
Submitted:
14 April 2023
Posted:
17 April 2023
You are already at the latest version
Abstract
Zika virus (ZIKV) is a mosquito-borne flavivirus and causes an infection that is associated with neurological manifestations, including microcephaly and Guillain-Barre syndrome. The mechanism of ZIKV-mediated neuropathogenesis is not well understood. In this study, we discovered that ZIKV induces the degradation of the Numb protein, which plays a crucial role in neurogenesis by allowing asymmetric cell division during embryonic development. Our data show that ZIKV reduced the Numb protein level in a time- and dose-dependent manner. However, ZIKV infection appears to have minimal effect on the Numb transcript. Treatment of ZIKV-infected cells with a proteasome inhibitor restores the Numb protein level, which suggests the involvement of the ubiquitin-proteasome pathway. In addition, ZIKV infection shortens the half-life of the Numb protein. Among the ZIKV proteins, the capsid protein significantly reduces the Numb protein level. Immunoprecipitation of the Numb protein co-precipitates the capsid protein, indicating the interaction between these two proteins. These results provide insights into the ZIKV-cell interaction that might contribute to its impact on neurogenesis.
Keywords:
Zika virus
; ZIKV
; the Numb protein
; the Capsid protein
1. Introduction
Zika virus (ZIKV) is an RNA virus of the genus Flavivirus, the Flaviviridae. ZIKV was first isolated in 1947 from a rhesus monkey in Africa [1,2]. The virus did not draw much attention until the first documented human outbreak in the Pacific islands in the late 2000s and the later epidemic in South America in 2015-2016 [3,4,5,6]. It is a mosquito-borne flavivirus associated with severe manifestations, including congenital Zika syndrome and Guillain-Barre syndrome (GBS) [6,7,8,9,10,11]. ZIKV infects neural precursor cells derived from pluripotent stem cells and causes apoptotic cell death and cell-cycle dysregulation [12,13,14]. ZIKV strains are phylogenetically grouped into African lineage and Asian lineage. An Asian lineage ZIKV strain infects embryonic mouse brains and causes microcephaly [15,16]. ZIKV is enveloped and has a positive-sense, single-stranded RNA genome of 10.7 kb in length. It has the common feature of Flavivirus that the genome encodes a single polyprotein that is cleaved into three structural proteins (capsid (C), precursor membrane (prM), and envelope (E)) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) [17]. Neither an effective treatment nor a vaccine is available to control and prevent ZIKV infection [18].
The Numb gene was initially discovered in Drosophila for its determination of cell fate in sensory neuron formation [19]. The function of the Numb protein in cell fate determination is best studied in Drosophila [20]. During neurogenesis, the Numb protein localizes to one side of the progenitor cell and selectively segregates into one daughter cell. The asymmetric division leads to one daughter cell generally differentiating into a neuron cell and the other becoming a progenitor for further proliferation. Its homolog in humans is encoded by the Numb gene, which is well-conserved from invertebrates to mammals. Knockout of the Numb gene is lethal to the mouse at the early embryo stage [21].
To date, ZIKV-cell interactions in the aspect of neuropathogenesis are not well understood. This study aimed to determine the ZIKV effect on Numb expression. We noted the Numb protein was decreased significantly in ZIKV-infected cells in a time and dose-dependent manner. Further study was conducted to examine the mechanism of the ZIKV-mediated reduction of the Numb protein. Our results provide insights into the ZIKV-cell interactions that may contribute to a better understanding of ZIKV-induced neuropathogenesis.
2. Materials and Methods
2.1. Viruses, cells, and chemicals
ZIKV PRVABC59 strain (ATCC VR-1843; GenBank Accession Number KX377337) and ZIKV MR766 strain (ATCC VR-1838; GenBank Accession Number NC012532.1) were used to infect Vero cells. Virus titers were determined in Vero cells by 10-fold serial dilutions and shown as the median tissue culture infectious dose (TCID50) as Log10/ml [52]. The multiplicity of infection was indicated in figure legends or results.
HEK293 (ATCC CRL-1573), HeLa (ATCC CCL-2), Vero (ATCC CCL-81), and SK-N-SH (ATCC HTB-11) cells were cultured in Dulbecco Modified Eagle Medium (DMEM; Corning Life Sciences, 10-013-CV) supplemented with 10% fetal bovine serum (Tissue Culture Biologicals, 101) at 37°C and 5% CO2.
MG132 (Enzo Life Sciences, BML-PI102-0025), a proteasome inhibitor, was used to treat cells at the final concentration of 10 µM for 6 hours before harvesting. A lysosome inhibitor, NH4Cl, was used to treat cells at a final concentration of 10 mM for 6 h prior to harvesting. A protein translation inhibitor, Cycloheximide (Fisher Scientific, AC357420010), was used at a final concentration of 50 μg/ml to ZIKV-infected and mock-infected cells to determine the half-life of Numb.
CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega, G7570) was used to determine cell viability by following the manufacturer's instructions.
2.2. Plasmids
The cloning of ZIKV genes for individual proteins was previously described [25]. The numb plasmid was constructed by cloning Numb into pCAGEN-Myc vector after PCR amplification of Numb with primers Numb-F1 and Numb-R1 (Table 1) on cDNA prepared from total RNA of Vero cells.
For shRNA against Numb, the oligos for the shRNA were cloned into the pSIREN-RetroQ-ZsGreen vector (Clontech, 632455) following the manufacturer's instruction. GP2-293 cells (Clontech, 631458) were used for retrovirus packaging, pSIREN-RetroQ-ZsGreen-shNumb and VSV-G vector (a gift from Akitsu Hotta (Addgene plasmid # 138479 ; http://n2t.net/addgene:138479 ; RRID:Addgene_138479) [53]) plasmids were co-transfected into GP2-293 cells. The preparation of control shRNA (C-shRNA) plasmid was described previously [25]. The culture supernatant containing the recombinant retrovirus was used to transduce Vero cells.
2.3. RNA isolation and real-time PCR
TRIzol™ reagent (Thermo Fisher Scientific, 15596026) was used for extracting total RNA from Vero cells following the manufacturer's instructions. Reverse transcription and real-time PCR (RT-qPCR) with SYBR Green detection (Thermo Fisher Scientific, 4334973) were performed as described previously [54,55,56]. The transcripts of RPL32 (ribosomal protein L32), a housekeeping gene, were also determined. The relative transcript levels were shown as folds compared to the control cells after RPL32 normalization. The real-time PCR primers used for Numb and ZIKV were Numb-RR-F1, Numb-RR-R1, ZIKV-RR-F, and ZIKV-RR-R (Table 1). All experiments were repeated at least three times, each conducted in triplicate.
2.4. Western blot (WB) analysis
The Laemmli sample buffer was used in cell lysis to prepare whole cell lysates for SDS-PAGE and WB as described previously [25,56,57]. The primary antibodies against Numb (Santa Cruz Biotechnology, sc-136554), Myc tag (Santa Cruz Biotechnology, sc- 40), GFP (Biolegend, 75818-584), ubiquitin (Santa Cruz Biotechnology, sc-8017), GAPDH (Santa Cruz Biotechnology, sc-365062), TUBB1/β-tubulin (Sigma, T7816), ZIKV C (GeneTex, GTX133317), ZIKV E (B.E.I. Resources, NR-50413), ZIKV NS4B (GeneTex, GTX133311), and ZIKV NS5 (GeneTex, GTX133329) were used in this study. Goat anti-mouse or anti-rabbit IgG conjugated with horseradish peroxidase (Bio-Rad, 170-5046 and 170-5047) were used as secondary antibodies in this study. For revealing the specific reactions, a chemiluminescence substrate was used, and the signal was recorded digitally using a ChemiDoc X.R.S. imaging system with the QuantityOne Program, version 4.6 (Bio-Rad Laboratories, Hercules, CA). Densitometry analysis of the WB images was done with the QuantityOne Program, version 4.6 (Bio-Rad). All WB images were acquired in the linear range of digital intensity without saturated pixels.
