Submitted:
18 April 2023
Posted:
19 April 2023
You are already at the latest version
Abstract
Gastric cancer is a challenging public health concern worldwide and remains a leading cause of cancer-related mortality. The primary risk factor implicated in gastric cancer development is infection with Helicobacter pylori. H. pylori induces chronic inflammation affecting the gastric epithelium, which can lead to DNA damage and promotion of precancerous lesions. Disease manifestations associated with H. pylori are attributed to virulence factors with multiple activities and its capacity to subvert host immunity. One of the most significant H. pylori virulence determinants is the cagPAI gene cluster, which encodes a type IV secretion system and the CagA toxin. This secretion system allows H. pylori to inject the CagA oncoprotein into host cells, causing multiple cellular perturbations. Despite the high prevalence of H. pylori infection, only a small percentage of affected individuals develop significant clinical outcomes, while most remain asymptomatic. Therefore, understanding how H. pylori triggers carcinogenesis and its immune evasion mechanisms is critical in preventing gastric cancer and miti-gating the burden of this life-threatening disease. This review aims to provide an overview of our current under-standing of H. pylori infection, its association with gastric cancer and other gastric diseases, and how it subverts the host immune system to establish persistent infection.
Keywords:
Gastric Cancer
; Helicobacter pylori
; cag Pathogenicity Island
; Cytotoxin-Associated Gene A
; Oncoprotein
; Vacuolating Toxin A
; Immune Evasion
1. Introduction
Gastric cancer (GC) remains among the deadliest cancers worldwide, taking close to one million lives annually. GC is the fourth leading cause of cancer-related deaths[1]. According to the American Cancer Society’s (ACS) estimates for 2022, in the United States, there are 26,380 new cases and 11,090 deaths attributed to GC[2]. Worldwide, in 2020 the incidence of GC was 1,089,103 and claimed 768,793 lives[3]. Although its incidence in the United States is declining, the prognosis for patients with GC is bleak since their five-year survival rate is low due to most GC cases (90%) being diagnosed at an advanced stage because of our inadequate understanding of the underlying mechanisms that regulate carcinogenesis and the lack of routine screening for GC.
The primary risk factor for GC is Helicobacter pylori, a micro-aerophilic, spiral-shaped Gram-negative bacterium classified as a class I carcinogen[4]. H. pylori is perhaps the most successful human pathogen since its infection is prevalent, infecting approximately one-half of the world’s population or approximately 4.4 billion individuals[5]. The human stomach is the only natural reservoir of H. pylori. In developing countries, the prevalence may reach as high as 90%[5]. In developed countries with the lowest infection rates, up to 40% of all adults are infected with H. pylori. Infection is generally acquired during childhood and, without appropriate treatment, can last the individual’s lifetime. Limiting infection is practically impossible due to antibiotic resistance and elevated reinfection rates. Although H. pylori infection is strongly associated with GC, only 1-3% of infected individuals develop GA, while most remain asymptomatic. It is not clear why only some H. pylori-infected individuals develop GA. However, various factors likely influence the outcome of the infection, including host susceptibility, environmental factors, and virulence of the infecting strain.
H. pylori was initially described by Australian scientists Barry Marshall and Robin Warren in 1982[6]. Their work with human gastric specimens identified H. pylori as the primary cause of chronic gastritis and peptic ulcer disease (PUD). Their research challenged the prevailing medical dogma that ulcers result from stress and lifestyle factors. Before their discovery, they treated peptic ulcers primarily using antacids and diet modification, such as avoiding spicy and acidic foods. However, these treatments were inadequate, and often vagotomy or surgery to remove the affected portion of the stomach or duodenum was the next step to reduce acid secretion. The work of Marshall and Warren transformed the treatment of PUD and has helped save many lives. Their efforts were recognized with the Nobel Prize in Physiology or Medicine in 2005. The infection is treated with antibiotics to eradicate the bacteria and acid-suppressing drugs.
2. H. pylori and Associated Diseases
2.1. Gastric Cancer
Although most persons infected with H. pylori are asymptomatic, infection with H. pylori may lead to several clinically significant disorders, including chronic gastritis, PUD, mucosal tissue-associated lymphoma, and GC. Chronic gastritis, if left untreated, may progress to atrophic gastritis and intestinal metaplasia, precancerous lesions in the sequence proposed by Pelayo Correa[7]. According to his model, H. pylori infection activates a cascade of histologic changes that progress through several phases, from chronic superficial gastritis to atrophic gastritis, intestinal metaplasia, and dysplasia, before ultimately resulting in GC. This model of GC progression stresses the importance of early detection and treatment of chronic gastritis, specifically in persons with a history of H. pylori infection.
The most common type of GC is adenocarcinoma, which represents 95% of cases. GC is usually regarded as one condition and is classified based on anatomic location within the stomach, histologic type of cancer, and stage[8]. Anatomically, GC is classified as either cardia/proximal (upper part of the stomach) or non-cardia/distal (antrum and pylorus). Histologically, it may be classified as either diffuse or intestinal. The GC stage is determined by the tumor size, spread to sentinel lymph nodes or whether it has metastasized to distant anatomical sites, such as the liver, lungs, or bones. Approximately 90% of non-cardia GC cases are due to H. pylori infection[9]. Because of the wealth of epidemiologic data linking H. pylori in the development of GC, in addition to observations in animal models, H. pylori was classified as a class I carcinogen by the International Agency for Research on Cancer (IARC) together with the World Health Organization (WHO)[10].
The incidence of GC varies geographically across different parts of the world. It is highest in Eastern Europe, Eastern Asia, and Latin America, while the lowest incidence is in North America, Western Europe, Australia, and Africa[11]. Not surprisingly, the highest incidence of GC occurs in developing countries, coinciding with high H. pylori carriage rates. An interesting observation was made with patients in Africa, where the prevalence of H. pylori infection is very high, but the incidence of GC is low. This apparent discrepancy between the high prevalence of H. pylori infection in Africa and the low incidence of GC in these regions was initially referred to as the "African Enigma" [12],[13]. To address this paradox, Fox and colleagues investigated the possibility that concurrent parasitic infection could influence the immune response to H. pylori, initially reported by us and others as skewed toward a Th1 immune response[14]. They hypothesized that helminth infections, which stimulate Th2-polarized responses, could modify the Th1 immune response induced by H. pylori and thus change the outcome of the infection. They studied mice infected with H. felis with and without simultaneous infection with the enteric nematode Heligmosomoides polygyrus, which has a strictly enteric life cycle[15]. They observed that the nematode infection prevented the development of gastric atrophy. This correlated with a significant decrease in mRNA for cytokines and chemokines associated with a gastric inflammatory response of Th1 cells[15]. These observations led to the conclusion that nematode infections ameliorate gastric atrophy, a precancerous lesion. However, subsequent studies in mice by the same group suggested that concomitant infection with other Helicobacter species could differentially affect gastric pathology[16]. They noted that H. muridarum coinfection significantly attenuated H. pylori-associated gastric pathology; however, coinfection with H. hepaticus promoted H. pylori-associated gastric disease[16]. Interestingly, the exacerbated pathology in mice coinfected with H. pylori and H. hepaticus was not due to increased Th1 responses since those mice had lower mRNA levels of gastric Th1 cytokines Tnf-α, Ifn-γ, and Il-1β than mice infected only with H. pylori. Instead, the dually infected mice had higher mRNA levels of gastric Il-17A than mice infected with H. pylori alone[16].
The concept of the "African enigma" was challenged by a study of the literature regarding PUD in the African continent[13]. As explained below, PUD is also linked to H. pylori infection, and similar observations have been made regarding its incidence in African populations; thus, PUD was considered a surrogate to examine its incidence in the context of H. pylori infection. The study concluded that the "African enigma" reflected inadequate data obtained from people lacking resources, healthcare access, and a comparatively short life expectancy[13]. However, a different set of H. pylori-related observations had been made with Asian populations with comparatively lower infection rates. One study reported the seroprevalence of H. pylori among Japanese and Chinese adults of approximately 50%, but the prevalence of GC in those populations is high[17]. One explanation for this higher prevalence of GC in East Asia could be differences in the H. pylori CagA virulence factor in East Asian strains compared to Western strains[18,19], which make those strains more virulent, as discussed in detail below. A more in-depth study to attempt to explain regional differences in GC prevalence in populations with similar rates of H. pylori infection was led by Correa’s team of investigators who examined GC rates among inhabitants in the state of Nariño, Colombia. The rates of GC among inhabitants of the high-altitude Andes Mountains were high (∼150 per 100,000), while the incidence rate of GC for those living at sea level was low (∼6 per 100,000)[20]. It is worth noting that the high-risk mountain (Tuquerres) population is only ~150 Mi away from the coastal low-risk populations (Tumaco). Although the prevalence of H. pylori infection is high in both groups (> 80% after age 10), there were significant differences in how their inhabitants were affected. Atrophy was more common, and the incidence of GC was higher in high versus low-altitude regions[20,21]. One study reported differences in the virulence genotypes (cagA positive and vacA s1 and m1) in both regions’ prevailing H. pylori strains [22]. It is important to note the differences between the inhabitants of those two regions. They differ in their ancestry, with primarily African origin in the coastal region (58%) and mostly Amerindian ancestry in the mountain region (67%)[23]. Also, dietary differences, the incidence of helminthiasis and toxoplasmosis, and, more recently, gastric microbiomes were reported to differ between both groups. Altogether, these observations in different regions led to the role of virulence of the infecting strains, the gastric microbiome, coinfections, environmental factors such as diet, and host genetics as factors influencing the outcome of the infection.
2.2. Peptic Ulcer Disease
PUD is another condition that most often involves H. pylori, accounting for 90–95% of duodenal ulcers and 70–85% of gastric ulcers[24]; the remainder of cases are due to non-steroidal anti-inflammatory drugs (NSAIDs). PUD is frequently defined as a rupture in the gastric or duodenal mucosa greater than 3-5 mm caused by an imbalance in mucosal protective and injurious factors[25]. PUD may have significant complications such as bleeding, perforation, penetration into adjacent organs, obstructions, and death may result from these complications. A systematic literature review reported an average mortality of 8.6% after bleeding and 23.5% after perforation 30 days later[26]. Mortality increases with age, comorbidities, shock, and treatment delays[26]. Although the incidence of PUD has decreased recently due to improved diagnosis and treatment of H. pylori infection, PUD still is a public health issue that causes significant distress and severe complications if left untreated. The annual incidence of PUD is 0.1-0.3%, affecting approximately 10% of the population worldwide[27]. H. pylori infection and the ensuing inflammation can alter gastric acid output resulting in either hypochlorhydria or hyperchlorhydria, which determines the type of peptic ulcer. Hypochlorhydria results from suppressed gastric acid secretion and may lead to pangastritis and the formation of gastric ulcers. These patients have an increased prevalence of corpus atrophy and intestinal metaplasia[28]. On the other hand, approximately 15% of patients infected with H. pylori develop hyperchlorhydria with predominant antral gastritis associated with duodenal ulcers.
2.3. Mucosa-Associated Lymphoid Tissue (MALT) Lymphoma
Another gastric condition involving H. pylori is extranodal marginal zone gastric mucosa-associated lymphoid tissue (MALT) lymphoma, a non-Hodgkin’s lymphoma, an indolent lymphoproliferative disease involving small heterogeneous B lymphocytes. Gastric MALT lymphoma accounts for 40 to 50% of primary gastric lymphomas and 1 to 6% of all gastric malignancies[29]. Although the stomach is the most common site of MALT lymphomas, it usually lacks organized lymphoid tissue. Still, active chronic inflammation of the H. pylori-infected gastric mucosa induces the organization of lymphoid tissue. Studies in vitro showed that gastric MALT lymphoma cells were stimulated by heat-killed H. pylori and involved H. pylori-specific T-cells via CD40 and CD40L interactions[30,31]. Interestingly, in those studies, the investigators noted that the T-cell clones from MALT lymphoma had reduced perforin-mediated cytotoxicity and Fas-mediated apoptosis[30]. The gastric mucosa of most cases of gastric MALT lymphoma contains H. pylori[32], and eradicating H. pylori with the corresponding treatment results in the complete remission of gastric MALT lymphoma in most cases[33,34].
