
Submitted:
17 April 2023
Posted:
18 April 2023
You are already at the latest version
Abstract
Viral diseases represent a major public health concern and an ever-present risk for developing into a future pandemic. Antiviral antibody therapeutics, either alone or in combination with other therapies, have emerged as valuable preventative and treatment options, including during a global emergency. Here we will discuss polyclonal and monoclonal antiviral antibody therapies, focusing on the unique biochemical and physiological properties that make them well suited as therapeutic agents. We will describe the methods of antibody characterization and potency assessment throughout development, highlighting similarities and differences between polyclonal and monoclonal products as appropriate. In addition, we will consider the benefits and challenges of antiviral antibodies when used in combination with other antibodies or other types of antiviral therapeutics. Lastly, we will discuss novel approaches to the characterization and development of antiviral antibodies and identify areas that would benefit from additional research.
Keywords:
antiviral therapy
; antiviral antibodies
; antibody combination therapy
; antibody potency
; potency assays
Introduction
Infectious diseases are a major global health burden with eight major diseases (HIV/AIDS, malaria, measles, hepatitis, dengue fever, rabies, tuberculosis and yellow fever) exacting a toll estimated at more than 156 million life years lost in 2016 alone1. The coronavirus disease 2019 (COVID-19) pandemic has further exacerbated the cost in human life and long-term health outcomes. Emerging and re-emerging viral diseases, such as Ebola, Zika, Hantavirus, Lassa fever, Hendra, highly pathogenic avian influenza, and others, continue to pose a risk not only for local/regional outbreaks but for becoming the next pandemic. The availability of safe and effective prophylaxis and treatment options for these and other infectious diseases is a top public health priority. Antibody therapeutics have long been used in communicable disease settings, for example post-exposure prophylaxis for rabies or hepatitis B with respective hyper- or specific- immune globulin (IG), or the use of monoclonal antibody (mAb) therapies for the prevention of respiratory syncytial virus (RSV) infection. Recent approvals of mAb therapies for human immunodeficiency virus type-1 (HIV-1) and Ebola virus (EBOV), as well as the rapid development and emergency use authorization of several mAbs against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) for prophylaxis and treatment of COVID-19, further highlight the potential of these molecules, either alone or in combination with other therapies, to make a significant impact on public health. In this review, we will discuss the biochemical and physiologic characteristics that render antibody molecules desirable therapeutics, preclinical assays that can be used to assess potency, and the benefits and challenges of antibody combination therapies, as well as highlighting areas in need of additional research.
Antibodies as therapeutics
With very few exceptions, antibody therapeutics approved to date are isotype G immunoglobulins (IgG). IgGs are protein macromolecules secreted in the blood of most vertebrates2 by differentiated plasma B cells that have a high affinity and specificity for their respective antigen. The IgG molecules can then be purified from human or animal plasma to produce polyclonal immune globulin products. These types of products, such as diphtheria antitoxin3, represent some of the first products to be licensed in the United States. In over a century of development, polyclonal products have undergone tremendous advances in the manufacturing process and characterization of safety and efficacy attributes. In the last few decades, antibody therapeutic development has shifted toward the development of monoclonal antibodies - IgG molecules that are produced in vitro after mature B cells have been isolated, immortalized, and cultured.
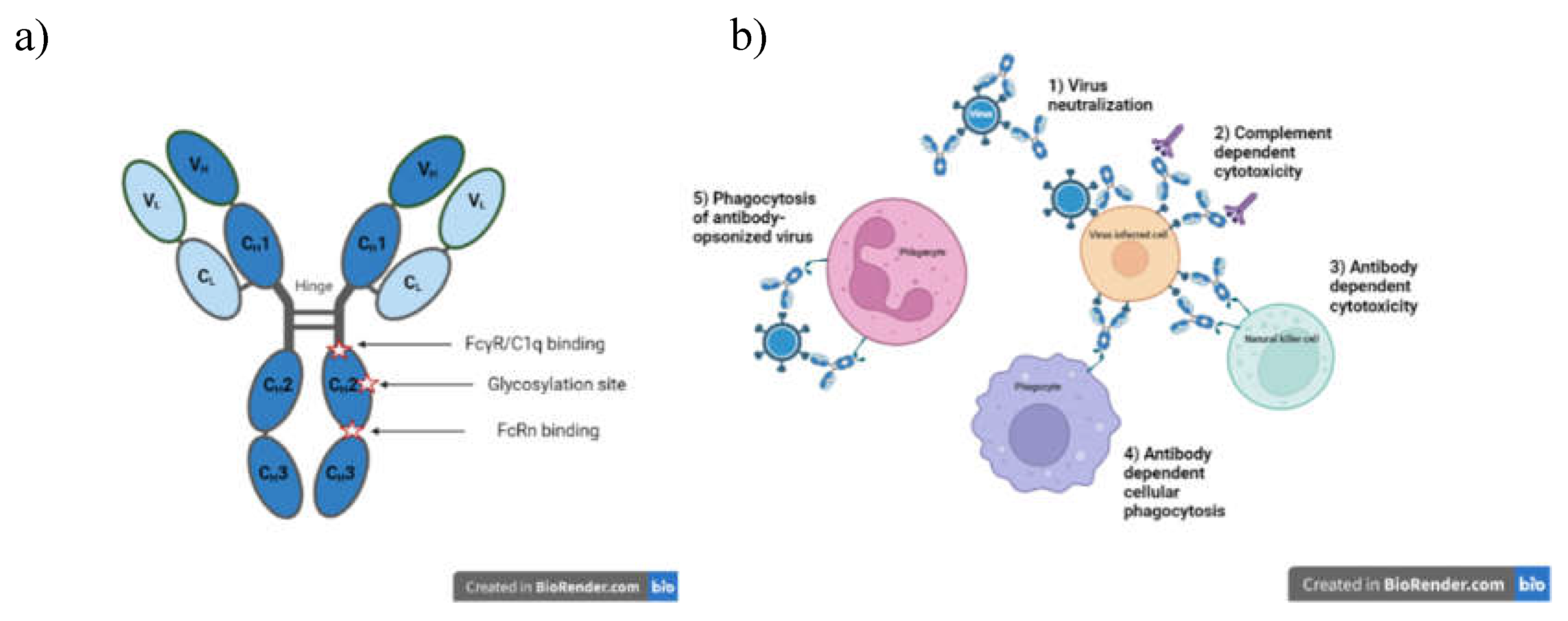
The structural and functional features of IgG antibodies render them well suited for use as therapeutics. Structurally, the molecule can be thought of as modular, with two identical heavy chains (HC) and two identical light chains (LC). The IgG HC is comprised of four domains, a variable (V) domain and three constant (CH1, CH2, and CH3) domains with a hinge region between the CH1 and CH2 domains (Figure 1a). The LC is comprised of two domains, a variable (V) domain and a constant (CL) domain. The fragment antigen binding (Fab) region in each chain contains both V and constant (CH1 or CL) domains, with the former housing the complementarity determining regions (CDR) responsible for epitope recognition and antibody specificity. When properly folded, the CDRs of the HC and LC come together to form the antigen-binding pocket. The fragment crystallizable (Fc) region, comprised of the HC CH2 and CH3 domains, is responsible for downstream processes (Fc effector functions) that result in immune activation and the ultimate destruction of the antigen. There are four different IgG isotypes (IgG1, IgG2, IgG3 and IgG4), with polymorphic variants for each isotype4. In addition, IgG antibodies have a single N-glycan in the constant region. These biochemical properties (i.e., sequence and glycan structures) play an important role in physicochemical (i.e., stability, shelf-life), pharmacokinetic, and pharmacodynamic properties of the antibody therapeutic, and thus should be well characterized during development.
The use of IgG products as prophylactic and therapeutic modalities for viral diseases is predicated on their ability to bind to one or more antigens on the surface of viral particles and/or infected cells via the antigen-binding pockets. They can neutralize the ability of viruses to enter cells by blocking attachment or fusion, inactivate virus particles, or trigger the killing of infected cells through Fc-mediated effector functions (Figure 1b)5. For the latter, the antigen-antibody complex is recognized by effector molecules, such as the C1q component of complement or Fc gamma receptors (FcγRs) present on the surface of effector cells, giving rise to immune signaling cascades that culminate with the clearance of viruses and/or infected cells. In some cases, Fc effector functions have been shown to enhance the antiviral activity of specific antibodies5-9.
On the other hand, antibody-dependent enhancement (ADE) of viral infection or disease can also occur, as has been documented in humans for dengue virus10. ADE can arise after natural infection, vaccination, or passive transfer of antibody therapies. It is widely thought that ADE occurs when antibodies of insufficient avidity or concentration are unable to neutralize the virus, but can facilitate the uptake of the virus-antibody complex by FcγR-bearing cells such as monocytes, dendritic cells, or macrophages11, resulting in increased viral production, enhanced immune activation (e.g., cytokine production), and more severe disease12. In addition to flaviviruses13, 14, ADE has been observed for mAbs against influenza virus, HIV-1, and EBOV in cell culture, but not typically when tested in animal models or in clinical trials, with a few exceptions11, 13. The risk of ADE can be reduced by engineering substitutions into the Fc region that disrupt FcγR binding, although these substitutions may also disrupt Fc effector functions that could contribute to clinical efficacy15, 16. Thus, when selecting antibodies best suited for use as an antiviral product, it is critical to optimize binding both to the antigen and FcγRs. For mAbs, IgG isotypes can be selected, Fc glycosylation patterns can be modified, or Fc regions can be engineered with substitutions that enhance or diminish select Fc effector functions. Although ADE in cell culture and animal studies has been observed with antiviral specific polyclonal immune globulins (IG)17, clinical ADE has not been reported for any FDA-approved specific IG products.
