
Submitted:
18 April 2023
Posted:
18 April 2023
You are already at the latest version
Abstract
Studies on the neurobiological causes of anxiety disorders suggest that the GABA system in-creases synaptic concentration and enhances the affinity of GABAA (type A) receptors for ben-zo-diazepine ligands. Flumazenil antagonizes the benzodiazepine binding site of the GABA (γ-aminobutyric acid) type /benzodiazepine receptor (BZR) complex in the central nervous sys-tem (CNS). The integration of the metabolites of flumazenil by LC-tandem mass spectrometry will complete understanding of the in vivo metabolism of flumazenil and accelerate radio-pharmaceutical inspection and registration. The main goal of our study was to investigate using reversed-phase HPLC (PR-HPLC) coupled with electrospray ionization triple quadrupole tan-dem mass spectrometry (ESI-QqQ MS) for the identification of flumazenil and its metabolites in a hepatic matrix. And a carrier-free nucleophilic fluorination with automatic synthesizer for [18F]flumazenil which applied to in vivo nano-positron emission tomography (Nano-PET)/computed tomography (CT) imaging and ex vivo bio-distribution used to analyze in normal rats. The study showed that 50% of the flumazenil was bio-transformed at 60 min by the rat liver homogenate, while one metabolite (M1) was a methyl transesterification product of flumazenil. In a rat liver microsomes system, two metabolites were identified (M2 and M3) as its carboxylic acid and hydroxylated ethylester forms, respectively. [18F]flumazenil in vivo nanoPET/CT imag-ing and ex vivo bio-distribution assay also showed significant effects on GABAA receptor availa-bility in the amygdala, prefrontal cortex, cortex, and hippocampus in the rat brain, worriless about the formation of metabolites. We showed completion of the bio-transformed course of flumazenil by the hepatic system; and [18F]flumazenil can be a good ligand and serve as a PET agent for determination of GABAA/BZR complex for multiplex neurological syndromes in the clinical stage.
Keywords:
Flumazenil
; LC-QqQ MS
; Metabolism
; r-aminobutyric acid receptor antagonist
; benzodiazepine receptors
1. Introduction
Anxiety is a common emotional disorder that involves worrying about future events, which is characterized by excessive and pointless worries, motor tension, and fatigue [1,2,3]. Anxiety is not an abnormal reaction. In contrast, appropriate level of anxiety enhances individual performance. Beyond the usual standards, it significantly impacts the actions of an individual, such as social activities, interpersonal relationships, and emotions, and is often referred to as “anxiety disorder” [4,5]. Several symptoms are indicative of anxiety, such as panic attacks, schizophrenia, stress, depression, insomnia, and anger. These are likely to intensify because of the nerve symptoms cause by signals generated [6,7]. Accordingly, abnormalities of the γ-aminobutyric acid (GABA) system in the brain are correlated with the onset of epilepsy, anxiety, and other psychiatric disorders [1,8,9,10]. The GABA receptor, which is present in abundance in the cortex of the brain, is highly sensitive and prone to accidental damage. There are several binding sites that contain the GABA receptor, including the benzodiazepine binding site. Upon receiving a signal from GABA, a chloride channel is opened that allows the entry of Cl- into the nerve cells, thereby reducing the intracellular potential of GABAA (type A) receptors [5,11,12]. GABA is the major inhibitory neurotransmitter in the central nervous system (CNS), and its receptors are widely distributed throughout the mammalian host. Substantial literature supports a link between altered GABAergic neurotransmission and numerous CNS disorders, including behavioral disorders, pain, and sleep[13,14]. The disruption of important functions of the CNS, such as intestinal motility, gastric emptying, nociception, and acid secretion are also affected in such scenarios [5,15]. In mammals, GABA receptors are almost only found in nervous tissues, such as the cerebral cortex, hippocampus, thalamus, basal ganglia, cerebellum, spinal cord, and retina (approximately 0.1-0.6 mg/g content of the brain tissue comprises the most important neurotransmitter substance in the CNS) [1,8,9,10]. About 30% of neurons rely on GABA to control signal transmission. Although growing evidence suggest that the GABA system plays a key role, the neurobiological reasons of anxiety disorders are yet to be fully understood [1,6,15].
Previous pre-clinical studies have suggested that an increased synaptic GABA concentration enhances the affinity of GABAA receptors for benzodiazepine ligands [16,17]. Flumazenil antagonizes the benzodiazepine binding site of the GABA/benzodiazepine receptor complex (BZR) in the CNS, thereby preventing chloride channel opening and inhibiting neuronal hyperpolarization [11,18]. Intravenous flumazenil treatment is used to recover from anesthesia and reduce the hypnotic and sedative effects of benzodiazepines via competitive inhibition at the benzodiazepine binding site on the GABAA receptor. The imidazo-benzodiazepine derivative flumazenil, or ethyl 8-fluoro-5-methyl-6-oxo-5,6-dihydro-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate (molecular formula: C15H14FN3O3; molecular weight: 303), was initially launched in 1987 by Roche under the trade name Anexate®. Later, it was confirmed as an antagonist, specific to benzene diazonium salts. Its mechanism is to compete with the benzene diazonium salt for GABAA receptors[11,19,20,21].
In animals, the metabolism of foreign chemicals mainly occurs in the liver [22]. Hepatic enzyme metabolism studies have been conducted using various hepatic components, including supersomes, microsomes, cytosol fractions, cell lines, hepatocytes, and liver slices[23]. The liver microsome is the most popular model because it is easy to apply, can be commercialized, and is affordable and well established, even though it is entirely comprised of cytochrome P450 (CYP) and uridine glucuronosyl transferase (UGT) enzymes. This gives an incomplete representation of the in vivo situation and renders the results unsuitable for quantitative evaluation. Most drug metabolites reduce biological activity, degrade into smaller molecules, and increase polarity and water solubility to facilitate excretion via urine[23]. Flumazenil is mainly metabolized and degraded by liver enzymes to reduce toxicity and is excreted by the kidneys[19]. Biotransformation of xenobiotics can also be conducted using whole liver homogenates to predict the types of reactions that may occur in vivo[24,25]. In this model, all enzymes present in the liver are conserved and mixed well with administer drug, thus resembling an in vivo model, wherein the parent ligand and metabolites are not lost from the biotransformation system. Gas chromatography and high-performance liquid chromatography (HPLC)-mass spectrometry-based methods have been used to determine flumazenil concentrations in the plasma and urine, as well as its bio-distribution in rodents and humans[26,27,28,29]. Earlier, Lavén studied the metabolic stability of flumazenil using a capillary liquid chromatography-mass spectrometry-based method[28]. Flumazenil is mostly metabolized in the liver, and the metabolites from liver microsomal enzyme catalysis of 11C-flumazenil and its 18F-analogue (fluoroethyl-flumazenil) were identified using ultra-high performance liquid chromatography (UHPLC)/quadrupole-time-of-flight-mass spectrometry (Q-ToF-MS) and HPLC/ (ESI-QqQ MS), respectively[26,29].
An increase in the binding capacity of GABAA benzodiazepine-receptor site-specific radio-ligand (18F-flumazenil), using molecular imaging tracers to visualize the distribution of GABAA receptors in the brain has been employed to help in the diagnosis of psychiatric/CNS disorders (such as epilepsy, anxiety, and insomnia)[1,20]. It can be traced by radioactive flumazenil derivatives that are more sensitive and accurate than 2-deoxy-2[18F]fluoro-D-glucose ([18F]F-FDG) imaging that is used for epileptic foci localization [19,21]. In the present study, a convenient non-carrier labeling F-18 with flumazenil with radio-HPLC unit for synthesis. [18F]flumazenil will employ on in vivo, ex vivo and in vitro analyze. The integration of the metabolites of flumazenil by rat liver homogenate and microsomes was analyzed by LC-tandem mass spectrometry. A complete understanding of the in vivo metabolism of [18F]flumazenil would accelerate the development of drugs for inspection and registration.
2. Results
2.1. High Performance Liquid Chromatography Analysis of Flumazenil
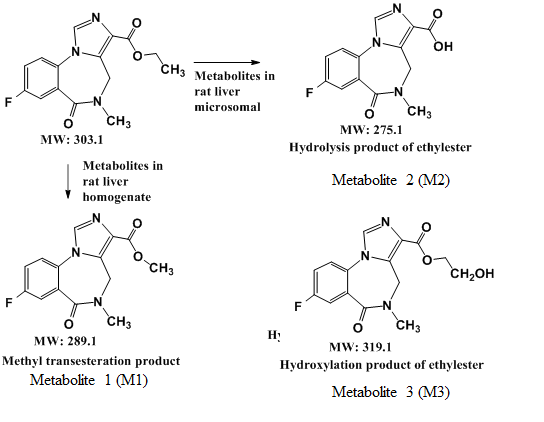
The ZORBAX Eclipse XDB-C18 column (100 × 4.6 mm, 5 μm) suitably separated bio-transformed flumazenil from the hydrophilic matrices of liver microsome and the liver homogenate solutions. The mobile phase composition was ammonium acetate aqueous buffer and ACN programmed gradient eluent (Table 1). After pretreatment, the typical flumazenil chromatogram exhibited a spike in the rat liver homogenate (Figure 1). The retention time (Rt) for flumazenil was 8.2 ± 0.1 min. The column efficiency exceeded 8,000 p/m with a tailing factor of 1.1 (calculated by Agilent ChemStation software 10.02) The peak of flumazenil was far from the hepatic matrix peaks, with a good resolution to avoid contamination. The dynamic range of flumazenil in the hepatic matrices was 1–100 ppm, with a linear least-squares regression equation of Y = 35.4 X-18. The calibration curve (six concentration points without being forced through zero) for flumazenil was sufficiently linear, with a correlation coefficient (r2) of 0.9992. The limits of detection (LOD) and quantification (LOQ) for flumazenil were 0.1 and 0.4 ppm, respectively, using DAD at λ of 250 nm. The incubated solutions were analyzed using HPLC to separate flumazenil and the metabolites from bio-matrices, and the peak area of flumazenil was measured in triplicate and averaged to investigate and determine the reaction trend of flumazenil in hepatic bio-systems.
