centre Charity for supporting this project. The authors would like to thank Rachel Scholey and Andy Hayes of the Bioinformatics and Genomic Technologies Core Facilities at the University of Manchester for providing support with regard to RNAseq. We also thank the Bioimaging Core Facility for guidance on confocal microscopy and sample preparation. In addition, we would thank the study participants for their contribution.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Supplemental information - Detailed experimental procedures
Cell culture and differentiation
IPSC lines SERU7, SOJD3 and EIPL1 were supplied by HipSci stem cell bank, Public Health England via ECACC. IPSC lines WT3 and WT4 were reprogrammed from adult human dermal fibroblasts reported in our previous publication [28]. IPSCs were maintained, feeder free, in Essential 8 Medium (Thermo Fisher Scientific, Loughborough, UK) and passaged at a 1:6 ratio, on Matrigel (Corning, Deeside, UK) coated plates, every 3-4 days. For anterior foregut endoderm (AFE) differentiation, iPSCs were dissociated with StemPro Accutase (Thermo Fisher Scientific, Loughborough, UK) for 5 minutes, 37°C, then plated on Matrigel coated plates at a density of 30,000 cells/cm2 in Essential 8 Medium with 10 µM Y27632 (Tocris, Bristol, UK). The following day the medium was changed to Dulbecco's Modified Eagle Medium (DMEM) containing 100x Glutamax (Thermo Fisher Scientific, Loughborough, UK), 100x non-essential amino acids (Sigma Aldrich, Gillingham, UK), 50x human serum replacement 3 (Sigma Aldrich, Gillingham, UK), 5 µM CHIR99021 (Tocris, Bristol, UK) and 100 ng/mL Activin A (Peprotech, London, UK), termed day 0. On days 1-2, cells were cultured in the same medium minus CHIR99021. On day 3, cells were cultured in the same base medium with the addition of Dorsomorphin (1 µM, Tocris, Bristol, UK) and SB43152 (2 µM, Tocris, Bristol, UK). Finally, on day 4, cells were cultured in the same base medium with the addition of endo-IWR1 (200 nM, Tocris, Bristol, UK) and SB43152 (2 µM). On day 5, anterior foregut endoderm cells were characterised for phenotypic markers or used for pulmonary organoid construction. For mesoderm differentiation, iPSCs were dissociated with StemPro Accutase for 5 minutes, 37°C, then plated on Matrigel coated plates at a density of 60,000 cells/cm2 in Essential 8 Medium with 10 µM Y27632. The following day the medium was changed to DMEM containing 100x Glutamax, 100x non-essential amino acids, 50x human serum replacement 3 and BMP4 (0.5 ng/mL, Peprotech, London, UK), termed day 0. On days 1-2, cells were cultured in the same base medium with the addition of Activin A (3 ng/mL), BMP4 (10 ng/mL) and FGF2 5 ng/mL. Finally, on days 3, cells were cultured in the same medium but with the addition on DAPT 2 µM (Tocris, Bristol, UK). On day 5, mesoderm cells were characterised for phenotypic markers or used for pulmonary organoid construction.
For pulmonary organoid construction, 12 mm cell culture inserts (Merck Millipore, Watford, UK) were first coated with a 60 µL layer of Matrigel and set for 2 hours at 37°C. AFE and mesoderm cells were dissociated in TrypLE (Thermo Fisher Scientific, Loughborough, UK), for 5 minutes at 37°C and cells counted. Cell suspensions containing 500,000 cells each of AFE and mesoderm cells per organoid were mixed with an equal volume of Matrigel and 200 µL pipetted on to the first layer of matrigel and set for 2 hours at 37°C. Following this, a final 60 µL layer of Matrigel was pipetted on to the hydrogel and set for 2 hours at 37°C. Finally, cell culture medium, 250µL DMEM containing 100x Glutamax, 100x non-essential amino acids, 50x human serum replacement 3, BMP4 (10 ng/mL), FGF2 (10 ng/mL), FGF7 (10 ng/mL) and FGF10 (10 ng/mL) was pipetted into the cell culture wells and inserts and incubated at 37°C. All work was performed with cooled liquids and tips to prevent premature Matrigel polymerisation. Organoids were maintained in culture for up to 14 days and the medium changed every 2-3 days.
