
Submitted:
12 April 2023
Posted:
18 April 2023
You are already at the latest version
Abstract
The Loropetalum chinense and Loropetalum chinense var. rubrum are typical and traditional ornamental and Chinese herbal medicine in Asia. However, more information is needed on the mechanisms underlying its flower coloring. Here, we profiled the flavonoid metabolome, full-length sequencing, and transcriptome analysis to investigate the flavonoid biosynthesis and global transcriptome changes among different petal coloring cultivars of L. chinense and L. chinense var. rubrum. The total anthocyanins and phenotypic of the petal were highly consistent with the petal color. Moreover, a total of 207 flavonoid components were identified. Of these, 12 flavonoid components were considered significantly different expression compounds among the four samples. Meanwhile, the first reference full-length transcriptome of L. chinense var. rubrum was being built, which had 171,783 high-quality non-redundant transcripts with correcting with next-generation sequencing (NGS). Among them, 52,851 transcripts were annotated in the seven database of NR, KOG, GO, NT, Pfam, Swiss-Port and KEGG. Combined with NGS analysis, the DETs involved in flavonoids and anthocyanins contributed greatest to the flower-coloring. Additionally, the different expressed of eight LcDFRs and four LcANS genes were positively correlated with flavonoid biosynthesis, and the four LcBZ1 and one Lc3Mat1 were positively correlated with the content of seven anthocyanins revealed by coupling with metabolomics and transcriptomics analysis. Together these results were used to mine candidate genes by analyzing flower coloring changes in a comprehensive metabolic and transcriptomic level in L. chinense and L. chinense var. rubrum.
Keywords:
Loropetalum chinense var. rubrum
; flavonoid biosynthesis
; anthocyanins
; flower coloring
; metabolomic
; transcriptomic.
1. Introduction
Loropetalum chinense var. rubrum belongs to the Hamamelidaceae family and is commonly used in landscaping plazas, neighborhoods, gardens, roads, and green area et al. [1,2]. It is first identified in Changsha, Hunan Province, and its wild resources are distributed in Luoxiao Mountains among Liuyang, Pingjiang, and Liling[3]. Moreover, L. chinense var. rubrum has gorgeous flower and leaf coloring and flowering 2-3 times per year[4]. The petal coloring of L. chinense and L. chinense var. rubrum could be divided into five groups: poly-chromatic, yellowish-white, green-white, purplish-pink, and purplish red[5]. The different distribution of anthocyanins in the petals of sponge tissue cells was considered the main reason for different petal coloring in L. chinense var. rubrum [5]. Furthermore, the different components and content of flavonoids in leaves lead to the different leaf coloring in L. chinense var. rubrum [6], while litter is known about the flavonoid biosynthesis in petals of L. chinense var. rubrum.
Flavonoids are the primary pigments that contribute to the millions of flower colors in the petals of plants [7]. The water-soluble flavonoids are responsible for the range of colors from yellow to red to violet to blue, including aurones, chalcones, flavones, flavonols, flavanones, flavanones, isoflavones and anthocyanins [7,8]. Flavones and flavonols are the co-pigments that contribute to flower colors and bluing [9]. The O-glycosides of anthocyanins are the colored flavonoids which are the main determinants of flower color, found in the vacuoles of petal cells [10,11]. It has a basic structure of 3,5,7-trihydroxy benzene-2-phenyl benzofuran, then further modification by glycosylation, acylation, and methylation depending on the plant species and varieties [8], which acquires diversified anthocyanidin structures and variable flower colors [12]. Hundreds of anthocyanins have been reported, and they are divided into six classes, including Pg (pelargonidin), Cy (cyanidin), Dp (delphinidin), Pn (peonidin), Pt (petunidin), and Mv (malvidin) [12,13]. Cyanidin derives peonidin, and delphinidin derives petunidin and malvidin formed by methylation, hydroxylation, glycosylation, and acylation. And then, this deriving of anthocyanins further creates orange-yellow, red, purple, and blue petals [14,15,16,17]. Thus, the flavonoid component and relative content are significant factors in determining the flower color, and the synthetic genes, regulatory genes, and modification genes create these.
The flavonoid biosynthetic pathway is well understood and conserved among seeds in plants [18]. Flavonoids (including anthocyanins) are generated through the phenylpropanoid pathway, which is divided into three stages [19,20]. The first stage is the general phenylpropanoid pathway catalyzed by phenylalanine ammonia-lyase (PAL), cinnamic acid 4-hydroxylase(C4H), and 4-coumarate-CoA ligase(4CL) [21]. The second stage involved in the production of colorless dihydro flavonols successively catalyzed by chalcone synthase (CHS) [22,23], chalcone isomerase (CHI) [24], flavanone 3-hydroxylase (F3H) [25], flavonoid-3'5'-hydroxylase (F3'5'H) [26,27] and flavonoid-3'-hydro flavonols (F3'H) [28]. The third stage is the anthocyanins derivates catalyzing by dihydro flavonol 4-reductase (DFR) [27,29] and anthocyanin synthase (ANS) [30,31]. And then, the anthocyanins catalyzed by glycosyltransferase (GT) [32] and methyltransferase (MT) [33], and acyltransferase (AT) [14] into stable anthocyanins and their further modifications. Of note, some of the flavonoid biosynthetic genes of LcvrCHS1 [34], LcCHS1, LcCHS2 [35], LcCHI [36], LcDFR1 and LcDFR2 [37] were reported in L. chinense var. rubrum.
Transcriptional control also plays a vital role in the modulation of flavonoid biosynthesis, and several transcription factors involved in the pathway have been elucidated [38,39,40]. MBW (MYB-bHLH-WD40) complex is well conserved and the central transcriptional regulator of activating structural genes to flavonoid pathway enzymes [41,42], which regulate the biosynthesis of flavan-3-ols [43]. WD40 is a 'master regulator' to activate the flavonoid pathway independently [44]. MYB transcription factor is the core component of the MBW complex, and its subgroup of R2R3-MYB is mainly involved in regulating flavonoid metabolism [45]. The overexpression of AN4 (a R2R3-MYB gene) promotes the expression of the anthocyanin biosynthesis gene of CHI, F3H, and DFR [46]. LhMYB12 activated the lily's CHS and DFR gene promoter directly and obstructed the anthocyanin biosynthesis under high temperatures [47]. Although MYB can stimulate its companion of bHLH, its expression depends on different pigmented tissues [43]. TT8 (a bHLH gene) accumulates the anthocyanin and proanthocyanin biosynthesis by expressing DFR in Arabidopsis thaliana [48]. The MADS-box genes of ScAGL11 and ScAG inhibit anthocyanin accumulation in the cineraria capitulum by down-regulated the expression of ScCHS2, ScDFR3, and ScF3H1[49]. To date, the molecular mechanisms of flavonoid biosynthesis are more demonstrated in plants, while it is no report on L. chinense var. rubrum.
Here, one L. chinense 'Xiangnong Xiangyun' and three L. chinense var. rubrum 'Huaye Jimu 2', 'Xiangnong Fenjiao', and 'Xiangnong Nichang' were studied (Figure 1A). We identified the components of the flavonoid biosynthesis pathway in the three L. chinense var. rubrum and one L. chinense cultivar with MRM (multiple reaction monitoring). Then, the full-length transcriptome data and the next-generation sequencing data were obtained by combining NGS and SMRT sequencing. Moreover, this approach, combined with the flavonoid metabolic data, was applied to explain the molecular mechanism of the flower coloring of Loropetalum cultivars, in which flavonoids are produced and accumulated. Accordingly, this study provides a valuable resource for further investigating flavonoid biosynthesis and ornamental flower coloring modification and breeding.
2. Materials and Methods
2.1. Plant materials
One Loropetalum chinense ('Xiangnong Xiangyun', XNXY) and three L. chinense var. rubrum ('Huaye Jimu 2', LDPF; 'Xiangnong Fenjiao', XNFJ; 'Xiangnong Nichang', XNNC) were planted in the germplasm garden of Loropetalum spp. at in Hunan Agricultural University, Changsha, Hunan province, China (E 113.08, N 28.17). XNXY, XNNC, and XNFJ were the naturally pollinated offspring of LDPF. The flowers (flowers in full bloom for one day), stems, leaves, and roots of LDPF were harvested, respectively, for Iso-Seq library construction. The flowers of XNXY, LDPF, XNFJ, and XNNC were also collected to total anthocyanin detection and sequencing library construction. We considered 50 flowers comprising one biological sample for each Loropetalum cultivar, and each sample was segregated as three independent biological replicates. All these samples were frozen in liquid nitrogen immediately and stored at -80 °C.
2.2. Petal color comparison
The RHS color chart (sixth edition 2015, Royal Horticultural Society, 80 Vincent Square, London SW1P 2 PE, UK) was used to describe the color differences among the four Loropetalum spps. A spectrophotometer (YS3020, Technology Co. Ltd., China) was used to measure the color-related value of fresh petals' L*, a*, and b*. The parameters of L*, a*, and b* were described in CIELAB (CIE 1976). In this system, the value of L* represented lightness (those had higher L* represented white or near white-color, while black or near-black color), a* was a positive or negative coordinate representing a purplish-red-bluish-green color, and b* was a positive or negative coordinate representing a yellow-blue color.
2.3. Total anthocyanins analysis
Total anthocyanin content was determined as described by Zhang et al. [50] and Dong et al. [51]. Briefly, 0.5 g of Loropetalum spp. petals was ground to a powder in liquid nitrogen. Then, these powders were extracted with 5 mL of a mixture solution of 0.05% HCl in methanol at 4 °C for 24 hours in darkness. The supernatant was transferred into a clean tube after centrifugation at 1000×g for 15 min. Draw 4 mL of buffer A (0.4 M KCl, pH 1.0) and buffer B (1.2 N citric acid, pH 4.5), respectively, mixed with one milliliter of supernatant (the mixture is 5 mL in total) and take absorbance measurement at 510 and 700 nm for A and B mixtures separately. The total anthocyanin content was calculated using the formula: TA= A × MW × 5 × 100 × V/e (Romero et al., 2008). TA represents the total anthocyanin content of the detected sample (mg/100 g, as cyanidin-3-O-glucose equivalent). V represents the final volume (ml). A= [A510 (pH 1.0)-A700 (pH1.0)] - [A510 (pH4.5)-A700 (pH 4.5)]. The value of 449.2 represents the molecular mass of cyanidin-3-O-glucose. Furthermore, the value of 26,900 reflects a molar absorptivity (e) at 510 nm [52]. Each biological replicate was repeated triplicate.
2.4. Flavonoids extraction and MRM
The freeze-dried flower was crushed using a mixer mill (MM 400, Retsch) with a zirconia bead for 1.5 min at 30 Hz. The 100 mg fine powder was extracted overnight at 4 °C with 1.0 mL 70% aqueous methanol. The extracts supernatant was absorbed (CNWBOND Carbon-GCB SPE Cartridge, 250 mg, 3 mL; ANPEL, Shanghai, China, www.anpel.com.cn/) after centrifugation at 10,000 g for 10 min before LC-MS analysis. The supernatant was analyzed using an LC-EMS-MS/MS (HPLC, Shim-pack UFLC SHIMADZU CBM30A system, www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q TRAP, www.appliedbiosystems.com.cn/). Moreover, the analytical conditions were composed of column of Waters ACQUITY UPLC HSS T3 C18 (1.8 µm, 2.1 mm*100 mm). Water and acetonitrile with 0.04 % acetic acid were used as solvent systems. The gradient program of HPLC was performed as 100:0V/V at 0min, 5:95V/V at 11.0min, 5:95V/V at 12.0min, 95:5V/V at 12.1min, 95:5V/V at 15.0 min. The flow rate was 0.40 mL/min with an injection volume of 5 μL, and the column temperature was 40 °C. The effluent was alternatively connected to an ESI-triple quadrupole-linear ion trap (Q TRAP)-MS [6,37].
A triple quadrupole-linear ion trap mass spectrometer (Q TRAP) API 4500 Q TRAP LC/MS/MS System equipped with LIT and triple quadrupole (QQQ) scans were used to get the information on flavonoid metabolites. Moreover, the analysis system was equipped with an ESI Turbo Ion-Spray interface, operating in a positive ion mode and controlled by Analyst 1.6.3 software (AB Sciex). The ESI system index sets were followed by Chen et al. [6] and Zhang et al. [37]. Instrument tuning and mass calibration were performed with 10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. QQQ scans were acquired as MRM experiments with collision gas (nitrogen) set to 5 psi. DP and CE for individual MRM transitions were done with further DP and CE optimization. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period [6,37].
