
Submitted:
18 April 2023
Posted:
18 April 2023
You are already at the latest version
Abstract
Cholestasis is a condition defined as an abnormal decrease of bile flow due to progressive pathological states or cholestatic liver diseases that affect the biliary tree at intrahepatic and extrahepatic level. It induces complications such as cirrhosis, liver failure, malignancies, pruritus, fatigue, bone disease and nutritional deficiencies that merit close follow-up and specific interventions to improve quality of life. Furthermore, cholestasis can progress to end-stage liver disease and represents an entity with high morbidity and mortality; and economic burden, Therefore, it is important that clinicians understand the treatment options for cholestatic liver diseases. This review addresses the pathophysiology of cholangiopathies, a general view of current treatments and their molecular targets, and a specific review of those groups of drugs. The objective is to provide clinicians with an overview of the current treatment of cholangiopathies based on evidence on its efficacy and safety.
Keywords:
Cholestasis
; Bile acid
; Ursodeoxycholic acid
; Fibrosis
; FXR agonist
; PXR agonist
; Fibreates
1. Introduction
Bile is a digestive fluid produced by the liver and released into the bile duct. It flows directionally from the liver into the duodenum via the bile duct as a passageway. Cholestasis is a condition defined as an abnormal decrease of bile flow due to progressive pathological states or cholestatic liver diseases (CLD) that affect the biliary tree at intrahepatic level and extrahepatic level [1].
CLD includes a wide variety of genetic, congenital, immunological, infectious, and idiopathic disorders but among them, the primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) embody the most impactful and frequent CLD with a pooled point prevalence rate of 22.27 cases per 100,000 inhabitants (IC95% 17.98–27.01), and a pooled annual incidence rate of 1.87 new cases per 100,000 inhabitants (IC95%: 1.46–2.34); and a incidence approximately of 0.5 to 1.3 cases per 100,000 person-years while prevalence range from 3.85 to 16.2 cases per 100,000 person-years, respectively [2,3].
Cholestasis induces complications such as cirrhosis, liver failure, malignancies, pruritus, fatigue, bone disease and nutritional deficiencies that merit close follow-up and specific interventions to improve quality of life [4]. CLD can progress to end-stage liver disease and are entities with high morbidity and mortality but also with an important economic burden which derivate from the lack of effective treatments. CLD represent global annual total costs equal to € 913,763 (€ 942 per patient) and € 459,506 (50.3%, € 474 per patient) deriving from hospitalizations, mostly due to liver transplantation (LT), 30.5%, and cirrhosis complications, 20.6%). Costs from outpatient activities were estimated in € 109,090 (11.9%, € 112 per patient) [5]. From 1988 to 2018, CLD, and particularly PBC and PSC accounted 14.2% of all liver transplants. About 10% to 40% of these patients will have a recurrence of primary disease after LT [6]. The aim of this review is to offer clinicians a review of the updated literature on the treatment of cholestasis and associated complications.
2. Pathophysiology of Cholangiopathies
The general sequence of events leading to cholestasis and its associated damage can be summarized as: 1). Ductular reaction, 2) Biliary stasis (intrinsic and/or extrinsic obstruction), 3) Modification of bile components to a cytotoxic profile and 4) Proinflammatory and profibrotic state. It is essential to know the general aspects to understand the therapeutic targets of the different pharmacological strategies (Figure 1).
2.1. Ductular Reaction
Cholangiocytes are the metabolically active epithelium that lines the bile ducts. It participates in the formation and modification of bile through a series of transmembrane channels, transporters and exchangers that are expressed in the apical or basolateral domain of cholangiocytes [7]. There are multiple triggers or promoters of activation (environmental, antigens, toxins, genetic and epigenetic factors) cholangiocytic or ductular reaction (DR) in which the number of ductules increases, accompanied by infiltration of leukocytes and lymphocytes, activation of liver progenitor cells, and an increase in matrix protein levels [8]. Initially, DR is a response to an aggressor stimulus, it tries to limit and eliminate the harmful factor. If DR is perpetuated, proinflammatory pathways (Notch and Hedgehog) induce cholangiocyte maturation, fibronectin deposition and cytokine transcription (interleukin-6, interleukin-8, tumor necrosis factor and various growth factors) that generate histological and structural changes that condition cholestasis. Furthermore, cholangiocytes recognize the presence of pathogens through pattern recognition receptors (PRR), and the activation of these PRRs triggers a signaling cascade which results in the expression of various cytokines, immunoglobulins, adhesion molecules and a strong cellular response (CD4+ T cells, CD8+ T cells, B cells, macrophages and natural killer cells) that can release pro- and anti-inflammatory molecules and angiogenic, fibrogenic and proliferative factor. This is the basis for consideration of immunomodulatory therapies for CLD [9].
2.2. Biliary Stasis
Impaired bile flow is due to intrahepatic or extrahepatic obstruction of bile ducts. Impaired secretion by hepatocytes transport is another predisposing factor for cholestatic status, RD, increase in bile acids (BAs) concentration and mitochondrial dysfunction [6]. The dysfunction of apical and basolateral cotransporters of the cholangiocytes generates cholestasis impairing secretion by the canalicular membrane. Primary BAs are synthesized in the liver and before secretion in the bile canaliculi, undergo conjugation to form bile salts that decrease passive reabsorption. Once in the intestine, those primary bile salts undergo dehydroxylation and deconjugation by the intestinal microbiota, forming secondary acids. BAs are passively reabsorbed in the upper intestine, but 90-95% of them are reabsorbed into the terminal ileum via the apical sodium-dependent bile acid transporter (ASBT). Then, BAs enter the portal circulation via transport by the organic solute transporter (OST) α/β and subsequently return to the hepatocyte via the sodium-taurocholate cotransporter polypeptide (NTCP) or the organic anion transporter polypeptide (OATP). This process of enterohepatic circulation is regulated by feedback mechanisms to protect hepatocytes from bile acid-induced cytotoxicity. The main negative feedback mechanism that regulates BAs homeostasis is through the nuclear farnesoid X receptor (FXR). In the enterocytes, BAs bind to FXR to decrease BAs synthesis via CYP7A1. This pathway can also be suppressed by the activity of fibroblast enteral growth factor 19 (FGF-19), that binds to its hepatocyte receptor, fibroblast growth factor 4/β-klotho complex, to inhibit transcription of the CYP7A1 gene. Fibroblast growth factor 19 (FGF19), an endocrine gastrointestinal hormone, controls BAs metabolism through its effects on CYP7A1, the first and rate-limiting enzyme in the classic pathway of BAs synthesis [10]. In cholestasis, high levels of BAs induce fibroblast growth factor 19 (FGF-19) expression. The increased concentration of FGF-19 in the gut stimulates activation of the FGFR4/β-klotho receptor in the liver and it is associated with malignancy risk [11]. In addition, FXR activation leads to downregulation of the intestinal bile acid transporter ASBT, the hepatic uptake transporters NTCP and OATP, and upregulation of bile salt export pump (BSEP) of hepatic efflux transporters (Figure 1) (Figure 2) [6-9].
2.3. Citotoxic Profile of Biliary Acids
BAs properties help with digestion, leakage, and accumulation of BAs in hepatocytes but in higher concentrations they can lead to the activation of cholangiocytes, causing chronic inflammation, proliferation, apoptosis ant with progression of damage; subsequent fibrosis [1-9]. BAs are composed of four steroid rings that form a hydrocarbon network that has hydrophobic and hydrophilic regions, whose balance is variable and explains differences in their biological properties, including their choleretic potency, solubilization properties and activation of BAs receptors. Hydrophobic BAs are potent detergents, hydrophilic BAs are not, and they lack membrane-disrupting properties, thus being non hepatotoxic, even in high concentrations. This is relevant for the therapeutic use of hydrophilic BAs, like ursodeoxycholic acid (UDCA), tauroursodeoxycholic acid (TUDCA) and new semisynthetic BAs derivatives such as 24-norursodeoxycholic acid (Nor-UDCA) in the treatment of liver diseases [12]. Defense mechanisms include alkalinization of bile via increased bicarbonate secretion and decreasing their ability to diffuse and lessening their cytotoxic effects; the cholehepatic shunting of BAs when recirculated through the periductular capillary plexus, increasing bile flow and augmenting bicarbonate-rich choleresis, and BAs can modulate inflammatory pathways by binding to a variety of nuclear and surface receptors (i.e. FXR, TGR5, and PXR) throughout the intestines and bile ducts [1,9].