2.5. Immunoprecipitation (IP)
Immunoprecipitation was done as described previously [25]. Cell lysates were clarified and incubated with specific antibodies indicated in results or figure legends, followed by incubation with protein A/G-magnetic beads (Bimake.com, B23202). The IP complexes were subjected to WB for the detection of target proteins indicated in results or figure legends.
2.6. Statistical analysis
Differences in gene expression between the treatment group and control were assessed by using the Student t-test in GraphPad Prism 5. A two-tailed P value of 0.05 was considered significant.
3. Results
3.1. ZIKV infection reduces the Numb protein level.
Vero cells were infected with the ZIKV PRVABC59 strain (Asian lineage) at a multiplicity of infection (MOI) of 10. Western blotting was performed to detect the Numb protein level. Results showed that the Numb protein level decreased by 90% in the ZIKV-infected cells at 40 hours post-infection (hpi) compared to the mock-infected cells (Figure 1A). To investigate if the African lineage ZIKV has a similar effect on the Numb protein level, we infected Vero cells with ZIKV MR766 strain. MR766 strain infection reduced the Numb protein level by 70% at 40 hpi (Figure 1B).
The results above showed Numb protein reduction in ZIKV-infected Vero cells. To exclude the possibility that Numb reduction is cell-line specific, we determined Numb protein levels in other cell lines, including HeLa cells and neuroblastoma SK-N-SH cells. The latter cell line is more physiologically relevant to ZIKV brain infection and its associated neurological disorders. Results showed the PR strain also reduced Numb in both HeLa cells and SK-N-SH cells (Figure 1C and 1D). These results demonstrate that ZIKV infection reduces the Numb protein level.
3.2. ZIKV reduces the Numb protein level in a temporal and dose-dependent manner.
Since ZIKV infection led to Numb protein reduction, we reasoned that the Numb protein reduction would be in a temporal manner. To test this, we infected Vero cells with PR strain at an MOI of 1 and harvested the cells for WB. The results showed ZIKV reduced the Numb protein level at 20 and 40 hpi by 30% and 80%, respectively, in comparison with mock-infected control (Figure 2A). In addition, Vero cells were infected with ZIKV PR strain at the MOIs of 0.1, 1, 5, and 10 and harvested for WB 24 hpi. The results showed that the Numb protein decreased further along with the incremental MOI (Figure 2B). ZIKV NS4B protein was detected (Figure 2B), and ZIKV RNA level was determined with reverse transcription and real-time PCR (RT-qPCR) (Figure 2C), which confirmed the increase of ZIKV replication along with the incremental inoculum.
3.3. ZIKV reduces the Numb protein via the ubiquitin-proteasome pathway.
The reason for a protein reduction in cells could be due to lower transcription, translation, or protein stability. For the reduction of the Numb protein level, we wondered if ZIKV had any effect on the Numb transcript. Total RNA was isolated from ZIKV-infected cells, and Numb mRNA was determined by RT-qPCR. The results showed that ZIKV PR infection had minimal effect on the Numb mRNA level in comparison with the mock-infected cells (Figure 3A). ZIKV RNA level was determined to confirm the infection (Figure 3B). These results indicate that the Numb protein reduction caused by ZIKV is not due to inhibition of transcription. There are two primary organelles involved in protein degradation in cells: the proteasomes and the lysosomes [22,23].
Next, we intended to determine which of the two organelles was responsible for the Numb protein degradation using two chemicals: MG132 and ammonium chloride (NH4Cl). MG132 is a proteasome inhibitor that blocks the degradation of ubiquitin-conjugated proteins in mammalian cells. NH4Cl is a lysosomotropic compound that raises the lysosomal pH to block proteolysis in the lysosomes [24]. MG132 or NH4Cl was added to the cells at 30 hpi, and the cells were harvested 6 h later for Western blotting. The treatment with MG132 resulted in the restoration of the Numb protein level in the ZIKV-infected Vero cells, while NH4Cl failed to do so (Figure 3C). The result suggests that ZIKV-mediated Numb reduction is via the ubiquitin-proteasome pathway instead of the lysosomal proteolysis. We then determined the Numb protein half-life in the ZIKV-infected cells, as degradation led to the Numb protein reduction. Cycloheximide, an inhibitor of protein translation, was added to both mock-infected and ZIKV-infected Vero cells. MG132 was used to treat the infected cells at 24 hpi for 6 h before the addition of cycloheximide to shore up the Numb protein level for this test. The cells were harvested at 0, 6, 12, 24, and 36 h after the cycloheximide treatment for WB. The result showed that the Numb protein level was reduced much faster in the infected cells than mock-infected cells and that densitometry analysis indicates ZIKV infection shortened the Numb half-life from 36 h to 12 h (Figure 3D). This result suggests that ZIKV reduces Numb protein stability by accelerating its degradation.
The results above showed the Numb protein reduction caused by ZIKV infection, possibly via the ubiquitin-proteasome pathway. We reasoned that the ZIKV infection would induce the Numb protein polyubiquitination, which is needed for target protein degradation via this pathway. To confirm this speculation, we conducted immunoprecipitation of the Numb protein, followed by WB with antibodies against ubiquitin and Numb. Results showed that the Numb ubiquitination level in the ZIKV-infected cells was significantly elevated by 2.75-fold in comparison to the mock-infected cells (Figure 3E). WB of input of the cell extract showed a similar total ubiquitination level between the infected and mock-infected cells. Together, these results indicate that ZIKV infection induced Numb polyubiquitination and degradation via the proteasomes.
3.4. ZIKV capsid protein induces the Numb reduction.
ZIKV genome encodes a single polyprotein that is produced and cleaved co-translationally into three structural proteins (C, prM, and E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) [17]. To determine which of these ZIKV proteins induces the Numb protein reduction, we transfected HEK293 cells with plasmids encoding the individual proteins of ZIKV [25] and a plasmid encoding the Numb protein. WB results showed that compared with the empty vector control, the ZIKV C protein induced a significant reduction of the Numb protein level by 99.5%, while the other proteins had much less or minimal effects (Figure 4A). So we selected the C protein to further study it.
Since ZIKV C induces the reduction of Numb, we speculated that the two proteins might have an interaction. To test the speculation, we infected Vero cells with ZIKV and conducted IP with the antibody against the Numb protein. The WB results showed that the Numb IP co-precipitated the ZIKV C protein (Figure 4B). To exclude the possibility of other viral proteins involved in the interaction, we transfected HEK293 cells with the plasmids encoding the C and the Numb protein and harvested the cells 24 h later for co-IP and WB The results showed that the ZIKV C protein was present in the Numb co-IP precipitates (Figure 4C). These results indicate that the ZIKV C protein induces the Numb reduction and interacts with the Numb protein.
3.5. The Numb knockdown has minimal effect on ZIKV replication.
The results above showed ZIKV-induced Numb reduction via the ubiquitin-proteasome pathway. We wondered if the Numb protein had any effect on ZIKV replication. To explore the role of the Numb protein in ZIKV proliferation, we conducted RNAi-mediated knockdown of Numb in Vero cells using recombinant retrovirus expressing shRNA against Numb. A control shRNA (C-shRNA) of an irrelevant sequence was included in the study. The Numb protein level in the cells with the shNumb was significantly reduced in comparison to the cells with the C-shRNA (Figure 5A). ZIKV infection further reduced the Numb protein level. To determine the ZIKV yield in the cells with the C-shRNA or shNumb, we collected cell culture supernatant samples at 24, 48, 72, 96, and 120 hpi for virus titration. The results showed that the ZIKV viral yields in the cells with the Numb knockdown were similar to the cells with the C-shRNA (Figure 5B). The Numb knockdown had minimal effect on the cell growth (Figure 5C). These results showed that the Numb knockdown had minimal effect on ZIKV replication in Vero cells.