2.5. RNA sample preparation
Total RNA was prepared by grinding tissues into powder in liquid nitrogen and mixing 0.5 g powder and 1 mL TRIzol reagent (GenStar P124-01). It is processed following the protocol provided by the manufacturer (#124-01, GenStar, Beijing, China, https://www.gene-star.com/pro_cont_10220.html#coming). All samples were replicated three times. Then 1% agarose gels and Nanodrop (NanoDrop products, USA) were used to detect RNA degradation and contamination. The quantified and quality of total RNA were assessed using Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA), respectively. Two experiments were conducted. One was the different organs (leaf, flower, root, and stem) of total RNA extracted and mixed with equal amounts to be a pool of L. chinense var. rubrum RNA. The other was four flower cultivars (LDPF, XNNY, XNFJ, and XNNC) with twelve libraries subjected to 2×150 paired-end RNA-seq using ILLUMINA novaseq 6000. All the qualified RNA samples were stored at -20℃ and used for SMRT sequencing, RNA-seq, and qRT-PCR analysis.
2.6. PacBio Iso-Seq library preparation and SMRT sequencing
A total of 3.9 μg of equal mixed RNA was sequenced on the PacBio Sequel platform (Pacific Bioscience, CA, USA) according to the manufacturer's instruction, as previously described (Hoang et al., 2017; Hoang et al., 2019). The Oligo (dT) magnetic beads were used to mRNA enriched for the cDNA libraries. The Iso-Seq library was prepared by the Clontech SMARTer PCR cDNA synthesis kit (#634926; Clontech, Takara Bio Inc., Japan) and the BluePippin Size Selection System protocol described by Pacific Biosciences (PN 100-092-800-03). The fraction of cDNA with a size over 4 kb was run on BluePippinTM System to remove the short SMRTbell templates. After size fraction, the optimal library was sequenced on a PacBio RS II instrument at Novogene technology company (Beijing, China).
The sequencguh reads using pbclassify.py with ignore polyA false and min Seq Length 200. Next, the non-full length and full-length fasta files were produced. At last, the isoform-level clustering (ICE) and final arrow polishing were fed into the clustering step following the parameter configuration of hq_quiver_min_accuracy 0.99, bin_by_primer false, bin_size_kb 1, qv_trim_5p 100, qv_trim_3q 30. Additionally, the nucleotide errors in consensus reads were corrected using the Illumina RNA-Seq data with the software LoRDEC [53]. Finally, the final transcripts were obtained in corrected consensus reads by removing the redundant sequences with software CD-HIT (-c0.95-T6 -G0 -Al 0.00 -aS 0.99) [54]. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in the National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA009285 and CRA009284) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
2.7. Illumina RAN-Seq library construction and sequencing
Twelve libraries of four cultivars of RNA samples were prepared and sequenced, respectively. A total of 2 μg RNA was used for shot reads sequencing on the HiSeq4000 platform, and 150 bp paired-end reads were generated at the Novogene technology company in Beijing, China. The NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, MA, USA) was used for sequencing libraries following the manufacturer's recommendations.
2.8. Gene functional annotation, coding sequences (CDS) prediction, transcription factor (TF), and long non-coding RNAs (lncRNA) identification
All final transcripts annotation were done by comparing against the following databases: NCBI non-redundant protein sequences (NR) [55], NCBI non-redundant nucleotide sequences (NT) [56], Protein family (Pfam) [57], Clusters of Orthologous Groups of proteins (KOG/COG) [58], A manually annotated and reviewed protein sequence database (Swiss-Port) [59], KEGG Ortholog database (KO) [60], Gene Ontology (GO) [61]. The software of BLAST with the set of e-value '1e-10' was used for NT database analysis [62]. The Diamond Blast with the option of e-value '1e-10' was applied to NR, KOG, Swiss-Port and KEGG database analysis [63]. Moreover, the software of Hmmscan was used for Pfam database analysis [64].
The ANGEL pipeline, a long-read ANGLE implementation, was used to determine cDNA protein-coding sequences. We used L. chinense var. rubrum and closely related species confident protein sequences for ANGEL training and then ran the ANGEL prediction for final transcripts [65]. The software of Itak [66] and Plant Transcription Factor database [67] was used to identify TFs, transcriptional regulators (TRs), and protein kinases (PKs) with final transcripts of L. chinense var. rubrum. The four tools were used to predict the lncRNAs of L. chinense var. rubrum: Coding-Non-Coding-Index (CNCI) [62], Coding Potential Calculator (CPC) [68], Pfam-scan [69], PLEK [53]. The detected transcripts predicted with coding potential by either/all of the four tools were filtered out, and those without coding potential were our candidate set of LncRNAs.
2.9. Quantitation and differential expression of transcripts and genes analysis
The twelves sample of raw data were subjected to quality control including adapters, lower reads, and ploy-N by fastQC v0.11.2 [70]. And then, the clean data were obtained and mapped onto the L. chinense var. rubrum full-length transcriptome of final transcripts using Bowtie2 (v2.2.5) [71]. Readcount for each transcript was estimated by the expected number of Fragments Per Kilobase of transcript sequence per Millions of base pairs sequenced (FPKM) [72] method by using RESM [73]. The correlation analysis between samples of each cultivar was used to test the repeatability of samples. The three biological samples with a correlation coefficient value more considerable than 0.92 were performed to differentially expressed genes (DEGs) and differentially expressed Transcripts (DETs) analyses. The DEGs and DETs between each paired sample were performed by the DESeq R packages [74]. Genes and transcripts with fold change ≥2 and adjusted p-value ≤0.05 were considered significant DEGs and DETs. The gene ontology (GO) enrichment of all the DEGs and DETs was performed using the GO-seq R packages [75]. KEGG database and KOBAS software were subjected to KEGG Pathway enrichment analysis, and significantly enriched metabolic or signal transduction pathways select with an adjusted p value of ≤0.05.
2.10. The correlation analysis between DETs and DCMs
The differentially content metabolites (DCMs) of flavonoid and anthocyanins and differentially expressed transcripts (DETs) based on KEGG enrichment analysis of flavonoid biosynthetic pathway and anthocyanins biosynthetic pathway genes were used for integrative analysis. Moreover, the spearman method was used to analyze the correlation coefficients for transcriptome and metabolome data integration. The connection between DETs and DCMs was shown by heatplot.
3. Results
3.1. Petal color phenotype and total anthocyanins content among the four Loropetalum cultivars
Four Loropetalum cultivars were selected to elucidate the mechanism of flavonoids biosynthesis in petals, a white flower petal control cultivar, namely, 'Xiangnong Xiangyun' (XNXY) (Royal Garden Color Card, NN155B), and three L. chinense var. rubrum of 'Huaye Jimu 2' (LDPF), 'Xingnong Fenjiao' (XNFJ) (Royal Garden Color Card, 61C) and 'Xiangnong Nichang' (XNNC) (Royal Garden Color Card, 63A) (Figure 1A). The a* value representing redness was 0.79, 1.5, 4.52, and 2.24 among XNXY, LDPF, XNFJ, and XNNC, respectively. The b* value of these four Loropetalum cultivars (XNXY, LDPF, XNFJ, and XNNC) was -5.89, -6.29, -4.36, and -4.74, which represented blueness. The L* value, representing lightness, showed at 80.43, 100.91, 96.49, and 98.91 in four Loropetalum cultivars, respectively.
The total anthocyanins in the blooming petals for two days were detective by a spectrophotometric pH differential method. Among these four Loropetalum cultivars, the lowest level of anthocyanins accumulation (23.03 mg/g of FW) was detected in XNXY. Among the three L. chinense var. rubrum cultivars, the flower color with chimera (Huaye Jimu 2, LDPF, 37.71 mg/g of FW) contains more anthocyanins than XNXY (Figure 1B). The results indicated that the highest level of anthocyanins content was in 'Xiangnong Nichang' (XNNC, 264.96 mg/g of FW) (Figure 1B). These results showed that L. chinense var. rubrum was a critical resource of anthocyanins compared to Asparagus officinalis (22.04 mg/g of FW) [51], Vaccinium corymbosum (about 1.8 mg/g of FW) [76], Lycoris longituba (1.25 mg/g of FW) [77] and other horticultural plants. Additionally, a surprising degree of anthocyanins accumulation was observed in the four Loropetalum cultivars. It was shown that the sharp differences in anthocyanins accumulation are due to genetic diversity and specificity.
3.2. Identification and qualification of flavonoid metabolite profiles from the petals of Loropetalum cultivars
To identify flavonoid metabolite profiles in Loropetalum cultivars, the extracts from the petals of XNXY, LDPF, XNFJ, and XNNC were analyzed by MRM. A total of 207 flavonoid metabolites were identified from the four samples, including 39 flavonol metabolites, seven isoflavone metabolites, 26 flavonoid metabolites, 78 flavone metabolites, 20 flavanone metabolites, 19 polyphenol metabolites, 15 anthocyanins metabolites, and three proanthocyanins metabolites, were isolated and identified in all the four Loropetalum cultivars' petal extracts (Table S1 and Figure 2A). The total content of each flavonoid component was significantly different. For example, in LDPF, XNNC, and XNFJ, the anthocyanins metabolites were the most abundant ones, followed by flavonol or polyphenol metabolites, while flavonol metabolites in XNXY and followed by polyphenol ones (Figure 2A). In addition, we performed principal component analysis (PCA), and these 207 flavonoid metabolites could be divided into four groups, which is consistent with the flower-color phenotypical characteristics of the four Loropetalum plants (Figure 2B). To sum up, the accumulation of flavonoid components in flowers is significantly correlated with variety specificity.
3.3. Differential accumulation of flavonoid metabolites in Loropetalum plant's petals
To investigate the different metabolites changes and expression levels of flavonoids involved in L. spps., the types and relative accumulation levels of flavonoid compounds were analyzed among the four selected cultivars (Supplementary table S1, Supplementary table S2, Supplementary table S3, Supplementary table S4, Supplementary table S5, Supplementary table S6, and Supplementary table S7). The results shown in Upset plot (Figure 2C) and Volcano plot (Figure 2D-I), demonstrate that metabolites significantly differed among them. In total, 168 flavonoid metabolites were commonly found in the four samples. Notably, eight flavonols (Kaempferide, Ayanin, Chrysoeriol 6-C-hexoside, 'di-C, C-hexosyl-apigenin', 8-C-hexosyl chrysoeriol O-hexoside, Tricin 7-O-hexosyl-O-hexoside, Acacetin O-glucuronic acid and Apigenin 6,8-C-diglucoside), one flavanone (Hesperetin O-Glucuronic acid), and three anthocyanins (Peonidin, Peonidin 3-sophoroside-5-glucoside, and Petunidin 3-O-glucoside) were coexisting in petals of LDPF, XNFJ, and XNNC (Figure 2C, Supplementary table S8). Furthermore, four flavones (Tricin O-rhamnosyl-O-malonylhexoside, C-hexosyl-chrysin O-feruloylhexoside, Chrysoeriol O-sinapoylhexoside, Apigenin 7-rutinoside (Isorhoifolin)), one flavonoid (Apiin), and one anthocyanin (Malvidin 3-acetyl-5-diglucoside) were common existed in XNFJ and XNNC (Figure 2C, Supplementary table S8). Additionally, one Anthocyanin (Cyanidin), one Flavone (Luteolin C-hexoside), and Flavone (Chrysoeriol 7-O-rutinoside) were found in LDPF, XNNC, and XNFJ, respectively. Moreover, one flavone (C-hexosyl-luteolin O-hexoside) and one Isoflavone (Glycitin) were detected in XNXY (Figure 2C, Supplementary table S8).
