
Submitted:
19 April 2023
Posted:
19 April 2023
You are already at the latest version
Abstract
Antibiotic resistance among bacterial pathogens is recognized as a major threat to human health worldwide. The emergence of multidrug-resistant bacteria can also be found in the community settings, apart from hospital environment, which indicates that reservoirs of antibiotic resistance genes do exist outside the hospital. The growth of antibiotic resistance is a consequence of bacterial adaptations in response to selective pressures. To survive in this hostile environment, bacteria develop defence mechanisms such as chemical modification of antibiotics, enzyme-catalysed antibiotic degradation, altered permeability, antibiotic efflux, mutation of target sites and biofilm formation, resulting in resistance to nearly all currently available antibiotics used in the clinical practice. The present review summarizes insights into the molecular mechanisms underlying the antibiotic resistance which is useful for planning strategies to combat antibiotic resistance and devise innovative therapeutic tools to fight against multidrug-resistant bacterial species.
Keywords:
antibiotics
; antibiotic resistance
; multidrug resistance genes
; molecular mechanisms
1. Introduction
The discovery of antibiotics, their commercialization and administration to treat infections has improved therapy and revolutionized modern medicine. Indeed, antibiotics administration have become one of the key medical procedures required for routine clinical interventions such as organ transplantation, surgery and cancer care. Unfortunately, this therapeutic accomplishment is now threatened by significantly increased antibiotic resistance among common bacterial pathogens, making it hard to treat critically ill patients [1]. In fact, resistance towards antibiotic has been described as one of the highest threats to public health in the 21st century [2].
Antibiotic resistance occurs when bacteria have the ability to survive or grow in the presence of an antibiotic concentration that is usually adequate to kill or inhibit their growth [3]. The lateral transfer of antibiotic resistance genes or spontaneous mutations may lead to the resistance trait. The transfer of genes that is resistant towards antibiotic can occur via various mechanisms, such as conjugation, transformation, transduction, nanotubes, gene transfer agents (GTA) and membrane vesicles [4]. In general, conjugation is a transfer of genetic material from a donor to a recipient and it requires a cell-to-cell contact. The production of nosocomial resistance usually involves conjugation strategy, which is a highly successful rate of gene transfer method involving mobile genetic element (MGEs) such as conjugative plasmids, transposon or integrons. MGEs play a vital role in the disseminations of antibiotic resistance genes between clinically relevant species [5]. Transformation is a direct uptake and expression of the extracellular DNA from the environment into a naturally competent recipient cell. Transformation can be the most basic kind of horizontal gene transfer (HGT), but only a few bacterial species may incorporate naked DNA "naturally" for the development of resistance [6]. As for transduction, the mechanism is phage-mediated [7].
In clinical practice, the terms 'resistant' and 'susceptible' are usually used to determine the possible failure or success of treatment. When the concentration required to kill or inhibit microorganisms failed to be achieved in a patient, resistance is more likely to occur [3]. In the 2019 AR Threats Report, the Centers for Disease Control and Prevention (CDC) states that there are over 2.8 million antibiotic-resistant infections occur in the US resulting in over 35,000 deaths each year. It was also highlighted that there are about 223,900 cases of Clostridioides difficile infections reported in 2017 which killed at least 12,800 people [8]. This condition is worsened by an inadequate supply of effective antibiotics, which leads to almost untreatable infections and leaves clinicians with no safe choices for treating infected patients.
It is critically important to understand the molecular basis of bacterial resistance mechanism in order to combat antibiotic resistance. Other than become intrinsically resistant to antibiotics, microorganisms also have the ability to develop resistance (acquired resistance) following exposure to the antibiotic [9]. Intrinsic resistance can be defined as the innate ability of a specific bacteria, to resist the actions of antibiotic due to their inherent functional or structural characteristics [10]. This can be achieved via extrusion of antibiotic molecules through efflux pumps and/or reduced permeability of the bacterial outer membrane. In contrast, acquired resistance occurs due to the acquisition of external resistance genetic determinants or mutations in chromosomal genes [11]. Acquired resistance caused the bacteria to resist a particular antibiotic’s activity to which it was previously susceptible.
