
Submitted:
18 April 2023
Posted:
19 April 2023
You are already at the latest version
Abstract
Metachromatic leukodystrophy (MLD) is a hereditary neurodegenerative disease characterized by demye-lination and motor and cognitive impairment due to the deficiency of the lysosomal enzyme arylsulfatase A (ARSA) or the saposin B activator protein (SapB). Current treatments are limited; however, gene therapy using adeno-associated virus (AAV) vectors for ARSA delivery has shown promising results. The main challenges for MLD gene therapy include optimizing AAV dosage, selecting the most effective serotype, and determining the best route of administration for ARSA delivery into the central nervous system. This study aims to evaluate the safety and efficacy of AAV serotype 9 encoding ARSA (AAV9-ARSA) gene therapy when administered intrave-nously or intrathecally in minipigs, a large animal model with anatomical and physiological similarities to humans. By comparing these two administration methods, this study contributes to the understanding of how to improve the effectiveness of MLD gene therapy and offers valuable insights for future clinical applications.
Keywords:
adeno-associated virus
; metachromatic leukodystrophy
; arylsulfatase A
; gene therapy
; central nervous system
; peripheral nervous system
; neurodegeneration
1. Introduction
Metachromatic leukodystrophy (MLD) is an autosomal recessive hereditary neurodegenerative disease that belongs to the group of lysosomal storage diseases (LSD). It is characterized by damage to the myelin sheath, which covers most of the nerve fibers in the central (CNS) and peripheral nervous system (PNS). MLD is caused by a deficiency in the lysosomal enzyme arylsulfatase A (ARSA) (OMIM: 250100) or the saposin B activator protein (SapB) (OMIM: 249900). Clinically, the disease manifests as progressive motor and cognitive impairment [1]. MLD is one of the most common leukodystrophies, with an incidence rate of 1:40,000. However, in some isolated populations, the incidence rate can be much higher, such as 1:75 among Habbani Jews and 1:2500 among the Navajo Indians [2].
The term MLD refers to the presence of metachromatic granules in affected cells, which form due to the accumulation of sulfatides and sphingolipids found in myelin. In MLD, sulfatides build up in oligodendrocytes, microglia, CNS neurons, Schwann cells, PNS macrophages, and cells in various internal organs [3,4,5,6,7,8,9].
Demyelination in MLD results in impaired motor function, spastic tetraparesis, ataxia, convulsions, optic nerve atrophy, and cognitive impairment [10,11]. The exact mechanisms of demyelination remain unknown, but it is believed that increased levels of sulfatides and decreased levels of their cleavage products cause instability in the myelin sheath, ultimately leading to demyelination [12]. Furthermore, the accumulation of sulfatides on the endoplasmic reticulum (ER) membrane triggers calcium release into the cytoplasm, causing changes in calcium homeostasis that lead to cellular stress and apoptosis [8].
Accumulation of sulfatides leads to neuronal degeneration, astrocyte dysfunction, and triggers an inflammatory response in MLD patients. Both plasma and cerebrospinal fluid (CSF) show elevated levels of monocyte chemoattractant protein 1 (MCP-1), interleukin 1 receptor antagonist (IL-1Ra), IL-8, macrophage inflammatory protein 1β (MIP-1β), and vascular endothelial growth factor (VEGF) [13]. In MLD, there may not be a correlation between demyelination and the presence of metachromatic material. Consequently, complement activation via an alternative pathway exacerbates myelin damage in MLD by inducing or enhancing an anti-myelin immune response [14]. Sulfatide accumulation and demyelination in the PNS can prompt the release of inflammatory cytokines, activate endoneurial macrophages, and recruit inflammatory myeloid cells and lymphocytes from the periphery. These processes contribute to apoptosis and can lead to the progression of demyelination and neuroinflammation, which are also observed in some other metabolic and neurodegenerative diseases [15].
A residual enzymatic activity of 10-15% can be sufficient for sulfatide degradation and maintenance of normal life in ARSA deficiency [16]. There is a pseudo-deficiency of ARSA, where the enzyme has low enzymatic activity (about 5-20%), but no disease phenotype is observed, as the residual activity is sufficient to catabolize its natural substrate [17]. In some instances, a shortened protein can be synthesized without a change in activity [18].
Based on residual enzymatic activity levels, MLD presents in various forms that differ in age of clinical manifestation and rate of disease progression: infantile (late infantile, from 0 to 4 years) [19,20], juvenile (from 4 to 15 years old) [21,22,23], and adult form (over 15 years old) [21,24,25,26]. The late infantile form is the most common, occurring in 50-60% of all patients, while 20-30% are diagnosed with the juvenile form, and the adult form is the rarest, affecting about 15-20% of MLD patients [1].
Currently, there is no effective treatment for MLD. Clinical cases involving bone marrow transplantation (BMT), hematopoietic stem cells (HSC), or umbilical cord blood transplantation have been reported, but the therapeutic efficacy of these approaches remains insufficient to prevent the worsening of neurological symptoms in patients [27,28]. Promising results have been achieved using gene therapy methods to deliver the wild-type ARSA gene within vectors based on various adeno-associated virus serotypes (AAV) [29,30], as well as using mesenchymal stem cells [31,32] and combined gene-cell therapy [33].
A primary challenge in treating diseases affecting the nervous system is the poor permeability of the blood-brain barrier (BBB), which limits drug access to the nervous system when administered systemically and consequently reduces the effectiveness of many therapeutic approaches [34,35]. Moreover, successful distribution of therapeutic drugs throughout the nervous system, including the PNS, is crucial for preventing the progression of MLD.
AAVs are promising vectors for ARSA delivery into neurons. AAVs can trans-synaptically transduce neurons over a wide range from the injection site through anterograde transport [36]. However, some studies have reported that anterograde transport down the axon is limited to specific serotypes, such as AAV5 [37], but not in AAV1, AAV2, AAV6, AAV8, or AAV9, while other studies have demonstrated anterograde transport of AAV1, AAV2, and AAV6 [36].
AAV5-ARSA administration into the brains of MLD mice resulted in prolonged expression of the ARSA gene (3-15 months), facilitating an almost complete disappearance of pathological changes in the brain and preventing sulfatide accumulation [38]. Promising results have also been obtained using AAV9 encoding ARSA and the green fluorescent protein reporter gene. Injecting the viral vector into the jugular veins of newborn MLD mice led to prolonged expression of the ARSA gene (up to 15 weeks), primarily in muscle and heart cells, with moderate expression also found in CNS cells. In treated mice, sulfatide accumulation was significantly reduced in the brain and spinal cord, and their levels showed no difference compared to wild-type mice [30].
Among new adeno-associated virus serotypes, AAVrh.10, isolated from non-human primates, migrated more efficiently from the intracerebral site compared to AAV1, AAV2, AAV5, AAV7, or AAV8 [39,40]. Injection of AAVrh.10-ARSA into the brains of 8-month-old MLD mice corrected the accumulation of certain sulfatides in oligodendrocytes, as this AAV serotype can apparently infect oligodendrocytes (up to 9%) [40]. In a phase I/II clinical trial investigating this virus (NCT01801709), four children with pre-symptomatic or very early symptomatic stages received 12 injections of 1 x 1012 or 4 x 1012 (depending on age) AAVrh.10-ARSA transducing units into the brain’s white matter. ARSA activity in the CSF, which had been undetectable before treatment, was reported to reach 20-70% of control values after injections. However, symptoms in early-stage patients continued to worsen, and asymptomatic patients developed MLD not significantly different from the natural course of the disease [29].
AAV-PHP.eB has been recently described as a highly efficient serotype for crossing the BBB following intravenous delivery, providing efficient transduction of the brain and spinal cord in GFAP-Cre mouse models expressing Cre under the control of the mouse GFAP promoter and C57Bl/6J mice [41,42]. A single intravenous administration of AAVPHP.eB-hARSA-HA into an MLD mouse model resulted in stable expression of the ARSA enzyme in the brain and spinal cord of experimental animals. Audouard et al. demonstrated a complete correction of sulfatide accumulation in the spinal cord of model animals [43], which was not observed when AAVrh10-ARSA was injected into the brain [40].
MLD is characterized by various clinical manifestations, and effective disease modeling is critical for studying its genetic basis, progression, diagnosis, and therapy [1]. However, there are no large animal models currently available to study MLD. Each model has its own advantages and limitations. Pigs possess several distinct advantages that make them suitable for research as an animal model. They have a large size, enabling easy puncture procedures, as well as highly developed CNS and PNS, in addition to sharing important anatomical and physiological similarities with the human body.
Consequently, MLD gene therapy faces several challenges; addressing these would significantly increase its effectiveness. Further optimization of the administered AAV dose is crucial, along with the selection of the most effective serotype and route of administration for ARSA delivery into the CNS. In this study, a comparative analysis was conducted to assess the safety and efficacy of gene therapy using AAV9-ARSA when administered intravenously or intrathecally into minipigs.
2. Results
2.1. Codon optimization
Codon optimization enhances the translation efficiency of mRNA into polypeptides, thus improving the effectiveness of expression vectors, including those used in gene therapy. Ribosomes translate codons with higher frequency faster, as they are recognized by tRNA molecules that are abundant in the cell. Therefore, the optimal codons used for the recipient organism’s genes are the most frequently occurring synonymous codons. This modification is believed to increase the expression efficiency of the therapeutic gene.
To create the plasmid vector, the codon composition of the cDNA nucleotide sequence of the ARSA gene was optimized using the OptimumGene™ algorithm (GenScript, USA). The mRNA nucleotide sequence of the human ARSA gene (NM_000487.6) was used as a template for codon optimization. Optimizing the codon composition allowed for the highest possible level of gene expression. The wild-type ARSA gene contains tandem rare codons that can reduce translation efficiency or even disable the translation mechanism. Codon optimization increased the codon adaptation index (CAI) of the ARSA gene from 0.83 to 0.90, with a CAI of 1.0 being considered desirable for the highest gene expression level. Additionally, the content of GC pairs was increased from 61.33 to 65.45 to enhance the stability of the ARSA gene mRNA. The optimization process also changed negative cis-acting sites, but the amino acid sequence of the ARSA gene remained the same at 481 amino acid residues.
The codon-optimized cDNA of the ARSA gene was synthesized and cloned into the pAAV-MCS plasmid vector (Agilent Technologies, USA) by GenScript (USA) (Figure 1A). The resulting plasmid construct was then transformed into E. coli (TOP10), and several clones were selected for isolation of plasmid DNA. To confirm the successful cloning, the isolated plasmid DNA was subjected to restriction analysis using BamHI restriction enzyme. The resulting plasmid, named pAAV-ARSA, was found to have the expected size of 6226 bp (Figure 1B).
To confirm the functionality of the pAAV-ARSA genetic construct, an immortalized line of primary human embryonic kidney cells HEK293T was transfected using TurboFect transfection agent (Thermo Fisher Scientific Inc., USA) according to the manufacturer’s recommended protocol. The transfection efficiency was assessed by measuring enzyme activity and western blot analysis. After 48 hours of transfection, HEK293T-pAAV-ARSA showed a 15-fold increase in ARSA enzymatic activity compared to native cells (Figure 2A). Additionally, the protein was detected at a molecular weight of approximately 33 kDa (Figure 2B).