During pharmaceutical development, mAb domains often undergo extensive biochemical engineering to optimize the properties of the antibody. For example, the CDRs can be grafted onto the framework regions of V domains from other mAbs and still retain their antigen binding properties in the context of a known protein fold. The Fc region can also be modified to alter pharmacokinetic properties and effector functions. On the other hand, although not subjected to Fc engineering, depending on the antigen or donor population, specific antiviral polyclonal IGs can be “enriched” for a particular isotype18, subclass, or glycosylation signature, leading to different Fc effector functions compared to other polyclonal IG products. For example, IgG1 and IgG4 are the most prevalent subclasses following measles infection or vaccination, with significant differences in titers in infected versus vaccinated individuals19. In addition, anti-SARS-CoV-2 antibodies from convalescent donors can have distinct glycosylation patterns depending on disease severity20. We will discuss some of the methods currently used to design, produce, and characterize antibody products, highlighting the differences between polyclonal and monoclonal antibody therapies.
Figure 1.
Structural and functional features of isotype G immunoglobulins (IgG). 1a. Structural features of IgG antibodies. The IgG macromolecule is a tetramer of two identical heavy (H) and light (L) chains depicted in dark and light blue, respectively, each containing variable (VH, VL) and constant (CH, CL) regions, as shown. The glycosylation site and the locations responsible for receptor and complement binding, are marked. These regions can be engineered to modulate downstream properties of IgG products. 1b. Antiviral functions of IgG antibodies. Antiviral pharmacologic properties of antibody therapies include 1) neutralization of viral entry to its cell target 2) complement and 3) antibody-mediated cytotoxicity of infected cells, 4) phagocytosis of infected cells and 5) clearing of opsonized virus by phagocytosis.
Figure 1.
Structural and functional features of isotype G immunoglobulins (IgG). 1a. Structural features of IgG antibodies. The IgG macromolecule is a tetramer of two identical heavy (H) and light (L) chains depicted in dark and light blue, respectively, each containing variable (VH, VL) and constant (CH, CL) regions, as shown. The glycosylation site and the locations responsible for receptor and complement binding, are marked. These regions can be engineered to modulate downstream properties of IgG products. 1b. Antiviral functions of IgG antibodies. Antiviral pharmacologic properties of antibody therapies include 1) neutralization of viral entry to its cell target 2) complement and 3) antibody-mediated cytotoxicity of infected cells, 4) phagocytosis of infected cells and 5) clearing of opsonized virus by phagocytosis.
Production and Characterization of Antibody Therapies
Specific Polyclonal Antibody Therapies
Specific polyclonal immune globulins (SpIG) are purified from pooled animal or human plasma. The first products were developed in 1898 and were comprised of little more than serum from horses vaccinated with live viruses, bacterial toxins, or snake venom. In 1903, diphtheria antitoxin made from vaccinated horses became the first licensed product in the United States. Research during World War II stimulated a major breakthrough in purification of IGs and other proteins from human plasma. IG purification methods are based on sequential alcohol precipitations, each with specific conditions of pH, ionic strength, temperature, protein concentration, and alcohol concentration21, 22.
For some products, purely chromatographic methods or caprylate precipitation methods have partially or completely supplanted alcohol precipitation. These changes are often driven by the need to increase yield of IgG (thus increasing availability)23. Nevertheless, alcohol-based fractionation remains as the backbone of early steps in production of IG products and is often combined with subsequent caprylate or polyethylene glycol precipitations. Modern IG products are further purified using column chromatography to remove unwanted plasma proteins. In addition, a minimum of two orthogonal, robust, dedicated viral clearance steps are performed, which often include solvent-detergent treatment and nanofiltration, as well as other virucidal (caprylate, heat treatment, low pH) and partitioning (chromatography, precipitations, depth filtration) steps. All viral clearance steps must be validated and found to be robust using scaled-down models of the manufacturing process and actual manufacturing intermediates spiked with virus as starting material. It should be emphasized that modern IG purification is highly complex with multiple steps, each of which must be controlled to result in a safe and intact product. Every manufacturing method is unique with respect to purification details and methodology (such as mixing speeds, equipment used, precipitation times, buffer types and concentrations, centrifugation vs. precipitation), and equipment. Thus, each product is also unique with respect to levels and types of plasma protein impurities and IG stability.
Specific polyclonal IG is used as the overarching term for all polyclonal preparations that are enriched for certain antiviral, antibacterial, or antitoxin antibodies. Antibody enrichment for human antibodies is achieved by either immunizing donors, or screening and selecting high-titer plasma from routine donations (as for Cytogam24) or convalescent donors (as for early versions of SARS-CoV-2 IG investigational products25, 26). “Hyperimmune” polyclonal antibodies are derived from animal or human donors who have been immunized intentionally for the purpose of obtaining high titer plasma (e.g., rabies, anthrax, and tetanus immune globulins). Nevertheless, convalescent plasma is often inaccurately referred to as “hyperimmune,” even though donors were not immunized. Under FDA-approved plasma center collection protocols, and after investigational safety studies are completed, hyperimmune plasma can be collected from consenting immunized donors.
For purposes of final product testing, a validated bioassay demonstrating neutralization in cell culture or in animals, is ideally performed for SpIG products. In special cases, adequate cell culture or animal models are not available at the time of licensure. In this situation, a binding assay has usually been selected and validated for product release contingent on discussions with FDA. Likewise, national or international IgG standards may be lacking. In these instances, an internal IgG standard is developed by the manufacturer.
Antiviral SpIG products licensed in the United States are shown in Table 1. A number of SpIGs (human, bovine, or equine plasma-derived) are under investigation for other viral infections, including SARS-CoV-2 (at least 12 studies in clinicaltrials.gov, e.g., including an International Network for Strategic Initiatives in Global HIV Trials study, NCT04910269) and influenza (NCT04850898). SpIGs from animal sources are produced by hyperimmunizing donor animals. Advantages of large animal donors (horses, sheep, or cattle) include the ability to immunize more frequently (which increases the yield and avidity of specific antibodies), to use experimental vaccinations, and to safely collect larger volumes of plasma. A major disadvantage includes potential allergic reactions in patients due to animal proteins, including the active ingredient. Animal-derived antibodies are often treated with pepsin or trypsin, to remove the Fc portion and reduce immunogenicity. These fragments lack effector functions that could be important for antibody activity, depending upon the virus. An interesting strategy has been developed using transchromosomic cattle that produce full-length human IgG antibodies. The cattle are knocked out for bovine antibody heavy and lambda light chains but contain an artificial chromosome encoding the respective human IgG chains. Chimeric antibodies consisting of human IgG heavy chains and bovine kappa light chains are removed during manufacturing42, thus the resulting IG product manufactured from these bovines contain only human IgGs, thus lowering the risk of immunogenicity. These transchromosomic bovines have been successfully hyperimmunized43.
Treatment timing and dosing for SpIG.
Treatment timing relative to infection depends on demonstrable efficacy of the product for pre- or post-exposure prophylaxis. Pre- and post-exposure prophylaxis can be effective (if adequately dosed) largely because viral burdens are relatively low. Even if an infection has been initiated, post-exposure prophylaxis attenuates disease severity of measles, HAV, and varicella zoster30, 36. When vaccines are given concomitantly with specific IG, such as for rabies, passive immunization provides a defensive “bridge” that acts immediately to neutralize the virus until vaccine responses arise. It is important that the dose of rabies IG (RIG) is not so high that it suppresses the vaccine response. In such contexts, both a minimum and maximum potency should be defined to assure optimal function of both RIG and the vaccine. Pharmacokinetic studies performed in healthy immunocompetent human subjects are used to define the dose of SpIG that is needed to avoid suppression of vaccine responses yet still be able to provide protection until vaccine responses are sufficiently developed.
Treatment of symptomatic viral disease with SpIG is much more challenging and often ineffective. In these cases, the viral burden may exceed the capacity of the IG, viruses may be relatively inaccessible within infected cells, and cellular immune responses may also be suppressed by the virus44. Notable lack of efficacy by specific IG for treatment of symptomatic infections such as rabies, influenza, HAV, HBV, measles, and varicella have been observed. The time windows for effective post-exposure prophylaxis of each infection have been established based on such failures. Treatment with CMVIG and HBVIG(IV) can prevent symptomatic disease in transplanted patients but are not curative. Vaccinia Immune Globulin is used to treat severe complications (eczema vaccinatum and progressive vaccinia) caused by live vaccinia virus vaccine (ACAM2000), which is used to prevent smallpox. Recently licensed replication-deficient vaccinia virus (Jynneos) generates an immune response but is thought to be incapable of causing eczema vaccinatum or progressive vaccinia. Both vaccines are indicated for prevention of smallpox. Jynneos is also licensed for prevention of monkeypox45.
Monoclonal Antibodies
To date, the FDA has approved four mAb therapies to prevent or treat viral diseases (Table 2): palivizumab for prevention of RSV in preterm infants and infants with other specific conditions, ibalizumab for treatment of HIV-1 in patients failing their current anti-retroviral regimen, and two products for treatment of Ebola virus disease caused by Zaire ebolavirus. One of these products, Inmazeb, consists of three mAbs that target non-overlapping epitopes on EBOV glycoprotein and represents the first co-formulated mAb cocktail approved by the FDA46.
Multiple mAbs are currently in advanced stages of clinical development or have been approved in other countries. Nirsevimab, a half-life extended mAb that targets the RSV fusion (F) protein 47, was recently approved by the European Medicines Agency for the prevention of RSV lower respiratory tract disease in neonates and infants during their first RSV season. In addition, three mAb products targeting the rabies virus glycoprotein have been approved in other countries: two in India (Rabishield, a single mAb, and TwinRab, a cocktail of two mAbs48), and one in China (ormutivimab49 ).
Several mAbs and mAb combinations that target the SARS-CoV-2 spike protein were rapidly developed after the onset of the COVID-19 pandemic and received emergency use authorization (EUA) from the FDA for the pre-exposure prophylaxis, post-exposure prophylaxis, and/or treatment of COVID-19. Although highly effective against early SARS-CoV-2 variants, these products are not currently authorized in the United States due to the emergence and widespread circulation of variants that are resistant to neutralization by these mAbs in cell culture50-56. However, if future variants emerge that are susceptible to these products, their authorization status may change. Refer to the FDA website for updated information on the status of EUAs for mAbs and other COVID-19 therapeutics57.