The flavonoid metabolites distribution could be divided into up- and down-regulated types. Based on the fold changes and the VIP value of OPLS-DA of flavonoid metabolites in the four samples, 59, 48, 50, 59, 56, and 48 differentially concentration of flavonoid metabolites (DCMs) (fold change ≥2 or fold change ≤0.5, and VIP ≥1) were detected in LDPF VS XNNC (Figure 2D, Supplementary table S9), LDPF VS XNFJ (Figure 2E, Supplementary table S10), LDPF VS XNXY (Figure 2F, Supplementary table S11), XNNC VS XNFJ (Figure 2G, Supplementary table S12), XNNC VS XNXY (Figure 2H, Supplementary table S13), and XNFJ VS XNXY (Figure 2I, Supplementary table S14), respectively. Then, these DCMs have introduced into the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and KEGG enrichment analysis to explore the potential metabolic pathway affected by the different coloring petals. The 'Biosynthesis of secondary metabolites' (ko01110), 'Flavonoid biosynthesis' (ko00941), 'Anthocyanin biosynthesis' (ko00942), 'Isoflavonoid biosynthesis' (ko00943), and 'Flavone and flavonol biosynthesis' (ko00944) were screened out to be the most relative metabolic pathway which related to the petals coloring (Figure S1, Supplementary table S15-S20). Notably, the different relative content of Kaempferide (Flavonol), Apiin (flavonoid), Glycitin (Isoflavone), Tricetin (Flavone), Naringenin 7-O-glucoside (Flavanone), Cyanidin 3,5-O-diglucoside (Anthocyanin), Cyanidin 3-O-glucoside (Anthocyanin), Delphinidin (Anthocyanin), Delphinidin 3-O-glucoside (Anthocyanin), Petunidin 3-O-glucoside (Anthocyanin), and Pelargonidin (Anthocyanin) were significantly different in the four cultivars of petals. Additionally, the Kaempferide, Malvidin 3-acetyl-5-diglucoside, Peonidin, Peonidin 3-sophoroside-5-glucoside, and Petunidin 3-O-glucoside, Cyanidin 3,5-O-diglucoside, Cyanidin 3-O-glucoside, Delphinidin 3-O-glucoside, and Pelargonidin were much higher content in XNNC, XNFJ, and LDPF, while the content of Delphinidin was highest in XNXY (Figure S2, Supplementary table S15-S20). The phenomenon mentioned above, flavonoid compounds from the three purple petals, indicated that these purple cultivars might differ in the expression of anthocyanin biosynthetic or regulatory gene expression from the white one.
3.4. Combined sequencing approach to tissues of L. chinensis var. rubrum
To identify and differentiate the flower transcriptome of L. chinense, two sequencing strategies were adopted by using the NGS and SMRT sequencing platforms. First, the twelve mRNA samples from four flower petals (LDPF, XNNC, XNFJ, and XNXY, each in triplicate) were performed 2×150 paired-end sequencing using HiSeq 4000 platform, with each sample yielding 8.14 Gb clean data on average, and the Q30 values of all the 12 samples were higher than 93.82 %. The average coverage of each Illumina transcriptome data mapping to the L. chinense var. rubrum full-length transcriptome was 82.92 % (80.36~86.77 %), which indicated that the data were comparable (Supplementary table 21). The second strategy was that full-length cDNAs from 12 pooled total RNA samples were normalized and performed SMRT sequencing using the PacBio platform (Supplementary table 22). In total, 46.16 Gb polymerase read bases (1,562,962 polymerase reads) were generated by PacBio sequence, with an average read length of 29,532 bp and N50 of 51,440 bp. After filtering using the RS Subreads.1 of PacBio RS, 35,173,249 subreads representing 43.50 Gb were obtained. After clustering and polished with ICE and arrow, 723,655 full-length non-chimeric reads were obtained. Finally, the clean Illumina reads were used to correct the SMRT reads of the polished consensus using the LoRDEC software, the redundant sequences were removed by CD-HIT software, and 171,783 high-quality non-redundant transcripts were produced, with a total length of 406,454,922 bp, and N50 length of 2,935 bp, and their lengths were in the range of 132 to 14,258 bp. The high-quality full-length transcriptome data was obtained.
The final transcripts were annotated with the NR, KOG, GO, NT, Pfam, Swiss-Port, and KEGG, and they were also analyzed involving metabolic pathways and functional classifications. In total, 144,893 transcripts (84.35% of all) were functionally annotated, and 52,851 transcripts (24.94 % of all) were subjected to all databases (Figure 3A). Furthermore, five public protein databases shared 43,776 transcripts (Figure 3B), with 131,741 transcripts (76.69 % of all) being assigned to NR database. The NR databases had the most matched sequences, and the top five species, including Vitis vinifera, Juglans regia, Theobroma cacao, Nelumbo nucifera, Ziziphus jujuba, which were with over 50 % best Blast hits ratio (Figure S3). In the KOG database, 85,328 transcripts were subjected to 26 functional categories (Figure S4), and the largest group was general function prediction only, with 19,255 transcripts (22.57 % of all). Especially in Q group, 3,830 transcripts were assigned to the category of 'Secondary metabolites biosynthesis, transport and catabolism'. A total of 76,974 transcripts were grouped into three GO categories of biological process (BPs), cellular components (CCs), and molecular functions (MFs). All the hit transcripts were divided into 54 subgroups, with many transcripts related to 'metabolic process' (35,888 of all) and 'biological regulation' (9,532 of all) (Figure S5). A total of 129,856 transcripts were annotated in six categories subjected to KEGG database (Figure S6). The most enriched was 'Metabolism' (40,595 of all), and these transcripts associated with the biosynthesis of carbohydrate metabolism (5,657 of all), phenylpropanoid biosynthesis (641 of all), flavonoid biosynthesis (267 of all), isoflavonoid biosynthesis (5 of all), and flavone and flavonol biosynthesis (62 of all) pathways.
The protein-coding and long non-coding RNA were obtained by full-length transcriptome sequencing. A total of 16,1387 CDSs predicted from the transcripts (Figure S7A). Furthermore, these CDSs were blasted and hit with PlnTFDB and iTAK software for TF annotation. In total, 5487 TFs were identified and divided into more than 28 families (Figure S7B). The most abundant family was C2H2 (397 of all), followed by C3H (341 of all), bHLH (324 of all), FAR1 (291 of all), and SNF2 (254 of all). In addition, 26,590 LncRNAs were predicted from the final transcripts, ranging from 200 to 11,293 bp (Figure S7C, Figure S7D). These data provided an abundant and gene and transcript pool for further study of metabolic process.
3.5. Global transcriptomic characteristics of flowers during full-bloom stage
To acquire insight into the molecular mechanism of L. chinense var. rubrum flowers, RNA samples from the four cultivars with dissimilar flower coloring were used for transcript quantification. The box histogram showed that all the transcripts' expression levels were significantly different in the four samples, and it was the lowest level in XNNC (Figure 4A). However, correlation analysis indicated that all the samples in each group had good reproducibility (Figure 4B). Moreover, the cluster dendrogram analysis and PCA analysis revealed that all the samples within each group had high consistency (Figure 4C, Figure 4D). A total of 8,713, 17,085, 11,818, 17,652, 9,803, 17,687, 13,412, 15,772, 1,412, 16,058, 7,854, and 7,521 differential expression transcripts (DETs) were identified in XNFJ VS LDPF, XNFJ VS XNNC, XNFJ VS XNXY, XNNC VS XNXY, and XNXY VS LDPF compared combinations, respectively (Figure 4E). Mainly, focusing on the pairwise comparisons group content of XNFJ and XNNC (petals with purl coloring), the number of upregulated DETs was higher than that of downregulated ones. While the downregulated DETs were higher than upregulated ones in the XNXY VS LDPF group (petals with white and mosaic coloring). Additionally, the number of upregulated differentials expressing LncRNA was higher than downregulated ones in the group of XNFJ VS LDPF and XNFJ VS XNXY (Figure S8).
Differences in transcripts were analyzed genes that may be involved in flower coloring formation among color-full petal cultivars. Furthermore, GO enrichment analysis was used to classify the function of the DETs. The results showed that the main enriched GO terms of DETs in the selected group belong to 'biological process' (BP), 'cellar component' (CC), and 'molecular function' (MF) of GO categories. In the group of XNNC VS LDPF (Figure S9, Supplementary table S23), XNNC VS XNXY (Figure S10, Supplementary table S24), XNFJ VS LDPF (Figure S11, Supplementary table S25), XNFJ VS XNNC (Figure S12, Supplementary table S26), XNFJ VS XNXY (Figure S13, Supplementary table S27), XNXY VS LDPF (Figure S14, Supplementary table S28) paired comparison, BP had 15, 15, 15, 15, 15, 15 significantly enriched GO terms, respectively; the CC had 11, 12, 3, 6, 8, 1 significant enriched GO terms, respectively; and MF had 15, 15, 16, 15, 15, 15 significant enriched GO terms, respectively. Moreover, the 'biding', 'catalytic activity', 'metabolic process', 'cellular process', and 'organic substance metabolic process' were the top five processes and molecular functions of significant enriched GO terms.
The KEGG enrichment analysis of the differential expressed transcripts was analyzed for their biological functions (Supplementary table S23-28, Figure S15). The results indicated that 'Phenylpropanoid biosynthesis', 'Flavonoid biosynthesis', 'Carotenoid biosynthesis', 'Monoterpenoid biosynthesis', 'Phenylalanine, tyrosine, and tryptophan biosynthesis', 'Photosynthesis-antenna proteins', and 'Plant hormone signal transduction' enrichment pathways were obtained in the six paired comparisons, which demonstrated that those DETs had significant effects on petal coloring. Furthermore, phenylpropanoids and flavonoids are the most significant contributors to flower and leaf coloring. Therefore, these DETs involved in 'Phenylpropanoid biosynthesis' and 'Flavonoid biosynthesis' might play critical roles in different petal coloring formations L. chinenses var. rubrum and L. chinenses.
3.6. Expression analysis indicates flavonoid compounds' biosynthesis and accumulation
Flavonoids are the primary pigment in plants, which can be classified into several subgroups, such as flavones, flavonols, anthocyanins, et al. In total, 433 DETs were detected in six compared subgroups (Figure S16). The KEGG enrichment analysis indicated that the "flavonoid biosynthesis" pathway was one of the most representative pathways in XNNC VS LDPF (Supplementary table S29), XNNC VS XNXY (Supplementary table S30), XNFJ VS LDPF (Supplementary table S31), XNFJ VS XNNC (Supplementary table S32), XNFJ VS XNXY (Supplementary table S33), and XNXY VS LDPF (Supplementary table S34). Based on the KEGG enrichment analysis of DCMs and DETs, the flavonoid biosynthetic pathway of L. chinense var. rubrum was constructed according to KEGG public database (Figure 5A). Eight flavonoid compounds were significantly different among the four samples (Figure 5A). The XNXY was with the highest relative content of myricetin, Naringenin-7-O-glucoside, Glycitin, Tricetin and Delphinidin. While the XNNC and XNFJ were with a higher relative content of Apiin, Kaempferol, and Pelargonidin. Furthermore, 73 DETs related to biosynthesis of flavonoid compounds, including eight LcCYP73A (encoding trans-cinnamate 4-monooxygenase), 20 LcCHS (encoding chalcone synthase), three LcCHI (encoding chalcone isomerase), 19 LcF3H (encoding naringenin 3-dioxygenase), eight LcDFR (encoding bifunctional dihydroflavonol 4-reductase or flavanone 4-reductase) and four LcANS (encoding anthocyanidin reductase) (Figure 5B). Additionally, all the different expressed LcDFR and LcANS genes were highly expressed in XNFJ or XNNC. Moreover, there was a strong correlation between the expression of flavonoid biosynthesis-related gene DETs and KEGG enrichment DCMs of flavonoids (Figure S17).
3.7. Co-expression analysis for the investigation of anthocyanin biosynthesis
Anthocyanins were the primary coloring pigments giving the colors orange, red, blue, and purple in flowers, leaves, fruits, seeds, and other tissues (Bueno et al., 2012). Anthocyanins were the essential products in the flavonoid pathway biosynthesis. Moreover, the colored unstable anthocyanidins (pelargonidin, cyanidin, and delphinidin) were converted to stable anthocyanins (pelargonidin-3-O-glucoside, cyanidin-3-O-glucoside, and delphinidin-3-glucoside) and their further modifications (such as acylation, glycosylation, and methylation) (Liu et al., 2021b). The KEGG enrichment analysis indicated that eight anthocyanins such as pelargonidin, delphinidin, cyanidin-3-O-glucoside, cyanidin-3,5-O-glucoside, petunidin-3-O-glucoside, peonidin-3-O-sophoroside-5-O-glucoside, malvidin-3-acetyl-5-O-diglucoside, and Delphinidin-3-O-glucoside were significantly different among the four samples (Supplementary table 1, Figure 6A). Furthermore, the relative concentration of pelargonidin, peonidin-O-3-sophoroside-5-O-glucoside, and Malvidin-3-acetyl-5-O-diglucoside was consistent with the petal coloring of all the samples (Supplementary table 1, Figure 6A). Twenty DETs-related genes of anthocyanins biosynthetic were identified (Figure 6B). The results indicated that the genes of UFOG1, UFOG7, UFOG9, UFOG12, and 3AT1_2 were relatively higher expressed in darker petals of samples.
