Submitted:
19 April 2023
Posted:
19 April 2023
You are already at the latest version
Abstract
Rapeseed (Brassica napus L.) is a globally important oilseed crop with various uses, including consumption of its succulent stems as a seasonal vegetable, but its uniaxial branching habit limits the stem yield. Therefore, developing a multi-stem rapeseed variety has become increasingly crucial. In this study, a nature mutant of wild type from germplasm resources with stable inheritance of the multi-stem trait (MS) was obtained and showed abnormal shoot apical meristem (SAM) development and increased main stem number compared to WT. Histological and scanning electron microscopy analyses revealed multiple SAMs in the MS mutant, while only a single SAM was found in WT. Compared to WT, the mutant exhibited increased accumulation of cytokinins (CKs), transcriptome and RT-qPCR analyses showed the expression of genes involved in CK biosynthesis and metabolism pathways were altered in MS mutant. These findings provide insight into the mechanism of multiple main stems formation in Brassica napus L. and lay a theoretical foundation for breeding multi-main stem rapeseed vegetable varieties.
Keywords:
Brassica napus L.
; Shoot apical meristem (SAM)
; shoot branching
; Transcriptome
; Cytokinin
1. Introduction
Rapeseed (Brassica napus L.) is one of the world's most important oil crops, accounting for approximately 15% of the world's vegetable oil production [1]. As the social economy develops, and consumers demand more diversified products, rapeseed is becoming increasingly versatile, serving as a vegetable, flower, honey, fodder, and fertilizer [1,2]. Although the crispy and succulent bolting stem of rapeseed is a seasonal vegetable commonly consumed in South China, its stem yield is lower than that of other crops, such as Chinese flowering cabbage (Brassica campestris L. ssp. chinensis var. utilis Tsen et Lee) and Chinese kale (Brassica alboglabra) due to its single main stem. Therefore, exploring or creating new multi-main branched rapeseed varieties could effectively enhance vegetable yield.
Branches and stems formation in plants is facilitated by the differentiation of axillary meristems (AM) at leaf axils, which elongate and differentiate into lateral branches [3,4]. The AM initiates from the shoot apical meristem (SAM), a group of pluripotent stem cells that generate leaf, axillary branch, flower primordia, and stem tissue [5,6]. Thus, the final number of branches or stems is determined by the number of SAMs. Phytohormones, including auxin, cytokinins, and strigolactones, play an important role in activating, initiating and regulating plant branching [7,8]. Among these, cytokinins (CKs), a group of N6 adenines that include isopentenyladenine (IP), trans-zeatin (tZ), cis-zeatin (cZ), and dihydrozeatin (DHZ), have been demonstrated to be positive regulators of branching and SAM development [9,10]. Nishimura et al. (2004) used the Arabidopsis ahk2 ahk3 ahk4 triple CK receptor mutant revealed that the functional of CK in the maintenance of SAM activity [11]. In Arabidopsis, the supershoot (sps) mutant forms multiple AMs in the leaf axils due to increased CK levels, promoting branching formation from rosette and cauline leaves [12]. Moreover, The loss of the negative feedback regulator of CK, type-A ARR homolog ABPH1, results in an increase in SAM size in maize [13].
The biosynthesis and catabolism of CK are dynamically balanced processes in plants. Cytokinin oxidase (CKX) can be irreversibly cleave the active CKs to decrease cytokinin levels [14]. CKX family genes have been identified in many plant species, such as Arabidopsis thaliana [15], Nicotiana tabacum [16], Zea mays [17], Oryza sativa [18], Triticum aestivum [19], and Glycine max [20]. In rapeseed (Brassica napus L.), 23 CKX genes were identified [21]. The CKX has been found that role in SAM development and plant branching. Werner et al. (2003) found that the Arabidopsis CKX gene family has seven members (AtCKX1 to AtCKX7), and CKX1 and CKX3 overexpression transgenic plants showed the decreased activity of the SAM [15]. LONELY GUY (LOG) encodes a cytokinin riboside 5’-monophosphate phosphoribohydrolase that works in the final step of bioactive CK synthesis, it was discovered through the analysis of rice (Oryza sativa) mutant lonely guy (log) that are termination of the shoot meristems [22]. In Arabidopsis, nine LOG genes (AtLOG1 to AtLOG9) were predicted as homologs of rice LOG [23]. Tokunaga et al. (2011) used multiple Atlog mutant, and found that the LOG genes were important for the maintenance of the SAM in Arabidopsis [24].
The multi-main stem (MMS) traits have been discovered in rice and wheat, which improve the growth potential and seed number and are therefore important for yield improvement [25,26]. Zhao et al. (2019) identified six candidate genes (RPT2A, HLR, CRK, LRR-RLK, AGL79, and TCTP) involved in SAM differentiation and axillary bud formation by QTL mapping and Gene-Fishing technique, which related to the formation of MMS phenotype in rapeseed [27]. However, the mechanism underlying MMS formation and the relationship between SAM and MMS formation remain unclear. In this study, we identified a rapeseed mutant with MMS from germplasm resources, and after six consecutive generations of self-selection, we generated lines with stable inheritance of the multiple main branching trait. The mutant, named Multiple Stem (MS) rapeseed, develops over six main stems from the base of the plant, with small branching angles. Our results showed that the development of SAM in MS mutant was abnormal compared to WT. Moreover, we found that the contents of CKs as well as the expression of CK pathway- related genes were altered in the SAM of MS mutant, indicating the involvement of CK signaling in MMS formation in rapeseed. Thus, this study lays the foundation for comprehending the role of CK in SAM development that is linked to the formation of main stems and provides references for selection and breeding of rapeseed varieties with multi-main stems.
2. Methods and materials
2.1. Plant materials and growth conditions
Wild type and MS mutant seeds were sterilized in 75% ethanol for 15 minutes and then rinsed repeatedly in sterile water. The seeds were germinated in an incubator at 25°C and sown in culture bottles once they germinated. The bottles were placed in a light incubator with 14 hours of light, 10 hours of dark, a day temperature of 22°C, a night temperature of 18°C, and a relative humidity of 60% at an intensity of 10,000 lux.
2.2. Tissue section
For paraffin sections, the SAM tissues of DAG20-old wild-type and MS mutant plants were cut using a scalpel and then placed in formaldehyde-acetic acid-alcohol (FAA) stationary liquid for 24 hours. The tissues were subsequently dehydrated with 50%, 70%, 85%, 95%, and 100% ethanol solutions for 40-60 minutes and then treated with xylene for decolorization and transparency. After embedding in paraffin, slices were deparaffinized, rehydrated, and stained for microscopic examination and photographed using the ECHO microscope (Revolve FL, San Diego, USA). Refer to [28,29] for further details.
2.3. Scanning electron microscopy
The SAM samples of DAG20-old wild type and MS mutant plants were used for scanning electron microscopy (SEM) analysis, which was performed by Wuhan Servicebio Technology CO., Ltd (Wuhan, China) with a Hitachi SU8100 SEM.
2.4. Quantification of cytokinins
Cytokinins determination of the SAM of wild type and MS mutant at DAG20 was performed on an HPLC-MS/MS (liquid chromatography-mass spectrometry) in Shanghai Applied Protein Technology Co., Ltd (Shanghai, China) following a previously described method [30]. The SAM from five individual replicated plants were pooled as one replicate, n=3.
2.5. RNA extraction, sequencing and analysis
The SAM of wild type and MS mutant plants at DAG15, 20, 25, 30, and 35 were collected, and samples from five individual replicated plants were pooled as one replicate, n=3. Samples were stored at −80 °C until use. Total RNA extraction and transcriptome sequencing were references as previously described. Briefly, total RNA was extracted using TRIZOL® Reagent (TRAN, Beijing, China) according to the manufacturer’s protocol and qualified using the QubitTM4 Fluorometer microvolume spectrophotometer (Thermo Fisher Scientific, Singapore). The cDNA libraries were sequenced on the Illumina HiSeq4000 sequencing platform by Lian Chuan Biotechnology Co., Ltd. (Hangzhou, China). Quality reads control of the raw RNA-Seq data off the machine was performed with the fastQC application v0.11.2 software. Low-quality reads and adapters were delete by the Trimmomatic (0.36.5) tool to acquisition the clean high-quality reads data [31]. The obtained clean reads are mapping to the published reference genome. (http://www.genoscope.cns.fr/brassicanapus/). StringTie (1.3.4) was employed to count the number of reads mapped onto each gene, and quantification of the gene expression level in the number of fragments per kilobase of the transcript sequence per million base pairs was sequenced (FPKM). Differential expression analysis performed using the DESeq2R package (2.11.38).