2.2. Mass Spectrometric Analysis of Flumazenil
Mass spectral analyses of flumazenil fragments (dissolved in methanol with 0.1% formic acid) were initially performed via syringe infusion (10 µL min-1) and injected into the instrument for the Q1 and MS/MS scans. Full Q1 spectra were scanned in positive ion modes, which were more suitable than the negative ion mode for obtaining a stronger signal and increased structural information about flumazenil. In the ESI spectrum of flumazenil, [M + H]+, and [M + Na]+ ions were observed at m/z values of 304 and 326, respectively. Product-ion scans were performed using different collision-activated dissociation conditions to optimize the de-clustering potential, entrance potential, collision energy, and collision cell exit potential. The 304 product ions were observed at m/z values of 276, 258, 229, 217, and 189 (Figure 2A). The fragment structures are an attempt to clarify the fragmentation of flumazenil and to assist with metabolite identification thereafter (Figure 2B). The most abundant product ions were 258 and 276, indicating metastable de-ethoxylation (Δm/z = −46) and de-ethylation (Δm/z = −28) from the parent molecular ion 304 to form acylium and protonated carboxylic acid, respectively.
2.3. Study of Flumazenil Biotransformation by Hepatic Enzyme Systems
2.3.1. Flumazenil Metabolism Study in Rat Liver Homogenate
After incubation in rat liver homogenate for 0 to 240 min, the broths were pre-treated to separate flumazenil concentrate and its derivatives from the hepatic matrices. This was followed by HPLC analysis of the overlapped chromatograms of flumazenil, before and after incubation in the liver homogenate, for various durations. The peak height of the parent drug was reduced, while a metabolite peak 1 (M1) appeared with (Rt = 7.45 ± 0.1 min) and gradually increased in the peak area (Figure 3A). The flumazenil biotransformation rate in the rat liver homogenate was evaluated based on trends in the peak area of flumazenil on chromatograms (Figure 3B). These trends showed that the rate of flumazenil biotransformation in rat liver homogenate was rapid during the first 30 min, but subsequently slowed down to approximately 50% at 60 min. After 4 h, approximately 65% of the parent drug was bio-transformed and the m/z of M1 was 290 amu less than that of flumazenil, while the core structures of M1 and flumazenil were identical (Figure 3C). Therefore, M1 is the methyl ester form of flumazenil. The metabolic pathway for flumazenil in rat liver homogenate was thought to be as methyl transesterification.
2.3.2. Flumazenil Metabolism Study in Human Liver Microsomal
Flumazenil biotransformation was also studied in rat hepatic microsomes. The 2 h reactions were conducted according to the directions with supplied microsomal. The established LC-DAD method was used to evaluate flumazenil in the reaction solutions. The time courses of flumazenil and its metabolites in rat hepatic microsomes were plotted based on the average chromatographic peak areas identified from experiments conducted in triplicate. Two metabolite peaks, 2 and 3 (M2, the major and M3, the minor) were identified in the rat liver microsomal system, with Rt at 6.5 and 7.7 min, respectively (Figure 4A, B). Flumazenil was detected at approximately 22% of the initial amount of rat liver microsomes, up to a 120 min incubation time; and the peak area of M2 was increasing 72%. And, no obvious increase in the peak area of M3 was observed (Figure 4C).
2.4. A summary of the Metabolic Pathways of Flumazenil
A summary of the metabolic pathways of flumazenil in rat liver homogenate and the microsomal enzymatic system were summarized shown in Figure 5. The flumazenil metabolism pathways identified in rat and human liver microsomes in the present study were feasible. In differences in metabolic pathways between liver microsomes and whole liver homogenates arise from differences in the enzyme expression levels, efficiencies, and reactants in the media. The biotransformation of the flumazenil ester into the carboxylic acid form occurred generally also for the another hepatoma radiopharmaceutical, 188Re-MN16ET. In addition to the flumazenil metabolites M2 and M3 had be identified, our study identified another metabolite, M1, in rat liver media; this appeared to be a methyl transesterification product of flumazenil and had not been discussed previously in any literatures. This methyl re-placement of the ethyl ester is through to be an undisclosed flumazenil metabolite.
2.5. In Vitro Serum Stability of [18F]flumazenil
3.7 MBq of [18F]flumazenil was incubation in SD rat exsanguinated serum. the in vitro serum stability of radiochemical purity (RCP%) values of [18F]flumazenil were 95.76%, 93.28%, 91.07%, 87.46%, 85.68%, 79.95% and 72.13% at 0, 5, 10, 30, 60, 120 and 240 minutes, respectively. Our in vitro serum stability result shows that intact [18F]flumazenil is >85% up to 1 hour under regular conditions (Table 2).
2.6. In Vivo NanoPET/CT Assay with [18F]flumazenil
On the [18F]flumazenil/NanoPET/CT images, radioactivity was detected with the GABAA receptor in all the brain regions of rats (Figure 6A). ROIs were extracted from a set of previously constructed regions, including the frontal cortex, cortex, striatum, hippocampus, amygdala, midbrain, cerebellum and pons (Figure 6B). This phenomenon was obtained in the 30–40 min post-injection frame, which displayed the significant differences in specific binding ratios (SBRs = target region-reference region)/reference region) like frontal cortex, cortex, amygdala and hippocampus. At this distribution time, the SBRs of these target regions were 2.07~3.32 times than radiation accumulated in the medulla (reference region).
2.7. Ex Vivo Bio-Distribution Assay with [18F]flumazenil
The results of bio-distribution study after i.v. injection of 37 MBq/200 μL [18F]flumazenil showed obviously reduce radioactivity in vivo blood biodistrbution. The %ID/g on the [18F]flumazenil/ blood were 5.47 ± 0.56, 0.99 ± 0.42, 0.07± 0.07, 0.10± 0.11, 0.03± 0.04 and 0.01 ± 0.01 at 5, 10, 30, 60, 120 and 240 minutes, respectively (Figure 7).
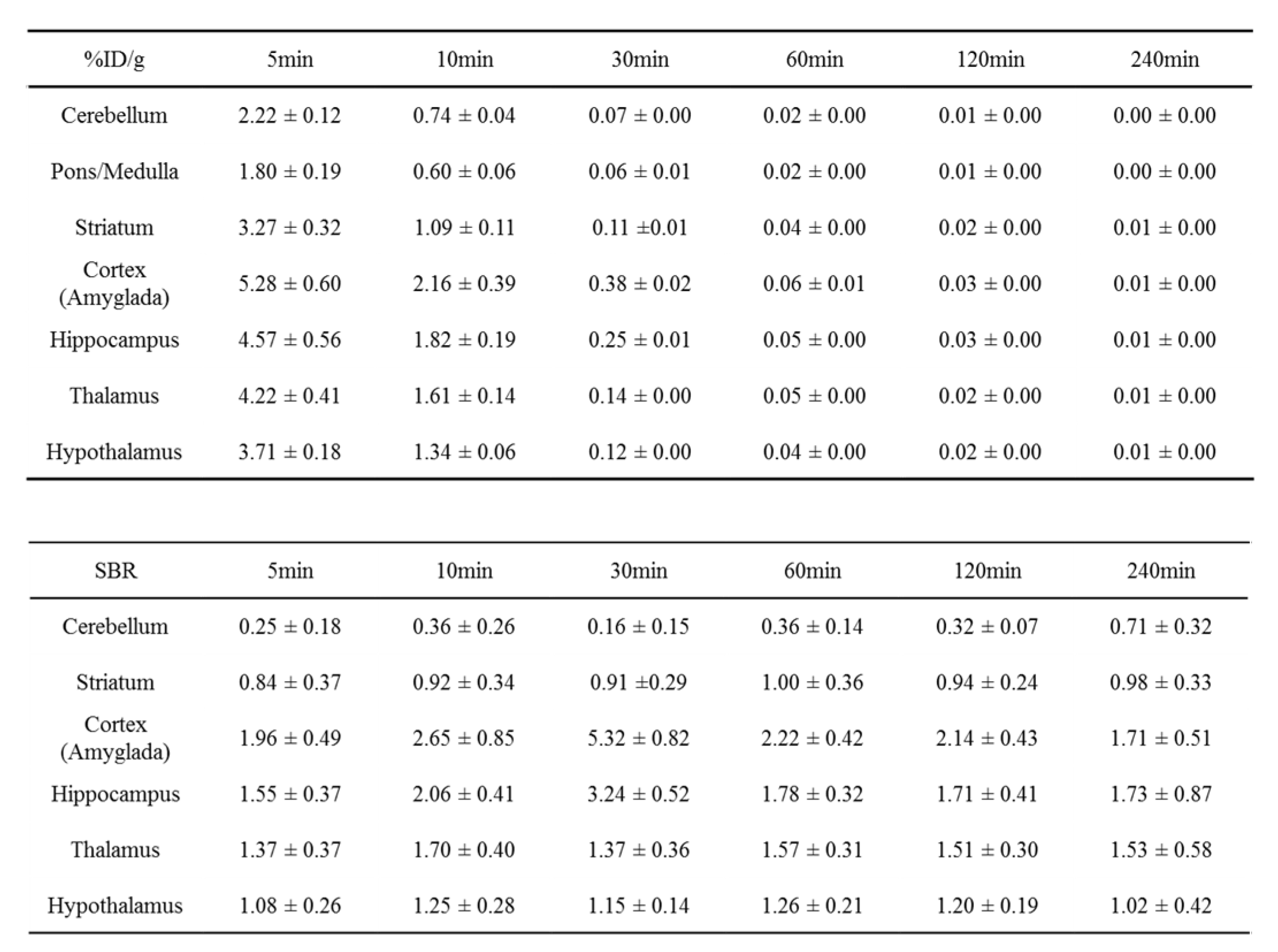
The different brain regions of [18F]flumazenil accumulated in the rat brain examined by bio-distribution. Post-injected [18F]flumazenil (37MBq/200 μL) at 5, 10, 30, 60, 120 and 240 minute. The rapid accumulated in brain regions will showed at 5 min. %ID/organ of brain were 1.92 ± 0.32, 0.39 ± 0.15, 0.03± 0.01, 0.03± 0.01, 0.001± 0.00 and 0.0004 ± 0.00 at 5, 10, 30, 60, 120 and 240 minutes, respectively (Supplementary Table S1). In the high density of GABAA in brain region as cortex, hippocampus and thalamus, the SBRs (pons/medulla as reference region) were 1.96 ± 0.49, 1.55 ± 0.37, 1.37 ± 0.37 (at 5 min), 2.65 ± 0.85, 2.06 ± 0.41, 1.70 ± 0.40 (at 10 min), 5.32 ± 0.82, 3.24 ± 0.52, 1.37 ± 0.36 (at 30min) and 2.22 ± 0.42, 1.78 ± 0.32, 1.57 ± 0.31 (at 60 min) as Table 3. Post-injection at 30 min reach the highest SBR than other internal times. The density pattern of GABAA visualized from the PET experiments was in good agreement with the results from the bio-distribution assay.