To explore the roles of DETs involved in the accumulation of anthocyanins, a correlation analysis among the relative expression profiles mentioned above DETs and the DCMs of anthocyanins was performed (Figure S18). As a result, the expression of UFOG1, UFOG7, UFOG9, UFOG12, and 3AT1_2 was positively correlated with the content of cyanindin-3-O-glucoside, delphinidin-3-O-glucoside, petunidin-3-O-glucoside, pelargonidin, malvidin-3-acetyl-5-O-diglucoside, peonidin and peonidin-3-sophoroside-5-O-glucoside, and negatively correlation with the content of delphinidin. In addition, the UFOG2, UFOG10, and GT1_2 positively correlated with the delphinidin content.
4. Discussion
What flowers do we like? Flower color is one of the most important factors influencing flowers' beauty [11]. Flavonoids are a common pigment component that presents a broad spectrum of colors from pale yellow to blue to pure, especially reported in petals. Flavonoids are a large group of plant-biosynthetic compounds which can be classified as anthocyanins, flavones, flavanols, flavanones, isoflavones, or other flavonoids compounds [78]. The anthocyanin in leaves is the primary coloring pigment in L. chinense var. rubrum [6]. In the present study, we found that the anthocyanins content of red-flowers L. chinense var. rubrum is higher than white-flowers L. chinense. Moreover, the content of anthocyanins is a vital factor influencing their flower coloring. XNFJ had the highest total anthocyanins and the redness color of a* value, while XNXY had the lowest total anthocyanins and lightness. It might be the main reason for the transformation from white to pink to purple.
Flavonoids are one of the essential pigments in many ornamental plant petals, and the composition of flavonoids' composition may vary among different color petals of the same species. The white flower chrysanthemum only contained flavones and flavonols, and the pink ones mainly contained anthocyanins, flavones, and flavonols [79]. The derivatives of delphinidin and cyanidin were more complicated in the red group of water lily than in others [80]. In total, 15 anthocyanin and 20 flavonols were identified in primula vulgaris cultivars, peonidin-type anthocyanins, cyanindi-3-O-glucoside, and dephininidin-3,5-di-O-glucoside-3'-caffeic ester were accumulated in pink flowers [35]. We found that 207 flavonoid metabolites were identified in the four cultivars (Table S1 and Figure 2A), and they were mainly involved in 39 flavonols, 7 isoflavones, 26 flavonoids, 78 flavones, 20 flavanones, 19 polyphenols, 15 anthocyanins and 3 proanthocyanins in L. chinense var. rubrum and L. chinense. About 168 flavonoids were common in the four samples, indicating no apparent alternatives of flavonoids among the samples of Loropetalum species. The same result was found in Nicotiana species [81], which might be the same species' close genetic background. Eight flavonols, one flavone, and three anthocyanins, reporting for the first time in Loropetalum species, were identified in petals of LDPF, XNFJ, and XNNC. These DCMs had a close correlation with the petals coloring of Loropetalum species. Furthermore, the enrichment analysis showed that these DCMs were identified to the metabolic pathway of 'Biosynthesis of secondary metabolites' (ko01110), 'Flavonoid biosynthesis' (ko00941), 'Anthocyanin biosynthesis' (ko00942), 'Isoflavonoid biosynthesis' (ko00943) and 'Flavone and flavonol biosynthesis' (ko00944).
The structural and functional genomic analysis provides a good insight into discovering the metabolic processing pathway in plants. The full-length transcripts, based on the SMRT sequencing stratagem that improves genome and transcriptome assembly, are essential for structural, functional, and comparative genomics studies [82,83,84]. The NGS strategies for transcriptomes were a convenient tool for correcting the SMRT sequencing errors reported in many plants [42,85]. In this study, we obtained 46.16 GB of raw data by PacBio sequencing with an average length of 29,523 bp and N50 of 51,440 bp. After correcting the SMRT reads with NGS data and CD-HIT software, 171,783 high-quality non-redundant transcripts were produced. It is much larger than Carthamus tincorius (79,926 transcripts) [86], Litchi chinense (50,808 trancripts) [87], Camellia oleifera (40,143 transcripts) [88], and Lolium multiflorum (72,722 transcripts) (Chen et al., 2018). Then, these transcripts were annotated with the NR, KOG, GO, NT, Pfam, Swiss-Port, and KEGG for gene function annotation and metabolic pathways analysis. As a result, 144,893 transcripts were functionally annotated, and the most enriched were 'Metabolism', including phenylpropanoid biosynthesis, flavonoid biosynthesis, isoflavonoid biosynthesis, and flavone and flavonol biosynthesis pathways (Figure 3A, Figure 3B, Figure S5-S8). Thus, SMRT sequencing combined with NGS sequencing strategy established an accurate abundant reference transcriptome of L. chinense var. rubrum for the first time.
Cultivars-specific flavonoids in L. chinense var. rubrum might be associated with the differential expression of essential biosynthetic genes. Flavonoid biosynthesis and anthocyanin biosynthesis genes had well summarized in some reviews [21,41,43,84]. The transcriptomes are based on NGS strategy as a convenient tool for quantitative gene expression data in plants. This study used the four cultivars with twelve samples for transcript qualification. All the differentially expressed genes were annotated with high-quality full-non-redundant reference transcriptomes. The DETs enrichment analyzed with KEGG annotation demonstrated that they were involved in 'Phenylpropanoid biosynthesis' and 'Flavonoid biosynthesis'. Combined with the DCMs analyzed, eight LcCYP73A (encoding trans-cinnamate 4-monooxygenase), 20 LcCHS (encoding chalcone synthase), three LcCHI (encoding chalcone isomerase), 19 LcF3H (encoding naringenin 3-dioxygenase), eight LcDFR (encoding bifunctional dihydroflavonol 4-reductase or flavanone 4-reductase) and four LcANS (encoding anthocyanidin reductase), and twenty genes of anthocyanins biosynthetic were identified in our study (Figure 5A, Figure 5B, Figure 6A, Figure 6B, Figure S19, Figure S20). Moreover, the correlation of DCMs and the flavonoid pathway (included of anthocyanin pathway) related genes were also discovered. The genes of UFOG1, UFOG7, UFOG9, UFOG12, and 3AT1_2 were relatively higher expressed in darker petals of samples. Furthermore, the UFOG2, UFOG10, and GT1_2 positively correlated with the delphinidin content.
5. Conclusion
For the first time, this study reports on the intergrade application of phenotypic, metabolomics, and transcriptomics to elucidate the flower coloring mechanism of L. chinense var. rubrum. The different pigment combination and their accumulations were the crucial reasons caused by the different petal coloration in L. chinense var. rubrum. Most flavonoids had significantly different content among the four cultivars for close genetic backgrounds. And the relative content of Kaempferide, Apiin, Glycitin, Tricetin, Naringenin 7-O-glucoside, pelargonidin, delphinidin, cyanidin-3-O-glucoside, cyanidin-3,5-O-glucoside, petunidin-3-O-glucoside, peonidin-3-O-sophoroside-5-O-glucoside, malvidin-3-acetyl-5-O-diglucoside and Delphinidin-3-O-glucoside were main differential content of components in L. chinense var. rubrum. An accurate abundant referent transcriptome was established by combining with SMRT and NGS strategy. The flavonoid biosynthesis genes, such as LcCYP73A, 20 LcCHS, three LcCHI, 19 LcF3H, eight LcDFR, four LcANS, four UFOGs and one 3AT1_2, correlated with the specific flavonoid's high determination the flower color of L. chinense var. rubrum. Together, these findings offer novel insights into the molecular basis for the flavonoid biosynthesis in L. chinense var. rubrum, and could serve as the basis for future research on the selective breeding of colorful ornamental plants.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: The metabolism view map of the significant metabolism pathways of the four different cultivars of L. chinense and L. chinense var. rubrum. (A). Significant changed pathways on the enrichment of LDPF VS XNNC. (B). Significant changed pathways on the enrichment of LDPF VS XNFJ. (C). Significant changed pathways on the enrichment of LDPF VS XNXY. (D). Significant changed pathways on the enrichment of XNNC VS XNFJ. (E). Significant changed pathways on the enrichment of XNNC VS XNXY. (F). Significant changed pathways on the enrichment of XNNC VS XNXY. (G). Significant changed pathways on the enrichment of XNFJ VS XNXY.; Figure S2: The top 20 differential metabolites among of different cultivars of L. chinense var. rubrum. (A). the top 20 change metabolite of LDPF VS XNNC. (B). the top 20 change metabolite of LDPF VS XNFJ. (C). the top 20 change metabolite of LDPF VS XNXY. D. the top 20 change metabolite of XNNC VS XNFJ. (E). the top 20 change metabolite of XNNC VS XNXY. (F). the top 20 change metabolite of XNFJ VS XNXY; Figure S3: The annotation static results of full-length nonredundant final transcripts to NR databases in different species; Figure S4: The classification map of full-length nonredundant final transcripts to GO database; Figure S5: The classification map of full-length nonredundant final transcripts to GO database; Figure S6:The classification chart of annotation of full-length nonredundant final transcripts to KEGG metabolic pathway; Figure S7: The annotation and prediction of CDS, transcription factors, and LncRNAs of L. chinense var. rubrum. (A). The number, percentage and length of CDS predicted by final transcripts of L. chinense var. rubrum. (B). The top 30 transcription factor family predicted by final transcripts of L. chinense var. rubrum. (C). Scatter diagram of LncRNAs and mRNA length distribution of L. chinense var. rubrum. (D). Annotation of full-length noncoding final transcripts predicted by cnci, pfam, plek and cpc software; Figure S8: The number of up- and down-regulated different expression LncRNA in the different compared combinations; Figure S9: GO enrichment analysis of DETs between XNNC VS LDPF group; Figure S10: GO enrichment analysis of DETs between XNNC VS XNXY group; Figure S11: GO enrichment analysis of DETs between XNFJ VS LDPF group; Figure S12: GO enrichment analysis of DETs between XNFJ VS XNNC group; Figure S13: GO enrichment analysis of DETs between XNFJ VS XNXY group; Figure S14: GO enrichment analysis of DETs between XNXY VS LDPF group; Figure S15: The top 20 enriched KEGG pathway of the differentially expressed genes (DEGs) among of different cultivars of L. chinense var. rubrum. (A). The enrichment pathway of XNNC VS LDPF. (B). The enrichment pathway of XNNC VS XNXY. (C). The enrichment pathway of XNFJ VS LDPF. (D). The enrichment pathway of XNFJ VS XNNC. (E). The enrichment pathway of XNFJ VS XNXY. (F). The enrichment pathway of XNXY VS LDPF; Figure S16: The Venn diagram of DETs between different comparisons; Figure S17: Correlation analyses of differentially expressed transcripts (DETs) involved in flavonoids; Figure S18: Correlation analyses of differentially expressed transcripts (DETs) involved in anthocyanins; Table S1: Type and content of flavonoids in four Loropetalum cultivars; Table S2: The differential accumulated metabolites in group of LDPF VS XNNC; Table S3: The differential accumulated metabolites in group of LDPF VS XNFJ; Table S4: The differential accumulated metabolites in group of LDPF VS XNFJ; Table S5: The differential accumulated metabolites in group of XNNC VS XNFJ; Table S6: The differential accumulated metabolites in group of XNNC VS XNXY; Table S7: The differential accumulated metabolites in group of XNFJ VS XNXY; Table S8: The differential accumulated flavonoid metabolites in all groups of L. chinense and L. chinense var. rubrum; Table S9: The differentially concentration of flavonoid metabolites analysis in group of LDPF VS XNNC; Table S10: The differentially concentration of flavonoid metabolites analysis in group of LDPF VS XNFJ; Table S11: The differentially concentration of flavonoid metabolites analysis in group of LDPF VS XNXY; Table S12: The differentially concentration of flavonoid metabolites analysis in group of XNNC VS XNFJ; Table S13: The differentially concentration of flavonoid metabolites analysis in group of XNNC VS XNXY; Table S14: The differentially concentration of flavonoid metabolites analysis in group of XNFJ VS XNXY; Table S15: The compared transcriptome of LDPF VS XNNC on KEGG metabolic pathway annotation; Table S16: The compared transcriptome of LDPF VS XNNC on KEGG metabolic pathway annotation; Table S17: The compared transcriptome of LDPF VS XNXY on KEGG metabolic pathway annotation; Table S18: The compared transcriptome of XNNC VS XNFJ on KEGG metabolic pathway annotation; Table S19: The compared transcriptome of XNNC VS XNXY on KEGG metabolic pathway annotation; Table S20: The compared transcriptome of XNFJ VS XNXY on KEGG metabolic pathway annotation; Table S21: Quality statistics of filtered reads; Table S22: Major indicators of the full-length transcriptome of Loropetalum chinense var. rubrum; Table S23: The GO enrichment analysis of DETs between XNNC VS LDPF group; Table S24: The GO enrichment analysis of DETs between XNNC VS XNXY group; Table S25: The GO enrichment analysis of DETs between XNFJ VS LDPF group; Table S26: The GO enrichment analysis of DETs between XNFJ VS XNNC group; Table S27: The GO enrichment analysis of DETs between XNFJ VS XNXY group; Table S28: The GO enrichment analysis of DETs between XNXY VS LDPF group; Table S29: The KEGG enrichment analysis of DETs between XNNC VS LDPF group; Table S30: The KEGG enrichment analysis of DETs between XNNC VS XNXY group; Table S31: The KEGG enrichment analysis of DETs between XNFJ VS LDPF group; Table S32: The KEGG enrichment analysis of DETs between XNFJ VS XNNC group; Table S33: The KEGG enrichment analysis of DETs between XNFJ VS XNXY group; Table S34: The KEGG enrichment analysis of DETs between XNXY VS LDPF group.