qRT-PCR
Total RNA from iPSCs, AFE cells or mesoderm progenitors was prepared using RNeasy Mini Kit (Qiagen, Hilden, Germany). For total RNA preparation from pulmonary organoids, the organoids-containing Matrigel was placed in 3 ml ice-cold Corning cell recovery solution (Corning, Deeside, UK), which was pipetted up and down to break the Matrigel and incubated on ice for 1 hour. The organoids was then collected by centrifugation at 300 g for 5 minutes and incubated with 3 ml 0.05% trypsin/EDTA at 37°C for 5 minutes to dissociate cells in the organoids. The cells were collected by centrifugation at 300 g for 5 minutes and then subjected to 1 ml TRIZOL (Sigma Aldrich, Gillingham, UK) and vortexed thoroughly followed by adding 0.2 ml phenol-chloroform (Sigma Aldrich, Gillingham, UK) and vortexing for 15 seconds. After incubation for 3 minute at room temperature, the sample was centrifuged at 12,000 g for 15 minutes at 4°C. The upper aqueous phase was carefully collected and transferred into a fresh tube. Half ml isopropanol (Sigma Aldrich, Gillingham, UK) was then added to the sample which was incubated at room temperature for 10 minutes followed by centrifugation at 12,000 g at 4°C for 10 minutes. The RNA pellet was washed twice with 1 ml 75% ethanol and collected by centrifugation at 7,500 for 5 minutes at 4°C. The RNA pellet was then air dried and resuspended in 20 μl RNase free H2O.
The cDNA was synthesised using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Watford, UK) following the manufacturer’s instructions. QRT-PCR reactions containing 5 ng cDNA per sample were prepared using SYBR Green chemistry and pre-designed primer sets (Bio-Rad Laboratories, Watford, UK) as listed in the Key resource table, and run on a CFX96 Real-Time PCR system (Bio-Rad Laboratories, Watford, UK). Expression was normalised to a housekeeping gene.
RNA sequencing (RNAseq)
Total RNA isolated using above method was submitted to the Genomic Technologies Core Facility (GTCF) at the University of Manchester. Quality and integrity of the RNA samples were assessed using a 4200 TapeStation (Agilent Technologies) and then libraries generated using the Illumina® Stranded mRNA Prep. Ligation kit (Illumina, Inc.) according to the manufacturer’s protocol. Briefly, total RNA (typically 0.025-1 μg) was used as input material from which polyadenylated mRNA was purified using poly-T, oligo-attached, magnetic beads. Next, the mRNA was fragmented under elevated temperature and then reverse transcribed into first strand cDNA using random hexamer primers and in the presence of Actinomycin D (thus improving strand specificity whilst mitigating spurious DNA-dependent synthesis). Following removal of the template RNA, second strand cDNA was then synthesized to yield blunt-ended, double-stranded cDNA fragments. Strand specificity was maintained by the incorporation of deoxyuridine triphosphate (dUTP) in place of dTTP to quench the second strand during subsequent amplification. Following a single adenine (A) base addition, adapters with a corresponding, complementary thymine (T) overhang were ligated to the cDNA fragments. Pre-index anchors were then ligated to the ends of the double-stranded cDNA fragments to prepare them for dual indexing. A subsequent PCR amplification step was then used to add the index adapter sequences to create the final cDNA library. The adapter indices enabled the multiplexing of the libraries, which were pooled prior to cluster generation using a cBot instrument. The loaded flow-cell was then paired-end sequenced (76 + 76 cycles, plus indices) on an Illumina HiSeq4000 instrument. Finally, the output data was demultiplexed and BCL-to-Fastq conversion performed using Illumina’s bcl2fastq software, version 2.20.0.422.
Flow cytometry
Cells were dissociated with Accutase for 5 minutes at 37°C then resuspended in HBSS containing Fixable Viability Dye – eFluor 780 (Thermo Fisher Scientific, Loughborough, UK) for 30 minutes at 4°C. A sample of cells were retained as an unstained control. Samples were then centrifuged at 300 g for 5 minutes and resuspended in 4% paraformaldehyde (PFA, Sigma-Aldrich, Gillingham, UK) for 4 minutes. Cells were then diluted 1:1 with PBS containing 2% foetal bovine serum (FBS) (Thermo Fisher Scientific, Loughborough, UK) and centrifuged at 300 g for 5 minutes. Staining for cell surface proteins was performed by resuspending cells in 100 µL PBS with 2% FBS and antibodies found in Table A1 for 30 minutes at 4°C. Samples were centrifuged and resuspended as before and permeablised in 100 µL PBS with 2% FBS 0.1% Triton X-100 for 20 minutes at 4°C. Following this staining for intracellular proteins was performed using and antibodies listed in the Key resource table 1 for 30 minutes at 4°C. Finally, samples were centrifuged and resuspended as before, filtered through a 40 µM cell strainer and flow cytometry performed on an LSR II Flow Cytometer (BD Biosciences, Wokingham, UK). Data was analysed with FACSDiva software (BD Biosciences, Wokingham, UK), cells were gated to exclude dead cells, debris and cell aggregates and histograms drawn to compare the intensity of each fluorophore in stained samples against and unstained control.

Table 1.