2.4. Extragastric Manifestations of H. pylori
Although the association of H. pylori infection with gastric diseases is well established, the infection is thought to exert systemic pathological effects leading to non-gastric clinical outcomes. Those conditions include type 2 diabetes mellitus[35], insulin resistance[36], myocardial infarction[37], iron deficiency anemia[38], primary immune thrombocytopenia[39], Parkinson disease [40], among others. However, it is unclear how infection with H. pylori is positively associated with these non-GI disorders.
As research into H. pylori disease associations and efforts to eradicate this common human pathogen have expanded, leading to increased prevalence of some conditions; a question that has emerged is whether H. pylori is a true pathogen or a commensal organism. Various studies have credited H. pylori with positive effects for the host because H. pylori infection was noted to be inversely associated with the development of some disorders. For instance, there seems to be an inverse relationship between the H. pylori infection and gastroesophageal reflux disease (GERD)[41]. A cross-sectional case-control study of 5,616 subjects undergoing both upper endoscopy and H. pylori serology reported an inverse relationship between the presence of H. pylori and GERD [41]. In that study, H. pylori prevalence was lower in cases with reflux esophagitis than in the controls (38.4% vs. 58.2%, P < 0.001). A meta-analysis showed that eradicating H. pylori could lead to erosive GERD[42].
Interestingly, a meta-analysis also showed an inverse correlation between H. pylori colonization and the risk of esophageal cancer[43]. The study suggested that the increase in esophageal cancer incidence may be linked to the decreased prevalence of H. pylori in Western countries. A likely mechanism underlying this outcome is H. pylori urease activity (described below in detail) which neutralizes gastric acidity and, in turn, decreases the risk of GERD.
Other studies have also attributed H. pylori colonization with protection against childhood asthma, inflammatory bowel disease (IBD), and celiac disease. The relationship between H. pylori and asthma has been the subject of active investigation. Evidence suggests that H. pylori infection may be associated with a reduced risk of developing asthma[44,45]. As the incidence of H. pylori infection decreases in developed countries and various developing areas, asthma in children and other atopic disorders are rising. Chen and colleagues found a compelling inverse relationship between H. pylori infection and the early onset of asthma[44]. A hospital-based case-control study of a pediatric population reported that children who were H. pylori seropositive had a reduced likelihood of developing asthma than seronegative children (adjusted OR, 0.31 [95% CI, 0.10-0.89])[46]. H. pylori infection may protect from asthma and atopy by promoting an immune response that reduces inflammation in the airways, a crucial feature of asthma. The inverse relationship between H. pylori infection and asthma may involve the H. pylori induction of a polarized Th1 response[14,47]. Several mechanisms contribute to the prevalent H. pylori-induced mucosal Th-1 response. One virulence factor discussed below is the H. pylori neutrophil-activating protein (HP-NAP), which strongly upregulates both IL-12 and IL-23 production, fostering polarized Th-1 response[48,49]. The resulting cytokines from those Th1 cells may inhibit the Th2 responses characteristic of atopy. Another possible mechanism underlying the inverse association between H. pylori and asthma is the induction of regulatory T cells (Tregs) by H. pylori infection[50,51,52], which may influence the prevention of allergic disease. In fact, H. pylori-positive persons have higher gastric and circulating Treg levels than H. pylori-negative individuals[53,54]. The remote regulation of the respiratory mucosa by immune responses in the gastrointestinal mucosa is consistent with the concept of the "common mucosal immune system[55]
IBD is another disease noted to have an inverse association with H. pylori infection. IBD is an umbrella term for chronic relapsing-remitting digestive disorders, including Crohn’s Disease (CD) and ulcerative colitis (UC). H. pylori infection was regarded as a possible IBD risk factor due to similarities in the immunobiology of H. pylori infection and IBD. However, multiple studies have suggested an inverse association between H. pylori infection and the prevalence of IBD. There are essential epidemiological differences between H. pylori and IBD. H. pylori infection is more prevalent in developing countries than in developed countries. In contrast, the opposite is true for IBD, which is more prevalent in developed than developing countries. The prevalence of IBD is steadily increasing in developed countries, while rates of H. pylori infection are decreasing. IBD is less frequent among individuals who are H. pylori seropositive when compared to seronegative subjects[56]. A meta-analysis of 80,789 subjects (6,130 patients with IBD and 74,659 non-IBD controls) revealed a significant negative correlation between IBD and H. pylori infection[57]. In that study, the investigators observed a consistent negative association between IBD and H. pylori infection regardless of age, ethnicity, and detection methods[57]. These observations suggest that H. pylori might exert a protective effect against IBD.
3. H. pylori Virulence Factors and Cancer Mechanisms
H. pylori possesses a panel of virulence mechanisms that allow it to survive in the harshest environment of the body, which is the stomach (pH between 1.5 and 3.5)[58], colonize the gastric mucosa, influence the integrity of the epithelium, and subvert the host response to establish persistent infection and induce pathology[59,60,61,62,63,64].
3.1. Urease
H. pylori urease is a critical virulence factor for H. pylori. It is the most abundant protein expressed by H. pylori, representing 10–15% of total protein by weight.[65]. H. pylori urease is a multimeric nickel-containing enzyme consisting of two subunits, α and β subunits, of 29.5 kDa and 66 kDa, which catalyzes the hydrolysis of urea into carbonic acid and ammonia. Six α and six β subunits aggregate to form a multimeric complex of 550 kDa.[65]. The released ammonia from urea hydrolysis increases the pH and provides a local protective environment for H. pylori.
H. pylori urease can play a role in pathogenesis beyond its enzymatic action. Urease influences the host response by activating monocytes and polymorphonuclear leucocytes, leading to inflammation and damage to the epithelium[66]. Approximately 30% of total urease is located on the bacterial surface[67,68]. The mechanism behind the presence of urease on the bacterial surface involves bacterial autolysis and association with remaining whole cells[69]. This surface localization allows urease to mediate additional effects via interactions with proteins on the surface of epithelial cells. We showed that urease binds to class II major histocompatibility complex (MHC) and triggers apoptosis of gastric epithelial cells[70,71], which express class II MC molecules[72,73]. This interaction is mediated by the urease A subunit[74]. Interestingly, the urease B subunit also contributes to the adhesion of H. pylori to gastric epithelial cells via association with CD74[75]. Using recombinant urease subunits and urease B knockout bacteria, H. pylori urease B was shown to bind CD74 on gastric epithelial cells and induce NF-κB activation and interleukin-8 (IL-8) production[75]. These responses decreased in the presence of blocking CD74 with monoclonal antibodies.
3.2. Adhesins
Adhesins are bacterial cell-surface proteins contributing to the bacterial attachment to host cells. The adhesion of H. pylori to the gastric epithelium is a key step for colonizing the gastric mucosa. A bacterial adhesin that contributes to this process and determines colonization density is encoded by the babA2 gene is the blood group antigen binding adhesin (BabA), which bind to Lewis B blood group antigens (Leb) on gastric epithelial cells[76,77]. One study classified H. pylori strains into BabA high producers (BabA-H) with Leb binding activity, BabA low producers (BabA-L) without Leb binding activity, and BabA-negative strain (lacked the babA gene)[78]. Studies by Sheu et al. noted that strains positive for the babA2 gene with lower levels of BabA expression appeared to be associated with the highest GC risk[79].
The H. pylori outer inflammatory protein A (OipA), or HopH, is an outer membrane protein that behaves as an adhesin and stimulates IL-8 secretion. Yamaoka’s group showed that the functional status (on or off) of Oipa is controlled by a slipped-strand mispairing mechanism that depends on the number of CT dinucleotide repeats in the 5′ region of the oipA gene[80]. H. pylori strains with the oipA gene on "on" status are associated with greater colonization density, higher IL-8 production, and, consequently, a higher neutrophil infiltration. Further, strains with the oipA gene on "on" status significantly associate with GC and PUD.
The adherence-associated lipoproteins A and B (AlpA/AlpB) are outer membrane proteins participating in H. pylori binding to gastric epithelial cells and enhancing colonization. They also induce the production of the inflammatory cytokines IL-6 and IL-8[81]. Proof of their role in adhesion came from studies in which antiserum against the AlpA fusion protein or knock-out of alpAB or alpA genes blocked or decreased the binding of H. pylori to epithelial cells[82,83]. Moreover, we reported that AlpAB mediated binding to live gastric epithelial cells and triggered cellular signaling pathways within the cells[81]. Despite the multiple adhesins employed by H. pylori to attach to gastric epithelial cells, deletion of alpAB alone reduced bacterial binding by 60–70% and reduced H. pylori colonization of mice.
Sialic acid-binding adhesin A (SabA) is another crucial virulence factor that facilitates adherence to the gastric epithelium and colonizing the gastric mucosa[84]. SabA is a sizeable outer membrane protein encoded by the sabA gene, which is present in the H. pylori cag pathogenicity island (described below)[84]. SabA is a pleiotropic protein with sialic acid-binding activity. It links the sialyl-Lewis A and X glycan antigens explicitly. SabA specifically binds to α2-3-linked sialic acids abundantly expressed on the gastric mucosa, especially in the gastric antrum, where H. pylori primarily colonize[85,86]. The SabA binding to sialic acids on surface gastric mucins aids H. pylori adherence to the epithelium, a crucial step in establishing infection. In addition to sialic acid binding, SabA has been shown to interact with other host molecules, such as laminin[85], fibronectin[85], and blood group antigens. These interactions enhance the ability of H. pylori to adhere to diverse types of gastric epithelial cells and promote colonization in different stomach regions.
3.3. CagA and the Pathogenicity Island
The H. pylori cag pathogenicity island (cag PAI) is a region of about 40 kb DNA that encode the cytotoxin-associated gene A (cagA) and approximately 27-30 additional genes that encode proteins that form a type IV secretion system (T4SS)[87]. The T4SS is a syringe-like pilus feature that facilitates the translocation of the effector protein CagA and other bacterial products into gastric epithelial cells, influencing the pathogenesis of H. pylori. CagA is a 120-140 kDa protein whose expression by H. pylori represents the most potent risk factor for GC[88]. The CagA N-terminal domain contains a binding site for α5β1 integrin which is a crucial interaction in the transfer of CagA into the gastric epithelial cells[89,90]. Once inside gastric epithelial cells, CagA binds to phosphatidyl serine (PS) in the inner surface of the cell membrane and is tyrosine-phosphorylated by Src/Abl tyrosine kinases at glutamate-proline-isoleucine-tyrosine-alanine (EPIYA) motifs located in the C-terminus of CagA. Phosphorylated CagA forms complexes with Src-homology 2 (SH2) domains in SHP2, Grb2, and CSK and modifies several signaling pathways in gastric epithelial cells resulting in anomalous cytoskeletal changes, cellular proliferation, and differentiation as well as induction of inflammatory cytokines[91]. The magnitude of activation of downstream pathways depends on the type of CagA EPIYA motif and the number of copies. EPIYA motifs vary widely among H. pylori strains and are classified into four types based on the variation of flanking regions and orders of spacers. The four EPIYA motif types are EPIYA-A, -B, -C, and -D. CagA in Western strains of H. pylori contains EPIYA-A, EPIYA-B, and EPIYA-C motifs. In contrast, CagA from East Asian H. pylori strains have EPIYA-A, EPIYA-B, and EPIYA-D but not the EPIYA-C motif[92]. Higashi and colleagues reported that the EPIYA-C segment comprises 34 amino-acid residues that variably repeat [92]. These motifs in the C-terminus of CagA enhance its polymorphism and are present as tandem repeats varying from one to seven[18]. The levels of phosphorylation and the effects seen in gastric epithelial cells are proportional to the number of EPIYA motifs[93]. Because CagA rarely contains both EPIYA-C and EPIYA-D motifs, EPIYA-C is considered characteristic of Western CagA, while EPIYA-D is a feature of East Asian CagA. The East Asian CagA has a stronger SHP-2 binding, which confers a stronger ability to perturb cellular functions[92], which may explain differences in GC incidence in East Asia versus Western countries.