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, M. C. and Y.X.; methodology, X.Z.; software, D.Z.; validation, Y.L., L.L. and L.Z.; formal analysis, L.Z.; investigation, X.Y.; resources, D.L. Z.; data curation, M.S.; writing—original draft preparation, Y. L.; writing—review and editing, Y.L.; visualization, M.C.; supervision, Y.L.; project administration, X.X.; funding acquisition, X.Y. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research was funded by “THE FORESTRY SCIENCE AND TECHNOLOGY INNOVATION FOUNDATION OF HUNAN PROVINCE FOR DISTINGUISHED YOUNG SCHOLARSHIP, grant number XLKJ202205”; “OPEN PROJECT OF HORTICULTURE DISCIPLINE OF HUNAN AGRICULTURAL UNIVERSITY, grant number 2021YYXK001”; “THE FOUND OF CHANGSHA MUNICIPAL SCIENCE AND TECHNOLOGY BUREAU, grant number KQ2202227”; “KEY PROJECT OF HUNAN PROVINCIAL EDUCATION DEPARTMENT, grant number 22A0155”; “THE FORESTRY BUREAU FOR INDUSTRIALIZATION MANAGEMENT OF HUNAN PROVINCE, grant number 2130221”; “HUNAN PROVINCIAL EDUCATION DEPARTMENT TEACHING REFORM PROJECT, grant number 2021JGYB101” and “HUNAN AGRICULTURAL UNIVERSITY TEACHING REFORM RESEARCH PROJECT, grant number XJJG-2020-071”.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- Xia, Z.; Damao, Z.; Li, Z.; Xiangfei, W.; Xingyao, X.; Dexin, G.; Xiaoying, Y.; and Yanlin, L. The Whole Genome Analysis of Loropetalum chinense var. Rubrum. Molecular Plant Breeding 2020, 18, 7023–7029. [Google Scholar]
- Shi, F. Comparison and Application of Lorpetalum Chinense Var.Rubrum and Photinia Fraseryin Landscape. Journal of Hengyang Normal University 2016, 37, 106–111. [Google Scholar]
- Shao, Z.; Hou, W.; Long, X.; Yang, G.; Chen, C.; Zeng, X.; Hou, B.; Gefei, Y.U.; Wenwen, W.U. The formation and development of the geography symbol product Loropetalum chinense var. rubrum in Liuyang City. Hunan Forestry Science & Technology 2007, 2, 71–73. [Google Scholar]
- Zhang, D.; Cai, W.; Zhang, X.; Li, W.; Zhou, Y.; Chen, Y.; Mi, Q.; Jin, L.; Xu, L.; Yu, X.; et al. Different pruning level effects on flowering period and chlorophyll fluorescence parameters of Loropetalum chinense var. rubrum. Peerj 2022, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, X.; Zhang, X.; Zhang, D.; Li, W.; Xiang, L.; Yang, Y.; Li, Y.; Xu, L. Phenotypic Diversity Analysis of the Progeny Variation of a ‘Mosaic Leaf’ Loropetalum chinense var. rubrum Based on Flower Organ Characteristics. Diversity 2022, 14. [Google Scholar] [CrossRef]
- Chen, Q.; Cai, W.; Zhang, X.; Zhang, D.; Li, W.; Xu, L.; Yu, X.; Li, Y. The Comparative Studies on Phytochemicals of Leaf Coloration of Loropetalum chinense var. rubrum. Acta Horticulturae Sinica 2021, 48, 1969–1982. [Google Scholar] [CrossRef]
- Iwashina, T. Contribution to Flower Colors of Flavonoids Including Anthocyanins: A Review. Natural Product Communications 2015, 10, 529–544. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F. Flower Colour. In Annual Plant Reviews 2018; Volume 20, pp. 201-239.
- Goto, T. Structure, stability and color variation of natural anthocyanins. In Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Springer: 1987; Volume 52, pp. 113-158.
- Koes, R.E.; Van Blokland, R.; Quattrocchio, F.; Van Tunen, A.J.; Mol, J.N. Chalcone synthase promoters in petunia are active in pigmented and unpigmented cell types. The Plant Cell 1990, 2, 379–392. [Google Scholar] [CrossRef]
- Hůla, M.; Flegr, J. What flowers do we like? The influence of shape and color on the rating of flower beauty. PeerJ 2016, 4, e2106. [Google Scholar] [CrossRef]
- Yin, X.; Wang, T.; Zhang, M.; Zhang, Y.; Irfan, M.; Chen, L.; Zhang, L. Role of core structural genes for flavonoid biosynthesis and transcriptional factors in flower color of plants. Biotechnology & Biotechnological Equipment 2021, 35, 1214–1229. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F.; Chandler, S. Recent progress of flower colour modification by biotechnology. International journal of molecular sciences 2009, 10, 5350–5369. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F. Flower colour and cytochromes P450. Philos Trans R Soc Lond B Biol Sci 2013, 368, 20120432. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant Journal 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Liang, C.-Y.; Rengasamy, K.P.; Huang, L.-M.; Hsu, C.-C.; Jeng, M.-F.; Chen, W.-H.; Chen, H.-H. Assessment of violet-blue color formation in Phalaenopsis orchids. BMC plant biology 2020, 20, 1–16. [Google Scholar] [CrossRef]
- Diretto, G.; Jin, X.; Capell, T.; Zhu, C.; Gomez-Gomez, L. Differential accumulation of pelargonidin glycosides in petals at three different developmental stages of the orange-flowered gentian (Gentiana lutea L. var. aurantiaca). PloS one 2019, 14, e0212062. [Google Scholar] [CrossRef]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The flavonoid biosynthesis network in plants. International journal of molecular sciences 2021, 22, 12824. [Google Scholar] [CrossRef]
- Haslam, E. Practical polyphenolics: from structure to molecular recognition and physiological action; Cambridge University Press: 1998.
- Winkel-Shirley, B. Molecular genetics and control of anthocyanin expression. Advances in Botanical Research 2002, 37, 75–94. [Google Scholar]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. The arabidopsis book 2011, 9, e0152. [Google Scholar] [CrossRef]
- Schröder, G. Stilbene and chalcone synthases: related enzymes with key functions in plant-specific pathways. Zeitschrift für Naturforschung C 1990, 45, 1–8. [Google Scholar] [CrossRef]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. The Plant Journal 1995, 8, 659–671. [Google Scholar] [CrossRef]
- van Tunen, A.J.; Mur, L.A.; Recourt, K.; Gerats, A.; Mol, J. Regulation and manipulation of flavonoid gene expression in anthers of petunia: the molecular basis of the Po mutation. The Plant Cell 1991, 3, 39–48. [Google Scholar]
- Wisman, E.; Hartmann, U.; Sagasser, M.; Baumann, E.; Palme, K.; Hahlbrock, K.; Saedler, H.; Weisshaar, B. Knock-out mutants from an En-1 mutagenized Arabidopsis thaliana population generate phenylpropanoid biosynthesis phenotypes. Proceedings of the National Academy of Sciences 1998, 95, 12432–12437. [Google Scholar] [CrossRef]
- Holton, T.A.; Brugliera, F.; Lester, D.R.; Tanaka, Y.; Hyland, C.D.; Menting, J.G.; Lu, C.-Y.; Farcy, E.; Stevenson, T.W.; Cornish, E.C. Cloning and expression of cytochrome P450 genes controlling flower colour. Nature 1993, 366, 276–279. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant physiology 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Schoenbohm, C.; Martens, S.; Eder, C.; Forkmann, G.; Weisshaar, B. Identification of the Arabidopsis thaliana flavonoid 3'-hydroxylase gene and functional expression of the encoded P450 enzyme. Bio. Chem. 2000, 381, 749–753. [Google Scholar] [CrossRef]
- Shirley, B.W.; Hanley, S.; Goodman, H.M. Effects of ionizing radiation on a plant genome: analysis of two Arabidopsis transparent testa mutations. The Plant Cell 1992, 4, 333–347. [Google Scholar]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. Journal of experimental botany 2011, 62, 2465–2483. [Google Scholar] [CrossRef]
- Zhang, Y.; Chu, G.; Hu, Z.; Gao, Q.; Cui, B.; Tian, S.; Wang, B.; Chen, G. Genetically engineered anthocyanin pathway for high health-promoting pigment production in eggplant. Molecular Breeding 2016, 36, 1–14. [Google Scholar] [CrossRef]
- Tohge, T.; Nishiyama, Y.; Hirai, M.Y.; Yano, M.; Nakajima, J.i.; Awazuhara, M.; Inoue, E.; Takahashi, H.; Goodenowe, D.B.; Kitayama, M. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. The Plant Journal 2005, 42, 218–235. [Google Scholar] [CrossRef]
- Veitch, N.C.; Grayer, R.J. Flavonoids and their glycosides, including anthocyanins. Natural product reports 2008, 25, 555–611. [Google Scholar] [CrossRef]
- Wei, X.; Yu, X.; Chen, J.; Fu, H.; Hu, B.; Chen, Y.; Da, L. Cloning and Sequence Analyzing of Chalcone Synthase Gene in Loropetalum chinense var.rubrum. Chinese Agricultural Science Bulletin 2013, 29, 24–28. [Google Scholar]
- Li, C.H.; Duo, D.Y.; Liao, X.S.; Zhang, Y.B. Cloning and Bioinformatic Analysis of Two Chalcone Synthases from Loropetalum Chinense var. Rubrum. Journal of Hunan University of Technology 2020, 34, 92–98. [Google Scholar]
- Rong, D.; Zhang, X.; Pan, T.; Wang, J.; Yang, G.; Zhang, B. Cloning,Expression and Transformation of LcFLS1 Gene fromLoropetalum chinense var.rubrum. Acta Botanica Boreali-Occidentalia Sinica 2019, 28, 1877–1887. [Google Scholar]
- Zhang, B.Y.; Li, C.H.; Liu, X.; Liao, X.S.; Rong, D.Y. Cloning and subcellular localization analysis of LcDFR1 and LcDFR2 in Loropetalum chinense var. rubrum. Journal of Southern Agriculture 2020, 51, 2865–2874. [Google Scholar]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant–environment interactions. Journal of integrative plant biology 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Ma, D.; Constabel, C.P. MYB repressors as regulators of phenylpropanoid metabolism in plants. Trends in Plant Science 2019, 24, 275–289. [Google Scholar] [CrossRef]
- Ohtani, M.; Demura, T. The quest for transcriptional hubs of lignin biosynthesis: beyond the NAC-MYB-gene regulatory network model. Current opinion in biotechnology 2019, 56, 82–87. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Šamec, D.; Tomczyk, M.; Milella, L.; Russo, D.; Habtemariam, S.; Suntar, I.; Rastrelli, L.; Daglia, M.; Xiao, J. Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnology advances 2020, 38, 107316. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends in plant science 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Applied Microbiology and Biotechnology 2022, 1–16. [Google Scholar] [CrossRef]
- Kiferle, C.; Fantini, E.; Bassolino, L.; Povero, G.; Spelt, C.; Buti, S.; Giuliano, G.; Quattrocchio, F.; Koes, R.; Perata, P. Tomato R2R3-MYB proteins SlANT1 and SlAN2: same protein activity, different roles. PLoS One 2015, 10, e0136365. [Google Scholar] [CrossRef]
- Cao, Y.; Jia, H.; Xing, M.; Jin, R.; Grierson, D.; Gao, Z.; Sun, C.; Chen, K.; Xu, C.; Li, X. Genome-wide analysis of MYB gene family in Chinese bayberry (Morella rubra) and identification of members regulating flavonoid biosynthesis. Frontiers in plant science 2021, 12, 1244. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, X.; Huang, R.; Yang, S.; Li, M.; Guo, Y. CRISPR/Cas9-mediated targeted mutation reveals a role for AN4 rather than DPL in regulating venation formation in the corolla tube of Petunia hybrida. Horticulture research 2021, 8. [Google Scholar] [CrossRef]
- Lai, Y.-S.; Shimoyamada, Y.; Nakayama, M.; Yamagishi, M. Pigment accumulation and transcription of LhMYB12 and anthocyanin biosynthesis genes during flower development in the Asiatic hybrid lily (Lilium spp.). Plant science 2012, 193, 136–147. [Google Scholar] [CrossRef]
- Nesi, N.; Debeaujon, I.; Jond, C.; Pelletier, G.; Caboche, M.; Lepiniec, L. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. The Plant Cell 2000, 12, 1863–1878. [Google Scholar] [CrossRef]
- Qi, F.; Liu, Y.; Luo, Y.; Cui, Y.; Lu, C.; Li, H.; Huang, H.; Dai, S. Functional analysis of the ScAG and ScAGL11 MADS-box transcription factors for anthocyanin biosynthesis and bicolour pattern formation in Senecio cruentus ray florets. Horticulture Research 2022. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Fang, Z.-Z.; Ye, X.-F.; Pan, S.-L. Identification of candidate genes involved in anthocyanin accumulation in the peel of jaboticaba (Myrciaria cauliflora) fruits by transcriptomic analysis. Gene 2018, 676, 202–213. [Google Scholar] [CrossRef]
- Dong, T.; Han, R.; Yu, J.; Zhu, M.; Li, Z. Anthocyanins Accumulation and Molecular Analysis of Correlated Genes by Metabolome and Transcriptome in Green and Purple Asparaguses ( Asparagus Officinalis, L.). Food Chemistry 2018, 271, 18–28. [Google Scholar] [CrossRef]
- Wrolstad, R.E.; Culbertson, J.D.; Cornwell, C.J.; Mattick, L.R. Detection of adulteration in blackberry juice concentrates and wines. Journal - Association of Official Analytical Chemists 1982, 65, 1417–1423. [Google Scholar] [CrossRef]
- Leena, S.; Eric, R. LoRDEC: accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Fu; L. ; Niu; B.; Zhu; Z.; Wu; S.; Li; W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics Oxford 2012. [Google Scholar]
- Yangyang, D.; Jianqi, L.I.; Songfeng, W.U.; Yunping, Z.H.U.; Yaowen, C.; Fuchu, H.E. Integrated nr database in protein annotation system and its localization. Comput Eng 2006, 32. [Google Scholar]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002. [Google Scholar] [CrossRef]
- Finn, R.D.; Alex, B.; Jody, C.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: the protein families database. Nucleic Acids Research 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003, 4, 41. [Google Scholar] [CrossRef]
- 59. Bairoch; Apweiler. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 2000.