Transcripts with p values < 0.05, |log2(fold change)| ≥ 1 were considered as differentially expressed genes (DEGs). Pearson correlation, Principal component analysis (PCA) and Venn diagram analysis and graphing was performed using the OmicShare tools (https://www.omicshare.com/tools). GO enrichment analysis was performed by AgriGO software (http://systemsbiology.cau.edu.cn/agriGOv2/index.php). KEGG analysis was reference Kyoto encyclopedia of genes and genomes (https://www.genome.jp/kegg/) database and used the KOBAS software to detect statistical enrichment of DEGs in the KEGG pathway. TBtools was employed to construct the heatmaps of transcriptome [32].
2.6. Real-time quantitative PCR (RT-qPCR)
To verify the reliability of transcriptomic data, we randomly selected some genes selected for RT-qPCR validation at different developmental stages to validate the RNA-seq data. performed in a 96-wells plate on an CFX96 Touch Real-Time PCR system (Bio-RAD, USA). The thermal conditions were 95 °C for 30 s, followed by 39 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 15 s. Then, the genes specific primers were designed based on multiple sequences alignment. The relative expression level of the selected DEGs was calculated with the 2−∆∆CT method [33], and specific primer information used in this study is listed in Table S5.
2.7. Statistical analysis
The data were analyzed and graphed using SPSS version 18.0.
3. Results
3.1. Phenotype investigation of MS mutant
In this study, we report the discovery of a natural mutant in the WT rapeseed background that exhibits a remarkable phenotype. Homozygote of the mutant was obtained by self-crossing for six generations. Unlike the WT, which develops a single main stem, the mutant produces more than two branches, typically six to seven (Figure 1A,B). Importantly, the branches in the MS mutant were formed from the basal of plant. Additionally, the branching architecture of the MS mutant is characterized by small branch angles and a compact structure (Figure 1C,D). Given its distinctive phenotype, this mutant was named MS (multi-main stems) rapeseed. To ensure the stable inheritance of this trait, we self-crossed the mutant for several generations to obtain homozygotes.
3.2. Abnormal development of the SAM in MS mutant
At the 20 days after germination (DAG), there are two axillary buds emerged in WT, while the MS mutant has multiple axillary buds at the basal of the plant (Figure 2A,B). Thus, the number of main stems in MS mutant significantly exceeds that of WT starting at DAG20. Shoot branching arises from the AM in the axils of leaves, which are initiated by the SAM [4]. Given that the MS mutant exhibits an increase in main stems, it is possible that SAM initiation is affected. To test this hypothesis, paraffin sectioning and scanning electron microscopy analysis of SAM from WT and MS mutant at DAG20 were performed. The SAM of WT exhibited a regular protuberance (Figure 2D,F). In contrast, the SAM in MS mutant exhibited an irregular shape and more than one SAM (Figure 2E,G). These results indicating a higher activity of the SAM in MS mutant, which leads to the generation of multiple main stems.
3.3. CK levels are elevated in the SAM of the MS mutant
CKs positively regulate the development of SAM [9]. Specifically, we investigated the CK levels in the SAMs of both WT and MS mutant at DAG20. HPLC-MS analysis revealed that the CKs content in the MS mutant was higher than that in the WT, with the most significant difference observed in the levels of cis-zeatin riboside (czR) and trans-zeatin riboside (tzR) (Figure 2H). Compared with the WT, the czR and tzR contents in MS mutant increased by 47% and 38%, respectively (Figure 2H). These findings suggest that the increased levels of CKs in the SAM of MS mutant may contribute to an increase in the number of SAM, ultimately leading to increased branching.
3.4. RNA-seq analysis of the SAM in MS mutant
In order to further elucidate the mechanism of development of SAM in MS mutant, transcriptomic analysis was performed. We selected the SAM of WT and mutant at five developmental stages (DAG15, 20, 25, 30, and 35) for transcriptome sequencing. Firstly, we analyzed the reliability of the RNA-seq data. By using the FPKM values of the average of three replicates of each sample, we plotted a heatmap of the correlation between samples (Figure 3A), which showed that the correlation coefficient between each sample was above 0.8, suggesting high data consistency. Additionally, principal component analysis (PCA) of the samples showed that the replicates exhibit good consistency among different samples, meeting the requirements for further data analysis (Figure 3B).
3.5. Differential transcriptional in different developmental stages in the SAM of MS mutant
The selection criteria for differential expressed genes (DEGs) were set as follows: p value < 0.05, and |log2(fold change) | ≥ 1. The transcriptome of the SAM of MS mutant was compared with that of the WT at five developmental stages, and DEGs were identified. At DAG15, a total of 2685 DEGs were identified, including 1553 genes were down-regulated, and 1132 genes were up-regulated. On the DAG20, a total of 4782 DEGs were identified, of which 3420 were down-regulated and 1362 were up-regulated. On the DAG25, a total of 5224 DEGs were identified, with 3058 down-regulated genes and 2166 up-regulated genes. On the DAG30, a total of 3590 DEGs were identified, with 2261 down-regulated genes and 1329 up-regulated genes. On the DAG35, a total of 5460 DEGs were identified, in which 2269 genes were down-regulated and 3193 genes were up-regulated (Figure 3C, Table S1). In addition, Venn diagram analysis of DEGs across these five developmental stages revealed that a total of 796 core genes were commonly regulated by both WT and MS mutant (Figure 3D).
3.6. GO and KEGG analysis of all DEGs
To further elucidate the functions of DEGs between the MS mutant and WT, Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed. For DEGs identified at DAG15, we observed 50 significantly enriched GO terms, with most DEGs being enriched in the molecular function “binding,” the cellular component “cell wall” and “apoplast,” and the biological process “translation” (Figure S1, Table S2). Similarly, 48, 50, 50, and 49 significant GO terms were identified for DEGs at DAG20, 25, 30, and 35, respectively. Most DEGs were involved in the molecular function “DAN-binding transcription factor activity” (DAG20 and 35), the biological process “response to chitin” (DAG20, 30, 35), “response to water deprivation” (DAG25), and the cellular component “ubiquitin ligase complex” (DAG20), “apoplast” (DAG25, 30, 35) (Figures S2–S5, Table S2).
Furthermore, we identified the top 10 KEGG pathways for DEGs at DAG15, 20, 25, 30, and 35. The metabolism of amino acids, such as “beta-Alanine metabolism,” “Valine, leucine and isoleucine degradation,” and “Alanine, aspartate and glutamate metabolism,” was significantly enriched, suggesting their potential involvement in the development of the MS mutant's SAM. Notably, the “Plant hormone signal transduction” was enriched from the DEGs at DAG20, 30, and 35, indicating that the DEGs involved in this pathway also participate in the formation of multi-main stems in the MS mutant (Figure 4, Table S3).
