Submitted:
18 April 2023
Posted:
19 April 2023
You are already at the latest version
Abstract
In recent years, the ecological and economic values of forest plants have been gradually recognized worldwide. However, the growing global demand for new forest plant varieties with higher wood production capacity and better stress tolerance cannot be satisfied by conventional phenotype-based breeding, marker-assisted selection, and genomic selection. In the recent past, diverse omics technologies, including genomics, transcriptomics, epigenomics, proteomics, and metabolomics, have been developed rapidly, providing powerful tools for the precision genetic breeding of forest plants. Genomics lays a solid foundation for understanding complex biological regulatory networks, while other omics technologies provide different perspectives at different levels. Multi-omics integration has combined the different omics technologies, becoming a powerful tool for genome-wide functional element identification in forest plant breeding. This review summarizes the recent progress of omics technologies and their applications in the genetic studies of forest plants. It will provide forest plant breeders with an elementary knowledge of multi-omics techniques for future breeding programs.
Keywords:
forest plants
; genetic breeding
; omics technologies
; multi-omics integration
; gene regulatory networks
1. Introduction
Forest ecosystems, the major component of the terrestrial ecosystems in the biosphere, cover more than 30% of the global land area [1] and provide multiple ecosystem services with high ecological and economic values [2]. Forest plants are the key players in the forest ecosystem, providing suitable habitats for forest biodiversity while playing critical roles in climate regulation, soil and water conservation, and hydrological and carbon cycles [3,4,5]. From an economic perspective, forest plants provide environmentally friendly products, including food, fiber, and fuel, to fulfill human needs [6,7]. However, in the past few decades, increasing human activities and dramatic climate changes have caused remarkable biodiversity losses and reductions in forest cover, especially in tropical areas [8,9,10], weakening the functions and ecosystem services of global forests [11]. Currently, the native forests and forest plantations are threatened by multiple human and natural stressors, while the restoration of forest services requires long periods [11]. Thankfully, the urgency to address forest health problems has been gradually recognized worldwide [10,11].
Since the middle of the twentieth century, numerous genetic breeding programs aiming to reduce deforestation and forest degradation rates have been conducted to develop new forest plant varieties with improved quality, productivity, and stress tolerance [12,13]. Conventional phenotype-based approaches, including hybrid, ploidy, and clonal breeding, have been successfully applied in several forest plants families, such as poplars [14], pines [15], spruces [16], eucalypts [17], and larches [18], which are the major sources of wood worldwide. However, the genetic improvement of forest plants by conventional breeding is time-consuming, expensive, and inconvenient due to the long reproductive cycle, large body size, and complex reproductive process [13,19]. A forest tree breeding program using the conventional approaches usually includes several cycles of breeding and phenotypic selection (Figure 1A), which may take tens of years. Moreover, some important traits of forest plants, such as drought tolerance, growth, and branching, have low heritability; thus, their assessment in the field is difficult [13]. Therefore, the growing global demand for new forest plant varieties cannot simply be satisfied by conventional breeding approaches.
With the rapid development of molecular genetics and sequencing technologies, scientists have invented “marker-assisted selection” (MAS), a more efficient and accurate approach, by integrating DNA markers into phenotypic selection [20,21]. MAS is an attractive tool for forest plant breeding as it allows individual selection based on genotypes instead of phenotypes (Figure 1B), largely accelerating the breeding cycles by eliminating the long waiting period until plant maturity [13]. In MAS-based breeding, markers in strong linkage disequilibrium (LD) with trait-related quantitative trait loci (QTLs) are used as molecular tools [22]. However, although MAS has been successfully implemented in the genetic improvement of simple traits controlled by few QTLs with large effects, it has not effectively improved complex traits controlled by multiple QTLs with minor effects [23]. Genomic selection (GS), an improved form of MAS, uses genome-wide dense markers, such as single-nucleotide polymorphisms (SNPs), to maximize the minor effect of QTLs in LD with at least one marker [24]. During GS, the genomic estimated breeding value (GEBV) for each genotyped individual is calculated using a prediction model trained from the genotyped and phenotyped population by a joint analysis of markers (Figure 1C), thereby avoiding potential bias in estimates and ensuring the overall accuracy of GS [25]. Compared to MAS, GS has significantly higher efficiency and accuracy for improving many traits in a wide range of species. However, the application of GS in forest plant breeding has been largely limited by the difficulty of genotyping extensive field plantations, non-universality among populations, and high genotyping cost due to the lack of reference genome, particularly in conifers [13,23,26].
Recent advances in high-throughput technologies using emerging omics approaches, including genomics, transcriptomics, epigenomics, proteomics, and metabolomics, have allowed a deeper dive into the molecular genetics of forest plant growth and development. These cutting-edge omics technologies provide powerful tools for understanding the underlying genetic mechanisms driving the complex architecture of various phenotypic traits, responses to biotic and abiotic stresses, and biosynthetic pathways of active compounds in forest plants. The increasing multi-omics data is also beneficial in the accurate genome-wide detection of functional elements using downstream bioinformatics analysis. Candidate functional elements identified through corroborating multi-omics evidence form a reliable basis for the genetic improvement of many growth traits and forest plant species. However, efficiently integrating large volumes of multi-omics data into forest plant genetic breeding programs is still challenging, especially for breeders with insufficient knowledge of omics and bioinformatics. This review discusses the recent progress of omics technologies, their applications in forest plant studies, and future application prospects of multi-omics approaches in the genetic improvement of forest plants.
2. Applications of omics technologies in forest plants
2.1. Genomics
The genome sequence of the model forest plant species, black cottonwood (Populus trichocarpa), was published in 2006 [27]. It was recently updated to version 4.1 (Phytozome 13, release date: 2022 Oct 5) with significantly improved accuracy and continuity compared to the initial version. The release of the black cottonwood genome spawned research in the functional genomics of forest trees, especially angiosperms. However, conifers, which belong to gymnosperms and dominate many temperate and boreal forests, have unusually large genome sizes ranging from 18 to 35 Gb with significantly different functional genomic characteristics from angiosperms [28]. In 2013, the first conifer genome belonging to Norway spruce (Picea abies) was published [29]. It was 4.3 Gb, which is only one-fifth of the Norway spruce actual genome size (19.6 Gb). However, the advent and development of next-generation sequencing (NGS) and third-generation sequencing (TGS) have greatly advanced the capacity to decode genomes for a wide range of forest plants. The emerging long-read and ultra-long-read sequencing and advanced assembly algorithms have provided useful tools for assembling complex plant genomes with high repeat content or heterozygosity (Figure 2) [30,31]. As a result, the difficulty of assembling a complete or near-complete genome of forest plants has been significantly alleviated in recent years [32]. To date, several high-quality forest plant genomes have been published, including gymnosperm species with genome sizes larger than 10 Gb, such as Ginkgo biloba [33], Taxus chinensis [34], T. wallichiana [35], Larix kaempferi [36], and Pinus tabuliformis [37].
A well-annotated reference-level genome provides a clear road map for downstream gene function and diversity studies, allowing an in-depth interpretation of genotype-phenotype relationships and functional DNA elements involved in various biological processes [38]. For example, the release of the black cottonwood genome has enabled studies on the genome-wide identification of regulatory genes and non-coding RNAs (ncRNAs) involved in several important biological processes, such as wood formation [39,40], annual growth cycle [41,42], flowering process [43,44], and responses to abiotic stresses [45,46,47] in Populus species. Moreover, a comprehensive phylogenetic analysis of functional elements based on the genomes of several model plant species, such as poplar, rice, and Arabidopsis, has been performed. Comparative genomic analysis among the different model species provides insights into the duplication history, selection pressure, and structural divergence of functional genes from an evolutionary aspect [48,49]. Therefore, the whole-genome scale discovery and cross-species comparison of insertion and distribution of transposable elements, especially the long terminal repeat retrotransposons (LTR-RTs), could help further investigate their effects on the adjacent gene expression and plant phenotype [50,51]. Overall, the accumulation of high-quality genome sequences will provide a comprehensive understanding of the contribution of functional DNA elements to phenotypic variation among forest plants.
Many forest plant species have a wide geographical distribution, large population size, and high genetic diversity at local and regional scales [52]. Thus, a single reference genome cannot simply represent the DNA sequence diversity within a species. However, population genomics studies using whole genome resequencing (WGRS) or reduced-representation genome sequencing (RRGS) can provide insights into the genetic diversity of forest plants at the SNPs level. SNPs can be detected by mapping reads to a reference genome and subsequent variant calling based on WGRS at the population level. The detected SNPs allow further genome-wide polymorphism analysis during adaptive population divergence. For example, the WGRS of 427 moso bamboos (Phyllostachys edulis) from multiple representative geographic regions and subsequent population genomic analysis revealed several candidate genes under balancing selection or related to several agriculturally important traits, such as clear culm height, node number, density, and compressive strength [53]. In addition, several candidate genes related to light response, growth-promoting cytokinin, and wood development were identified by genome sequencing and WGRS of 80 silver birch (Betula pendula) with clear evidence of recent natural selection [54]. However, despite the continuously decreasing sequencing cost, the WGRS of many plant samples/species is still expensive. As an alternative or complementary approach to WGRS, RRGS consisting of reduced-representation libraries and restriction-site-associated DNA sequencing, has been developed by integrating restriction enzymes into high-throughput sequencing to obtain a reduced genome representation [55]. Compared to WGRS, RRGS has apparent advantages of high efficiency and low cost and does not require a reference genome [55]. RRGS enables genome-wide SNP discovery for non-model species lacking genome sequence information or species with large and complex genomes [56,57]. Furthermore, the genetic linkage map can be constructed using RRGS data, and QTL mapping and genome-wide association analysis (GWAS) can be applied to identify the phenotype-genotype relationships across genomes of forest plants [58,59,60]. However, RRGS can also result in missing information possibly related to population differentiation, limiting its application scope.
WGRS and RRGS easily detect SNPs and short insertions and deletions. However, structural variations (SVs), including the presence/absence variants (PAVs), copy number variants, and chromosomal rearrangements, are rarely detected by short-read sequencing [61]. SVs genetically control the phenotypic variability within and between plant species [62,63]. Recent advances in long-read sequencing have enabled the generation of high-quality assemblies for several individuals per species across many plant species, providing a solid foundation for accurately identifying SVs by pan-genomic analysis at the species level [61,64]. Therefore, a pan-genome represents a more comprehensive DNA sequence diversity of a plant species or taxonomic group. As a result, pan-genomic studies are carried out to comprehensively understand the genetic diversity of several model plant species or economically important crops, including Arabidopsis, barley, rice, tomato, and soybean [65]. However, pan-genome research has been conducted on very few forest plant species, including poplar [66] and pecan [67]. Given the universality of sequencing technologies, assembly algorithms, and pan-genomic analysis pipelines, pan-genome will soon become a routine analysis tool for mining genetic variation and functional DNA elements of forest plant species.
In addition to genome sequences, population genomics, and pan-genomics, the three-dimensional genome structure, chloroplast genome, and mitochondrial genome are also useful for gene discovery and genetic engineering of plants [68,69,70]. With the increased different genomic resources, integrating the existing genomic data will facilitate an efficient and accurate functional element identification of forest plants, beneficial for further breeding processes. Genomic data from different sources have been submitted to publicly available databases, such as the National Center of Biotechnology Information, Ensembl, CoGe, GigaDB, and BIG Data Center. The development of a web-based, comprehensive genomic database, similar to BRAD [71] and Gramene [72], could also largely accelerate the genomic data integration for the genetic breeding of forest plant species.
2.2. Transcriptomics
Transcriptomics is one of the most commonly used omics approaches in plant biology research. It involves studying the transcriptome, the complete set of transcripts generated by a cell or tissue [73]. Understanding the transcriptome is crucial in elucidating the structural and functional organization of the genome [74]. Several hybridization- and sequence-based approaches [74], such as microarray, expressed sequence tag (EST) sequencing, serial analysis of gene expression, and RNA sequencing (RNA-seq), have been developed for transcriptome profiling. Among these approaches, RNA-seq, which captures all transcripts by high-throughput sequencing, is a revolutionary tool for accurate high-resolution transcriptome analysis [75]. For example, NGS-based RNA-seq generates millions of short reads, 25 to 300 bp in length.