2.4. Profibrotic State
Ultimately, proliferation can lead to cell cycle disruption, senescence, and apoptosis, ductopenia, local angiogenesis, fibrosis, and recruitment of innate and adaptive immune cells, mesenchymal cells, and endothelial cells. Cholangiocyte dysfunction can lead to biliary fibrosis, through increased intracellular cyclic monophosphate adenosine (cAMP) levels, β-catenin-mediated secretion of pro-inflammatory cytokines and chemokines. Those process induce to early recruitment of macrophages, activation of latent Transforming growth factor beta (TGFβ) and accumulation of myofibroblasts (Figure 2) [13]. Unless reversed, these events lead to periportal fibrosis, ductopenia, eventually biliary cirrhosis and sometimes malignant transformation [3].
3. Therapeutic Options
Based on pathophysiology, the treatment of cholestasis should pursue the following objectives: limit BAs injury by modulating the hydrophobicity of the pool or reduce the size of the pool by interfering with the intestinal absorption, induce choleresis to deload hepatocytes from BAs and to limit cholangiocyte damage, but also modulate inflammation.
3.1. Hydrophilic Bile Acids (BA): UDCA
UDCA (3α,7β-dihydroxy-5β-cholanoic acid) accounts for 1 – 3% of human bile, most of it is conjugated with glycine. When administered orally, it turns the BAs pool more hydrophilic (up to 40%) thus reducing the toxic effect of hydrophobic BAs [12]. This has been shown it patients with PSC, reaching a plateau at doses of 22-25 mg/kg [14]. Although a previous study showed an unchanged hydrophobic BAs pool [15]. The discrepancies in the evidence led to a deeper investigation regarding the effects of UDCA in CLD. Since then, many other mechanisms of action have been described as an intracellular signaling molecule (Ca2+ agonist), activator of protein kinases (MAPK: Erk1/2, p38MAPK) and α5β1 integrins in hepatocytes, and a signaling molecule that stimulates vesicular exocytosis and, therefore causing a choleretic effect. There is also the “The biliary HCO3− umbrella” hypothesis, which states that the alkaline pH near the apical surface, due to HCO3− secretion by hepatocytes and colongiocytes, prevents the permeation of hydrophobic BAs (Figure 1) [16].
The clinical effect of UDCA was first proven in patients with PBC in a prospective study that showed improvement in liver function tests and in pruritus [17]. Later, it was shown, in placebo-controlled trials, that it improves liver histology and delays progression to cirrhosis and LT [18,19,20]. Because of its efficacy and safety, UDCA is recommended for all patients with PBC as a first-line therapy [21].
In patients with PSC the use of UDCA as treatment has not shown such positive results. A 2009 meta-analysis of randomized controlled trials showed that UDCA can improve liver biochemistry but has no effect on liver histology or survival free of transplantation [22]. Another meta-analysis comprising 567 patients found no significant difference in mortality [OR, 0.6 (95% CI, 0.4–1.4)], main symptoms such as pruritus [OR,1.5 (95% CI, 0.3–7.2)], or fatigue [OR, 0.0 (95% CI, 0.1–7.7)], and no difference in the risk of cholangiocarcinoma [OR, 1.7 (95% CI, 0.6–5.1)] and in histology stage progression [OR, 0.9 (95% CI, 0.34–2.44)] [23]. A 5-year Scandinavian multicenter trial with high UDCA doses (17 - 23 mg/kg/d) vs. placebo could not show a significant difference in symptoms, liver biochemistry, risk of cholangiocarcinoma, death, or LT. [24]. Studies abour testing higher UDCA doses (28 – 30 mg/kg/d) had to be stopped prematurely because of severe adverse events [25]. There is few evidence about worsening of liver biochemistry when UDCA is discontinued in patients with PSC. In this regard one study showed improvement when therapy was reinstated [26].
UDCA, in patients with PSC, has also been studied as chemoprophylaxis for colorectal cancer and cholangiocarcinoma, showing conflicting data. Some small studies (52 and 59 patients) have shown lower incidence of colorectal dysplasia in patients with PSC treated with UDCA [27,28]. A randomized clinical trial of 98 patients with UDCA 17 – 23 mg/kg/d, showed no difference in the rate of colorectal neoplasia at 5 nor 15 years follow up [29], and higher doses (28 – 30 mg/kg/d) have been associated with more risk of low-grade colonic dysplasia [30]. One meta-analysis found a protective effect of UDCA for colorectal cancer and/or high-grade dysplasia, which did not hold for early lesions or for overall risk [31]. Another meta-analysis showed no significant effect of UDCA on risk of colorectal neoplasia [32].
Due to the shown evidence, the recommendation made by EASL and AASLD, regarding UDCA in PSC patients, is that UDCA can be used at doses of 13 – 23 mg/kg/d, because of its effect in liver biochemistry. Both associations recommend against using doses of 28 – 30 mg/kd/d [21,33]. Regarding UDCA in PBC, as stated above it is the first-line therapy in main guidelines. Yet, an important number of patients are unresponsive or intolerant to UDCA. Besides, the efforts to develop therapeutic options with better impact on quality-of-life or progression of CLD has led to produce new drugs or redirect old ones to other molecular targets.
3.2. FXR Agonists: Tropifexor (LJN-452), Cilofexor (GS-9674) and EDP-305
This group of molecules are compounds without the classical BAs structure, but able to bind and activate FXR, inducing beneficial effects such as reducing hepatic fibrosis since it prevents toxicity due to accumulation of BAs. This pharmaceutical approach is potentially used like the second therapeutic line in patients who have an inadequate response to UDCA (approximately 40% of PBC) [11,34].
Tropifexor is a non-bile acid FXR agonist that has been optimized for enhanced fit within the ligand-binding domain of FXR. In animal models, potently regulated FXR target genes in the liver and intestine showed superior efficacy to obeticholic acid in non-alcoholic steatohepatitis (NASH) and cholestasis. Tropifexor has a pharmacokinetic profile suitable for once daily dosing in humans and has shown effective FXR target engagement via transient and dose-dependent increases in FGF19 in healthy volunteers and patients with NASH and primary bile acid diarrhea (pBAD).
Tropifexor was found to be safe and well tolerated, with improved levels of markers of bile duct injury at very low doses. Itch of mild to moderate severity was observed in all groups including placebo but was more frequent at the highest tropifexor dose. A phase 2 study investigated tropifexor efficacy in PBC patients with inadequate UDCA response. Patients were randomized in receiving once daily doses of 30 μg, 60 μg, or 90 μg of tropifexor or placebo for 4 weeks. In the 90 μg group, gamma-glutamyl transferase (GGT) levels showed 72% reduction. HDL was reduced by 33% and 26% at the doses of 60 and 90 μg, respectively. No increase was observed in LDL or total cholesterol [34]. In a more recent study, 61 enrolled patients received 30, 60, 90, and 150 μg tropifexor, respectively and 21 received placebo. By day 28, tropifexor caused 26–72% reduction in GGT from baseline at 30 to 150 μg doses (p <0.001 at 60, 90, and 150 μg tropifexor vs. placebo). Pruritus was the most frequent adverse event in tropifexor group with most events of mild to moderate severity. It was also observed decreases seen in LDL, HDL, and total cholesterol levels. Tropifexor showed improvement in cholestatic markers when compared to placebo [35].
Cilofexor is another non-steroidal FXR agonist tested in a phase II double-blind, placebo- controlled study in patients without cirrhosis with large-duct PSC. They were randomized to receive cilofexor 100 mg, 30 mg, or placebo orally once daily for 12 weeks. 52 patients were randomized to dose-dependent reductions. In the group of 100 mg, was observed at week 12 a significant reduction in serum alkaline phosphatase (ALP) (median reduction −21%; P = 0.029 versus placebo), GGT (−30%; P < 0.001), alanine aminotransferase (ALT) (−49%; P = 0.009), and aspartate aminotransferase (AST) (−42%; P = 0.019). Adverse events were similar between cilofexor and placebo-treated patients, the more frequent adverse effect was pruritus [36], particularly in patients treated with the higher dose. In a more recent study, a phase II trial, preliminary efficacy of cilofexor in a 96-week was evaluated. At week 96, reductions in liver biochemistry parameters occurred, including serum ALP (median, −8.3% [IQR, −25.9% to 11.0%]; P = .066), GGT (−29.8% [IQR, −42.3% to −13.9%]; P < .001), ALT (−29.8% [IQR, −43.7% to −6.6%]; P = .002), and aspartate aminotransaminase AST (−16.7% [IQR, −35.3% to 1.0%]; P = .010) [37].