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Research 2004, 32, D277. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: tool for the unification of biology. Nature Genetics 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schffer, A.A.; Zhang, J.; Zhang, Z.; Webb, M.; Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 1997, 25, 3389. [Google Scholar] [CrossRef]
- Benjamin; Buchfink; Chao; Xie; Daniel, H.; Huson. Fast and sensitive protein alignment using DIAMOND. Nature methods 2015.
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Research 2013. [Google Scholar] [CrossRef]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J Bioinform Comput Biol 2006, 4, 649–664. [Google Scholar] [CrossRef]
- Zheng; Jiao; Sun; HH; Rosli; HG; Pombo; MA; Zhang; PF. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. MOL PLANT 2016.
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic acids research 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Gao, G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research 2007, 35, W345–349. [Google Scholar] [CrossRef] [PubMed]
- Robert; Finn; Penelope; Coggill; Ruth; Eberhardt; Sean. The Pfam protein families database: towards a more sustainable future. Nucleic acids research 2016.
- Freire-Pritchett, P.; Ray-Jones, H.; Della Rosa, M.; Eijsbouts, C.Q.; Orchard, W.R.; Wingett, S.W.; Wallace, C.; Cairns, J.; Spivakov, M.; Malysheva, V. Detecting chromosomal interactions in Capture Hi-C data with CHiCAGO and companion tools. Nature protocols 2021, 16, 4144–4176. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. BIOINFORMATICS -OXFORD- 2010.
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology 2010, 11, R14. [Google Scholar] [CrossRef]
- Spinardi, A.; Cola, G.; Gardana, C.S.; Mignani, I. Variation of Anthocyanin Content and Profile Throughout Fruit Development and Ripening of Highbush Blueberry Cultivars Grown at Two Different Altitudes. Frontiers in Plant Science 2019, 10. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Shi, T.; Chen, M.; Wang, L. Integrating Transcriptomic and GC-MS Metabolomic Analysis to Characterize Color and Aroma Formation during Tepal Development in Lycoris longituba. Plants 2019, 8, 53. [Google Scholar] [CrossRef]
- Qiu, X.H.; Zhang, Y.J.; Yin, G.F.; Shi, C.Y.; Yu, X.Y.; Zhao, N.J.; Liu, W.Q. [Photosynthetic Parameters Inversion Algorithm Study Based on Chlorophyll Fluorescence Induction Kinetics Curve]. Guang pu xue yu guang pu fen xi = Guang pu 2015, 35, 2194–2197. [Google Scholar]
- Chen, S.M.; Li, C.H.; Zhu, X.R.; Deng, Y.M.; Sun, W.; Wang, L.S.; Chen, F.D.; Zhang, Z. The identification of flavonoids and the expression of genes of anthocyanin biosynthesis in the chrysanthemum flowers. Biologia Plantarum 2012, 56, 458–464. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, X.; Shu, Q.; Li, H.; Zhong, P.; Zhang, H.; Xu, Y.; Wang, L.; Wang, L. Relationship between the composition of flavonoids and flower colors variation in tropical water lily (Nymphaea) cultivars. PLoS One 2012, 7, e34335. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhu, Y.; Cui, G.; Zhang, X.; Hu, R.; Deng, Z.; Lei, L.; Wu, L.; Mei, L. A Comparative Study of Flavonoids and Carotenoids Revealed Metabolite Responses for Various Flower Colorations Between Nicotiana tabacum L. and Nicotiana rustica L. Front Plant Sci 2022, 13, 828042. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nature communications 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nature communications 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, L.; Chen, C.; An, X.; Zhang, Y.; Wang, Y.; Li, Q. Full-length transcriptome analysis of Phytolacca americana and its congener P. icosandra and gene expression normalization in three Phytolaccaceae species. BMC Plant Biol 2020, 20, 396. [Google Scholar] [CrossRef]
- Zhou, C.; Zhu, C.; Tian, C.; Xu, K.; Huang, L.; Shi, B.; Lai, Z.; Lin, Y.; Guo, Y. Integrated volatile metabolome, multi-flux full-length sequencing, and transcriptome analyses provide insights into the aroma formation of postharvest jasmine (Jasminum sambac) during flowering. Postharvest Biology and Technology 2022, 183, 111726. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genomics 2018, 19, 548. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Z.; He, M.; Gao, H.; Zhu, H.; Yun, Z.; Qu, H.; Jiang, Y. Unveiling the complexity of the litchi transcriptome and pericarp browning by single-molecule long-read sequencing. Postharvest Biology and Technology 2020, 168, 111252. [Google Scholar] [CrossRef]
- Gong, W.; Qiling, S.; Ji, K.; Gong, S.; Wang, L.; Chen, L.; Zhang, J.; Yuan, D. Full-Length Transcriptome from Camellia oleifera Seed Provides Insight into the Transcript Variants Involved in Oil Biosynthesis. Journal of Agricultural and Food Chemistry 2020, 68, 14670–14683. [Google Scholar] [CrossRef]
Figure 1.
Phenotypes of Loropetalum cultivars and total anthocyanin content. (A). Phenotypes of four Loropetalum cultivars, namely, 'Xiangnong Xiangyun' (XNXY), 'Huye Jimu 2' (LDPF), 'Xiangnong Fenjiao' (XNFJ) and 'Xiangnong Nichang' (XNNC) of 2 days after the flowering stage. (B). Total anthocyanin content in the petals of four Loropetalum cultivars, namely, 'Xiangnong Xiangyun' (XNXY), 'Huye Jimu 2' (LDPF), 'Xiangnong Fenjiao' (XNFJ) and 'Xiangnong Nichang' (XNNC). Error bars indicate standard error (+SE) of the mean. Different letters indicated significant differences at p≤0.05 level basing on Duncan's test.
Figure 1.
Phenotypes of Loropetalum cultivars and total anthocyanin content. (A). Phenotypes of four Loropetalum cultivars, namely, 'Xiangnong Xiangyun' (XNXY), 'Huye Jimu 2' (LDPF), 'Xiangnong Fenjiao' (XNFJ) and 'Xiangnong Nichang' (XNNC) of 2 days after the flowering stage. (B). Total anthocyanin content in the petals of four Loropetalum cultivars, namely, 'Xiangnong Xiangyun' (XNXY), 'Huye Jimu 2' (LDPF), 'Xiangnong Fenjiao' (XNFJ) and 'Xiangnong Nichang' (XNNC). Error bars indicate standard error (+SE) of the mean. Different letters indicated significant differences at p≤0.05 level basing on Duncan's test.
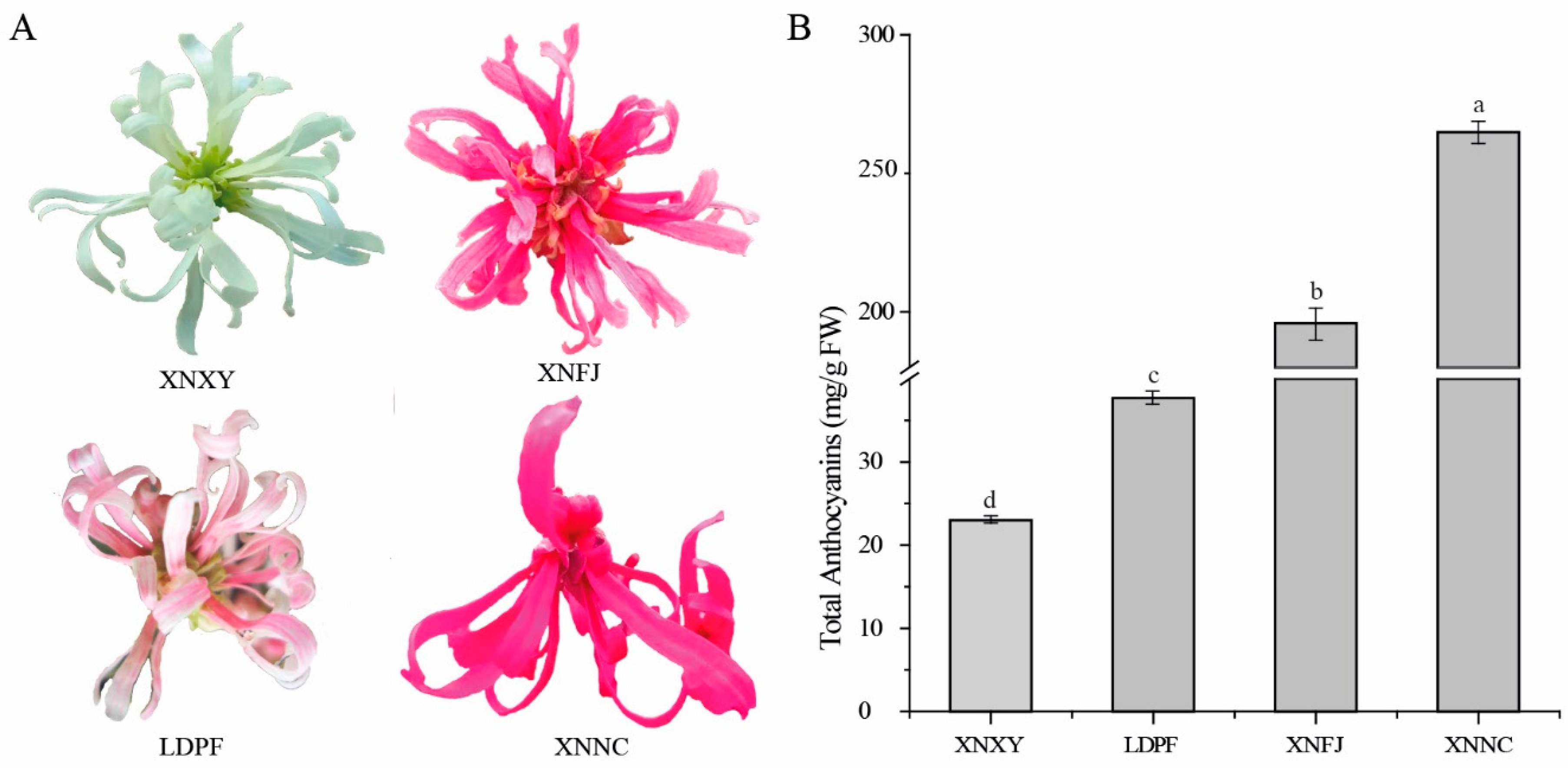
Figure 2.