Many computational tools have been developed to interpret the RNA-seq short reads data. For example, a transcriptome assembly is reconstructed by aligning RNA-seq reads to a known genome assembly using assemblers such as Cufflinks [76], StringTie [77], and Scripture [78]. Even without a reference genome, RNA-seq reads are de novo assembled into transcripts using assemblers such as Trinity [79], SOAPdenovo-Trans [80], and Oases-Velvet [81]. These reference-based or de novo strategies have been successfully applied in constructing a reference transcriptome with RNA-seq reads for many plant species. However, although NGS short reads have high accuracy, they rarely span multiple exons. Besides, assembling NGS short reads into full-length transcripts is complicated by alternative splicing events frequently occurring in the genome [82]. Luckily, this challenge is alleviated by TGS-based transcriptome sequencing approaches, such as full-length isoform sequencing (Iso-Seq) and nanopore-based direct RNA sequencing, which allows the direct sequencing of full-length transcripts without assembly [83,84]. However, given the relatively high error rate of TGS reads, highly accurate NGS RNA-seq reads are required to improve TGS-based transcriptome assembly accuracy [85]. At present, the high-quality and full-length transcriptome of forest plant species, including Larix kaempferi [86], Chosenia arbutifolia [87], Fritillaria cirrhosa [88], and Alsophila spinulosa has been obtained [89].
The reference-level transcriptome assembly is crucial for the downstream analysis of gene expressions under multiple conditions (Figure 3). The gene expression levels are usually evaluated based on the RNA-seq reads. By mapping the RNA-seq reads to a reference transcriptome or genome, the number of reads matching each gene, and gene expression levels are quantified by normalizing the read counts using algorithms, such as fragments per kilobase of mapped reads, transcripts per million and counts per million [90]. Subsequently, the differential expression (DE) analysis is performed using either non-parametric or parametric tools, such as DESeq2 [91], edgeR [92], and SAMseq [93]. DE analysis is widely used to analyze the forest plant responses to biotic and abiotic stresses, including drought, heat, salinity, flooding, cold, ultraviolet radiation, diseases, and insects [94,95]. For instance, the gene expression analysis in poplar root under polyethylene glycol-induced drought stress by TGS and NGS transcriptomic sequencing revealed several differentially expressed genes related to plant responses to drought in the biosynthesis and metabolism pathways [96]. In addition, the dual RNA-seq analysis of the interactions between Norway spruce and Heterobasidion revealed that several genes involved in the abscisic acid signaling were differentially expressed in Norway spruce, which might contribute to the Norway spruce response to the pathogen attack [97]. DE analysis also outlines the transcriptome dynamics across different tissues and plant developmental stages [98]. Notably, DE analysis under multiple conditions may identify too many or too few differentially expressed genes, requiring the analysts to integrate biological data from other sources or change the software parameters.
The gene expression data can also be used to construct the gene co-expression network (GCN), a powerful tool for further elucidation of the gene regulatory relationships and identification of candidate functional genes [99]. The weighted gene co-expression network analysis (WGCNA) is a widely used pipeline to construct GCN by clustering genes into modules based on their expression patterns and hub genes within each module [100]. The gene co-expression analysis has been successfully applied in detecting key functional genes regulating various biological processes in forest plant species, such as Pinus tabuliformis [101], Populus trichocarpa [102], Hevea brasiliensis [103], and Zanthoxylum armatum [104]. As a result, the cross-species GCN comparison effectively deduces the origin of new phenotypes and conserved gene functions at the species level [105]. Although this approach is rarely applied in forest plants, it has great potential in mining hub genes encoding important traits of forest plants. Moreover, transcriptome sequence and RNA-seq data can also be applied in the genome-wide detection of SNP and simple sequence repeat markers (Figure 3), which are valuable molecular tools in forest plant breeding [106].
With the advances in RNA-seq and tissue processing approaches, single-cell RNA-seq (scRNA-seq) has become a revolutionary tool for studying plant functional genomics at the cellular level [107]. scRNA-seq can analyze any tissue in any plant. This emerging approach obtains transcripts of thousands of cells per sample, providing new insights into gene expression heterogeneity across cells. Using the scRNA-seq data, cells can be clustered into different categories using dimensionality reduction and clustering, which allows for the reconstruction of the cell differentiation trajectories [107]. Distinct expression patterns of different cell clusters provide further insights into the gene regulatory networks and candidate genes related to plant organ development. For instance, the scRNA-seq of 6,796 poplar stem cells predicted the cell differentiation trajectories involved in phloem and xylem development and candidate genes related to vascular development in poplars [108]. Another newly developed technique, spatial transcriptomics, quantify and localize gene expression within the tissue by combing histological imaging and RNA sequencing [109]. Its derivative, spatial single-cell transcriptomics, also recently developed, integrates the spatial information from spatial transcriptomics and cellular gene expression based on cRNA-seq, clearly elucidating the complex spatial gene regulatory networks related to plant organ development [110]. Despite its high costs, spatial single-cell transcriptomics has great potential in precisely breeding forest plants.
2.3. Epigenomics
Epigenomics studies the epigenome, which consists of the biochemical modifications in the nuclear DNA, histone proteins, and ncRNAs [111]. Although these epigenomic modifications do not alter the nucleotide sequences, they are inherited across generations through mitosis or imprinting. Epigenetic changes, such as DNA methylation, histone modifications and variants, and ncRNA regulation, are frequently induced by environmental stresses or endogenous signals during plant development, which alters the chromatin structure and gene expression [112]. As a result, epigenetics is a powerful driving force in the environmental adaptation of plants by altering their phenotypic plasticity [111]. Therefore, epigenomic studies can provide important insights into the epigenetic basis underlying complex phenotypes and local adaptation of forest plants, which cannot be deduced using DNA sequence variants.
The availability of high-quality reference genomes facilitates the genome-wide detection of epigenetic variants at the single-nucleotide level in forest plant species [113]. DNA methylation has been extensively studied in plants by detecting base modifications using bisulfite or long-read sequencing [114]. In plants, DNA methylation occurs as CG, CHG, and CHH in gene bodies and transposable elements. More importantly, the methylation levels substantially vary across plant species, tissues, and cells [115]. In addition, DNA methylation is a highly dynamic process during plant growth and development, including the establishment, maintenance, and active removal of methylation sites [115]. The dynamics of DNA methylation play important roles in the epigenetic regulation of plant growth, development, and response to environmental stresses [116]. For instance, several studies have revealed the potential role of DNA methylation in flower development [117], drought tolerance [118], wood formation [119], and immune response to pathogen infection [120] in Populus species. Since DNA methylation alters gene expression, transcriptome analysis is usually combined with methylation analysis to investigate the functional relationship between the epigenome and transcriptome [114].
Moreover, the recently developed single-cell methylation profiling approaches have allowed the tracing of DNA methylation dynamics at the single-cell level [121]. In addition to the DNA methylation marks, histone marks, including histone modifications and variants, also transcriptionally silence or activate genes [122,123]. Histone modifications include histone methylation, acetylation, phosphorylation, ubiquitylation, and sumoylation, reversible amino acid modifications at the N-terminal tail of histone proteins within the nucleosome core [112]. Among these modifications, histone methylation and acetylation are the most studied modifications regulating plant development and environmental stress response [124,125]. Histone variants are sequence variants of core histones H2A, H2B, H3, and H4, which regulate nucleosome structure and function [126]. Histone marks are detected by DNA/RNA-protein interactions across the genome using chromatin immunoprecipitation sequencing (ChIP-seq) or the transposase-accessible chromatin assay with high-throughput sequencing (ATAC-seq) [127].
Furthermore, regulatory ncRNAs, including the long non-coding and small RNAs, are important epigenetic marks with diverse functions in response to abiotic stress in forest plants [128]. For instance, several micro RNAs are differentially expressed during Norway spruce embryo development, potentially contributing to epigenetic memory and climatic adaptation [129]. Overall, the functional analysis of different epigenetic marks has extended the scope of plant biology.
Understanding the epigenetic mechanisms and variants is beneficial for the epigenetic improvement of forest plants. Naturally or artificially-induced epigenetic variants serve as a novel genetic resource for plant epi-breeding [112]. Since the naturally occurring epigenetic variations are greatly limited, several laboratory-based approaches, including chemical treatment, biotic and abiotic stress treatment, tissue culture, grafting, RNA interference, and direct epigenome editing by CRISPR/Cas9 have been applied to manually modify the plant epigenome [130]. These artificial methods induce a wider range of phenotypic variation while increasing the transgenerationally inherited epialleles; hence, they are powerful tools in epi-breeding programs (Figure 4). Natural and artificially-induced epialleles can be employed as epigenetic markers in quantitative epigenetics based on the epigenetic quantitative trait loci and epigenome-wide association, which are important steps at the early stage of epi-breeding [131]. Moreover, epigenome editing methods using advanced CRISPR/Cas9 or CRISPR off technologies directly increase the stress resilience of plants through epigenome engineering [132,133]. In recent years, quantitative epigenetics and epigenome engineering have been successfully applied in the epigenetic breeding of crop plants, including rice, tomato, potato, and soybean [134,135,136,137]. Compared to crops, the epigenetic mechanisms and variants in forest plants are relatively understudied. With increased knowledge of the epigenetics of forest plants, epigenetic breeding will play an important role in improving more complex traits, promoting the forest plants adaptation to the changing climate.
2.4. Proteomics
Proteins are large biological molecules, the main undertakers of life activities and important components of plant cells and tissues, which form the physical basis of life. Proteomics studies the proteome, including the composition, localizations, modifications, and interactions of all the proteins expressed in a genome [138]. The plant proteome significantly varies across the cells and under different developmental and environmental conditions [139]. Most eukaryotic proteins undergo post-translational modifications, altering protein expressions and functions [140]. Thus, proteomics is a powerful omics tool for comprehensively understanding the biological processes in the post-genomic era [141]. Genome sequencing provides DNA sequences of protein-coding genes, laying a solid foundation for proteomics research. The proteome contains much more complex functional gene information than that provided by the genome [142]. As a result, the accurate identification and quantification of the complete proteome are highly challenging. So far, several sequencing technologies have been developed for proteomics-based analysis (Figure 5), including protein microarrays, gel-based approaches, quantitative approaches (isobaric tags for absolute and relative quantification (iTRAQ), isotope-coded affinity tag, and stable isotope labeling with amino acids), and high-throughput approaches (mass spectrometry and nuclear magnetic resonance spectroscopy) [138]. Among these approaches, mass spectrometry with liquid chromatography tandem-mass spectrometry and matrix-assisted laser desorption ionization time-of-flight widely monitors plant proteome dynamics. A comprehensive plant proteome profiling provides valuable insights into the molecular mechanisms underlying plant growth, development, and stress response [139]. For example, the tandem mass tag-based proteome sequencing of Masson pine (Pinus massoniana) with different resin yields revealed several differentially expressed proteins related to resinosis [143]. In addition, the Picea asperata somatic embryo proteome profiling using iTRAQ and comparative proteomics analysis under partial desiccation treatment provided novel insights into stress-related proteins and metabolic pathways in P. asperata [144]. Integrating proteomics, including single-cell and spatial proteomics, with computational approaches and other omics will enhance the proteomics potential in deepening our understanding of the functions and interactions of proteins [145,146].