In addition to the approved and previously authorized mAbs and those directed against SARS-CoV-2, many other mAbs have been or are under development against existing and emerging diseases (see 58-61 for some examples).
Historically, therapeutic mAbs were derived from immunized mice or rats and engineered as chimeric or humanized mAbs to reduce the immunogenicity due the “foreignness” of rodent mAbs in humans. Currently, most mAbs are of human origin, derived from “humanized mice” that express human germline V(D)J region genes, or from phage display libraries generated from human donor lymphocytes. However, many antiviral mAbs are isolated directly from previously infected patients (see58, 62-64). Regardless of the source, many considerations inform the selection and engineering of candidate mAbs.
Fc engineering approaches: Most mAbs developed for viral diseases are selected first for their ability to neutralize virus entry. However, Fc effector functions play a major role in the immune system’s response to infectious diseases8. For mAbs, the contribution of Fc effector functions to disease protection has been demonstrated for several viruses including Ebola virus65, HIV-16,66, 67, influenza68, SARS-COV-269, and Rift Valley fever virus70. However, ADE of infection or disease is a possible negative consequence of FcγR binding71-73. Therefore, depending on what is known about specific viral diseases, different approaches are used to engineer the Fc region of mAbs to either enhance or diminish FcγR binding. Amino acid residues have been identified in the IgG Fc region that contact the complement component C1q, FcγRs, or the neonatal Fc receptor (FcRn), which is responsible for the long half-life of IgG (reviewed in74, 75). Substitutions can be engineered at these residues to alter Fc effector functions or extend the half-life of a mAb, which allows less frequent dosing76.
In addition to Fc engineering, there is a better understanding of specific Fc glycan structures and their association with different effector functions, e.g., afucosylated mAbs have better antibody dependent cellular cytotoxicity (ADCC) compared to highly fucosylated antibodies, and galactosylation is associated with complement dependent cytotoxicity (CDC) and can influence ADCC activity77 . Therefore, cell lines have been engineered to produce mAbs with up to 100% afucosylation to enhance ADCC activity74, 78. The understanding of the relationship between antibody glycan structures and Fc effector functions is ongoing and additional strategies may be developed to further engineer mAb glycan structures. For example, the effect of galactosylation on ADCC activity may depend on the specific linkage of the galactose monosaccharide79. Fc effector functions can be reduced by introducing substitutions at the glycosylation site (N297) in the CH2 domain to prevent the addition of a glycan80, 81, thus providing another glycoengineering approach for antiviral mAbs.
Other approaches for the development of mAbs: Three of the four approved monoclonal antiviral products are single mAbs, but the anti-Ebola virus mAb cocktail of atoltivimab, maftivimab, odesivimab-ebgn was the first fixed dose co-formulated mAb combination product approved by the FDA. Many other mAbs to treat viral diseases and for other indications are used in combination, but only a few to date are co-formulated82. The advantage of antibody cocktails over a single mAb is that they might be less susceptible to escape, depending on the different targeted epitopes. As seen for the anti-SARS-CoV-2 mAb combinations previously authorized for the prophylaxis or treatment of COVID-19, they all target the SARS-CoV-2 receptor binding domain of the SARS-CoV-2 spike protein but have little neutralization activity against current variants. MAbs that target regions outside the receptor binding domain could neutralize virus or mediate Fc effector functions and might be less susceptible to escape. For example, a recent report demonstrated that mAbs targeting the conserved fusion peptide region adjacent to the S2′ cleavage site of the spike protein are broadly neutralizing against betacoronaviruses83.
Advantages and Disadvantages of Polyclonal and Monoclonal Antibodies
There are advantages and disadvantages when selecting a product for treatment or prophylaxis of viral diseases, some of which are summarized in Table 3. For approved products, the choice is often based on which products are available for a specific viral disease. For example, currently only SpIG products are approved in the United States to treat rabies, CMV, HBV, varicella or vaccinia, whereas only mAb therapies are approved to prevent or treat RSV, HIV-1, and EBOV disease. There are other considerations that also play a role in development or deployment of antibody therapies in an infectious disease setting. Although resistance to polyclonal antibodies has been reported84, polyclonal antiviral products are less likely to result in treatment-emergent resistance, or the formation of anti-drug antibodies, whereas both issues are a larger concern for mAb products. On the other hand, given the relative ease of engineering, development, and production of mAbs, they are well suited for rapid development, especially in an emerging infectious disease setting. Both types of products can have drawbacks that include the potential to interfere with the immune response to the vaccine or natural infection, as well as specific diagnostics, and the potential to result in enhanced infection or disease, as already described. Despite these limitations, the benefit-to-risk ratio for these approved products is favorable, as demonstrated in clinical trials and by routine clinical use in viral disease settings.
Future Directions and Conclusions
As the applications of antiviral antibody therapies expand, there are several areas that can benefit from further development. The international standardization of assays and reagents (e.g., viruses and cell lines) for measuring antibody activity could help address the variability in potency often observed for the same antibody in different assays or laboratories (e.g., 10-100-fold range in EC50 values for anti-SARS-CoV-2 mAbs)231. In addition, more work is needed to better understand the role of Fc effector functions in viral diseases using cell culture, animal models, and clinical studies. Furthermore, preclinical assays are being developed that are more physiologically relevant, especially potency assays that capture multiple functions of the antibody. For example, organ-on-a chip and microphysiological systems can incorporate multiple cell types including immune cells, simulate blood flow and organ perfusion, and provide data that serves as a bridge between standard cell culture assays and clinical studies. Although still early in development and not commonly used in regulatory applications, such technologies are expected to become increasingly powerful and more widely used. These systems can also help address ethical concerns and societal pressures to replace, reduce, and refine animal research, and they have the potential to provide information that is more predictive of clinical efficacy.
Another area with unharnessed potential, especially for SpIG therapies, is the selection of specific glycosylation signatures to modulate downstream immune responses232. These strategies have been proposed for use in the setting of autoimmune disease, but they could potentially be applied to viral diseases as well. When combined with other novel technologies, such as the production of recombinant IG preparations233, such methods have the potential to result in antiviral antibody preparations with improved properties.
Production of SpIG from convalescent plasma in a pandemic setting remains time-consuming and challenging. Convalescent plasma is often the earliest available antibody-containing treatment that could be effective for prevention of severe disease. Advances in technologies that can be used to rapidly and inexpensively select donations containing high titers of neutralizing antibodies (from among thousands of donations) are needed both for direct use of convalescent plasma and manufacturing of SpIG. Biosensor-based methods that reliably measure neutralizing potency in plasma donations and products, and that can be used in a low biocontainment (BSL-2) setting are promising. In addition to improved donor screening, technical advances in manufacturing that would maximize the yield of SpIG during a pandemic could include affinity matrices or changes in manufacturing steps to allow virus-specific IgM to copurify with IgG.
For mAbs, large volumes of product are usually infused intravenously for several hours. The length of infusion time may depend on the amount of mAb needed per body weight and whether a patient is experiencing infusion-related reactions typical of mAbs. One strategy to reduce the time of infusion is to co-formulate a high concentration of the mAb with recombinant human hyaluronidase234. The recombinant human hyaluronidase degrades hyaluronic acid in the extracellular matrix, facilitating rapid delivery of large volume subcutaneous injections and bioavailability of the product. This has already been accomplished with several mAbs for oncology, including the combination of rituximab, trastuzumab, daratumumab or trastuzumab and pertuzumab with recombinant human hyaluronidase235. This approach is being studied with an anti-HIV mAb (clinicaltrials.gov #NCT03538626). These formulations provide more convenient dosing for patients.
Other developments for anti-viral mAbs include bispecific antibodies, single domain antibodies derived from camelids, and other scaffold proteins such as DARPIns and Adnectins that are engineered in the loop regions between more structured regions of the core domain to mimic antibody CDRs236-239. Some of these technologies may be able to target epitopes that are difficult for traditional antibodies to recognize. Furthermore, these novel constructs may be more cost effective to manufacture than mAb cocktails and lower doses may be as effective as higher doses of a mAb cocktail. However, clinical studies are needed to determine efficacy and safety and to see if there are issues, such as immunogenicity, related to these novel products.
Whether alone or in combination, antiviral antibody therapies can provide important prophylaxis and treatment options to help relieve the burden of viral diseases. This space is rapidly evolving, and, as more experience is gained through successful clinical applications, the products of the future have the potential to overcome many of the challenges we describe, while continuing to fulfill the promise of safety and effectiveness.
References
- Armitage C. The high burden of infectious disease. Nature. 2021;598(S9).
- Parra D, Takizawa F, Sunyer JO. Evolution of B cell immunity. Annu Rev Anim Biosci. Jan 2013;1:65-97. [CrossRef]
- Science and the Regulation of Biological Products. Center for Biologics Evaluation and Research; 2002. https://www.fda.gov/about-fda/histories-product-regulation/science-and-regulation-biological-products.
- Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. [CrossRef]
- Gunn BM, Yu WH, Karim MM, et al. A Role for Fc Function in Therapeutic Monoclonal Antibody-Mediated Protection against Ebola Virus. Cell Host Microbe. Aug 8 2018;24(2):221-233 e5. [CrossRef]
- Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. Sep 11 2014;158(6):1243-1253. [CrossRef]
- DiLillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J Clin Invest. Feb 2016;126(2):605-10. [CrossRef]
- Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. Jan 2018;18(1):46-61. [CrossRef]
- Phelps M, Balazs AB. Contribution to HIV Prevention and Treatment by Antibody-Mediated Effector Function and Advances in Broadly Neutralizing Antibody Delivery by Vectored Immunoprophylaxis. Front Immunol. 2021;12:734304. [CrossRef]
- Halstead SB. Dengue Antibody-Dependent Enhancement: Knowns and Unknowns. Microbiol Spectr. Dec 2014;2(6). [CrossRef]
- Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. Oct 2020;20(10):633-643. [CrossRef]
- Screaton G, Mongkolsapaya J, Yacoub S, Roberts C. New insights into the immunopathology and control of dengue virus infection. Nat Rev Immunol. Dec 2015;15(12):745-59. [CrossRef]
- Brown JA, Singh G, Acklin JA, et al. Dengue Virus Immunity Increases Zika Virus-Induced Damage during Pregnancy. Immunity. Mar 19 2019;50(3):751-762.e5. [CrossRef]
- Martín-Acebes MA, Saiz JC, Jiménez de Oya N. Antibody-Dependent Enhancement and Zika: Real Threat or Phantom Menace? Front Cell Infect Microbiol. 2018;8:44. [CrossRef]
- Kotaki T, Kurosu T, Grinyo-Escuer A, et al. An affinity-matured human monoclonal antibody targeting fusion loop epitope of dengue virus with in vivo therapeutic potency. Sci Rep. Jun 21 2021;11(1):12987. [CrossRef]
- Lu J, Chen L, Du P, et al. A human monoclonal antibody to neutralize all four serotypes of dengue virus derived from patients at the convalescent phase of infection. Virology. Nov 2022;576:74-82. [CrossRef]
- Pinto AK, Hassert M, Han X, et al. The Ability of Zika virus Intravenous Immunoglobulin to Protect From or Enhance Zika Virus Disease. Front Immunol. 2021;12:717425. [CrossRef]
- Wilson CS, Hoopes EM, Falk AC, Moore DJ. A human IgM enriched immunoglobulin preparation, Pentaglobin, reverses autoimmune diabetes without immune suppression in NOD mice. Sci Rep. Jul 11 2022;12(1):11731. [CrossRef]
- Isa MB, Martinez LC, Ferreyra LJ, et al. Measles virus-specific IgG4 antibody titer as a serologic marker of post-vaccinal immune response. Viral Immunol. Summer 2006;19(2):335-9. [CrossRef]
- Siekman SL, Pongracz T, Wang W, et al. The IgG glycome of SARS-CoV-2 infected individuals reflects disease course and severity. Front Immunol. 2022;13:993354. [CrossRef]
- Cohn EJ, Strong LE, Hughes WL, et al. Preparation and properties of serum and plasma proteins; a system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. J Am Chem Soc. March 1 1946;68(3):459-75. [CrossRef]
- Oncley JL, Melin M, Richert DA, Cameron JW, Gross PM. The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta1-lipoprotein into subfractions of human plasma. J Am Chem Soc. Feb 1949;71(2):541-50. [CrossRef]
- Lebing W, Remington KM, Schreiner C, Paul HI. Properties of a new intravenous immunoglobulin (IGIV-C, 10%) produced by virus inactivation with caprylate and column chromatography. Vox Sang. Apr 2003;84(3):193-201. [CrossRef]
- CytoGam Prescribing Information. https://www.accessdata.fda.gov/spl/data/2a40733c-106b-41cf-94f0-f10a03180ac8/2a40733c-106b-41cf-94f0-f10a03180ac8.xml.
- Vandeberg P, Cruz M, Diez JM, et al. Production of anti-SARS-CoV-2 hyperimmune globulin from convalescent plasma. Transfusion. Jun 2021;61(6):1705-1709. [CrossRef]
- Burnouf T, Gathof B, Bloch EM, et al. Production and Quality Assurance of Human Polyclonal Hyperimmune Immunoglobulins Against SARS-CoV-2. Transfus Med Rev. Jul 2022;36(3):125-132. [CrossRef]
- HyperRAB prescribing information. https://www.accessdata.fda.gov/spl/data/f993778d-01fb-4670-af67-a0e08d6b258b/f993778d-01fb-4670-af67-a0e08d6b258b.xml.
- Imogam prescribing information. https://www.accessdata.fda.gov/spl/data/8026005f-7587-47fe-bb78-ec6247a3434b/8026005f-7587-47fe-bb78-ec6247a3434b.xml.
- Kedrab prescribing information. https://www.accessdata.fda.gov/spl/data/5e5c130a-693b-47f9-b44a-3d8f9cde3f98/5e5c130a-693b-47f9-b44a-3d8f9cde3f98.xml.
- VariZIG prescribing information. https://www.accessdata.fda.gov/spl/data/272379b7-f0e7-4560-8d79-3fd0024c3010/272379b7-f0e7-4560-8d79-3fd0024c3010.xml.
- Levin MJ, Duchon JM, Swamy GK, Gershon AA. Varicella zoster immune globulin (VARIZIG) administration up to 10 days after varicella exposure in pregnant women, immunocompromised participants, and infants: Varicella outcomes and safety results from a large, open-label, expanded-access program. PLoS One. 2019;14(7):e0217749. [CrossRef]
- Vaccinia Immune Globulin prescribing information. https://www.fda.gov/media/78174/download.
- Centers for Disease C, Prevention. Household transmission of vaccinia virus from contact with a military smallpox vaccinee--Illinois and Indiana, 2007. MMWR Morb Mortal Wkly Rep. May 18 2007;56(19):478-81.
- Centers for Disease C, Prevention. Progressive vaccinia in a military smallpox vaccinee - United States, 2009. MMWR Morb Mortal Wkly Rep. May 22 2009;58(19):532-6.
- Razonable RR, Humar A. Cytomegalovirus in solid organ transplant recipients-Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin Transplant. Sep 2019;33(9):e13512. [CrossRef]
- GamaSTAN prescribing information. https://www.accessdata.fda.gov/spl/data/38a323af-7c25-42d1-9c29-532ef61999b8/38a323af-7c25-42d1-9c29-532ef61999b8.xml.
- HyperHEP B prescribing information. https://www.accessdata.fda.gov/spl/data/391b2218-8a15-4e5e-8717-aa49efcc2210/391b2218-8a15-4e5e-8717-aa49efcc2210.xml.
- Nabi-HB prescribing information. https://www.accessdata.fda.gov/spl/data/ee1560c0-18e1-b617-e053-2a95a90aa1af/ee1560c0-18e1-b617-e053-2a95a90aa1af.xml.
- HepaGAM B prescribing information. https://www.accessdata.fda.gov/spl/data/56525de0-f47d-11eb-85b4-0800200c9a66/56525de0-f47d-11eb-85b4-0800200c9a66.xml.
- Te H, Doucette K. Viral hepatitis: Guidelines by the American Society of Transplantation Infectious Disease Community of Practice. Clin Transplant. Sep 2019;33(9):e13514. [CrossRef]
- FDA. Letter to Immune Globulin (Human) Licensed Manufacturers: Option to Lower Lot Release Specification for Required Measles Antibody Potency Testing. https://www.fda.gov/media/118428/download.
- Gardner CL, Sun C, Luke T, et al. Antibody Preparations from Human Transchromosomic Cows Exhibit Prophylactic and Therapeutic Efficacy against Venezuelan Equine Encephalitis Virus. J Virol. Jul 15 2017;91(14). [CrossRef]
- Saied AA, Nascimento MSL, do Nascimento Rangel AH, et al. Transchromosomic bovines-derived broadly neutralizing antibodies as potent biotherapeutics to counter important emerging viral pathogens with a special focus on SARS-CoV-2, MERS-CoV, Ebola, Zika, HIV-1, and influenza A virus. J Med Virol. Oct 2022;94(10):4599-4610. [CrossRef]
- Stauft CB, Tegenge M, Khurana S, et al. Pharmacokinetics and Efficacy of Human Hyperimmune Intravenous Immunoglobulin Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 Infection in Adult Syrian Hamsters. Clin Infect Dis. Aug 24 2022;75(1):e459-e465. [CrossRef]
- Rao AK, Petersen BW, Whitehill F, et al. Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for Preexposure Vaccination of Persons at Risk for Occupational Exposure to Orthopoxviruses: Recommendations of the Advisory Committee on Immunization Practices - United States, 2022. MMWR Morb Mortal Wkly Rep. Jun 3 2022;71(22):734-742. [CrossRef]
- Tshiani Mbaya O, Mukumbayi P, Mulangu S. Review: Insights on Current FDA-Approved Monoclonal Antibodies Against Ebola Virus Infection. Front Immunol. 2021;12:721328. [CrossRef]
- Hammitt LL, Dagan R, Yuan Y, et al. Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N Engl J Med. Mar 3 2022;386(9):837-846. [CrossRef]
- de Melo GD, Hellert J, Gupta R, Corti D, Bourhy H. Monoclonal antibodies against rabies: current uses in prophylaxis and in therapy. Curr Opin Virol. Apr 2022;53:101204. [CrossRef]
- Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. MAbs. Jan-Dec 2023;15(1):2153410. [CrossRef]
- Cao Y, Wang J, Jian F, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. Feb 2022;602(7898):657-663. [CrossRef]
- Dejnirattisai W, Huo J, Zhou D, et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. Feb 3 2022;185(3):467-484 e15. [CrossRef]
- Planas D, Saunders N, Maes P, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. Feb 2022;602(7898):671-675. [CrossRef]
- Sheward DJ, Kim C, Fischbach J, et al. Omicron sublineage BA.2.75.2 exhibits extensive escape from neutralising antibodies. Lancet Infect Dis. Nov 2022;22(11):1538-1540. [CrossRef]
- Group AC--TfIwC-S. Tixagevimab-cilgavimab for treatment of patients hospitalised with COVID-19: a randomised, double-blind, phase 3 trial. Lancet Respir Med. Oct 2022;10(10):972-984. [CrossRef]
- Imai M, Ito M, Kiso M, et al. Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N Engl J Med. Jan 5 2023;388(1):89-91. [CrossRef]
- Wang Q, Iketani S, Li Z, et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell. Dec 14 2022; [CrossRef]
- Coronavirus Disease 2019 (COVID-19) EUA Information. Accessed 02/17/2023, https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs.