3.7. The expression of CK-related genes was affected in the SAM of MS mutant
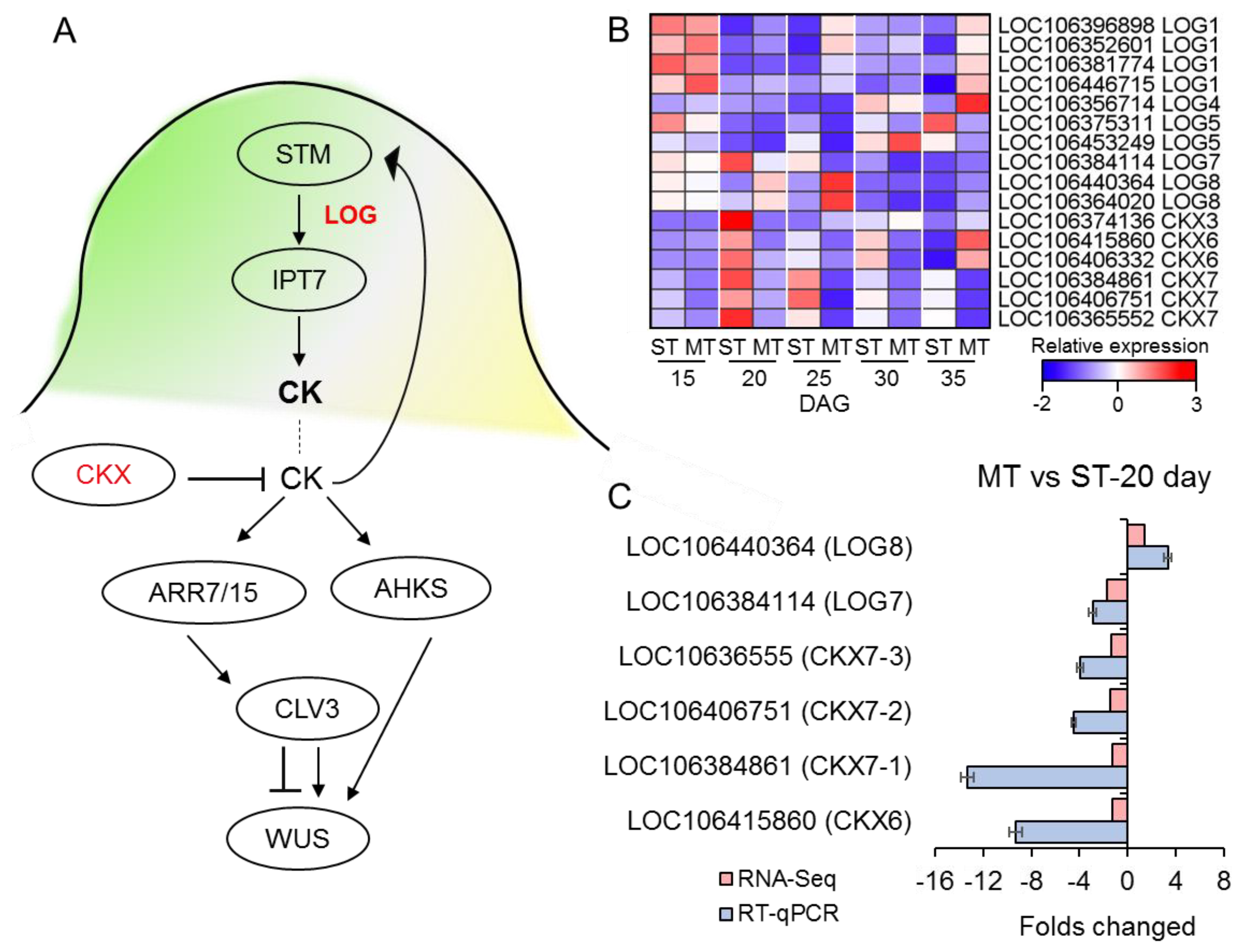
Given the crucial role of CKs in regulating development of SAM and the significantly higher CKs level in the SAM of MS mutant compared to WT (Figure 2C), we focused on the transcriptional levels of genes involved in CK synthesis and metabolism pathways. LONELY GUY (LOG) activates the CK signaling by releasing the free bases from the nucleotide forms, while destruction of CK is carried out by cytokinin oxidase/dehydrogenase (CKX) (Figure 5A). The CK signaling pathway is activated by LONELY GUY (LOG), which releases free bases from nucleotide forms, while cytokinin oxidase/dehydrogenase (CKX) degrades CKs (Figure 5A). In this study, we detected 14 LOG (LOC106396898 (LOG1), LOC106352601 (LOG1), LOC106446715 (LOG1), LOC106356714 (LOG4), LOC106375311 (LOG5), LOC106453249 (LOG5), LOC106384114 (LOG7), LOC106440364 (LOG8), LOC106364020 (LOG8)) and CKX (LOC106415860 (CKX6), LOC106406332 (CKX6), LOC106384861 (CKX7), LOC106406751 (CKX7), LOC10636555 (CKX7)) genes with different expression in the MS mutant at different DAGs.
During the DAG15, before the formation of multi-stem in the MS mutant, and there was no significant change in the expression of the LOG and CKX genes in the MS mutant compared to the WT. However, during the 20 days when the multi-stem of the MS mutant started to form, we found that the expression of the LOG8 was upregulated in the mutant but not in the WT. Conversely, the transcript levels of CKX6 and CKX7 were downregulated in the MS mutant, but upregulated in the WT. The same trends were observed for these genes as DAG25. However, at the DAG30 and 35, the expression of LOG and CKX genes shown different patterns (Figure 5B, Table S4). Based on our previous research findings of paraffin sectioning, CKs content, and KEGG enrichment analysis, it is suggested that the formation of the multi-stem in the MS mutant mainly initiates on the DAG20.
Therefore, following DAG20, we selected LOG7 (LOC106384114), LOG8 (LOC106440364) and CKX6 (LOC106415860), CKX7-1 (LOC106384861), CKX7-2 (LOC106406751), and CKX7-3 (LOC10636555) were selected for RT-qPCR validation and further analysis. Both the transcriptome and RT-qPCR results showed that the expressions of LOG8 were up-regulated in MS mutant compared to WT, while there was no significant difference in the expression of LOG7. In contrast, the expression of CKX6 and CKX7-1, CKX7-2, CKX7-3 was down-regulated in MS mutant relative to WT (Figure 5C). Taken together, the dramatic down-regulation of the CKX genes and the up-regulation of LOG8 in the MS mutant could lead to a significant increase in the CK content, which would result in the abnormal formation of SAM and shoot branching.
4. Discussion
Multiple main stem formation is a crucial determinant of stem architecture in rapeseed and plays an essential role in improving vegetable yield. Despite its importance, the mechanism underlying the formation of multi-main stems in rapeseed remains largely unknown. In this study, we identified the MS mutant that exhibits 6-7 main stems (Figure 1). We hypothesized that the increased main branching was caused by abnormal development of the SAM. Further investigation revealed that the MS mutant had a greater number of SAMs with an irregular shape and increased axillary buds at the basal of the MS mutant than WT at DAG20 (Figure 2A–G). These results provide evidence that the increased number of main stems in the MS mutant is a result of an increased number of SAMs.
As the development of SAM in the MS mutant was distinct from that of the WT by DAG20, we chose to perform transcriptome analysis at different time points, including the early germination stage (DAG15), the day when SAM development started to produce abnormalities (DAG20), and later time points (DAG25, 30, and 35), to gain a comprehensive understanding of the SAM development in the MS mutant compared to the WT. In the DAG15, there were a total of 2685 DEGs, which was much lower than those observed on DAG20 (4782), DAG25 (5224), DAG30 (3590), and DAG35 (5460) (Figure 3C, Table S1). Therefore, it is reasonable to conclude that at least by DAG20, the development and gene expression of SAM in the MS mutant begin to undergo significant changes compared to the WT, which ultimately lead to the formation of multiple main stems in the DAG60 and DAG120 (Figure 1).
Furthermore, we found that the "Linoleic acid metabolism" and "alpha-Linoleic acid metabolism" pathways were enriched in the MS mutant at DAG20, 25, 30, and 35 (Figure 4). These pathways are associated with jasmonic acid (JA) synthesis, which may be related to the wounding caused by collecting the samples. Additionally, we observed that the "plant hormone signal transduction" pathway were enriched at least three time points (DAG20, 30, 35) suggesting that phytohormones also play a crucial role in the development of SAM in the MS mutant (Figure 4).
CK has been recognized as a critical regulator of SAM development in plants [35]. In this study, we observed that the content of cis-zeatin riboside (czR) and trans-zeatin riboside (tzR) was higher in the SAM of the MS mutant than in the WT (Figure 2H). We proposed that the altered CKs content in MS mutant at least partly affected the expression of CK-related genes. Using transcriptome and RT-qPCR analysis, we show that the CK synthesis gene LOG8 (LOC106440364) was elevated in the MS mutant compared to the WT, while the CK degradation genes CKX6 (LOC106415860), CKX7-1 (LOC106384861), CKX7-2 (LOC106406751), and CKX7-3 (LOC10636555) were downregulated in the MS mutant (Figure 5B,C). This suggest that the abnormal development of the SAM in the MS mutant caused by the altered expression of CK-related genes, which leads to elevated levels of CK.
In addition to CK, shoot branching is also regulated by auxin and strigolactones (SLs) [36,37,38,39,40]. Previous studies have shown that CK, auxin, and SL signals can interact with each other [7,41,42]. In this study, we found that at the DAG20, the GO enrichment analysis revealed that 45 up-regulated and 12 down-regulated DEGs in the MS mutant was involved in “auxin-activated signaling pathway”, and “response to auxin” was among the pathways enriched from the 2 up-regulated and 43 down-regulated DEGs in MS mutant (Figure S2, Table S2), indicating that the auxin pathway might involve in the formation of multiple main stems in the MS mutant, and whether auxin and SL are involved in the regulation of branching in the MS mutant by CK signaling needs further study. It is known that CK activates the WUSCHEL (WUS) expression, which is a positive regulator of the size of meristem [43]. Therefore, it would be interesting to investigate the mechanisms by which CK regulates the development of the SAM in the MS mutant.
Besides phytohormones, sugar also promoting cell division, and play a role in plant branching. Salam et al. (2017) reported that the branching of potato (Solanum tuberosum) was induced by the application of exogenous sugar (sucrose, fructose, glucose) [44]. Our KEGG analysis revealed that at the DAG20 and DAG25, when the SAM was abnormal in MS mutant, the DEGs between the MS and WT were enriched in “Amino sugar and nucleotide sugar metabolism”, and “starch and sucrose metabolism” pathways (Figure 4B,C, Table S3), suggesting that the sugar signaling may be involved in the formation of multi-main stems in the MS mutant. Thus, shoot branching is a complex quantitative trait that is under polygenic control and is regulated by phytohormones and external environmental factors, much research on CK, SLs, auxin, as well as sugar is needed to fully understand the increased branching in the MS mutant.