Proteomics also identifies the candidate proteins underlying complex traits by linking protein expression to genetic maps through QTL analysis at the protein level [147]. The identified proteins serve as powerful biomarkers for the precision breeding of quantitative traits in plants [148]. In crops, proteomics and associated QTL mapping have successfully identified functional proteins and genes related to production or stress tolerance [149]. For instance, the large-scale proteome sequencing of 102 barley genotypes revealed drought-sensitive proteins in the different genotypes [150]. Further genetic linkage analysis of these proteins identified several proteomic QTLs (pQTLs) with potential breeding value for drought-tolerant barley. The label-free proteome sequencing of 148 recombinant inbred lines of pepper (Capsicum annuum) also revealed several candidate hotspot regions encoding functional proteins related to fruit development by pQTL analysis [151].
Despite its great potential in plant biology research and genetic breeding, proteomics is limited by several challenges compared to genomic and transcriptomic approaches [152]. First, the identification and quantification of the whole proteome are still challenging due to the limitations of the different proteome sequencing methods. Second, the precision and reproducibility of proteome sequencing and existing proteomics pipelines are unsatisfactory. Furthermore, deciphering the complex proteomic networks is still challenging since proteomics is far more complex than genomics. To date, proteomics has mainly served as an ancillary strategy for functional studies in the system biology of plants. However, the large-scale applications of plant proteomics are still a long way off [153].
2.5. Metabolomics
Metabolomics is an emerging post-genomics tool for comprehensive qualitative and quantitative studies of small-molecule metabolites with molar masses below 1000 in the cells or tissues [154]. Plants produce various metabolites, including primary and secondary metabolites. Primary metabolites are essential for plant growth and development, while secondary metabolites play a major role in the plant responses to environmental factors [155]. Metabolites are the end products of gene transcription and protein expression within an organism (Figure 6) and act as links between genotypes and phenotypes [156]. Among the omics tools, metabolomics has the closest relationship with the phenotype [157]. Since metabolomics was invented, it has become an increasingly popular system biology tool for deciphering plant science [158].
The plant metabolome is highly dynamic and complex, with many small-molecule metabolites of diverse structures and content, making accurate metabolome profiling challenging [159]. To date, a few analytical techniques have been developed for high-throughput quantitative metabolomics, including nuclear magnetic resonance, liquid chromatography-mass spectrometry, capillary electrophoresis-mass spectrometry, and gas chromatography-mass spectrometry [160]. Metabolomics is classified into targeted and untargeted based on the study subject [161]. Targeted metabolomics performs target analysis of known metabolites with high sensitivity and accuracy, revealing the fluctuations in specific metabolic pathways. In contrast, untargeted metabolomics performs non-biased detection of all metabolites while identifying the differential metabolites with significant changes for further screening analysis. Like other omics, single-cell sequencing can be integrated into metabolomics to unravel the cellular metabolism dynamics under environmental changes [162]. Given the metabolic complexity in plants and the limitations in each analytical platform/method, combined approaches are increasingly employed in plant metabolome profiling studies.
The metabolite variation modulates diverse biological processes and may alter plant phenotypes [163]. As a result, transcriptomics and metabolomics-based correlation analyses have enabled the genome-wide discovery of key genes controlling known and new metabolic pathways [164]. This strategy has been regularly applied in forest plants, including Zanthoxylum armatum [104], Phyllostachys edulis [165], Populus tomentosa [166], and Hevea brasiliensis [167]. Given that most metabolic traits are heritable across generations [168], combining metabolomics with QTL and GWAS can establish direct links between metabolites and phenotypes based on large-scale population metabolomic and phenotypic data [158]. To date, most metabolome QTL (mQTL) and metabolome-based GWAS (mGWAS) have been applied in hunting genes related to metabolic traits in crop plants [158], with a few success cases in poplars and apple trees. For example, mQTL analysis of the untargeted metabolic profiling data and genetic linkage maps revealed mQTL hotspots with many peel- and flesh- related metabolites [169]. In addition, the mGWAS analysis for flavonoid features in Popuous tomentosa using targeted metabolomics data revealed more than 1,500 significant associations accounted for phenotypic variation [170].
Moreover, metabolic markers can be determined using metabolic profiling data under various stress conditions. The selected metabolites can help plant breeders accurately identify stress levels [158]. Metabolomics has proven to be an effective tool in plant genetic breeding programs. However, a single approach to identifying and quantifying all metabolites within a plant species is still lacking [160]. Therefore, future advances in metabolomics approaches should discover more interesting gene and metabolic pathways beneficial for further plant breeding.
2.6. Multi-omics integration
Genomics is the most used omics discipline. The whole-genome DNA sequence informs the basic properties of a plant species but cannot solely determine the final phenotype. At the same time, not all DNA sequence variants lead to phenotypic variation. Instead, phenotypic plasticity is shaped by many molecular mechanisms, including epigenetic modification, gene expression and silencing, post-translational protein modification, and metabolite accumulation. Therefore, a single omics cannot sufficiently and comprehensively unravel the complex biological regulatory networks controlling the various phenotypic traits [171].
The continuous and rapid progress in developing various high-throughput omics technologies has facilitated the integration of different omics data for plant system biology studies [172]. Transcriptomics, proteomics, and metabolomics are the most frequently used omics technologies in multi-omics integration (MOI) studies of plants, as they are the core of system biology [172]. MOI studies are accelerated by genomic information provided by well-annotated genomes and associated genomic analysis epigenomics and other omics approaches. However, multiple omics platforms produce much high-throughput data, greatly challenging the subsequent MOI analysis [173]. The MOI analysis mainly involves the establishment of associations between different omics data sets [174]. Therefore, analysts require a good understanding of the formats and characteristics of various omics data sources and a good background in software operations, statistical modeling, and data interpretation.
The most simple and intuitive analysis strategy in MOI studies is the correlation analysis of two or more omics data sets using various models, such as Pearson, Spearman, and Kendall rank correlation analyses [175]. These correlation analyses can be applied to differentially expressed or specific biochemical pathway-related transcripts, proteins, and metabolites. However, various biological factors along with experimental errors, may cause weak correlations between different omics data sets [172,176]. For example, transcriptome and proteome sequencing of Quercus ilex under severe drought conditions recorded a poor correlation (r = 0.11) between mRNA and protein [177]. Such inconsistencies are alleviated by further sequencing and analysis. Another strategy for analyzing MOI-related data is clustering analysis based on the similarity of various omics data using hierarchical or partition clustering methods [178]. Several statistical approaches, including similarity matrices, canonical correlation and co-inertia analysis, and matrix factorization, have been successfully applied in grouping forest plant multi-omics data [178]. For instance, Pascual et al. (2017) used a k-means clustering approach to integrate proteomic, metabolomic, and physiological data of Pinus radiata based on their quantitative trends during different periods of ultra violet treatment, obtaining 30 clusters [179]. The clustering results can be further correlated with specific scientific questions. Moreover, multivariate-based analysis has enabled the integration of multi-omics data using multi-variant data analysis approaches, such as principal components analysis, partial least squares, and orthogonal projections to latent structures (OPLS) [180]. For example, the OPLS analysis of the transcriptome, proteome, and metabolome data from transgenic poplars identified several proteins related to wood formation [181].
A common weakness of correlation, clustering, and multivariate-based analyses is that they are based on statistical methods rather than prior knowledge of molecular mechanisms. To integrate known pathway information into MOI analysis, bioinformatics analysts have developed several pathway-based approaches, such as pathway mapping and co-expression analyses [172]. Pathway mapping analysis maps various omics data sets against publicly available pathway databases, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) and MetaCyc [182]. For example, López-Hidalgo et al. (2018) reconstructed and visualized 123 of the 127 known KEGG pathways at the transcriptome, proteome, and metabolome level for Quercus ilex using MapMan software [183]. At the same time, multiple omics data sets could be employed to annotate certain plant metabolic pathways using known pathway information as the reference. For example, Wang et al. (2021) reconstructed the biosynthesis of two alkaloids, sanshools and wgx-50, using transcriptome and metabolome data [104].
Alternatively, the co-expression analysis can be integrated into existing pathway databases. WGCNA approach, the most popular gene co-expression analysis tool, can detect regulatory networks for each omics layer, construct a consensus correlation network [184,185], and identify hub elements, such as hub genes, proteins, and metabolites. Besides, the multi-omics WGCNA approach can perform efficient gene and module clustering and provide key regulatory network information through MOI.
However, neither statistical-based nor pathway-based MOI methods are independent. Instead, these methods are often combined to answer biological questions precisely. Several complementary MOI approaches exist, such as top-down differential analysis and bottom-up genome-scale modeling [172]. These MOI approaches have been widely used in the in-depth analysis of complex metabolic pathways and other biological processes in forest plants. However, the heterogeneity in signal-to-noise ratio across multiple omics layers, the adverse effects of missing values, the limitation in the interpretation ability of multi-omics models, and data sharing difficulties, such as metadata annotation, data storage, and computing resources remain universal challenges across all-pervading MOI studies [186]. In any case, MOI analysis is a powerful tool for genome-wide functional element identification and forest plant breeding. Besides, the development of single-cell sequencing technology provides an exciting opportunity for single-cell MOI analysis.
3. Conclusions and prospects
Forest plants, including overstory trees and understory shrubs and herbs, are recalcitrant and orphan plant species. Most forest plants, except several woody plant models, are less studied than crop plants since most are inedible. With the improvement of people’s living standards and the increasing global demand for wood, forest plant breeding for cultivars with higher wood production and greater resilience to climate change has received worldwide attention. Compared to conventional phenotype-based breeding, MAS, and GS, plant breeding in the omics era provides a more efficient tool for genetically improving many objective traits across many plant species. Forest plant breeding using omics technologies is greatly promoted by the rapid development of high-throughput platforms and successful omics-based breeding in crop plants. Moreover, most forest plant breeding programs do not result in edible products. Therefore, forest plant breeders do not usually have to worry about the safety of genetically modified foods. The omics technologies, including genomics, transcriptomics, epigenomics, proteomics, and metabolomics, are useful tools for the genome-wide identification of functional elements contributing to various phenotypes. The availability of a complete or nearly complete genome sequence of model and non-model forest plants facilitates downstream analysis based on the various omics technologies. Epigenomics, transcriptomics, proteomics, and metabolomics, are useful tools for understanding the gene regulatory networks of forest plants at different levels. At the same time, MOI analysis has become a popular method for forest plant breeding as it combines the advantages of different omics technologies, accurately identifying key functional elements by establishing associations between different omics layers. The identified functional elements can be used as powerful biomarkers for the precision breeding of forest plants. They also form an important foundation for functional verification and genetic engineering studies. However, although we are in the big data era, the current MOI analysis technique is under developed. Establishing effective links between different omics layers is still challenging for forest plant breeders, who require a solid omics background, big data processing, mining skills, and systems biology knowledge. This review provides elementary knowledge on applying multi-omics in forest plant breeding. Multi-omics techniques are highly innovative and still evolving, leading to higher resolution and a deeper understanding of complex traits in forest plants. Therefore, as the population grows and climate change intensifies, forest plants provide a better ecosystem and economic services.