3. Discussion
The metabolic path and rate of a drug are closely related to the distribution of drugs in the body, the rate of elimination, and the period of efficacy[11,17,19]. A study of drug metabolites is of great significance for the proper application of drugs [18]. The mixed reaction of a test drug solution with animal tissue slices, homogeneous solution, separation and extraction components, liver cells, etc., can be used for metabolic reaction research[11,17,19]. Tandem mass spectrometry was used to determine the identity of the flumazenil relative derivatives in the liver homogenate and hepatic microsomal bio-systems[23,24]. After a metabolic reaction, the solution was separated by HPLC-tandem MS analysis and analyzed by tandem MS to identify the metabolites[30]. Tandem mass spectral analyses of flumazenil fragments were consistent with the results reported by Amini and Leveque, [26,31,32] and fragment structures were also in line with Leveque’s work [32]. However, the metastable fragmented ion structures of m/z 229, 217, and 189 corresponded with 5-ring delocalized imidazolium, 4-ring delocalized hydrazoium, and the losing N2 fragments, respectively. The fragmentation pathway has not been previously reported.
For metabolism, a drug molecule is gradually transformed into more polar substances by metabolic enzymes, so the Rt chromatography times of the metabolites are faster than those of the original drug molecule[11,17,19]. In a rat liver homogenate solution, flumazenil was bio-transformed to M1, a methyl transesterification product, with a molecular weight of 289, which less 14 molecular weight of the parent drug (Figure 3). Its substructures were equal to the core fragment ions of flumazenil. On the other hand, the status of the metabolites M2 and M3 in the rat liver microsomal bio-system were determined (Figure 4). The parent ion of M2 had an m/z of 276, with fragmentation ions of 258 (Δm/z = -18, -H2O) and 217, both of which were identical to the core fragmented ions of flumazenil. The mass spectra demonstrated that M2 had a molecular weight of 275 and the same substructure as flumazenil. M2 was, therefore, suggested to be the carboxylic acid form of flumazenil or the hydrolyzed product of the ethyl ester. For the other metabolite, M3, the m/z of the parent ion was 320 (Δm/z = +16, +O than 304), with mass spectrum fragments of 276 and 258. These mass data indicated that M3 likely resulted from the hydroxylation of flumazenil as its ethyl ester group to the hydroxyethyl ester. In the flumazenil metabolism, M1 (in rat liver homogenate solution) and M3 (in microsomal hepatic metabolic systems), biotransformation might occur in its nitrogen-methyl group to undergo demethylation (-CH3) and N-methanol products cause the molecular weights of 289 and 319, respectively. It is the difference in the tandem mass spectra from the parent drug.
In this study, fresh rat livers were homogenized and mixed with the test drug flumazenil solution to conduct metabolic reactions, analyze its metabolites, and compare the differences in metabolic reactions between liver homogenized liquid and liver microparticles. The flumazenil metabolism pathways identified in rat and human liver microsomes were similar to those identified by Amini in monkey microsome [26] but differed from those identified in a rat liver homogenate. The flumazenil metabolic pathways in three hepatic enzymatic systems were summarized in Figure 5. Because the homogenized solution of rat liver retains all liver enzyme functions, the influence of a liver matrix on the matrix liver disturbance of trace metabolite analysis may also be significant[13]. The metabolic rate and products of the rat liver microsomes and rat liver homogenates to flumazenil were compared to determine the metabolic mechanism of liver enzymes to flumazenil[11,17].
Synthesis of [18F]flumazenil was conducted via a fast and convenient non-carrier nucleophilic substitution combined with 18F- ion to nitro-flumazenil as the precursor, using an automatic synthetic technique and solid extraction purification to replace the semi-preparative HPLC. In In vitro serum stability, [18F]flumazenil showed a stable radiopurifityfor stored at 30% ethanol showed the RCP (%) values could >85% up until 1 hour at regular conditions (Table 2). By utilizing [18F]flumazenil NanoPET/CT imaging, we observed a global accumulated in GABAA receptor availability in rats brain regions, although at the same time had flumazenil metabolite compound[31,32]. There will not influence in [18F]flumazenil in GABAA receptor by PET imaging (Figure 6). According by bio-distribution of [18F]flumazenil, whatever in blood or brain regions showed a rapidly decline trend from 5−10 min (Figure 7). Evaluate the SBR ratio from GABAA receptor abundant areas (like cortex, amyglada, hippocampus) with post-injected times, at 30 min reach the highest SBR ratio at cortex (amyglada) and hippocampus relatively to pons/medulla (SBRs value 5.32 ± 0.82 and 3.24 ± 0.52, respectively) (Table 3). Post-injected at 30 min also supported the PET imaging findings.
Limitations remain in this study: 1. This is a relatively novel experiment, we used tradition method for identify flumazenil metabolism, but we wish could direct analysis on human or primates with [18F]flumazenil; 2. the radiochemical purity under regular condition only 10 min >90%, the radio-purity need to extend at less for 60min for clinical used. Advantages of the present study included: 1. In the flumazenil metabolism, M1−3 biotransformation might occur in its nitrogen-methyl group to undergo demethylation; 2. Post-admitted of [18F]flumazenil for 30 min, we accumulated static PET images at 30−40 minutes, enough to provide clarity brain images, although its rapidly decline at short time; 3. we demonstrated that the specific binding ratios of the GABAA receptor are profoundly influenced in brain specific areas, including the amygdala, prefrontal cortex, cortex, and hippocampus, suggesting that these brain regions may be involved in the processing and storage of conditioned fear memory [1,31,33]
Now, an increase in the binding capacity of GABAA benzodiazepine-receptor site-specific radio-ligand (18F-flumazenil), using molecular imaging tracers to visualize the distribution of GABAA receptors in the brain has been employed to help in the diagnosis of psychiatric/CNS disorders (such as epilepsy, anxiety, and insomnia) [1,20]. It can be traced by radioactive flumazenil derivatives that are more sensitive and accurate than 2-deoxy-2[18F]fluoro-D-glucose ([18F]FDG) imaging that is used for epileptic foci localization [19,21]. In the present study, the integration of the metabolites of flumazenil by rat liver homogenate and microsomes was analyzed by LC-tandem mass spectrometry. A complete understanding of the in vivo metabolism of flumazenil would accelerate the development of drugs for inspection and registration. And the final product with [18F]flumazenil, we showed completion of the biology characteristically by in vitro, ex vivo and in vivo image. [18F]flumazenil can be a good ligand and serve as a PET agent for determination of GABAA/BZR complex for multiplex neurological syndromes in the future clinical stage.
4. Materials and Methods
4.1. Materials and Reagents
Analytical-grade laboratory chemicals for LC/MS were used as received, without further purification. Methanol, acetonitrile (ACN) (both HPLC and MS grade), ammonium acetate, ethyl acetate (EA), phosphate buffer pellets, sucrose, and bovine serum albumin (BSA) were purchased from Merck (Darmstadt, Germany). Flumazenil was purchased from Futurechem (Seoul, Korea). Deionized water was prepared using a Smart DQ3 reverse osmosis reagent water system (Merck Millipore, Billerica, MA, USA) with a 0.22 μm polyvinylidene fluoride (PVDF) filter and a UV light source to produce ultra-pure water (total organic carbon <5 ppb, resistivity ≧18.2 MΩ-cm). A ZORBAX Eclipse XDB-C18 reversed phase HPLC column (100 × 4.6 mm, particle size: 5 μm) from Agilent Technologies (Palo Alto, CA, USA) was used to study flumazenil and its metabolites.
4.2. Animals
All experiments were carried out on Sprague Dawley (SD) rats (3-4 weeks) purchased from a licensed breeder (BioLASCO Taiwan Co., Taipei, Taiwan). All animal procedures and experimental protocols were approved by the Ethical Animal Use Committee of the Institute of Nuclear Energy Research (INER), Atomic Energy Council in Taiwan. And performed in compliance with Taiwan’s laws for the care and use of laboratory animals (LAC-2019-0253). The rats were maintained at 21 ± 2°C with 50 ± 20% relative humidity under an alternative 12 h light and 12 h dark cycle. The rats identified as the normal condition then for NanoPET/CT imaging, serum stability and bio-distribution of their brains was performed. Fresh rat livers were obtained from healthy male SD rats and frozen at –70°C. Rat liver microsomes and co-enzymes (NADPH A and B) were purchased from BD Biosciences (Bedford, MA, USA) and stored at -70°C.
4.3. Apparatus and Equipment
4.3.1. High Performance Liquid Chromatography Instrumentation
Quantification of flumazenil metabolism by hepatic enzymes was performed using HPLC (Agilent 1100 series, Palo Alto, CA, USA), which is comprised of an on-line degasser, binary pump, auto sampler, thermostat column oven (maintained at 25°C), and a diode array detector (DAD, 250 nm). Data were acquired and processed using Agilent ChemStation software (ed. 10.02). Separation was achieved using a C18 column with a guard column and ammonium acetate buffer-ACN gradient elution. The mobile phase consisted of eluent A (5 mM ammonium acetate aqueous buffer with 1% ACN, pH 7.0) and B (ACN only) at a flow rate of 0.6 mL min-1. These were mixed according to the gradient program shown in Table 1 to separate metabolites and flumazenil from the sample matrices. The overall LC time for the flumazenil biotransformation study was 15 min. An analyte solution of 5 µL was injected using an auto-sampler thermostat at 4°C.
4.3.2. Liquid Chromatography-Mass Spectrometry/Mass Spectrometry Instrumentation
Mass spectrometric analysis of flumazenil in ACN was initially conducted on a 4,000 QTrap LC-MS/MS system with API Analyst software 1.4.1 (AB Sciex, Concord, ON, Canada) to obtain the appropriate MS parameters and m/z for flumazenil fragments. Samples were introduced into the spectrometer, either by the HPLC system (Agilent 1100) or by a syringe pump, at a flow rate of 10 µL min-1 (Harvard Apparatus Inc., Holliston, MA, USA). Chromatographic separation was conducted as described in the section on HPLC conditions. The analytes were ionized by a turbo spray ion source (electrospray ionization, [ESI]) in the positive-ion mode at 5,000 V and 350°C. Mass spectra were recorded within a range of 100–1000 amu, with unit resolutions in Q1 and Q3. The other experimental parameters are listed in Table 1. LC-tandem MS determination of the flumazenil concentration was conducted in the multiple reaction monitoring positive-ion mode with transition ion pairs of 304 → 258 and 304 → 276. Vaporized liquid nitrogen gas was used as the nebulizer, curtain, and collision gas in all 4,000 QTrap LC–MS/MS studies.