CagA has also been reported to impact epithelial cells in a tyrosine phosphorylation-independent manner. CagA has a conserved repeat responsible for phosphorylation-independent activity (CRPIA: FPLKRHDKVDDLSKVG) motif in its C-terminal region, different from the EPIYA motifs. The CRPIA motif in non-phosphorylated CagA interacts with c-Met, the hepatocyte growth factor scatter factor receptor, which plays a role in the invasive growth of neoplastic cells. CagA binds c-Met, which then binds phospholipase Cγ (PLCγ) and turns on phosphatidylinositol 3-kinase/Akt signaling. This initiates β-catenin and NF-κB signaling, leading to proliferation and inflammation[94,95]. Growth factor receptor-bound protein 2 (Grb2) also interacts with CagA independent of tyrosine phosphorylation, and this interaction stimulates the Ras/MEK/ERK pathway to cause cell scattering and proliferation[96].
Support for the role of CagA as an oncoprotein was obtained from multiple observations. A study by Peek’s group that included Mongolian gerbils infected with the carcinogenic strain 7.13 resulted in gastric dysplasia and cancer in >50% of gerbils infected with the wild-type strain. In that study, none of the gerbils infected with the cagA− mutant strain developed these preneoplastic lesions[97]. Various studies in which the H. pylori cagA gene alone was transfected into cells have shown that its expression significantly affects the transfected cells. In one report, CagA transfected into human gastric epithelial cells revealed that CagA targets partitioning-defective 1 (PAR1). The CagA-PAR1 interaction causes junctional and polarity defects that release cells from growth-inhibitory signals and promote neoplasia[98]. Using the transfection approach, another group demonstrated that CagA interacted with E-cadherin and perturbed the formation of E-cadherin/-catenin complexes, leading to the accumulation of β-catenin in the cytoplasm and nucleus and activating β-catenin signaling[99]. In that study, CagA-transfected cells expressed intestinal-specific molecules as an indication of intestinal transdifferentiation of gastric epithelial cells. Another process whereby CagA promotes gastric cancer was recently reported in which CagA phosphorylation affects the ubiquitin-proteasome system by binding the E3 ubiquitin ligases SIVA1 and ULF[100]. That interaction caused the activation of ULF and degradation of SIVA1 and the tumor suppressor p14ARF. The suppression of ARF results in the inhibition of apoptosis and oncogenic stress response, advancing cancer. Perhaps the most convincing study was reported by Ohnishi et al., who engineered transgenic mice expressing wild-type or phosphorylation-resistant CagA[101]. The mice expressing wild-type CagA developed gastric epithelial hyperplasia, and some developed gastric polyps and adenocarcinomas. Remarkably, some wild-type CagA transgenic mice developed hematological malignancies such as myeloid leukemias and B-cell lymphomas. In contrast, mice expressing phosphorylation-resistant CagA did not have evidence of these pathological abnormalities[101]. These observations led to CagA being regarded as a bacterial oncoprotein of importance in human neoplasia.
Another bacterial product translocated by the T4SS into gastric epithelial cells is peptidoglycan, which is recognized by NOD1 (Nucleotide Binding Oligomerization Domain Containing 1), a cytoplasmic pattern recognition receptor[102,103,104], abundantly expressed in gastric epithelial cells[104]. NOD1 activation results in NF-κB activation and inflammatory cytokine production[105].
3.4. Vacuolating Toxin (VacA)
The H. pylori vacuolating toxin (VacA) is a major secreted virulence factor without known homologs in other bacterial species except for H. cetorum, a Helicobacter species found in the stomachs of marine mammals[106]. The VacA toxin is a pore-forming toxin secreted by H. pylori and has pleiotropic effects on host cells. The pores induced by VacA in the membranes of the gastric epithelial lining lead to the leakage of small molecules and ions from the cells. This damage to the cells disrupts the epithelium’s barrier function, facilitating invasion and colonization of the stomach. VacA can also induce apoptosis in immune cells and gastric epithelial cells. This contributes to gastric inflammation and the progression of H. pylori-associated diseases. VacA also modulates the host immune response to H. pylori by suppressing the activation of specific immune cells and inhibiting the production of cytokines. This permits H. pylori to evade immune-mediated clearance and establish a persistent infection in the stomach.
VacA is first synthesized as a 140 kDa pro-toxin with an N-terminal signal peptide, a central region representing the toxin, and a C-terminus that mediates transport function. The central region (about 88 kDa), the mature virulent form of the toxin, is secreted after processing and is further cleaved into an A subunit and B subunit of 33 and 55 kDa, respectively. The p33 form was initially regarded as the pore-forming subunit, and the p55 form was ascribed to the cell binding function[107,108]. However, both subunits bind and form vacuoles [109,110]. H. pylori VacA binds to sphingomyelin on lipid rafts[111] and is then endocytosed via a clathrin-independent route[112]. VacA is delivered to early endosomes, which are routed by F-actin comets to become integrated into motile vesicles that fuse with mitochondria or late endosomes, where VacA induces apoptosis or vacuolation, respectively[113,114]. VacA has multiple damaging effects on mitochondria, including activation of the proapoptotic proteins Bax and Bak[115], cytochrome c release[116], and mitochondrial fragmentation[117], ultimately resulting in cell death.
Even though all strains of H. pylori have the vacA gene, there is substantial diversity in the gene, which includes five heterogenic regions with considerable sequence differences. The first three regions identified correspond to the signal (s) sequence, middle (m), and intermediate (i)[118]. There are two genotypes for s (s1a-c, s2), four for m (m1a, m1b, m1c, and m2), and three for the i region (subdivided into i1a, i1b, and i2). More recently, the deletion d-region (d1 and d2) and the c-region (c1 and c2), which corresponds to a 15 bp deletion at the 3′ end of the p55 domain of the vac A gene, were described[119,120]. The signal peptide region of VacA provides vacuolating activity and target specificity. The s2 region contains an additional 12-amino-acid N-terminal sequence which abolishes the formation of anion-selective channels and cell vacuolation[121]. While s1/m1 and s1/m2 VacA genotypes cause severe chronic inflammation when compared to the other genotypes, H. pylori strains with m1 VacA represent a higher risk factor for gastric ulcers[122], and VacA s1 and m1 genotypes are associated with higher pathology, including substantial infiltrates of neutrophils and lymphocytes, gastric atrophy, and intestinal metaplasia[123,124]. Yamaoka’s group showed that the vacAs1i1m1 allelic combination is strongly associated with the existence of cagA[125], and a recent study by Chang and colleagues demonstrated that strains expressing both vacAs1m1 and cagA concurrently have a 4.8-fold greater risk of inducing preneoplastic lesions than other H. pylori strains[126]. The i region is found between the s and m regions and is associated with polymorphism and GC[127]. The m region has a 148 amino acid segment that defines VacA’s cell binding specificity[128]. The roles of the c and d regions in the biology of VacA are currently unknown.
VacA is crucial in H. pylori’s ability to evade host immunity since it can act on several immune cells affecting innate and adaptive immunity. VacA impairs functions of macrophages[129,130], eosinophils[131], mast cells[132], dendritic cells[133], and lymphocytes[134,135]. Due to its effects on endosomal trafficking, VacA interferes with antigen processing by B cells. Molinari and colleagues showed that VacA disrupts the proteolytic processing of antigens and inhibits the Ii-dependent antigen presentation pathway, which involves newly synthesized major histocompatibility complex (MHC) class II in endosomes[136]. Macrophages, another professional antigen-presenting cell type, are also disrupted by VacA in their ability to process antigens as the toxin induces the formation of large vesicles termed megasomes, impairing the maturation and function of endosomal compartments[137]. VacA also impairs T cells as it efficiently prevents the proliferation of T cells by inducing a G1/S cell cycle arrest. VacA was shown to block the T cell receptor/interleukin-2 (IL-2) signaling pathway at the Ca++-calmodulin-dependent phosphatase calcineurin[135]. These effects of VacA on immune cells undoubtedly contribute to immune avoidance by H. pylori and highlight its important role in the pathogenesis of H. pylori-associated diseases.
3.5. H. pylori Neutrophil activating protein (HP-NAP)
HP-NAP, or NapA, is another critical virulence factor of H. pylori[49]. HP-NAP is a highly conserved protein encoded by the napA gene expressed by most H. pylori strains. It is a 164-amino acid protein secreted by H. pylori and can be found in both the bacterial cell wall and the extracellular milieu. NAP’s three-dimensional structure was characterized by Zanotti et al. as having a quaternary structure consisting of a spherical dodecamer containing ∼17 kDa identical subunits with a four-helix bundle structure similar to bacterial ferritins[138]. HP-NAP is a multifunctional protein that has been shown to interact with various cells and modulate host immunity.
Among the critical functions of HP-NAP is its ability to activate neutrophils to generate reactive oxygen species (ROS)[139] and reactive nitrogen species (RNS) and to adhere to endothelial cells[140]. ROS and RNS induced by H. pylori can cause DNA damage and mutations that lead to the initiation of cancer[141]. HP-NAP activates neutrophils by binding to specific receptors on their surface, such as the formyl peptide receptor 1 (FPR1) and Toll-like receptor 2 (TLR2)[48,142], which stimulate the release of ROS, RNS, and inflammatory cytokines[48,143,144]. A recent report showed that HP-NAP could induce the formation of neutrophil extracellular traps (NETs)[145], a novel mechanism of neutrophil-mediated host defense[146]. Cytokine production by HP-NAP-activated neutrophils leads to further neutrophil recruitment to the site of infection. The immune responses are meant to clear H. pylori. Still, they can also cause collateral damage to the gastric mucosa, leading to inflammation and tissue injury[147], which are outcomes that implicate HP-NAP in GC. Overall, the chronic inflammation of the gastric mucosa is a pivotal contributor to the development of H. pylori-related diseases, including GC.
In addition to its influence on innate immune cells, HP-NAP affects the adaptive immune response. HP-NAP crosses the epithelial barrier and promotes the skewing of CD4+ T helper (Th) cell responses to Th1 responses by inducing the production of IL-12 and IL-23 by neutrophils, monocytes, and dendritic cells[48,148]. Those cytokines promote the differentiation of monocytes toward matured dendritic cells (DCs). DCs respond to HP-NAP with increased expression of major histocompatibility complex (MHC) class II molecules and produce IL-12 to further the polarization of Th1 cells[148]. It is important to note that IL-23 produced in response to HP-NAP may promote the development of Th17 and the production of interleukin-17 (IL-17). IL-17 is a pro-inflammatory cytokine critical in the immune response to bacterial infections. IL-17 may contribute to the recruitment of neutrophils and the clearance of H. pylori infection. However, excessive production of IL-17 can also contribute to chronic inflammation and tissue damage.
3.6. H. pylori γ-glutamyltranspeptidase (GGT)
GGT is an enzyme that plays a crucial role in H. pylori’s survival and pathogenesis. H. pylori GGT (HpGGT) is a type I membrane protein anchored to the bacterial outer membrane. HpGGT is highly conserved and present in all strains[149]. It involves several critical functions, including glutathione detoxification, nutrient acquisition, and host immune response modulation. HpGGT catalyzes transpeptidation and hydrolysis of the gamma-glutamyl group of glutathione and similar compounds[150]. HpGGT hydrolyzes glutamine into glutamate, ammonia, and glutathione into glutamate and cysteinylglycine[151]. Since H. pylori cannot take up extracellular glutamine and glutathione directly, this enzyme allows H. pylori to use extracellular glutamine and glutathione as sources of glutamate for subsequent use in the tricarboxylic acid cycle[152]. HpGGT is vital for the survival of H. pylori in acidic conditions as H. pylori GGT- isogenic mutant strains cannot colonize the gastric mucosa in animal models of infection or do so less efficiently[153,154]. A study by Chevalier et al. reported that H. pylori GGT- mutants could not be recovered from the stomachs of mice at 3-60 days post-infection[154].