Reagent details.
| Reagent | Supplier | Catalogue Number |
|---|---|---|
| ACE2 primary antibody | R&D Systems | MAB933-SP |
| AQP5 primary antibody | ThermoFisher Scientific | PA5-99403 |
| SFTPA1 primary antibody | Universal Biologics | A3133-50ul |
| SFTPB primary antibody | Biomatik | CAU25609 |
| SFTPC primary antibody | ThermoFisher Scientific | PA571680 |
| T1α primary antibody | ThermoFisher Scientific | BMS1105 |
| TTF1 primary antibody | Caltag Medsystems | H00007080-M01-100ug |
| Donkey anti Mouse A488 secondary antibody | Abcam | ab150105 |
| Donkey anti Rabbit A594 secondary antibody | Abcam | ab150076 |
| FOXA2 e660 conjugated antibody | eBioscience | 50-4778 |
| GATA4 A647 conjugated antibody | Bioss | bs-1778R-A647 |
| PDGFRa A594 conjugated antibody | Bioss | bs-0231R-A594 |
| SOX17 PerCP CY5.5 conjugated antibody | BD Pharmingen | 562387 |
| TBX1 A488 conjugated antibody | Bioss | bs-8257R-A488 |
| VEGFR2 PE conjugated antibody | Bioss | bs-10412R-PE |
| FOXA2 primer | BioRad | qHsaCID0014658 |
| GATA4 primer | BioRad | qHsaCID0012121 |
| PDGFRa primer | BioRad | qHsaCID0007202 |
| SOX17 primer | BioRad | qHsaCED0046246 |
| TBX1 primer | BioRad | qHsaCID0013392 |
| VEGFR2 primer | BioRad | qHsaCID0006310 |
| B2M | Forward: 5′ GAGGCTATCCAGCGTACTCCA 3′ | Reverse: 5′ CGGCAGGCATACTCATC TTTT 3′ |
| GAPDH | Forward: 5′ CATGTTCGTCATGGGTGTGAACCA 3′ | Reverse: 5′ ATGGCATGGACTGTGGTCATGAGT 3′ |
| Recombinant SARS-CoV-2 Spike S1 protein | R&D Systems | 10522-CV |
| SARS-CoV-2 Spike S1 Subunit Antibody | R&D Systems | MAB105403-SP |
Binding of SARS-CoV2 spike protein S1 to organoids
For the SARS-CoV-2 Spike S1 protein binding to organoids, 5 uL (1 μg) of recombinant SARS-CoV-2 Spike S1 protein (R&D Systems, Abingdon, UK) was added to each well of the organoids culture and incubated in the CO2 incubator at 37°C for 3 hours. The organoids were then harvested and imaged by confocal microscopy as described below.
3. D Confocal imaging
Pulmonary organoids were harvested for immunofluorescent staining and confocal imaging at day 9 after the co-culture of AFE and mesoderm cell populations. Sample preparation was performed as described by Dekkers et al [32]. The culture medium was first removed and the organoids were washed with 1 mL of PBS without disrupting the 3D matrix. The entire 3D matrix containing the organoids in the 12 mm cell culture insert was then gently scraped and transferred into a new 24-well plate and plated on ice. One mL of ice-cold cell recovery solution was added to each well and incubate on a horizontal shaker at 4°C (60 r.p.m.) for 60 min to dissolve the 3D matrix. One-mL tips were pre-coated by dipping the full length of the tips in 1% BSA in PBS. Before using, the tips were used to pipet 1 mL of the BSA solution two times to prevent the organoids from sticking to the tips. The tip was then used to gently resuspend the dissolved organoids-containing matrix five to ten times before transferring the organoids to a 15-mL tube pre-coated with the 1% BSA. The culture well was rinsed with 1 mL of ice-cold 1% BSA to collect all organoids. The 15-mL tube that contains the organoids was top up to 10 mL with cold (4°C) PBS and centrifuge at 70g for 3 min at 4°C to collect the organoids.
For organoid fixation and blocking, the organoids pellet was gently resuspend in 1 mL of 4% Paraformaldehyde using a 1-mL tip precoated with 1% BSA, then incubate at 4°C for 45 min with gently resuspending the organoids halfway through the incubation period. The tube was then topped up to 10 mL with cold (4°C) 0.1% (vol/vol) PBS-Tween (PBT), gently swirling to mix the sample. After incubation at 4°C for 10 min, the sample was centrifuged at 70g for 5 min at 4°C to pellet the organoids. To block the nonspecific binding sites of the organoids, the pellet was first resuspend in cold (4°C) organoid washing buffer (OWB) that contains 0.1% (vol/vol) Triton X-100 and 0.2% BSA in PBS, then an appropriate amount (200 μL or more) of organoids suspension per staining were transferred to a low-adherence 24-well plate and leave at 4 °C for 15 min.