The identification of a mutation site in a newly discovered natural mutant typically requires the use of various tools and approaches. One of the most powerful tools in identifying loci or key genes associated with agronomic traits is the genome-wide association study (GWAS) [45,46]. In rapeseed, as with many other crop plants, most agronomic traits are quantitatively inherited and controlled by quantitative trait loci (QTL) [47,48,49,50]. QTLs related to branch number have been identified in rapeseed [51,52,53,54]. Based on GWAS, numerous loci related to branch number have been identified on almost all rapeseed chromosomes, including A01, A03, A07, C04, C07, C09 [55,56,57,58,59]. In addition to GWAS, Li er al. (2020) identified a major QTL related to branching (shoot branching 9, qSB.A09) on the chromosome A09 in Brassica rapa L. ssp. Chinensis by integrating QTL mapping with BSA-seq (bulked segregant analysis) [60]. Given that 43 QTLs were identified for MMS in rapeseed, and six candidate genes related to the formation of MMS were obtained from QTL mapping and Gene-Fishing technique [27], it is worthwhile to explore the major QTLs or candidate genes associated with multi-main stems in the MS mutant using these methods in the future.
5. Conclusions
Our study sheds light on the mechanism underlying the formation of multiple main stems in rapeseed. Specifically, we found that the abnormal development of the SAM in the MS mutant is closely linked to the increased number of main stems. By combining the LC-MS and transcriptome analysis, we found that the expression of CK-related genes was differently altered, which promoted the CK synthesis in the SAM, leading to the increased number of SAM in MS mutant. These findings highlight the importance of CK in regulating SAM development during multi-main stem formation in rapeseed. This study not only provide insights for breeding new rapeseed varieties with multiple main stems but also provide valuable resources for future research on SAM development and shoot branching in rapeseed. However, the loci responsible for the multi-main stems phenotype in the MS mutant remain unknown, and further GWAS and genetic fine mapping will be necessary to identify the major QTLs or candidate genes associated with multi-main stems in the MS mutant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: The GO enrichment analysis of DEGs between MS mutant and WT at 15 days after germination (DAG). Figure S2: The GO enrichment analysis of DEGs between MS mutant and WT at 20 days after germination (DAG). Figure S3: The GO enrichment analysis of DEGs between MS mutant and WT at 25 days after germination (DAG). Figure S4: The GO enrichment analysis of DEGs between MS mutant and WT at 30 days after germination (DAG). Figure S5: The GO enrichment analysis of DEGs between MS mutant and WT at 35 days after germination (DAG). Table S1: The DEGs between MS mutant and WT at different DAG; Table S2: GO analysis of DEGs between MS mutant and WT at different DAG; Table S3: KEGG analysis of DEGs between MS mutant and WT at different DAG; Table S4: FPKM values of CK-related genes WT and MS mutant at different DAG; Table S5: The primers used for RT-qPCR.
Author Contributions
Conceived and designed the research work: Q.W., S.L. and L.L. Performed the experiments: Q.W., N.X., C.S., J.T., C.M., Y.Y. and X.P. Analyzed the data and wrote the manuscript: Q.W., N.X. and C.S. Perform the phenotype analysis: M.G., R.L., and R.D. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (No. 32060681), and the Major Science and Technology Projects in Yunnan Province (No. 202205AR070001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data were included in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mänd M., Williams I.H., Viik E., Karise R.: Oilseed rape, bees and integrated pest management. In: Biocontrol-Based Integrated Management of Oilseed Rape Pests. Edited by Williams IH. Dordrecht: Springer Netherlands; 2010: 357-379.
- Raboanatahiry N., Li H., Yu L., Li M.: Rapeseed (Brassica napus): Processing, Utilization, and Genetic Improvement. In: Agronomy. vol. 11; 2021.
- Shimizu-Sato, S.; Mori, H. Shimizu-Sato, S.; Mori, H. Control of outgrowth and dormancy in axillary buds. Plant physiology 2001, 127, 1405–1413.
- McSteen, P.; Leyser, O. McSteen, P.; Leyser, O. Shoot branching. Annual review of plant biology 2005, 56, 353–374. [CrossRef]
- Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Plant stem cell niches. Annual review of plant biology 2012, 63, 615–636. [CrossRef]
- Barton, M.K. Barton, M.K. Twenty years on: the inner workings of the shoot apical meristem, a developmental dynamo. Dev Biol 2010, 341, 95–113. [CrossRef]
- Barbier, F.F.; Dun, E.A.; Kerr, S.C.; Chabikwa, T.G.; Beveridge, C.A. Barbier, F.F.; Dun, E.A.; Kerr, S.C.; Chabikwa, T.G.; Beveridge, C.A. An Update on the Signals Controlling Shoot Branching. Trends Plant Sci 2019, 24, 220–236. [CrossRef]
- Wang, B.; Smith, S.M.; Li, J. Wang, B.; Smith, S.M.; Li, J. Genetic Regulation of Shoot Architecture. Annual review of plant biology 2018, 69, 437–468. [CrossRef]
- Shani, E.; Yanai, O.; Ori, N. Shani, E.; Yanai, O.; Ori, N. The role of hormones in shoot apical meristem function. Current opinion in plant biology 2006, 9, 484–489. [CrossRef]
- Sakakibara, H. Sakakibara, H. Cytokinins: activity, biosynthesis, and translocation. Annual review of plant biology 2006, 57, 431–449. [CrossRef]
- Nishimura, C.; Ohashi, Y.; Sato, S.; Kato, T.; Tabata, S.; Ueguchi, C. Nishimura, C.; Ohashi, Y.; Sato, S.; Kato, T.; Tabata, S.; Ueguchi, C. Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. The Plant cell 2004, 16, 1365–1377. [CrossRef]
- Tantikanjana, T.; Yong, J.W.; Letham, D.S.; Griffith, M.; Hussain, M.; Ljung, K.; Sandberg, G.; Sundaresan, V. Tantikanjana, T.; Yong, J.W.; Letham, D.S.; Griffith, M.; Hussain, M.; Ljung, K.; Sandberg, G.; Sundaresan, V. Control of axillary bud initiation and shoot architecture in Arabidopsis through the SUPERSHOOT gene. Genes & development 2001, 15, 1577–1588. [CrossRef]
- Giulini, A.; Wang, J.; Jackson, D. Giulini, A.; Wang, J.; Jackson, D. Control of phyllotaxy by the cytokinin-inducible response regulator homologue ABPHYL1. Nature 2004, 430, 1031–1034. [CrossRef]
- Werner, T.; Köllmer, I.; Bartrina, I.; Holst, K.; Schmülling, T. Werner, T.; Köllmer, I.; Bartrina, I.; Holst, K.; Schmülling, T. New insights into the biology of cytokinin degradation. Plant biology (Stuttgart, Germany) 2006, 8, 371–381. [CrossRef]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Van Onckelen, H.; Schmülling, T. Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Van Onckelen, H.; Schmülling, T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. The Plant cell 2003, 15, 2532–2550. [CrossRef]
- Motyka, V.; Faiss, M.; Strand, M.; Kaminek, M.; Schmulling, T. Motyka, V.; Faiss, M.; Strand, M.; Kaminek, M.; Schmulling, T. Changes in Cytokinin Content and Cytokinin Oxidase Activity in Response to Derepression of ipt Gene Transcription in Transgenic Tobacco Calli and Plants. Plant physiology 1996, 112, 1035–1043. [CrossRef]
- Morris, R.O.; Bilyeu, K.D.; Laskey, J.G.; Cheikh, N.N. Morris, R.O.; Bilyeu, K.D.; Laskey, J.G.; Cheikh, N.N. Isolation of a gene encoding a glycosylated cytokinin oxidase from maize. Biochemical and biophysical research communications 1999, 255, 328–333. [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [CrossRef]
- Mameaux, S.; Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Jack, P.; Werner, P.; Gray, J.C.; Greenland, A.J.; et al. Mameaux, S.; Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Jack, P.; Werner, P.; Gray, J.C.; Greenland, A.J.; et al. Molecular, phylogenetic and comparative genomic analysis of the cytokinin oxidase/dehydrogenase gene family in the Poaceae. Plant Biotechnol J 2012, 10, 67–82. [CrossRef]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Vankova, R.; Tanaka, M.; Seki, M.; Ham le, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Le, D.T.; Nishiyama, R.; Watanabe, Y.; Vankova, R.; Tanaka, M.; Seki, M.; Ham le, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Identification and expression analysis of cytokinin metabolic genes in soybean under normal and drought conditions in relation to cytokinin levels. PLoS One 2012, 7, e42411. [CrossRef]