Global metabolic changes among of different cultivars of L. chinense var. rubrum. (A). Classification and proportion of 207 flavonoid components in different cultivars of XNXY, XNFJ, XNNC and LDPF. (B). PCA of metabolites in different cultivars of XNXY, XNFJ, XNNC and LDPF. (C). Upset plot showing overlap of metabolites in different cultivars of XNXY, XNFJ, XNNC and LDPF. (D). Volcano map of metabolites between LDPF and XNNC. (E). Volcano map of metabolites between LDPF and XNFJ. (F). Volcano map of metabolites between LDPF and XNXY. (G). Volcano map of metabolites between XNNC and XNFJ. (H). Volcano map of metabolites between XNNC and XNXY. (I). Volcano map of metabolites between XNFJ and XNXY.
Figure 2.
Global metabolic changes among of different cultivars of L. chinense var. rubrum. (A). Classification and proportion of 207 flavonoid components in different cultivars of XNXY, XNFJ, XNNC and LDPF. (B). PCA of metabolites in different cultivars of XNXY, XNFJ, XNNC and LDPF. (C). Upset plot showing overlap of metabolites in different cultivars of XNXY, XNFJ, XNNC and LDPF. (D). Volcano map of metabolites between LDPF and XNNC. (E). Volcano map of metabolites between LDPF and XNFJ. (F). Volcano map of metabolites between LDPF and XNXY. (G). Volcano map of metabolites between XNNC and XNFJ. (H). Volcano map of metabolites between XNNC and XNXY. (I). Volcano map of metabolites between XNFJ and XNXY.
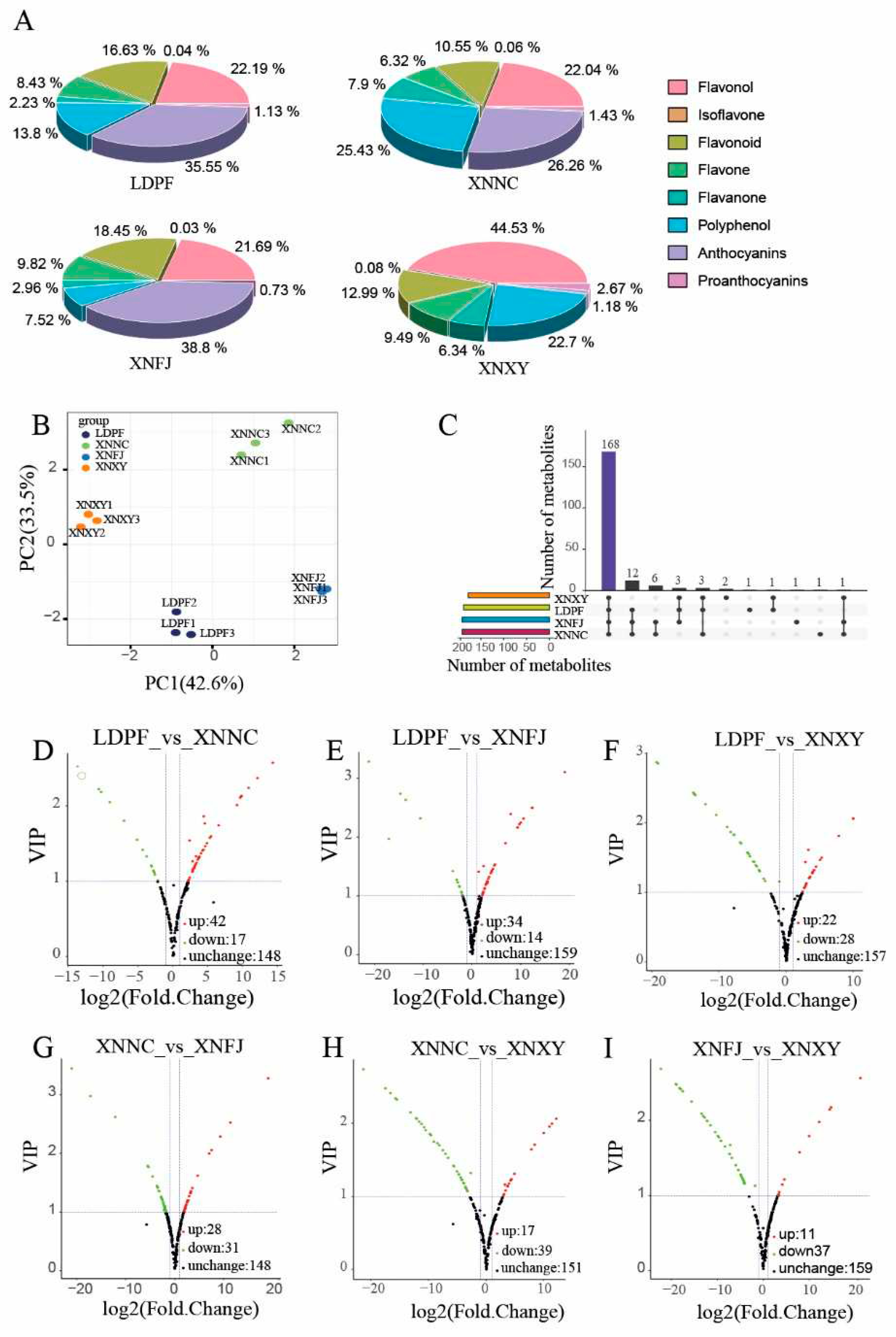
Figure 3.
The annotation results of full-length nonredundant final transcripts to public databases. (A). The static annotation results of full-length nonredundant final transcripts to seven databases. (B). Annotation of full-length non-redundant final transcripts to NR, NT, KOG, KEGG and GO.
Figure 3.
The annotation results of full-length nonredundant final transcripts to public databases. (A). The static annotation results of full-length nonredundant final transcripts to seven databases. (B). Annotation of full-length non-redundant final transcripts to NR, NT, KOG, KEGG and GO.
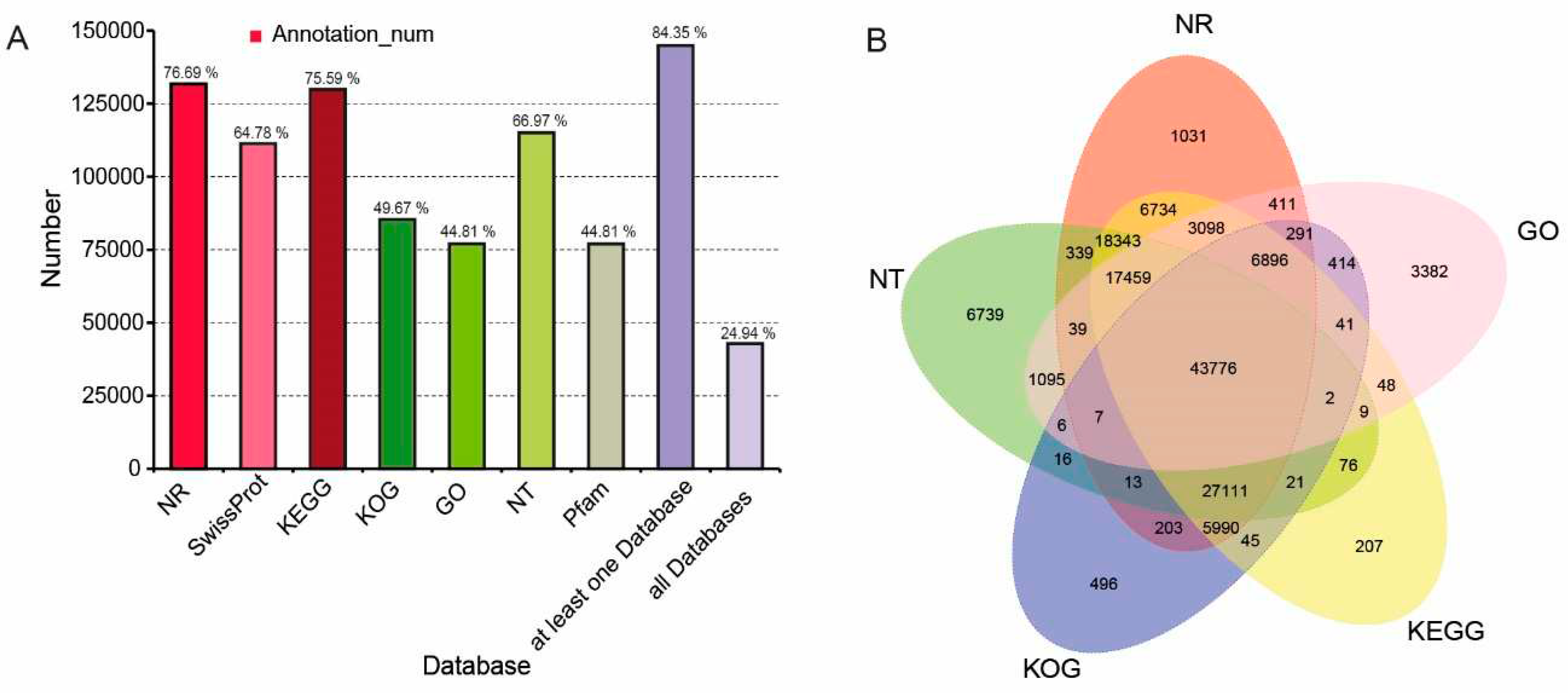
Figure 4.
RNA-seq data profiles of L. chinense var. rubrum. Note: XNFJ1-XNFJ3 belong to XNFJ, XNNC1-XNNC3 belong to XNNC, XNXY1-XNXY3 belong to XNXY, and LDPF1-LDPF3 belong to LDPF. (A). Box histogram of all transcript's expression among all samples. Note, FPKM value means Fragments per Kilobase of transcript per Million mapped reads. (B). Heatmap of Pearson's correlation coefficient of all transcript's expression among all samples. (C). Clustering tree of transcripts expression of all samples which shows the distance between samples. (D). PCA of transcripts expression of all samples which shows principal components 1 and 2 represent high cohesion within groups and good separation among different cultivars. (E). The number of up- and down-regulated different expression transcripts in the different compared combinations. Note, DEG_number: the number of differential expressed genes and transcripts.
Figure 4.
RNA-seq data profiles of L. chinense var. rubrum. Note: XNFJ1-XNFJ3 belong to XNFJ, XNNC1-XNNC3 belong to XNNC, XNXY1-XNXY3 belong to XNXY, and LDPF1-LDPF3 belong to LDPF. (A). Box histogram of all transcript's expression among all samples. Note, FPKM value means Fragments per Kilobase of transcript per Million mapped reads. (B). Heatmap of Pearson's correlation coefficient of all transcript's expression among all samples. (C). Clustering tree of transcripts expression of all samples which shows the distance between samples. (D). PCA of transcripts expression of all samples which shows principal components 1 and 2 represent high cohesion within groups and good separation among different cultivars. (E). The number of up- and down-regulated different expression transcripts in the different compared combinations. Note, DEG_number: the number of differential expressed genes and transcripts.