To confirm the functionality of the AAV9-ARSA genetic construct, HEK293T cells were transfected with protamine sulfate. The efficiency of recombinant protein expression in vitro was confirmed by activity testing and western blot analysis. The enzymatic activity in the genetically modified HEK293T cells was found to be 2-fold higher compared to the native and modified AAV9-GFP cells, indicating a successful transduction and overexpression of the ARSA enzyme in the modified HEK293T cells (Figure 3A). Western blot analysis confirmed the presence of the ARSA protein in the modified HEK293T cells, with a molecular weight of approximately 33 kDa (Figure 3B). Furthermore, the purity analysis of the vector through SDS-PAGE electrophoresis indicated that the AAV9-ARSA construct was pure without any impurities. The viral proteins were shown to be viral protein 1 (VP1) with a molecular weight of 81.4 kDa, viral protein 2 (VP2) with a molecular weight of 66.3 kDa, and viral protein 3 (VP3) with a molecular weight of 59.8 kDa, with VP3 predominating as expected (Figure 3C).
2.2. Analysis of ARSA enzymatic activity
Animals were divided into three groups. The first group received intrathecal administration of AAV9-ARSA at a dose of 1 × 1012 genomic copies/kg, the second group received intravenous administration at a dose of 3.77 × 1013 genomic copies/kg, and the third group served as the control with no AAV9-ARSA administration. The functionality of the recombinant AAV9-ARSA was tested by measuring ARSA activity in the plasma, CSF, and various CNS structures.
No significant differences in ARSA enzymatic activity were observed in the plasma of animals from the different groups. However, following intrathecal administration of AAV9-ARSA, ARSA enzymatic activity in porcine CSF increased. Specifically, on days 7, 14, 21, and 35, a significant increase in ARSA enzymatic activity in the CSF was observed by 146%, 169%, 153%, and 138%, respectively (Figure 4).
An increase in ARSA enzymatic activity was found, compared to control group, in the cerebellum, cervical, and lumbar spinal cord, following intravenous administration of AAV9-ARSA by 154%, 255%, and 357%, respectively (Figure 5). These results indicate that neuronal cells begin to express functional enzyme in vivo, following genetic modification by AAV9-ARSA, and the increase was observed in both groups of animals.
In the first group of experimental animals, mRNA copies of ARSA gene were detected in different regions of the central nervous system following intrathecal injection. Specifically, 91774, 11181, 3883, and 15144 copies of ARSA gene mRNA per μg of total RNA were detected in the cerebellum, cervical, thoracic, and lumbar spinal cord, respectively (Figure 6A).
In the second group of experimental animals, after intravenous administration, codon-optimized ARSA gene overexpression was only observed in the thoracic spinal cord and dorsal root ganglia of the thoracic region. Specifically, 1104 copies of ARSA gene mRNA were detected per µg of total RNA in the thoracic spinal cord, while in the ganglia of dorsal roots of cervical, thoracic, and lumbar regions, 203, 666, and 185 copies of ARSA gene mRNA per μg of total RNA, respectively, were detected (Figure 6B).
2.3. Biochemical blood analysis
Biochemical parameters of minipigs’ blood sera were assessed after AAV9-ARSA administration. ALT and creatinine-J levels did not show any changes after AAV9-ARSA administration. A statistically significant decrease in AST levels was observed on day 35 in the second group of animals that received intravenous administration. However, in the first group of animals that received intrathecal injection, AST levels remained unchanged (Figure 7). In the second group of animals, bilirubin levels were significantly lower on days 7 and 35 following intravenous administration of the drug, while no changes in bilirubin total levels were observed in the first group of animals that received intrathecal administration of the drug.
The inflammatory cytokine profile of porcine serum and CSF was also studied. No statistically significant difference was detected between the levels of inflammatory cytokines and chemokines in different animal groups in the minipigs’ blood serum. In CSF, an increase in IL-1ra levels was detected following both intrathecal (0.288 ± 0.257 pg/mL) and intravenous (0.078 ± 0.057 pg/mL) AAV9-ARSA administration, while no IL-1ra was detected in the CSF of animals in the control group (Figure 8).
2.4. Pathomorphological analysis
A comparative pathomorphological analysis of kidney, heart, spleen, lungs, and liver tissues was performed in both control and experimental groups. The results did not reveal any significant pathomorphological changes in the studied organs as a result of intrathecal or intravenous administration of AAV9-ARSA (Figure 9). Microscopic examination of the kidney and heart in the study groups did not reveal any foci of hemorrhage or other circulatory disorders, deformation of renal glomeruli or muscle fibers, stromal edema, or leukocyte infiltration. Pathological analysis of the spleen did not reveal any signs of hyperimmune response, such as hyperplasia or plethora, or immunosuppression, such as atrophy of lymphoid follicles, desolation of the spleen pulp, or splenic anemia. Microscopic examination of lung tissue showed that the parenchyma of lung alveoli was lined with flattened alveolar epithelium, thin partitions were located between them without signs of edema, and no erythrostasis or diapedesis hemorrhages were found in the capillaries. Unfortunately, microscopic sections of the liver could not be obtained due to technical reasons.
2.5. Assessment of ARSA expression in nervous tissue
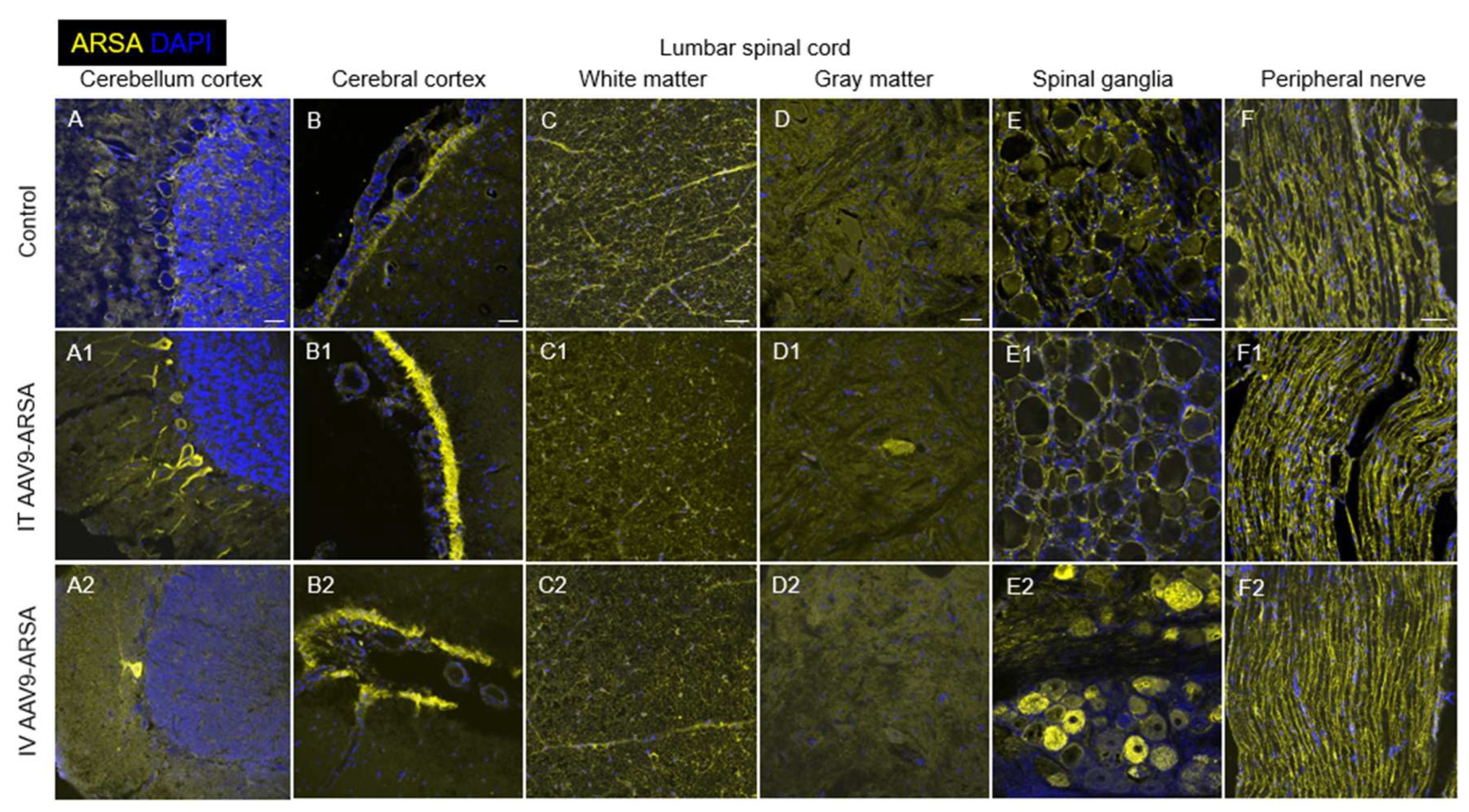
As a result of immunofluorescent analysis, Purkinje neurons overexpressing ARSA were found in the cerebellar cortex in both experimental groups of animals, but not in the control group (Figure 10A, A1-2). The number of these cells was higher in the group of intrathecal drug administration [45 ± 40.8], compared to intravenous administration group [10.5 ± 4.3]. It should be noted that in the second experimental group, Purkinje neurons overexpressing ARSA were mostly localized singly within one gyrus (Figure A2), while, in control group, ARSA expression was lower and mainly localized on the periphery of the cytoplasm only (Figure 10A).
Analysis of the occipital cortex showed increased expression of ARSA in the subarachnoid space in minipigs from both experimental groups, compared to control group (Figure 10B, B1-2). However, analysis of average ARSA glow intensity in this area did not reveal significant difference between experimental groups. In minipigs of both experimental and control groups, ARSA+ cells were also found in cerebral cortex. Specific luminescence in these cells was localized on cytoplasm periphery of cell body, and partially in the processes.
Analysis of transverse sections of cervical, thoracic, and lumbar spinal cord showed no significant differences in ARSA expression in the white and gray matter in animals of the experimental and control groups. In the white matter, ARSA expression was markedly higher, compared to gray matter, and predominantly localized in the processes and bodies of glial cells (Figure 10C, C1-2). The big picture of ARSA expression in the lumbar spinal cord of experimental group (with intrathecal AAV9-ARSA administration) was similar to that described above. However, single ARSA-overexpressing neurons were found in the ventral horns, which was not observed in the group of intravenous AAV9-ARSA administration, or control group (Figure 10D, D1 2).
Immunohistochemical analysis of spinal ganglia at the level of cervical, thoracic, and lumbar spinal cord revealed the presence of ARSA overexpressing neurons only in the experimental group of AAV9-ARSA intravenous administration (Figure 10E, E1-2). The number of ARSA-overexpressing neurons varied greatly within this group, however, no significant difference in this indicator was found between spinal ganglia of different levels (cervical, thoracic, and lumbar regions). Analysis of the spinal roots and lateral femoral cutaneous nerve showed no difference in ARSA expression in experimental and control animals (Figure 10F, F1-2). It is worth to mention that ARSA expression in the porcine PNS is, originally, significantly higher than the CNS. Accordingly, the possibility of AAV9-ARSA penetration into the PNS from the subarachnoid space could not be assessed under given conditions.
3. Discussion
Metachromatic leukodystrophy (MLD) is a group of hereditary monogenic diseases characterized by lysosomal dysfunctions resulting from the accumulation of uncleaved substrate. Currently, gene and gene-cell therapy are considered promising approaches for the treatment of MLD. Gene-cell therapy utilizes genetically modified cells transduced with retro- and lentiviruses [44,45]. Clinical studies (NCT01560182) have shown that transplantation of CD34+ hematopoietic stem cells (HSCs) with ARSA overexpression to patients with pre-symptomatic or very early symptomatic MLD resulted in remyelination and normalization of motor activity. However, one of the nine patients who already had symptoms of the disease at the time of transplantation did not experience an improvement in motor activity [33]. Despite these promising results, there are still a number of challenges, including long-term monitoring of efficacy and safety in patients, as retro- and lentivirus-based drugs are at risk of insertional mutagenesis [46]. Genetically modified HSCs are also known to have a therapeutic effect using cross-correction mechanisms [47].