- Corti D, Lanzavecchia A. Efficient Methods To Isolate Human Monoclonal Antibodies from Memory B Cells and Plasma Cells. Microbiol Spectr. Oct 2014;2(5). [CrossRef]
- Dibo M, Battocchio EC, Dos Santos Souza LM, et al. Antibody Therapy for the Control of Viral Diseases: An Update. Curr Pharm Biotechnol. 2019;20(13):1108-1121. [CrossRef]
- Hastie KM, Cross RW, Harkins SS, et al. Convergent Structures Illuminate Features for Germline Antibody Binding and Pan-Lassa Virus Neutralization. Cell. Aug 8 2019;178(4):1004-1015 e14. [CrossRef]
- Li H, Buck T, Zandonatti M, et al. A cocktail of protective antibodies subverts the dense glycan shield of Lassa virus. Sci Transl Med. Oct 26 2022;14(668):eabq0991. [CrossRef]
- Corti D, Misasi J, Mulangu S, et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science. Mar 18 2016;351(6279):1339-42. [CrossRef]
- Karuna ST, Corey L. Broadly Neutralizing Antibodies for HIV Prevention. Annu Rev Med. Jan 27 2020;71:329-346. [CrossRef]
- Wrammert J, Smith K, Miller J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. May 29 2008;453(7195):667-71. [CrossRef]
- !!! INVALID CITATION !!! 5;
- Lewis GK. Role of Fc-mediated antibody function in protective immunity against HIV-1. Immunology. May 2014;142(1):46-57. [CrossRef]
- Asokan M, Dias J, Liu C, et al. Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc Natl Acad Sci U S A. Aug 4 2020;117(31):18754-18763. [CrossRef]
- Vanderven HA, Kent SJ. The protective potential of Fc-mediated antibody functions against influenza virus and other viral pathogens. Immunol Cell Biol. Apr 2020;98(4):253-263. [CrossRef]
- Zhang A, Stacey HD, D'Agostino MR, Tugg Y, Marzok A, Miller MS. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat Rev Immunol. Dec 19 2022:1-16. [CrossRef]
- Cartwright HN, Barbeau DJ, McElroy AK. Isotype-Specific Fc Effector Functions Enhance Antibody-Mediated Rift Valley Fever Virus Protection In Vivo. mSphere. Oct 27 2021;6(5):e0055621. [CrossRef]
- Taylor A, Foo SS, Bruzzone R, Dinh LV, King NJ, Mahalingam S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev. Nov 2015;268(1):340-64. [CrossRef]
- Smatti MK, Al Thani AA, Yassine HM. Viral-Induced Enhanced Disease Illness. Front Microbiol. 2018;9:2991. [CrossRef]
- doi:10.1016/b978-0-12-809468-6.00033-4.
- Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front Immunol. 2017;8:1751. [CrossRef]
- Liu R, Oldham RJ, Teal E, Beers SA, Cragg MS. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies (Basel). Nov 17 2020;9(4). [CrossRef]
- Ko S, Jo M, Jung ST. Recent Achievements and Challenges in Prolonging the Serum Half-Lives of Therapeutic IgG Antibodies Through Fc Engineering. BioDrugs. Mar 2021;35(2):147-157. [CrossRef]
- Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. Dec 2015;25(12):1325-34. [CrossRef]
- Golay J, Andrea AE, Cattaneo I. Role of Fc Core Fucosylation in the Effector Function of IgG1 Antibodies. Front Immunol. 2022;13:929895. [CrossRef]
- Hatfield G, Tepliakova L, Gingras G, et al. Specific location of galactosylation in an afucosylated antiviral monoclonal antibody affects its FcgammaRIIIA binding affinity. Front Immunol. 2022;13:972168. [CrossRef]
- Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. Oct 15 1989;143(8):2595-601.
- Bolt S, Routledge E, Lloyd I, et al. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol. Feb 1993;23(2):403-11. [CrossRef]
- Liu D, Shameem M. Antiviral monoclonal antibody cocktails as a modern weapon in combating pandemics. Ther Deliv. Feb 2022;13(2):67-69. [CrossRef]
- Dacon C, Tucker C, Peng L, et al. Broadly neutralizing antibodies target the coronavirus fusion peptide. Science. Aug 12 2022;377(6607):728-735. [CrossRef]
- Tarafdar S, Virata ML, Yan H, et al. Multiple epitopes of hepatitis B virus surface antigen targeted by human plasma-derived immunoglobulins coincide with clinically observed escape mutations. J Med Virol. Feb 2022;94(2):649-658. [CrossRef]
- Center for Drug Evaluation and Research OoPQ. Potency Assay Considerations for Monoclonal Antibodies and Other Therapeutic Proteins Targeting Viral Pathogens, Guidance for Industry (Draft). https://www.fda.gov/media/165746/download.
- Huber M, Trkola A. Humoral immunity to HIV-1: neutralization and beyond. J Intern Med. Jul 2007;262(1):5-25. [CrossRef]
- Klasse PJ. Neutralization of Virus Infectivity by Antibodies: Old Problems in New Perspectives. Adv Biol. 2014;2014. [CrossRef]
- Reading SA, Dimmock NJ. Neutralization of animal virus infectivity by antibody. Arch Virol. 2007;152(6):1047-59. [CrossRef]
- Jiang XR, Song A, Bergelson S, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. Feb 2011;10(2):101-11. [CrossRef]
- Schmidt F, Weisblum Y, Muecksch F, et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J Exp Med. Nov 2 2020;217(11). [CrossRef]
- Clapham PR. Vesicular stomatitis virus pseudotypes of retroviruses. Methods Mol Biol. 1992;8:95-102. [CrossRef]
- Kim Y, Zheng X, Eschke K, et al. MCMV-based vaccine vectors expressing full-length viral proteins provide long-term humoral immune protection upon a single-shot vaccination. Cell Mol Immunol. Feb 2022;19(2):234-244. [CrossRef]
- Racine T, Kobinger GP, Arts EJ. Development of an HIV vaccine using a vesicular stomatitis virus vector expressing designer HIV-1 envelope glycoproteins to enhance humoral responses. AIDS Res Ther. Sep 12 2017;14(1):55. [CrossRef]
- Takada A, Feldmann H, Stroeher U, et al. Identification of protective epitopes on ebola virus glycoprotein at the single amino acid level by using recombinant vesicular stomatitis viruses. J Virol. Jan 2003;77(2):1069-74. [CrossRef]
- Takada A, Robison C, Goto H, et al. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A. Dec 23 1997;94(26):14764-9. [CrossRef]
- Bannert N, Farzan M, Friend DS, et al. Human Mast cell progenitors can be infected by macrophagetropic human immunodeficiency virus type 1 and retain virus with maturation in vitro. J Virol. Nov 2001;75(22):10808-14. [CrossRef]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. Feb 1 1995;206(2):935-44. [CrossRef]
- Freed EO, Englund G, Martin MA. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol. Jun 1995;69(6):3949-54. [CrossRef]
- Louder MK, Sambor A, Chertova E, et al. HIV-1 envelope pseudotyped viral vectors and infectious molecular clones expressing the same envelope glycoprotein have a similar neutralization phenotype, but culture in peripheral blood mononuclear cells is associated with decreased neutralization sensitivity. Virology. Sep 1 2005;339(2):226-38. [CrossRef]
- Lundquist CA, Zhou J, Aiken C. Nef stimulates human immunodeficiency virus type 1 replication in primary T cells by enhancing virion-associated gp120 levels: coreceptor-dependent requirement for Nef in viral replication. J Virol. Jun 2004;78(12):6287-96. [CrossRef]
- Sarzotti-Kelsoe M, Daniell X, Todd CA, et al. Optimization and validation of a neutralizing antibody assay for HIV-1 in A3R5 cells. J Immunol Methods. Jul 2014;409:147-60. [CrossRef]
- Matsuura Y, Tani H, Suzuki K, et al. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology. Aug 1 2001;286(2):263-75. [CrossRef]
- Renelt S, Schult-Dietrich P, Baldauf HM, et al. HIV-1 Infection of Long-Lived Hematopoietic Precursors In Vitro and In Vivo. Cells. Sep 23 2022;11(19). [CrossRef]
- Riepler L, Rossler A, Falch A, et al. Comparison of Four SARS-CoV-2 Neutralization Assays. Vaccines (Basel). Dec 28 2020;9(1). [CrossRef]
- Chikere K, Webb NE, Chou T, et al. Distinct HIV-1 entry phenotypes are associated with transmission, subtype specificity, and resistance to broadly neutralizing antibodies. Retrovirology. Jun 23 2014;11:48. [CrossRef]
- Mann AM, Rusert P, Berlinger L, Kuster H, Gunthard HF, Trkola A. HIV sensitivity to neutralization is determined by target and virus producer cell properties. AIDS. Aug 24 2009;23(13):1659-67. [CrossRef]
- Miyamoto F, Kawaji K, Oishi S, Fujii N, Kaku M, Kodama EN. Anti-HIV-1 activity determined by beta-galactosidase activity in the multinuclear activation of an indicator assay is comparable with that by a conventional focus counting method. Antivir Chem Chemother. Apr 2015;24(2):77-82. [CrossRef]
- Spenlehauer C, Gordon CA, Trkola A, Moore JP. A luciferase-reporter gene-expressing T-cell line facilitates neutralization and drug-sensitivity assays that use either R5 or X4 strains of human immunodeficiency virus type 1. Virology. Feb 15 2001;280(2):292-300. [CrossRef]
- Sarzotti-Kelsoe M, Bailer RT, Turk E, et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods. Jul 2014;409:131-46. [CrossRef]
- Bentley EM, Mather ST, Temperton NJ. The use of pseudotypes to study viruses, virus sero-epidemiology and vaccination. Vaccine. Jun 12 2015;33(26):2955-62. [CrossRef]
- Comas-Garcia M, Colunga-Saucedo M, Rosales-Mendoza S. The Role of Virus-Like Particles in Medical Biotechnology. Mol Pharm. Dec 7 2020;17(12):4407-4420. [CrossRef]
- Du R, Cui Q, Caffrey M, Rong L. Ebola Virus Entry Inhibitors. Adv Exp Med Biol. 2022;1366:155-170. [CrossRef]
- Kaku Y, Noguchi A, Marsh GA, et al. Second generation of pseudotype-based serum neutralization assay for Nipah virus antibodies: sensitive and high-throughput analysis utilizing secreted alkaline phosphatase. J Virol Methods. Jan 2012;179(1):226-32. [CrossRef]
- Kaku Y, Noguchi A, Marsh GA, et al. A neutralization test for specific detection of Nipah virus antibodies using pseudotyped vesicular stomatitis virus expressing green fluorescent protein. J Virol Methods. Sep 2009;160(1-2):7-13. [CrossRef]
- Khetawat D, Broder CC. A functional henipavirus envelope glycoprotein pseudotyped lentivirus assay system. Virol J. Nov 12 2010;7:312. [CrossRef]
- Nooraei S, Bahrulolum H, Hoseini ZS, et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J Nanobiotechnology. Feb 25 2021;19(1):59. [CrossRef]
- Rudometova NB, Shcherbakov DN, Rudometov AP, Ilyichev AA, Karpenko LI. Model systems of human immunodef iciency virus (HIV-1) for in vitro eff icacy assessment of candidate vaccines and drugs against HIV-1. Vavilovskii Zhurnal Genet Selektsii. Mar 2022;26(2):214-221. [CrossRef]
- Steeds K, Hall Y, Slack GS, et al. Pseudotyping of VSV with Ebola virus glycoprotein is superior to HIV-1 for the assessment of neutralising antibodies. Sci Rep. Aug 31 2020;10(1):14289. [CrossRef]
- Steffen I, Simmons G. Pseudotyping Viral Vectors With Emerging Virus Envelope Proteins. Curr Gene Ther. 2016;16(1):47-55. [CrossRef]
- Wang B, Meng XJ. Structural and molecular biology of hepatitis E virus. Comput Struct Biotechnol J. 2021;19:1907-1916. [CrossRef]
- Ryu W-S. Virus Vectors. Molecular Virology of Human Pathogenic Viruses. 1st ed. Elsevier Inc.; 2017:263-275:chap 19.