4. Discussion
Our data demonstrate that ZIKV infection reduces the Numb protein level via the ubiquitin-proteasome pathway and that the ZIKV capsid protein induces Numb degradation. Intriguingly, the Numb knockdown has minimal effect on ZIKV replication, which suggests that the ZIKV-mediated Numb reduction is probably related to ZIKV pathogenesis, especially considering the Numb's role in embryonic neurogenesis.
To determine the ZIKV effect on Numb expression, we used three cell lines and two strains of the virus to confirm the virus-mediated reduction. The Asian-lineage PR strain appears to induce more and quicker Numb reduction than the African-lineage MR766 strain, which is potentially consistent with the clinical manifestation that the Asian-lineage strains are associated with neurological manifestations. The cell lines tested include SK-N-SH, derived from neuroblastoma cells, which is more physiologically relevant to ZIKV infection of neural cells. Our results show that ZIKV infection reduces the Numb protein in SK-N-SH cells at 72 hpi, which may be due to the slower cell growth than Vero and HeLa cells and subsequently lower ZIKV replication in this cell line. The ZIKV-mediated reduction of Numb is shown to be temporal and dose-dependent, which indicates the virus-specific effect.
ZIKV induces Numb degradation via the ubiquitin-proteasome pathway, shown by the restoration by MG132 but not NH4Cl treatment. This test eliminates the potential involvement of the lysosomes as MG132 also targets certain hydrolases in the lysosomes, whereas NH4Cl only targets this organelle [24]. We further showed the half-life reduction of the Numb protein and its elevated polyubiquitination by ZIKV infection, which provides further evidence that ZIKV reduces Numb via the ubiquitin-proteasome pathway. Our data shows the Numb half-life is 36 hours in Vero cells, which is much longer than 10 hours in C2C12 myoblasts [26]. This discrepancy is possibly caused by the different cell types, as C2C12 myoblasts are stem cells that differentiate into muscular cells, while Vero cells are derived from the kidney epithelial cells of an African green monkey.
Our screening of ZIKV proteins showed that the C protein is the primary one responsible for Numb reduction. We noted that NS2B reduced the Numb protein level by 50%, suggesting it may play a minor role in the Numb reduction. Intriguingly, NS2B3, NS4A, NS4B, and NS5 could induce an increase in the Numb level in transient expression. This observation does not corroborate with the results of the whole virus infection, and we did not pursue it further. Multiple bands were observed for several ZIKV proteins, including prM, NS3, and NS4A, in the transiently transfected cells, which suggests that there might have post-translational cleavage. In determining the mechanism of the C-mediated Numb reduction mechanism, we noted that Numb IP could precipitate the C protein in both ZIKV-infected and transiently co-transfected cells. This result suggests that the Numb protein interacts with the C protein in the absence of other viral proteins. The ZIKV C protein is known to bind the viral genomic RNA and is involved in virion assembly [27]. Besides virion assembly, the C protein is also known to interact with cellular proteins to play roles in assisting ZIKV replication, modulating cellular metabolism, and antiviral response. The C protein induces the loss of peroxisomes, which have an important role in innate immunity [28]. It targets the nonsense-mediated mRNA decay (NMD) pathway, a cellular mRNA surveillance mechanism, via interacting with up-frameshift protein 1 (UPF1), a central NMD regulator, and targeting it for degradation [29]. The disruption of the NMD pathway may contribute to neuropathology. In a mosquito cell line stably expressing C, 157 interactors were identified, and eight have proviral activity during ZIKV infection in the cells, including transitional endoplasmic reticulum protein TER94 [30]. ZIKV inhibits the formation of stress granules (SG), and C interacts with SG components G3BP1 and Caprin-1 [31]. The ZIKV C, but not other flaviviruses, antagonizes endoribonuclease Dicer and consequently inhibits miRNA biogenesis in neural stem cells, which leads to the disruption of corticogenesis [32]. Mutant C (H41R) loses interaction with Dicer, and ZIKV-H41R does not inhibit neurogenesis. These data demonstrate that the C protein plays an important role in ZIKV replication and invasion/pathogenesis. Our finding of C-mediated Numb reduction further contributes to the literature on the functions of this essential protein.
The effect of the Numb protein on virus infection is rarely studied and appears to be virus dependent. The Numb protein is needed for hepatitis C virus entry as RNAi-mediated knockdown of the Numb inhibits the virus entry [33]. However, Numb inhibition activates Notch signaling, which assists the transcription of covalently closed circular DNA (cccDNA) of the hepatitis B virus [34]. Our results of RNAi-mediated knockdown of Numb demonstrated that the Numb protein has no effect on ZIKV replication or cell growth of Vero cells. Taken together, our data suggest that ZIKV-mediated Numb reduction might be relevant to its neuropathogenesis instead of viral replication. This is possible because Vero cells are differentiated and Numb appears not to contribute to the machinery needed for ZIKV replication. However, we cannot exclude the possibility that Numb might be needed for ZIKV replication in neural stem cells because Numb reduction affects the fate of neural stem cell differentiation. So, if the total number of differentiated cells is reduced due to Numb downregulation, ZIKV proliferation might be affected consequently.
Numb functions in cell fate determination by antagonizing several developmental pathways, including Notch, Hedgehog, and WNT signaling, which are essential for regulation in cell proliferation, differentiation, cell fate determination, and self-renewal of stem cells and progenitor cells during embryonic development and in adult organs [35,36,37]. Numb inhibits Notch signaling via interaction with the Notch E3 ligase Itch and the Notch intracellular domain (NICD), leading to the degradation of NICD [20,35,38]. Numb suppresses the Hedgehog signaling pathway, a master regulator of embryonic development [39], by targeting the Hedgehog Gli1 transcription factor for Itch-dependent degradation [40]. Numb also inhibits the WNT pathway, another regulator of embryonic development, by promoting the degradation of β-catenin [41] and enhances p53 tumor suppressor activity by interacting with its E3 ligase MDM2 to block p53 degradation [42,43]. For these functions, Numb has been characterized as a tumor suppressor [20,44,45]. Indeed, a lower level of Numb correlates with a worse prognosis for several types of cancers.
Numb is finely regulated at transcriptional, translational, and post-translational levels. There are six functionally distinct isoforms of Numb ranging from 54-72 kDa in mammals due to alternative splicing of Numb mRNA [46]. The Numb translation is suppressed by Musashi-1, an RNA-binding protein abundant in neural progenitor cells [47]. MicroRNA miR-146a inhibits Numb expression to regulate the proliferation and differentiation of muscle stem cells [48]. The Numb protein level is controlled by ubiquitin-dependent proteolytic degradation that is mediated by E3 ligase L.N.X. [49], MDM2 [43], and Siah-1 [50]. We assume that C potentially interacts with one of these E3 ligases and enhances its activity in Numb polyubiquitination. Indeed, a bioinformatic analysis of the ZIKV-host protein interaction network implies that the C protein interacts with MDM2 [51]. The mechanistic details of the ZIKV C-mediated Numb reduction will be examined in future studies.
In conclusion, our results demonstrate that ZIKV induces Numb reduction via the ubiquitin-proteasome pathway and that the C protein is responsible for the downregulation. The ZIKV-mediated reduction of the Numb protein is expected to interfere with its functions, which is potentially relevant to ZIKV neuropathogenesis. This study provides insight into ZIKV-cell interactions and may contribute to our better understanding of the molecular mechanism of ZIKV-induced neuropathogenesis.