- Liu, P.; Zhang, C.; Ma, J.Q.; Zhang, L.Y.; Yang, B.; Tang, X.Y.; Huang, L.; Zhou, X.T.; Lu, K.; Li, J.N. Liu, P.; Zhang, C.; Ma, J.Q.; Zhang, L.Y.; Yang, B.; Tang, X.Y.; Huang, L.; Zhou, X.T.; Lu, K.; Li, J.N. Genome-Wide Identification and Expression Profiling of Cytokinin Oxidase/Dehydrogenase (CKX) Genes Reveal Likely Roles in Pod Development and Stress Responses in Oilseed Rape (Brassica napus L.). Genes (Basel) 2018, 9. [CrossRef]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [CrossRef]
- Kuroha, T.; Tokunaga, H.; Kojima, M.; Ueda, N.; Ishida, T.; Nagawa, S.; Fukuda, H.; Sugimoto, K.; Sakakibara, H. Kuroha, T.; Tokunaga, H.; Kojima, M.; Ueda, N.; Ishida, T.; Nagawa, S.; Fukuda, H.; Sugimoto, K.; Sakakibara, H. Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. The Plant cell 2009, 21, 3152–3169. [CrossRef]
- Tokunaga, H.; Kojima, M.; Kuroha, T.; Ishida, T.; Sugimoto, K.; Kiba, T.; Sakakibara, H. Tokunaga, H.; Kojima, M.; Kuroha, T.; Ishida, T.; Sugimoto, K.; Kiba, T.; Sakakibara, H. Arabidopsis lonely guy (LOG) multiple mutants reveal a central role of the LOG-dependent pathway in cytokinin activation. The Plant journal : for cell and molecular biology 2012, 69, 355–365. [CrossRef]
- Lu, Z.; Shao, G.; Xiong, J.; Jiao, Y.; Wang, J.; Liu, G.; Meng, X.; Liang, Y.; Xiong, G.; Wang, Y.; et al. Lu, Z.; Shao, G.; Xiong, J.; Jiao, Y.; Wang, J.; Liu, G.; Meng, X.; Liang, Y.; Xiong, G.; Wang, Y.; et al. MONOCULM 3, an ortholog of WUSCHEL in rice, is required for tiller bud formation. Journal of genetics and genomics = Yi chuan xue bao 2015, 42, 71–78. [CrossRef]
- Hu, Y.S.; Ren, T.H.; Li, Z.; Tang, Y.Z.; Ren, Z.L.; Yan, B.J. Hu, Y.S.; Ren, T.H.; Li, Z.; Tang, Y.Z.; Ren, Z.L.; Yan, B.J. Molecular mapping and genetic analysis of a QTL controlling spike formation rate and tiller number in wheat. Gene 2017, 634, 15–21. [CrossRef]
- Zhao, W.; Chao, H.; Zhang, L.; Ta, N.; Zhao, Y.; Li, B.; Zhang, K.; Guan, Z.; Hou, D.; Chen, K.; et al. Zhao, W.; Chao, H.; Zhang, L.; Ta, N.; Zhao, Y.; Li, B.; Zhang, K.; Guan, Z.; Hou, D.; Chen, K.; et al. Integration of QTL Mapping and Gene Fishing Techniques to Dissect the Multi-Main Stem Trait in Rapeseed (Brassica napus L.). Front Plant Sci 2019, 10, 1152. [CrossRef]
- Xu, A.; Wei, C. Xu, A.; Wei, C. Comprehensive comparison and applications of different sections in investigating the microstructure and histochemistry of cereal kernels. Plant methods 2020, 16, 8. [CrossRef]
- Kong, Y.; Ebrahimpour, P.; Liu, Y.; Yang, C.; Alonso, L.C. Kong, Y.; Ebrahimpour, P.; Liu, Y.; Yang, C.; Alonso, L.C. Pancreatic Islet Embedding for Paraffin Sections. J Vis Exp 2018, 10.3791/57931. [CrossRef]
- Tarkowski, P.; Ge, L.; Yong, J.W.H.; Tan, S.N. Tarkowski, P.; Ge, L.; Yong, J.W.H.; Tan, S.N. Analytical methods for cytokinins. TrAC Trends in Analytical Chemistry 2009, 28, 323–335. [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Molecular Plant 2020, 13, 1194–1202. [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 2001, 25, 402–408. [CrossRef]
- Shi, B.; Vernoux, T. Shi, B.; Vernoux, T. Hormonal control of cell identity and growth in the shoot apical meristem. Current opinion in plant biology 2022, 65, 102111. [CrossRef]
- Skylar, A.; Wu, X. Skylar, A.; Wu, X. Regulation of meristem size by cytokinin signaling. Journal of integrative plant biology 2011, 53, 446–454. [CrossRef]
- Skoog, F.; Thimann, K.V. Skoog, F.; Thimann, K.V. Further Experiments on the Inhibition of the Development of Lateral Buds by Growth Hormone. Proc Natl Acad Sci U S A 1934, 20, 480–485. [CrossRef]
- Stirnberg, P.; van De Sande, K.; Leyser, H.M. Stirnberg, P.; van De Sande, K.; Leyser, H.M. MAX1 and MAX2 control shoot lateral branching in Arabidopsis. Development 2002, 129, 1131–1141. [CrossRef]
- Zou, J.; Chen, Z.; Zhang, S.; Zhang, W.; Jiang, G.; Zhao, X.; Zhai, W.; Pan, X.; Zhu, L. Zou, J.; Chen, Z.; Zhang, S.; Zhang, W.; Jiang, G.; Zhao, X.; Zhai, W.; Pan, X.; Zhu, L. Characterizations and fine mapping of a mutant gene for high tillering and dwarf in rice (Oryza sativa L.). Planta 2005, 222, 604–612. [CrossRef]
- Beveridge, C.A. Beveridge, C.A. Long-distance signalling and a mutational analysis of branching in pea. Plant Growth Regulation 2000, 32, 193–203. [CrossRef]
- Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pages, V.; Dun, E.A.; Pillot, J.P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.C.; et al. Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pages, V.; Dun, E.A.; Pillot, J.P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.C.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [CrossRef]
- Hayward, A.; Stirnberg, P.; Beveridge, C.; Leyser, O. Hayward, A.; Stirnberg, P.; Beveridge, C.; Leyser, O. Interactions between auxin and strigolactone in shoot branching control. Plant physiology 2009, 151, 400–412. [CrossRef]
- Domagalska, M.A.; Leyser, O. Domagalska, M.A.; Leyser, O. Signal integration in the control of shoot branching. Nature reviews. Molecular cell biology 2011, 12, 211–221. [CrossRef]
- Gordon, S.P.; Chickarmane, V.S.; Ohno, C.; Meyerowitz, E.M. Gordon, S.P.; Chickarmane, V.S.; Ohno, C.; Meyerowitz, E.M. Multiple feedback loops through cytokinin signaling control stem cell number within the Arabidopsis shoot meristem. Proc Natl Acad Sci U S A 2009, 106, 16529–16534. [CrossRef]
- Salam, B.B.; Malka, S.K.; Zhu, X.; Gong, H.; Ziv, C.; Teper-Bamnolker, P.; Ori, N.; Jiang, J.; Eshel, D. Salam, B.B.; Malka, S.K.; Zhu, X.; Gong, H.; Ziv, C.; Teper-Bamnolker, P.; Ori, N.; Jiang, J.; Eshel, D. Etiolated Stem Branching Is a Result of Systemic Signaling Associated with Sucrose Level. Plant physiology 2017, 175, 734–745. [CrossRef]
- Xiao, Q.; Bai, X.; Zhang, C.; He, Y. Xiao, Q.; Bai, X.; Zhang, C.; He, Y. Advanced high-throughput plant phenotyping techniques for genome-wide association studies: A review. J Adv Res 2022, 35, 215–230. [CrossRef]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: the missing heritability is in the field. Genome biology 2011, 12, 232. [CrossRef]
- Khan, S.U.; Saeed, S.; Khan, M.H.U.; Fan, C.; Ahmar, S.; Arriagada, O.; Shahzad, R.; Branca, F.; Mora-Poblete, F. Khan, S.U.; Saeed, S.; Khan, M.H.U.; Fan, C.; Ahmar, S.; Arriagada, O.; Shahzad, R.; Branca, F.; Mora-Poblete, F. Advances and Challenges for QTL Analysis and GWAS in the Plant-Breeding of High-Yielding: A Focus on Rapeseed. Biomolecules 2021, 11. [CrossRef]
- Sun, F.; Liu, J.; Hua, W.; Sun, X.; Wang, X.; Wang, H. Sun, F.; Liu, J.; Hua, W.; Sun, X.; Wang, X.; Wang, H. Identification of stable QTLs for seed oil content by combined linkage and association mapping in Brassica napus. Plant Sci 2016, 252, 388–399. [CrossRef]
- Wang, T.; Wei, L.; Wang, J.; Xie, L.; Li, Y.Y.; Ran, S.; Ren, L.; Lu, K.; Li, J.; Timko, M.P.; et al. Wang, T.; Wei, L.; Wang, J.; Xie, L.; Li, Y.Y.; Ran, S.; Ren, L.; Lu, K.; Li, J.; Timko, M.P.; et al. Integrating GWAS, linkage mapping and gene expression analyses reveals the genetic control of growth period traits in rapeseed (Brassica napus L.). Biotechnol Biofuels 2020, 13, 134. [CrossRef]
- Zhao, C.; Xie, M.; Liang, L.; Yang, L.; Han, H.; Qin, X.; Zhao, J.; Hou, Y.; Dai, W.; Du, C.; et al. Zhao, C.; Xie, M.; Liang, L.; Yang, L.; Han, H.; Qin, X.; Zhao, J.; Hou, Y.; Dai, W.; Du, C.; et al. Genome-Wide Association Analysis Combined With Quantitative Trait Loci Mapping and Dynamic Transcriptome Unveil the Genetic Control of Seed Oil Content in Brassica napus L. Front Plant Sci 2022, 13, 929197. [CrossRef]
- Li, Y.; Shen, J.; Wang, T.; Chen, Q.; Zhang, X.; Fu, T.; Meng, J.; Tu, J.; Ma, C.J.C. Li, Y.; Shen, J.; Wang, T.; Chen, Q.; Zhang, X.; Fu, T.; Meng, J.; Tu, J.; Ma, C.J.C. QTL analysis of yield-related traits and their association with functional markers in Brassica napus L. Crop and Pasture Science 2007, 58, 759–766.