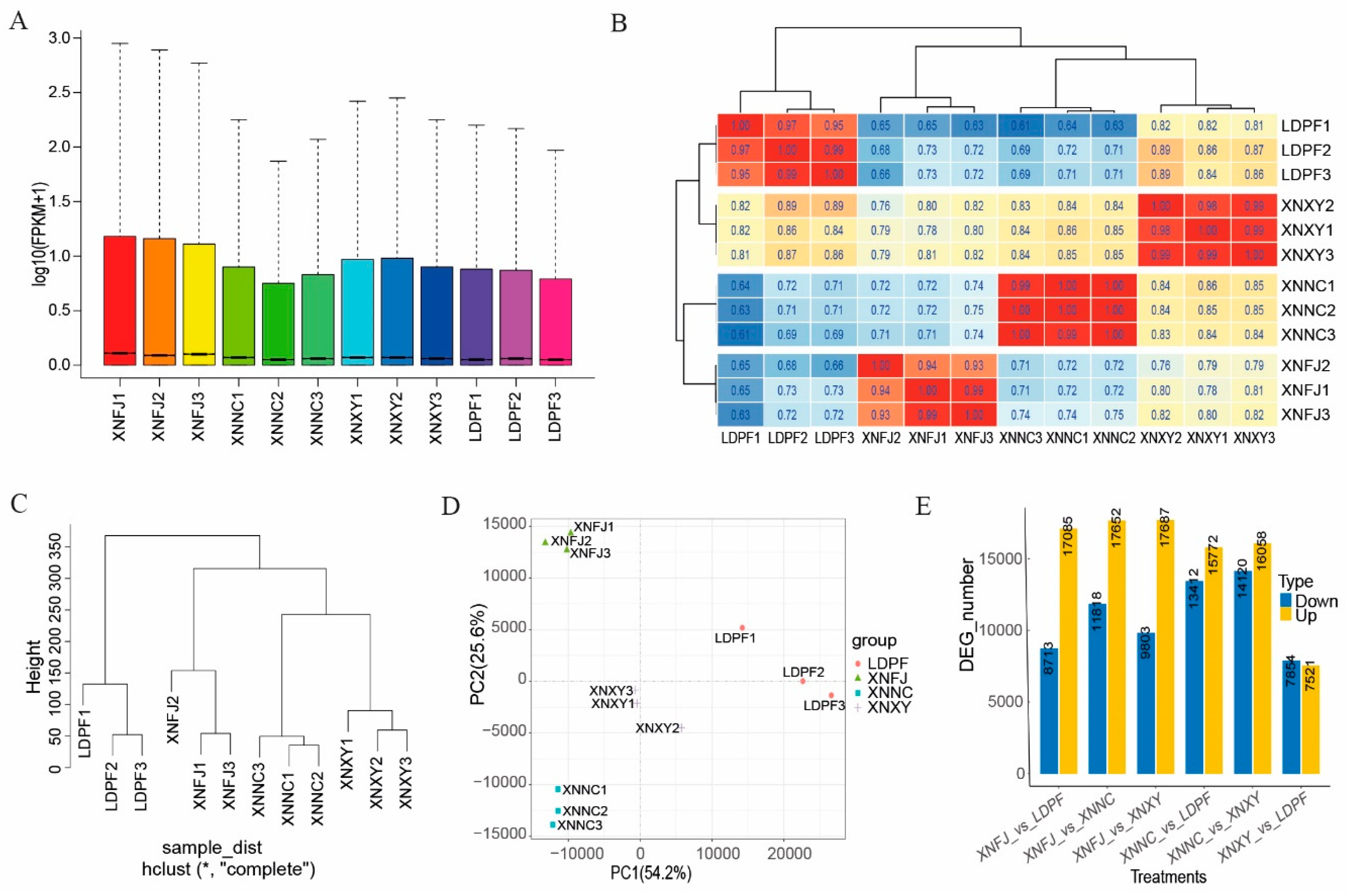
Figure 5.
Simplified representation of flavonoid metabolism and heat map produced by significant different gene related to flavonoid synthetic of L. chinense var. rubrum. (A). Simplified representation of flavonoid metabolism. Red rectangular boxes represented significant changed metabolism according to KEGG enrichment analysis. (B). Heat map produced by significant different gene related to flavonoid synthetic. And the gray rectangular boxes represented the enzyme coded by related genes. The orange square represented upregulated, and purple square represented downregulated. White square represented the gene related to flavonoid synthetic that hadn't significant changed.
Figure 5.
Simplified representation of flavonoid metabolism and heat map produced by significant different gene related to flavonoid synthetic of L. chinense var. rubrum. (A). Simplified representation of flavonoid metabolism. Red rectangular boxes represented significant changed metabolism according to KEGG enrichment analysis. (B). Heat map produced by significant different gene related to flavonoid synthetic. And the gray rectangular boxes represented the enzyme coded by related genes. The orange square represented upregulated, and purple square represented downregulated. White square represented the gene related to flavonoid synthetic that hadn't significant changed.
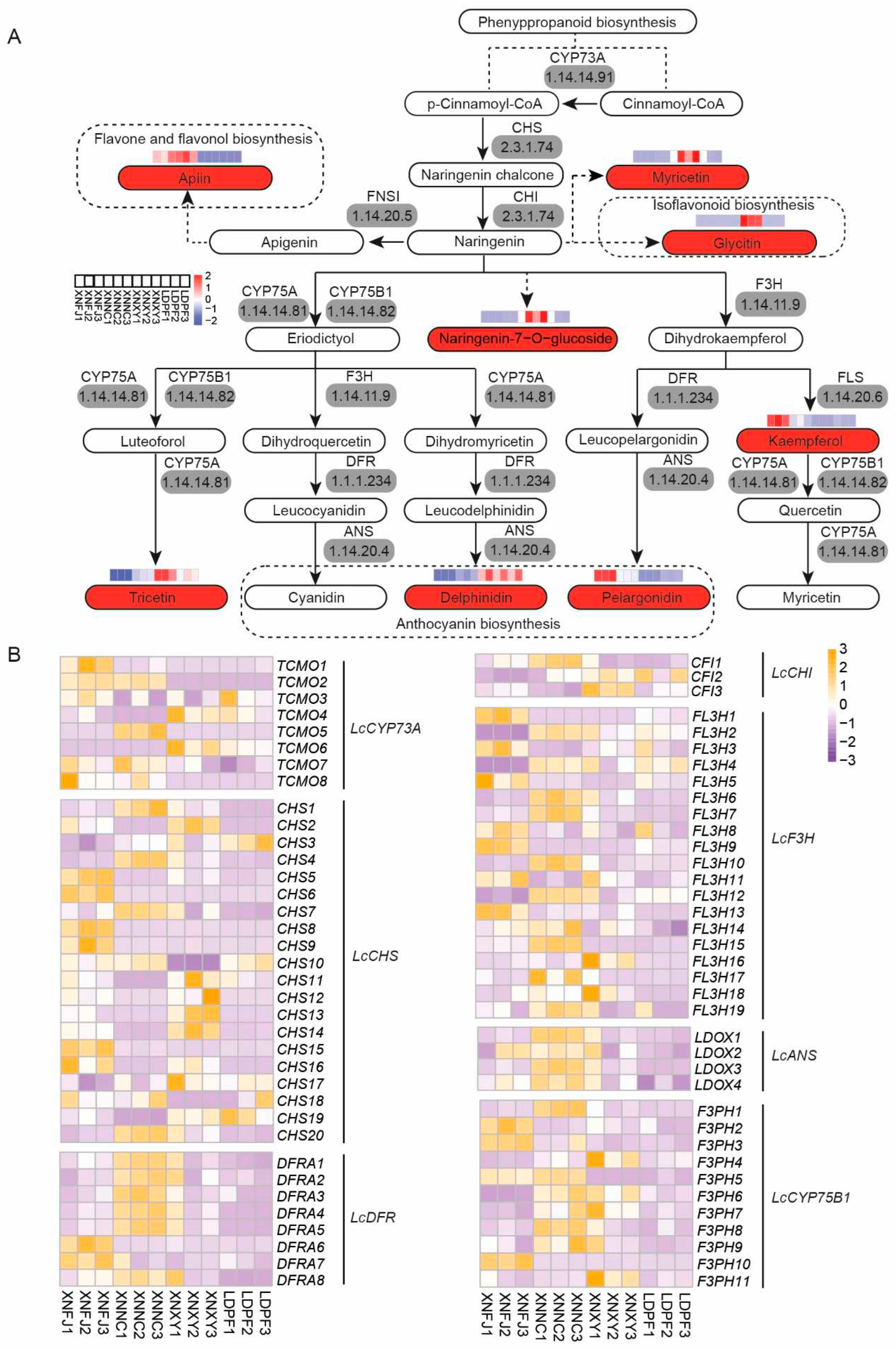
Figure 6.
Simplified representation of anthocyanins metabolism and heat map of related genes of L. chinense var. rubrum. (A). Simplified representation of flavonoid metabolism. (B). Heat map produced by significant different gene related to flavonoid synthetic. And the gray rectangular boxes represented the enzyme coded by related genes. The orange square represented upregulated, and purple square represented downregulated. White square represented the gene related to flavonoid synthetic that hadn't significant changed.
Figure 6.
Simplified representation of anthocyanins metabolism and heat map of related genes of L. chinense var. rubrum. (A). Simplified representation of flavonoid metabolism. (B). Heat map produced by significant different gene related to flavonoid synthetic. And the gray rectangular boxes represented the enzyme coded by related genes. The orange square represented upregulated, and purple square represented downregulated. White square represented the gene related to flavonoid synthetic that hadn't significant changed.
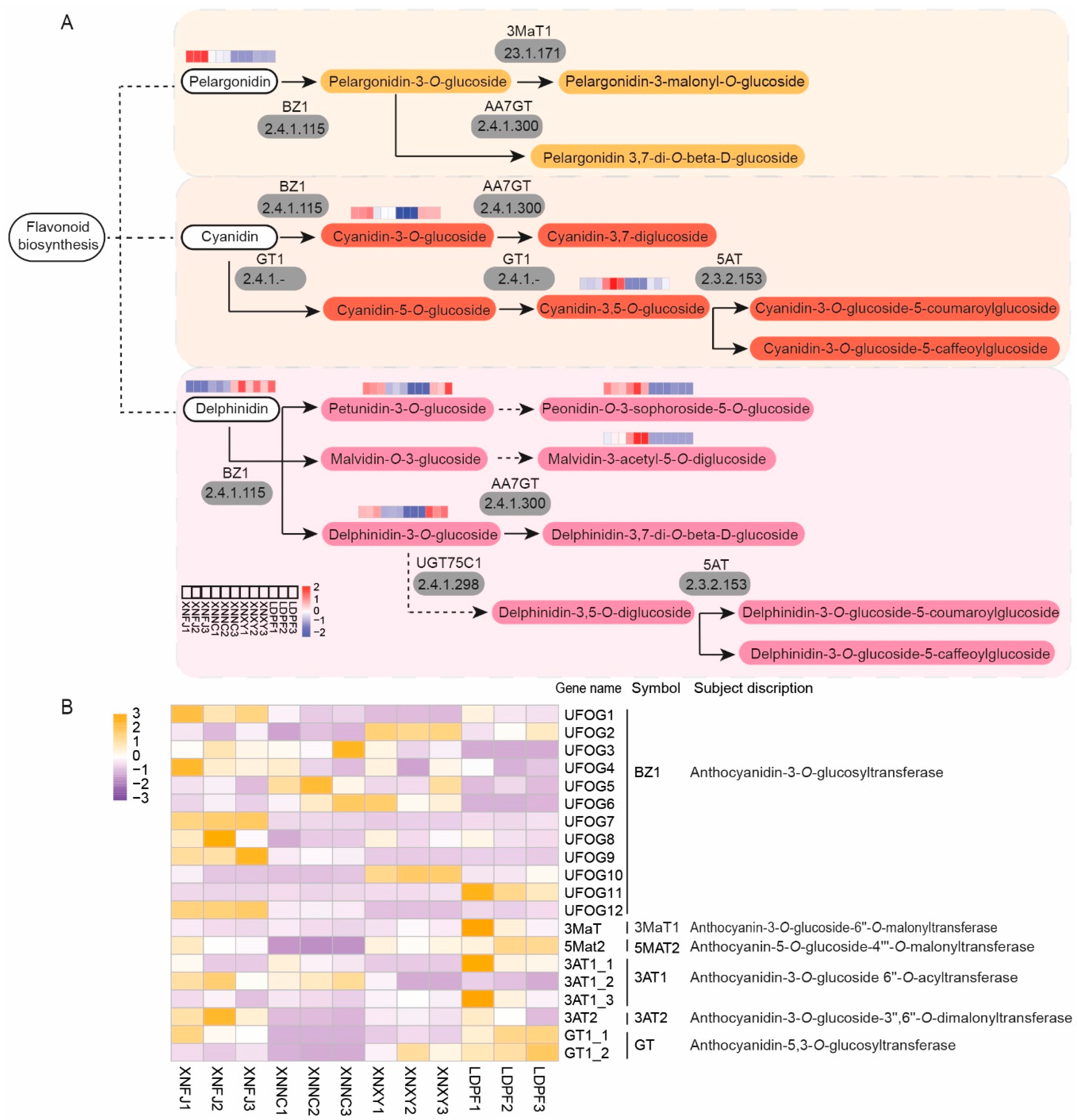
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