Author Contributions
J.H. and Y.Z. contributed to the study design. J.H., L.Y., P.C., S.Y., Y.W., S.L., Q.T., and Y.Z. were involved in data acquisition and analysis and drafting of the manuscript. All authors reviewed the manuscript.
Funding
This study was partially funded by a seed grant from the University of Maryland.
Acknowledgments
The following reagent was obtained through BEI Resources, NIAID, N.I.H.: Monoclonal Anti-Zika Virus Envelope (E) Protein, Clone ZV-16 (immunoglobulin G, Mouse), NR-50413.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans R Soc Trop Med Hyg 1952, 46, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika Virus Outbreak, Bahia, Brazil. Emerg Infect Dis 2015, 21, 1885–1886. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.; Roy-Burman, A.; Tuholske, C.; Busch, M.P.; Bakkour, S.; Stone, M.; Linnen, J.M.; Gao, K.; Coleman, J.; Bloch, E.M. Real-Time Evolution of Zika Virus Disease Outbreak, Roatan, Honduras. Emerg Infect Dis 2017, 23, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Soto, L.A. Zika Virus in the Americas: An Environmental Health Perspective. P R Health Sci J 2018, 37, S5–S14. [Google Scholar] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N Engl J Med 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Panchaud, A.; Stojanov, M.; Ammerdorffer, A.; Vouga, M.; Baud, D. Emerging Role of Zika Virus in Adverse Fetal and Neonatal Outcomes. Clin Microbiol Rev 2016, 29, 659–694. [Google Scholar] [CrossRef]
- Plourde, A.R.; Bloch, E.M. A Literature Review of Zika Virus. Emerg Infect Dis 2016, 22, 1185–1192. [Google Scholar] [CrossRef]
- Raposo-Amaral, C.E. Microcephaly: Consequence of the Zika Virus Global Outbreak. J Craniofac Surg 2016, 27, 1383–1384. [Google Scholar] [CrossRef]
- Crisanto-Lopez, I.E.; Jesus, P.L.; Lopez-Quecho, J.; Flores-Alonso, J.C. Congenital Zika syndrome. Bol Med Hosp Infant Mex 2023, 80, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Herrlinger, S.; Yang, S.L.; Lai, F.; Moore, J.M.; Brindley, M.A.; Chen, J.F. Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 2016, 143, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.S.; Sampaio, G.L.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep 2016, 6, 39775. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol 2016, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Abraham, R.; Shim, B.S.; Choe, H.; Page, D.T. Zika virus infection during the period of maximal brain growth causes microcephaly and corticospinal neuron apoptosis in wild type mice. Sci Rep 2016, 6, 34793. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J Virol 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ling, L.; Zhang, Z.; Marin-Lopez, A. Current Advances in Zika Vaccine Development. Vaccines (Basel) 2022, 10. [Google Scholar] [CrossRef]
- Uemura, T.; Shepherd, S.; Ackerman, L.; Jan, L.Y.; Jan, Y.N. numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell 1989, 58, 349–360. [Google Scholar] [CrossRef]
- Flores, A.N.; McDermott, N.; Meunier, A.; Marignol, L. NUMB inhibition of NOTCH signalling as a therapeutic target in prostate cancer. Nat Rev Urol 2014, 11, 499–507. [Google Scholar] [CrossRef]
- Zilian, O.; Saner, C.; Hagedorn, L.; Lee, H.Y.; Sauberli, E.; Suter, U.; Sommer, L.; Aguet, M. Multiple roles of mouse Numb in tuning developmental cell fates. Curr Biol 2001, 11, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Hewitt, E.W. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem 2016, 60, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yang, L.; Chang, P.; Yang, S.; Lin, S.; Tang, Q.; Wang, X.; Zhang, Y.J. Zika virus NS2A protein induces the degradation of KPNA2 (karyopherin subunit alpha 2) via chaperone-mediated autophagy. Autophagy 2020, 16, 2238–2251. [Google Scholar] [CrossRef]
- Liu, X.H.; Yao, S.; Levine, A.C.; Kirschenbaum, A.; Pan, J.; Wu, Y.; Qin, W.; Collier, L.; Bauman, W.A.; Cardozo, C.P. Nandrolone, an anabolic steroid, stabilizes Numb protein through inhibition of mdm2 in C2C12 myoblasts. J Androl 2012, 33, 1216–1223. [Google Scholar] [CrossRef]
- Tan, T.Y.; Fibriansah, G.; Kostyuchenko, V.A.; Ng, T.S.; Lim, X.X.; Zhang, S.; Lim, X.N.; Wang, J.; Shi, J.; Morais, M.C.; et al. Capsid protein structure in Zika virus reveals the flavivirus assembly process. Nat Commun 2020, 11, 895. [Google Scholar] [CrossRef]
- Farelo, M.A.; Korrou-Karava, D.; Brooks, K.F.; Russell, T.A.; Maringer, K.; Mayerhofer, P.U. Dengue and Zika Virus Capsid Proteins Contain a Common PEX19-Binding Motif. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The Cellular N.M.D. Pathway Restricts Zika Virus Infection and Is Targeted by the Viral Capsid Protein. MBio 2018, 9. [Google Scholar] [CrossRef]
- Gestuveo, R.J.; Royle, J.; Donald, C.L.; Lamont, D.J.; Hutchinson, E.C.; Merits, A.; Kohl, A.; Varjak, M. Analysis of Zika virus capsid-Aedes aegypti mosquito interactome reveals proviral host factors critical for establishing infection. Nat Commun 2021, 12, 2766. [Google Scholar] [CrossRef]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Gotte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J Virol 2017, 91. [Google Scholar] [CrossRef]
- Zeng, J.; Dong, S.; Luo, Z.; Xie, X.; Fu, B.; Li, P.; Liu, C.; Yang, X.; Chen, Y.; Wang, X.; et al. The Zika Virus Capsid Disrupts Corticogenesis by Suppressing Dicer Activity and miRNA Biogenesis. Cell Stem Cell 2020, 27, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Neveu, G.; Ziv-Av, A.; Barouch-Bentov, R.; Berkerman, E.; Mulholland, J.; Einav, S. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol 2015, 89, 4387–4404. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kawaguchi, K.; Honda, M.; Hashimoto, S.; Shirasaki, T.; Okada, H.; Orita, N.; Shimakami, T.; Yamashita, T.; Sakai, Y.; et al. Notch signaling facilitates hepatitis B virus covalently closed circular DNA transcription via cAMP response element-binding protein with E3 ubiquitin ligase-modulation. Sci Rep 2019, 9, 1621. [Google Scholar] [CrossRef] [PubMed]