- Shi, J.; Li, R.; Qiu, D.; Jiang, C.; Long, Y.; Morgan, C.; Bancroft, I.; Zhao, J.; Meng, J. Shi, J.; Li, R.; Qiu, D.; Jiang, C.; Long, Y.; Morgan, C.; Bancroft, I.; Zhao, J.; Meng, J. Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 2009, 182, 851–861. [CrossRef]
- Ding, G.; Zhao, Z.; Liao, Y.; Hu, Y.; Shi, L.; Long, Y.; Xu, F. Ding, G.; Zhao, Z.; Liao, Y.; Hu, Y.; Shi, L.; Long, Y.; Xu, F. Quantitative trait loci for seed yield and yield-related traits, and their responses to reduced phosphorus supply in Brassica napus. Annals of botany 2012, 109, 747–759. [CrossRef]
- Chen, W.; Zhang, Y.; Liu, X.; Chen, B.; Tu, J.; Tingdong, F. Chen, W.; Zhang, Y.; Liu, X.; Chen, B.; Tu, J.; Tingdong, F. Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F(2) populations. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 2007, 115, 849–858. [CrossRef]
- Luo, X.; Ma, C.; Yue, Y.; Hu, K.; Li, Y.; Duan, Z.; Wu, M.; Tu, J.; Shen, J.; Yi, B.; et al. Luo, X.; Ma, C.; Yue, Y.; Hu, K.; Li, Y.; Duan, Z.; Wu, M.; Tu, J.; Shen, J.; Yi, B.; et al. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genomics 2015, 16, 379. [CrossRef]
- Li, F.; Chen, B.; Xu, K.; Gao, G.; Yan, G.; Qiao, J.; Li, J.; Li, H.; Li, L.; Xiao, X.; et al. Li, F.; Chen, B.; Xu, K.; Gao, G.; Yan, G.; Qiao, J.; Li, J.; Li, H.; Li, L.; Xiao, X.; et al. A genome-wide association study of plant height and primary branch number in rapeseed (Brassica napus). Plant Sci 2016, 242, 169–177. [CrossRef]
- He, Y.; Wu, D.; Wei, D.; Fu, Y.; Cui, Y.; Dong, H.; Tan, C.; Qian, W. He, Y.; Wu, D.; Wei, D.; Fu, Y.; Cui, Y.; Dong, H.; Tan, C.; Qian, W. GWAS, QTL mapping and gene expression analyses in Brassica napus reveal genetic control of branching morphogenesis. Sci Rep 2017, 7, 15971. [CrossRef]
- Zheng, M.; Peng, C.; Liu, H.; Tang, M.; Yang, H.; Li, X.; Liu, J.; Sun, X.; Wang, X.; Xu, J.; et al. Zheng, M.; Peng, C.; Liu, H.; Tang, M.; Yang, H.; Li, X.; Liu, J.; Sun, X.; Wang, X.; Xu, J.; et al. Genome-Wide Association Study Reveals Candidate Genes for Control of Plant Height, Branch Initiation Height and Branch Number in Rapeseed (Brassica napus L.). Front Plant Sci 2017, 8, 1246. [CrossRef]
- Li, B.; Gao, J.; Chen, J.; Wang, Z.; Shen, W.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; et al. Li, B.; Gao, J.; Chen, J.; Wang, Z.; Shen, W.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; et al. Identification and fine mapping of a major locus controlling branching in Brassica napus. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 2020, 133, 771–783. [CrossRef]
- Li, P.; Su, T.; Zhang, B.; Li, P.; Xin, X.; Yue, X.; Cao, Y.; Wang, W.; Zhao, X.; Yu, Y.; et al. Li, P.; Su, T.; Zhang, B.; Li, P.; Xin, X.; Yue, X.; Cao, Y.; Wang, W.; Zhao, X.; Yu, Y.; et al. Identification and fine mapping of qSB.A09, a major QTL that controls shoot branching in Brassica rapa ssp. chinensis Makino. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 2020, 133, 1055–1068. [CrossRef]
Figure 1.
Morphological features of the stem. (A) The phenotype of wild-type (WT) and multi-stems (MS) mutants after germination 60 days. Bar =5 cm. (B) The number of the main stems of WT and MS mutant. Values are the mean (±SD), n = 10. (C) The phenotype of WT and (D) MS mutant after germination 120 days. Bar = 10 cm. The yellow lines indicated the enlargement of the basal of stem in WT and MS mutant plants.
Figure 1.
Morphological features of the stem. (A) The phenotype of wild-type (WT) and multi-stems (MS) mutants after germination 60 days. Bar =5 cm. (B) The number of the main stems of WT and MS mutant. Values are the mean (±SD), n = 10. (C) The phenotype of WT and (D) MS mutant after germination 120 days. Bar = 10 cm. The yellow lines indicated the enlargement of the basal of stem in WT and MS mutant plants.
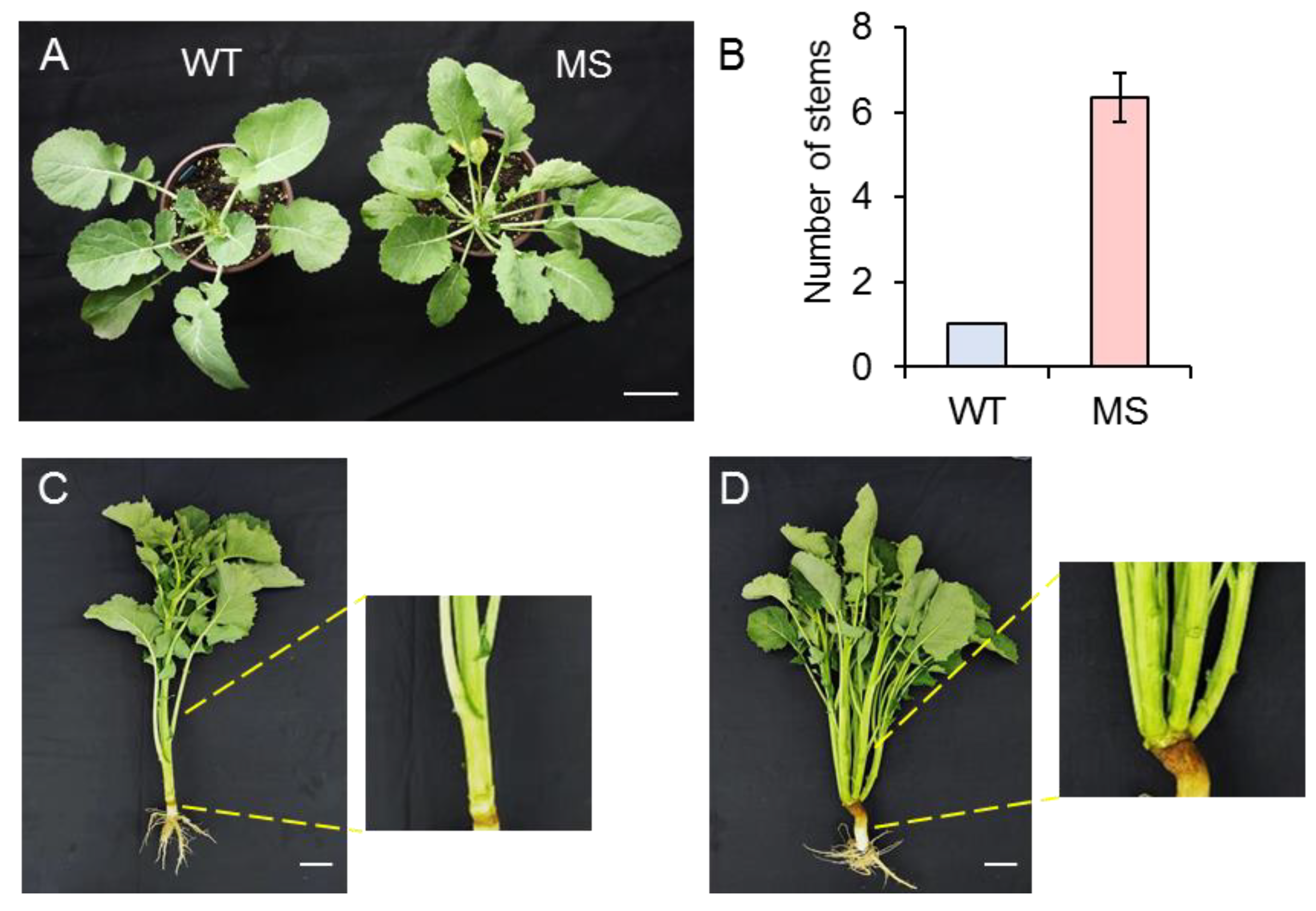
Figure 2.