Author Contributions
M.W. and Q.Z. designed the study. M.W. and R.L. collected the references and drafted the manuscript. Q.Z. reviewed and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Sichuan Province Central Committee Guide Local Science and Technology Development Project (grant no. 2022ZYD0095) and the start-up funds provided by Chengdu University (2081921039).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Keenan, R.J.; Reams, G.A.; Achard, F.; de Freitas, J.V.; Grainger, A.; Lindquist, E. Dynamics of global forest area: Results from the FAO Global Forest Resources Assessment 2015. For. Ecol. Manage. 2015, 352, 9–20. [Google Scholar] [CrossRef]
- García-Nieto, A.P.; García-Llorente, M.; Iniesta-Arandia, I.; Martín-López, B. Mapping forest ecosystem services: from providing units to beneficiaries. Ecosyst. Serv. 2013, 4, 126–138. [Google Scholar] [CrossRef]
- Smith, P.; Ashmore, M.R.; Black, H.I.; Burgess, P.J.; Evans, C.D.; Quine, T.A.; Thomson, A.M.; Hicks, k.; Orr, H.G. The role of ecosystems and their management in regulating climate, and soil, water and air quality. J. Appl. Ecol. 2013, 50, 812–829. [Google Scholar] [CrossRef]
- Brockerhoff, E.G.; Barbaro, L.; Castagneyrol, B.; Forrester, D.I.; Gardiner, B.; González-Olabarria, J.R.; Lyver, P.O.; Meurisse, N.; Oxbrough, A.; Taki, H; et al. Forest biodiversity, ecosystem functioning and the provision of ecosystem services. Biodiversity Conserv. 2017, 26, 3005–3035. [Google Scholar] [CrossRef]
- Anderegg, W.R.; Trugman, A.T.; Badgley, G.; Anderson, C.M.; Bartuska, A.; Ciais, P.; Cullenward, D.; Field, C.B.; Freeman, J.; Goetz, S.J.; et al. Climate-driven risks to the climate mitigation potential of forests. Science 2020, 368, eaaz7005. [Google Scholar] [CrossRef]
- Griscom, B.W.; Adams, J.; Ellis, P.W.; Houghton, R.A.; Lomax, G.; Miteva, D.A.; Schlesinger, W.H.; Shoch, D.; Siikamäki, J.V.; Smith, P.; et al. Natural climate solutions. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11645–11650. [Google Scholar] [CrossRef] [PubMed]
- Isabel, N.; Holliday, J.A.; Aitken, S.N. Forest genomics: Advancing climate adaptation, forest health, productivity, and conservation. Evol. Appl. 2020, 13, 3–10. [Google Scholar] [CrossRef]
- Kirilenko, A.P.; Sedjo, R.A. Climate change impacts on forestry. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19697–19702. [Google Scholar] [CrossRef]
- Betts, M.G.; Wolf, C.; Ripple, W.J.; Phalan, B.; Millers, K.A.; Duarte, A.; Butchart, S.H.M.; Levi, T. Global forest loss disproportionately erodes biodiversity in intact landscapes. Nature 2017, 547, 441–444. [Google Scholar] [CrossRef]
- Curtis, P.G.; Slay, C.M.; Harris, N.L.; Tyukavina, A.; Hansen, M.C. Classifying drivers of global forest loss. Science 2018, 361, 1108–1111. [Google Scholar] [CrossRef]
- Trumbore, S.; Brando, P.; Hartmann, H. Forest health and global change. Science 2015, 349, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Koskela, J.; Vinceti, B.; Dvorak, W.; Bush, D.; Dawson, I.K.; Loo, J.; Kjaer, E.D.; Navarro, C.; Padolina, C.; Bordács, S.; et al. Utilization and transfer of forest genetic resources: A global review. For. Ecol. Manage. 2014, 333, 22–34. [Google Scholar] [CrossRef]
- Lebedev, V.G.; Lebedeva, T.N.; Chernodubov, A.I.; Shestibratov, K.A. Genomic selection for forest tree improvement: Methods, achievements and perspectives. Forests 2020, 11, 1190. [Google Scholar] [CrossRef]
- Dickmann, D; Isebrands, J. ; Eckenwalder, J.; Richardson, J. Poplar culture in North America. NRC Research Press: Ottawa, ON, Canada, 2001. [Google Scholar]
- Dorman, K.W. The genetics of breeding southern pines. Agricultural handbook 471. USDA Forest Service, Washington, DC, 1976.
- Lee, S.J. Improving the timber quality of Sitka spruce through selection and breeding. Forestry 1999, 72, 123–146. [Google Scholar] [CrossRef]
- Eldridge, K.; Davidson, J.; Harwood, C.; van Wyk, G. Eucalypt domestication and breeding. Clarendon Press: Oxford, UK, 1994.
- Lelu-Walter, M.A.; Pâques, L.E. Simplified and improved somatic embryogenesis of hybrid larches (Larix × eurolepis and Larix × marschlinsii). Perspectives for breeding. Ann. For. Sci. 2009, 66, 104. [Google Scholar] [CrossRef]
- Ahmar, S.; Ballesta, P.; Ali, M.; Mora-Poblete, F. Achievements and challenges of genomics-assisted breeding in forest trees: From marker-assisted selection to genome editing. Int. J. Mol. Sci. 2021, 22, 10583. [Google Scholar] [CrossRef]
- Ribaut, J.M.; Hoisington, D. Marker-assisted selection: new tools and strategies. Trends Plant Sci. 1998, 3, 236–239. [Google Scholar] [CrossRef]
- Collard, B.C.; Mackill, D.J. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B 2008, 363, 557–572. [Google Scholar] [CrossRef]
- Collard, B.C.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de Los Campos, G.; Burgueño, J.; González-Camacho, J. M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: methods, models, and perspectives. In Trends Plant Sci.; 2017; Volume 22, pp. 961–975. [Google Scholar]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.A.; Ortiz, R. Genomic selection: genome-wide prediction in plant improvement. Trends Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed]
- Mackay, J.; Dean, J.F.; Plomion, C.; Peterson, D.G.; Cánovas, F.M.; Pavy, N.; Ingvarsson, P.K.; Savolainen, O.; Guevara, M.A.; Fluch, S.; et al. Towards decoding the conifer giga-genome. Plant Mol. Biol. 2012, 80, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Winefield, C.; Bombarely, A.; Prentis, P.; Waterhouse, P. Tools and strategies for long-read sequencing and de novo assembly of plant genomes. Trends Plant Sci. 2019, 24, 700–724. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef]
- Sun, Y.; Shang, L.; Zhu, Q.H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Wang, G.; Cui, P.; Wu, S.; Ai, C.; Hu, N.; Li, A.; He, B.; Shao, X.; et al. The nearly complete genome of Ginkgo biloba illuminates gymnosperm evolution. Nat. Plants 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Xiong, X.; Gou, J.; Liao, Q.; Li, Y.; Zhou, Q.; Bi, G.; Li, C.; Du, R.; Wang, X.; Sun, T.; et al. The Taxus genome provides insights into paclitaxel biosynthesis. Nat. Plants 2021, 7, 1026–1036. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, X.; Liu, X.; Zhu, X.; Li, Z.; Chu, H.; Wang, Q.; Lou, Q.; Cai, B.; Yang, Y.; et al. Chromosome-level genome of Himalayan yew provides insights into the origin and evolution of the paclitaxel biosynthetic pathway. Mol. Plant 2021, 14, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xie, Y.H.; Li, Z.; Liu, Y.J.; Sun, X.M.; Li, J.J.; Quan, W.P.; Zeng, Q.Y.; Van de Peer, Y.; Zhang, S.G. The Larix kaempferi genome reveals new insights into wood properties. J. Integr. Plant Biol. 2022, 64, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y.; et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J. Plant genome sequences: past, present, future. Curr. Opin. Plant Biol. 2019, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, J.P.; Liu, H.; Li, H.; Lin, Y.C.J.; Shi, R.; Yang, C.; Gao, J.; Zhou, C.; Li, Q.; et al. Hierarchical transcription factor and chromatin binding network for wood formation in Populus trichocarpa. Plant Cell 2019, 31, 602–626. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, C.; Li, D.; Li, S.; Lin, Y.C.J.; Wang, J.P.; Chiang, V.L.; Li, W. A PtrLBD39-mediated transcriptional network regulates tension wood formation in Populus trichocarpa. Plant Communications 2022, 3, 100250. [Google Scholar] [CrossRef]
- Lian, C.; Li, Q.; Yao, K.; Zhang, Y.; Meng, S.; Yin, W.; Xia, X. Populus trichocarpa PtNF-YA9, a multifunctional transcription factor, regulates seed germination, abiotic stress, plant growth and development in Arabidopsis. Front. Plant Sci. 2018, 9, 954. [Google Scholar] [CrossRef]
- André, D.; Marcon, A.; Lee, K.C.; Goretti, D.; Zhang, B.; Delhomme, N.; Schmid, M.; Nilsson, O. FLOWERING LOCUS T paralogs control the annual growth cycle in Populus trees. Curr. Biol. 2022, 32, 2988–2996. [Google Scholar] [CrossRef]
- Mohamed, R.; Wang, C.T.; Ma, C.; Shevchenko, O.; Dye, S.J.; Puzey, J.R.; Etherington, E.; Sheng, X.; Meilan, R.; Strauss, S.H.; et al. Populus CEN/TFL1 regulates first onset of flowering, axillary meristem identity and dormancy release in Populus. Plant J. 2010, 62, 674–688. [Google Scholar] [CrossRef]
- Ye, Y.; Xin, H.; Gu, X.; Ma, J.; Li, L. Genome-wide identification and functional analysis of the Basic Helix-Loop-Helix (bHLH) transcription family reveals candidate PtFBH genes involved in the flowering process of Populus trichocarpa. Forests 2021, 12, 1439. [Google Scholar] [CrossRef]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wang, S.; Zhao, X.; Gao, N.; Wang, Y.; Yang, Y.; Wu, E.; Jiang, C.; Cheng, Y.; Wu, W.; et al. Role of lncRNAs in cis- and trans-regulatory responses to salt in Populus trichocarpa. Plant J. 2022, 110, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lin, Y.C.J.; Wang, P.; Zhang, B.; Li, M.; Chen, S.; Shi, R.; Tunlaya-Anukit, S.; Liu, X.; Wang, Z.; et al. The AREB1 transcription factor influences histone acetylation to regulate drought responses and tolerance in Populus trichocarpa. Plant Cell 2019, 31, 663–686. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Han, Y.; Jin, Q.; Lin, Y.; Cai, Y. Comparative genomic analysis of the GRF genes in Chinese pear (Pyrus bretschneideri Rehd), poplar (Populous), grape (Vitis vinifera), Arabidopsis and rice (Oryza sativa). Front. Plant Sci. 2016, 7, 1750. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Movahedi, A.; Liu, G.; Li, Y.; Liu, S.; Yu, C.; Chen, Y.; Zhong, F.; Zhang, J. Comprehensive analysis of carotenoid cleavage dioxygenases gene family and its expression in response to abiotic stress in poplar. Int. J. Mol. Sci. 2022, 23, 1418. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2020, 42, 251–269. [Google Scholar] [CrossRef]
- Du, J.; Tian, Z.; Hans, C.S.; Laten, H.M.; Cannon, S.B.; Jackson, S.A.; Shoemaker, R.C.; Ma, J. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: Insights from genome-wide analysis and multi-specific comparison. Plant J. 2010, 63, 584–598. [Google Scholar] [CrossRef]
- Ingvarsson, P.K.; Hvidsten, T.R.; Street, N.R. Towards integration of population and comparative genomics in forest trees. New Phytol. 2016, 212, 338–344. [Google Scholar] [CrossRef]
- Zhao, H.; Sun, S.; Ding, Y.; Wang, Y.; Yue, X.; Du, X.; Wei, Q.; Fan, G.; Sun, H.; Lou, Y.; et al. Analysis of 427 genomes reveals moso bamboo population structure and genetic basis of property traits. Nat. Commun. 2021, 12, 5466. [Google Scholar] [CrossRef]
- Salojärvi, J.; Smolander, O.P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J.; et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 2017, 49, 904–912. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Caballero, M.; Lauer, E.; Bennett, J.; Zaman, S.; McEvoy, S.; Acosta, J.; Jackson, C.; Townsend, L.; Eckert, A.; Whetten, R.W.; et al. Toward genomic selection in Pinus taeda: Integrating resources to support array design in a complex conifer genome. Appl. Plant Sci. 2021, 9, e11439. [Google Scholar] [CrossRef] [PubMed]
- Varas-Myrik, A.; Sepúlveda-Espinoza, F.; Fajardo, A.; Alarcón, D.; Toro-Núñez, Ó.; Castro-Nallar, E.; Hasbún, R. Predicting climate change-related genetic offset for the endangered southern South American conifer Araucaria araucana. For. Ecol. Manage. 2022, 504, 119856. [Google Scholar] [CrossRef]