For immunolabeling, 200 μL of OWB was first added to one of the empty wells, which was served as the reference well. The plates containing organoids were tilted to 45° and the OWB were carefully aspirated until 200 μL of OWB with the organoids was left in the plate (use the reference well to estimate 200 μL). Two hundred μL primary antibodies in OWB (2× concentration) was added to each well which was incubated overnight at 4 °C with mild rocking (60 r.p.m on a horizontal shaker). The following day, 1 mL of OWB was added to each well to wash the organoids. After 3 min when all organoids were settled at the bottom, the OWB was removed leaving 200 μL in each well. One mL of OWB per well was then added to each well which were incubated for 2 h with mild rocking. The washing steps were repeated for two more times, then 200 μL secondary antibodies in OWB (2× concentration) were added per well and incubated at 4 °C overnight with mild rocking. The washing steps were repeated at least three times. The organoids were then counterstained by incubating with 0.1 μg/ml DAPI (Thermo Fisher Scientific, Loughborough, UK) for 15 min under dark at RT, then the above washing steps were repeated three times with a final rinsed with PBS (containing 0.1% BSA). The organoids were then ready for imaging.
For 3D imaging, the organoids were transfer to a 35 mm µ-Dish (ibidi, Gräfelfing, Germany) pre-filled with 3 mL 1% BSA in PBS. The organoid suspension was gently swirled to collect the organoids to the centre of the dish and allowed to settle for 5 minutes before been imaged on a Leica SP8 confocal microscope with 20× water immersion objective for multiphoton imaging. The acquisition settings were: scan mode = sequential scan, format = 1,024 × 1,024, speed = 600 HZ, line average = 3.0, line accumulation 1.0, frame average 1.0, frame accumulation1.0, rotation 0.0, pinhole 1AU, zoom factor 2.0~3.0, zoom factor 2.0~3.0. Seq.1 was assigned to detect DAPI and AF647, using wavelengths of 410-478 nm and 657-800 nm, respectively. Seq.2 was assigned to detect AF488 (498-637nm) and the white light. The Hybrid detector (HyD) was chosen to detect fluorescent signals. Smart gain for PMT was set to 800-960, smart gain for HyD was set to 10%-40%, and the smart offset was 0.0%. The laser% for all fluorescence is ranging from 5% to 15% to gain an optimum imaging.
Histology
Human lung sections were obtained following informed consent and ethical approval (National Research Ethics Service, REC 20/NW/0302) from the Manchester Allergy, Respiratory and Thoracic Surgery (ManARTS) Biobank (study number M2014-18). Immunofluorescent staining of 5 µm sections of human lung was performed as follows. Samples were deparaffinised and rehydrated through sequential washes with xylene and 100% and 90% ethanol, 3 minutes each. Antigen retrieval was performed in boiling citrate buffer, pH6, for 20 minutes, after which samples were incubated with primary antibodies listed in the Key resource table, overnight at 4°C. The next day samples were washed 3 times with PBS then incubated with secondary antibodies, Table A1, for 2 hours at room temperature. Finally, samples were washed 3 times with PBS, dehydrated through 70%, 90% and 100% ethanol and mounted with ProLong Diamond Antifade Mountant containing DAPI (Thermo Fisher Scientific, Loughborough, UK). Slides were imaged on an Olympus IX83 inverted microscope and images processed with Olympus CellSens software.
Statistics and data analysis
Statistical analysis – Statistical significance was determined by unpaired Student’s t-test using GraphPad Prism. Data are presented as mean ± SEM. A minimum significance value of p<0.05 were considered significant.
RNAseq analysis – Unmapped paired-end sequences from the HiSeq 4000 were assessed by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence adapters were removed, and reads were quality trimmed to quality q20 using Trimmomatic_0.36 (PMID: 24695404). The reads were mapped against the reference human (hg38) genome and counts per gene were calculated using annotation from GENCODE 30 (http://www.gencodegenes.org/) using STAR_2.5.3a (PMID: 23104886). Normalisation, Principal Components Analysis (PCA), and differential expression were calculated in DESeq2_1.20.0 using default settings (PMID:25516281) and independent filtering significance level (alpha) 0.05. Gene ontology enrichment analysis were conducted by clusterProfiler software package (version 3.16.0) to visualize biological functions enriched in the differentially expressed and notable upregulated (>2-fold mean increase) or downregulated (>2-fold mean decrease) genes of the dataset. The lung differentiation related gene were re-identified and presented through heatmap plot with pheatmap software package (version 1.0.12).
References
- Mitchell, A.; Drinnan, C.T.; Jensen, T.; Finck, C. Production of high purity alveolar-like cells from iPSCs through depletion of uncommitted cells after AFE induction. Differentiation 2017, 96, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Morrisey, E.E.; Hogan, B.L.M. Preparing for the First Breath: Genetic and Cellular Mechanisms in Lung Development. Developmental Cell 2010, 18, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Green, M.D.; Chen, A.; Nostro, M.-C.; d'Souza, S.L.; Schaniel, C.; Lemischka, I.R.; Gouon-Evans, V.; Keller, G.; Snoeck, H.-W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.L.; Islam, M.N.; O'Neill, J.; Hu, Z.; Yang, Y.-G.; Chen, Y.-W.; Mumau, M.; Green, M.D.; Vunjak-Novakovic, G.; Bhattacharya, J.; et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hayden, P.J.; Harbell, J.W. Special review series on 3D organotypic culture models: Introduction and historical perspective. In Vitro Cellular & Developmental Biology-Animal.