- Frise, E.; Knoblich, J.A.; Younger-Shepherd, S.; Jan, L.Y.; Jan, Y.N. The Drosophila Numb protein inhibits signaling of the Notch receptor during cell-cell interaction in sensory organ lineage. Proc Natl Acad Sci U S A 1996, 93, 11925–11932. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Feder, J.N.; Jiang, M.M.; Jan, L.Y.; Jan, Y.N. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron 1996, 17, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat Rev Mol Cell Biol 2016, 17, 722–735. [Google Scholar] [CrossRef]
- McGill, M.A.; McGlade, C.J. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem 2003, 278, 23196–23203. [Google Scholar] [CrossRef]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev Cell 2008, 15, 801–812. [Google Scholar] [CrossRef]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat Cell Biol 2006, 8, 1415–1423. [Google Scholar] [CrossRef]
- Hwang, W.L.; Jiang, J.K.; Yang, S.H.; Huang, T.S.; Lan, H.Y.; Teng, H.W.; Yang, C.Y.; Tsai, Y.P.; Lin, C.H.; Wang, H.W.; et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat Cell Biol 2014, 16, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008, 451, 76–80. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Shifman, O.; Unger, T.; Elkeles, A.; Haupt, Y.; Oren, M. The Mdm2 oncoprotein interacts with the cell fate regulator Numb. Mol Cell Biol 1998, 18, 3974–3982. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 2004, 167, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Dong, M.; Chen, C.; Li, Y.; Liu, Q.; Dong, Q. Musashi2 promotes the development and progression of pancreatic cancer by down-regulating Numb protein. Oncotarget 2017, 8, 14359–14373. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Seok, J.; Kang, G.H.; Lim, K.M.; Cho, S.G. The role of NUMB/NUMB isoforms in cancer stem cells. BMB Rep 2021, 54, 335–343. [Google Scholar] [CrossRef]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol Cell Biol 2001, 21, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.; Tan, J.; Duan, Y.; Duan, J.; Wang, W.; Jin, F.; Jin, Z.; Yuan, X.; Liu, Y. Cyclic stretch induced miR-146a upregulation delays C2C12 myogenic differentiation through inhibition of Numb. Biochem Biophys Res Commun 2009, 378, 259–263. [Google Scholar] [CrossRef]
- Nie, J.; McGill, M.A.; Dermer, M.; Dho, S.E.; Wolting, C.D.; McGlade, C.J. L.N.X. functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO J 2002, 21, 93–102. [Google Scholar] [CrossRef]
- Susini, L.; Passer, B.J.; Amzallag-Elbaz, N.; Juven-Gershon, T.; Prieur, S.; Privat, N.; Tuynder, M.; Gendron, M.C.; Israel, A.; Amson, R.; et al. Siah-1 binds and regulates the function of Numb. Proc Natl Acad Sci U S A 2001, 98, 15067–15072. [Google Scholar] [CrossRef]
- Teng, Y.; Liu, S.; Guo, X.; Liu, S.; Jin, Y.; He, T.; Bi, D.; Zhang, P.; Lin, B.; An, X.; et al. An Integrative Analysis Reveals a Central Role of P53 Activation via MDM2 in Zika Virus Infection Induced Cell Death. Front Cell Infect Microbiol 2017, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Stein, D.A.; Fan, S.M.; Wang, K.Y.; Kroeker, A.D.; Meng, X.J.; Iversen, P.L.; Matson, D.O. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet Microbiol 2006, 117, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun 2020, 11, 1334. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, R.; Ma, Z.; Xiao, Y.; Nan, Y.; Wang, Y.; Lin, S.; Zhang, Y.J. Porcine Reproductive and Respiratory Syndrome Virus Antagonizes JAK/STAT3 Signaling via nsp5, Which Induces STAT3 Degradation. J Virol 2017, 91, e02087–02016. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Opriessnig, T.; Stein, D.A.; Halbur, P.G.; Meng, X.J.; Iversen, P.L.; Zhang, Y.J. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res 2008, 77, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, R.; Yang, S.; Ma, Z.; Lin, S.; Nan, Y.; Li, Q.; Tang, Q.; Zhang, Y.J. Karyopherin Alpha 6 Is Required for Replication of Porcine Reproductive and Respiratory Syndrome Virus and Zika Virus. J Virol 2018, 92, e00072–00018. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Wang, K.Y.; Stein, D.A.; Patel, D.; Watkins, R.; Moulton, H.M.; Iversen, P.L.; Matson, D.O. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res 2007, 73, 12–23. [Google Scholar] [CrossRef]
Figure 1.
ZIKV infection reduces Numb protein level. A. Infection of Vero cells with ZIKV PRVABC59 (PR) strain leads to lower Numb protein level. The cells were inoculated with an MOI of 10 of the ZIKV PR strain and harvested for Western blotting 40 hours post-infection (hpi). Relative levels of Numb are shown below the images after normalization with tubulin. B. Infection with ZIKV MR766 strain also induces reduction of Numb protein level. Vero cells were infected at an MOI of 10 and harvested at 40 hpi for WB. Relative levels of the Numb protein are shown below the images after normalization with GAPDH. C. Infection of HeLa cells with ZIKV PR strain reduces the Numb protein. The cells were inoculated at an MOI of 10 and harvested for WB 36 hpi. D. Numb protein level decreased in SK-N-SH cells with ZIKV infection. The cells were inoculated at an MOI of 10 and harvested for WB 72 hpi.
Figure 1.
ZIKV infection reduces Numb protein level. A. Infection of Vero cells with ZIKV PRVABC59 (PR) strain leads to lower Numb protein level. The cells were inoculated with an MOI of 10 of the ZIKV PR strain and harvested for Western blotting 40 hours post-infection (hpi). Relative levels of Numb are shown below the images after normalization with tubulin. B. Infection with ZIKV MR766 strain also induces reduction of Numb protein level. Vero cells were infected at an MOI of 10 and harvested at 40 hpi for WB. Relative levels of the Numb protein are shown below the images after normalization with GAPDH. C. Infection of HeLa cells with ZIKV PR strain reduces the Numb protein. The cells were inoculated at an MOI of 10 and harvested for WB 36 hpi. D. Numb protein level decreased in SK-N-SH cells with ZIKV infection. The cells were inoculated at an MOI of 10 and harvested for WB 72 hpi.
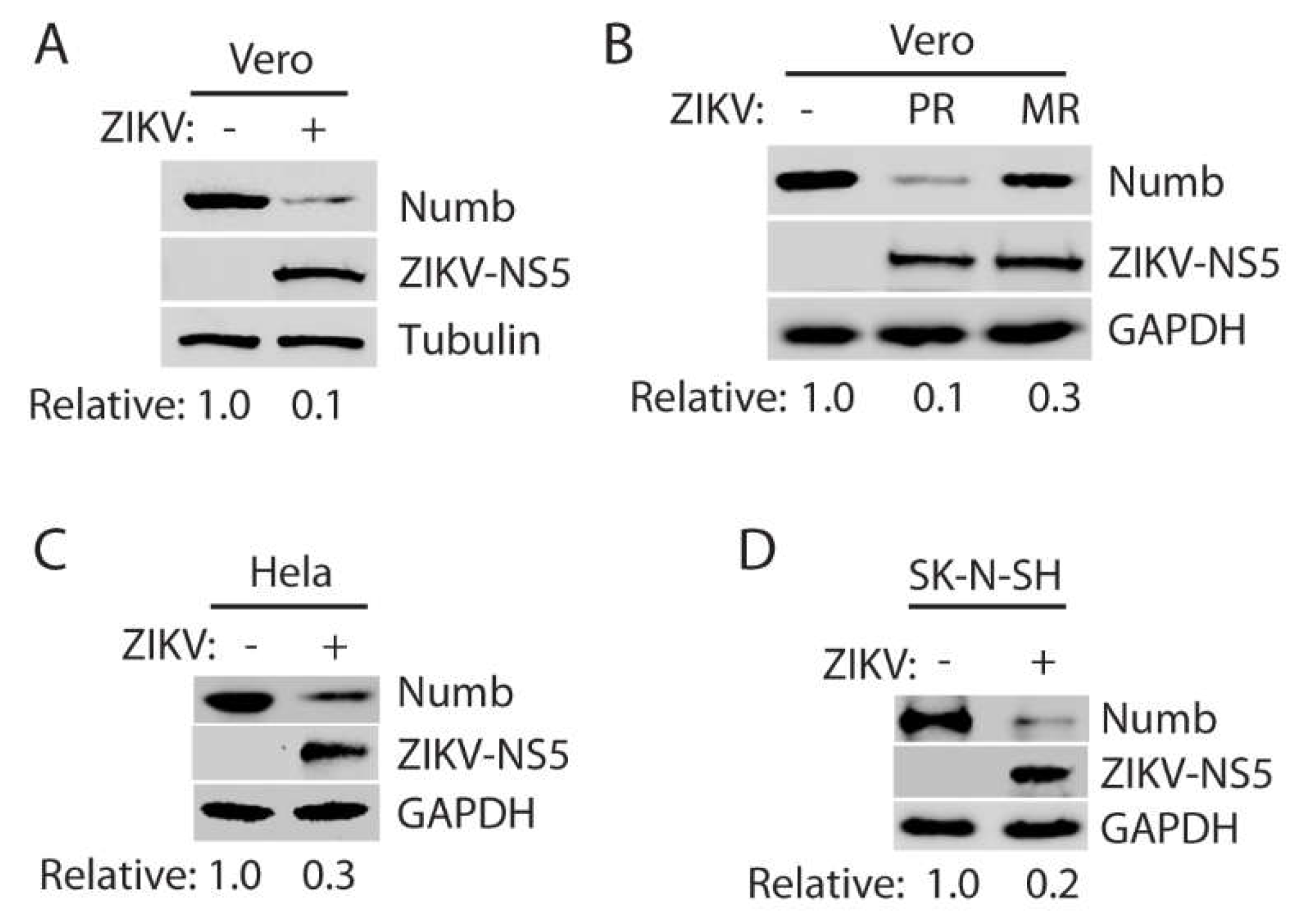
Figure 2.