The development of the shoot apical meristem in WT and MS mutant. (A) Phenotypes of the wild-type (WT) and multi-stem (MS) mutant after germination 20 days, bar = 2 cm. (B) Close-up views of the WT and (C) MS mutant red panels in (A), respectively. bar = 0.5 cm. (D) Paraffin section of the shoot apical meristem (SAM) of WT and (E) MS mutans after germination 20 days, and × 100 magnification. n = 10. (F) Scanned images of the SAM in WT (voltage 5kV, magnification 2000 and cross section 17 mm) and (G) MS mutant after germination 20 days (voltage 15kV, magnification 2000 and cross section 10.7 mm), bar = 20 µm. n = 10. (H) The cytokinins content in the SAM of WT and MS mutants after germination 20 days. tZ: trans-zeatin, cZ: cis-zeatin, iPR: isopentenyladenine riboside, czR: cis-zeatin riboside, tzR: trans-zeatin riboside, IP: isopentenyladenine. Values are the mean (±SD). Significant differences between WT and MS mutant were indicated. (Two-tailed student’s; *P < 0.05, NS, not significant, n=3).
Figure 2.
The development of the shoot apical meristem in WT and MS mutant. (A) Phenotypes of the wild-type (WT) and multi-stem (MS) mutant after germination 20 days, bar = 2 cm. (B) Close-up views of the WT and (C) MS mutant red panels in (A), respectively. bar = 0.5 cm. (D) Paraffin section of the shoot apical meristem (SAM) of WT and (E) MS mutans after germination 20 days, and × 100 magnification. n = 10. (F) Scanned images of the SAM in WT (voltage 5kV, magnification 2000 and cross section 17 mm) and (G) MS mutant after germination 20 days (voltage 15kV, magnification 2000 and cross section 10.7 mm), bar = 20 µm. n = 10. (H) The cytokinins content in the SAM of WT and MS mutants after germination 20 days. tZ: trans-zeatin, cZ: cis-zeatin, iPR: isopentenyladenine riboside, czR: cis-zeatin riboside, tzR: trans-zeatin riboside, IP: isopentenyladenine. Values are the mean (±SD). Significant differences between WT and MS mutant were indicated. (Two-tailed student’s; *P < 0.05, NS, not significant, n=3).
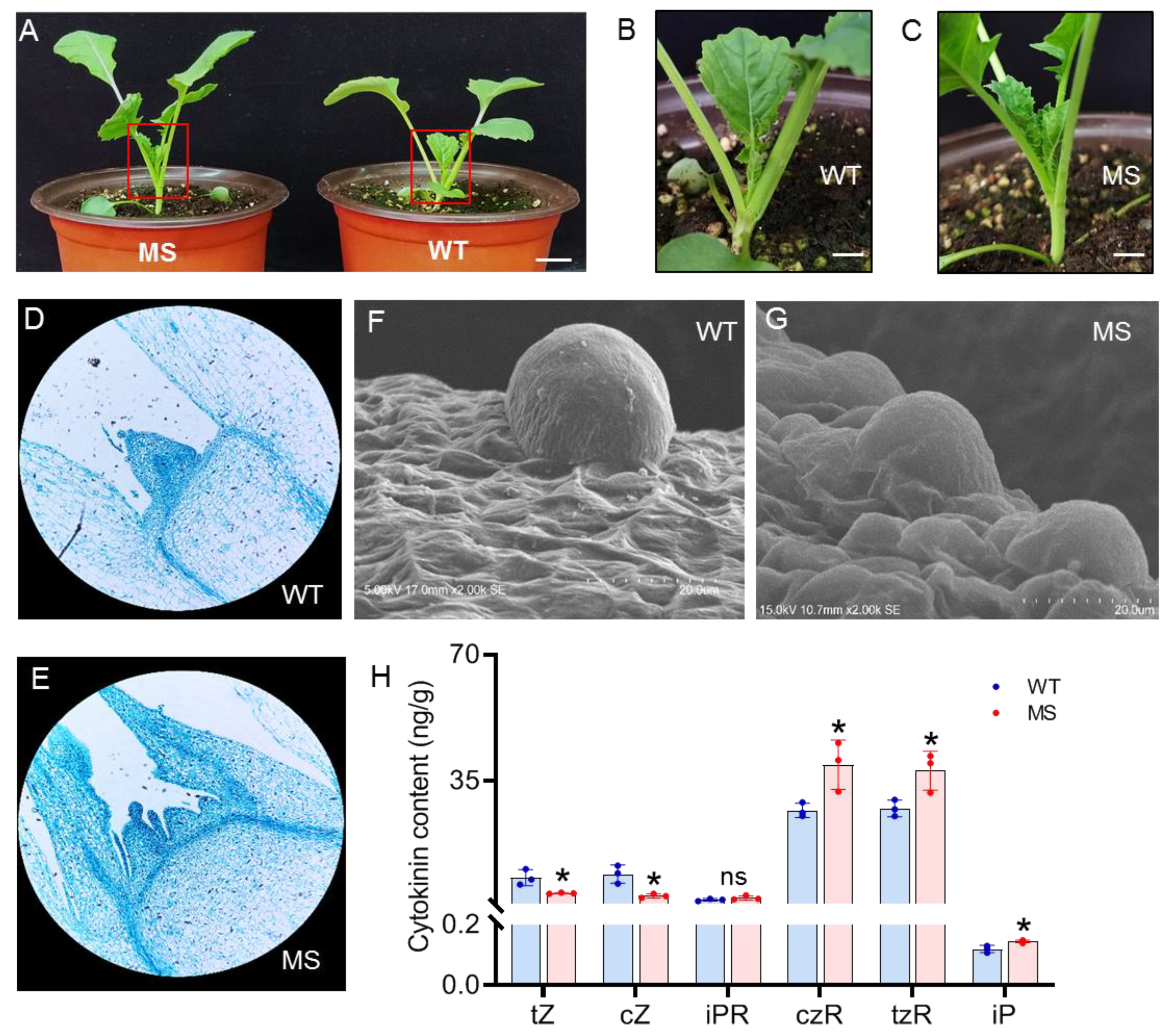
Figure 3.
Global transcript analysis of WT compared with MS mutant. The SAM of WT and MS mutant were collected at 15, 20, 25, 30, 35 days after germination (DAG). (A) Heatmap of Pearson correlation between samples according to gene expression values. (B) Principal component analysis (PCA) of the 10 groups of transcriptome data. (C) The number of different expression genes (DEGs) between MS mutant (M) and WT (S) at different DAG. (D) Venn diagrams indicating the numbers of common and specific DEGs identified between MS mutant (M) and WT (S) at different DAG. ST: Time after germination of single-stem plants (WT), MT: Time after germination of multi-stems (MS) mutants. Complete data can be found in Table S1.
Figure 3.
Global transcript analysis of WT compared with MS mutant. The SAM of WT and MS mutant were collected at 15, 20, 25, 30, 35 days after germination (DAG). (A) Heatmap of Pearson correlation between samples according to gene expression values. (B) Principal component analysis (PCA) of the 10 groups of transcriptome data. (C) The number of different expression genes (DEGs) between MS mutant (M) and WT (S) at different DAG. (D) Venn diagrams indicating the numbers of common and specific DEGs identified between MS mutant (M) and WT (S) at different DAG. ST: Time after germination of single-stem plants (WT), MT: Time after germination of multi-stems (MS) mutants. Complete data can be found in Table S1.