- Tran, F.; Klein, C.; Arlt, A.; Imm, S.; Knappe, E.; Simmons, A.; Rosenstiel, P.; Seibler, P. Stem Cells and Organoid Technology in Precision Medicine in Inflammation: Are We There Yet? Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef]
- Ohata, K.; Ott, H.C. Human-scale lung regeneration based on decellularized matrix scaffolds as a biologic platform. Surgery Today 2020, 50, 633–643. [Google Scholar] [CrossRef]
- Gotoh, S.; Ito, I.; Nagasaki, T.; Yamamoto, Y.; Konishi, S.; Korogi, Y.; Matsumoto, H.; Muro, S.; Hirai, T.; Funato, M.; et al. Generation of Alveolar Epithelial Spheroids via Isolated Progenitor Cells from Human Pluripotent Stem Cells. Stem Cell Reports 2014, 3, 394–403. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. The Journal of Clinical Investigation 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.H.; Tsai, Y.-H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Dye, B.R.; Dedhia, P.H.; Miller, A.J.; Nagy, M.S.; White, E.S.; Shea, L.D.; Spence, J.R. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. Elife 2016, 5, e19732. [Google Scholar] [CrossRef]
- Leeman, K.T.; Pessina, P.; Lee, J.H.; Kim, C.F. Mesenchymal Stem Cells Increase Alveolar Differentiation in Lung Progenitor Organoid Cultures. Sci. Rep. 2019, 9, 6479. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gordon, J.; Manley, N.R.; Litingtung, Y.; Chiang, C. Bmp4 is required for tracheal formation: a novel mouse model for tracheal agenesis. Dev. Biol. 2008, 322, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.; Liu Y-h Xing, Y.; Hu, L.; Borok, Z.; Kwong, K.Y.C.; Minoo, P. Mesodermal deletion of transforming growth factor-beta receptor II disrupts lung epithelial morphogenesis: cross-talk between TGF-beta and Sonic hedgehog pathways. J. Biol. Chem. 2008, 283, 36257–36264. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Danilenko, D.M.; Scully, S.A.; Bolon, B.; Ring, B.D.; Tarpley, J.E.; DeRose, M.; Simonet, W.S. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes. Dev. 1998, 12, 3156–3161. [Google Scholar] [CrossRef]
- Ackerman, K.G.; Wang, J.; Luo, L.; Fujiwara, Y.; Orkin, S.H.; Beier, D.R. Gata4 is necessary for normal pulmonary lobar development. Am. J. Respir. Cell Mol. Biol. 2007, 36, 391–397. [Google Scholar] [CrossRef]
- Golzio, C.; Havis, E.; Daubas, P.; Nuel, G.; Babarit, C.; Munnich, A.; Vekemans, M.; Zaffran, S.; Lyonnet, S.; Etchevers, H.C. ISL1 directly regulates FGF10 transcription during human cardiac outflow formation. PloS one 2012, 7, e30677–e30677. [Google Scholar] [CrossRef]
- Gong, H.; Yan, Y.; Fang, B.; Xue, Y.; Yin, P.; Li, L.; Zhang, G.; Sun, X.; Chen, Z.; Ma, H.; et al. Knockdown of Nucleosome Assembly Protein 1-Like 1 Induces Mesoderm Formation and Cardiomyogenesis Via Notch Signaling in Murine-Induced Pluripotent Stem Cells. Stem Cells 2014, 32, 1759–1773. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Huang, S.X.; de Carvalho, A.L.R.T.; Ho, S.-H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.