ZIKV reduces the Numb protein in a temporal and dose-dependent manner. A. Temporal reduction of Numb by ZIKV infection. The cells were inoculated with ZIKV PR strain at an MOI of 1 and harvested for WB 20 and 40 hpi. Relative levels of the Numb protein are shown below the images. B. Dose-dependent reduction of the Numb protein by ZIKV PR strain. The cells were harvested for WB 24 hpi. Relative levels of the Numb protein are shown below the images. C. ZIKV RNA levels in the infected cells detected by RT-qPCR. Error bars represent the standard errors of the means of three repeated experiments. Asterisks denote significant differences in RNA level from an MOI of 0.1 (**, P < 0.01; ***, P < 0.001).
Figure 2.
ZIKV reduces the Numb protein in a temporal and dose-dependent manner. A. Temporal reduction of Numb by ZIKV infection. The cells were inoculated with ZIKV PR strain at an MOI of 1 and harvested for WB 20 and 40 hpi. Relative levels of the Numb protein are shown below the images. B. Dose-dependent reduction of the Numb protein by ZIKV PR strain. The cells were harvested for WB 24 hpi. Relative levels of the Numb protein are shown below the images. C. ZIKV RNA levels in the infected cells detected by RT-qPCR. Error bars represent the standard errors of the means of three repeated experiments. Asterisks denote significant differences in RNA level from an MOI of 0.1 (**, P < 0.01; ***, P < 0.001).
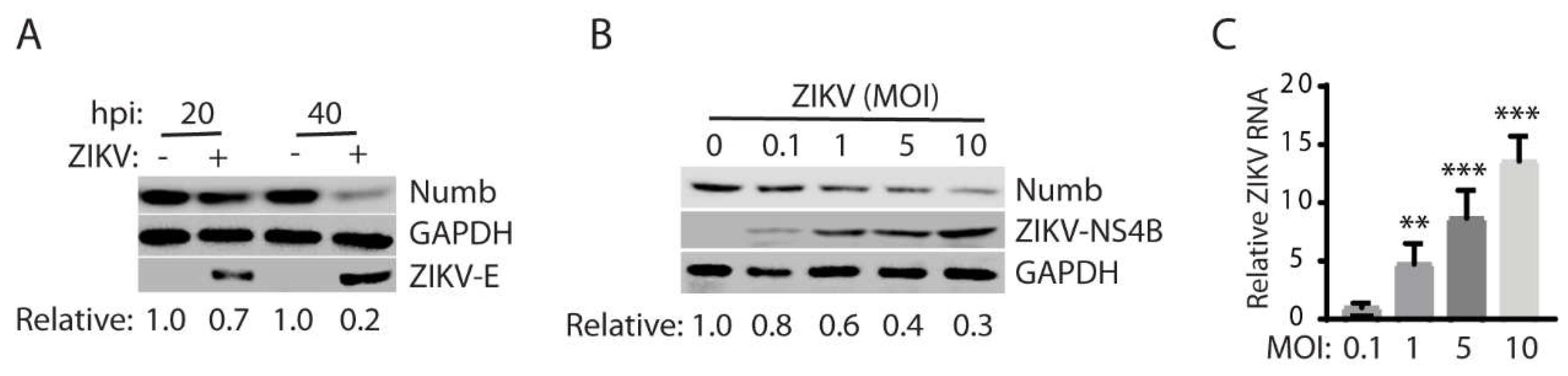
Figure 3.
Numb was decreased by ZIKV infection through the ubiquitin-proteasome pathway. A. ZIKV infection has minimum effect on Numb mRNA level detected by RT-qPCR. The relative Numb mRNA level is shown compared to the mock-infected Vero cells. The cells were infected with the ZIKV PR strain at an MOI of 1 and harvested at 30 hpi. B. ZIKV RNA level in the infected cells. A standard curve was included for calculating the absolute RNA level. Total ZIKV RNA is shown as copies in Log10 per well of a 24-well plate. C. MG132 treatment restores the Numb protein levels in ZIKV-infected cells. Vero cells were infected with ZIKV at an MOI of 1 and 30 hpi, and treated with MG132 or NH4Cl at a final concentration of 10 µM and 10 mM, respectively. The cells were harvested 6 h post-treatment. DMSO was included as a solvent control in the first two lanes. D. The Numb protein half-life shortened by ZIKV infection. Vero cells were infected with ZIKV at an MOI of 10 for 24 h and treated with MG132 for 6 h, before being treated with cycloheximide. Mock-infected cells were included for control. E. ZIKV infection has a significant effect on the Numb protein ubiquitination. Vero cells were infected with ZIKV and harvested for immunoprecipitation with Numb antibody. The IP precipitate was subjected to Western blotting with antibodies against ubiquitin (Ub) and Numb. The input of cell lysate was included as a control.
Figure 3.
Numb was decreased by ZIKV infection through the ubiquitin-proteasome pathway. A. ZIKV infection has minimum effect on Numb mRNA level detected by RT-qPCR. The relative Numb mRNA level is shown compared to the mock-infected Vero cells. The cells were infected with the ZIKV PR strain at an MOI of 1 and harvested at 30 hpi. B. ZIKV RNA level in the infected cells. A standard curve was included for calculating the absolute RNA level. Total ZIKV RNA is shown as copies in Log10 per well of a 24-well plate. C. MG132 treatment restores the Numb protein levels in ZIKV-infected cells. Vero cells were infected with ZIKV at an MOI of 1 and 30 hpi, and treated with MG132 or NH4Cl at a final concentration of 10 µM and 10 mM, respectively. The cells were harvested 6 h post-treatment. DMSO was included as a solvent control in the first two lanes. D. The Numb protein half-life shortened by ZIKV infection. Vero cells were infected with ZIKV at an MOI of 10 for 24 h and treated with MG132 for 6 h, before being treated with cycloheximide. Mock-infected cells were included for control. E. ZIKV infection has a significant effect on the Numb protein ubiquitination. Vero cells were infected with ZIKV and harvested for immunoprecipitation with Numb antibody. The IP precipitate was subjected to Western blotting with antibodies against ubiquitin (Ub) and Numb. The input of cell lysate was included as a control.