- Gasmi M, Glynn J, Jin MJ, Jolly DJ, Yee JK, Chen ST. Requirements for efficient production and transduction of human immunodeficiency virus type 1-based vectors. J Virol. Mar 1999;73(3):1828-34. [CrossRef]
- Salmon P, Trono D. Lentiviral vectors for the gene therapy of lympho-hematological disorders. Curr Top Microbiol Immunol. 2002;261:211-27. [CrossRef]
- Todd CA, Greene KM, Yu X, et al. Development and implementation of an international proficiency testing program for a neutralizing antibody assay for HIV-1 in TZM-bl cells. J Immunol Methods. Jan 31 2012;375(1-2):57-67. [CrossRef]
- Wei X, Decker JM, Liu H, et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. Jun 2002;46(6):1896-905. [CrossRef]
- Seaman MS, Janes H, Hawkins N, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. Feb 2010;84(3):1439-52. [CrossRef]
- Hoenen T. Minigenome Systems for Filoviruses. Methods Mol Biol. 2018;1604:237-245. [CrossRef]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. Nov 2002;20(11):1151-4. [CrossRef]
- Li J, Bentsman G, Potash MJ, Volsky DJ. Human immunodeficiency virus type 1 efficiently binds to human fetal astrocytes and induces neuroinflammatory responses independent of infection. BMC Neurosci. May 12 2007;8:31. [CrossRef]
- Saeed MF, Kolokoltsov AA, Davey RA. Novel, rapid assay for measuring entry of diverse enveloped viruses, including HIV and rabies. J Virol Methods. Aug 2006;135(2):143-50. [CrossRef]
- Tobiume M, Lineberger JE, Lundquist CA, Miller MD, Aiken C. Nef does not affect the efficiency of human immunodeficiency virus type 1 fusion with target cells. J Virol. Oct 2003;77(19):10645-50. [CrossRef]
- Tscherne DM, Manicassamy B, Garcia-Sastre A. An enzymatic virus-like particle assay for sensitive detection of virus entry. J Virol Methods. Feb 2010;163(2):336-43. [CrossRef]
- Wyma DJ, Jiang J, Shi J, et al. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol. Apr 2004;78(7):3429-35. [CrossRef]
- Leroy H, Han M, Woottum M, et al. Virus-Mediated Cell-Cell Fusion. Int J Mol Sci. Dec 17 2020;21(24). [CrossRef]
- Bossart KN, Broder CC. Viral glycoprotein-mediated cell fusion assays using vaccinia virus vectors. Methods Mol Biol. 2004;269:309-32. [CrossRef]
- Moulard M, Phogat SK, Shu Y, et al. Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120-CD4-CCR5 complexes. Proc Natl Acad Sci U S A. May 14 2002;99(10):6913-8. [CrossRef]
- Saw WT, Matsuda Z, Eisenberg RJ, Cohen GH, Atanasiu D. Using a split luciferase assay (SLA) to measure the kinetics of cell-cell fusion mediated by herpes simplex virus glycoproteins. Methods. Nov 15 2015;90:68-75. [CrossRef]
- Wang L, Zhao J, Nguyen LNT, et al. Blockade of SARS-CoV-2 spike protein-mediated cell-cell fusion using COVID-19 convalescent plasma. Sci Rep. Mar 10 2021;11(1):5558. [CrossRef]
- Oguntuyo KY, Stevens CS, Hung CT, et al. Quantifying Absolute Neutralization Titers against SARS-CoV-2 by a Standardized Virus Neutralization Assay Allows for Cross-Cohort Comparisons of COVID-19 Sera. mBio. Feb 16 2021;12(1). [CrossRef]
- Schendel SL, Saphire EO. Assay Standardization for Neutralizing Antibody. Why Different Labs Can Get Different Results and A Path Forward. In "Therapeutic Neutralizing Monoclonal Antibodies: Report of a Summit sponsored by Operation Warp Speed and the National Institutes of Health". August 20, 2020:52-66.
- Li Y, O'Dell S, Walker LM, et al. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J Virol. Sep 2011;85(17):8954-67. [CrossRef]
- Lorenzi JCC, Mendoza P, Cohen YZ, et al. Neutralizing Activity of Broadly Neutralizing anti-HIV-1 Antibodies against Primary African Isolates. J Virol. Mar 1 2021;95(5). [CrossRef]
- Sanders DA. No false start for novel pseudotyped vectors. Curr Opin Biotechnol. Oct 2002;13(5):437-42. [CrossRef]
- Li Q, Liu Q, Huang W, Li X, Wang Y. Current status on the development of pseudoviruses for enveloped viruses. Rev Med Virol. Jan 2018;28(1). [CrossRef]
- Chen RE, Zhang X, Case JB, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. Apr 2021;27(4):717-726. [CrossRef]
- Cho A, Muecksch F, Schaefer-Babajew D, et al. Anti-SARS-CoV-2 receptor-binding domain antibody evolution after mRNA vaccination. Nature. Dec 2021;600(7889):517-522. [CrossRef]
- Dong J, Zost SJ, Greaney AJ, et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat Microbiol. Oct 2021;6(10):1233-1244. [CrossRef]
- Lusvarghi S, Pollett SD, Neerukonda SN, et al. SARS-CoV-2 BA.1 variant is neutralized by vaccine booster-elicited serum but evades most convalescent serum and therapeutic antibodies. Sci Transl Med. May 18 2022;14(645):eabn8543. [CrossRef]
- Shi R, Shan C, Duan X, et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. Aug 2020;584(7819):120-124. [CrossRef]
- Yamasoba D, Kimura I, Nasser H, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. Jun 9 2022;185(12):2103-2115 e19. [CrossRef]
- Farrell AG, Dadonaite B, Greaney AJ, et al. Receptor-Binding Domain (RBD) Antibodies Contribute More to SARS-CoV-2 Neutralization When Target Cells Express High Levels of ACE2. Viruses. Sep 16 2022;14(9). [CrossRef]
- Lempp FA, Soriaga LB, Montiel-Ruiz M, et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature. Oct 2021;598(7880):342-347. [CrossRef]
- VanBlargan LA, Errico JM, Halfmann PJ, et al. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med. Mar 2022;28(3):490-495. [CrossRef]
- FDA. Integrated Review Application Number 761172. Accessed 2/17/2023, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761172Orig1s000IntegratedR.pdf.
- FDA. Multi-Discipline Review, Application Number 761169. Accessed 2/17/2023, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761169Orig1s000MultidisciplineR.pdf.
- Hsieh YT, Aggarwal P, Cirelli D, Gu L, Surowy T, Mozier NM. Characterization of FcγRIIIA effector cells used in in vitro ADCC bioassay: Comparison of primary NK cells with engineered NK-92 and Jurkat T cells. J Immunol Methods. Feb 2017;441:56-66. [CrossRef]
- Parekh BS, Berger E, Sibley S, et al. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs. May-Jun 2012;4(3):310-8. [CrossRef]
- de Taeye SW, Rispens T, Vidarsson G. The Ligands for Human IgG and Their Effector Functions. Antibodies (Basel). Apr 25 2019;8(2). [CrossRef]
- Goncalvez AP, Engle RE, St Claire M, Purcell RH, Lai CJ. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc Natl Acad Sci U S A. May 29 2007;104(22):9422-7. [CrossRef]
- Huang X, Yue Y, Li D, et al. Antibody-dependent enhancement of dengue virus infection inhibits RLR-mediated Type-I IFN-independent signalling through upregulation of cellular autophagy. Sci Rep. Feb 29 2016;6:22303. [CrossRef]
- Littaua R, Kurane I, Ennis FA. Human IgG Fc receptor II mediates antibody-dependent enhancement of dengue virus infection. J Immunol. Apr 15 1990;144(8):3183-6.
- Stettler K, Beltramello M, Espinosa DA, et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science. Aug 19 2016;353(6301):823-6. [CrossRef]
- Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. Aug 21 2020;369(6506):1014-1018. [CrossRef]
- Copin R, Baum A, Wloga E, et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell. Jul 22 2021;184(15):3949-3961 e11. [CrossRef]
- Starr TN, Greaney AJ, Addetia A, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. Feb 19 2021;371(6531):850-854. [CrossRef]
- Weisblum Y, Schmidt F, Zhang F, et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. Oct 28 2020;9. [CrossRef]
- Ruiz SI, Zumbrun EE, Nalca A. Animal Models of Human Viral Diseases. Animal Models for the Study of Human Disease. 2017:853-901.