4.4. Procedures for Metabolism Study
4.4.1. Biotransformation and Pre-Treatment for High Performance Liquid Chromatography of Flumazenil in Rat Liver Homogenate
A normal rat liver was taken from a healthy male SD rat, weighing 400–450 g. It was homogenized, as described in the following procedures. The rats were sacrificed via CO2 inhalation for 5 min in a closed box. After confirmation of euthanization, the entire liver (approximately 14 g) was removed, washed with ice-cold normal saline solution, and immediately minced in a 0.25 M sucrose solution (30 mL) in an ice bath. The rat liver tissue solution (40 mL) was homogenized in a motor-driven homogenizer, aliquoted (1.5 mL) into sample tubes, and stored at −70°C. Samples were used for biotransformation studies within 30 days and were thawed in an ice bath before use. For the biotransformation, the liver homogenate (1 mL) was placed in a 15 mL test tube and mixed well with a 1:1 (v/v) solution of 0.25% BSA in 0.01 M phosphate-buffered saline solution (1 mL, pH 7.4). The flumazenil solution intended for study (10 μL volume; 1,000 μg mL-1 in dimethyl sulfoxide [DMSO]) was added to the tube, which was swirled completely and placed in a preheated water bath (37°C, 70 rpm) to initiate the enzymatic reaction. After specified incubation periods (0, 10, 20, 30, 45, 60, 90, 120, and 240 min), ACN (2 mL) was added to each tube to stop the reaction and precipitate proteins, which were then centrifuged (10,000 rpm, 5 min, 4°C) to separate the supernatants. Liquid-liquid extraction with EA (400 μL) was performed in triplicate to concentrate flumazenil in the supernatant and desalt the reaction. The EA fraction (1.2 mL) was collected in a microtube and purged with N2 (thermostat set at 30°C) to vaporize the EA until the volume was reduced to less than 50 μL. The samples were again dissolved in ACN to generate a final volume of 1.0 mL and filtered through a 0.22 µm PVDF disk filter for LC-DAD/tandem MS analysis. The liver homogenate (2 mL) was mixed well with a buffer solution (2 mL) and DMSO (10 µL) to generate a blank background solution.
4.4.2. Flumazenil Metabolization in Rat Liver Microsomes
Incubation solutions were prepared according to the liver microsomes package directions. First, 2 μL of a 5-mM flumazenil solution in DMSO was added to a reaction mixture buffered with 0.5 M potassium phosphate pH 7.4 (200 μL) containing co-enzyme NADPH solution A (50 μL) and B (10 μL), and 25 μL of liver microsome protein solution (0.5 mg). The final volume of the incubated solution was adjusted to 1.0 mL with deionized water (713 μL). The reaction vials were capped, mixed well, and placed in a pre-heated water bath (37°C, 70 rpm) to initiate the enzymatic reaction. After incubating the reactions for specified durations (0, 10, 20, 30, 45, 60, 90, and 120 min), 100 μL of each of the incubated solutions was mixed well with 100 μL of ACN in microcentrifuge tubes to terminate the enzymatic reactions. This was followed by centrifugation to precipitate proteins (10,000 g, 4 min, 4°C). The supernatants were filtered through a 0.22 µm PVDF disk filter and analyzed using LC-DAD and tandem MS. The liver microsome solution (25 μL) was also mixed with DMSO (2 μL without flumazenil), buffer, and the co-enzyme solution to generate a blank solution.
4.5. Synthesis of [18F]flumazenil
4.6. In Vitro Serum Stability
Serum preparation: Health SD rat was deeply anesthetized with isoflurane gas (3% isoflurane in 50% oxygen, 1 mL/min). 1 mL whole blood of health SD rat was withdrawnthrough tail vein at room temperature and centrifuged at 3000 rpm/min for 10 minutes to achieve a prepared serum for stability test.
Stability in serum. The stability was studied by incubation of 3.7MBq of [18F]flumazenil in 200uL of prepared serum at 37°C. At different time points (0, 5, 10, 30, 60, 120 and 240 minutes), each sample was treated with 1 mL of acetonitrile for precipitating the serum proteins. After mixing and centrifuging for 2 minutes at 5000 rpm, the radiochemical purity of [18F]flumazenil was determined using radio-TLC methods. Radio-TLC was performed on a silica gel plate TLC-SG (1.5 × 6 cm), using ethyl acetate: ethanol (80/20) as the developing agent.
4.7. In Vivo NanoPET/CT Neuroimaging Studies
Rats were injected with approximately 37 MBq /200 μL of [18F]flumazenil via the lateral tail vein. After being distributed for 30 min, isoflurane gas (3%) was used to anesthetize the animals, which were then placed inside the NanoPET/CT PLUS system (Bioscan Europe, Ltd. Paris, France) for statistical analysis (post-injection 30-40 min). The InVivoScope software was used to capture the NanoPET images. Pmod 3.3 software was used to merge NanoPET/CT images, and then set them in the brain template. A region of interest (ROI) (cerebellum, medulla, pons, midbrain, striatum, frontal cortex, cortex, amygdala and hippocampus) was defined in each brain region on the NanoPET images with coronal sections. The average radioactivity concentration within each brain section was calculated by averaging the pixel values within multiple ROIs. When analyzing the NanoPET data, the medulla was used as the reference region to determine the specific binding ratios for all target regions.
4.8. Ex Vivo Bio-Distribution Assay
While the SD rats were under isoflurane anesthesia, [18F]flumazenil (37 MBq/200 μL) was injected directly into the tail vein. The rats (n=3 for each time point) were sacrificed by the administration of carbon dioxide at 5, 10, 30, 60, 120 and 240-minute post injection. The different brain regions and blood were removed and weighed, and the radioactivity was counted with an automatic gamma counter (WIZARD2 2480, PerkinElmer Instruments Inc.). The percentage dose per organ was calculated by a comparison of the tissue counts with suitably diluted aliquots of the injected material. Different brain regions corresponding to cerebellum, pons/medulla, striatum, cortex/amyglada, hippocampus, thalamus and hypothalamus were dissected out from the brain. The % dose/g and specific binding ratio (SBR) of samples was calculated by comparing the sample counts with the count of the diluted initial dose.
5. Conclusions
In conclusion, flumazenil metabolism pathways were identified in the liver microsomes and homogenates of rats by HPLC combined with ESI-QqQ MS, revealing the mechanism of biotransformation of flumazenil. And a new metabolite compound of methyl replacement at the ethyl ester is thought to be a novel flumazenil metabolite. Through an automatic synthesizer process, success radio-fluorination of [18F]flumazenil and its utilization in PET showed evidences of an animal model expressing the GABAA/BZR receptor. Distribution of [18F]flumazenil for 30 min, although in metabolism analyze showed about 50% bio-transformed metabolite, but in ex vivo bio-distribution and in vivo image studies still showed significant different in target region. The remaining complete compound structure could be continuously accumulated at the target regions of brain to achieve the purpose by molecular nuclear medicine imaging. Those complete analysis of flumazenil biotransformation by hepatocytes may provide evidence of [18F]flumazenil effectivity in patients with anxiety at the clinical stage in the future.
Author Contributions
Wei-Hsi Chen; methodology of HPLC/MS, writing-original draft preparation and results explantation, Chuang-Hsin Chiu; resources, supervision, Shiou-Shiow Farn; resources and formal analysis, Kai-Hung Cheng, Yuan-Ruei Huang, Shih-Ying Lee; investigation, formal analysis, validation, Yao-Ching Fang; data curation, Yu-Hua Lin; article editing and Kang-Wei Chang; resources, supervision, project administration and writing-review and editing. All authors have read and agreed to the published version of the manuscript.”.
Funding
This study was financially supported by Tri-Service General Hospital, National Defense Medical Center [Grant: TSGH-E-111219].
Conflicts of Interest
“The authors declare no conflict of interest.”.
References
- Nutt, D.J. and A.L. Malizia, New insights into the role of the GABA(A)-benzodiazepine receptor in psychiatric disorder. Br J Psychiatry, 2001. 179: p. 390-6.
- Nutt, D.J., Overview of diagnosis and drug treatments of anxiety disorders. CNS Spectr, 2005. 10(1): p. 49-56.
- Babaev, O., C. Piletti Chatain, and D. Krueger-Burg, Inhibition in the amygdala anxiety circuitry. Exp Mol Med, 2018. 50(4): p. 1-16.
- Crocq, M.A., A history of anxiety: from Hippocrates to DSM. Dialogues Clin Neurosci, 2015. 17(3): p. 319-25.
- Lee, H.J., et al., Profiling of gene expression in the brain associated with anxiety-related behaviors in the chronic phase following cranial irradiation. Sci Rep, 2022. 12(1): p. 13162.
- Kalueff, A.V. and D.J. Nutt, Role of GABA in anxiety and depression. Depress Anxiety, 2007. 24(7): p. 495-517.
- Collins, S.A. and I. Ninan, Development-Dependent Plasticity in Vasoactive Intestinal Polypeptide Neurons in the Infralimbic Cortex. Cereb Cortex Commun, 2021. 2(1): p. tgab007.
- Kraguljac, N.V., et al., Neuroimaging Biomarkers in Schizophrenia. Am J Psychiatry, 2021. 178(6): p. 509-521.
- Heiss, W.D., Radionuclide imaging in ischemic stroke. J Nucl Med, 2014. 55(11): p. 1831-41.
- Ishibashi, K., et al., PET Imaging of (18)F-FDG, (11)C-methionine, (11)C-flumazenil, and (11)C-4DST in Progressive Multifocal Leukoencephalopathy. Intern Med, 2017. 56(10): p. 1219-1223.
- Ghosh, K.K., et al., An In Vivo Study of a Rat Fluid-Percussion-Induced Traumatic Brain Injury Model with [(11)C]PBR28 and [(18)F]flumazenil PET Imaging. Int J Mol Sci, 2021. 22(2).
- Leech, R. and D.J. Sharp, The role of the posterior cingulate cortex in cognition and disease. Brain, 2014. 137(Pt 1): p. 12-32.
- Valentine, K.E., et al., THE EFFICACY OF HYPNOSIS AS A TREATMENT FOR ANXIETY: A META-ANALYSIS. Int J Clin Exp Hypn, 2019. 67(3): p. 336-363.