Gene therapy shows great promise for treating LSD. ARSA encoding different AAV serotypes can be a potential therapy for MLD as AAVs can transduce neurons in a wide range from the injection site through anterograde neuronal transport [48]. AAVs have several attractive features as vectors, such as low immunogenicity compared to other vectors, broad tropism, and the ability to transduce neurons over a wide range from the injection site. AAV can be easily constructed as a gene delivery vector by replacing the viral genome with the therapeutic cassette. Different AAV serotypes have different tropisms for different tissues. Among all the identified and characterized serotypes, AAV9 has the highest tropism for CNS and can cross the BBB [49,50].
AAV9 efficacy has been demonstrated in various preclinical models for CNS disorders, and in some clinical studies [51]. Intrathecal administration of AAV9 ensures transgenes distribution throughout the nervous system. When administered intravenously, AAV9 can cross the BBB and enter the CNS [52]. The effectiveness of intravenous administration of rAAV9 has been shown in a mouse model [53]. In our study, intravenous and intrathecal administration of recombinant AAV9, containing a unique codon-optimized sequence of the human ARSA gene, were compared and analyzed for their efficacy and safety in minipigs.
We produced recombinant AAV9-ARSA and confirmed in vitro ARSA overexpression using enzyme activity assay and western blot analysis. We then evaluated the ability of AAV9-ARSA to synthesize a functionally active enzyme after intrathecal or intravenous administration in minipigs. Following intrathecal administration of AAV9-ARSA, we observed an increase in ARSA enzymatic activity in the CSF, which is consistent with other studies [54,55]. RT-PCR results showed overexpression in the cerebellum, cervical, thoracic, and lumbar spinal cord following intrathecal administration. IHC also showed overexpression in the cerebellum and ventral horns of the lumbar region, as well as in neurons in the spinal cord, but no statistically significant difference was found between the groups. Other researchers have shown transduction of CNS and PNS neurons following intrathecal administration of AAV9-GFP to mice [56,57]. In our study, overexpression was not observed in the PNS.
We hypothesize that the differences in promoter usage, injection site, anatomical variations among experimental animals, and AAV vector design may have contributed to the observed discrepancies. Rachel M. Bailey et al. used a self-complementary AAV vector [56] and reported increased enzymatic activity in the cerebellum, cervical, and thoracic spinal cord following intravenous administration of AAV9-ARSA. RT-PCR analysis demonstrated ARSA overexpression in the thoracic spinal cord and spinal ganglia of the cervical, thoracic, and lumbar regions. IHC analysis also revealed overexpression in the cerebellum and spinal ganglia. Our findings are supported by studies conducted by Yingqi Lin et al., where they demonstrated broad expression of the transgene in various organs of minipigs following intravenous administration of AAV9-GFP, with overexpression primarily observed in various regions of the brain, and without any adverse inflammatory reactions [58].
In order to evaluate the safety of our research, we conducted a biochemical blood test, cytokine profiling in blood serum, and a pathomorphological analysis. High-dose intravenous administration of AAV9 has been reported to cause systemic and sensory neuron toxicity in non-human primates [59]. However, in our study, biochemical analysis showed no increase in the levels of AST, ALT, total bilirubin, or creatinine-J, following both intrathecal and intravenous administration of AAV9-ARSA. Cytokine profiling analysis of the minipigs’ CSF revealed an increase in IL-1ra levels following both intrathecal and intravenous administration of AAV9-ARSA. IL-1ra is induced in response to various forms of stress, such as excess excitatory neurotransmitters during seizures, infections and inflammations, and neurotrauma [60]. It can be synthesized in the CNS by neurons, microglia, and infiltrating macrophages [61]. The increase in IL-1ra levels observed in our study could be due to the weekly lumbar punctures performed for CSF sampling, which may have caused inflammation and increased IL-1ra levels to provide an anti-inflammatory response. Pathomorphological analysis did not reveal any abnormalities, and although it was not performed for the liver, biochemical analysis showed no evidence of hepatotoxicity.
We have demonstrated that both intravenous and intrathecal administration of AAV9-ARSA into minipigs results in an increase in enzymatic activity in the CNS. Intrathecal administration leads to transduction of CNS cells and expression of a functionally active ARSA enzyme. Intravenous administration, on the other hand, results in overexpression in the cerebellum, suggesting virus penetration into the CNS from the periphery. While the increase in enzymatic activity of ARSA in various parts of the CNS following intravenous administration of AAV9-ARSA has been demonstrated, immunocytochemical analysis did not confirm these findings. In addition, intravenous administration of AAV9-ARSA resulted in transduction of spinal ganglia neurons in the PNS, which was not observed with intrathecal administration of the same construct. This suggests that intravenous delivery of AAV9 may be useful for the treatment of PNS, but it requires higher doses of the vector compared to intrathecal administration, which is more invasive but uses lower doses.
4. Materials and Methods
4.1. Genetic Construct Design and Analysis
The OptimumGene algorithm was employed for the optimization of the ARSA gene codon composition, taking into consideration various factors that could potentially affect gene expression levels. These factors include codon shift, GC composition, CpG dinucleotide content, mRNA secondary structure, tandem repeats, restriction sites interfering with cloning, premature polyadenylation sites, and additional minor ribosome binding sites. The nucleotide sequence of the CDS mRNA of the human ARSA gene (GeneBank #NM_001085425.3, 1530 bp) was utilized as a matrix for codon optimization. Following codon optimization, the CDS mRNA of the ARSA gene was cloned into the pAAV-MCS plasmid vector (Agilent Technologies, USA) using recombinase at EcoRI/BamHI restriction sites. Codon optimization, de novo synthesis of the CDS mRNA nucleotide sequence of the ARSA gene, and its cloning into the pAAV-MCS plasmid vector were performed by GenScript (USA). The correct assembly of the genetic construct was verified through restriction analysis using BamHI restrictase (#ER0051, Thermo Fisher Scientific Inc., USA) and sequencing.
4.2. Production of preparative amounts of plasmid constructs required for AAV assembly
Escherichia coli TOP10 strain (Invitrogen, USA) was utilized for generating preparative amounts of plasmids through transformation. Cells were incubated in LS-LB medium without antibiotics. Competent cells were prepared using the CaCl2 method. Genetic transformation of competent cells was performed using heat shock, after which the transformed bacterial cells were incubated on a selective medium containing ampicillin. Plasmid DNA (pAAV-ARSA, pAAV-RC, and pHelper) was isolated from the resulting bacterial biomass (GeneJET Plasmid Miniprep Kit, #K0502, Thermo Fisher Scientific Inc., USA).
4.3. Preparation and purification of recombinant AAV
AAV viruses were produced using the standard co-transfection of three plasmids into AAV293 cells via the calcium phosphate method. AAV293 cells were cultured at 37°C in humid conditions with 5% CO2 in complete DMEM medium (PanEco, Russia), containing 10% fetal calf serum, L-glutamine, and 1% antibiotics such as penicillin and streptomycin. Cells were centrifuged 72 hours post-transfection, after which lysis buffer (NaCl, Tris-HCl pH 8.5, MgCl2, dH2O), DNase (Benzonase® Nuclease, Sigma-Aldrich, USA), and 25% sodium deoxycholate were added to the pellet. The lysates were purified and subjected to an iodixanol density gradient of 60%, 40%, 25%, and 15%. In the final stage of virus purification, a concentrator suitable for 50 kDa proteins (Vivaspin 20, membrane 50 kDa, Sartorius, UK) was utilized. Viral titer was determined through quantitative PCR using primers (forward 5’-3’ - GGAACCCCTAGTGATGGAGTT, reverse 5’-3’ - CGGCCTCAGTGAGCGA) and a probe targeting ITRs (5’-3’ (FAM) CACTCCCTCTCTGCGCGCTCG (BBQ).
4.4. Determination of the overall purity of virus particles
Overall purity of viral particles was determined by sodium dodecyl sulfate–polyacrylamide gel protein electrophoresis (SDS-PAGE). A polyacrylamide gel was prepared with an acrylamide concentration gradient (4% for the concentrating gel, and 10% for the separating gel). AAV9-ARSA was incubated at 70°C for 15 minutes prior to loading onto the gel. Protein marker (Thermo Scientific™, Cat. No. 26616) and the sample were added to their corresponding wells, and electrophoresis was performed at a constant voltage of 200 V.
After electrophoresis, the gel was stained with Coomassie blue (Thermo Fisher, Cat. No. LC6065) for 2-3 hours with gentle shaking. The gel was then washed in a solution of 40% methanol and 10% acetic acid until protein bands were visible. Finally, the image was captured using the ChemiDocXRS+ gel documentation system (BioRad, USA).
4.5. Western blot analysis
For transfection of HEK293T with recombinant AAV9-ARSA, cells were seeded in 6-well plate 24 hours prior. A mixture was prepared considering a multiplicity of infection (MOI) of 100 (100 viral particles per cell), with protamine sulfate added to achieve a final concentration of 10 μg/ml. Cell growth medium was replaced with the prepared transduction mixture. After 6 hours, the medium was switched to fresh. Transgene expression was assessed using Western blot analysis.
Western blot analysis was performed using the standard Laemmli method under denaturing conditions (SDS-PAGE). Protein transfer from gel to membrane (PVDF) was conducted using a Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell (BioRad, USA). Non-specific binding was prevented by incubating the membrane in blocking buffer. Membranes were then incubated with primary rabbit anti-ARSA monoclonal antibodies (#PAG619Hu01, Cloud-Clone Corp., USA), followed by incubation with secondary antibodies (polyclonal goat antibodies to human immunoglobulin G conjugated with horseradish peroxidase, Sigma, #A6154, USA). Visualization of the immunoprecipitate was carried out using ECL Western Blotting Substrate (#W1001, Promega, USA). The membrane was examined using the ChemiDocXRS+ gel documentation system (BioRad, USA).
4.6. Animals
All experimental procedures on animals were reviewed and approved by the local ethics committee of Kazan Federal University (Protocol No. 23, June 30, 2020). Healthy female minipigs at the age of 4 months (weighing 9-12 kg) were used in the study. Experimental animals (15 minipigs) were divided into 3 groups: (1) intrathecal administration of AAV9-ARSA, at a dose of 1 × 1012 genomic copies (gc)/kg (n=5); (2) intravenous administration of AAV9-ARSA, at a dose of 3.77 × 1013 gc/kg (n=5); (3) control group with no AAV9-ARSA administration (n=5). Animals were housed in specialized animal care facilities of Kazan State Academy of Veterinary Medicine named after N.E. Bauman (KGAVM), under the supervision of qualified personnel. Animal euthanasia was performed in strict compliance with the recommendations for euthanasia of experimental animals of the European Commission.
4.7. Material Sampling
Prior to the virus injection, CSF and blood samples were taken as time point zero. Later, CSF and blood samples were taken 7, 14, 21, 28, 35 days following the virus injection, in order to analyze ARSA enzymatic activity in dynamics. On the 35th day after the virus injection, experimental animals were euthanized. To determine ARSA expression levels, quantitative RT-PCR was performed, as well as a test for ARSA enzymatic activity in homogenates of various parts of the nervous system.