HpGGT has wide-ranging effects in gastric epithelial cells and plays a significant role in the pathogenesis of Hp-induced GC. HpGGT functions through multiple pathways to damage the gastric epithelial barrier. HpGGT induces epithelial cell apoptosis by a mitochondria-dependent pathway and reduces cell viability[155]. HpGGT also promotes cell death by reducing survivin levels[156], prompting cell cycle arrest at the G1-S phase transition[157], and ROS production, resulting in glutathione depletion and DNA damage[158]. HpGGT stimulates the expression of heparin-binding epidermal growth factor-like growth factor (HB-EGF), a ligand of the epidermal growth factor receptor (EGFR)[159]. HB-EGF binding to EGFR initiates the Raf/Ras/MEK/Erk and PI3K/Akt pathways, reducing apoptosis and promoting proliferation[160]. The expression of HB-EGF is increased in various types of cancer, including GC[161]. HB-EGF also contributes to GC progression by potentiating the epithelial-mesenchymal transition[162]. H. pylori infection also affects mesenchymal stem cells (MSCs) by inducing their migration to the gastric mucosa, where they may contribute to GC development by differentiating into epithelial cells or assisting in angiogenesis[163]. HpGGT was recently shown to disturb MSCs by obstructing alpha-ketoglutarate to boost trimethylation of histones H3K9 and H3K27, triggering PI3K/AKT signaling, and helping proliferation, migration, self-renewal, and pluripotency in cancerogenesis[164]. Another recent report showed that HpGGT might help GC development by activating the Wnt/β-catenin signaling pathway through up-regulation of ten-eleven translocation 1 (TET1)[165], which is a crucial DNA demethylase and is overexpressed in GC.
HpGGT also affects immune cells directly. One of the earliest studies on the immune response effects of HpGGT showed that the enzyme could dampen T-cell proliferation[166,167]. It was reported to induce cell cycle arrest at the G1 phase due to interference with the Ras-dependent signaling pathway[167]. This effect of HpGGT on T cell proliferation is thought to mediate immunosuppression which assists in the persistence of H. pylori infection. Another study showed that HpGGT-induced microRNA-155 (miR-155) expression in both CCRF-CEM cells (a human T lymphoblast cell line) and primary human peripheral blood mononuclear cells[168]. This response depends on forkhead box P3 (Foxp3), the master regulator of regulatory T cells (Treg) development[169], and requires activation of the cyclic adenosine monophosphate cascade. Treg cells have an immune suppressive activity, often found in the H. pylori-infected gastric mucosa[170], promoting higher H. pylori colonization and infiltrating tumors[171]. In support of the role of HpGGT in promoting Treg development, mice infected with ggt- isogenic H. pylori mutant strains were reported to have lower Treg counts than wild-type-infected mice[133]. These observations suggested that HpGGT is influential in regulating the immune system.
4. Immune Checkpoints in H. pylori Infection as Immune Escape Mechanisms
In addition to the various immune evasion mechanisms associated with virulence factors described above, H. pylori stealthily direct the expression of multiple receptors that influence T cell activity. The optimal balance between protective immunity and immune tolerance controls immune responses. T-cell activity is determined by the combination of signals initiated by T cell receptors (TCR) recognition of antigen/MHC complexes and co-stimulatory molecules on APCs, divided into co-stimulatory and co-inhibitory molecules (Figure 1). While the co-stimulatory receptors are critical in initiating immune responses, co-inhibitory molecules are essential for avoiding immune-driven pathology but can also restrain immune-mediated clearance of pathogens. Many pathogens and cancers promote inhibitory interactions via immune checkpoint proteins to evade immune clearance. H. pylori induces the expression of gastric epithelial cells of the checkpoint inhibitor B7-H1 (aka PD-L1), which inhibits effector T cells via PD-1 engagement on their surface and fosters Treg cell development[172,173,174]. This contributes to the overall T cell supression elicited by the various virulence factors described above (Figure 2). Further, the H. pylori-infected gastric epithelial cells simultaneously downregulate their expression of the co-stimulator B7-H2, thus, not only reducing effector T cell responses but also altering T cell sub-sets balances by increasing Treg and decreasing Th17 cell numbers[173,174,175]. This imbalance favors bacterial persistence. We demonstrated that H. pylori uses cagPAI to induce these effects on gastric epithelial cells and activates the mTOR/p70 S6 kinase pathway to downregulate B7-H2 expression[172,175]. B7-H1 expression is also increased in GC[176,177,178]. In a subsequent report, we demonstrated that H. pylori T4SS-mediated transfer of CagA and cell wall peptidoglycan (PG) fragment upregulated B7-H3 expression by gastric epithelial cells via activation of the p38MAPK signaling pathway[179]. Thus, H. pylori effectively hijack the regulated expression of co-inhibitory molecules to avoid immunity and may favor immune escape by developing cancer cells through these mechanisms.
5. Conclusions and Future Directions
Over the past 40 years, significant advances were made in our comprehension of H. pylori’s properties since its initial identification as a human pathogen. While infection with H. pylori was initially linked with chronic gastritis and the development of peptic ulcer disease, research by multiple laboratories worldwide has established the bacterium as the most potent known risk factor for GC, one of the world’s deadliest cancers.
Although antibiotic therapies are available to treat H. pylori infection, the high prevalence of the bacterium and the escalation of antibiotic resistance highlights the need for a protective vaccine. H. pylori has an extensive array of virulence factors that mediate pathogenesis and could represent potential vaccine targets to prevent the morbidity and mortality associated with the infection. Nevertheless, vaccine development efforts have encountered various challenges. The variability among H. pylori strains and the bacterium’s immune escape mechanisms create significant obstacles to developing an effective vaccine. Clinical trials of candidate vaccines have yielded mixed or disappointing results.
Although an H. pylori vaccine is not currently available, continued research is promising and may eventually lead to an avenue to help decrease the burden of H. pylori-associated diseases. In summary, H. pylori is the foremost risk factor for developing GC, and understanding how to override the bacterium’s immune evasion strategies is crucial for developing new approaches to treat and prevent the conditions associated with the infection.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Stomach (Gastric) Cancer Key Statistics.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Infection with Helicobacter pylori. IARC Monogr Eval. Carcinog. Risks Hum 61, 177–240.
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. A human model of gastric carcinogenesis. 1988, 48, 3554–60.
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren classification and individualized chemotherapy in gastric cancer. Oncol. Lett. 2016, 11, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Plummer, M. , Franceschi, S., Vignat, J., Forman, D. and De Martel, C. (2015) Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer, Int J Cancer 136, 487–490.
- Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-. IARC Monogr Eval Carcinog Risks Hum, Various 61, 1. 14 June.
- Jemal, A. , Bray, F. ( CA Cancer J Clin 61, 69–90.
- Holcombe, C. Helicobacter pylori: the African enigma. Gut 1992, 33, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Agha, A. and Graham, D. Y. (2005) Evidence-based examination of the African enigma in relation to Helicobacter pylori infection. Scand J Gastroenterol 40.
- Bamford, K.B.; Fan, X.; Crowe, S.E.; Leary, J.F.; Gourley, W.K.; Luthra, G.K.; Brooks, E.G.; Graham, D.Y.; Reyes, V.E.; Ernst, P.B. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 1998, 114, 482–492. [Google Scholar] [CrossRef]
- Fox, J.G.; Beck, P.; Dangler, C.A.; Whary, M.T.; Wang, T.C.; Shi, H.N.; Nagler-Anderson, C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000, 6, 536–542. [Google Scholar] [CrossRef]
- Ge, Z. , Feng, Y., Muthupalani, S., Eurell, L. L., Taylor, N. S., Whary, M. T. and Fox, J. G. (2011) Coinfection with enterohepatic helicobacter species can aeliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 Mice. Infect Immun 79, 3861–3871.
- Singh, K.; Ghoshal, U.C. Causal role of Helicobacter pylori infection in gastric cancer: An Asian enigma. World J. Gastroenterol. 2006, 12, 1346–51. [Google Scholar] [CrossRef]
- Xia, Y.; Yamaoka, Y.; Zhu, Q.; Matha, I.; Gao, X. A Comprehensive Sequence and Disease Correlation Analyses for the C-Terminal Region of CagA Protein of Helicobacter pylori. PLOS ONE 2009, 4, e7736. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y. , Kodama, T., Kashima, K., Graham, D. Y. and Sepulveda, A. R. (1998) Variants of the 3’ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J. Clin. Microbiol 36, 2258–2263.
- Correa, P.; Cuello, C.; Duque, E.; Burbano, L.C.; Garcia, F.T.; Bolanos, O.; Brown, C.; Haenszel, W. Gastric Cancer in Colombia. III. Natural History of Precursor Lesions 2. JNCI J. Natl. Cancer Inst. 1976, 57, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.; de Sablet, T.; Asim, M.; Piazuelo, M.B.; Barry, D.P.; Verriere, T.G.; Sierra, J.C.; Hardbower, D.M.; Delgado, A.G.; Schneider, B.G.; et al. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene 2014, 34, 3429–3440. [Google Scholar] [CrossRef] [PubMed]
- Bravo, L. E. , Van Doorn, L. J., Realpe, J. L. and Correa, P. (2002) Virulence-associated genotypes of Helicobacter pylori: Do they explain the African enigma? American Journal of Gastroenterology 97.
- Kodaman, N.; Pazos, A.; Schneider, B.G.; Piazuelo, M.B.; Mera, R.; Sobota, R.S.; Sicinschi, L.A.; Shaffer, C.L.; Romero-Gallo, J.; de Sablet, T.; et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc. Natl. Acad. Sci. 2014, 111, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Bakhti, S. Z. , Raei, N., Latifi-Navid, S., Zahri, S. and Yazdanbod, A. (2019) Inverse relationship between cagG-positive Helicobacter pylori status and risk of gastric ulcer. Br J Biomed Sci 76.
- Sverdén, E. , Agréus, L., Dunn, J. M. and Lagergren, J. (2019) Peptic ulcer disease. BMJ, British Medical Journal Publishing Group 367.
- Lau, J. Y. , Sung, J., Hill, C., Henderson, C., Howden, C. W. and Metz, D. C. (2011) Systematic review of the epidemiology of complicated peptic ulcer disease: incidence, recurrence, risk factors and mortality. Digestion, Digestion 84, 102–113.
- Lanas, A. and Chan, F. K. L. (2017, ) Peptic ulcer disease. The Lancet, Lancet Publishing Group. 5 August.
- El-Omar, E.; Oien, K.; El-Nujumi, A.; Gillen, D.; Wirz, A.; Dahill, S.; Williams, C.; Ardill, J.; McColl, K. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology 1997, 113, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, P.G.; Du, M. Gastrointestinal lymphoma: where morphology meets molecular biology. J. Pathol. 2005, 205, 255–274. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Amedei, A.; Manghetti, M.; Costa, F.; Baldari, C.T.; Quazi, A.S.; Telford, J.L.; Romagnani, S.; del Prete, G. Impaired T-cell regulation of B-cell growth in Helicobacter pylori–related gastric low-grade MALT lymphoma. Gastroenterology 1999, 117, 1105–1112. [Google Scholar] [CrossRef]
- Hussell, T. , Isaacson, P. G., Crabtkee, J. E. and Spencer, J. O. (1996) Helicobacter p ylori-specific tumour-infiltrating T cells provide contact-dependent help for the growth of malignant B cells in low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Journal of Pathology 178, 122–127.