- Agatonovic-Kustrin, S., et al., Anxiolytic Terpenoids and Aromatherapy for Anxiety and Depression. Adv Exp Med Biol, 2020. 1260: p. 283-296.
- Zhu, S., et al., Structure of a human synaptic GABA(A) receptor. Nature, 2018. 559(7712): p. 67-72.
- Kim, W., et al., PET measurement of "GABA shift" in the rat brain: A preclinical application of bolus plus constant infusion paradigm of [(18)F]flumazenil. Nucl Med Biol, 2017. 45: p. 30-34.
- Mizuno, J., [Flumazenil]. Masui, 2013. 62(1): p. 10-8.
- Rodnick, M.E., et al., Novel fluorine-18 PET radiotracers based on flumazenil for GABAA imaging in the brain. Nucl Med Biol, 2013. 40(7): p. 901-5.
- Moon, B.S., et al., Routine production of [(18)f]flumazenil from iodonium tosylate using a sample pretreatment method: a 2.5-year production report. Mol Imaging Biol, 2014. 16(5): p. 619-25.
- Vivash, L., et al., In vivo measurement of hippocampal GABAA/cBZR density with [18F]-flumazenil PET for the study of disease progression in an animal model of temporal lobe epilepsy. PLoS One, 2014. 9(1): p. e86722.
- Vivash, L., et al., 18F-flumazenil: a γ-aminobutyric acid A-specific PET radiotracer for the localization of drug-resistant temporal lobe epilepsy. J Nucl Med, 2013. 54(8): p. 1270-7.
- Kaplan, M.M. and R.D. Utiger, Iodothyronine metabolism in rat liver homogenates. J Clin Invest, 1978. 61(2): p. 459-71.
- Dunn, W.B. and D.I. Ellis, Metabolomics: Current analytical platforms and methodologies. TrAC Trends in Analytical Chemistry, 2005. 24(4): p. 285-294.
- Chen, W.H., et al., High performance liquid chromatography-tandem mass spectrometry method for ex vivo metabolic studies of a rhenium-labeled radiopharmaceutical for liver cancer. Eur J Mass Spectrom (Chichester), 2014. 20(5): p. 375-82.
- Marwah, A., P. Marwah, and H. Lardy, Ergosteroids. VI. Metabolism of dehydroepiandrosterone by rat liver in vitro: a liquid chromatographic-mass spectrometric study. J Chromatogr B Analyt Technol Biomed Life Sci, 2002. 767(2): p. 285-99.
- Amini, N., et al., Identification of PET radiometabolites by cytochrome P450, UHPLC/Q-ToF-MS and fast radio-LC: applied to the PET radioligands [11C]flumazenil, [18F]FE-PE2I, and [11C]PBR28. Anal Bioanal Chem, 2013. 405(4): p. 1303-10.
- Lavén, M., et al., Determination of flumazenil in human plasma by liquid chromatography-electrospray ionisation tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2004. 808(2): p. 221-7.
- Lavén, M., K. Markides, and B. Långström, Analysis of microsomal metabolic stability using high-flow-rate extraction coupled to capillary liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2004. 806(2): p. 119-26.
- Levêque, P., et al., Assessment of [18F]fluoroethylflumazenil metabolites using high-performance liquid chromatography and tandem mass spectrometry. J Chromatogr B Biomed Sci Appl, 2001. 754(1): p. 35-44.
- Pascual, B., et al., Decreased carbon-11-flumazenil binding in early Alzheimer's disease. Brain, 2012. 135(Pt 9): p. 2817-25.
- Brambilla, P., et al., GABAergic dysfunction in mood disorders. Mol Psychiatry, 2003. 8(8): p. 721-37, 715.
- Müller Herde, A., et al., GABA(A) receptor subtypes in the mouse brain: Regional mapping and diazepam receptor occupancy by in vivo [(18)F]flumazenil PET. Neuroimage, 2017. 150: p. 279-291.
- Toth, I. and I.D. Neumann, Animal models of social avoidance and social fear. Cell Tissue Res, 2013. 354(1): p. 107-18.
- Farn, S.-S., et al., Automated Synthesis of [18F]Flumazenil Application in GABAA Receptor Neuroimaging Availability for Rat Model of Anxiety. Pharmaceuticals, 2023. 16(3): p. 417.
Figure 1.
Typical chromatogram of flumazenil exhibited a spiked in the rat liver homogenate. Analyzed HPLC using ZORBAX Eclipse XDB-C18 column separated flumazenil from liver microsome and the liver homogenate solutions (DAD at λ of 250 nm). The dynamic range of flumazenil in the hepatic matrices was 1–100 ppm, and the limits of detection (LOD) and quantification (LOQ) for flumazenil were 0.1 and 0.4 ppm, respectively.
Figure 1.
Typical chromatogram of flumazenil exhibited a spiked in the rat liver homogenate. Analyzed HPLC using ZORBAX Eclipse XDB-C18 column separated flumazenil from liver microsome and the liver homogenate solutions (DAD at λ of 250 nm). The dynamic range of flumazenil in the hepatic matrices was 1–100 ppm, and the limits of detection (LOD) and quantification (LOQ) for flumazenil were 0.1 and 0.4 ppm, respectively.
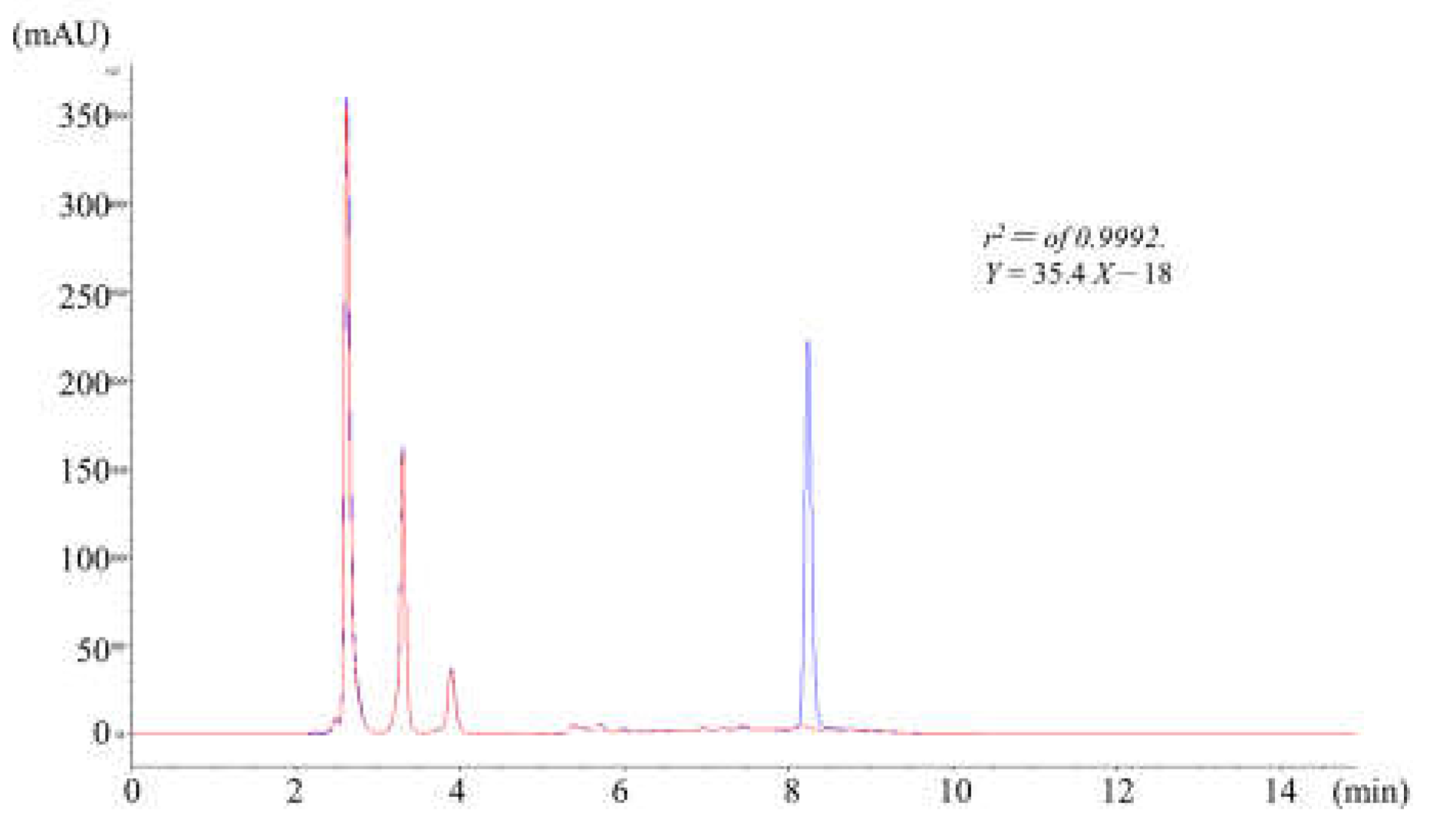
Figure 2.
Mass spectrum of fragmentation ions from m/z = 304 (dissolved in methanol with 0.1% formic acid, 10 µL min-1) injected into the instrument for the Q1 and MS/MS scans. (A) product-ion scans using different collision-activated dissociation conditions to optimize the de-clustering potential fragmentation scheme of flumazenil based on the tandem mass spectrum. (B)The fragment structures of flumazenil with metabolite identification, the most abundant product ions were 258 and 276, from the parent molecular ion 304 and were also observed at m/z values of 229, 217, and 189, respectively.
Figure 2.
Mass spectrum of fragmentation ions from m/z = 304 (dissolved in methanol with 0.1% formic acid, 10 µL min-1) injected into the instrument for the Q1 and MS/MS scans. (A) product-ion scans using different collision-activated dissociation conditions to optimize the de-clustering potential fragmentation scheme of flumazenil based on the tandem mass spectrum. (B)The fragment structures of flumazenil with metabolite identification, the most abundant product ions were 258 and 276, from the parent molecular ion 304 and were also observed at m/z values of 229, 217, and 189, respectively.