Parts of the following organs were sampled from each animal: cerebellum, occipital lobe of the brain, cervical (C6-7), thoracic (Th6-7) and lumbar (L2-3) sections of the spinal cord with spinal roots and ganglia, lateral femoral cutaneous nerve, heart (left ventricle), lungs, kidneys, spleen, and liver. Each of the organ parts was placed in a 10% formalin solution. After 48 hours of fixation, each organ part was divided into 2 fragments; one was embedded in paraffin, and the other was transferred consecutively into 15%, and then 30% sucrose. Samples obtained from minipigs’ nervous tissue (in 30% sucrose) were placed in tissue freezing medium (Tissue-Tek O.C.T. Compound, Sakura). On a Microm HM 560 cryostat (Thermo Scientific), transverse or sagittal sections of the studied nervous system organs 20 μm thick were obtained and used for subsequent immunofluorescent analysis. Samples of internal organs embedded in paraffin were cut on a Minus S700A (RWD) microtome 5–7 µm thick, and then stained with hematoxylin and eosin.
4.8. Determination of ARSA enzymatic activity
To determine ARSA enzymatic activity in organ homogenates, total protein concentration in samples was determined using the Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific Inc., USA). Samples were normalized according to total protein concentration. Next, 50 µl of cell lysate or tissue homogenate sample was incubated with a solution of nitrocatechol sulfate substrate (0.01 M p-Nitrocatechol sulfate dipotassium salt (#N7251, Sigma-Aldrich), 0.5 M sodium acetate, 5 × 10-4 Na4P2O7, 10% sodium chloride, pH = 5) for 1 hour at 37°C. The reaction was then stopped by adding 1 N sodium hydroxide. Sulfatase dilutions (#S9626, Sigma-Aldrich) were used as standards, and optical density was measured at a wavelength of 515 nm.
4.9. Quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from animal organ parts using the TRIzol Reagent (Invitrogen, USA) following the manufacturer’s protocol. Primers and probes specific to human ARSA were designed using the GenScript Online Real-time PCR (TaqMan) Primer Design Tool (GenScript, USA) and synthesized by Evrogen (Russia). The primer sequences were as follows: forward 5’-CAAGGTACATGGCATTCGCA-3’ and reverse 5’-CTGTGGATAGTGGGTGTGGT-3’. The probe sequence was 5’-CCTGCCGCTGTGCATCTGCCA-3’ labeled with FAM (6-carboxyfluorescein) on the 5’ end and BHQ-1 (Black Hole Quencher 1) on the 3’ end. Isolated RNA was reverse transcribed into cDNA using the GoScript™ Reverse Transcription System (Promega, USA) according to the manufacturer’s instructions. Real-time PCR based on TaqMan was performed in 96-well MicroAmp plates (BioRad, USA), using a qPCR mix that contained 1 µl of cDNA template, 0.3 µl of primer and probe mixture (with a final concentration of 300 nM for each primer), 4.7 µl of MilliQ H2O, and 4 µl of 10x TaqMan-buffer (Lytech, Russia), with a final volume of 10 µl. PCR amplification was carried out using the CFX96 Touch™ Real-Time PCR Detection System (BioRad, USA) under the following temperature cycling conditions: preheating at 95°C for 3 min, 45 denaturation cycles at 95°C for 10 sec, and annealing at 55°C for 30 sec.
4.10. Biochemical blood analysis and cytokine profile analysis
To evaluate safety, biochemical blood analysis and cytokine profiling were performed. Whole blood was collected from animals into test tubes with gel and blood clotting activator. The samples were then centrifuged at 1900 rpm for 20 minutes to separate the serum, and levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, and creatinine were measured using a ChemWell 2900 biochemical analyzer (USA).
Porcine serum and CSF samples were analyzed using the MILLIPLEX MAP Porcine Cytokine/Chemokine Magnetic Bead Panel - Immunology Multiplex Assay (PCYTMG-23K-13PX, Merck), which included GM-CSF (colony-stimulating factor 2 (granulocyte-macrophage)), IFNγ (interferon gamma), IL-1α (interleukin 1 alpha), IL-1β, IL-1ra (IL-1 antagonist), IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-18, and TNF-α (tumor necrosis factor-alpha). For each sample, 50 microliters were used to determine analyte concentrations. Data were analyzed using a Luminex 200 analyzer with MasterPlex CT control software and MasterPlex QT analysis software (MiraiBio division of Hitachi Software, USA).
4.11. Immunofluorescence Analysis
To analyze the tissue expression of the ARSA protein, cryostat transverse or sagittal sections of central (CNS) and peripheral (PNS) nervous system organs obtained from minipigs were used. For immunofluorescent labeling, sections were blocked with 5% normal goat serum, then incubated with primary antibody (anti-ARSA, Cloud-Clone), and subsequently with secondary antibodies (Alexa 546, Abcam). Following successive washes in PBS, sections were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (10 μg/ml in PBS, Sigma-Aldrich) to visualize nuclei. Sections were mounted with medium (ImmunoHistoMount, Santa Cruz) and examined using a LSM 700 confocal scanning microscope (Carl Zeiss).
4.12. Statistical Analysis
Obtained data were analyzed using GraphPad Prism 8 software (GraphPad Software). The Shapiro-Wilk test and one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test were applied to determine statistically significant differences, which were designated as * for p < 0.05, ** for p < 0.01, *** for p < 0.001, and **** for p < 0.0001.
Author Contributions
AM and AS contributed to the investigation and writing of the original draft. VS, AR contributed to the reviewing, and editing of the manuscript. YM, AK, DS, SI contributed to the investigation. All authors have read and agreed to the published version of the manuscript.
Funding
The study was funded by the subsidy allocated to Kazan Federal University for the state assignment No FZSM-2023-0011 in the sphere of scientific activities.
Institutional Review Board Statement
The study was conducted in accordance with the local ethics committee of Kazan Federal University (Protocol No. 23, June 30, 2020).
Acknowledgments
We thank the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, IGB RAS for rAAV production and purification. The rAAV production was performed using the equipment of IGB RAS facilities supported by the Ministry of Science and Higher Education of the Russian Federation. This paper has been supported by the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030).
Conflicts of interest
The authors declare no conflict of interest.
References
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Mullagulova, A.I.; Kitaeva, K.V.; Allegrucci, C.; Rizvanov, A.A. Metachromatic leukodystrophy: Diagnosis, modeling, and treatment approaches. Frontiers in Medicine 2020, 7, 576221. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ospina, N. Arylsulfatase a deficiency. In Genereviews((r)), Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Stephens, K.; Amemiya, A., Eds. Seattle (WA), 1993.
- Wanner, M.R.; Karmazyn, B.; Fan, R. Multidetector ct diagnosis of massive hemobilia due to gallbladder polyposis in a child with metachromatic leukodystrophy. Pediatr Radiol 2015, 45, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Almarzooqi, S.; Quadri, A.; Albawardi, A. Gallbladder polyps in metachromatic leukodystrophy. Fetal and pediatric pathology 2018, 37, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sun, Z.; Ezekian, B.; Schooler, G.R.; Prasad, V.K.; Kurtzberg, J.; Rice, H.E.; Tracy, E.T. Gallbladder abnormalities in children with metachromatic leukodystrophy. J Surg Res 2017, 208, 187–191. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Ranganathan, S. Pathology of the gallbladder in a child with metachromatic leukodystrophy. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 2015, 18, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Kurian, J.J.; Jacob, T.J.K. An unusual presentation of gall bladder papillomatosis in association with metachromatic leukodystrophy. BMJ Case Rep 2018, 2018. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Bugiani, M. Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 2017, 134, 351–382. [Google Scholar] [CrossRef]
- Hyde, T.M.; Ziegler, J.C.; Weinberger, D.R. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Archives of neurology 1992, 49, 401–406. [Google Scholar] [CrossRef]
- Ali Mallick, M.S.; Godil, A.; Khetpal, A.; Rizvi, A.H.; Khan, F. Infantile metachromatic leukodystrophy in an 18 month old girl. J Pak Med Assoc 2016, 66, 1197–1200. [Google Scholar]
- Liaw, H.R.; Lee, H.F.; Chi, C.S.; Tsai, C.R. Late infantile metachromatic leukodystrophy: Clinical manifestations of five taiwanese patients and genetic features in asia. Orphanet journal of rare diseases 2015, 10, 144. [Google Scholar] [CrossRef]
- Barkovich, A.J. Concepts of myelin and myelination in neuroradiology. AJNR Am J Neuroradiol 2000, 21, 1099–1109. [Google Scholar] [PubMed]
- Thibert, K.A.; Raymond, G.V.; Tolar, J.; Miller, W.P.; Orchard, P.J.; Lund, T.C. Cerebral spinal fluid levels of cytokines are elevated in patients with metachromatic leukodystrophy. Sci Rep 2016, 6, 24579. [Google Scholar] [CrossRef]
- Koski, C.L.; Vanguri, P.; Shin, M.L. Activation of the alternative pathway of complement by human peripheral nerve myelin. J Immunol 1985, 134, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Beerepoot, S.; Nierkens, S.; Boelens, J.J.; Lindemans, C.; Bugiani, M.; Wolf, N.I. Peripheral neuropathy in metachromatic leukodystrophy: Current status and future perspective. Orphanet journal of rare diseases 2019, 14, 240. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.; Frazier, S.B.; Clark, M.; Childers, A.; Pruthi, S.; Wenger, D.A.; Duis, J. A closer look at arsa activity in a patient with metachromatic leukodystrophy. Molecular genetics and metabolism reports 2019, 19, 100460. [Google Scholar] [CrossRef] [PubMed]
- Fluharty, A.L.; Meek, W.E.; Kihara, H. Pseudo arylsulfatase a deficiency: Evidence for a structurally altered enzyme. Biochem Biophys Res Commun 1983, 112, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Gieselmann, V.; Polten, A.; Kreysing, J.; von Figura, K. Arylsulfatase a pseudodeficiency: Loss of a polyadenylylation signal and n-glycosylation site. Proc Natl Acad Sci U S A 1989, 86, 9436–9440. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.A.; Khaliq, S.; Amar, A.; Mahmood, S. Metachromatic leukodystrophy (mld): A pakistani family with novel arsa gene mutation. Journal of molecular neuroscience : MN 2017, 63, 84–90. [Google Scholar] [CrossRef]