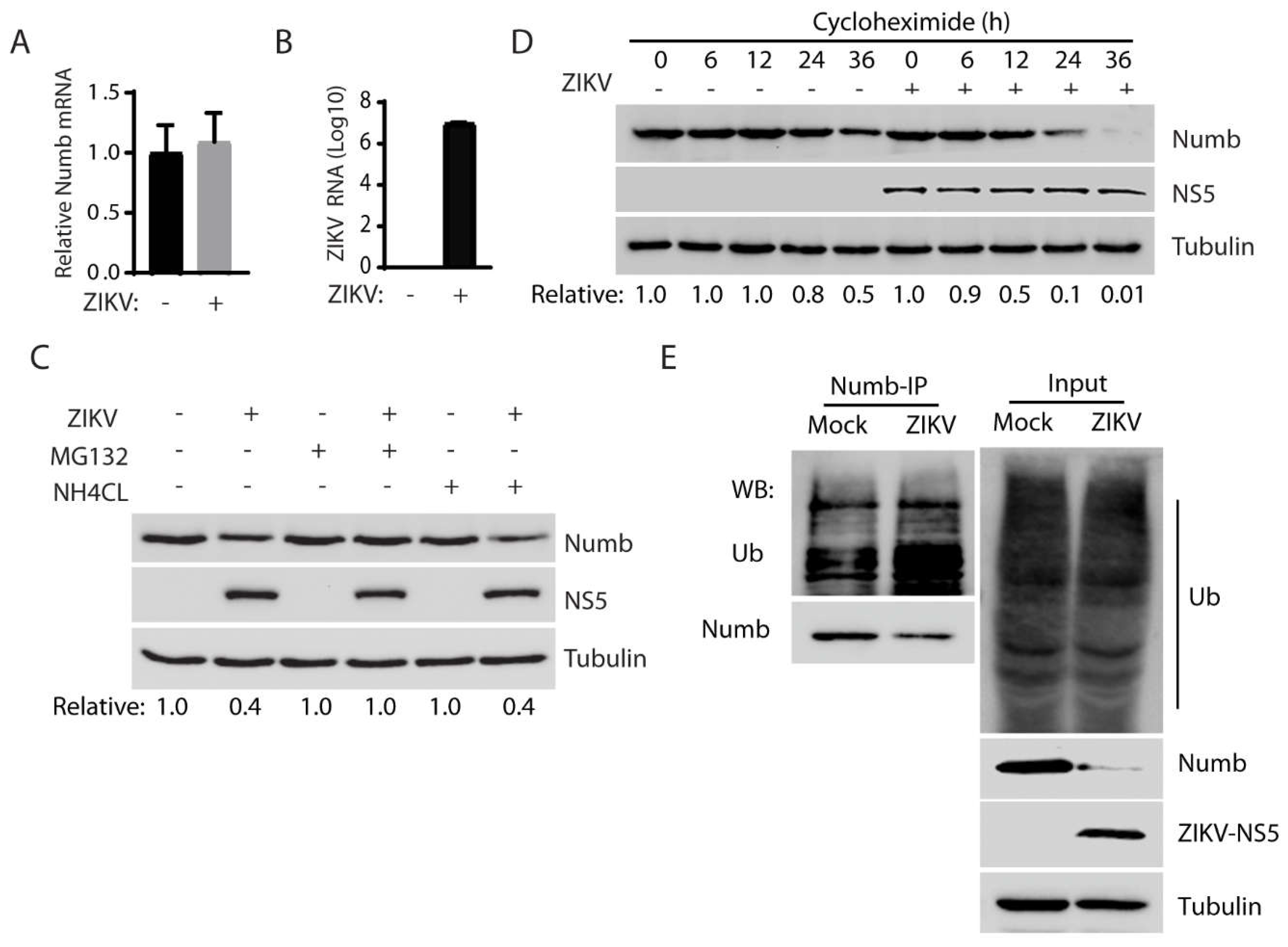
Figure 4.
ZIKV Capsid protein induces Numb reduction. A. Identification of the ZIKV protein that induces Numb reduction. HEK293 cells were co-transfected with the plasmids encoding the individual ZIKV proteins and MYC-Numb. The cells were harvested for WB at 48 h post-transfection. Molecular weight markers are indicated on the left. Relative Numb levels are indicated below the images. E.V.: empty vector. B. ZIKV-Capsid is present in Numb co-IP precipitates. The input of cell lysate is included for control. C. Capsid protein is present in MYC co-IP precipitates. HEK293 cells were transfected with MYC-Numb and YFP-C plasmids. The input of cell lysate is included for control. E.V.: empty vector.
Figure 4.
ZIKV Capsid protein induces Numb reduction. A. Identification of the ZIKV protein that induces Numb reduction. HEK293 cells were co-transfected with the plasmids encoding the individual ZIKV proteins and MYC-Numb. The cells were harvested for WB at 48 h post-transfection. Molecular weight markers are indicated on the left. Relative Numb levels are indicated below the images. E.V.: empty vector. B. ZIKV-Capsid is present in Numb co-IP precipitates. The input of cell lysate is included for control. C. Capsid protein is present in MYC co-IP precipitates. HEK293 cells were transfected with MYC-Numb and YFP-C plasmids. The input of cell lysate is included for control. E.V.: empty vector.
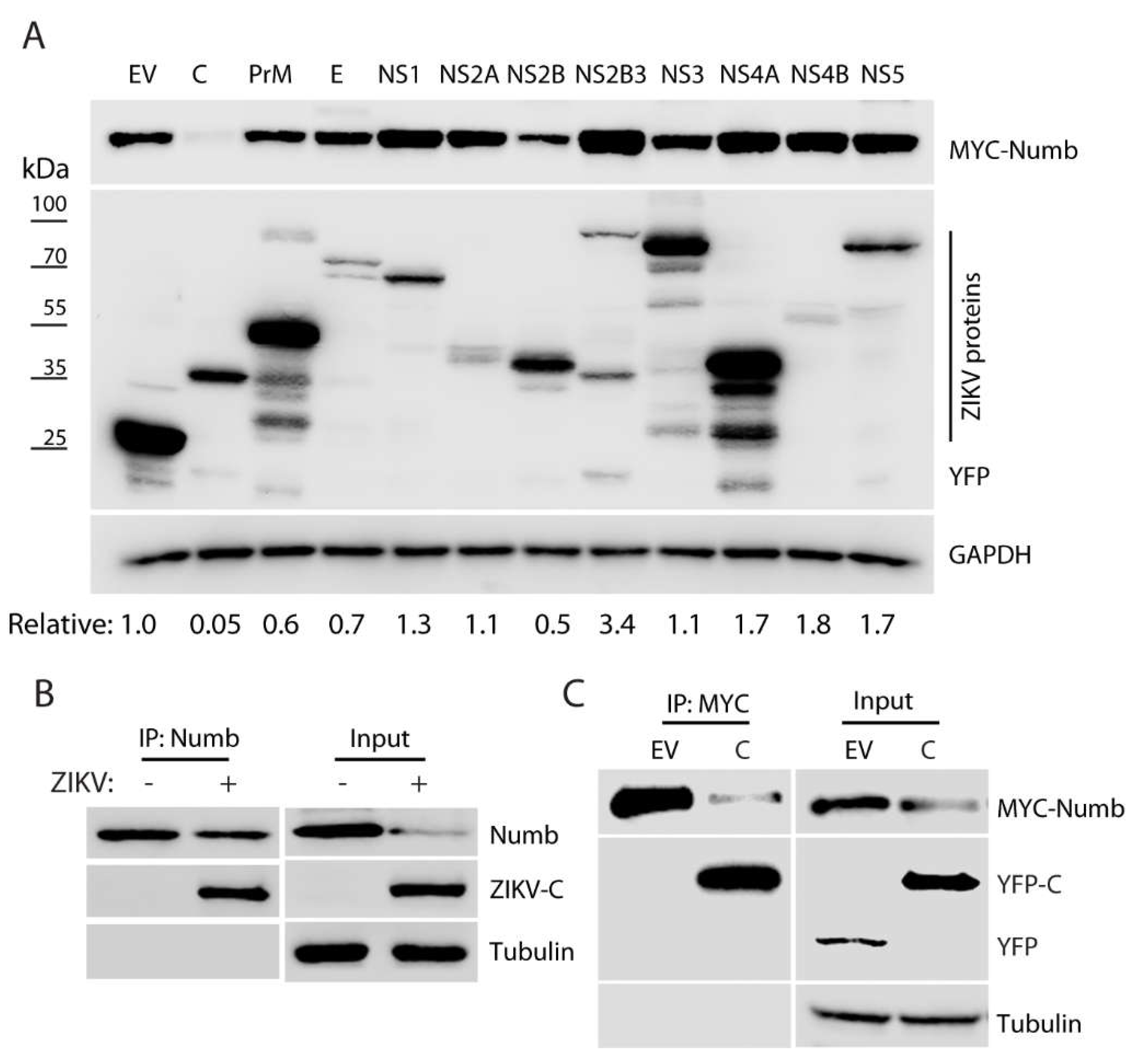
Figure 5.