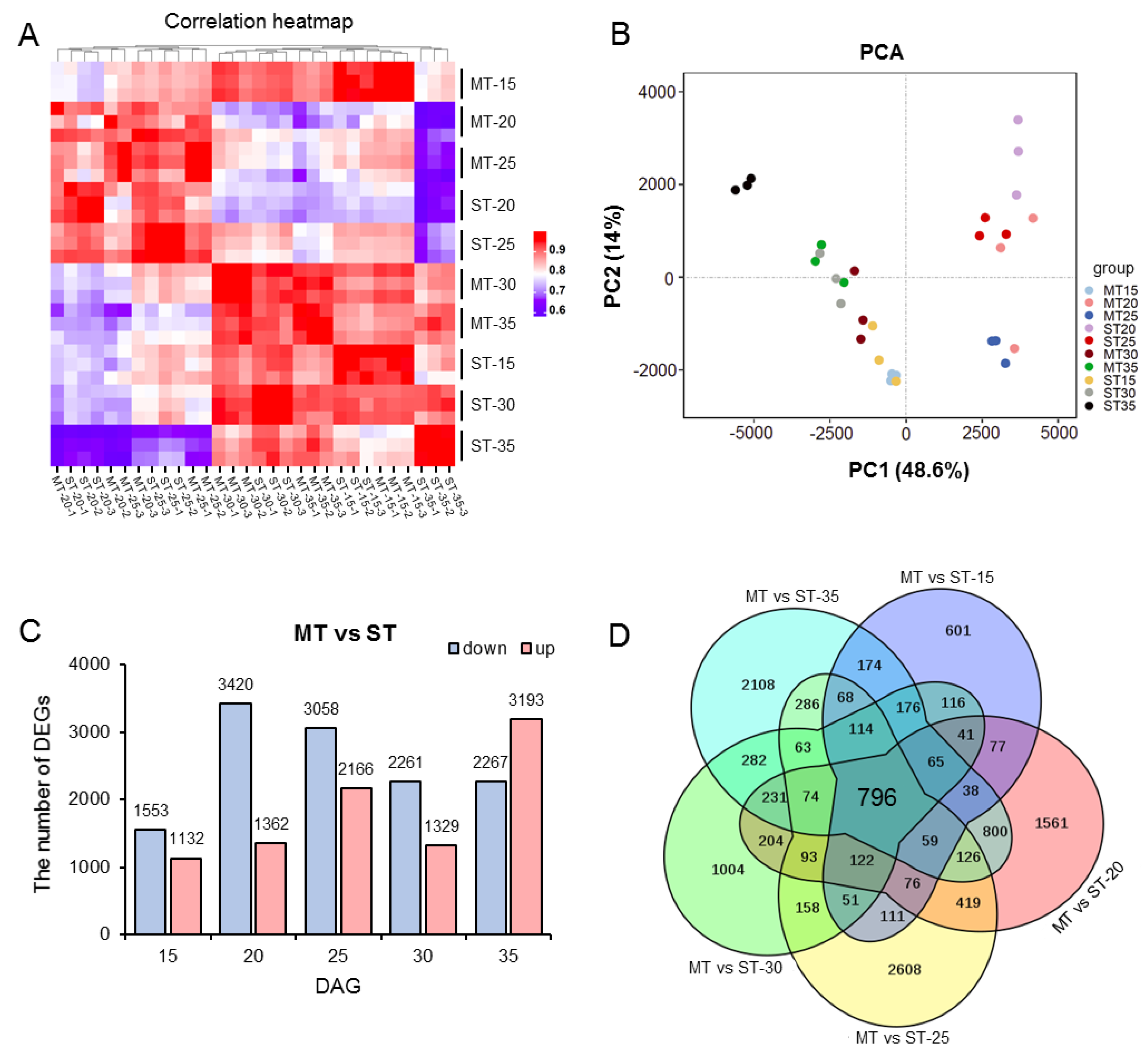
Figure 4.
KEGG enrichment analysis of DEGs between MS mutant and WT. Top 10 KEGG pathways of DEGs in MS mutant compared with WT at (A) 15, (B) 20, (C) 25, (D) 30, (E) 35 days after germination. Complete data can be found in Table S3.
Figure 4.
KEGG enrichment analysis of DEGs between MS mutant and WT. Top 10 KEGG pathways of DEGs in MS mutant compared with WT at (A) 15, (B) 20, (C) 25, (D) 30, (E) 35 days after germination. Complete data can be found in Table S3.
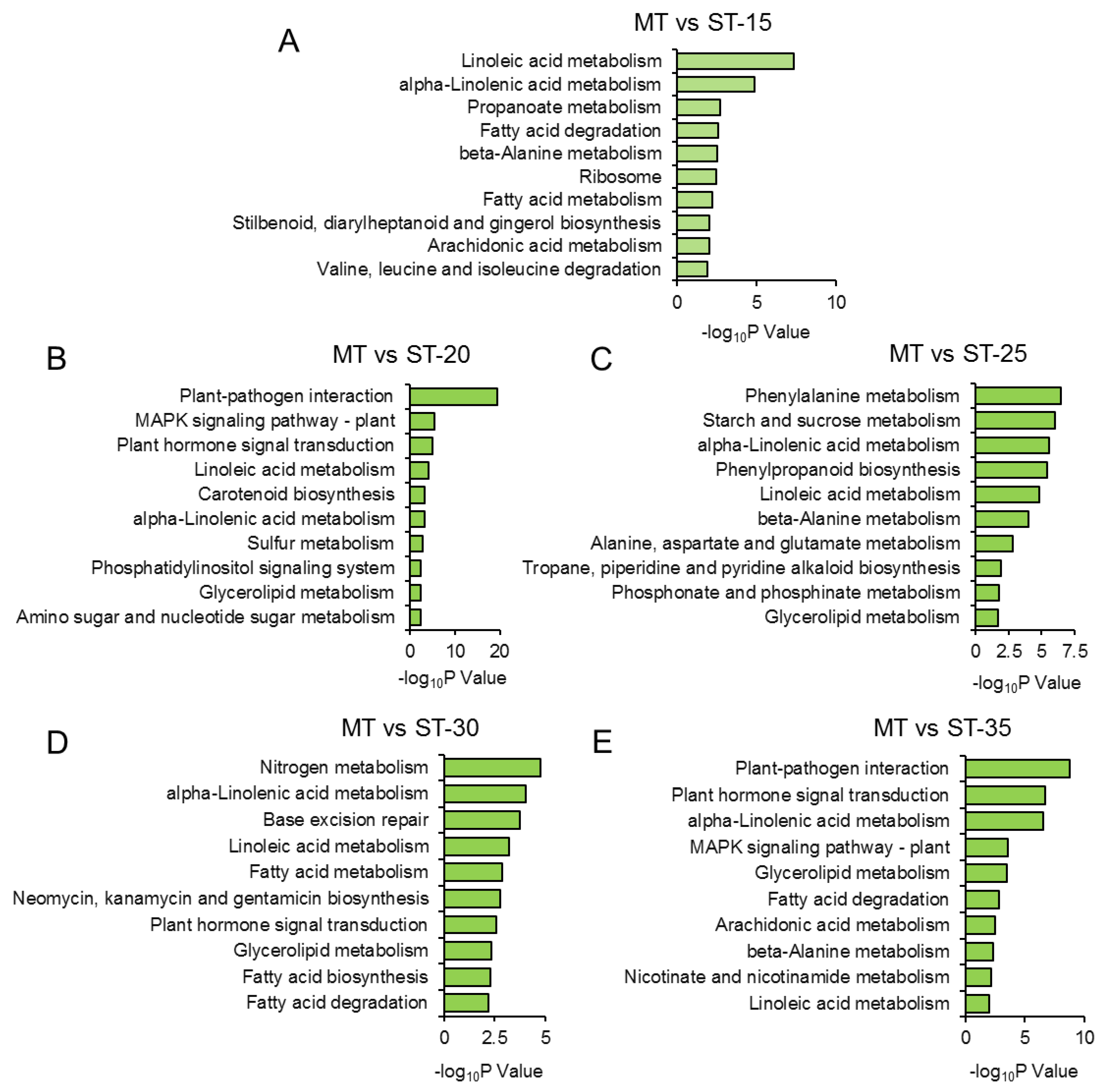
Figure 5.
The expression of genes involved in cytokinin signaling. (A) The gene networks involved in the cytokinin (CK) signaling pathway. LOG positively regulates the CK synthesis, CKX negatively regulates the synthesis of CK. Adapted from the study by [34]. (B) Heatmap of the LOG and CKX gene family in WT (S) and MS mutant (M) at 15, 20, 25, 30 and 35 days after germination (DAG). The colors correspond to the average of FPKM of three replicates, ranging from blue (low expression) to red (high expression). Complete data can be found in Table S4. (C) RT-qPCR and the RNA-Seq indicated that the relative expression (fold change) of LOG and CKX genes in mutant (M) relative to WT (S) at 20 days after germination. ST: Time after germination of single-stem plants (WT), MT: Time after germination of multi-stems (MS) mutants.
Figure 5.
The expression of genes involved in cytokinin signaling. (A) The gene networks involved in the cytokinin (CK) signaling pathway. LOG positively regulates the CK synthesis, CKX negatively regulates the synthesis of CK. Adapted from the study by [34]. (B) Heatmap of the LOG and CKX gene family in WT (S) and MS mutant (M) at 15, 20, 25, 30 and 35 days after germination (DAG). The colors correspond to the average of FPKM of three replicates, ranging from blue (low expression) to red (high expression). Complete data can be found in Table S4. (C) RT-qPCR and the RNA-Seq indicated that the relative expression (fold change) of LOG and CKX genes in mutant (M) relative to WT (S) at 20 days after germination. ST: Time after germination of single-stem plants (WT), MT: Time after germination of multi-stems (MS) mutants.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