- Alam, S.T.; Akhter, S.; Rahman, M.M.; Islam, K.A.; Siddique, R.; Helaly, L.; Ahmed, S. A rare case of metachromatic leukodystrophy confirmed by arylsulfatase a. Mymensingh medical journal : MMJ 2015, 24, 864–867. [Google Scholar]
- van Rappard, D.F.; de Vries, A.L.C.; Oostrom, K.J.; Boelens, J.J.; Hollak, C.E.M.; van der Knaap, M.S.; Wolf, N.I. Slowly progressive psychiatric symptoms: Think metachromatic leukodystrophy. Journal of the American Academy of Child and Adolescent Psychiatry 2018, 57, 74–76. [Google Scholar] [CrossRef]
- Brown, T.M.; Martin, S.; Fehnel, S.E.; Deal, L.S. Development of the impact of juvenile metachromatic leukodystrophy on physical activities scale. Journal of patient-reported outcomes 2017, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Kumperscak, H.G.; Paschke, E.; Gradisnik, P.; Vidmar, J.; Bradac, S.U. Adult metachromatic leukodystrophy: Disorganized schizophrenia-like symptoms and postpartum depression in 2 sisters. Journal of psychiatry & neuroscience : JPN 2005, 30, 33–36. [Google Scholar]
- Black, D.N.; Taber, K.H.; Hurley, R.A. Metachromatic leukodystrophy: A model for the study of psychosis. The Journal of neuropsychiatry and clinical neurosciences 2003, 15, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Espejo, L.M.; de la Espriella, R.; Hernandez, J.F. [metachromatic leukodystrophy. Case presentation]. Revista colombiana de psiquiatria 2017, 46, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Bostantjopoulou, S.; Katsarou, Z.; Michelakaki, H.; Kazis, A. Seizures as a presenting feature of late onset metachromatic leukodystrophy. Acta neurologica Scandinavica 2000, 102, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Boucher, A.A.; Miller, W.; Shanley, R.; Ziegler, R.; Lund, T.; Raymond, G.; Orchard, P.J. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: The largest single-institution cohort report. Orphanet journal of rare diseases 2015, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Groeschel, S.; Kuhl, J.S.; Bley, A.E.; Kehrer, C.; Weschke, B.; Doring, M.; Bohringer, J.; Schrum, J.; Santer, R.; Kohlschutter, A.; et al. Long-term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol 2016, 73, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Sevin, C.; Roujeau, T.; Cartier, N.; Baugnon, T.; Adamsbaum, C.; Piraud, M.; Martino, S.; Mouiller, P.; Couzinie, C.; Bellesme, C.; et al. , Intracerebral gene therapy in children with metachromatic leukodystrophy: Results of a phase i/ii trial. Molecular genetics and metabolism 2018, 123, S129–S129. [Google Scholar] [CrossRef]
- Miyake, N.; Miyake, K.; Asakawa, N.; Yamamoto, M.; Shimada, T. Long-term correction of biochemical and neurological abnormalities in mld mice model by neonatal systemic injection of an aav serotype 9 vector. Gene Ther 2014, 21, 427–433. [Google Scholar] [CrossRef]
- Koc, O.N.; Day, J.; Nieder, M.; Gerson, S.L.; Lazarus, H.M.; Krivit, W. Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (mld) and hurler syndrome (mps-ih). Bone Marrow Transplant 2002, 30, 215–222. [Google Scholar] [CrossRef]
- Meuleman, N.; Vanhaelen, G.; Tondreau, T.; Lewalle, P.; Kwan, J.; Bennani, J.; Martiat, P.; Lagneaux, L.; Bron, D. Reduced intensity conditioning haematopoietic stem cell transplantation with mesenchymal stromal cells infusion for the treatment of metachromatic leukodystrophy: A case report. Haematologica 2008, 93, e11–13. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Bellettato, C.M.; Scarpa, M. Possible strategies to cross the blood-brain barrier. Italian journal of pediatrics 2018, 44, 131. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current strategies for brain drug delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Zingg, B.; Chou, X.L.; Zhang, Z.G.; Mesik, L.; Liang, F.X.; Tao, H.W.; Zhang, L.I. Aav-mediated anterograde transsynaptic tagging: Mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 2017, 93, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, D.F.; Kreuz, S.; Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of aav serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. Plos One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Sevin, C.; Benraiss, A.; Van Dam, D.; Bonnin, D.; Nagels, G.; Verot, L.; Laurendeau, I.; Vidaud, M.; Gieselmann, V.; Vanier, M.; et al. Intracerebral adeno-associated virus-mediated gene transfer in rapidly progressive forms of metachromatic leukodystrophy. Hum Mol Genet 2006, 15, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Cearley, C.N.; Wolfe, J.H. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and rh10 in the mouse brain. Mol Ther 2006, 13, 528–537. [Google Scholar] [CrossRef]
- Piguet, F.; Sondhi, D.; Piraud, M.; Fouquet, F.; Hackett, N.R.; Ahouansou, O.; Vanier, M.T.; Bieche, I.; Aubourg, P.; Crystal, R.G.; et al. Correction of brain oligodendrocytes by aavrh.10 intracerebral gene therapy in metachromatic leukodystrophy mice. Hum Gene Ther 2012, 23, 903–914. [Google Scholar] [CrossRef]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields aav variants for widespread gene transfer to the adult brain. Nature Biotechnology 2016, 34, 204–+. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sanchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered aavs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nature Neuroscience 2017, 20, 1172–+. [Google Scholar] [CrossRef] [PubMed]
- Audouard, E.; Oger, V.; Meha, B.; Cartier, N.; Sevin, C.; Piguet, F. Complete correction of brain and spinal cord pathology in metachromatic leukodystrophy mice. Front Mol Neurosci 2021, 14, 677895. [Google Scholar] [CrossRef] [PubMed]
- Matzner, U.; Hartmann, D.; Lullmann-Rauch, R.; Coenen, R.; Rothert, F.; Mansson, J.E.; Fredman, P.; D’Hooge, R.; De Deyn, P.P.; Gieselmann, V. Bone marrow stem cell-based gene transfer in a mouse model for metachromatic leukodystrophy: Effects on visceral and nervous system disease manifestations. Gene Ther 2002, 9, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Capotondo, A.; Fasano, S.; del Carro, U.; Marchesini, S.; Azuma, H.; Malaguti, M.C.; Amadio, S.; Brambilla, R.; Grompe, M.; et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest 2006, 116, 3070–3082. [Google Scholar] [CrossRef]
- Tucci, F.; Scaramuzza, S.; Aiuti, A.; Mortellaro, A. Update on clinical ex vivo hematopoietic stem cell gene therapy for inherited monogenic diseases. Mol Ther 2021, 29, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New approaches to tay-sachs disease therapy. Front Physiol 2018, 9, 1663. [Google Scholar] [CrossRef] [PubMed]
- Zingg, B.; Chou, X.L.; Zhang, Z.G.; Mesik, L.; Liang, F.; Tao, H.W.; Zhang, L.I. Aav-mediated anterograde transsynaptic tagging: Mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 2017, 93, 33–47. [Google Scholar] [CrossRef]
- Kantor, B.; Bailey, R.M.; Wimberly, K.; Kalburgi, S.N.; Gray, S.J. Methods for gene transfer to the central nervous system. Adv Genet 2014, 87, 125–197. [Google Scholar]
- Liu, D.; Zhu, M.; Zhang, Y.; Diao, Y. Crossing the blood-brain barrier with aav vectors. Metab Brain Dis 2021, 36, 45–52. [Google Scholar] [CrossRef]
- Saraiva, J.; Nobre, R.J.; Pereira de Almeida, L. Gene therapy for the cns using aavs: The impact of systemic delivery by aav9. J Control Release 2016, 241, 94–109. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular aav9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Li, H.; Cao, C.; Sikoglu, E.M.; Denninger, A.R.; Su, Q.; Eaton, S.; Liso Navarro, A.A.; Xie, J.; Szucs, S.; et al. A single intravenous raav injection as late as p20 achieves efficacious and sustained cns gene therapy in canavan mice. Mol Ther 2013, 21, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Bell, P.; Gurda, B.L.; Wang, Q.; Louboutin, J.P.; Zhu, Y.; Bagel, J.; O’Donnell, P.; Sikora, T.; Ruane, T.; et al. Intrathecal gene therapy corrects cns pathology in a feline model of mucopolysaccharidosis i. Mol Ther 2014, 22, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Hinderer, C.; Buza, E.L.; Louboutin, J.P.; Jahan, T.; Bell, P.; Chichester, J.A.; Tarantal, A.F.; Wilson, J.M. Safe and sustained expression of human iduronidase after intrathecal administration of adeno-associated virus serotype 9 in infant rhesus monkeys. Hum Gene Ther 2019, 30, 957–966. [Google Scholar] [CrossRef]
- Bailey, R.M.; Rozenberg, A.; Gray, S.J. Comparison of high-dose intracisterna magna and lumbar puncture intrathecal delivery of aav9 in mice to treat neuropathies. Brain Res 2020, 1739, 146832. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.J.; Dykstra, J.A.; Riedl, M.S.; Kitto, K.F.; Belur, L.R.; McIvor, R.S.; Elde, R.P.; Fairbanks, C.A.; Vulchanova, L. Biodistribution of adeno-associated virus serotype 9 (aav9) vector after intrathecal and intravenous delivery in mouse. Front Neuroanat 2014, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, C.; Wang, W.; Li, J.; Huang, C.; Zheng, X.; Liu, Z.; Song, X.; Chen, Y.; Gao, J.; et al. Intravenous aav9 administration results in safe and widespread distribution of transgene in the brain of mini-pig. Front Cell Dev Biol 2022, 10, 1115348. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human smn. Hum Gene Ther 2018, 29, 285–298. [Google Scholar] [CrossRef]
- Yue, Y.; Shang, C.; Dong, H.; Meng, K. Interleukin-1 in cerebrospinal fluid for evaluating the neurological outcome in traumatic brain injury. Biosci Rep 2019, 39. [Google Scholar] [CrossRef]
- Bartfai, T.; Sanchez-Alavez, M.; Andell-Jonsson, S.; Schultzberg, M.; Vezzani, A.; Danielsson, E.; Conti, B. Interleukin-1 system in cns stress: Seizures, fever, and neurotrauma. Ann N Y Acad Sci 2007, 1113, 173–177. [Google Scholar] [CrossRef]
Figure 1.
Characterization and analysis of the expression plasmid vector pAAV-ARSA. (a) pAAV-ARSA plasmid map; (b) Analysis of restriction results for plasmid DNA, M – GeneRullerTM DNA Ladder 1kb marker, 1 – pAAV-ARSA restriction with BamHI enzyme (Cat. No. ER0051, Thermo Fisher Scientific Inc., USA), 2 – pAAV-ARSA cDNA.
Figure 1.
Characterization and analysis of the expression plasmid vector pAAV-ARSA. (a) pAAV-ARSA plasmid map; (b) Analysis of restriction results for plasmid DNA, M – GeneRullerTM DNA Ladder 1kb marker, 1 – pAAV-ARSA restriction with BamHI enzyme (Cat. No. ER0051, Thermo Fisher Scientific Inc., USA), 2 – pAAV-ARSA cDNA.