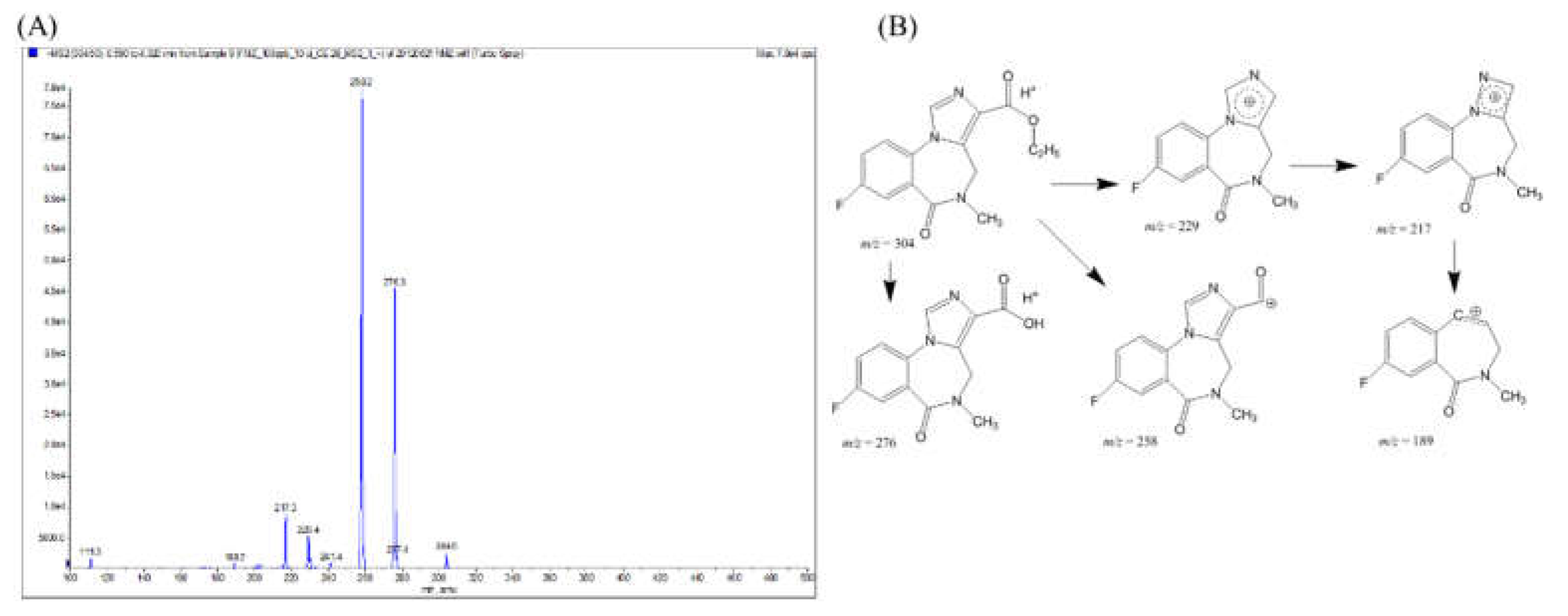
Figure 3.
(A) After pre-treated flumazenil concentrate for 0 to 240 min, the derivatives from the hepatic matrices by HPLC analysis. (B) Overlapped chromatograms of flumazenil incubation in rat liver homogenate for various duration. (C) The flumazenil biotransformation in the rat liver homogenate was evaluated based on trends in the peak area of flumazenil on chromatograms. In the first 30 min subsequently slowed down, and approximately 50% at 60 min, 4 h approximately 65%.
Figure 3.
(A) After pre-treated flumazenil concentrate for 0 to 240 min, the derivatives from the hepatic matrices by HPLC analysis. (B) Overlapped chromatograms of flumazenil incubation in rat liver homogenate for various duration. (C) The flumazenil biotransformation in the rat liver homogenate was evaluated based on trends in the peak area of flumazenil on chromatograms. In the first 30 min subsequently slowed down, and approximately 50% at 60 min, 4 h approximately 65%.
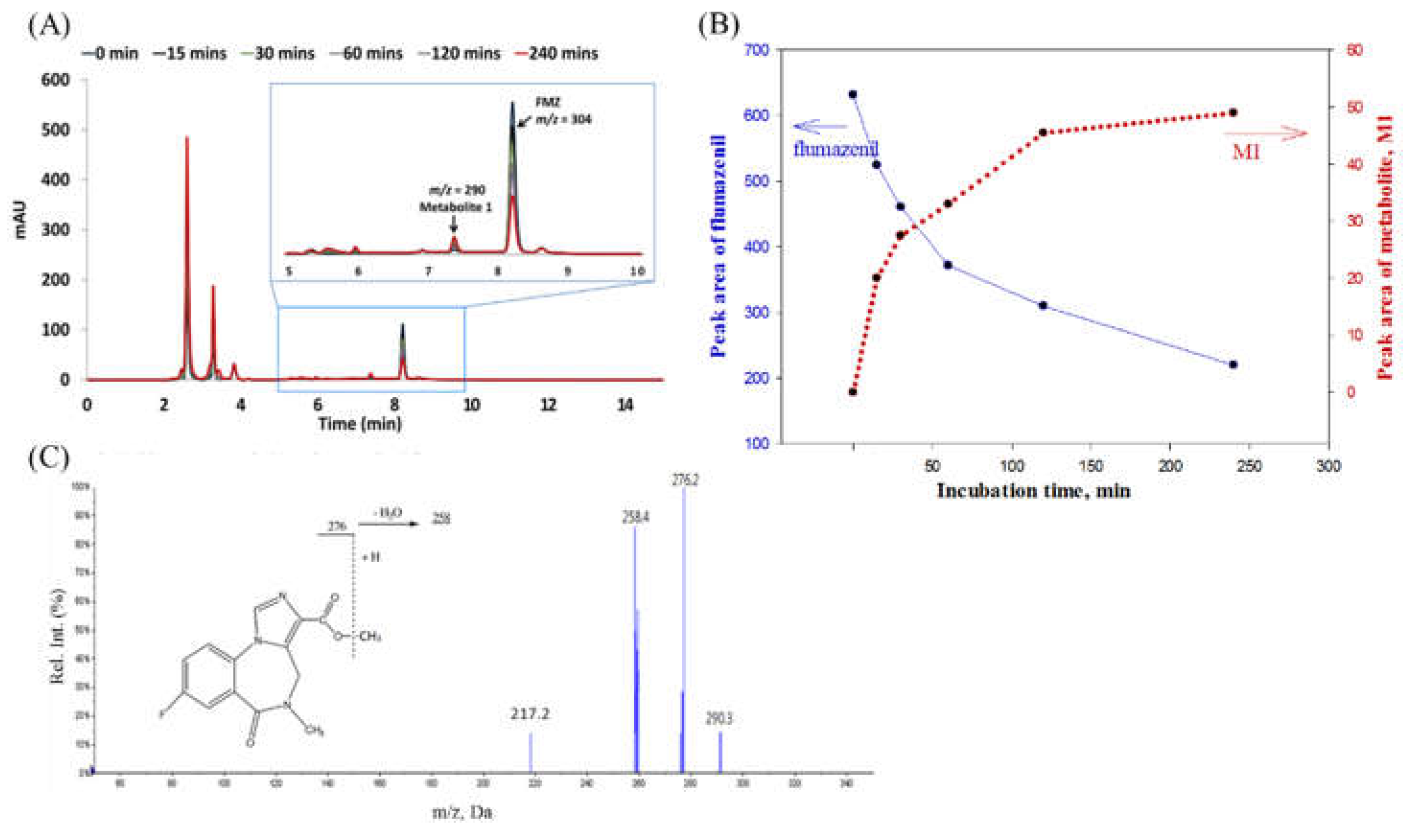
Figure 4.
Flumazenil metabolism study in human liver microsomal. Tandem mass spectra of M2 and M3, m/z = 276 and 320 and proposed metabolites of flumazenil in rat microsomal (A) and (B), respectively. (C) Time course of biotransformation of flumazenil in rat liver microsomal for various duration.
Figure 4.
Flumazenil metabolism study in human liver microsomal. Tandem mass spectra of M2 and M3, m/z = 276 and 320 and proposed metabolites of flumazenil in rat microsomal (A) and (B), respectively. (C) Time course of biotransformation of flumazenil in rat liver microsomal for various duration.
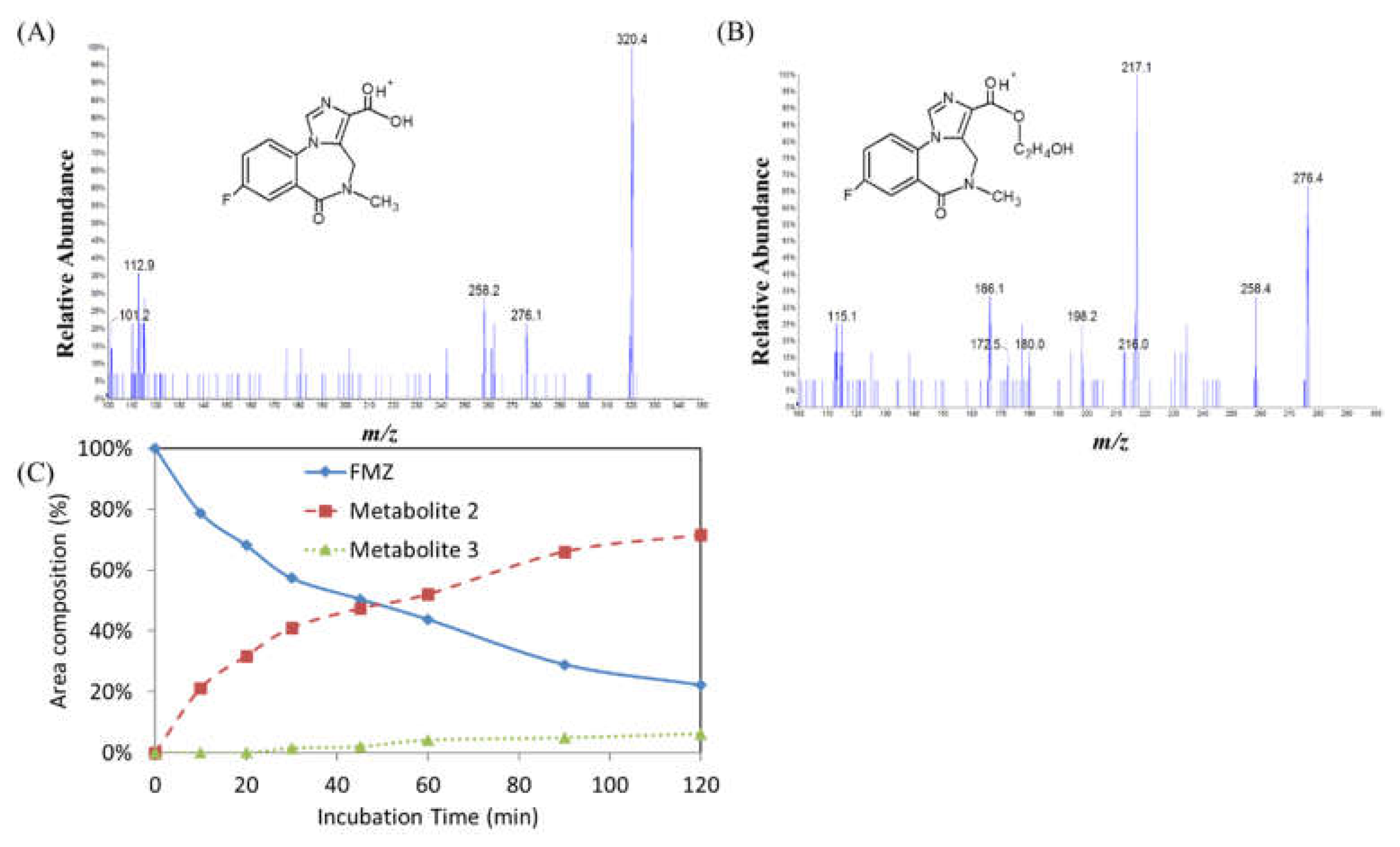
Figure 5.