In the INTREPID Study, 68 patients with PBC were randomized to 12-week treatment with EDP-305, which has been shown to suppress liver injury and fibrosis in animal models vs. placebo. The intent-to-treat analysis found that a dose of EDP-305 1 mg, 45% of patients experienced a response in ALP levels vs. with the dose of 2 mg. However, the study did not meet its primary endpoint of a 20% reduction in ALP levels [38].
3.3. Fibroblast Growth Factor 19 (FGF-19) Analogs
NGM282 (also known as M70) is the first engineered non-tumorigenic endocrine hormone FGF19 analogue, that potently regulates CYP7A1-mediated BAs homeostasis. This molecule was designed to retain CYP7A1 suppression to reduce bile acid-associated biliary injury. In NGM282, a 5-amino acid deletion (P24-S28) coupled with the substitution of 3 amino acids at critical positions (A30S, G31S, H33L) within the amino terminus, enable biased FGFR4 signaling so that NGM282 does not activate signal transducer and activator of transcription 3, a signaling pathway essential for FGF19-mediated hepatocarcinogenesis.
In animal models of PSC, treatment with NGM282 resulted in a rapid and robust reduction in ALP, ALT and AST concentrations and improvement in histological features associated with PSC. In a phase II, international multicenter, randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of NGM282 vs. placebo; 62 patients with PSC were randomly assigned 1:1:1 to receive NGM282 1 mg, 3 mg, or placebo once daily for 12 weeks. The primary outcome was changes in ALP levels from baseline to week 12. There were none significant differences in the mean change from baseline in ALP between the NGM282 and placebo groups. In contrast, NGM282 significantly reduced hepatic CYP7A1 activity (compared with placebo), and BAs levels. In addition, fibrosis biomarkers that predict transplant-free survival were significantly improved. Adverse events were mainly gastrointestinal symptoms and more frequent in the treatment groups [10]. In another 28-day, double-blind, placebo-controlled phase 2 trial, 45 PBC patients who had an inadequate response to UDCA, were randomly assigned 1:1:1 to receive subcutaneous daily doses of either NGM282 at 0.3 mg, 3 mg, or placebo. At day 28, 50% of patients receiving NGM282 0.3 mg and 46% of those receiving NGM282 3 mg achieved 15% or a greater reduction in ALP levels from baseline, compared with 7% of patients receiving placebo. NGM282 also significantly reduced serum concentrations of transaminases and immunoglobulins [39]. There is evidence with NGM282 and another engineered analog of FGF-19 (Aldafermin), on safety and tolerability in healthy volunteers and in patients with NASH. In was found a rapid and significant reductions in liver fat content with an acceptable safety profile [40,41].
3.4. PPAR Agonists: Fibrates, Seladelpar, Elafibranor
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors involved in the regulation of metabolic homeostasis. There are three main PPARs; PPARα thar mainly influences fatty acid metabolism, PPARβ/δ which participates in fatty acid oxidation and regulates blood glucose and cholesterol levels, while PPARγ regulates adipogenesis, energy balance, and lipid biosynthesis. Hence, they have been studied and used for a long time as therapeutic targets in metabolic diseases, for example thiazolidinediones in diabetes or fibrates in hypertriglyceridemia [42,43]. Furthermore, fenofibrate a PPARα agonist which is highly expressed in muscle, heart, and liver, is used to treat patients with CLD who are refractory to UDCA monotherapy. When activated, PPARα downregulates BAs synthesis through inhibition of the BAs-synthesizing enzymes, cytochrome P450, cholesterol 7A1-hydroxylase (CYP7A1) and cytochrome sterol 27-hydroxylase (CYP27A1). Moreover, ligands of PPARα may play a role in the regulation of bile excretory function trough up-regulation human MDR3 expression, which is localized to the canalicular membrane of hepatocytes, where it is the major determinant of biliary salts secretion, thereby reducing cholestasis [44].
Despite several trials, there is insufficient data to establish the safety and efficacy of fibrates on PBC. To the date, most studies involve the combination of UDCA and bezafibrate (400 mg daily) or fenofibrate (150-200 mg daily). The first studies about the use of bezafibrate found an improvement in ALP, GGT and IgM levels but also pruritus [45-49]. A study by Ohmoto et al, reported an improvement in serum markers of hepatic fibrosis (7S domain of type IV collagen, the triple-helix do-main of type IV collagen, hyaluronic acid, and procollagen type III N-terminal propeptide) [49]. On the other hand, few studies have addressed the fibrotic effects of fibrates through histological assessment and remains unclear.
Regarding the use of fenofibrate, at least six trials were conducted in the first decade of 2000s, showing an improvement in transaminases and IgM levels [50-55]. In this matter, a metanalysis reported that the combination therapy with UDCA and fenofibrate was more effective in reducing ALP than UDCA monotherapy, but it did not improve clinical symptoms [56].
More recently the BEZURSO trial showed up to 60% reduction in ALP after only 3 months of add-on bezafibrate therapy [57], while another trial in 2021 conclude that combined therapy was associated with a significant decrease in all-cause and liver-related mortality or need for LT (adjusted hazard ratios: 0.3253, 95% CI 0.1936-0.5466 and 0.2748, 95% CI 0.1336-0.5655, respectively; p <0.001 for both) [58].
Another novel molecule is MBX-8025 also known as seladelpar. It is PPARδ agonist that demonstrated potent anti-cholestatic effects in clinical studies. The first study published in 2017 was a 12-week, double-blind and placebo-controlled trial. It included 70 patients that were randomly assigned to placebo, seladelpar 50 mg/day, or seladelpar 200 mg/day while UDCA was continued. Despite seladelpar normalized ALP levels in patients who completed 12 weeks of treatment; the study was stopped early because it was associated with increase in aminotransferases levels. Other side effects included nausea and diarrhea [59].
The ENHANCE trial on safety and efficacy of seladelpar in patients with PBC included 112 patients urresponsive to UDCA. They receive seladelpar at doses of 2 mg, 5 mg, and 10 mg for 1 year. The dosage was increased up to 10 mg after 12 weeks, depending on biochemical response. In its phase 3, The investigators reported an absolute reduction in ALP of nearly 45% with the 10 mg dose, a decrease of approximately 122 units. Other serum liver tests reflected a similar benefit from seladelpar. Adverse events were mild to moderate. The most common problem was pruritus [60]. More recently, an open-label study in patients with PBC, evaluated the effects of 1-year of seladelpar treatment on quality life and pruritus. It was carried out by self-reported experiences of 101 patients with PBC using the pruritus visual analog scale (VAS), 5D-itch scale, and PBC-40 questionnaires along with BAs profiles. They received a daily dose of 5 and 10 mg of seladelpar. After one year of treatment, the patients improved pruritus and therefore sleep disturbances and had significant reductions in serum BAs [61].
The therapeutic effect of seladelpar relies on downregulation of BAs synthesis and modulation in their transport and metabolism. Studies in rodents reported that seladelpar reduces hepatocyte CYP7A1 via the fibroblast growth factor 21 signaling pathway. CYP7A1 is a rate-limiting enzyme in the classic pathway of BAs synthesis [62,63].
Whether seladelpar should be used in the first-line or as an add-on to UDCA for patients with PBC should be assessed. Several clinical trials assessing either efficacy or safety of seladelpar are currently ongoing.
Another PPAR agonist is elafibranor which initially emerged as therapy for NASH due to its effects over metabolic syndrome and expression of proinflammatory genes that may result in antifibrotic properties [64]. In regard of cholestasis, elafibranor activates PPARα which inhibits CYP7A1 expression leading to a reduction in BAs synthesis while the induction of CYP3A4, SULT2A1 and UGT2B4; and the expression of BSEP and MRP2 decrease BAs output and toxicity [65]. Moreover, in the same spectrum of its antifibrotic effects, elafibranor may have beneficial effects in the progression of PBC and PSC. The reduction fibrosis is achieved by the activation of PPARα and PPARδ that lead to the inhibition of NF-κB and AP-1 pathways. These two pathways are related to liver damage and inflammation [66,67].