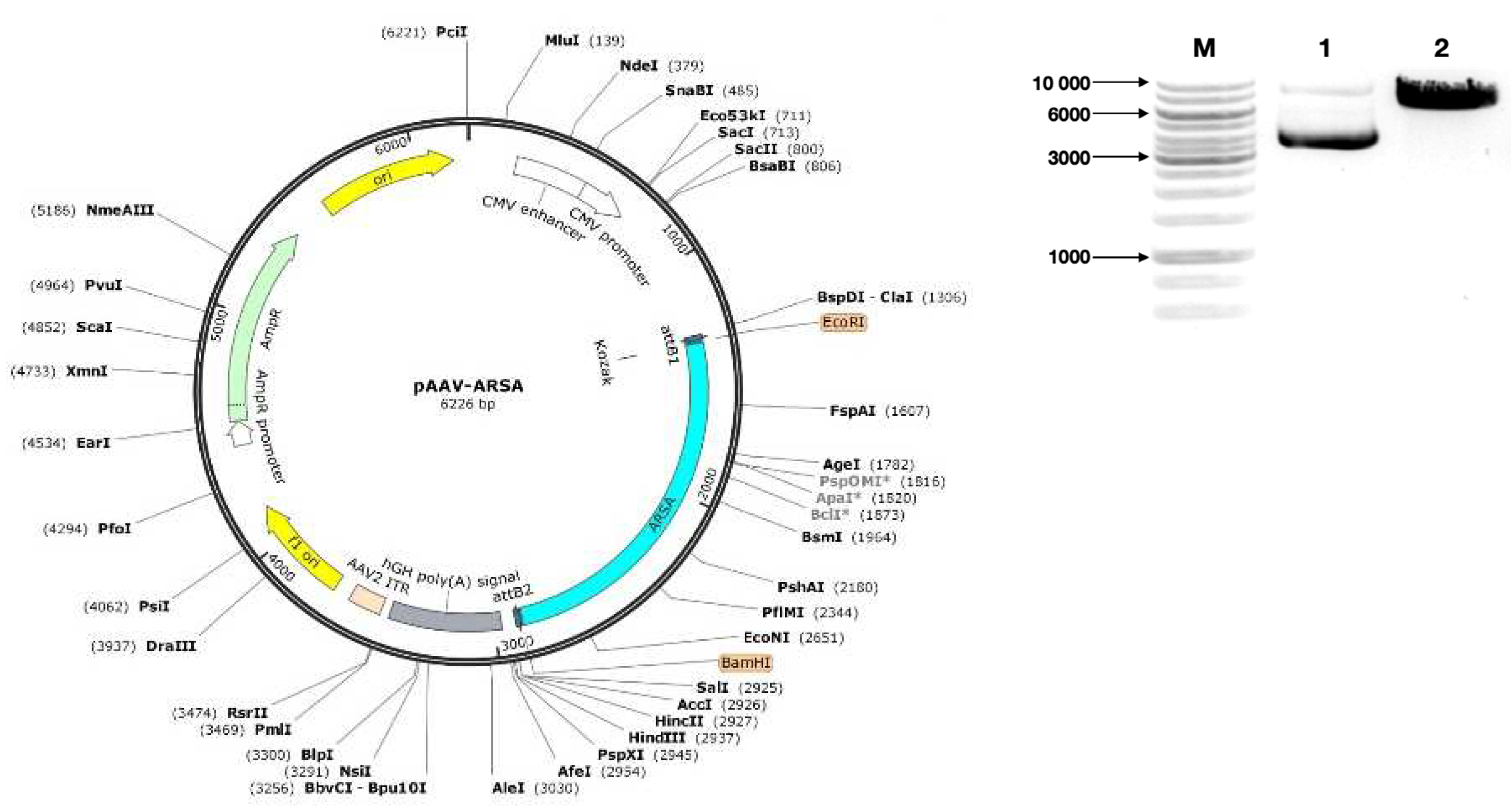
Figure 2.
(a) Analysis of ARSA enzymatic activity in HEK293T cell lysate after pAAV-ARSA transfection; (b) Western blot analysis of ARSA proteins in native and genetically modified HEK293T pAAV-ARSA.
Figure 2.
(a) Analysis of ARSA enzymatic activity in HEK293T cell lysate after pAAV-ARSA transfection; (b) Western blot analysis of ARSA proteins in native and genetically modified HEK293T pAAV-ARSA.
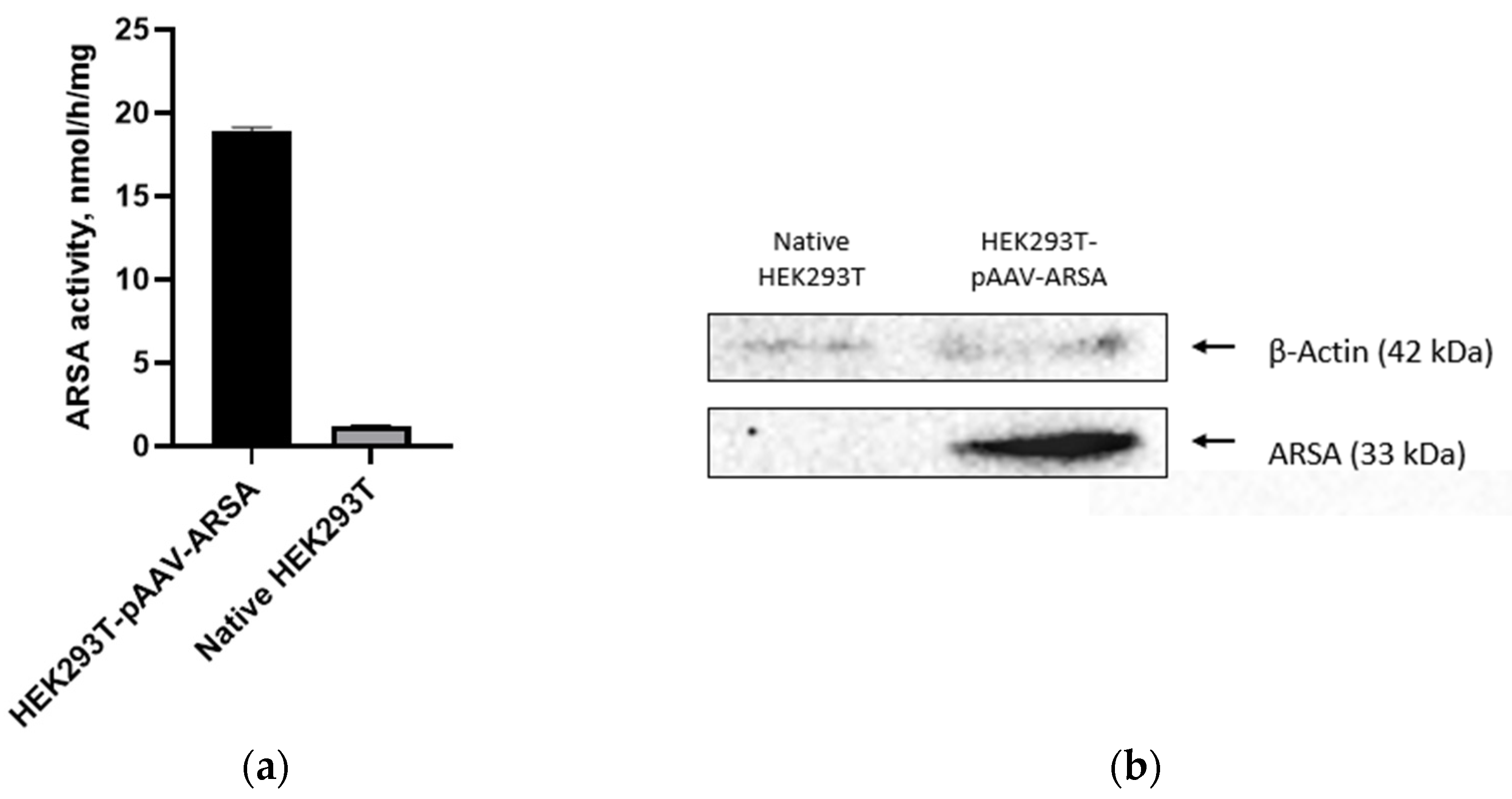
Figure 3.
(a) Analysis of ARSA enzymatic activity in HEK293T cells lysate following AAV9-ARSA transduction. (b) Western blot analysis of ARSA proteins in native and genetically modified HEK293T AAV9-ARSA. (c) Determination of overall purity of AAV-ARSA by SDS-PAGE protein electrophoresis, where VP1, VP2, VP3 are viral proteins.
Figure 3.
(a) Analysis of ARSA enzymatic activity in HEK293T cells lysate following AAV9-ARSA transduction. (b) Western blot analysis of ARSA proteins in native and genetically modified HEK293T AAV9-ARSA. (c) Determination of overall purity of AAV-ARSA by SDS-PAGE protein electrophoresis, where VP1, VP2, VP3 are viral proteins.
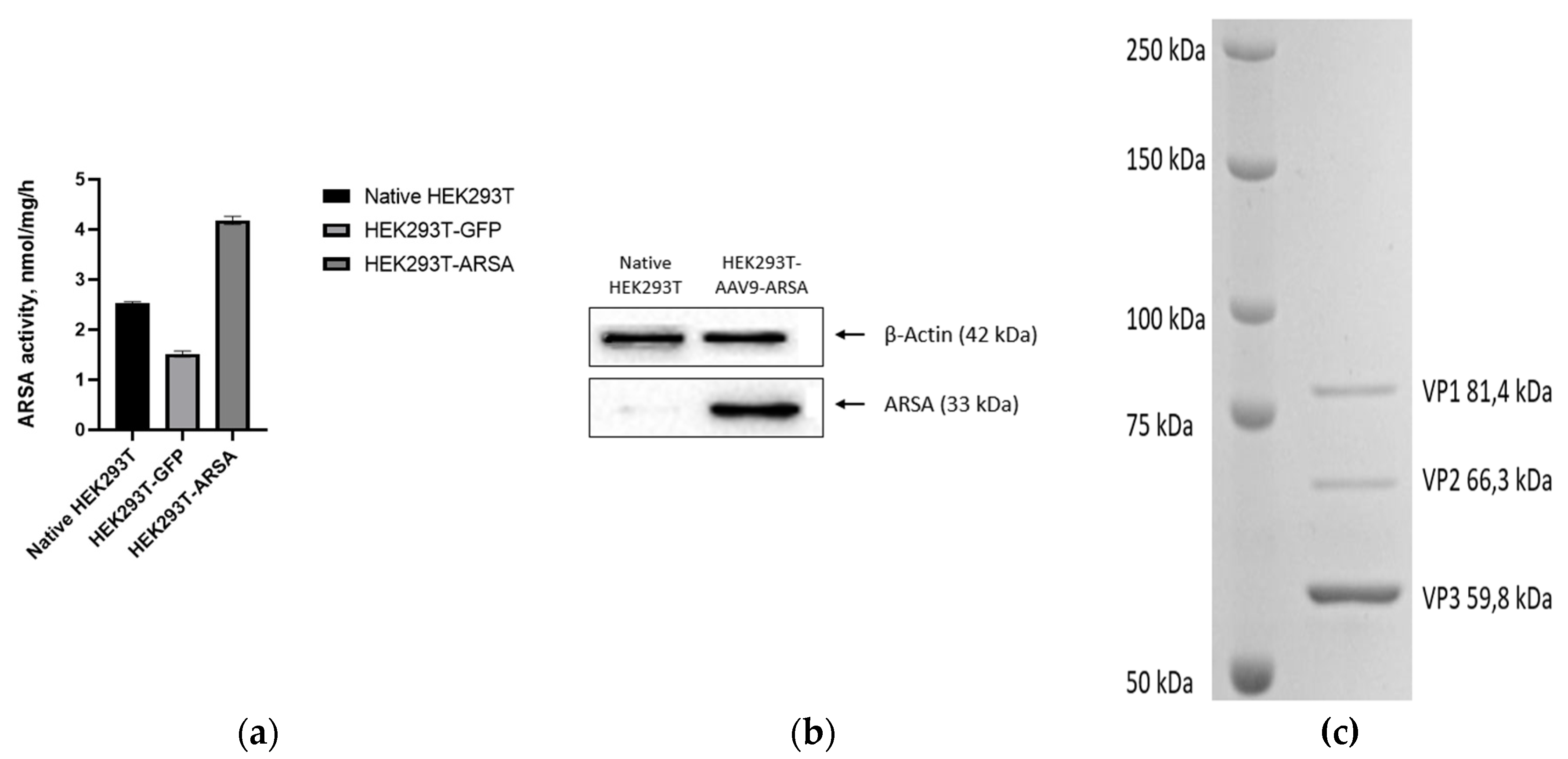
Figure 4.
Analysis of ARSA enzymatic activity in plasma and CSF of minipig gc following AAV9-ARSA administration. Data are presented as mean ± S.D. * – p < 0.05, ** – p < 0.01, *** – p < 0.001. IT AAV9-ARSA – intrathecal administration of AAV9-ARSA; IV AAV9-ARSA – intravenous administration of AAV9-ARSA.