-L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nature Cell Biology 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Suezawa, T.; Kanagaki, S.; Moriguchi, K.; Masui, A.; Nakao, K.; Toyomoto, M.; Tamai, K.; Mikawa, R.; Hirai, T.; Murakami, K.; et al. Disease modeling of pulmonary fibrosis using human pluripotent stem cell-derived alveolar organoids. Stem Cell Reports 2021, 16, 2973–2987. [Google Scholar] [CrossRef]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Surendran, H.; Nandakumar, S.; Pal, R. Human Induced Pluripotent Stem Cell-Derived Lung Epithelial System for SARS-CoV-2 Infection Modeling and Its Potential in Drug Repurposing. Stem Cells Dev. 2020, 29, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Wang, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Revealing Tissue-Specific SARS-CoV-2 Infection and Host Responses using Human Stem Cell-Derived Lung and Cerebral Organoids. Stem Cell Reports 2021, 16, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, N.; Fan, S.; Zhao, L.; Song, W.; Gong, Y.; Shen, Q.; Zhang, C.; Ren, P.; Lin, C.; et al. Establishment of human distal lung organoids for SARS-CoV-2 infection. Cell Discov. 2021, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Alieva, M.; Wellens, L.M.; Ariese, H.C.R.; Jamieson, P.R.; Vonk, A.M.; Amatngalim, G.D.; Hu, H.; Oost, K.C.; Snippert, H.J.G.; et al. High-resolution 3D imaging of fixed and cleared organoids. Nature Protocols 2019, 14, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, J.; Zhao, S.; Han, L.; Yang, H.; Lin, Y.; Rong, Z. Long-Term Engraftment Promotes Differentiation of Alveolar Epithelial Cells from Human Embryonic Stem Cell Derived Lung Organoids. Stem Cells Dev. 2018, 27, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; McCauley, K.B.; Kotton, D.N.; Hawkins, F. Differentiation of human airway-organoids from induced pluripotent stem cells (iPSCs). Methods Cell Biol. 2020, 159, 95–114. [Google Scholar] [PubMed]
- Kelleher, J.; Dickinson, A.; Cain, S.; Hu, Y.; Bates, N.; Harvey, A.; Ren, J.; Zhang, W.; Moreton, F.C.; Muir, K.W.; et al. Patient-Specific iPSC Model of a Genetic Vascular Dementia Syndrome Reveals Failure of Mural Cells to Stabilize Capillary Structures. Stem Cell Reports 2019, 13, 817–831. [Google Scholar] [CrossRef]
- Porotto, M.; Ferren, M.; Chen, Y.-W.; Siu, Y.; Makhsous, N.; Rima, B.; Briese, T.; Greninger, A.L.; Snoeck, H.-W.; Moscona, A. Authentic Modeling of Human Respiratory Virus Infection in Human Pluripotent Stem Cell-Derived Lung Organoids. MBio 2019, 10, e00723–e00719. [Google Scholar] [CrossRef]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kuhnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nature Communications 2018, 9. [Google Scholar] [CrossRef]
- Trump, L.R.; Nayak, R.C.; Singh, A.K.; Emberesh, S.; Wellendorf, A.M.; Lutzko, C.M.; Cancelas, J.A. : Neutrophils Derived from Genetically Modified Human Induced Pluripotent Stem Cells Circulate and Phagocytose Bacteria In Vivo. Stem Cells Translational Medicine 2019, 8, 557–567. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Characterisation of endodermal and mesodermal progenitors prior to co-culture. A. Diagram illustrating the entire lung organoids differentiation process. B-C, Gene expression analysis of iPSCs differentiated to AFE (B) and mesoderm (C) prior to co-culture. N=3, data displayed as mean ± SEM. Statistical significance determined by unpaired t-test, *** p < 0.001, **** p < 0.0001. D-E, Flow cytometry analysis of iPSCs differentiated to AFE (D) and mesoderm (E) prior to co-culture. N=3, data displayed as representative histograms with the mean ± SEM.