A phase 2 clinical trial about the effects of elafibranor in patients with PBC was carried out during a 12-week administration of 80 mg of elafibranor in 45 patients with PBC and incomplete response to UDCA. The study found that the intervention reduced the levels of ALP, total bilirubin, and inflammatory marker such as C-reactive protein. There was no effect on pruritus [65]. Despite the promising molecular targets of elafibranor there is still insufficient information about the efficacy and safety of this drug. In 2020, was announced the phase II of ELATIVE clinical trial about the efficacy and safety of elafibranor in patients with PBC and inadequate response or intolerance to UDCA [68].
3.5. ASBT Inhibitors
About 90-95% of BAs are reabsorbed into the terminal ileum via ASBT. Current evidence explains that the inhibition of ASBT reduces the overload BAs in the liver. The decrease of ASBT expression increases the excretion of fecal BAs and the total concentration of BAs in liver [69]. On the other hand, the interruption of BAs enterohepatic circulation which lowers their concentrations in the liver also leads to bile salt spilling over to the colon resulting in diarrhea, irregular bowel movement, and abdominal pain [70,71]. A recent study in mice found that inhibition of ASBT in the kidneys stimulates the renal bile salts excretion and consequently improving intestinal side effects [71]. Yet, new ASBT inhibitors like Odevixibat, Lopixibat and GSK2330672 are restricted to intestinal ASBT blockading.
The first ASBT inhibitor approved by the FDA was Lopixbat (Maralixibat) but only for children with Alagille syndrome which cause severe cholestasis and pruritus due to deletion or mutation of JAG1 gene, leading to defective bile duct development. The use of Maralixibat 380 μg/kg once per day and the subsequent increase to 380 μg/kg twice per day reduced serum BAS and pruritus according to a placebo-controlled study [72].
Regarding GSK2330672 (Linerixibat), in 2017 a double-blind, randomised, placebo-controlled trial with 22 patients reported that the administration of GSK2330672 for two weeks improved pruritus (assessed by itch scores) and total serum BAs concentrations declined by 50% from baseline after received the intervention. Despite intestinal adverse effected, the drug was well tolerated [73]. In this matter, a later study in healthy Japanese volunteers investigated the safety and tolerability of GSK2330672 versus placebo. No serious adverse events were present and the main side effects was diarrhea [74]. Another, but larger placebo-controlled trial found that GSK2330672 was not significantly different versus placebo [75].
3.6. Immune-Modulation Drugs: Corticosteroids and Biological Therapies
The use of corticosteroids in cholestasis remains an area of active investigation. In vitro studies and animal models support the beneficial effects of corticosteroids. The mechanism of these drugs in cholestasis are related with the reduction of cell edema and relieving the inflammation of the bile duct cells and liver cells. Moreover, the immunomodulatory effect over Kupffer cells and the release of reactive oxygen species (ROS) protects from liver damage and thereby improving transaminases levels and the biochemical markers of cholestasis (Figure 2) [78,79], but clinical evidence is still controversial. Two metanalyses about the combination of corticosteroids with UDCA for patients with PBC conclude that combination therapy of those two drugs did not differ significantly from the monotherapy in improving fatigue, jaundice, mortality, death/LT, or adverse events, but was significantly superior to the monotherapy in reducing serum biochemical liver markers [80,81]. Most of studies on steroids in PCB are for overlap syndrome.
Budesonide is a corticosteroid that has been studied for long time for patients with PBC because of its high glucocorticoid receptor binding affinity in the liver when compared to other steroids. It is also a receptor/pregnane X receptor (PXR) agonist which is involved in BAs synthesis, metabolism, and transport [82]. Nevertheless, its efficacy and safety are anecdotical. The first study was randomised placebo-controlled trial; the intervention was budesonide 9 mg/day and UDCA for two years. Patients showed some improve in liver histology [83]. A phase 3 trial of combine therapy with budesonide and UDCA in patients with PBC and incomplete response to UDCA failed to meet its primary endpoint of histological improvement [84]. Moreover, long-term use of budesonide is related with important side effect, especially as liver disease progresses [21,85].
Biological agents are a novel therapy for cholestasis and there still ongoing trials to evaluate their efficacy in PBC, some of those drugs are Ustekinumab or Rituximab. The latter is anti-CD20 chimeric monoclonal antibody; that is use deplete B cells to decrease autoantibody production and antigen presentation. This is important since the hallmark of PBC is the presence of anti-mitochondrial autoantibodies and high amounts of IgM [86,87]. The same mechanism of Rituximab applies to PSC and clinical trials show beneficial effects, even for PSC recurrence after LT, though studies are still limited [88,89]. The results of two open-label studies on the efficacy of Rituximab in patients with PBC and incomplete UDCA response, suggested a limited efficacy of rituximab in PBC patients, even though an impressive reduction in ALP levels was observed [86, 87].
Other biological therapies include Abatacept, a modified antibody to cytotoxic T-lymphocyte antigen 4. It was studied in an open-label trial for 24 weeks, has demonstrated the inefficacy of this protein in achieving the required clinical outcomes [90]. In the case of Baricitinib, a selective JAK inhibitor (JAK1 and JAK2), was evaluated in a small randomized, double-blinded placebo-controlled trial in patients with PBC and inadequate to UDCA. Only two patients were enrolled and completed the trial (one received baricitinib and the other placebo). The patient treated with baricitinib demonstrated a 30% decrease in ALP and a 7-point improvement in itch score [91]. The effect of Baricitinib over JAK and STAT proteins is to regulate interleukins, interferons, and the switch toward T helper (Th) 1, 2, or 17 of naïve T cells. Regarding interleukins, Ustekimumab, a monoclonal antibody has been investigated in a multicentric, open-label study including PBC patients with an inadequate response to UDCA. Unfortunately, the results of this study failed to demonstrate the efficacy of this antibody in achieving a decrease, even moderate, in ALP levels [92]. Another monoclonal antibody that did not show efficacy in patients with cholestasis is Vedolizumab. It was studied for patients with PSC and inflammatory bowel disease [93].
Finally, the novel NI-0801 monoclonal antibody, which is an anti-CXCL10 (a chemokine secreted in response to interferon-γ-stimulation implicated in the hepatic recruitment of inflammatory T cells) was evaluated in a phase 2 study enrolling 29 UDCA-non-responder patients with PBC. In the 3-month follow-up performed after 6 doses of NI-0801 10 mg/Kg every 2 weeks the trial was terminated due to no significant therapeutic benefits obtained. Side effects included headaches, pruritus, fatigue, and diarrhea [94]
Biological agents showed promising results in certain scenarios, but conflicting results have been produced and these molecules are not part of the current therapeutic options. In addition, there are few studies and with a small population. More studies are required to establish long-term beneficial effects.
4. Conclusions
CLD includes a wide variety of genetic, congenital, immunological, infectious, and idiopathic disorders that may induce complications such as cirrhosis, liver failure, malignancies, pruritus, fatigue, bone disease and nutritional deficiencies that merit close follow-up and specific interventions to improve quality of life. According to guidelines, UDCA is the first line of treatment in patients with CLD, especially for PBC and PSC. Nevertheless, an important proportion of those patients are intolerant or unresponsive to UDCA. Moreover, quality of life may improve as symptoms like pruritus decrease but liver histology may not improve. In this matter several drugs have been tested in CLD with a variety of results (Figure 2). PPAR agonist seems to be the best options in general, especially when combined with UDCA. Besides, they are a good option for patients with metabolic syndrome. The case of ASBT inhibitors is a good example of success, they have a good safety profile despite intestinal side effect and have FDA approbation. Yet, there is insufficient evidence to use in other CLD besides Alagille syndrome. FXR agonist have shown promising results and can be combine with UDCA or (FGF-19) analogs. Finally, biological therapies and corticosteroids in theory and in vitro model are effective targets but its efficacy has not been established in clinical trials.