- Beddingfield BJ, Maness NJ, Fears AC, et al. Effective Prophylaxis of COVID-19 in Rhesus Macaques Using a Combination of Two Parenterally-Administered SARS-CoV-2 Neutralizing Antibodies. Front Cell Infect Microbiol. 2021;11:753444. [CrossRef]
- Haagmans BL, Noack D, Okba NMA, et al. SARS-CoV-2 Neutralizing Human Antibodies Protect Against Lower Respiratory Tract Disease in a Hamster Model. J Infect Dis. Jun 15 2021;223(12):2020-2028. [CrossRef]
- Jha A, Barker D, Lew J, et al. Efficacy of COVID-HIGIV in animal models of SARS-CoV-2 infection. Sci Rep. Oct 10 2022;12(1):16956. [CrossRef]
- Kim C, Ryu DK, Lee J, et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. Jan 12 2021;12(1):288. [CrossRef]
- Maisonnasse P, Aldon Y, Marc A, et al. COVA1-18 neutralizing antibody protects against SARS-CoV-2 in three preclinical models. Nat Commun. Oct 20 2021;12(1):6097. [CrossRef]
- Winkler ES, Gilchuk P, Yu J, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell. Apr 1 2021;184(7):1804-1820 e16. [CrossRef]
- Yadav PD, Mendiratta SK, Mohandas S, et al. ZRC3308 Monoclonal Antibody Cocktail Shows Protective Efficacy in Syrian Hamsters against SARS-CoV-2 Infection. Viruses. Dec 3 2021;13(12). [CrossRef]
- Zost SJ, Gilchuk P, Case JB, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. Aug 2020;584(7821):443-449. [CrossRef]
- FDA. S6(R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals, Guidance for Industry. Accessed 2/17/2023, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s6r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals.
- Mahmood I, Tegenge MA. Prediction of tissue concentrations of monoclonal antibodies in mice from plasma concentrations. Regul Toxicol Pharmacol. Aug 2018;97:57-62. [CrossRef]
- Keeler SP, Fox JM. Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections. Viruses. May 31 2021;13(6). [CrossRef]
- Schmaljohn AL, Orlandi C, Lewis GK. Deciphering Fc-mediated Antiviral Antibody Functions in Animal Models. Front Immunol. 2019;10:1602. [CrossRef]
- Robbie GJ, Criste R, Dall'acqua WF, et al. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. Dec 2013;57(12):6147-53. [CrossRef]
- Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci U S A. Apr 17 2012;109(16):6181-6. [CrossRef]
- Proetzel G, Roopenian DC. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods. Jan 1 2014;65(1):148-53. [CrossRef]
- 8862; FDA. Product Development Under the Animal Rule, Guidance for Industry. Accessed 2/17/2023, https://www.fda.gov/media/88625/download.
- FDA. Animal Rule Approvals. Accessed 2/17/2023, https://www.fda.gov/drugs/nda-and-bla-approvals/animal-rule-approvals.
- Sharp JC, Fletcher WB. Experience of anti-vaccinia immunoglobulin in the United Kingdom. Lancet. Mar 24 1973;1(7804):656-9. [CrossRef]
- Bahmanyar M, Fayaz A, Nour-Salehi S, Mohammadi M, Koprowski H. Successful protection of humans exposed to rabies infection. Postexposure treatment with the new human diploid cell rabies vaccine and antirabies serum. JAMA. Dec 13 1976;236(24):2751-4.
- Nelson NP, Weng MK, Hofmeister MG, et al. Prevention of Hepatitis A Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices, 2020. MMWR Recomm Rep. Jul 3 2020;69(5):1-38. [CrossRef]
- Fisher RW, Reed JL, Snoy PJ, et al. Postexposure prevention of progressive vaccinia in SCID mice treated with vaccinia immune globulin. Clin Vaccine Immunol. Jan 2011;18(1):67-74. [CrossRef]
- Bray M, Wright ME. Progressive vaccinia. Clin Infect Dis. Mar 15 2003;36(6):766-74. [CrossRef]
- 1456; FDA. REGEN-COV (casirivimab and imdevimab). 2/17/2023. https://www.fda.gov/media/145611/download.
- 1458; FDA. Bamlanivimab and etesevimab. Accessed 2/17/2023, https://www.fda.gov/media/145802/download.
- 1547; FDA. EVUSHELD™ (tixagevimab co-packaged with cilgavimab). Accessed 2/17/2023, https://www.fda.gov/media/154701/download.
- Ejemel M, Smith TG, Greenberg L, et al. A cocktail of human monoclonal antibodies broadly neutralizes North American rabies virus variants as a promising candidate for rabies post-exposure prophylaxis. Sci Rep. Jun 7 2022;12(1):9403. [CrossRef]
- Koszalka P, Subbarao K, Baz M. Preclinical and clinical developments for combination treatment of influenza. PLoS Pathog. May 2022;18(5):e1010481. [CrossRef]
- Maertens J, Logan AC, Jang J, et al. Phase 2 Study of Anti-Human Cytomegalovirus Monoclonal Antibodies for Prophylaxis in Hematopoietic Cell Transplantation. Antimicrob Agents Chemother. Mar 24 2020;64(4). [CrossRef]
- Mahomed S, Garrett N, Baxter C, Abdool Karim Q, Abdool Karim SS. Clinical Trials of Broadly Neutralizing Monoclonal Antibodies for Human Immunodeficiency Virus Prevention: A Review. J Infect Dis. Feb 13 2021;223(3):370-380. [CrossRef]
- FDA. Codevelopment of Two or More New Investigational Drugs for Use in Combination, Guidance for Industry. Accessed 2/17/2023, https://www.fda.gov/media/80100/download.
- Li A, Katinger H, Posner MR, et al. Synergistic neutralization of simian-human immunodeficiency virus SHIV-vpu+ by triple and quadruple combinations of human monoclonal antibodies and high-titer anti-human immunodeficiency virus type 1 immunoglobulins. J Virol. Apr 1998;72(4):3235-40. [CrossRef]
- Miglietta R, Pastori C, Venuti A, Ochsenbauer C, Lopalco L. Synergy in monoclonal antibody neutralization of HIV-1 pseudoviruses and infectious molecular clones. J Transl Med. Dec 13 2014;12:346. [CrossRef]
- ter Meulen J, van den Brink EN, Poon LL, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. Jul 2006;3(7):e237. [CrossRef]
- Zhong L, Haynes L, Struble EB, Tamin A, Virata-Theimer ML, Zhang P. Antibody-mediated synergy and interference in the neutralization of SARS-CoV at an epitope cluster on the spike protein. Biochem Biophys Res Commun. Dec 18 2009;390(3):1056-60. [CrossRef]
- Patel HD, Nikitin P, Gesner T, et al. In Vitro Characterization of Human Cytomegalovirus-Targeting Therapeutic Monoclonal Antibodies LJP538 and LJP539. Antimicrob Agents Chemother. Aug 2016;60(8):4961-71. [CrossRef]
- Copin R, Baum A, Wloga E, et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell. Jul 22 2021;184(15):3949-3961.e11. [CrossRef]
- Gottlieb RL, Nirula A, Chen P, et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. Jama. Feb 16 2021;325(7):632-644. [CrossRef]
- Spencer DA, Shapiro MB, Haigwood NL, Hessell AJ. Advancing HIV Broadly Neutralizing Antibodies: From Discovery to the Clinic. Front Public Health. 2021;9:690017. [CrossRef]
- Cox M, Peacock TP, Harvey WT, et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat Rev Microbiol. Feb 2023;21(2):112-124. [CrossRef]
- Julg B, Stephenson KE, Wagh K, et al. Safety and antiviral activity of triple combination broadly neutralizing monoclonal antibody therapy against HIV-1: a phase 1 clinical trial. Nat Med. Jun 2022;28(6):1288-1296. [CrossRef]
- Chan CEZ, Seah SGK, Chye H, et al. The Fc-mediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. PLoS One. 2021;16(6):e0253487. [CrossRef]
- Schäfer A, Muecksch F, Lorenzi JCC, et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J Exp Med. Mar 1 2021;218(3). [CrossRef]
- Winkler ES, Gilchuk P, Yu J, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell. Apr 1 2021;184(7):1804-1820.e16. [CrossRef]
- Yamin R, Jones AT, Hoffmann HH, et al. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature. Nov 2021;599(7885):465-470. [CrossRef]
- Chigutsa E, Jordie E, Riggs M, et al. A Quantitative Modeling and Simulation Framework to Support Candidate and Dose Selection of Anti-SARS-CoV-2 Monoclonal Antibodies to Advance Bamlanivimab Into a First-in-Human Clinical Trial. Clin Pharmacol Ther. Mar 2022;111(3):595-604. [CrossRef]
- Chigutsa E, O'Brien L, Ferguson-Sells L, Long A, Chien J. Population Pharmacokinetics and Pharmacodynamics of the Neutralizing Antibodies Bamlanivimab and Etesevimab in Patients With Mild to Moderate COVID-19 Infection. Clin Pharmacol Ther. Nov 2021;110(5):1302-1310. [CrossRef]
- Magyarics Z, Leslie F, Bartko J, et al. Randomized, Double-Blind, Placebo-Controlled, Single-Ascending-Dose Study of the Penetration of a Monoclonal Antibody Combination (ASN100) Targeting Staphylococcus aureus Cytotoxins in the Lung Epithelial Lining Fluid of Healthy Volunteers. Antimicrob Agents Chemother. Aug 2019;63(8). [CrossRef]
- General Chapter: USP. General Tests and Assays, Biological Tests and Assays, <85> Bacterial Endotoxins Test. USP–NF Rockville, MD: USP; DOI: https://doiorg/1031003/USPNF_M98830_02_01.
- Murga JD, Franti M, Pevear DC, Maddon PJ, Olson WC. Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob Agents Chemother. Oct 2006;50(10):3289-96. [CrossRef]
- Vir Biothechnology, Inc. Press Release, 11/6/2022. Accessed 2/17/2023, https://investors.vir.bio/news-releases/news-release-details/vir-biotechnology-presents-new-data-evaluating-potential-vir-0.