Figure 4.
Analysis of ARSA enzymatic activity in plasma and CSF of minipig gc following AAV9-ARSA administration. Data are presented as mean ± S.D. * – p < 0.05, ** – p < 0.01, *** – p < 0.001. IT AAV9-ARSA – intrathecal administration of AAV9-ARSA; IV AAV9-ARSA – intravenous administration of AAV9-ARSA.
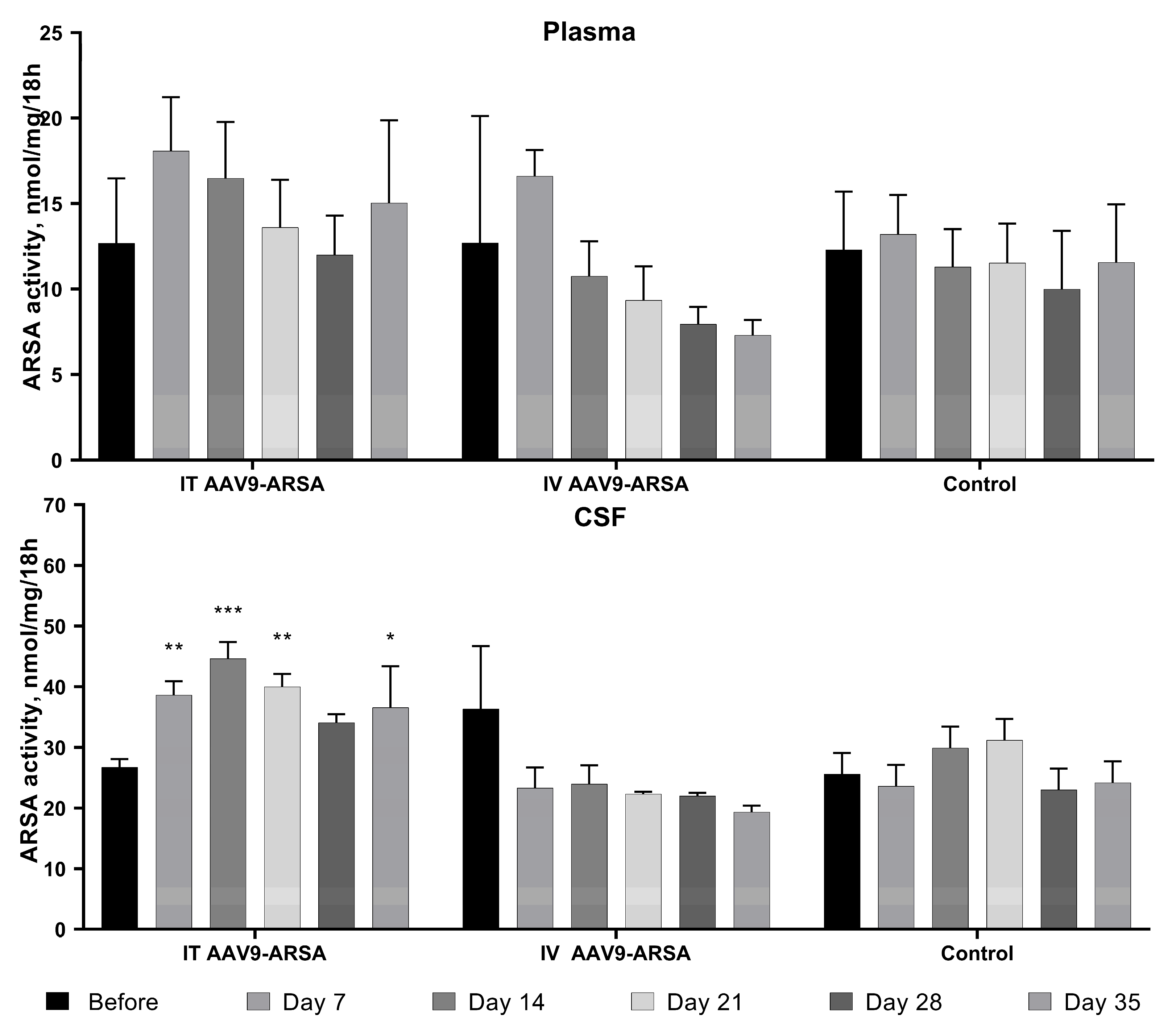
Figure 5.
Analysis of ARSA enzymatic activity in homogenates of various organ parts of the minipigs’ nervous system following administration of AAV9-ARSA. Data are presented as mean ± S.D. * – p < 0.05, ** – p < 0.01. Control – control group of animals; IT AAV9-ARSA – intrathecal administration of AAV9-ARSA; IV AAV9-ARSA – intravenous administration of AAV9-ARSA.
Figure 5.
Analysis of ARSA enzymatic activity in homogenates of various organ parts of the minipigs’ nervous system following administration of AAV9-ARSA. Data are presented as mean ± S.D. * – p < 0.05, ** – p < 0.01. Control – control group of animals; IT AAV9-ARSA – intrathecal administration of AAV9-ARSA; IV AAV9-ARSA – intravenous administration of AAV9-ARSA.

Figure 6.
Copy number of ARSA gene mRNA in different parts of the nervous system of minipigs on the 35th day following AAV9-ARSA administration. Data obtained by quantitative PCR. Data are presented as mean ± S.D. (a) following intrathecal administration of AAV9-ARSA; (b) following intravenous administration of AAV9-ARSA.
Figure 6.
Copy number of ARSA gene mRNA in different parts of the nervous system of minipigs on the 35th day following AAV9-ARSA administration. Data obtained by quantitative PCR. Data are presented as mean ± S.D. (a) following intrathecal administration of AAV9-ARSA; (b) following intravenous administration of AAV9-ARSA.
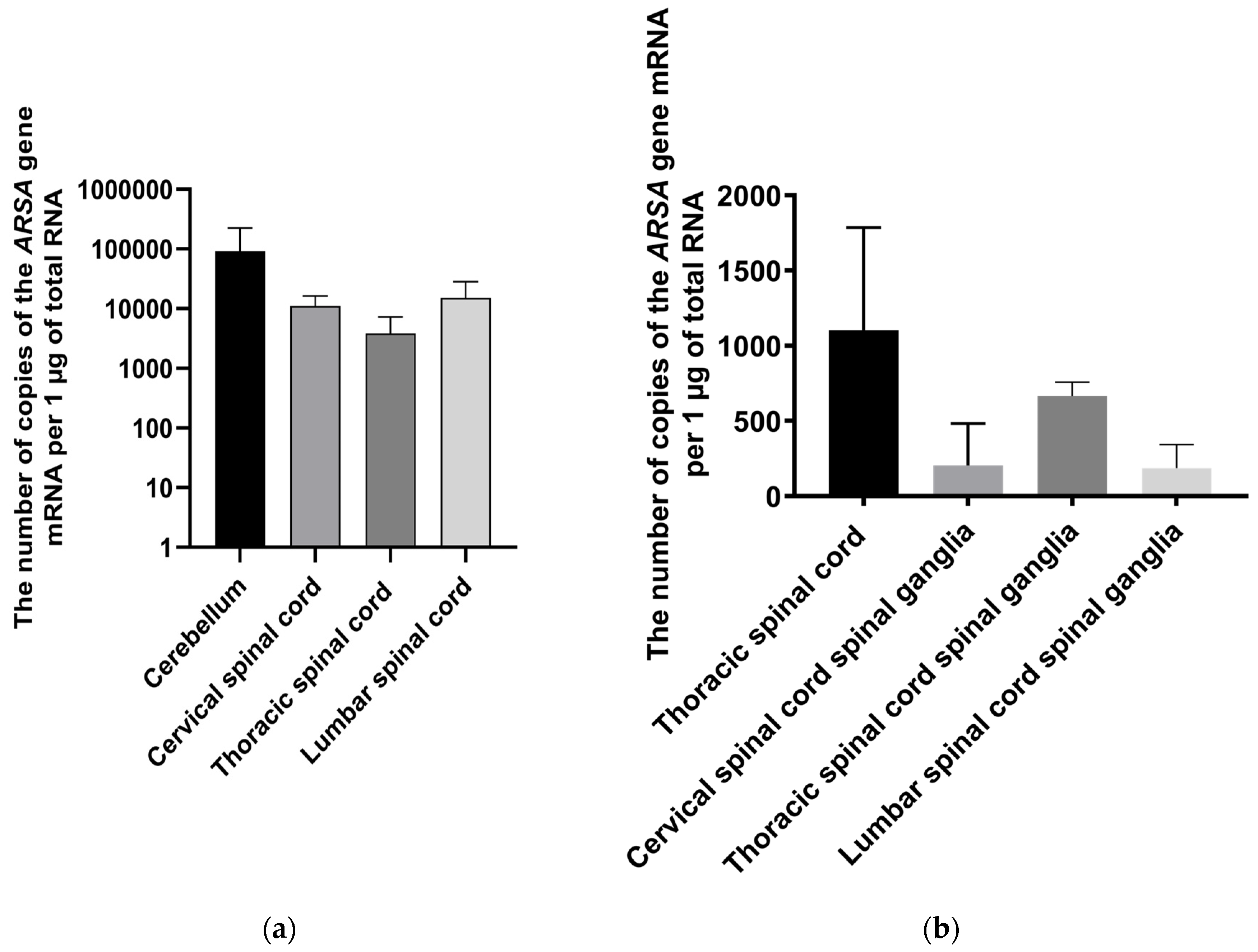
Figure 7.
Biochemical parameters of the minipigs’ blood sera following AAV9-ARSA administration. IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of the AAV9-ARSA vector. * p <0.05, ** p < 0.01.
Figure 7.
Biochemical parameters of the minipigs’ blood sera following AAV9-ARSA administration. IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of the AAV9-ARSA vector. * p <0.05, ** p < 0.01.