Author Contributions
Conceptualization, Nahum Mendez-Sanchez and Ana Leticia Ordoñez-Vazquez; validation and formal analysis, Nahum Mendez-Sanchez; data curation, Ana Leticia Ordoñez-Vazquez and Carlos Esteban Coronel-Castillo; writing—original draft preparation, Nahum Mendez-Sanchez, Ana Leticia Ordoñez-Vazquez and Carlos Esteban Coronel-Castillo.; writing—review and editing, Carlos Esteban Coronel-Castillo and Nahum Mendez-Sanchez; visualization, Carlos Esteban Coronel-Castillo.; supervision, Nahum Mendez-Sanchez; project administration, Nahum Mendez-Sanchez.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sanjel, B.; Shim, W.S. Recent advances in understanding the molecular mechanisms of cholestatic pruritus: A review. Biochim Biophys Acta Mol Basis 2020, 1;1866(12):165958. [CrossRef]
- Gazda, J.; Drazilova, S.; Janicko, M.; et al. The Epidemiology of Primary Biliary Cholangitis in European Countries: A Systematic Review and Meta-Analysis. Can J Gastroenterol Hepatol. 2021, 19; 2021:9151525. [CrossRef]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary Sclerosing Cholangitis, Part 1: Epidemiology, Etiopathogenesis, Clinical Features, and Treatment. 2018. [Google Scholar]
- Malik, A.; Kardashian, A.A.; Zakharia, K.; et al. Preventative care in cholestatic liver disease: Pearls for the specialist and subspecialist. Liver Res 2019, 3(2):118-127. [CrossRef]
- Gerussi, A.; Restelli, U.; Croce, D. ; et tal. Cost of illness of Primary Biliary Cholangitis - a population-based study. Dig Liver Dis 2021, 53(9):1167-1170. [CrossRef]
- Yokoda, R.T.; Rodriguez, E.A. Review: Pathogenesis of cholestatic liver diseases. World J Hepatol 2020, 12(8): 423–435. [CrossRef]
- Méndez-Sánchez, N. Bile Acids in Health and Disease Foreword. Ann Hepatol 2017;16(Suppl. 1: s3-105.):s3. [CrossRef]
- Desmet, V.J. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows Arch 2011 Mar;458(3):251-9. [CrossRef]
- Goldstein, J.; Levy, C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018;38(9):1520-1535. [CrossRef]
- Hirschfield, G.M.; Chazouillères, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J Hepatol 2019;1;70(3):483–93. [CrossRef]
- Méndez-Sánchez, N. Management of primary biliary cholangitis: the importance to identify patients' non-responders to standard treatment. Minerva Med 2018;109(6):407-409. [CrossRef]
- Cabrera, D.; Arab, J.P.; Arrese, M. UDCA, NorUDCA, and TUDCA in Liver Diseases: A Review of Their Mechanisms of Action and Clinical Applications. Handb Exp Pharmacol 2019; 256:237-264. [CrossRef]
- Fabris, L.; Fiorotto, R.; Spirli, C.; et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol 2019; 16(8): 497–511. [CrossRef]
- Hartl, L.; Haslinger, K.; Angerer, M.; Semmler, G.; Schneeweiss-Gleixner, M.; Jachs, M.; Simbrunner, B.; Bauer, D.J.M.; Eigenbauer, E.; Strassl, R.; et al. Progressive cholestasis and associated sclerosing cholangitis are frequent complications of COVID-19 in patients with chronic liver disease. Hepatology 2022;76(6):1563-1575. [CrossRef]
- Beuers, U.; Spengler, U.; Kruis, W.; et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatology 1992;16(3):707-14. [CrossRef]
- Beuers, U.; Trauner, M.; Jansen, P.; et al. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol 2015;62(1 Suppl):S25-37. [CrossRef]
- Poupon, R.; Chrétien, Y.; Poupon, R.E.; et al. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis?. Lancet 1987; 11;1(8537):834-6. [CrossRef]
- Poupon, R.E.; Balkau, B.; Eschwège, E.; et al. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med 1991; 30;324(22):1548-54. [CrossRef]
- Poupon, R.E.; Balkau, B.; Guéchot, J.; et al. Predictive factors in ursodeoxycholic acid-treated patients with primary biliary cirrhosis: role of serum markers of connective tissue. Hepatology 1994; 19(3):635-40. [CrossRef]
- Poupon, R.E.; Bonnand, A.M.; Chrétien, Y.; et al. Ten-year survival in ursodeoxycholic acid-treated patients with primary biliary cirrhosis. The UDCA-PBC Study Group.Hepatology. 1999; 29(6):1668-71. [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017 Jul;67(1):145-172.
- Shi, J.; Li, Z.; Zeng, X.; et al. Ursodeoxycholic acid in primary sclerosing cholangitis: meta-analysis of randomized controlled trials. Hepatol Res. 2009; 39(9):865-73. [CrossRef]
- Triantos, C.K.; Koukias, N.M.; Nikolopoulou, V.N.; et al. Meta-analysis: ursodeoxycholic acid for primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011; 34(8):901-10. [CrossRef]
- Olsson, R.; Boberg, K.M.; de Muckadell, O.S.; et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005; 129(5):1464-72. [CrossRef]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009; 50(3):808-14. [CrossRef]
- Black, D.D.; Mack, C.; Black, D.D.; et al. A Prospective Trial of Withdrawal and Reinstitution of Ursodeoxycholic Acid in Pediatric Primary Sclerosing Cholangitis. Hepatol Commun.2019; 29;3(11):1482-1495. [CrossRef]
- Pardi, D.S.; Loftus, E.V. Jr.; Kremers, W.K.; et al. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003; 124(4):889-93. [CrossRef]
- Tung, B.Y.; Emond, M.J.; Haggitt, R.C.; et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med.2001; 16;134(2):89-95. [CrossRef]
- Lindström, L.; Boberg, K.M.; Wikman, O.; et al. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Aliment Pharmacol Ther.2012; 35(4):451-7. [CrossRef]
- Eaton, J.E.; Silveira, M.G.; Pardi, D.S.; et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. 2011; 106(9):1638-45. [CrossRef]
- Singh, S.; Khanna, S.; Pardi, D.S.; et al. Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: a systematic review and meta-analysis. Inflamm Bowel Dis.2013; 19(8). [CrossRef]
- Hansen, J.D.; Kumar, S.; Lo, W.K. Ursodiol and colorectal cancer or dysplasia risk in primary sclerosing cholangitis and inflammatory bowel disease: a meta-analysis. Dig Dis Sci. 2013; 58(11):3079-87. [CrossRef]
- Bowlus, C.L.; Arrivé, L.; Bergquist, A.; et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology.2023; 1;77(2):659-702. [CrossRef]
- Floreani, A.; Gabbia, D.; de Martin, S. Update on the Pharmacological Treatment of Primary Biliary Cholangitis. Vol. 10, Biomedicines. MDPI; 2022. [CrossRef]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K. v.; et al. Farnesoid X receptor agonist tropifexor attenuates cholestasis in a randomised trial in patients with primary biliary cholangitis. JHEP Reports. 2022; 1;4(11). [CrossRef]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology. 2019; 1;70(3):788–801. [CrossRef]
- Trauner, M.; Bowlus, C.L.; Gulamhusein, A.; et al. Safety and sustained efficacy of the farnesoid X receptor (FXR) agonist cilofexor over a 96-week open-label extension in patients with PSC. Clin Gastroenterol Hepatol. 2022; 4;S1542-3565(22)00720-0. [CrossRef]
- Kowdley, K.V.; Bonder, A.; Heneghan, M.A.; et al. Final data of the phase 2a intrepid study with EDP-305, a non-bile acid farnesoid x receptor (FXR) agonist. Hepatology (Baltimore, Md.) 2020, 72(1 SUPPL), 746A-747A. [CrossRef]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J. ; Weltman M, et al. NGM282 for Treatment of Patients With Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol Commun 2018 Aug 30;2(9):1037-1050. [CrossRef]
- Harrison, SA.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H. L, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet. 2018; 24;391(10126):1174–85. [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology. 2021; 1;160(1):219-231.e1. [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; et al. PPAR Agonists and Metabolic Syndrome: An Established Role?. Int J Mol Sci. 2018; 19(4): 1197. [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications- a review. Nutr J.2014; 14; 13:17. [CrossRef]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. On Fibrates and Cholestasis: A review. Hepatology. 2015; 62(2): 635–643. [CrossRef]
- Kita, R.; Takamatsu, S.; Kimura, T.; et al. Bezafibrate may attenuate biliary damage associated with chronic liver diseases accompanied by high serum biliary enzyme levels. J Gastroenterol 2006;41:686–692. [CrossRef]
- Hazzan, R.; Tur-Kaspa, R. Bezafibrate treatment of primary biliary cirrhosis following incomplete response to ursodeoxycholic acid. J Clin Gastroenterol 2010;44:371–373. [CrossRef]
- Takeuchi, Y.; Ikeda, F.; Fujioka, S.; et al. Additive improvement induced by bezafibrate in patients with primary biliary cirrhosis showing refractory response to ursodeoxycholic acid. J Gastroenterol Hepatol 2011;26:1395–1401. [CrossRef]
- Iwasaki, S.; Ohira, H.; Nishiguchi, S.; et al. The efficacy of ursodeoxycholic acid and bezafibrate combination therapy for primary biliary cirrhosis: a prospective, multicenter study. Hepatol Res 2008;38:557–564. [CrossRef]