- Parsonnet, J.; Hansen, S.; Rodriguez, L.; Gelb, A.B.; Warnke, R.A.; Jellum, E.; Orentreich, N.; Vogelman, J.H.; Friedman, G.D. Helicobacter pylori Infection and Gastric Lymphoma. New Engl. J. Med. 1994, 330, 1267–1271. [Google Scholar] [CrossRef]
- Ruskone-Fourmestraux, A.; Fischbach, W.; Aleman, B.M.P.; Boot, H.; Du, M.Q.; Megraud, F.; Montalban, C.; Raderer, M.; Savio, A.; Wotherspoon, A.; et al. EGILS consensus report. Gastric extranodal marginal zone B-cell lymphoma of MALT. Gut 2011, 60, 747–758. [Google Scholar] [CrossRef]
- Zullo, A.; Hassan, C.; Cristofari, F.; Andriani, A.; De Francesco, V.; Ierardi, E.; Tomao, S.; Stolte, M.; Morini, S.; Vaira, D. Effects of Helicobacter pylori Eradication on Early Stage Gastric Mucosa–Associated Lymphoid Tissue Lymphoma. Clin. Gastroenterol. Hepatol. 2010, 8, 105–110. [Google Scholar] [CrossRef]
- Li, J. Z. , Li, J. Y., Wu, T. F., Xu, J. H., Huang, C. Z., Cheng, D., Chen, Q. K. and Yu, T. (2017) Helicobacter pylori Infection Is Associated with Type 2 Diabetes, Not Type 1 Diabetes: An Updated Meta-Analysis. Gastroenterol Res Pract, Hindawi Limited 2017.
- Upala, S. , Sanguankeo, A., Saleem, S. A. and Jaruvongvanich, V. (2017) Effects of Helicobacter pylori eradication on insulin resistance and metabolic parameters: A systematic review and meta-analysis. Eur J Gastroenterol Hepatol, Lippincott Williams and Wilkins 29, 153–159.
- Liu, J. , Wang, F. and Shi, S. (2015) Helicobacter pylori Infection Increase the Risk of Myocardial Infarction: A Meta-Analysis of 26 Studies Involving more than 20,000 Participants. Helicobacter, John Wiley & Sons, Ltd 20, 176–183.
- Xu, M.-Y.; Cao, B.; Yuan, B.-S.; Yin, J.; Liu, L.; Lu, Q.-B. Association of anaemia with Helicobacter pylori infection: a retrospective study. Sci. Rep. 2017, 7, 13434–13434. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T. , Pandey, J. P., Okazaki, Y., Asahi, A., Kawakami, Y., Ikeda, Y. and Kuwana, M. (2009) Single nucleotide polymorphism of interleukin-1beta associated with Helicobacter pylori infection in immune thrombocytopenic purpura. Tissue Antigens, Tissue Antigens 73, 353–357.
- Shen, X. , Yang, H., Wu, Y., Zhang, D. and Jiang, H. (2017) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter, John Wiley & Sons, Ltd 22, e12398.
- Chung, S.J.; Lim, S.H.; Choi, J.; Kim, D.; Kim, Y.S.; Park, M.J.; Yim, J.Y.; Kim, J.S.; Cho, S.-H.; Jung, H.C.; et al. Helicobacter pylori Serology Inversely Correlated With the Risk and Severity of Reflux Esophagitis inHelicobacter pylori Endemic Area: A Matched Case-Control Study of 5,616 Health Check-Up Koreans. J. Neurogastroenterol. Motil. 2011, 17, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Hu, J.; Wang, X.; Ren, M.; Lu, G.; Lu, X.; Zhang, D.; He, S. The Effect of Helicobacter pylori Eradication in Patients with Gastroesophageal Reflux Disease: A Meta-Analysis of Randomized Controlled Studies. Dig. Dis. 2020, 38, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Islami, F. and Kamangar, F. (2008) Helicobacter pylori and Esophageal Cancer Risk -- A Meta-Analysis. Cancer Prev Res (Phila), NIH Public Access 1, 329.
- Chen, Y.; Blaser, M.J. Helicobacter pyloriColonization Is Inversely Associated with Childhood Asthma. J. Infect. Dis. 2008, 198, 553–560. [Google Scholar] [CrossRef] [PubMed]
- D’Elios, M.M.; Amedei, A.; Codolo, G.; Del Prete, G.; de Bernard, M. The effect of Helicobacter pylori on asthma and allergy. J. Asthma Allergy 2010, 3, 139–147. [Google Scholar] [CrossRef]
- Elias, N.; Nasrallah, E.; Khoury, C.; Mansour, B.; Abu Zuher, L.; Asato, V.; Muhsen, K. Associations of Helicobacter pylori seropositivity and gastric inflammation with pediatric asthma. Pediatr. Pulmonol. 2020, 55, 2236–2245. [Google Scholar] [CrossRef]
- Karttunen, R.; Andersson, G.; Poikonen, K.; Kosunen, T.U.; Karttunen, T.; Juutinen, K.; Niemelä, S. Helicobacter pylori induces lymphocyte activation in peripheral blood cultures. Clin. Exp. Immunol. 1990, 82, 485–488. [Google Scholar] [CrossRef]
- Amedei, A.; Cappon, A.; Codolo, G.; Cabrelle, A.; Polenghi, A.; Benagiano, M.; Tasca, E.; Azzurri, A.; D’elios, M.M.; Del Prete, G.; et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J. Clin. Investig. 2006, 116, 1092–1101. [Google Scholar] [CrossRef]
- Reyes, V. E. and Beswick, E. J. (2007) Helicobacter pylori neutrophil activating protein’s potential as tool in therapeutic immune modulation. Expert Opin Ther Pat 17.
- Lundgren, A. , Suri-Payer, E., Enarsson, K., Svennerholm, A. M. and Lundin, B. S. (2003) Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun 71, 1755–1762.
- Beswick, E.J.; Pinchuk, I.V.; Earley, R.B.; Schmitt, D.A.; Reyes, V.E. Role of Gastric Epithelial Cell-Derived Transforming Growth Factor β in Reduced CD4 + T Cell Proliferation and Development of Regulatory T Cells during Helicobacter pylori Infection. Infect. Immun. 2011, 79, 2737–2745. [Google Scholar] [CrossRef]
- Beswick, E. J. , Pinchuk, I. V, Earley, R. B., Schmitt, D. A. and Reyes, V. E. (2011) The Role of Gastric Epithelial Cell-Derived TGF-{beta} in Reduced CD4+ T Cell Proliferation and Development of Regulatory T Cells during Helicobacter pylori Infection 1. Infect. Immun 79, 2737–2745.
- Lundgren, A. , Stromberg, E., Sjoling, A., Lindholm, C., Enarsson, K., Edebo, A., Johnsson, E., Suri-Payer, E., Larsson, P., Rudin, A., et al. (2005) Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun 73, 523–531.
- Wang, S. K. , Zhu, H. F., He, B. S., Zhang, Z. Y., Chen, Z. T., Wang, Z. Z. and Wu, G. L. (2007) CagA+ H. pylori infection is associated with polarization of T helper cell immune responses in gastric carcinogenesis. World Journal of Gastroenterology : WJG, Baishideng Publishing Group Inc 13, 2923.
- Mestecky, J. The common mucosal immune system and current strategies for induction of immune responses in external secretions. J. Clin. Immunol. 1987, 7, 265–276. [Google Scholar] [CrossRef]
- Sayar, R. , Shirvani, J. S., Hajian -Tilaki, K., Vosough, Z. and Ranaei, M. (2019) The negative association between inflammatory bowel disease and Helicobacter pylori seropositivity. Caspian J Intern Med, Caspian J Intern Med 10, 217–222.
- Castanõ-Rodríguez, N. , Kaakoush, N. O., Lee, W. S. and Mitchell, H. M. (2017) Dual role of Helicobacter and Campylobacter species in IBD: a systematic review and meta-analysis. Gut, BMJ Publishing Group 66, 235–249.
- Ewe, K.; Schwartz, S.; Petersen, S.; Press, A.G. Inflammation Does Not Decrease Intraluminal pH in Chronic Inflammatory Bowel Disease. Dig. Dis. Sci. 1999, 44, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V. E. , Almanza, R. J. and Ye, G. (1998) Helicobacter pylori: A Trojan horse in the gastric environment? Mucosal Immunology Update 5, 68–70.
- Beswick, E. J. , Suarez, G. and Reyes, V. E. (2006) H. pylori and host interactions that influence pathogenesis. World J Gastroenterol 12.
- Suarez, G. , Reyes, V. E. and Beswick, E. J. (2006) Immune response to H. pylori. World J Gastroenterol 12.
- Reyes, V. E. , Suárez, G., Sierra, J. C. and Beswick, E. J. (2009) Helicobacter pylori. Vaccines for Biodefense and Emerging and Neglected Diseases.
- Alzahrani, S. , Lina, T. T., Gonzalez, J., Pinchuk, I. V., Beswick, E. J. and Reyes, V. E. (2014) Effect of Helicobacter pylori on gastric epithelial cells. World J Gastroenterol 20.
- Lina, T. T. , Alzahrani, S., Gonzalez, J., Pinchuk, I. V., Beswick, E. J. and Reyes, V. E. (2014) Immune evasion strategies used by Helicobacter pylori. World J Gastroenterol 20.
- Ha, N.-C.; Oh, S.-T.; Sung, J.Y.; Cha, K.A.; Lee, M.H.; Oh, B.-H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 2001, 8, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10, 57–64. [Google Scholar] [CrossRef] [PubMed]
- E Dunn, B.; Phadnis, S.H. Structure, function and localization of Helicobacter pylori urease. . 1999, 71, 63–73. [Google Scholar]
- E Dunn, B.; Vakil, N.B.; Schneider, B.G.; Miller, M.M.; Zitzer, J.B.; Peutz, T.; Phadnis, S.H. Localization of Helicobacter pylori urease and heat shock protein in human gastric biopsies. Infect. Immun. 1997, 65, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Phadnis, S.H.; Parlow, M.H.; Levy, M.; Ilver, D.; Caulkins, C.M.; Connors, J.B.; E Dunn, B. Surface localization of Helicobacter pylori urease and a heat shock protein homolog requires bacterial autolysis. Infect. Immun. 1996, 64, 905–12. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Gunasena, H.; Cheng, Z.; Espejo, R.; Crowe, S.E.; Ernst, P.B.; Reyes, V.E. Helicobacter pylori Urease Binds to Class II MHC on Gastric Epithelial Cells and Induces Their Apoptosis. J. Immunol. 2000, 165, 1918–1924. [Google Scholar] [CrossRef]
- Fan, X.; Crowe, S.E.; Behar, S.; Gunasena, H.; Ye, G.; Haeberle, H.; Van Houten, N.; Gourley, W.K.; Ernst, P.B.; Reyes, V.E. The Effect of Class II Major Histocompatibility Complex Expression on Adherence of Helicobacter pylori and Induction of Apoptosis in Gastric Epithelial Cells: A Mechanism for T Helper Cell Type 1–mediated Damage. J. Exp. Med. 1998, 187, 1659–1669. [Google Scholar] [CrossRef]
- Barrera, C.; Espejo, R.; E Reyes, V. Differential glycosylation of MHC class II molecules on gastric epithelial cells: Implications in local immune responses. Hum. Immunol. 2002, 63, 384–393. [Google Scholar] [CrossRef]
- Fan, X. J. , Gunasena, H., Gonzales, M., Almanza, R., Cheng, Z., Ye, G., Barrera, C., Crowe, S. E., Ernst, P. B. and Reyes, V. E. (1998) Class II MHC molecules on gastric epithelial cells act as receptors for Helicobacter pylori urease and signal apoptosis of the epithelium. FASEB Journal 12.
- Beswick, E. J. J. , Pinchuk, I. V. V, Minch, K., Suarez, G., Sierra, J. C. C., Yamaoka, Y. and Reyes, V. E. E. (2006) The Helicobacter pylori Urease B Subunit Binds to CD74 on Gastric Epithelial Cells and Induces NF-{kappa}B Activation and Interleukin-8 Production. Infect. Immun 74, 1148–1155.