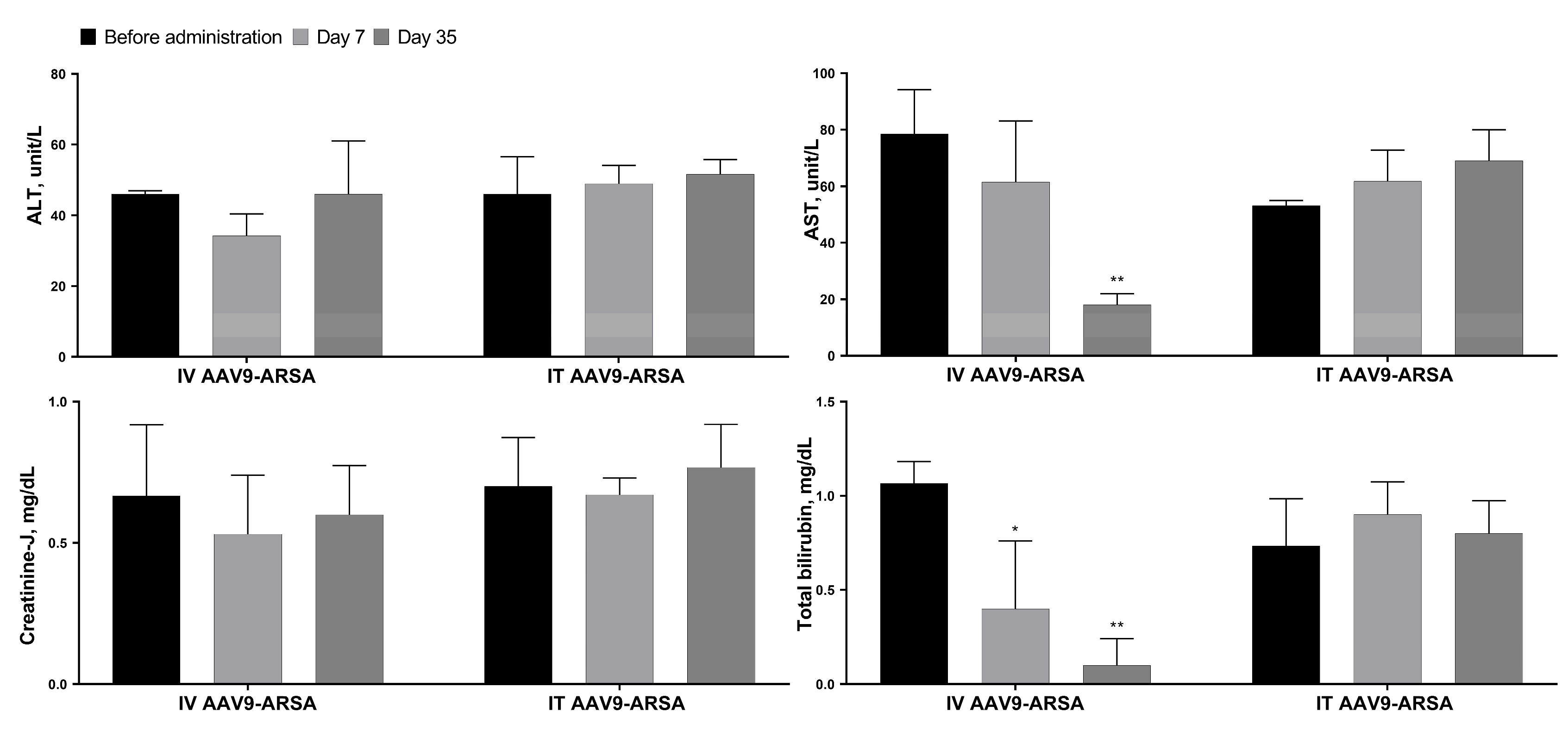
Figure 8.
Analysis of inflammatory cytokines and chemokines levels following intrathecal and intravenous administration of AAV9-ARSA. (a) Cytokine profile of porcine blood serum. (b) Cytokine profile of porcine CSF. IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of AAV9-ARSA.
Figure 8.
Analysis of inflammatory cytokines and chemokines levels following intrathecal and intravenous administration of AAV9-ARSA. (a) Cytokine profile of porcine blood serum. (b) Cytokine profile of porcine CSF. IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of AAV9-ARSA.
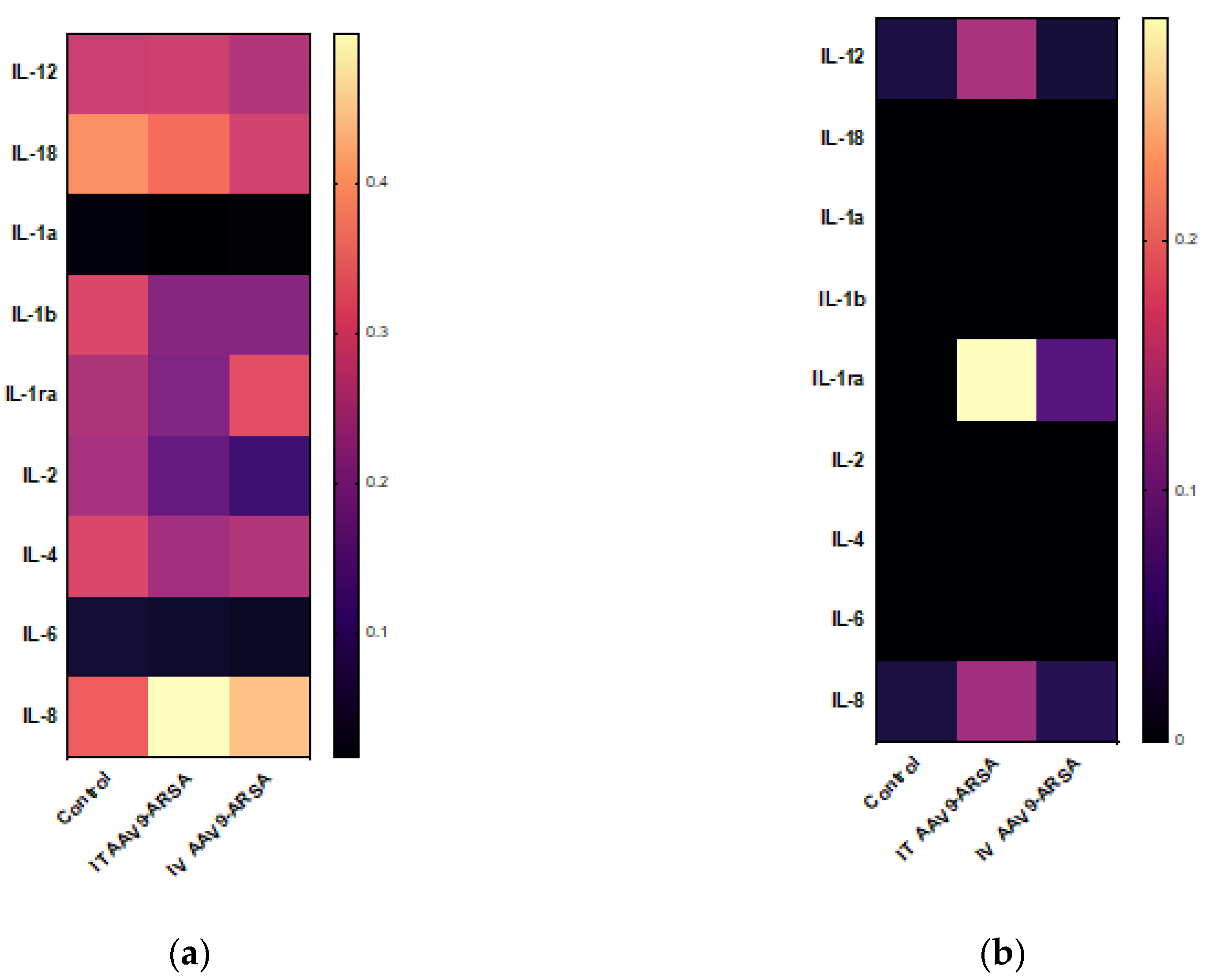
Figure 9.
Pathological analysis of kidney, heart, spleen, and lungs tissues in the control and experimental groups on the 35th day following AAV9-ARSA administration. Staining with hematoxylin and eosin. Control – control group of animals, IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of the AAV9-ARSA vector.
Figure 9.
Pathological analysis of kidney, heart, spleen, and lungs tissues in the control and experimental groups on the 35th day following AAV9-ARSA administration. Staining with hematoxylin and eosin. Control – control group of animals, IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of the AAV9-ARSA vector.
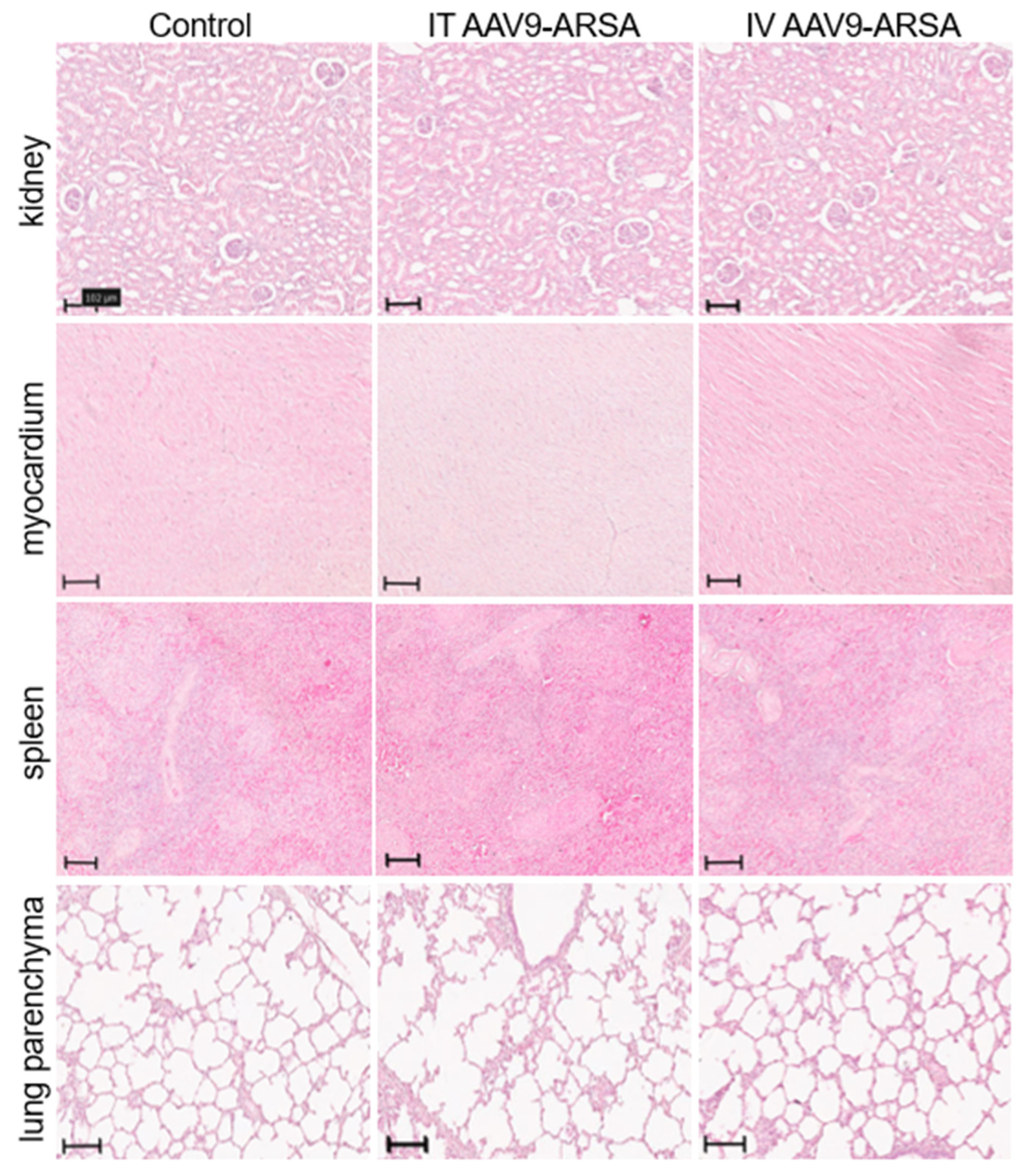
Figure 10.
Assessment of ARSA (yellow) expression in different regions of nervous tissue in the control group of animals (A-F), and in experimental groups 1 and 2 on day 35 after intrathecal (A1-F1), and intravenous (A2-F2) administration of AAV9-ARSA, respectively. Nuclei are stained with DAPI (blue). Scale bar: 50 µm. Control – control group of animals; IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of AAV9-ARSA vector.
Figure 10.
Assessment of ARSA (yellow) expression in different regions of nervous tissue in the control group of animals (A-F), and in experimental groups 1 and 2 on day 35 after intrathecal (A1-F1), and intravenous (A2-F2) administration of AAV9-ARSA, respectively. Nuclei are stained with DAPI (blue). Scale bar: 50 µm. Control – control group of animals; IT AAV9-ARSA - intrathecal administration of AAV9-ARSA, IV AAV9-ARSA - intravenous administration of AAV9-ARSA vector.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