- Ohmoto, K.; Yoshioka, N.; Yamamoto, S. Long-term effect of bezafibrate on parameters of hepatic fibrosis in primary biliary cirrhosis. J Gastroenterol 2006; 41(5):502-3. [CrossRef]
- Ohira, H.; Sato, Y.; Ueno, T.; Sata, M. Fenofibrate treatment in patients with primary biliary cirrhosis. Am J Gastroenterol 2002; 97:2147–2149. [CrossRef]
- Dohmen, K.; Mizuta, T.; Nakamuta, M.; et al. Fenofibrate for patients with asymptomatic primary biliary cirrhosis. World J Gastroenterol 2004;10:894–898. [CrossRef]
- Levy C, Peter JA, Nelson DR, Keach J, Petz J, Cabrera R, Clark V, et al. Pilot study: fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment Pharmacol Ther 2011;33:235–242. [CrossRef]
- Han, X.F.; Wang, Q.X.; Liu, Y.; et al. Efficacy of fenofibrate in Chinese patients with primary biliary cirrhosis partially responding to ursodeoxycholic acid therapy. J Dig Dis 2012;13:219–224. [CrossRef] [PubMed]
- Liberopoulos, E.N.; Florentin, M.; Elisaf, M.S.; et al. Fenofibrate in primary biliary cirrhosis: a pilot study. Open Cardiovasc Med J. 2010; 4:120–126. [CrossRef]
- Zhang, H.; Li, S.; Feng, Y.; et al. Efficacy of fibrates in the treatment of primary biliary cholangitis: a meta-analysis. Clin Exp Med 2022 Nov 1. [CrossRef]
- Zhang, Y.; Li, S.; He, L.; et al. Combination therapy of fenofibrate and ursodeoxycholic acid in patients with pri mary biliary cirrhosis who respond incompletely to UDCA monotherapy: a meta-analysis. Drug Des Devel Ther. 2015; 9: 2757–2766. [CrossRef]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N Engl J Med. 2018; 7;378(23):2171-2181. [CrossRef]
- Tanaka, A.; Hirohara, J.; Nakano, T.; et al. Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J Hepatol.2021; 75(3):565-571. [CrossRef]
- Jones, D.; Boudes, PF.; Sawin, MG.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: a double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study.Lancet Gastroenterol Hepatol.2017; 2(10):716-726. [CrossRef]
- Bowlus, C.L.; Galambos, M.R.; Aspinall, R.J.; et al. A phase II, randomized, open-label, 52-week study of seladelpar in patients with primary biliary cholangitis. J Hepatol.2022; 77(2):353-364. [CrossRef]
- Kremer, A.E.; Mayo, M.J.; Hirschfield, G.; et al. Seladelpar improved measures of pruritus, sleep, and fatigue and decreased serum bile acids in patients with primary biliary cholangitis. Liver Int. 2022; 42(1):112-123. [CrossRef]
- Zhang, L.; Huang, X.; Meng, Z.; et al. Significance and mechanism of CYP7a1 gene regulation during the acute phase of liver regeneration. Mol Endocrinol. 2009; 23(2):137-45. [CrossRef]
- Colapietro, F.; Gershwin, M.E.; Lleo, A. PPAR agonists for the treatment of primary biliary cholangitis: old and new tales. J Transl Autoimmun. 2023; 6: 100188. [CrossRef]
- Westerouen Van Meeteren, M.J.; Drenth, J.P.H.; Tjwa, E.T.T.L. Elafibranor: a potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. 2020; 29(2):117-123. [CrossRef]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; et al. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J Hepatol. 2021; 74(6):1344-1354. [CrossRef]
- Tao, Li.; Ren, X.; Zhai, W.; et al. Progress and Prospects of Non-Canonical NF-κB Signaling Pathway in the Regulation of Liver Diseases. Molecules. 2022; 2;27(13):4275.
- Li, F.; Patterson, A.D.; Krausz, K.W.; et al. Metabolomics reveals an essential role for peroxisome proliferator-activated receptor α in bile acid homeostasis. J Lipid Res. 2012; 53(8):1625-35. [CrossRef]
- Trial ELATIVE.
- Yang, N.; Dong, Y.Q.; Jia, G.X.; et al. ASBT(SLC10A2): A promising target for treatment of diseases and drug discovery. Biomed Pharmacother.2020; 132:110835. [CrossRef]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; et al. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr-/-.Leiden mice. PLoS One. 2019; 24;14(6): e0218459. [CrossRef]
- Kunst, R.F.; de Waart, D.R.; Wolters, F.; et al. Systemic ASBT inactivation protects against liver damage in obstructive cholestasis in mice. JHEP Rep. 2022; 27;4(11):100573. [CrossRef]
- Gonzales, E.; Hardikar, W.; Stormon, M.; et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. The Lancet. 2021; 398(10311):1581-1592. [CrossRef]
- Hegade, V.S.; Kendrick, S.F.; Dobbins, R.L.; et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet. 2017; 18;389(10074):1114-1123. [CrossRef]
- Ino, H.; Endo, A.; Wakamatsu, A.; et al. Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Evaluations Following Single Oral Doses of GSK2330672 in Healthy Japanese Volunteer. Clin Pharmacol Drug Dev. 2019; 8(1):70-77. [CrossRef]
- Levy, C.; Kendrick, S.; Bowlus, C.L.; et al. GLIMMER: A Randomized Phase 2b Dose-Ranging Trial of Linerixibat in Primary Biliary Cholangitis Patients with Pruritus. Clin Gastroenterol Hepatol. 2022; 4; S1542-3565(22)01021-7. [CrossRef]
- Thompson, R.; Arnell, H.; Artan, R.; et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022; 7(9):830-842. [CrossRef]
- Deeks, E.M. Odevixibat: First Approval. Drugs. 2021; 81(15): 1781–1786. [CrossRef]
- Hempfling, W.; Grunhage, F.; Dilger, K.; et al. Pharmacokinetics and pharmacodynamic action of budesonide in early- and late-stage primary biliary cirrhosis. Hepatology. 2003; 38(1):196-202. [CrossRef]
- Al-Aqil, F.A.; Monte, M.J.; Peleteiro-Vigil, A.; et al. Interaction of glucocorticoids with FXR/FGF19/FGF21-mediated ileum-liver crosstalk. Biochim Biophys Acta Mol Basis Dis. 2018; 1864(9 Pt B):2927-2937. [CrossRef]
- Zhang, Y. ; Lu. J.; Wang, F.; et al. Combination Therapy of Ursodeoxycholic Acid and Corticosteroids for Primary Biliary Cirrhosis with Features of Autoimmune Hepatitis: A Meta-Analysis. Gastroenterol Res Pract. 2013; 2013: 490731. [CrossRef]
- Zhang, H.; Li, S.; Yang, J.; et al. A meta-analysis of ursodeoxycholic acid therapy versus combination therapy with corticosteroids for PBC-AIH-overlap syndrome: evidence from 97 monotherapy and 117 combinations. Prz Gastroenterol.2015; 10(3):148-55. [CrossRef]
- Silveira, M.G.; Lindor, K.D. Obeticholic acid and budesonide for the treatment of primary biliary cirrhosis. Expert Opin Pharmacother. 2014; 15(3):365-72. [CrossRef]
- Hempfling, W.; Grunhage, F.; Dilger, K.; et al. Pharmacokinetics and pharmacodynamic action of budesonide in early and late-stage primary biliary cirrhosis. Hepatology 2003; 38:196–202. [CrossRef]
- Hirschfield, G.M.; Beuers, U.; Kupcinskas, L.; et al. A placebo-controlled randomised trial of budesonide for PBC following an insufficient response to UDCA. J. Hepatol. 2021; 74, 321–329. [CrossRef]
- Geier, A.; Gartung, C.; Dietrich, C.G.; et al. Side effects of budesonide in liver cirrhosis due to chronic autoimmune hepatitis: influence of hepatic metabolism versus portosystemic shunts on a patients complicated with HCC. World J Gastroenterol 2003; 9: 2681–85. [CrossRef]
- Myers, R.P.; Swain, M.G.; Lee, S.S.; et al. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol. 2013; 108(6):933-941. [CrossRef]
- Tsuda, M.; Moritoki, Y.; Lian, Z.X.; et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and incomplete response to ursodeoxycholic acid. Hepatology. 2012; 55(2):512-21. [CrossRef]
- Hart, P.A.; Topazian, M.D.; Witzig, T.E.; et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: The Mayo Clinic experience. Gut. 2012; 62,1607-1615. [CrossRef]
- Yamada, Y.; Hoshino, K.; Fuchimoto, Y.; et al. Rituximab induction to prevent the recurrence of PSC after liver transplantation – The lessons learned from ABO-Incompatible living donor liver transplantation. Transplant Direct. 2018; 4(2): e342. [CrossRef]
- Bowlus, C.L. ; Yang, G-X.; Liu, C.H.; et al. Therapeutic trials of biologics in primary biliary cholangitis: an open label study of abatacept and review of the literature. J Autoimmun. 2019; 101:26–34. [CrossRef]
- Gordon, S.C; Trudeau, S.; Regev, A.; et al. Baricitinib and primary biliary cholangitis. J. Transl. Autoimmun. 2021; 4, 100107. [CrossRef]
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-concept study. Hepatology. (2016); 64:189–99. [CrossRef]
- Lynch, K.D.; Chapman, R.W.; Keshav, S.; et al. Effects of vedolizumab in patients with primary sclerosing cholangitis and inflammatory bowel diseases. Clin Gastroenterol Hepatol. (2019); 18:179–87. e6. [CrossRef]
- De, Graaf K.L.; Lapeyre, G.; Guilhot, F.; et al. NI-0801, an anti-chemokine (C-X-C motif) ligand 10 antibody, in patients with primary biliary cholangitis and an incomplete response to ursodeoxycholic acid. Hepatol. Commun. 2018; 2, 492–503. [CrossRef]
Figure 1.