- Goto, S.; Kajiya-Kanegae, H.; Ishizuka, W.; Kitamura, K.; Ueno, S.; Hisamoto, Y.; Kudoh, H.; Yasugi, M.; Nagano, A.J.; Iwata, H. Genetic mapping of local adaptation along the altitudinal gradient in Abies sachalinensis. Tree Genet. Genomes 2017, 13, 1–13. [Google Scholar] [CrossRef]
- Fuentes-Utrilla, P.; Goswami, C.; Cottrell, J.E.; Pong-Wong, R.; Law, A.; A’Hara, S.W.; Lee, S.J.; Woolliams, J.A. QTL analysis and genomic selection using RADseq derived markers in Sitka spruce: the potential utility of within family data. Tree Genet. Genomes 2017, 13, 1–12. [Google Scholar] [CrossRef]
- Lu, N.; Zhang, M.; Xiao, Y.; Han, D.; Liu, Y.; Zhang, Y.; Yi, F.; Zhu, T.; Ma, W.; Fan, E.; et al. Construction of a high-density genetic map and QTL mapping of leaf traits and plant growth in an interspecific F1 population of Catalpa bungei × Catalpa duclouxii Dode. BMC Plant Biol. 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Schreiber, M.; Stein, N.; Mascher, M. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 2021, 28, dsaa030. [Google Scholar] [CrossRef]
- Saxena, R.K.; Edwards, D.; Varshney, R.K. Structural variations in plant genomes. Briefings Funct. Genomics 2014, 13, 296–307. [Google Scholar] [CrossRef]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Current status of structural variation studies in plants. Plant Biotechnol. J. 2021, 19, 2153–2163. [Google Scholar] [CrossRef]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 1–19. [Google Scholar] [CrossRef]
- Shi, J.; Tian, Z.; Lai, J.; Huang, X. Plant pan-genomics and its applications. Mol. Plant 2023, 16, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Pinosio, S.; Giacomello, S.; Faivre-Rampant, P.; Taylor, G.; Jorge, V.; Le Paslier, M.C.; Zaina, G.; Bastien, C.; Cattonaro, F.; Marroni, F.; et al. Characterization of the poplar pan-genome by genome-wide identification of structural variation. Mol. Biol. Evol. 2016, 33, 2706–2719. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.T.; Bentley, N.B.; Bhattarai, G.; Jenkins, J.W.; Sreedasyam, A.; Alarcon, Y.; Bock, C.; Beth Boston, L.; Carlson, J.; Cervantes, K.; et al. Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding. Nat. Commun. 2021, 12, 4125. [Google Scholar] [CrossRef] [PubMed]
- Doğan, E.S.; Liu, C. Three-dimensional chromatin packing and positioning of plant genomes. Nat. Plants 2018, 4, 521–529. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 1–29. [Google Scholar] [CrossRef]
- Maliga, P. Engineering the plastid and mitochondrial genomes of flowering plants. Nat. Plants 2022, 8, 996–1006. [Google Scholar] [CrossRef]
- Chen, H.; Wang, T.; He, X.; Cai, X.; Lin, R.; Liang, J.; Wu, J.; King, G.; Wang, X. BRAD V3.0: an upgraded Brassicaceae database. Nucleic Acids Res. 2022, 50, D1432–D1441. [Google Scholar] [CrossRef]
- Tello-Ruiz, M.K.; Naithani, S.; Gupta, P.; Olson, A.; Wei, S.; Preece, J.; Jiao, Y.; Wang, B.; Chougule, K.; Garg, P.; et al. Gramene 2021: harnessing the power of comparative genomics and pathways for plant research. Nucleic Acids Res. 2021, 49, D1452–D1463. [Google Scholar] [CrossRef]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Waern, K.; Snyder, M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr. Protoc. Mol. Biol. 2010, 89, 4–11. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J. Gnirke, A.; Nusbaum, C. et al. Ab initio reconstruction of cell type–specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef]
- An, D.; Cao, H.X.; Li, C.; Humbeck, K.; Wang, W. Isoform sequencing and state-of-art applications for unravelling complexity of plant transcriptomes. Genes 2018, 9, 43. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.; Gu, L.; Reddy, A.S. Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and nanopore-based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Shumate, A.; Wong, B.; Pertea, G.; Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022, 18, e1009730. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Hirakawa, H.; Iki, T.; Fukuda, Y.; Hirao, T.; Tamura, A.; Takahashi, M. Comprehensive collection of genes and comparative analysis of full-length transcriptome sequences from Japanese larch (Larix kaempferi) and Kuril larch (Larix gmelinii var. japonica). BMC Plant Biol. 2022, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, Y.; Zheng, J.; Zhou, J.; Jiao, Z.; Wang, B.; Zhuge, Q. Full-length transcriptome characterization and comparative analysis of Chosenia arbutifolia. Forests 2022, 13, 543. [Google Scholar] [CrossRef]
- Li, R.; Xiao, M.; Li, J.; Zhao, Q.; Wang, M.; Zhu, Z. Transcriptome Analysis of CYP450 Family Members in Fritillaria cirrhosa D. Don and Profiling of Key CYP450s Related to Isosteroidal Alkaloid Biosynthesis. Genes 2023, 14, 219. [Google Scholar] [CrossRef]
- Hong, Y.; Wang, Z.; Li, M.; Su, Y.; Wang, T. First multi-organ full-length transcriptome of tree fern Alsophila spinulosa highlights the stress-resistant and light-adapted genes. Front. Genet. 2022, 12, 2812. [Google Scholar] [CrossRef]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of differential gene expression results of RNA-seq data: review and integration. Briefings Bioinf. 2019, 20, 2044–2054. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Li, J.; Tibshirani, R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Stat. Methods Med. Res. 2013, 22, 519–536. [Google Scholar] [CrossRef]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R. Abiotic and biotic stress combinations. New Phytol. 2014, 203, 32–43. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.K. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Z.; Feng, H.; Yang, J.; Li, C. Characterization of the gene expression profile response to drought stress in Populus ussuriensis using PacBio SMRT and Illumina Sequencing. Int. J. Mol. Sci. 2022, 23(7), 3840. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, A.; Zeng, Z.; Ghimire, R.P.; Kivimäenpää, M.; Raffaello, T.; Liu, M.; Mukrimin, M.; Kasanen, R.; Sun, H.; Julkunen-Tiitto, R.; et al. Dual RNA-seq analysis provides new insights into interactions between Norway spruce and necrotrophic pathogen Heterobasidion annosum sl. BMC Plant Biol. 2019, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Klepikova, A.V.; Penin, A.A. Gene expression maps in plants: Current state and prospects. Plants 2019, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Serin, E.A.; Nijveen, H.; Hilhorst, H.W.; Ligterink, W. Learning from co-expression networks: possibilities and challenges. Front. Plant Sci. 2016, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.J.; Chen, X.; Song, Y.T.; Zhang, G.F.; Zhou, X.Q.; Que, S.P.; Mao, F.; Pervaiz, T.; Lin, J.X.; Li, Y.; et al. MADS-box transcription factors MADS11 and DAL1 interact to mediate the vegetative-to-reproductive transition in pine. Plant Physiol. 2021, 187, 247–262. [Google Scholar] [CrossRef]
- Wang, R.; Xie, M.; Zhao, W.; Yan, P.; Wang, Y.; Gu, Y.; Jiang, T.; Qu, G. WGCNA reveals genes associated with lignification in the secondary stages of wood formation. Forests 2023, 14, 99. [Google Scholar] [CrossRef]
- Francisco, F.R.; Aono, A.H.; da Silva, C.C.; Gonçalves, P.S.; Scaloppi Junior, E.J.; Le Guen, V.; Fritsche-Neto, R.; Souza, L.M.; de Souza, A.P. Unravelling rubber tree growth by integrating GWAS and biological network-based approaches. Front. Plant Sci. 2021, 12, 768589. [Google Scholar] [CrossRef]
- Wang, M.; Tong, S.; Ma, T.; Xi, Z.; Liu, J. Chromosome-level genome assembly of Sichuan pepper provides insights into apomixis, drought tolerance, and alkaloid biosynthesis. Mol. Ecol. Resour. 2021, 21, 2533–2545. [Google Scholar] [CrossRef]
- Ovens, K.; Eames, B.F.; McQuillan, I. Comparative analyses of gene co-expression networks: Implementations and applications in the study of evolution. Front. Genet. 2021, 12, 695399. [Google Scholar] [CrossRef]
- Younessi-Hamzekhanlu, M.; Gailing, O. Genome-wide SNP markers accelerate perennial forest tree breeding rate for disease resistance through marker-assisted and genome-wide selection. Int. J. Mol. Sci. 2022, 23, 12315. [Google Scholar] [CrossRef]
- Rich-Griffin, C.; Stechemesser, A.; Finch, J.; Lucas, E.; Ott, S.; Schäfer, P. Single-cell transcriptomics: a high-resolution avenue for plant functional genomics. Trends Plant Sci. 2020, 25, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tong, S.; Jiang, Y.; Ai, F.; Feng, Y.; Zhang, J.; Gong, J.; Qin, J.; Zhang, Y.; Zhu, Y.; et al. Transcriptional landscape of highly lignified poplar stems at single-cell resolution. Genome Biol. 2021, 22, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019, 20, 317–317. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, S. A new era for plant science: spatial single-cell transcriptomics. Curr. Opin. Plant Biol. 2021, 60, 102041. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Epigenomics of plant responses to environmental stress. Epigenomes 2018, 2, 6. [Google Scholar] [CrossRef]
- Singh, D.; Chaudhary, P.; Taunk, J.; Singh, C.K.; Sharma, S.; Singh, V. J.; Singh, D.; Chinnusamy, V.; Yadav, R.; Pal, M. Plant epigenomics for extenuation of abiotic stresses: challenges and future perspectives. J. Exp. Bot. 2021, 72, 6836–6855. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.; Ribeyre, Z.; Vigneaud, J.; Sow, M.D.; Fichot, R.; Messier, C.; Pinto, J.; Nolet, P.; Maury, S. Advances and promises of epigenetics for forest trees. Forests 2020, 11, 976. [Google Scholar] [CrossRef]
- Gouil, Q.; Keniry, A. Latest techniques to study DNA methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.Q.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mohapatra, T. Dynamics of DNA methylation and its functions in plant growth and development. Front. Plant Sci. 2021, 12, 596236. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Ma, K.; Bo, W.; Zhang, Z.; Zhang, D. Sex-specific DNA methylation and gene expression in andromonoecious poplar. Plant Cell Rep. 2012, 31, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Sow, M.D.; Le Gac, A.L.; Fichot, R.; Lanciano, S.; Delaunay, A.; Le Jan, I.; Lesage-Descauses, M.C.; Citerne, S.; Caius, J.; Brunaud, V.; et al. RNAi suppression of DNA methylation affects the drought stress response and genome integrity in transgenic poplar. New Phytol. 2021, 232, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Cheng, H.; Tian, S.; Liu, Y.; Wang, S.; Zhang, H.; Saqib, M.; Wei, H.; Wei, Z. DNA methylation and its effects on gene expression during primary to secondary growth in poplar stems. BMC genomics 2020, 21, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhou, K.; Yang, X.; Yang, Y.; Ma, Y.; Wang, Y. Crosstalk of DNA methylation triggered by pathogen in poplars with different resistances. Front. Microbiol. 2021, 12, 4098. [Google Scholar] [CrossRef]
- Karemaker, I.D.; Vermeulen, M. Single-cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef]
- Weber, C.M.; Henikoff, S. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014, 28, 672–682. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Kumar, V.; Thakur, J.K.; Prasad, M. Histone acetylation dynamics regulating plant development and stress responses. Cell. Mol. Life Sci. 2021, 78, 4467–4486. [Google Scholar] [CrossRef]