- Beswick, E.J.; Bland, D.A.; Suarez, G.; Barrera, C.A.; Fan, X.; Reyes, V.E. Helicobacter pyloriBinds to CD74 on Gastric Epithelial Cells and Stimulates Interleukin-8 Production. Infect. Immun. 2005, 73, 2736–2743. [Google Scholar] [CrossRef]
- Boren, T. , Falk, P., Roth, K. A., Larson, G. and Normark, S. (1993) Attachment of Helicobater pylori to human gastric epithelium mediated by blood group antigens. Science (1979) 262, 1892–1895.
- Sheu, B.-S.; Sheu, S.-M.; Yang, H.-B.; Huang, A.-H.; Wu, J.-J. Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut 2003, 52, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S. , Olaniyi Ojo, O., Arnqvist, A., Yih Wu, J., Odenbreit, S., Haas, R., Graham, D. Y. and Yamaoka, Y. (2007) Helicobacter pylori BabA Expression, Gastric Mucosal Injury, and Clinical Outcome. Clin Gastroenterol Hepatol, NIH Public Access 5, 49.
- Sheu, S.-M.; Sheu, B.-S.; Chiang, W.-C.; Kao, C.-Y.; Wu, H.-M.; Yang, H.-B.; Wu, J.-J. H. pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol. 2012, 12, 89–89. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y. , Kwon, D. H. and Graham, D. Y. (2000) A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A 97, 7533–7538.
- Lu, H.; Wu, J.Y.; Beswick, E.J.; Ohno, T.; Odenbreit, S.; Haas, R.; Reyes, V.E.; Kita, M.; Graham, D.Y.; Yamaoka, Y. Functional and Intracellular Signaling Differences Associated with the Helicobacter pylori AlpAB Adhesin from Western and East Asian Strains. J. Biol. Chem. 2007, 282, 6242–6254. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Till, M.; Hofreuter, D.; Faller, G.; Haas, R. Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol. Microbiol. 1999, 31, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Faller, G.; Haas, R. Role of the AlpAB proteins and lipopolysaccharide in adhesion of Helicobacter pylori to human gastric tissue. Int. J. Med Microbiol. 2002, 292, 247–256. [Google Scholar] [CrossRef]
- Mahdavi, J. , Sondén, B. ( NIH Public Access 297, 573.
- Walz, A.; Odenbreit, S.; Mahdavi, J.; Borén, T.; Ruhl, S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology 2005, 15, 700–708. [Google Scholar] [CrossRef]
- Hirmo, S. , Kelm, S., Schauer, R., Nilsson, B. and Wadström, T. (1996) Adhesion of Helicobacter pylori strains to α-2,3-linked sialic acids. Glycoconj J, Kluwer Academic Publishers 13, 1005–1011.
- Backert, S. , Tegtmeyer, N. and Fischer, W. (2015) Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol 10.
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B 2017, 93, 196–219. [Google Scholar] [CrossRef]
- Kaplan-Türköz, B. , Jiménez-Soto, L. F., Dian, C., Ertl, C., Remaut, H., Louche, A., Tosi, T., Haas, R. and Terradot, L. (2012) Structural insights into Helicobacter pylori oncoprotein CagA interaction with β1 integrin. Proc Natl Acad Sci U S A 109.
- Jiménez-Soto, L.F.; Kutter, S.; Sewald, X.; Ertl, C.; Weiss, E.; Kapp, U.; Rohde, M.; Pirch, T.; Jung, K.; Retta, S.F.; et al. Helicobacter pylori Type IV Secretion Apparatus Exploits β1 Integrin in a Novel RGD-Independent Manner. PLOS Pathog. 2009, 5, e1000684. [Google Scholar] [CrossRef]
- Hatakeyama, M. Helicobacter pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. 2002, 99, 14428–14433. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C. , Machado, J. C. and Yamaoka, Y. (2005) Pathogenesis of Helicobacter pylori Infection. Helicobacter 10 Suppl 1, 14–20.
- Churin, Y.; Al-Ghoul, L.; Kepp, O.; Meyer, T.F.; Birchmeier, W.; Naumann, M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J. Cell Biol. 2003, 161, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mimuro, H.; Kiga, K.; Fukumatsu, M.; Ishijima, N.; Morikawa, H.; Nagai, S.; Koyasu, S.; Gilman, R.H.; Kersulyte, D.; et al. Helicobacter pylori CagA Phosphorylation-Independent Function in Epithelial Proliferation and Inflammation. Cell Host Microbe 2009, 5, 23–34. [Google Scholar] [CrossRef]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 Is a Key Mediator of Helicobacter pylori CagA Protein Activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Franco, A. T. , Johnston, E., Krishna, U., Yamaoka, Y., Israel, D. A., Nagy, T. A., Wroblewski, L. E., Piazuelo, M. B., Correa, P. and Peek Jr., R. M. (2008) Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res 68, 379–387.
- Saadat, I. , Higashi, H., Obuse, C., Umeda, M., Murata-Kamiya, N., Saito, Y., Lu, H., Ohnishi, N., Azuma, T., Suzuki, A., et al. (2007) Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447, 330–333.
- Murata-Kamiya, N. , Kurashima, Y., Teishikata, Y., Yamahashi, Y., Saito, Y., Higashi, H., Aburatani, H., Akiyama, T., Peek Jr., R. M., Azuma, T., et al. (2007) Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene.
- Palrasu, M.; Zaika, E.; Paulrasu, K.; Gokulan, R.C.; Suarez, G.; Que, J.; El-Rifai, W.; Peek, R.M.; Garcia-Buitrago, M.; Zaika, A.I. Helicobacter pylori pathogen inhibits cellular responses to oncogenic stress and apoptosis. PLOS Pathog. 2022, 18, e1010628. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; E Girardin, S.; Moran, A.P.; Athman, R.; Mémet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef]
- Grubman, A. , Kaparakis, M. ( regulates direct killing of Helicobacter pylori by antimicrobial peptides 94. Cell Microbiol 12, 626–639.
- Suarez, G.; Romero-Gallo, J.; Piazuelo, M.B.; Wang, G.; Maier, R.J.; Forsberg, L.S.; Azadi, P.; Gomez, M.A.; Correa, P.; Peek, R.M. Modification of Helicobacter pylori Peptidoglycan Enhances NOD1 Activation and Promotes Cancer of the Stomach. Cancer Res 2015, 75, 1749–1759. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The Protein ATG16L1 Suppresses Inflammatory Cytokines Induced by the Intracellular Sensors Nod1 and Nod2 in an Autophagy-Independent Manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef]
- Harper, C. G. , Feng, Y., Xu, S., Taylor, N. S., Kinsel, M., Dewhirst, F. E., Paster, B. J., Greenwell, M., Levine, G., Rogers, A., et al. (2002) Helicobacter cetorum sp. nov., a Urease-positive Helicobacter species isolated from dolphins and whales. J Clin Microbiol 40.
- Reyrat, J.-M.; Lanzavecchia, S.; Lupetti, P.; de Bernard, M.; Pagliaccia, C.; Pelicic, V.; Charrel, M.; Ulivieri, C.; Norais, N.; Ji, X.; et al. 3D imaging of the 58 kda cell binding subunit of the Helicobacter pylori cytotoxin. J. Mol. Biol. 1999, 290, 459–470. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.S.; Iwamoto, H.; Cao, P.; Vinion-Dubiel, A.D.; Li, Y.; Szabo, G.; Shao, Z.; Cover, T.L. Essential Role of a GXXXG Motif for Membrane Channel Formation by Helicobacter pylori Vacuolating Toxin. J. Biol. Chem. 2003, 278, 12101–12108. [Google Scholar] [CrossRef] [PubMed]
- González-Rivera, C.; Gangwer, K.A.; McClain, M.S.; Eli, I.M.; Chambers, M.G.; Ohi, M.D.; Lacy, D.B.; Cover, T.L. Reconstitution of Helicobacter pylori VacA Toxin from Purified Components. Biochemistry 2010, 49, 5743–5752. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Functional Properties of the p33 and p55 Domains of the Helicobacter pylori Vacuolating Cytotoxin. J. Biol. Chem. 2005, 280, 21107–21114. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.R.; Patel, H.K.; Kostolansky, S.S.; Ballivian, R.A.; Eichberg, J.; Blanke, S.R. Sphingomyelin Functions as a Novel Receptor for Helicobacter pylori VacA. PLOS Pathog. 2008, 4, e1000073. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V. , Galmiche, A., Doye, A., Necchi, V., Solcia, E. and Boquet, P. (2000) High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol Biol Cell 11.
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O.; Canonici, A.; Sciullo, A.; Sommi, P.; Fabbri, A.; Ricci, V.; et al. Helicobacter pylori Counteracts the Apoptotic Action of Its VacA Toxin by Injecting the CagA Protein into Gastric Epithelial Cells. PLOS Pathog. 2009, 5, e1000603. [Google Scholar] [CrossRef]
- Gauthier, N. C. , Monzo, P., Gonzalez, T., Doye, A., Oldani, A., Gounon, P., Ricci, V., Cormont, M. and Boquet, P. (2007) Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. Journal of Cell Biology 177.
- Yamasaki, E. , Wada, A., Kumatori, A., Nakagawa, I., Funao, J., Nakayama, M., Hisatsune, J., Kimura, M., Moss, J. and Hirayama, T. (2006) Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem, J Biol Chem 281, 11250–11259.
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef]
- Jain, P.; Luo, Z.-Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. 2011, 108, 16032–16037. [Google Scholar] [CrossRef]
- Atherton, J. C. , Cao, P., Peek, R. M., Tummuru, M. K. R., Blaser, M. J. and Cover, T. L. (1995) Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem, J Biol Chem 270, 17771–17777.
- Bakhti, S. Z. , Latifi-Navid, S., Mohammadi, S., Zahri, S., Bakhti, F. S., Feizi, F., Yazdanbod, A. and Siavoshi, F. (2016) Relevance of Helicobacter pylori vacA 3’-end Region Polymorphism to Gastric Cancer. Helicobacter, Helicobacter 21, 305–316.
- Soyfoo, D.M.; Doomah, Y.H.; Xu, D.; Zhang, C.; Sang, H.-M.; Liu, Y.-Y.; Zhang, G.-X.; Jiang, J.-X.; Xu, S.-F. New genotypes of Helicobacter Pylori VacA d-region identified from global strains. BMC Cell Biol. 2021, 22, 1–12. [Google Scholar] [CrossRef]
- McClain, M. S. , Cao, P., Iwamoto, H., Vinion-Dubiel, A. D., Szabo, G., Shao, Z. and Cover, T. L. (2001) A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J Bacteriol 183.