A summary of the metabolic pathways of flumazenil in rat liver homogenate and the microsomal enzymatic system.
Figure 5.
A summary of the metabolic pathways of flumazenil in rat liver homogenate and the microsomal enzymatic system.
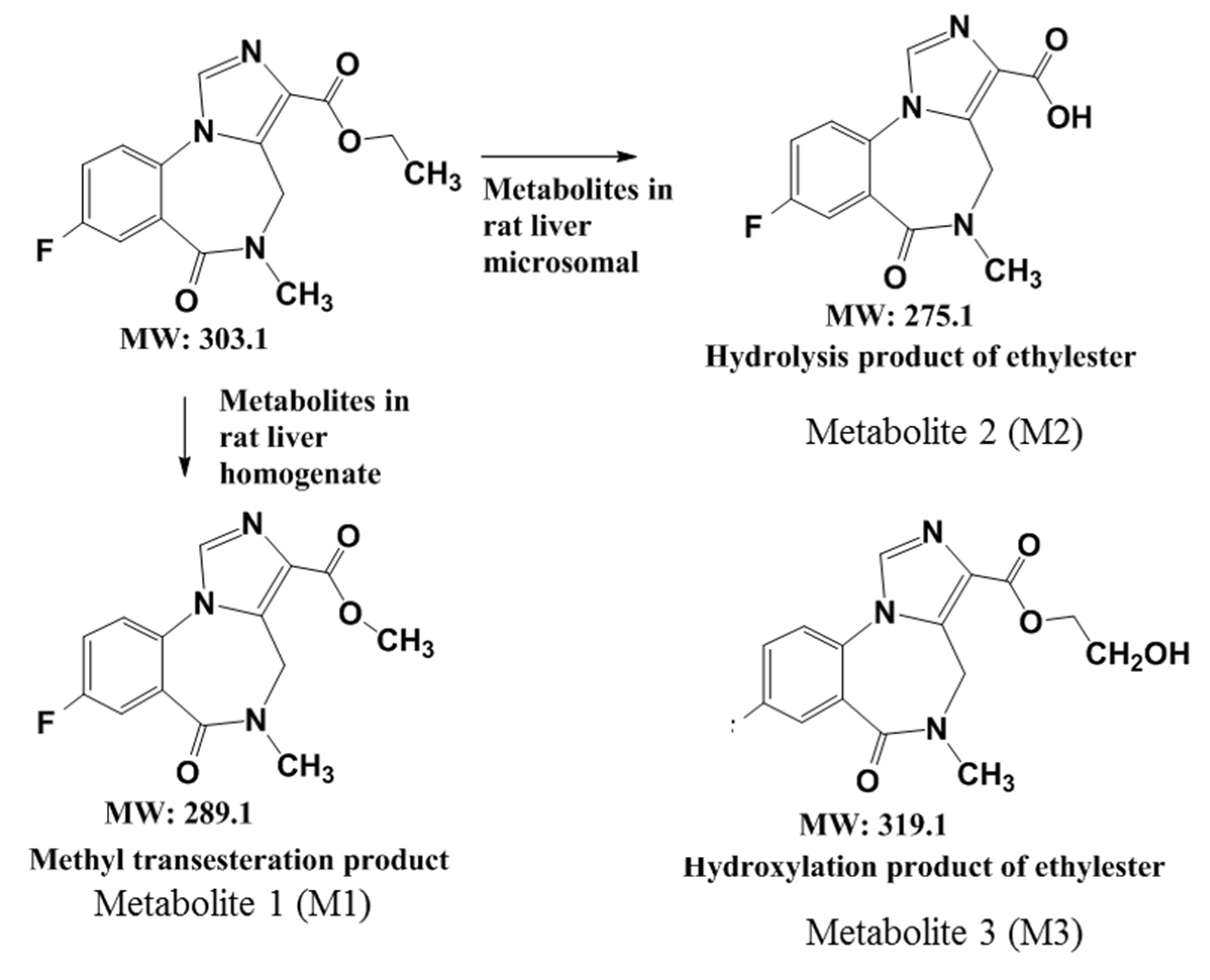
Figure 6.
The brain regional distribution of GABAA receptors as imaged by [18F]flumazenil Nano-PET/CT in normal SD rats. Acquired NanoPET/CT static images at 30-40 min. The rats displayed markedly higher uptake of [18F]flumazenil in prefrontal cortex, cortex, amygdala and hippocampus, medulla was delineated as ROIs and used as a reference region. In Vivo NanoPET/CT of coronal views of [18F]flumazenil showing 30–40 min (A). The specific uptake ratio (SBR) in different brain regions at post-injected for 30−40 minutes were significantly cerebral uptake of [18F]flumazenil (B).
Figure 6.
The brain regional distribution of GABAA receptors as imaged by [18F]flumazenil Nano-PET/CT in normal SD rats. Acquired NanoPET/CT static images at 30-40 min. The rats displayed markedly higher uptake of [18F]flumazenil in prefrontal cortex, cortex, amygdala and hippocampus, medulla was delineated as ROIs and used as a reference region. In Vivo NanoPET/CT of coronal views of [18F]flumazenil showing 30–40 min (A). The specific uptake ratio (SBR) in different brain regions at post-injected for 30−40 minutes were significantly cerebral uptake of [18F]flumazenil (B).
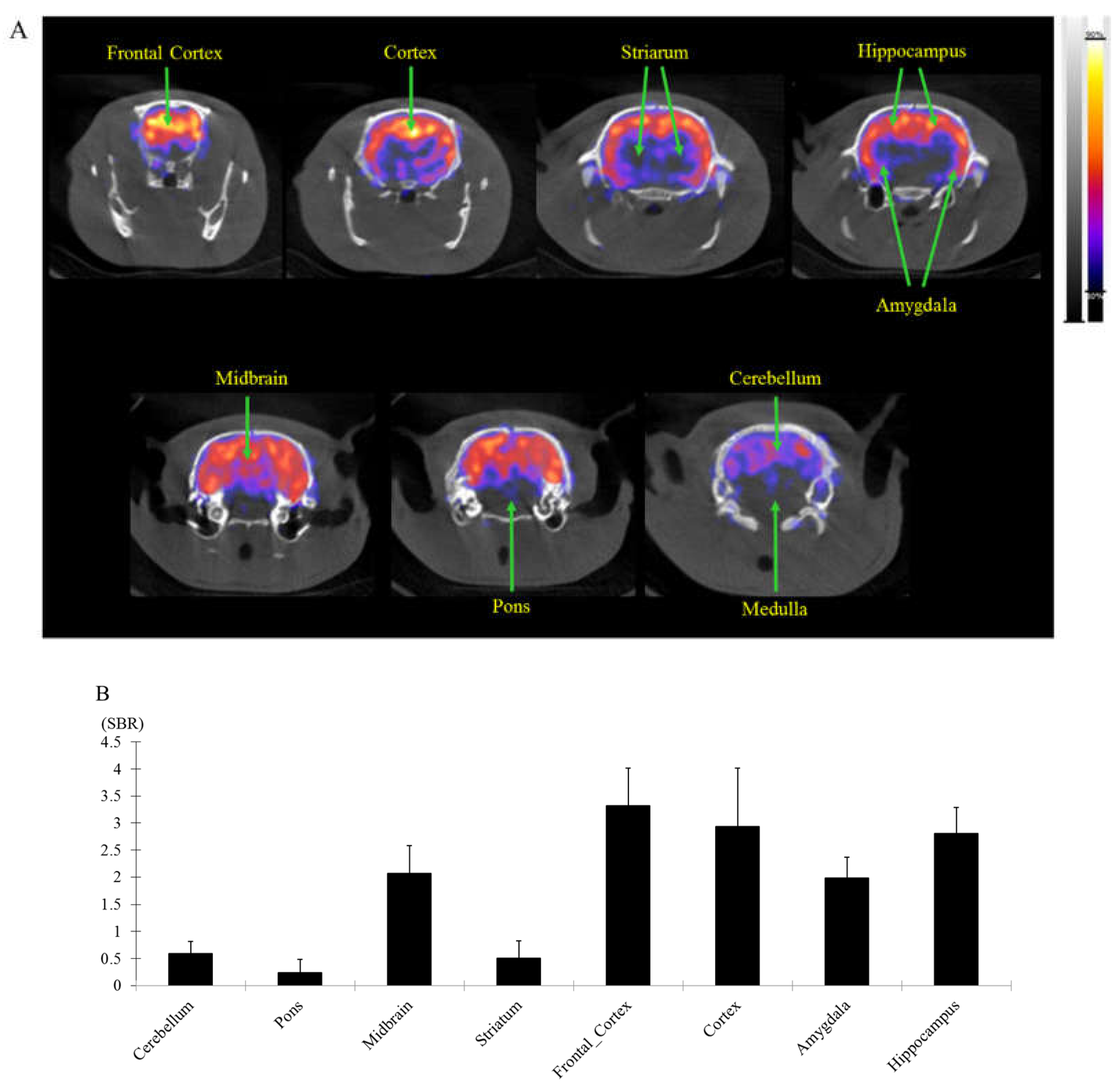
Figure 7.