Figure 1.
Characterisation of endodermal and mesodermal progenitors prior to co-culture. A. Diagram illustrating the entire lung organoids differentiation process. B-C, Gene expression analysis of iPSCs differentiated to AFE (B) and mesoderm (C) prior to co-culture. N=3, data displayed as mean ± SEM. Statistical significance determined by unpaired t-test, *** p < 0.001, **** p < 0.0001. D-E, Flow cytometry analysis of iPSCs differentiated to AFE (D) and mesoderm (E) prior to co-culture. N=3, data displayed as representative histograms with the mean ± SEM.
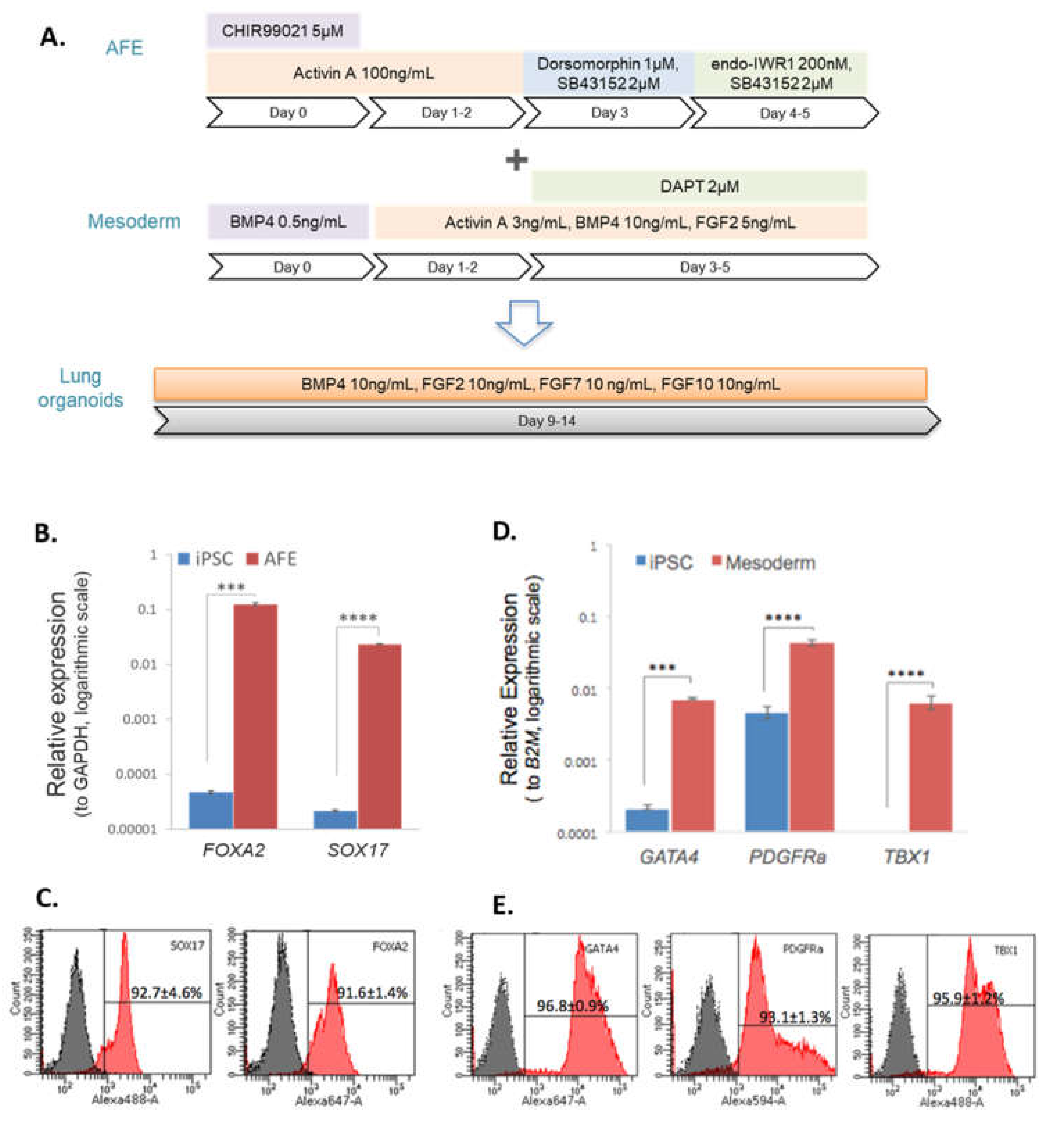
Figure 2.
Immunofluorescent staining of alveolar cell markers in iPSC derived pulmonary organoids. After 9 days of co-culture of AFE and mesoderm progenitors, the organoids were stained for alveolar epithelial type I and type II markers including AQP5, T1α SPA, SPB, SPC, and NKX2.1. A, representative confocal images of organoids derived from 3 different iPSC lines. Scale bars 100 µm. B, staining of the same markers in normal human lung sections. Scale bars 50 µm.
Figure 2.
Immunofluorescent staining of alveolar cell markers in iPSC derived pulmonary organoids. After 9 days of co-culture of AFE and mesoderm progenitors, the organoids were stained for alveolar epithelial type I and type II markers including AQP5, T1α SPA, SPB, SPC, and NKX2.1. A, representative confocal images of organoids derived from 3 different iPSC lines. Scale bars 100 µm. B, staining of the same markers in normal human lung sections. Scale bars 50 µm.

Figure 3.
Gene expression profiling of iPSC derived pulmonary organoids. Fourteen days into the pulmonary organoid generation following co-culturing of AFE and mesoderm progenitors, total RNA from the organoids and the undifferentiated iPSCs were analysed by RNAseq (A-E) and candidate genes were confirmed by RT-qPCR (F-J). A, PCA plot comparing RNAseq data between organoids differentiated from 2 independent iPSC lines and the undifferentiated iPSCs. B-E, Heatmap showing differentially expressed genes relating to lung development. F-I. RT-qPCR for expression of alveolar epithelial type I and type II marker genes including SPA, SPC, AQP5 and NKX2.1 from samples of 3 independent differentiations of 2 iPSC lines. Data are presented as mean ± SE, n=3. Statistical significance was determined by an unpaired t-test, **p<0.01, ****p<0.0001.
Figure 3.
Gene expression profiling of iPSC derived pulmonary organoids. Fourteen days into the pulmonary organoid generation following co-culturing of AFE and mesoderm progenitors, total RNA from the organoids and the undifferentiated iPSCs were analysed by RNAseq (A-E) and candidate genes were confirmed by RT-qPCR (F-J). A, PCA plot comparing RNAseq data between organoids differentiated from 2 independent iPSC lines and the undifferentiated iPSCs. B-E, Heatmap showing differentially expressed genes relating to lung development. F-I. RT-qPCR for expression of alveolar epithelial type I and type II marker genes including SPA, SPC, AQP5 and NKX2.1 from samples of 3 independent differentiations of 2 iPSC lines. Data are presented as mean ± SE, n=3. Statistical significance was determined by an unpaired t-test, **p<0.01, ****p<0.0001.