- Wang, H.-J.; Kuo, C.-H.; Yeh, A.A.M.; Chang, P.C.L.; Wang, W.-C. Vacuolating Toxin Production in Clinical Isolates of Helicobacter pylori with Different vacA Genotypes. J. Infect. Dis. 1998, 178, 207–212. [Google Scholar] [CrossRef]
- Basso, D.; Zambon, C.; Letley, D.P.; Stranges, A.; Marchet, A.; Rhead, J.L.; Schiavon, S.; Guariso, G.; Ceroti, M.; Nitti, D.; et al. Clinical Relevance of Helicobacter pylori cagA and vacA Gene Polymorphisms. Gastroenterology 2008, 135, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Figueiredo, C.; Carneiro, F.; Gomes, A.T.; Barreira, R.; Figueira, P.; Salgado, C.; Belo, L.; Peixoto, A.; Bravo, J.C.; et al. Helicobacter pylori Genotypes May Determine Gastric Histopathology. Am. J. Pathol. 2001, 158, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Zali, M.R.; Yamaoka, Y. The association of vacA genotypes and Helicobacter pylori-related gastroduodenal diseases in the Middle East. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-L.; Yeh, Y.-C.; Sheu, B.-S. The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J. Biomed. Sci. 2018, 25, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Olivares, A.; Torres, E.; Yilmaz, O.; Cohen, H.; Perez-Perez, G. Diversity of VacA Intermediate Region among Helicobacter pylori Strains from Several Regions of the World. J. Clin. Microbiol. 2010, 48, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Fernandez, T.; Burroni, D.; Pagliaccia, C.; Atherton, J.C.; Reyrat, J.-M.; Rappuoli, R.; Telford, J.L. Cell Specificity of Helicobacter pylori Cytotoxin Is Determined by a Short Region in the Polymorphic Midregion. Infect. Immun. 2000, 68, 3754–3757. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Forster, S.; Irving, A.; Tate, M.; Ferrero, R.L.; Hertzog, P.; Frøkiær, H.; Kaparakis-Liaskos, M. Helicobacter pylori VacA Suppresses Lactobacillus acidophilus-Induced Interferon Beta Signaling in Macrophages via Alterations in the Endocytic Pathway. Mbio 2013, 4, e00609–12. [Google Scholar] [CrossRef]
- Zheng, P.-Y.; Jones, N.L. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell. Microbiol. 2003, 5, 25–40. [Google Scholar] [CrossRef]
- Jung, M. K. , Joo, S. K., Jin, Y. L., Kim, Y. J., Youn, H. J., In, Y. K., Young, J. C., Oh, Y. K., Kim, N., Hyun, C. J., et al. (2007) Vacuolating Cytotoxin in Helicobacter pylori Water-Soluble Proteins Upregulates Chemokine Expression in Human Eosinophils via Ca2+ Influx, Mitochondrial Reactive Oxygen Intermediates, and NF-κB Activation. Infect Immun, American Society for Microbiology (ASM) 75, 3373.
- Supajatura, V.; Ushio, H.; Wada, A.; Yahiro, K.; Okumura, K.; Ogawa, H.; Hirayama, T.; Ra, C. Cutting Edge: VacA, a Vacuolating Cytotoxin of Helicobacter pylori, Directly Activates Mast Cells for Migration and Production of Proinflammatory Cytokines. J. Immunol. 2002, 168, 2603–2607. [Google Scholar] [CrossRef]
- Oertli, M. , Noben, M., Engler, D. B., Semper, R. P., Reuter, S., Maxeiner, J., Gerhard, M., Taube, C. and Muller, A. (2013) Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc. Natl. Acad. Sci. U. S. A 110, 3047–3052.
- Boncristiano, M.; Paccani, S.R.; Barone, S.; Ulivieri, C.; Patrussi, L.; Ilver, D.; Amedei, A.; D’Elios, M.M.; Telford, J.L.; Baldari, C.T. The Helicobacter pylori Vacuolating Toxin Inhibits T Cell Activation by Two Independent Mechanisms. J. Exp. Med. 2003, 198, 1887–1897. [Google Scholar] [CrossRef]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pyloriVacuolating Cytotoxin Inhibits T Lymphocyte Activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Salio, M.; Galli, C.; Norais, N.; Rappuoli, R.; Lanzavecchia, A.; Montecucco, C. Selective Inhibition of Ii-dependent Antigen Presentation by Helicobacter pylori Toxin VacA. J. Exp. Med. 1998, 187, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.-A.H.; Schlesinger, L.S.; Kang, B. Virulent Strains of Helicobacter pylori Demonstrate Delayed Phagocytosis and Stimulate Homotypic Phagosome Fusion in Macrophages. J. Exp. Med. 2000, 191, 115–128. [Google Scholar] [CrossRef]
- Zanotti, G.; Papinutto, E.; Dundon, W.G.; Battistutta, R.; Seveso, M.; Del Giudice, G.; Rappuoli, R.; Montecucco, C. Structure of the Neutrophil-activating Protein from Helicobacter pylori. J. Mol. Biol. 2002, 323, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Satin, B. , Del, G. G., Della V, B., Dusi, S., Laudanna, C., Tonello, F., Kelleher, D., Rappuoli, R., Montecucco, C. and Rossi, F. (2000) The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J. Exp. Med 191, 1467–1476.
- Evans Jr., D. J., Evans, D. G., Takemura, T., Nakano, H., Lampert, H. C., Graham, D. Y., Granger, D. N. and Kvietys, P. R. (1995) Characterization of a Helicobacter pylori neutrophil-activating protein. Infect. Immun 63, 2213–2220.
- Marnett, L. J. (2000) Oxyradicals and DNA damage. Carcinogenesis.
- Fu, H. W. (2014) Helicobacter pylori neutrophil-activating protein: From molecular pathogenesis to clinical applications. World J Gastroenterol 20.
- Montemurro, P.; Nishioka, H.; Dundon, W.G.; de Bernard, M.; Del Giudice, G.; Rappuoli, R.; Montecucco, C. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a potent stimulant of mast cells. Eur. J. Immunol. 2002, 32, 671–6. [Google Scholar] [CrossRef] [PubMed]
- Polenghi, A.; Bossi, F.; Fischetti, F.; Durigutto, P.; Cabrelle, A.; Tamassia, N.; Cassatella, M.A.; Montecucco, C.; Tedesco, F.; de Bernard, M. The Neutrophil-Activating Protein of Helicobacter pylori Crosses Endothelia to Promote Neutrophil Adhesion In Vivo. J. Immunol. 2007, 178, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.-H.; Hong, Z.-W.; Chen, C.-C.; Chang, H.-W.; Fu, H.-W. Helicobacter pylori Neutrophil-Activating Protein Directly Interacts with and Activates Toll-like Receptor 2 to Induce the Secretion of Interleukin-8 from Neutrophils and ATRA-Induced Differentiated HL-60 Cells. Int. J. Mol. Sci. 2021, 22, 11560. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Oleastro, M.; Ménard, A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology 2013, 2, 1110–1134. [Google Scholar] [CrossRef]
- Ramachandran, M.; Jin, C.; Yu, D.; Eriksson, F.; Essand, M. Vector-Encoded Helicobacter pylori Neutrophil-Activating Protein Promotes Maturation of Dendritic Cells with Th1 Polarization and Improved Migration. J. Immunol. 2014, 193, 2287–2296. [Google Scholar] [CrossRef]
- Rossi, M. , Bolz, C., Revez, J., Javed, S., El-Najjar, N., Anderl, F., Hyytiäinen, H., Vuorela, P., Gerhard, M. and Hänninen, M. L. (2012) Evidence for conserved function of γ-glutamyltranspeptidase in Helicobacter genus. PLoS One 7.
- Suzuki, H.; Kumagai, H. [Gamma-glutamyltranspeptidase from Escherichia coli K-12]. . 1989, 61, 491–6. [Google Scholar] [PubMed]
- Shibayama, K. , Wachino, J. I., Arakawa, Y., Saidijam, M., Rutherford, N. G. and Henderson, P. J. F. (2007) Metabolism of glutamine and glutathione via gamma-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: possible significance in the pathophysiology of the organism. Mol Microbiol, Mol Microbiol 64, 396–406.
- Shibayama, K. , Wachino, J. I., Arakawa, Y., Saidijam, M., Rutherford, N. G. and Henderson, P. J. F. (2007) Metabolism of glutamine and glutathione via γ-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: Possible significance in the pathophysiology of the organism. Mol Microbiol 64, 396–406.
- McGovern, K. J. , Blanchard, T. G., Gutierrez, J. A., Czinn, S. J., Krakowka, S. and Youngman, P. (2001) γ-glutamyltransferase is a Helicobacter pylori virulence factor but is not essential for colonization. Infect Immun 69.
- Chevalier, C. , Thiberge, J. M., Ferrero, R. L. and Labigne, A. (1999) Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol Microbiol, Mol Microbiol 31, 1359–1372.
- Kim, K. M. , Lee, S. G., Park, M. G., Song, J. Y., Kang, H. L., Lee, W. K., Cho, M. J., Rhee, K. H., Youn, H. S. and Baik, S. C. (2007) Gamma-glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem Biophys Res Commun, Biochem Biophys Res Commun 355, 562–567.
- Valenzuela, M.; Bravo, D.; Canales, J.; Sanhueza, C.; Díaz, N.; Almarza, O.; Toledo, H.; Quest, A.F.G. Helicobacter pylori–Induced Loss of Survivin and Gastric Cell Viability Is Attributable to Secreted Bacterial Gamma-Glutamyl Transpeptidase Activity. J. Infect. Dis. 2013, 208, 1131–1141. [Google Scholar] [CrossRef]
- Kim, K.-M.; Lee, S.-G.; Kim, J.-M.; Kim, D.-S.; Song, J.-Y.; Kang, H.-L.; Lee, W.-K.; Cho, M.-J.; Rhee, K.-H.; Youn, H.-S.; et al. Helicobacter pylori γ-glutamyltranspeptidase induces cell cycle arrest at the G1-S phase transition. J. Microbiol. 2010, 48, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Ling, S.S.M.; Lui, S.Y.; Yeoh, K.G.; Ho, B. Helicobacter pylori γ-Glutamyl Transpeptidase Is a Pathogenic Factor in the Development of Peptic Ulcer Disease. Gastroenterology 2010, 139, 564–573. [Google Scholar] [CrossRef]
- Busiello, I. , Acquaviva, R., Di Popolo, A., Blanchard, T. G., Ricci, V., Romano, M. and Zarrilli, R. (2004) Helicobacter pyloriγ-glutamyltranspeptidase upregulates COX-2 and EGF-related peptide expression in human gastric cells. Cell Microbiol, John Wiley & Sons, Ltd 6, 255–267.
- Nagy, T.A.; Frey, M.R.; Yan, F.; Israel, D.A.; Polk, D.B.; Peek, R.M. Helicobacter pyloriRegulates Cellular Migration and Apoptosis by Activation of Phosphatidylinositol 3-Kinase Signaling. J. Infect. Dis. 2009, 199, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Miyagawa, J.; Shinomura, Y.; Kanayama, S.; Isozaki, K.; Yamamori, K.; Mizuno, H.; Ishiguro, S.; Kiyohara, T.; Miyazaki, Y.; et al. Significance of the association between heparin-binding epidermal growth factor-like growth factor and CD9 in human gastric cancer. Int. J. Cancer 2002, 98, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Yin, F. , Grabowska, A. M., Clarke, P. A., Whelband, E., Robinson, K., Argent, R. H., Tobias, A., Kumari, R., Atherton, J. C. and Watson, S. A. (2010) Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut 59.
- Shi, H. , Qi, C., Meng, L., Yao, H., Jiang, C., Fan, M., Zhang, Q., Hou, X. and Lin, R. (2021) Bone marrow-derived mesenchymal stem cells promote Helicobacter pylori-associated gastric cancer progression by secreting thrombospondin-2. Cell Prolif 54.
- Wang, Z.; Wang, W.; Shi, H.; Meng, L.; Jiang, X.; Pang, S.; Fan, M.; Lin, R. Gamma-glutamyltransferase of Helicobacter pylori alters the proliferation, migration, and pluripotency of mesenchymal stem cells by affecting metabolism and methylation status. J. Microbiol. 2022, 60, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Shi, H.; Wang, Z.; Fan, M.; Pang, S.; Lin, R. The Gamma-glutamyltransferase gene of Helicobacter pylori can promote gastric carcinogenesis by activating Wnt signal pathway through up-regulating TET1. Life Sci. 2021, 267, 118921. [Google Scholar] [CrossRef]
- Schmees, C. , Prinz, C., Treptau, T., Rad, R., Hengst, L., Voland, P., Bauer, S., Brenner, L., Schmid, R. M. and Gerhard, M. (2007) Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology 132, 1820–1833.
- Gerhard, M.; Schmees, C.; Voland, P.; Endres, N.; Sander, M.; Reindl, W.; Rad, R.; Oelsner, M.; Decker, T.; Mempel, M.; et al. A Secreted Low--Molecular-Weight Protein From Helicobacter pylori Induces Cell-Cycle Arrest of T Cells. Gastroenterology 2005, 128, 1327–1339. [Google Scholar] [CrossRef]
- Fassi Fehri, L. , Koch, M., Belogolova, E., Khalil, H., Bolz, C., Kalali, B., Mollenkopf, H. J., Beigier-Bompadre, M., Karlas, A., Schneider, T., et al. (2010) Helicobacter pylori induces miR-155 in T cells in a cAMP-Foxp3-dependent manner. PLoS One 5.