Pathophysiology of cholangiopathies.Following the model of patients with PBC. Patients with CLD are at proinflammatory state. On a permissive genetic background, patients are to expose to a microbial or environment similar to immunogenic form of pyruvate dehydrogenase complex (PDC)–E2, mitochondrial autoantigen, in apoptotic blebs that will induce an immune response. 2) Dysregulation of the innate and adaptive results in a targeted immune response directed at cholangiocytes. There is an increased expression of the effector cytokines IL-6 and IL-8, and the leukocyte chemoattractant CXCL10. Dysregulated over-production of IFNγ results in T-cell infiltration of periductular areas, the production of anti-mitochondrial antibody (AMA) in conjunction with B cells and the upregulation of bile acid production. 3) Attacked by cytotoxic T cells and mitochondrial damage with secondary failure of biliary transporters (AE2, BSEP and the HCO3 umbrella) cholangiocytes loss their protections against from toxic hydrophobic bile acids. 4) Inflammation and injury lead to ductular reaction, accompanied by infiltration of leukocytes and lymphocytes, activation of liver progenitor cells, and an increase in matrix protein levels through different proteins such as TGFβ1/2. Bile salts will be acidified and become hydrophobic, eventually cross the membrane leading to cellular apoptosis. 5) In the inflamed portal tracts, chronic bile duct damage leads to bile leakage, retention of hydrophobic bile acids that cause local bile salt injury, interruption of the enterohepatic circulation and bile salt absorption in the ileum and FXR-mediated production of FGF19 with significant reduction of de novo bile salt synthesis. 6) Apoptosis and cell injury promotes formation of reactive oxygen species (ROS) both in liver cells and cholagiocytes. This contributes to a profibrotic state. Furthermore, apoptosis releases apoptotic blebs that perpetuates a proinflammatory state. This stimulation induces cells to evolve toward an exhausted, profibrogenic phenotype which can contribute to the development of hepatic stellate cell-mediated liver fibrosis.
Figure 1.
Pathophysiology of cholangiopathies.Following the model of patients with PBC. Patients with CLD are at proinflammatory state. On a permissive genetic background, patients are to expose to a microbial or environment similar to immunogenic form of pyruvate dehydrogenase complex (PDC)–E2, mitochondrial autoantigen, in apoptotic blebs that will induce an immune response. 2) Dysregulation of the innate and adaptive results in a targeted immune response directed at cholangiocytes. There is an increased expression of the effector cytokines IL-6 and IL-8, and the leukocyte chemoattractant CXCL10. Dysregulated over-production of IFNγ results in T-cell infiltration of periductular areas, the production of anti-mitochondrial antibody (AMA) in conjunction with B cells and the upregulation of bile acid production. 3) Attacked by cytotoxic T cells and mitochondrial damage with secondary failure of biliary transporters (AE2, BSEP and the HCO3 umbrella) cholangiocytes loss their protections against from toxic hydrophobic bile acids. 4) Inflammation and injury lead to ductular reaction, accompanied by infiltration of leukocytes and lymphocytes, activation of liver progenitor cells, and an increase in matrix protein levels through different proteins such as TGFβ1/2. Bile salts will be acidified and become hydrophobic, eventually cross the membrane leading to cellular apoptosis. 5) In the inflamed portal tracts, chronic bile duct damage leads to bile leakage, retention of hydrophobic bile acids that cause local bile salt injury, interruption of the enterohepatic circulation and bile salt absorption in the ileum and FXR-mediated production of FGF19 with significant reduction of de novo bile salt synthesis. 6) Apoptosis and cell injury promotes formation of reactive oxygen species (ROS) both in liver cells and cholagiocytes. This contributes to a profibrotic state. Furthermore, apoptosis releases apoptotic blebs that perpetuates a proinflammatory state. This stimulation induces cells to evolve toward an exhausted, profibrogenic phenotype which can contribute to the development of hepatic stellate cell-mediated liver fibrosis.