The %ID/g on the [18F]flumazenil/ blood distribution analysis was counted with an automatic gamma counter. The percentage dose per gram was calculated by a comparison of the blood counts with suitably diluted aliquots of the injected material. The blood %ID/g of [18F]flumazenil were 5.47 ± 0.56, 0.99 ± 0.42, 0.07± 0.07, 0.10 ± 0.11, 0.03 ± 0.04 and 0.01 ± 0.01 at 5, 10, 30, 60, 120 and 240 minutes, respectively.
Figure 7.
The %ID/g on the [18F]flumazenil/ blood distribution analysis was counted with an automatic gamma counter. The percentage dose per gram was calculated by a comparison of the blood counts with suitably diluted aliquots of the injected material. The blood %ID/g of [18F]flumazenil were 5.47 ± 0.56, 0.99 ± 0.42, 0.07± 0.07, 0.10 ± 0.11, 0.03 ± 0.04 and 0.01 ± 0.01 at 5, 10, 30, 60, 120 and 240 minutes, respectively.

Table 1.
High performance liquid chromatography (HPLC) and tandem mass spectrometry instrumental parameters for flumazenil biotransformation.
Table 1.
High performance liquid chromatography (HPLC) and tandem mass spectrometry instrumental parameters for flumazenil biotransformation.
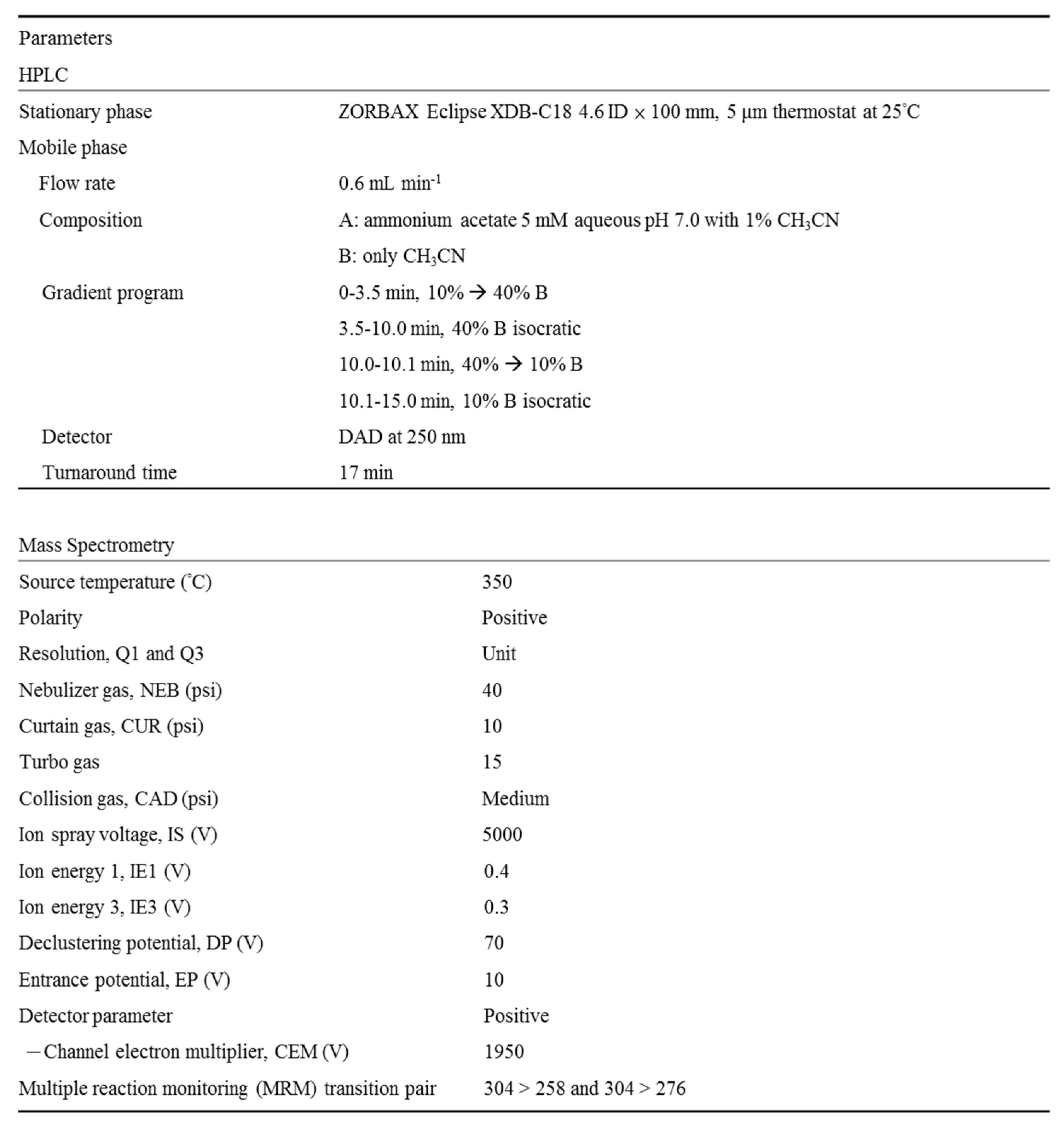 |
Table 2.
In vitro Serum Stability of [18F]flumazenil. Prepared serum for incubation of 3.7MBq of [18F]flumazenil at 37°C. At different time points (0, 5, 10, 30, 60, 120 and 240 minutes), treated with 1 mL of acetonitrile for precipitating the serum proteins. The radio-chemical purity of [18F]flumazenil was determined using radio-TLC methods. Radio-TLC was performed on a silica gel plate TLC-SG (1.5 × 6 cm), using ethyl acetate: ethanol (80/20) as the developing agent.
Table 2.
In vitro Serum Stability of [18F]flumazenil. Prepared serum for incubation of 3.7MBq of [18F]flumazenil at 37°C. At different time points (0, 5, 10, 30, 60, 120 and 240 minutes), treated with 1 mL of acetonitrile for precipitating the serum proteins. The radio-chemical purity of [18F]flumazenil was determined using radio-TLC methods. Radio-TLC was performed on a silica gel plate TLC-SG (1.5 × 6 cm), using ethyl acetate: ethanol (80/20) as the developing agent.
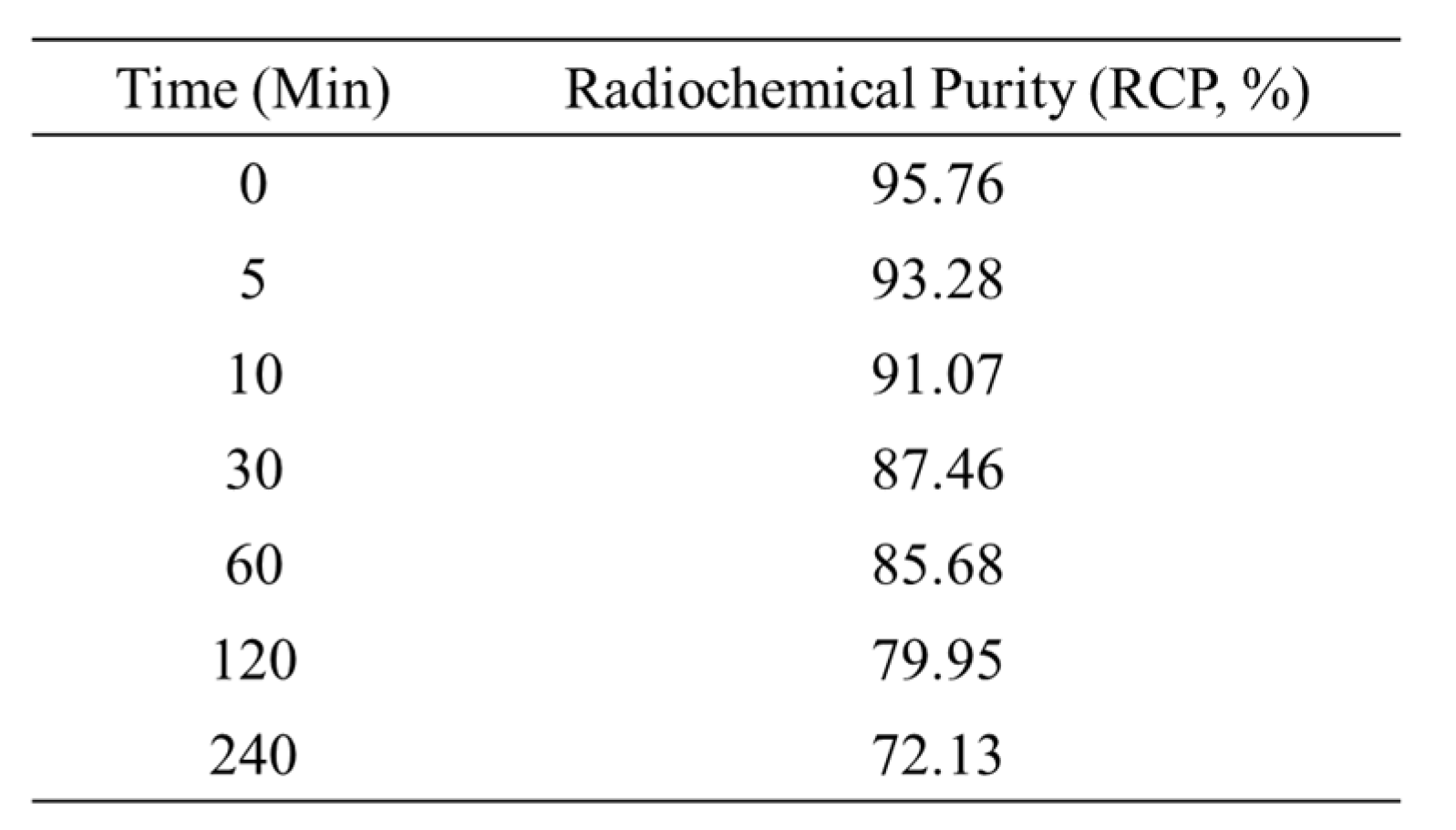 |
Table 3.
Ex-vivo biodistribution of [18F]flumazenil at 5, 10, 30, 60, 120, and 240 min after injection in SD rats. The percentage dose per gram (%ID/g) was calculated by comparing the sample counts with the count of the diluted initial dose. In SBR calculated, different brain regions will be corresponding to pons/medulla.
Table 3.
Ex-vivo biodistribution of [18F]flumazenil at 5, 10, 30, 60, 120, and 240 min after injection in SD rats. The percentage dose per gram (%ID/g) was calculated by comparing the sample counts with the count of the diluted initial dose. In SBR calculated, different brain regions will be corresponding to pons/medulla.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