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Kandulski, A. , Wex, T., Kuester, D., Peitz, U., Gebert, I., Roessner, A. and Malfertheiner, P. (2008) Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter, Helicobacter 13, 295–303.
- Ohue, Y. and Nishikawa, H. (2019) Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci., Division of Cancer Immunology, Research Institute/Exploratory Oncology Research & Clinical Trial Center (EPOC), National Cancer Center, Tokyo, Japan Division of Cancer Immunology, Research Institute/Exploratory Oncology Research & Clinical Trial Center (E 110, 2080–2089.
- Lina, T. T. , Alzahrani, S., House, J., Yamaoka, Y., Sharpe, A. H., Rampy, B. A., Pinchuk, I. V. and Reyes, V. E. (2015) Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS One 10.
- Das, S.; Suarez, G.; Beswick, E.J.; Sierra, J.C.; Graham, D.Y.; Reyes, V.E. Expression of B7-H1 on Gastric Epithelial Cells: Its Potential Role in Regulating T Cells during Helicobacter pylori Infection. J. Immunol. 2006, 176, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E. J. J. , Pinchuk, I. V. V, Das, S., Powell, D. W. W. and Reyes, V. E. E. (2007) B7-H1 Expression on Gastric Epithelial Cells after Helicobacter pylori Exposure Promotes the Development of CD4+ CD25+ FoxP3+ Regulatory T Cells. Infect. Immun 75, 4334–4341.
- Lina, T. T. , Pinchuk, I. V., House, J., Yamaoka, Y., Graham, D. Y., Beswick, E. J. and Reyes, V. E. (2013) CagA-dependent downregulation of B7-H2 expression on gastric mucosa and inhibition of Th17 responses during Helicobacter pylori infection. Journal of Immunology 191.
- Hou, J.; Yu, Z.; Xiang, R.; Li, C.; Wang, L.; Chen, S.; Li, Q.; Chen, M.; Wang, L. Correlation between infiltration of FOXP3+ regulatory T cells and expression of B7-H1 in the tumor tissues of gastric cancer. Exp. Mol. Pathol. 2014, 96, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Shigemori, T.; Toiyama, Y.; Okugawa, Y.; Yamamoto, A.; Yin, C.; Narumi, A.; Ichikawa, T.; Ide, S.; Shimura, T.; Fujikawa, H.; et al. Soluble PD-L1 Expression in Circulation as a Predictive Marker for Recurrence and Prognosis in Gastric Cancer: Direct Comparison of the Clinical Burden Between Tissue and Serum PD-L1 Expression. Ann. Surg. Oncol. 2018, 26, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Böger, C. , Behrens, H. M., Krüger, S., Röcken, C., Boger, C., Behrens, H. M., Kruger, S. and Rocken, C. (2017) The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology, Taylor and Francis Inc., Department of Pathology, Christian-Albrechts-University, Kiel, Germany Department of Pathology, Christian-Albrechts-University, Kiel, Germany Department of Pathology, Christian-Albrechts-University, Kiel, Germany Department of Pathology, Christian-Albrech 6, e1293215.
- Lina, T.T.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Helicobacter pylori Elicits B7-H3 Expression on Gastric Epithelial Cells: Implications in Local T Cell Regulation and Subset Development During Infection. Clin. Oncol. Res. 2019, 2, 1–12. [Google Scholar] [CrossRef]
Figure 1.
Immune Checkpoints Regulators of T cell Activity. In red are immune checkpoints whose expression is altered by H. pylori. Black arrows represent positive signals. Red arrows represent inhibitory signals.
Figure 1.
Immune Checkpoints Regulators of T cell Activity. In red are immune checkpoints whose expression is altered by H. pylori. Black arrows represent positive signals. Red arrows represent inhibitory signals.
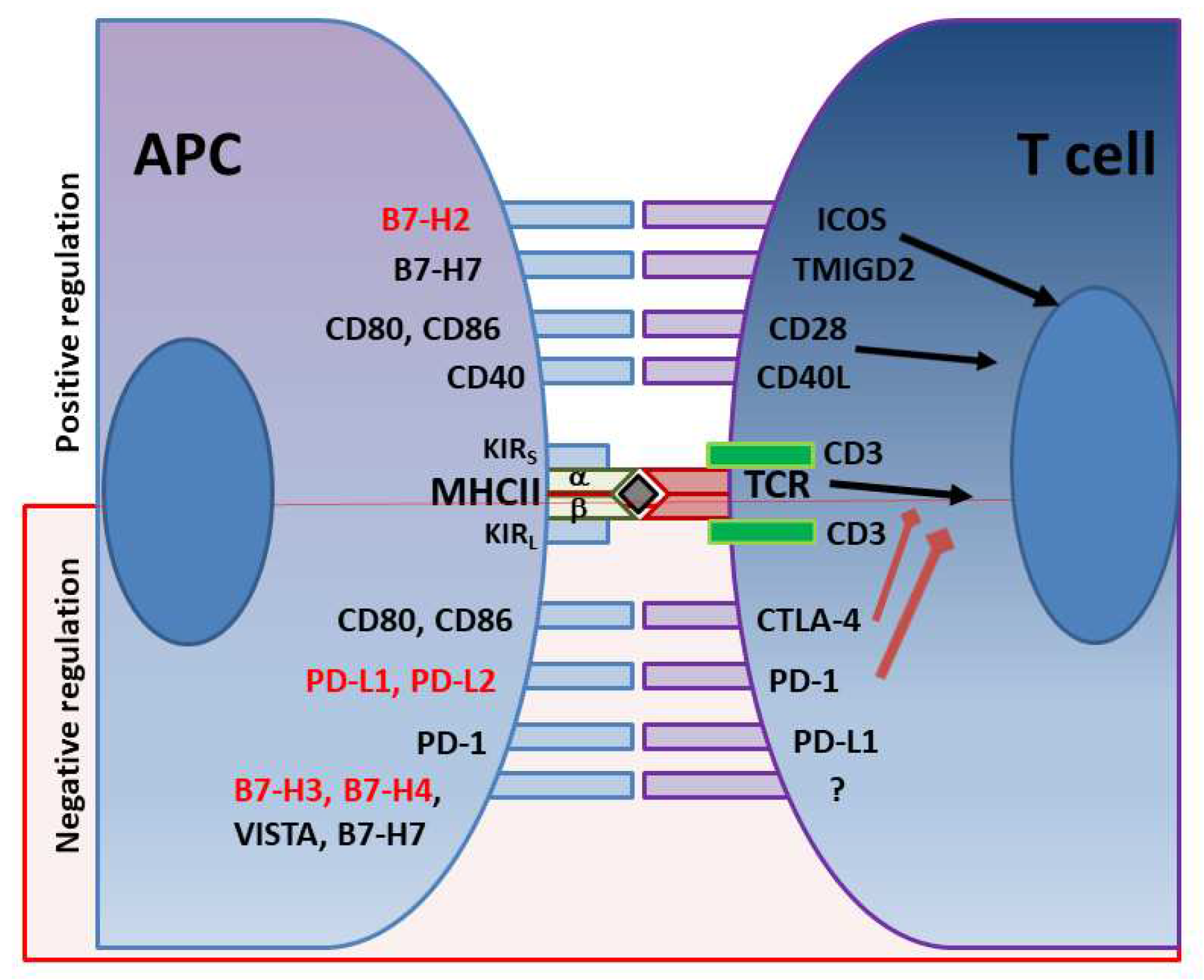
Figure 2.
H. pylori sets Immunosuppressive Environment. H. pylori interferes with effector T cells functions using an array of virulence factors and by manipulating the expression by the epithelium of immune checkpoint regulatory receptors.
Figure 2.
H. pylori sets Immunosuppressive Environment. H. pylori interferes with effector T cells functions using an array of virulence factors and by manipulating the expression by the epithelium of immune checkpoint regulatory receptors.
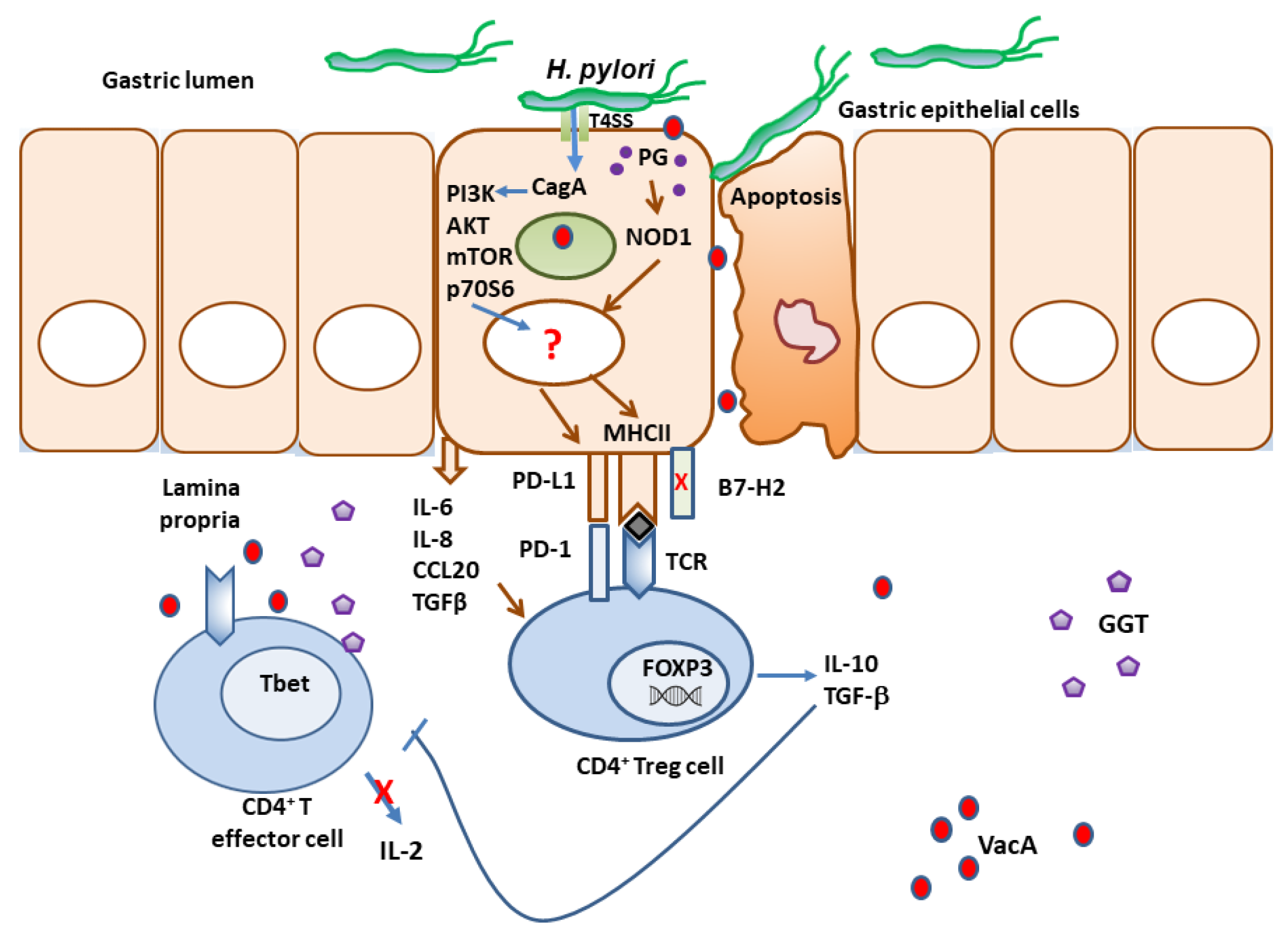
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