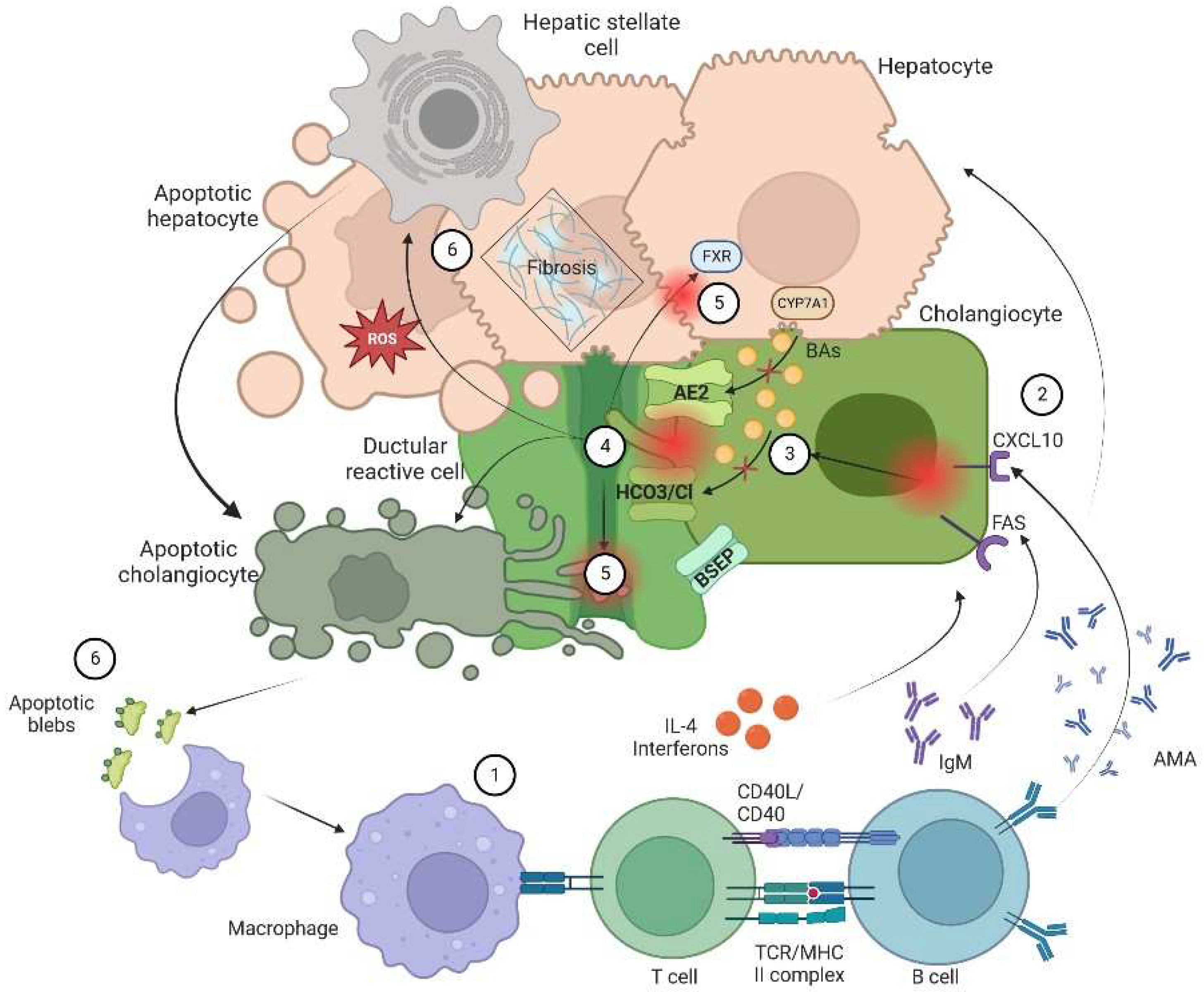
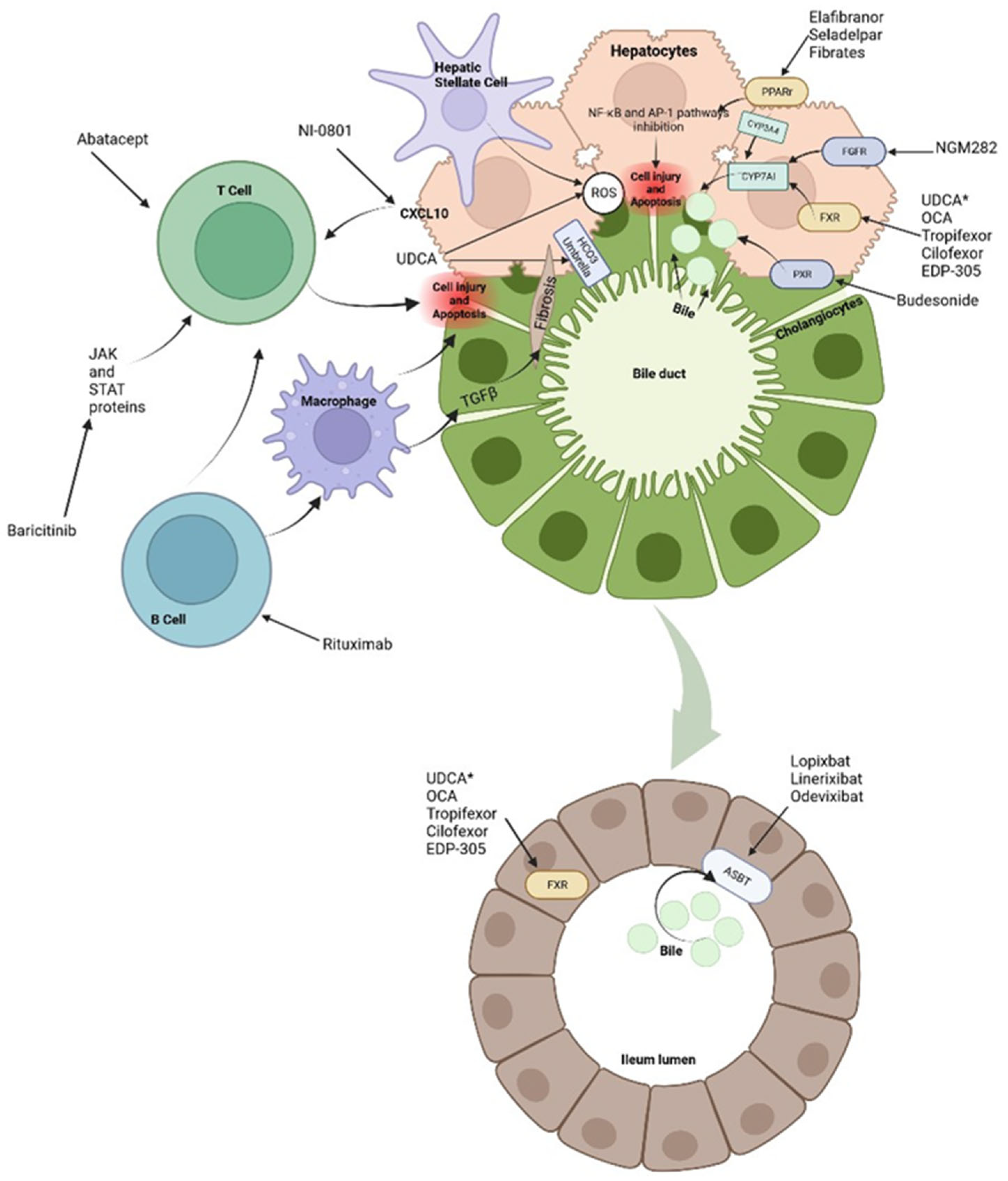
Figure 2.
Treatments and molecular targets.In the liver, FXR agonists, PPAr agonist and FGR agonist improve cholestasis by decreasing BAs production and output via CYP7A1 inhibition. PPAr agonist and FRX also modulate proinflammatory state by suppression inflammatory pathways such as NF-kB. This results in ROS and interleukin reduction there, a decrease in hepatic stellate cells activation and immune cells activation. Immunomodulatory therapies, like Rituximab or Baricitinib are aimed at blocking antigen presentation and then direct damage to liver cells It is important to notice that apoptosis in liver cells releases apoptotic blebs that perpetuates a proinflammatory state and formation ROS both in liver cells and cholagiocytes. This contributes to a profibrotic state. *UDCA serve as a partial agonist of FXR but also improve BAs flow and enhances ions channels. Its effects on the biliary HCO3− umbrella, near the apical surface, prevents the permeation of hydrophobic BAs; hence contributing to the reduction in cell injury. In the gut, intestinal FXR agonist, improves cholestasis due to the reduced BAs pool total size. FXR protects the liver from high BAs by regulating the expression levels of BAs transporters. The events triggered by FXR in the gut liver axis probably represent a therapeutic option through the decrease in the excess of BAs in the liver. About 90-95% of BAs are reabsorbed into the terminal ileum via ASBT. the inhibition of ASBT reduces the overload BAs in the liver. The decrease of ASBT ex-pression increases the excretion of fecal BAs and the total concentration of BAs in liver.
Figure 2.
Treatments and molecular targets.In the liver, FXR agonists, PPAr agonist and FGR agonist improve cholestasis by decreasing BAs production and output via CYP7A1 inhibition. PPAr agonist and FRX also modulate proinflammatory state by suppression inflammatory pathways such as NF-kB. This results in ROS and interleukin reduction there, a decrease in hepatic stellate cells activation and immune cells activation. Immunomodulatory therapies, like Rituximab or Baricitinib are aimed at blocking antigen presentation and then direct damage to liver cells It is important to notice that apoptosis in liver cells releases apoptotic blebs that perpetuates a proinflammatory state and formation ROS both in liver cells and cholagiocytes. This contributes to a profibrotic state. *UDCA serve as a partial agonist of FXR but also improve BAs flow and enhances ions channels. Its effects on the biliary HCO3− umbrella, near the apical surface, prevents the permeation of hydrophobic BAs; hence contributing to the reduction in cell injury. In the gut, intestinal FXR agonist, improves cholestasis due to the reduced BAs pool total size. FXR protects the liver from high BAs by regulating the expression levels of BAs transporters. The events triggered by FXR in the gut liver axis probably represent a therapeutic option through the decrease in the excess of BAs in the liver. About 90-95% of BAs are reabsorbed into the terminal ileum via ASBT. the inhibition of ASBT reduces the overload BAs in the liver. The decrease of ASBT ex-pression increases the excretion of fecal BAs and the total concentration of BAs in liver.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