- Vir Biothechnology, Inc. Press Release, 06/25/22. Accessed 2/22/2022, https://investors.vir.bio/news-releases/news-release-details/vir-biotechnology-announces-new-clinical-data-its-broad.
- Xiao F, Fofana I, Thumann C, et al. Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut. Mar 2015;64(3):483-94. [CrossRef]
- Caskey M, Klein F, Lorenzi JC, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. Jun 25 2015;522(7557):487-91. [CrossRef]
- Mouquet H, Scharf L, Euler Z, et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci U S A. Nov 20 2012;109(47):E3268-77. [CrossRef]
- Drug Database: Leronlimab. Accessed 2/17/2023, https://clinicalinfo.hiv.gov/en/drugs/leronlimab/patient.
- TROGARZO® (ibalizumab-uiyk). Accessed 2/17/2023, https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761065s013lbl.pdf.
- Kiso M, Yamayoshi S, Kawaoka Y. Triple combination therapy of favipiravir plus two monoclonal antibodies eradicates influenza virus from nude mice. Commun Biol. May 7 2020;3(1):219. [CrossRef]
- Cross RW, Bornholdt ZA, Prasad AN, et al. Combination therapy protects macaques against advanced Marburg virus disease. Nat Commun. Mar 25 2021;12(1):1891. [CrossRef]
- Cross RW, Bornholdt ZA, Prasad AN, et al. Combination therapy with remdesivir and monoclonal antibodies protects nonhuman primates against advanced Sudan virus disease. JCI Insight. May 23 2022;7(10). [CrossRef]
- Nakamura G, Chai N, Park S, et al. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe. Jul 17 2013;14(1):93-103. [CrossRef]
- Paules CI, Lakdawala S, McAuliffe JM, et al. The Hemagglutinin A Stem Antibody MEDI8852 Prevents and Controls Disease and Limits Transmission of Pandemic Influenza Viruses. J Infect Dis. Aug 1 2017;216(3):356-365. [CrossRef]
- Tharakaraman K, Subramanian V, Viswanathan K, et al. A broadly neutralizing human monoclonal antibody is effective against H7N9. Proc Natl Acad Sci U S A. Sep 1 2015;112(35):10890-5. [CrossRef]
- Yi KS, Choi JA, Kim P, et al. Broader neutralization of CT-P27 against influenza A subtypes by combining two human monoclonal antibodies. PLoS One. 2020;15(7):e0236172. [CrossRef]
- Cox M, Peacock TP, Harvey WT, et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat Rev Microbiol. Feb 2023;21(2):112-124. [CrossRef]
- Washburn N, Schwab I, Ortiz D, et al. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc Natl Acad Sci U S A. Mar 17 2015;112(11):E1297-306. [CrossRef]
- Keating SM, Mizrahi RA, Adams MS, et al. Generation of recombinant hyperimmune globulins from diverse B-cell repertoires. Nat Biotechnol. Aug 2021;39(8):989-999. [CrossRef]
- Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. Jul 2007;4(4):427-40. [CrossRef]
- Pitiot A, Heuze-Vourc'h N, Secher T. Alternative Routes of Administration for Therapeutic Antibodies-State of the Art. Antibodies (Basel). Aug 26 2022;11(3). [CrossRef]
- Sroga P, Safronetz D, Stein DR. Nanobodies: a new approach for the diagnosis and treatment of viral infectious diseases. Future Virology. 2020;15(3):195-205. [CrossRef]
- Nyakatura EK, Soare AY, Lai JR. Bispecific antibodies for viral immunotherapy. Hum Vaccin Immunother. Apr 3 2017;13(4):836-842. [CrossRef]
- Walser M, Mayor J, Rothenberger S. Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors. Viruses. Oct 12 2022;14(10). [CrossRef]
- Wensel D, Sun Y, Davis J, et al. A Novel gp41-Binding Adnectin with Potent Anti-HIV Activity Is Highly Synergistic when Linked to a CD4-Binding Adnectin. J Virol. Jul 15 2018;92(14). [CrossRef]

Table 1.
FDA-approved polyclonal antibodies for the prevention or treatment of viral diseases.
| Target | Trade Name(s) | Donors | Indications | Used With |
|---|---|---|---|---|
| Rabies27-29 | HyperRAB, Imogam, KedRab |
Vaccinated | Post-exposure prophylaxis | Rabies vaccine (required, see prescribing information for rabies IG) |
| Varicella30 | VARIZIG | Donations selected from high-titer donors after natural infection* | Post-exposure prophylaxis in patients at risk for severe infection | Concomitant use of acyclovir reported to occur in clinical practice31 |
| Vaccinia32 |
Vaccinia Immune Globulin (Human) | Vaccinated | Treatment of severe complications after smallpox vaccination | Investigational antiviral drugs and/or cidofovir33, 34 |
| Cytomegalovirus (CMV)24 |
CytoGam | Donations selected from source plasma | Prevention of CMV disease in patients receiving organ transplants from CMV donors | Ganciclovir recommended in prescribing information; other drugs recommended in practice guidelines35 |
| Hepatitis A (HAV)36 | GamaSTAN | Regular donors | Pre- and Post-exposure prophylaxis | None |
| Hepatitis B (HBVIG/IGIV)37-39 | HyperHEP B, Nabi-HB |
Vaccinated | Post-exposure prophylaxis | None |
| HepaGam-B | Vaccinated | Post-exposure prophylaxis Prevention of HBV recurrence in HBsAg+ liver transplant recipients |
Concomitant treatment with other drugs recommended in practice guidelines40 | |
| Measles36,† |
GamaSTAN | Regular donors | Prevention or attenuation of measles in susceptible individuals | None |
| Rubella28 | GamaSTAN | Regular donors | To modify rubella in exposed pregnant women who will not be undergoing a therapeutic abortion | None |
*GamaSTAN may be used only if VariZIG is not available36. †In patients receiving IG products to correct antibody deficiencies, doses of intravenous IG (IVIG) that should prevent measles infections for travelers to measles-endemic areas are suggested41.
Table 2.
FDA-approved monoclonal antibodies for the prevention or treatment of viral diseases.
| Non-proprietary name | Trade name | Target | Indication |
|---|---|---|---|
| palivizumab | Synagis | RSV F protein | for the prevention of serious lower respiratory tract disease caused by RSV in pediatric patients (specific conditions and age limitations) |
| ibalizumab | Trogarzo | CD4 (post attachment HIV-1 inhibitor) | in combination with other antiretroviral(s), for the treatment of HIV-1 infection in heavily treatment-experienced adults with multidrug resistant HIV-1 infection failing their current antiretroviral regimen |
| atoltivimab, maftivimab, odesivimab-ebgn | Inmazeb | Ebola virus glycoprotein | for the treatment of infection caused by Zaire ebolavirus in adult and pediatric patients, including neonates born to a mother who is RT-PCR positive for Zaire ebolavirus infection |
| ansuvimab-zykl | Ebanga | Ebola virus glycoprotein | for the treatment of infection caused by Zaire ebolavirus in adult and pediatric patients, including neonates born to a mother who is RT-PCR positive for Zaire ebolavirus infection |
Table 3.
Advantages and disadvantages of specific polyclonal and monoclonal antibodies for the prophylaxis or treatment of viral diseases.
Table 3.
Advantages and disadvantages of specific polyclonal and monoclonal antibodies for the prophylaxis or treatment of viral diseases.
| Specific Polyclonal Antibodies | Monoclonal Antibodies | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
Table 4.
Examples of Combinations of mAbs and Other Types of Antivirals.
| Virus | mAb(s) (Target) | Antiviral(s) | Stage | References |
|---|---|---|---|---|
| HBV/HDV | VIR-3434 (HBsAg) | VIR-2218±NrtI±pegIFN | phase 2 | NCT04856085*, also see217, 218 |
| HCV | various (HCV receptors)† | Various† | preclinical | 219 |
| HIV-1 | teropavimab+zinlirvimab (gp120) | various‡ | phase 1-2 | NCT04811040*Also see220, 221 |
| HIV-1 | leronlimab (CCR5) | approved antiretrovirals | phase 2-3 | 216, 222 |
| HIV-1 | ibalizumab (CD4) | approved antiretrovirals | approved | 223 |
| IAV | various (HA) § | oseltamivir | phase 2 | 194 |
| IAV | CR9114+F3A19 (HA) | favipiravir | preclinical | 224 |
| MARV | MR186-YTE (GP) | remdesivir | preclinical | 225 |
| SUDV | ADI-15878+ADI-23774 (GP) | remdesivir | preclinical | 226 |
*Clinicaltrials.gov study number †The mAbs tested were OM-7D3-B3 (anti-CLDN1 mAb), NK-8H5-E3 (anti-SR-BI mAb), and QV-6A8-F2C4 (anti-CD81 mAb). The antivirals tested were HCV NS3/4A protease inhibitors (simeprevir, danoprevir, boceprevir, telaprevir), NS5A inhibitors (daclatasvir), and NS5B nucleotide analog polymerase inhibitors (sofosbuvir). ‡The therapeutics being tested in combination with these mAbs in clinical trials include FDA-approved antiretrovirals (e.g., the HIV-1 capsid inhibitor lenacapavir) and investigational drugs, such as peptide fusion inhibitors, therapeutic vaccines, latency-reversing agents, and immunomodulators (e.g., pegylated interferon). §The mAbs tested in combination with oseltamivir in clinical trials include CT-P27 (a combination of two mAbs), MEDI8852, MHAA4549A, and VIS410. Abbreviations: GP, glycoprotein; HA, hemagglutinin; HBsAg, hepatitis B virus surface antigen; HBV, hepatitis B virus; HCV, hepatitis C virus; HDV, hepatitis delta virus; HIV-1, human immunodeficiency virus type-1; IAV, influenza A virus; mAb, monoclonal antibody; MARV, Marburg virus; NrtI, nucleos(t)ide analog reverse transcriptase inhibitor; pegIFN, pegylated interferon-α; SUDV, Sudan virus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