Numb knockdown has minimal effect on ZIKV replication. A. Numb knockdown in Vero cells has no significant effect on ZIKV replication than control shRNA. Vero cells were transduced with recombinant retrovirus expressing control shRNA (C-shRNA) or shRNA against Numb (shNumb) three times, followed by inoculation with ZIKV and harvested 48 hpi. B. ZIKV titers from infected Vero cells at different time points. C. Numb knockdown has minimal effect on cell viability. NS: no significant difference.
Figure 5.
Numb knockdown has minimal effect on ZIKV replication. A. Numb knockdown in Vero cells has no significant effect on ZIKV replication than control shRNA. Vero cells were transduced with recombinant retrovirus expressing control shRNA (C-shRNA) or shRNA against Numb (shNumb) three times, followed by inoculation with ZIKV and harvested 48 hpi. B. ZIKV titers from infected Vero cells at different time points. C. Numb knockdown has minimal effect on cell viability. NS: no significant difference.
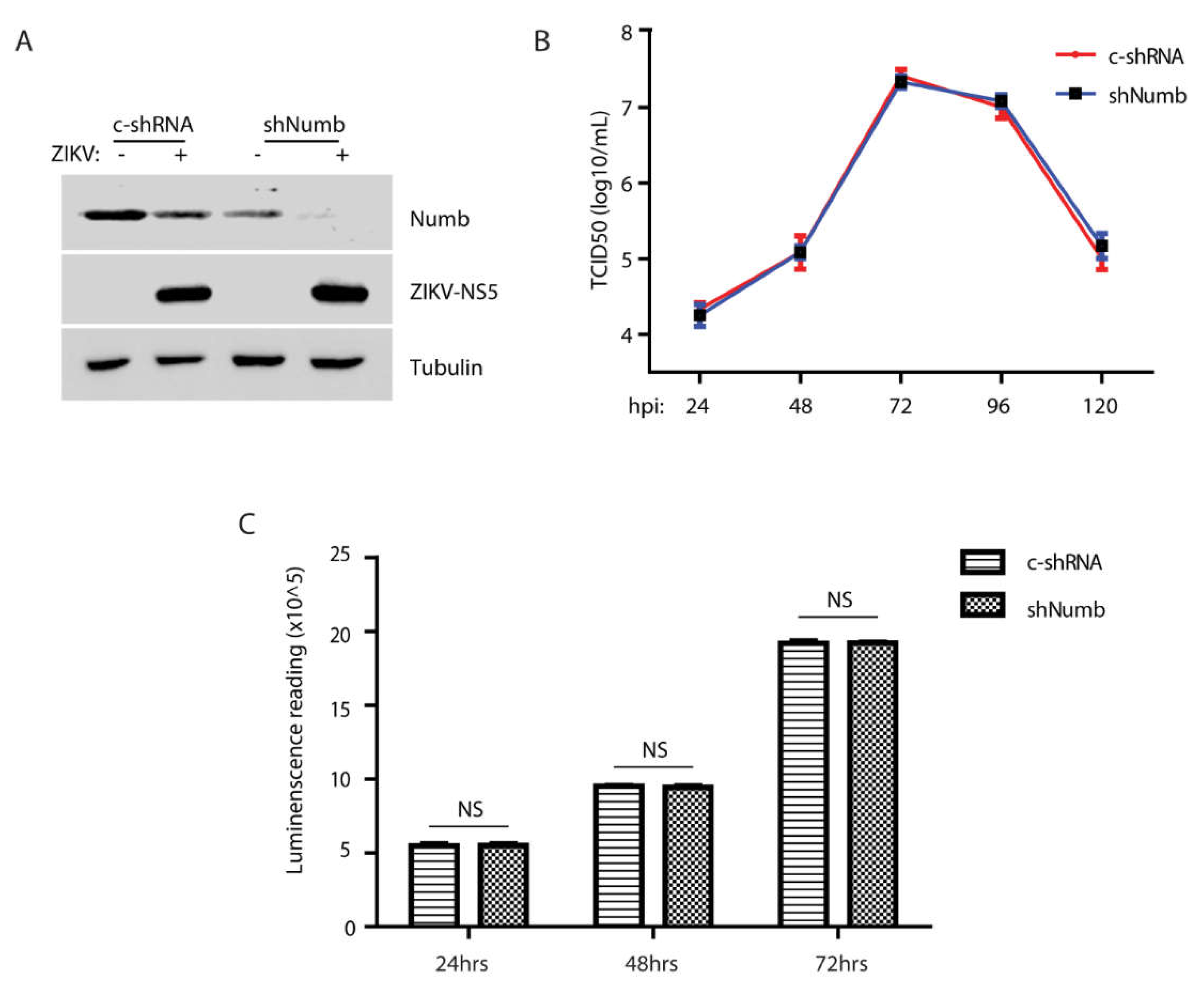

Table 1.
List of primers used in this study .
| Primera | Sequences (5' to 3')b | Target gene/vector |
| ZIKV-RR-F | AARTACACATACCARAACAAAGTGGT | NS5 |
| ZIKV-RR-R | TCCRCTCCCYCTYTGGTCTTG | NS5 |
| Numb-F1 | CGAATTCAACAAATTACGGCAAAGTTT | Numb |
| Numb-R1 | GCTCGAGTTAAAGTTCAATTTCAAACG | Numb |
| Numb-RR-F1 | GCTACCACCAGTCCCTTCTT | Numb |
| Numb-RR-R1 | GTGCCTGTAGGAACCTCTGT | Numb |
| shNUMB1-F | GATCCGGAATAAATATTATATATATTCAAGAGATATATATAATATTTATTCCTTTTTTG | shNumb |
| shNUMB1-R | AATTCAAAAAAGGAATAAATATTATATATATCTCTTGAATATATATAATATTTATTCCG | shNumb |
| shNUMB2-F | GATCCGCTCTATAGAGAATATATATTCAAGAGATATATATTCTCTATAGAGCTTTTTTG | shNumb |
| shNUMB2-R | AATTCAAAAAAGCTCTATAGAGAATATATATCTCTTGAATATATATTCTCTATAGAGCG | shNumb |
| shNUMB3-F | GATCCGAATAAATATTATATATAATTCAAGAGATTATATATAATATTTATTCTTTTTTG | shNumb |
| shNUMB3-R | AATTCAAAAAAGAATAAATATTATATATAATCTCTTGAATTATATATAATATTTATTCG | shNumb |
a. F: forward primer, R: reverse primer. b. The italicized alphabets indicate restriction enzyme cleavage sites for cloning.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