- He, K.; Cao, X.; Deng, X. Histone methylation in epigenetic regulation and temperature responses. Curr. Opin. Plant Biol. 2021, 61, 102001. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.; Jiang, D.; Berger, F. Histone variants take center stage in shaping the epigenome. Curr. Opin. Plant Biol. 2021, 61, 101991. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhang, Y. Profiling chromatin regulatory landscape: Insights into the development of ChIP-seq and ATAC-seq. Mol. Biomed. 2020, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Chen, M.; Yang, X.; Bao, H.; Yang, Y.; Wang, Y. The intersection of non-coding RNAs contributes to forest trees’ response to abiotic stress. Int. J. Mol. Sci. 2022, 23, 6365. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, I.A.; Fossdal, C.G.; Johnsen, Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010, 187, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Dalakouras, A.; Vlachostergios, D. Epigenetic approaches to crop breeding: current status and perspectives. J. Exp. Bot. 2021, 72, 5356–5371. [Google Scholar] [CrossRef] [PubMed]
- 131. Gahlaut, V; Zinta, G; Jaiswal, V; Kumar, S. Quantitative epigenetics: a new avenue for crop improvement. Epigenomes 2020, 4, 25. [CrossRef] [PubMed]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519. [Google Scholar] [CrossRef]
- Deng, X.; Song, X.; Wei, L.; Liu, C.; Cao, X. Epigenetic regulation and epigenomic landscape in rice. Natl. Sci. Rev. 2016, 3, 309–327. [Google Scholar] [CrossRef]
- Raju, S.K.K.; Shao, M.R.; Sanchez, R.; Xu, Y.Z.; Sandhu, A.; Graef, G.; Mackenzie, S. An epigenetic breeding system in soybean for increased yield and stability. Plant Biotechnol. J. 2018, 16, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Liu, S.; Yu, L.; Xiao, Y.; Zhang, S.; Wang, X.; Xu, Y.; Yu, H.; Li, Y.; Yang, J.; et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat. Biotechnol. 2021, 39, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- García-Murillo, L.; Valencia-Lozano, E.; Priego-Ranero, N.A.; Cabrera-Ponce, J.L.; Duarte-Aké, F.P.; Vizuet-de-Rueda, J.C.; Rivera-Toro, D.M.; Herrera-Ubaldo, H.; de Folter, S.; Alvarez-Venegas, R. CRISPRa-mediated transcriptional activation of the SlPR-1 gene in edited tomato plants. Plant Sci. 2023, 329, 111617. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: technologies and their applications. J. Chromatogr. Sci. 2016, 55, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Mergner, J.; Kuster, B. Plant proteome dynamics. Annu. Rev. Plant Biol. 2022, 73, 67–92. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C. Deciphering protein post-translational modifications using chemical biology tools. Nat. Rev. Chem. 2020, 4, 674–695. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, D.; Thaminy, S.; Hottiger, M.O.; Stagljar, I. The post-genomic era of interactive proteomics: Facts and perspectives. Proteomics 2002, 2, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Harmon, A.C. Advances in plant proteomics. Proteomics 2006, 6, 5504–5516. [Google Scholar] [CrossRef]
- Traversari, S.; Giovannelli, A.; Emiliani, G. Wood formation under changing environment: Omics approaches to elucidate the mechanisms driving the early-to-latewood transition in Conifers. Forests 2022, 13, 608. [Google Scholar] [CrossRef]
- Jing, D.; Zhang, J.; Xia, Y.; Kong, L.; OuYang, F.; Zhang, S.; Zhang, H.; Wang, J. Proteomic analysis of stress-related proteins and metabolic pathways in Picea asperata somatic embryos during partial desiccation. Plant Biotechnol. J. 2017, 15, 27–38. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Yang, J.; Zhou, W.; Dai, S. Exploring the diversity of plant proteome. J. Integr. Plant Biol. 2021, 63, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Mund, A.; Brunner, A.D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Zivy, M.; de Vienne, D. Proteomics: a link between genomics, genetics and physiology. Plant Mol. Biol. 2000, 44, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Kage, U.; Kumar, A.; Dhokane, D.; Karre, S.; Kushalappa, A.C. Functional molecular markers for crop improvement. Crit. Rev. Biotechnol. 2016, 36, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Agregán, R.; Echegaray, N.; López-Pedrouso, M.; Aadil, R.M.; Hano, C.; Franco, D.; Lorenzo, J.M. Proteomic advances in cereal and vegetable crops. Molecules 2021, 26, 4924. [Google Scholar] [CrossRef]
- Rodziewicz, P.; Chmielewska, K.; Sawikowska, A.; Marczak, Ł.; Łuczak, M.; Bednarek, P.; Mikołajczak, K.; Ogrodowicz, P.; Kuczyńska, A.; Krajewski, P.; et al. Identification of drought responsive proteins and related proteomic QTLs in barley. J. Exp. Bot. 2019, 70, 2823–2837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, B.; Huang, R.; Suo, H.; Zhang, Z.; Chen, W.; Dai, X.; Zou, X.; Ou, L. Transcriptome- and proteome-wide association of a recombinant inbred line population revealed twelve core QTLs for four fruit traits in pepper (Capsicum annuum L.). Hortic. Res 2022, 9, uhac015. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: a powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 1–20. [Google Scholar] [CrossRef]
- Jorrin Novo, J.V. Proteomics and plant biology: contributions to date and a look towards the next decade. Expert Rev. Proteomics 2021, 18, 93–103. [Google Scholar] [CrossRef]
- Schauer, N.; Fernie, A.R. Plant metabolomics: towards biological function and mechanism. Trends Plant Sci. 2006, 11, 508–516. [Google Scholar] [CrossRef]
- Bourgaud, F.; Gravot, A.; Milesi, S.; Gontier, E. Production of plant secondary metabolites: a historical perspective. Plant Sci. 2001, 161, 839–851. [Google Scholar] [CrossRef]
- Macel, M.; Van Dam, N.M.; Keurentjes, J.J. Metabolomics: the chemistry between ecology and genetics. Mol. Ecol. Resour. 2010, 10, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Montenegro-Burke, J.R.; Warth, B.; Spilker, M.E.; Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 2018, 36, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant metabolomics: an indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Plant metabolomics: analytical platforms and integration with functional genomics. Phytochem. Rev. 2008, 7, 479–497. [Google Scholar] [CrossRef]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef] [PubMed]
- Ribbenstedt, A.; Ziarrusta, H.; Benskin, J.P. Development, characterization and comparisons of targeted and non-targeted metabolomics methods. PLoS One 2018, 13, e0207082. [Google Scholar] [CrossRef] [PubMed]
- Zenobi, R. Single-cell metabolomics: analytical and biological perspectives. Science 2013, 342, 1243259. [Google Scholar] [CrossRef]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the diversity of plant metabolism. Trends Plant Sci. 2019, 24, 83–98. [Google Scholar] [CrossRef]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for plant improvement: status and prospects. Front. Plant Sci. 2017, 8, 1302. [Google Scholar] [CrossRef]
- Wang, H. , Guo, L. , Zha, R., Gao, Z., Yu, F., & Wei, Q. (2022). Histological, metabolomic and transcriptomic analyses reveal mechanisms of cold acclimation of the Moso bamboo (Phyllostachys edulis) leaf. Tree Physiology 2022, 42, 2336–2352. [Google Scholar] [PubMed]
- Ren, S.; Ma, K.; Lu, Z.; Chen, G.; Cui, J.; Tong, P.; Wang, L.; Teng, N.; Jin, B. Transcriptomic and metabolomic analysis of the heat-stress response of Populus tomentosa Carr. Forests 2019, 10, 383. [Google Scholar] [CrossRef]
- Mao, C.; Li, L.; Yang, T.; Gui, M.; Li, X.; Zhang, F.; Zhao, Q.; Wu, Y. Transcriptomics integrated with widely targeted metabolomics reveals the cold resistance mechanism in Hevea brasiliensis. Front. Plant Sci. 2023, 13, 1092411. [Google Scholar] [CrossRef] [PubMed]
- Soltis, N.E.; Kliebenstein, D.J. Natural variation of plant metabolism: genetic mechanisms, interpretive caveats, and evolutionary and mechanistic insights. Plant Physiol. 2015, 169, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Chibon, P.Y.; de Vos, R.C.; Schipper, B.A.; Walraven, E.; Beekwilder, J.; van Dijk, T.; Finkers, R.; Visser, R.G.F.; van de Weg, E.W.; et al. Genetic analysis of metabolites in apple fruits indicates an mQTL hotspot for phenolic compounds on linkage group 16. J. Exp. Bot. 2012, 63, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Du, Q.; Xiao, L.; Lv, C.; Quan, M.; Li, P.; Yao, L.; Song, F.; Zhang, D. Multi-omics analysis provides insights into genetic architecture of flavonoid metabolites in Populus. Ind. Crops Prod. 2021, 168, 113612. [Google Scholar] [CrossRef]
- Yang, L.; Yang, Y.; Huang, L.; Cui, X.; Liu, Y. From single-to multi-omics: future research trends in medicinal plants. Briefings Bioinf. 2023, 24, bbac485. [Google Scholar] [CrossRef] [PubMed]
- Jamil, I.N.; Remali, J.; Azizan, K.A.; Nor Muhammad, N.A.; Arita, M.; Goh, H.-H.; Aizat, W.M. Systematic multi-omics integration (MOI) approach in plant systems biology. Front. Plant Sci. 2020, 11, 944. [Google Scholar] [CrossRef]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinf. Biol. Insights 2020, 14, 1–24. [Google Scholar] [CrossRef]
- Yu, S.; Drton, M.; Promislow, D.E.; Shojaie, A. CorDiffViz: an R package for visualizing multi-omics differential correlation networks. BMC bioinf. 2021, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 2016, 16, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Sánchez, V.M.; Castillejo, M.Á.; López-Hidalgo, C.; Alconada, A.M.M.; Jorrín-Novo, J.V.; Rey, M.-D. Changes in the transcript and protein profiles of Quercus ilex seedlings in response to drought stress. J. Proteomics 2021, 243, 104263. [Google Scholar] [CrossRef] [PubMed]
- Pierre-Jean, M.; Deleuze, J.-F.; Le Floch, E.; Mauger, F. Clustering and variable selection evaluation of 13 unsupervised methods for multi-omics data integration. Briefings Bioinf. 2020, 21, 2011–2030. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Cañal, M.J.; Escandon, M.; Meijon, M.; Weckwerth, W.; Valledor, L. Integrated physiological, proteomic, and metabolomic analysis of ultra violet (UV) stress responses and adaptation mechanisms in Pinus radiata. Mol. Cell. Proteomics 2017, 16, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Saito, K.; Yamazaki, M. Integrated omics analysis of specialized metabolism in medicinal plants. Plant J. 2017, 90, 764–787. [Google Scholar] [CrossRef]
- Obudulu, O.; Mähler, N.; Skotare, T.; Bygdell, J.; Abreu, I.N.; Ahnlund, M.; Gandla, M.L.; Petterle, A.; Moritz, T.; Hvidsten, T.R.; et al. A multi-omics approach reveals function of Secretory Carrier-Associated Membrane Proteins in wood formation of Populus trees. BMC genomics 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Altman, T.; Travers, M.; Kothari, A.; Caspi, R.; Karp, P.D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC bioinf. 2013, 14, 1–15. [Google Scholar] [CrossRef]
- López-Hidalgo, C.; Guerrero-Sanchez, V.M.; Gómez-Gálvez, I.; Sánchez-Lucas, R.; Castillejo-Sánchez, M.A.; Maldonado-Alconada, A.M.; Valledor, L.; Jorrin-Novo, J.V. A multi-omics analysis pipeline for the metabolic pathway reconstruction in the orphan species Quercus ilex. Front. Plant Sci. 2018, 9, 935. [Google Scholar] [CrossRef]
- Zoppi, J.; Guillaume, J.-F.; Neunlist, M.; Chaffron, S. MiBiOmics: an interactive web application for multi-omics data exploration and integration. BMC bioinf. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Hu, H.; Campbell, M.T.; Yeats, T.H.; Zheng, X.; Runcie, D.E.; Covarrubias-Pazaran, G.; Broeckling, C.; Yao, L.; Caffe-Treml, M.; Gutiérrez, L.; et al. Multi-omics prediction of oat agronomic and seed nutritional traits across environments and in distantly related populations. Theor. Appl. Genet. 2021, 134, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef]
Figure 1.