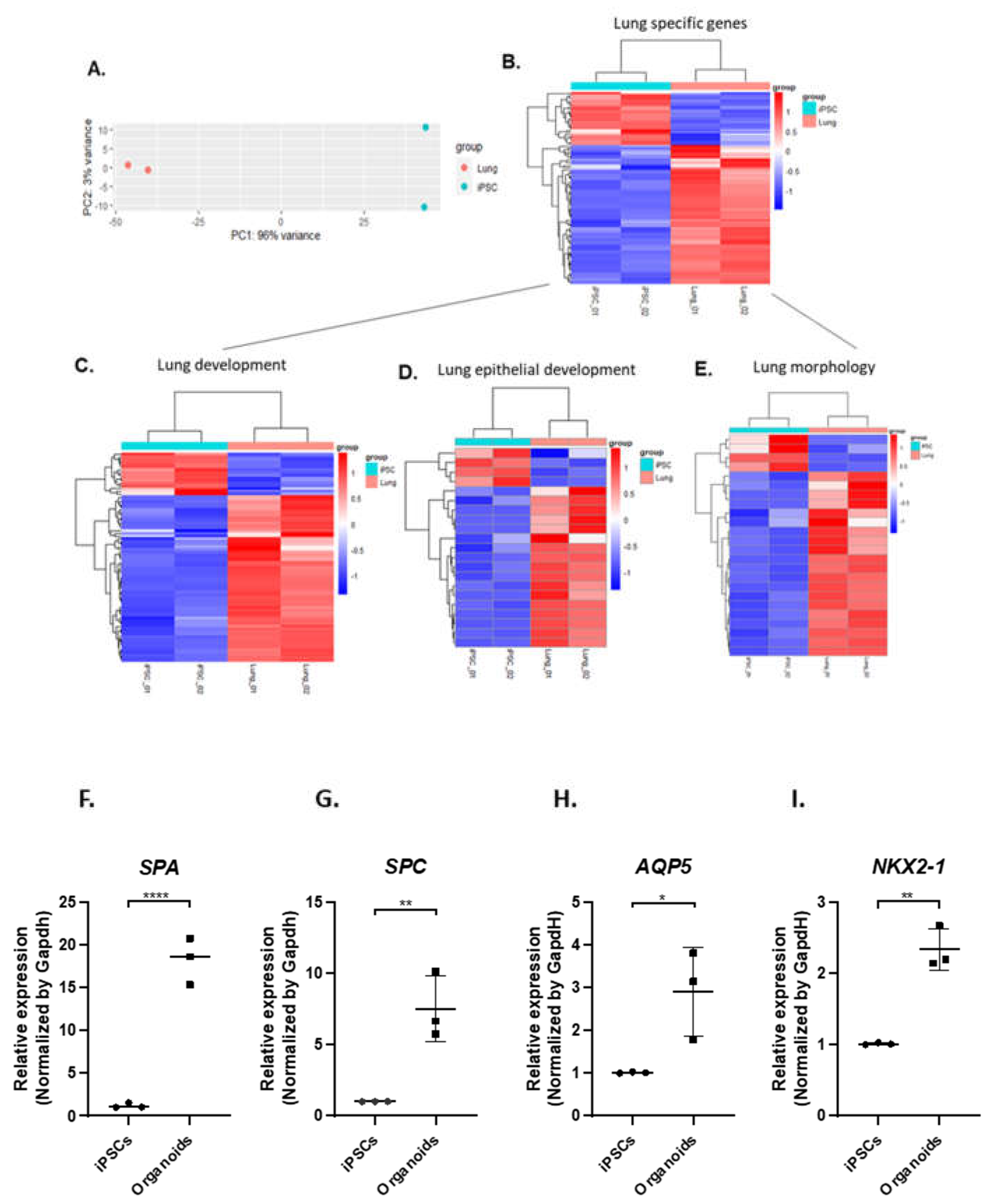
Figure 4.
IPSC derived pulmonary organoids express ACE2 capable of binding SARS-CoV2 spike protein. Experiments were carried out on day 14 pulmonary organoids post co-culture of AFE and mesoderm progenitors. A–D, Organoids were immunostained for ACE2, as well as the alveolar epithelial marker SPC. E–H, Organoids were incubated with recombinant SARS-CoV2 spike protein followed by immunofluorescent staining using antibodies specific to the SARS-CoV2 spike protein, as well as the alveolar epithelial marker SPC. Figures are representative images of organoids derived from 3 different iPSC lines. Scale bars 100 µm.
Figure 4.
IPSC derived pulmonary organoids express ACE2 capable of binding SARS-CoV2 spike protein. Experiments were carried out on day 14 pulmonary organoids post co-culture of AFE and mesoderm progenitors. A–D, Organoids were immunostained for ACE2, as well as the alveolar epithelial marker SPC. E–H, Organoids were incubated with recombinant SARS-CoV2 spike protein followed by immunofluorescent staining using antibodies specific to the SARS-CoV2 spike protein, as well as the alveolar epithelial marker SPC. Figures are representative images of organoids derived from 3 different iPSC lines. Scale bars 100 µm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