Basic procedures of (A), conventional phenotype-based breeding; (B), marker-assisted selection; and (C), genomic selection.
Figure 1.
Basic procedures of (A), conventional phenotype-based breeding; (B), marker-assisted selection; and (C), genomic selection.
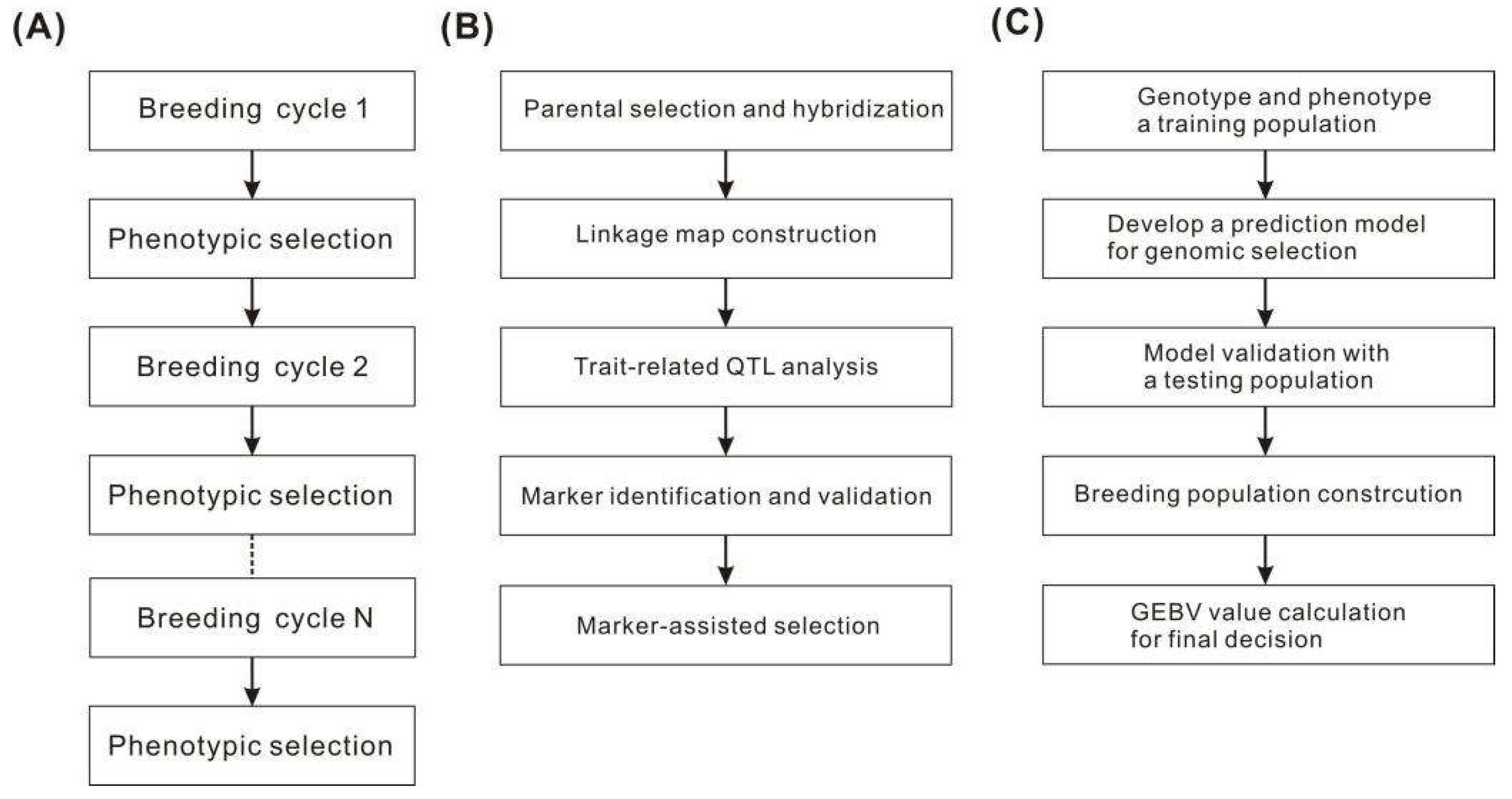
Figure 2.
A schematic diagram showing the advantage of TGS reads in the assembly of highly complex genomic regions over NGS reads.
Figure 2.
A schematic diagram showing the advantage of TGS reads in the assembly of highly complex genomic regions over NGS reads.
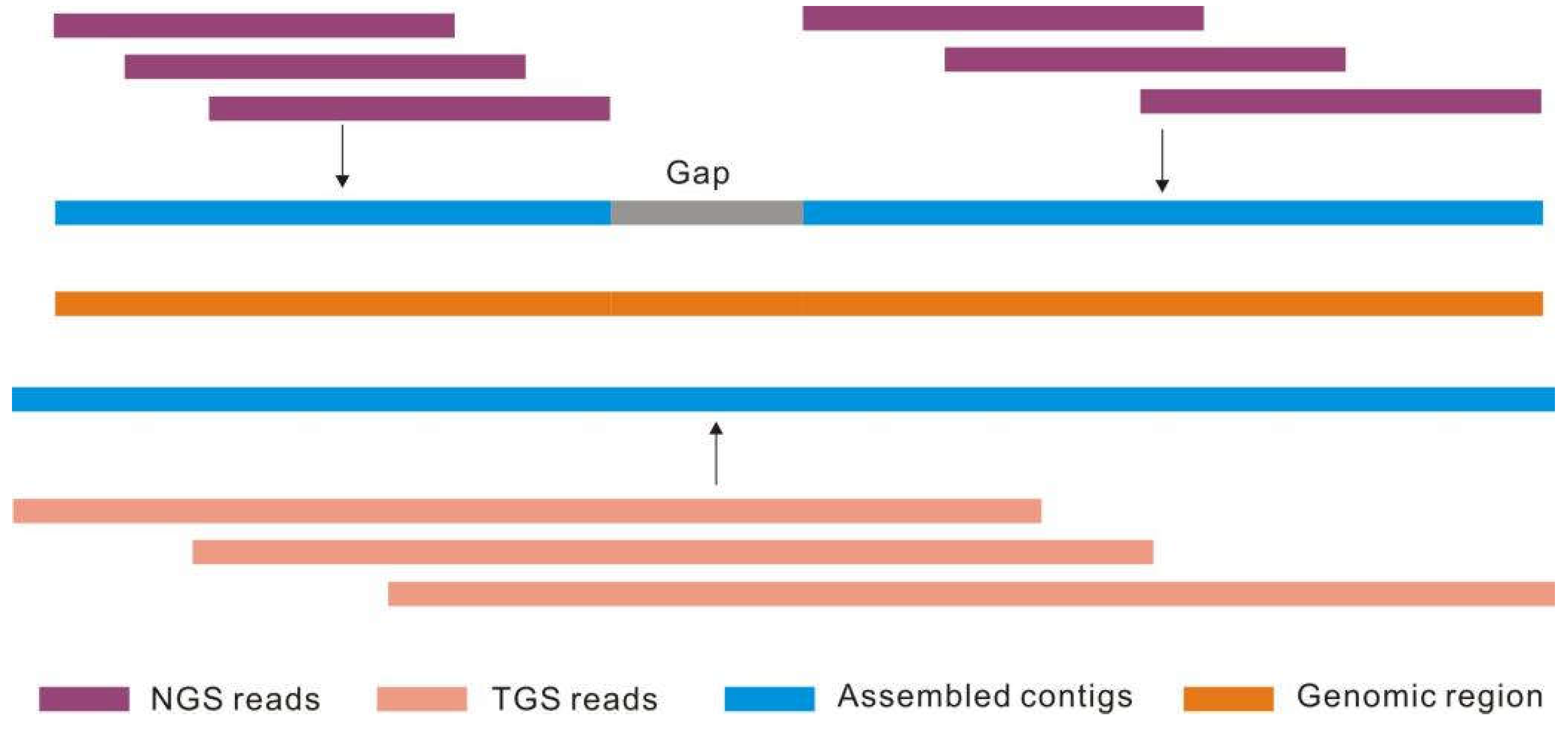
Figure 3.
A commonly used workflow for the analysis of whole transcriptome sequencing data.
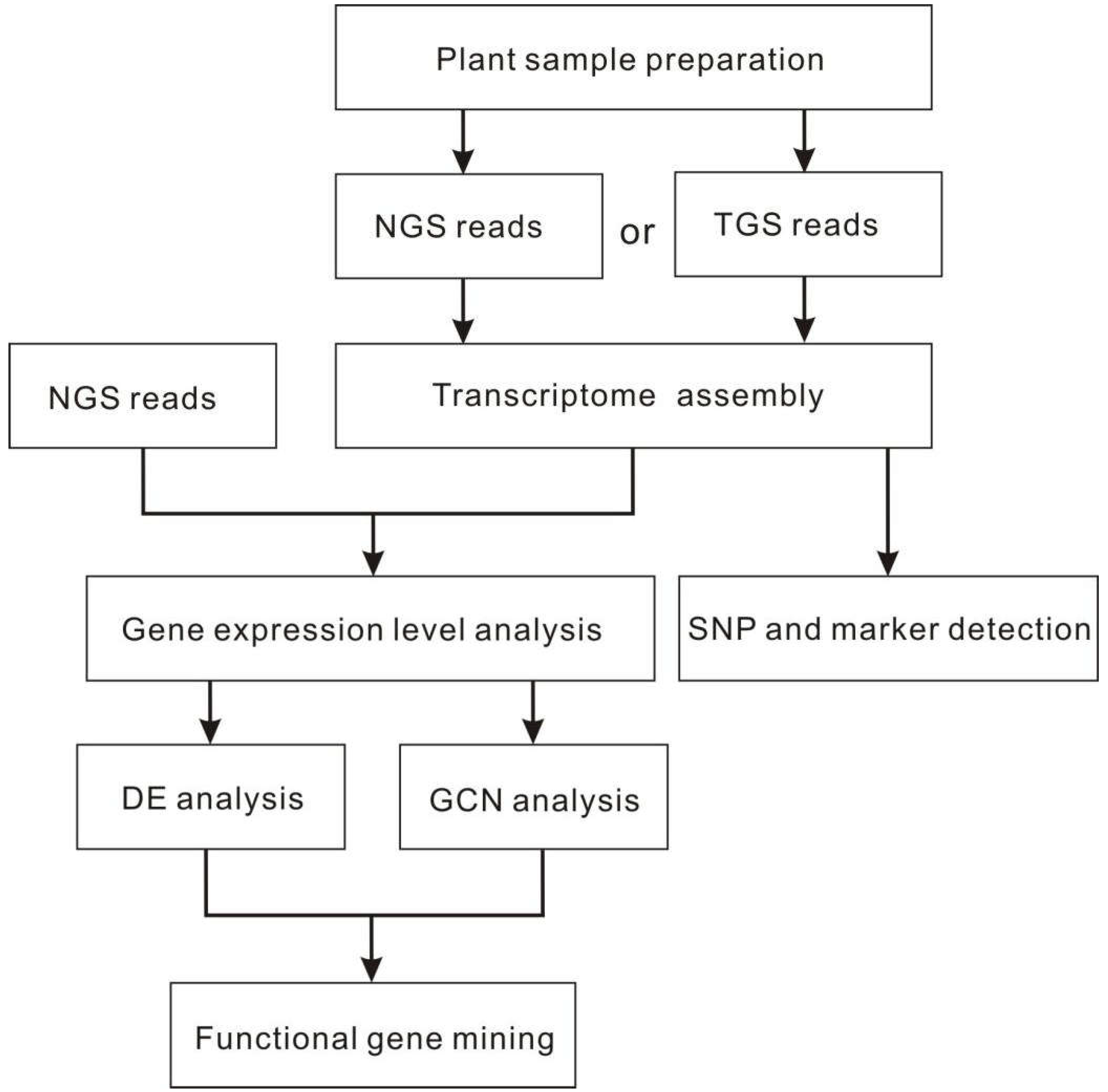
Figure 4.
Basic procedure of epi-breeding using epigenetic markers.
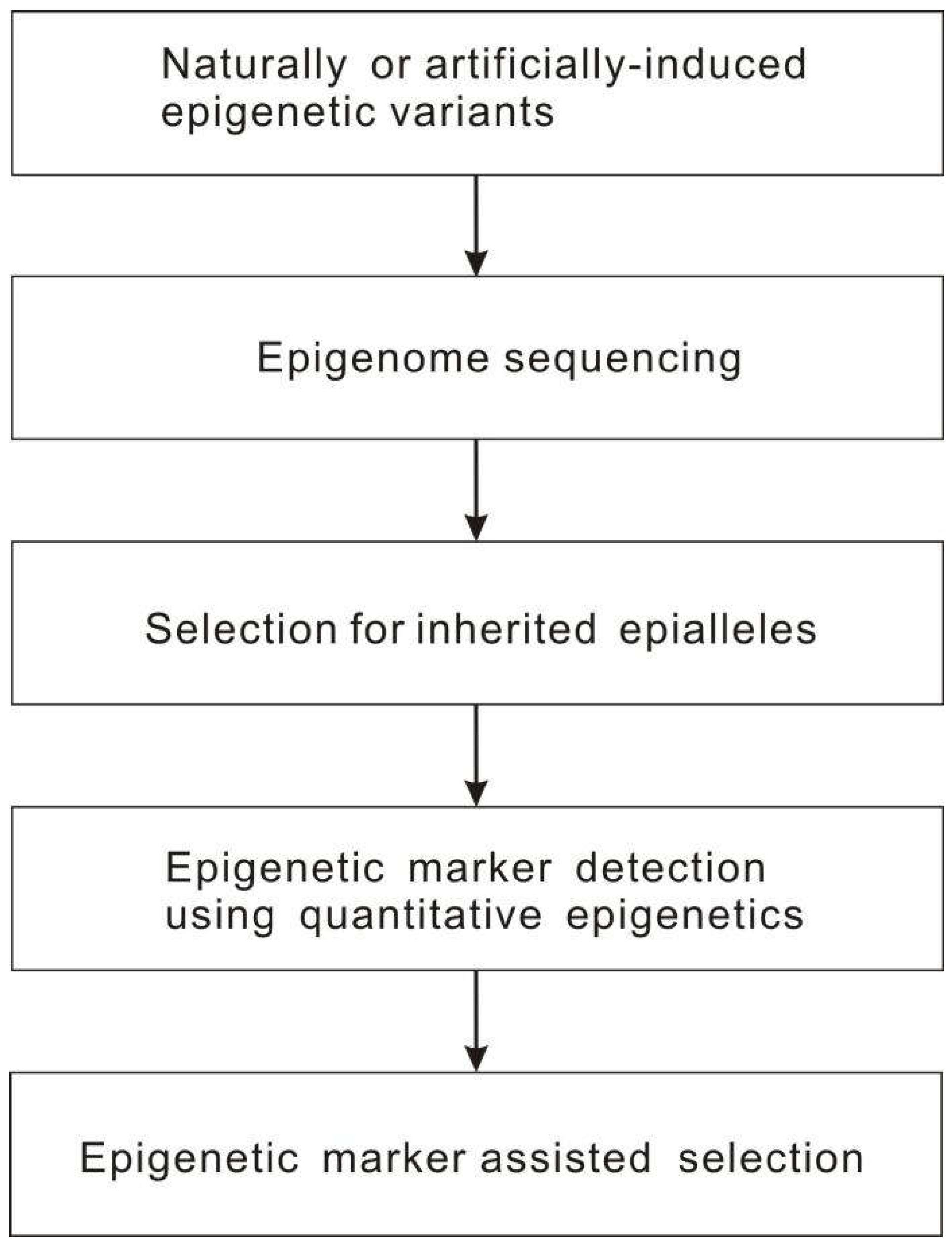
Figure 5.
Two most common pipelines for quantitative proteome analysis.
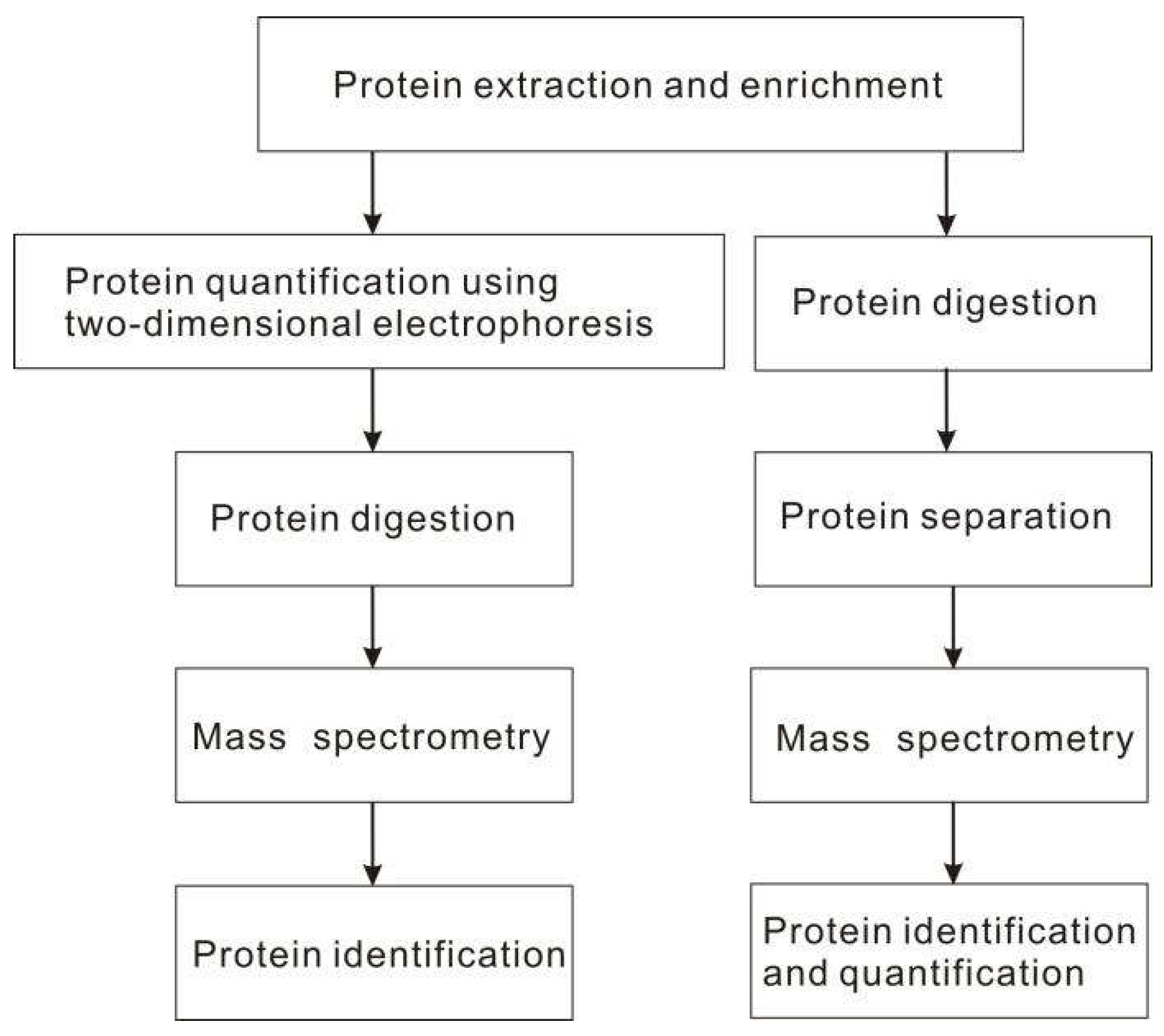
Figure 6.
Relationships among various omics technologies.
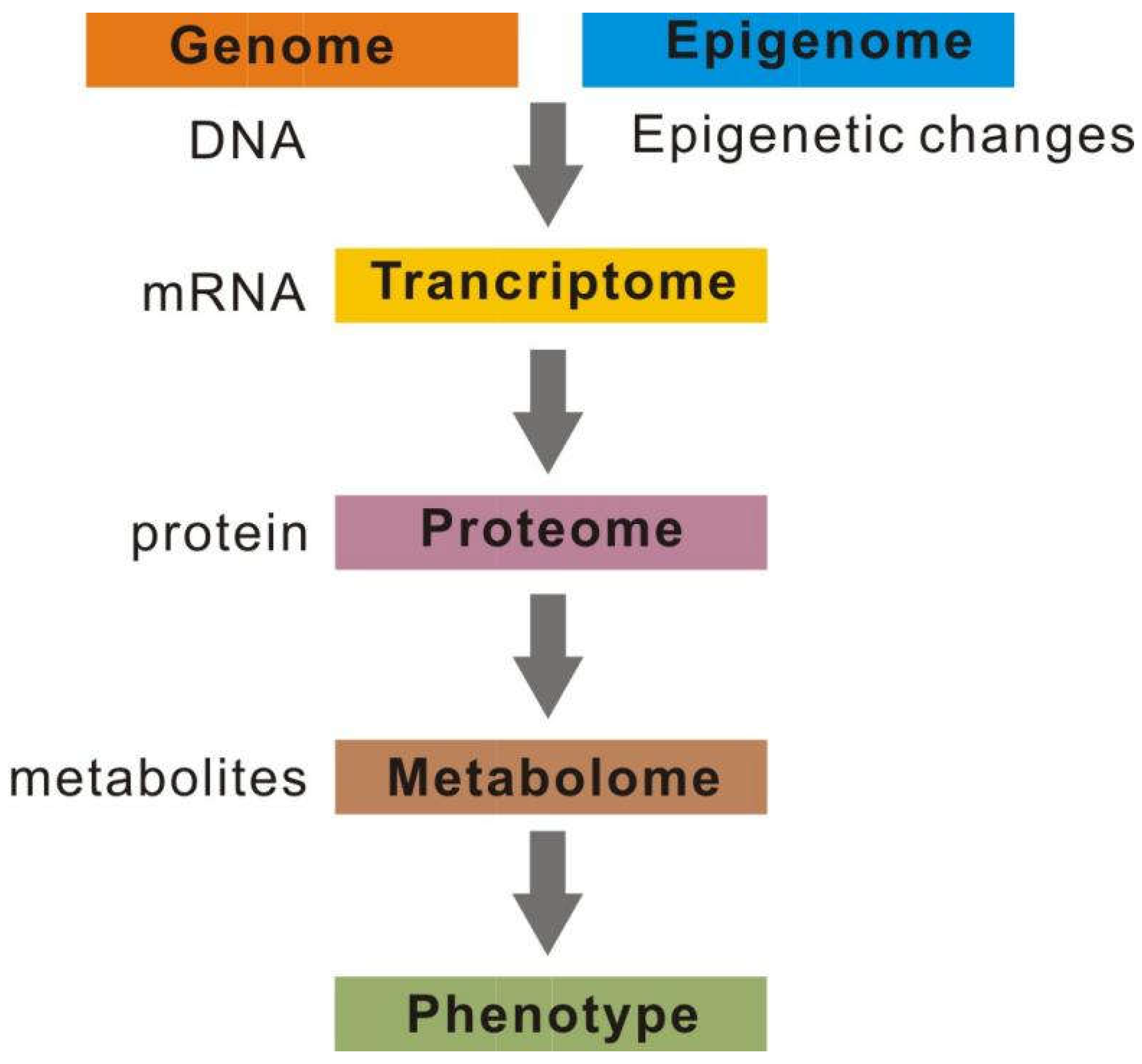
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