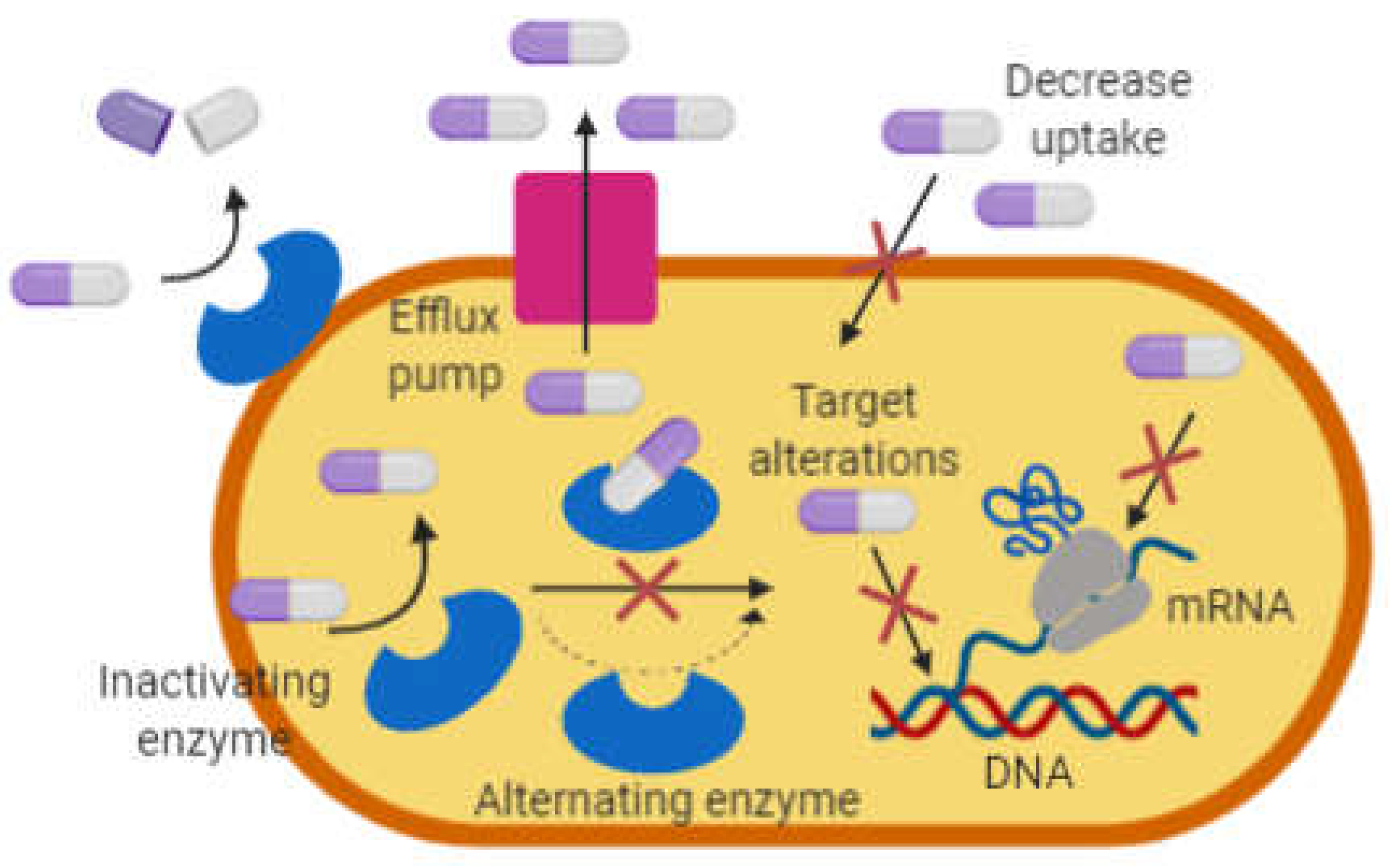
In general, bacteria undergo mutations making it resistant to antibiotics via one of these mechanisms; i) modification and degradation of antibiotics molecules, ii) decrease permeability and efflux pumps, and iii) changes in target sites (Figure 1).
2. Modification and Degradation of Antibiotics
Chemical modification and degradation of antibiotics are effective mechanisms used by Gram-negative and Gram-positive bacteria to survive in the presence of antibiotics [9]. The chemical modification of the antibiotic is widely seen to occur in the group of antibiotic that inhibits synthesis of protein at the ribosome level [13].
2.1. Chemical modification
Aminoglycosides are one of the broad-spectrum antibiotics that is used to treat serious infections such as tuberculosis that are caused by Mycobacterium tuberculosis [14]. Other than that, aminoglycosides are also effective against members of Enterobacteria family including Escherichia coli and Klebsiella pneumoniae [15]. Aminoglycoside binds effectively to the A-site located in 16S rRNA of 30S ribosomal subunit resulting in the inhibition of protein synthesis [16]. From the interaction, aminoglycoside encourages mistranslation by codon misreading thus, results in a protein synthesis error that leads to cell damage [17]. By disrupting protein synthesis, it has the ability to kill bacteria [18].
Chemical modification of antibiotics is facilitated by various modifying enzymes also known as transferases. These enzymes are known as aminoglycoside-modifying enzyme (AMEs) and these enzymes’ production are capable of introducing chemical changes to the molecular structure of antibiotics [19]. AMEs covalently modify the amino (-NH2) or hydroxyl (-OH) group of aminoglycoside molecule and genes encoding AMEs are typically carried by MGEs [19]. These modifying enzymes have different biochemical reactions with various catalytic activity such as phosphorylation (O-phosphotransferases), adenylation (O-adenyltransferases) and acetylation (N-acetyltransferases) [17]. Regardless of various biochemical reactions, the addition of functional groups to the aminoglycosides by AMEs leads to conformational changes that decrease the binding affinity of antibiotics to their target sites and consequently lead to a loss in antibacterial potency [20].
Antibiotic inactivation via phosphorylation is a biochemical process that transfers phosphate group to the hydroxyl group of antibiotic by the action of aminoglycoside phosphotransferases (APHs) enzyme and is widely seen in aminoglycoside and chloramphenicol [21]. An example of this is the phosphorylation of kanamycin and neomycin by APH(3’) causing the inability of the antibiotics to bind to their ribosomal target site [19]. Adenylation is the process of transferring the AMP molecule to the hydroxyl group of antibiotic, catalyzes by aminoglycoside nucleotidyltransferases (ANTs) and lincosamide nucleotidyltransferases (LNU) [22]. For example, in E. coli and Staphylococcus aureus, clindamycin (a class of lincosamides) can be inactivated by lincosamide nucleotidyltransferase encoded by linB genes [23]. As for aminoglycosides, ANT(2″) is the example of an enzyme that affects the activity of 4,6-di-substituted aminoglycosides that was first described in K. pneumoniae [16]. On another note, acetylation is a process of transferring functional group of acetyl to the amino group of antibiotic catalysed by aminoglycoside acetyltransferases (AACs) enzyme that usually occurred to amikacin, netilmicin, and tobramycin by AAC(6′)-1 enzyme [24,25].
Another example of resistance through chemical modification of antibiotic can be seen in chloramphenicol. By interacting with the peptidyl-transferase center of the 50S ribosomal subunit, chloramphenicol inhibits bacterial protein synthesis [9]. The production of acetyltranferases enzyme known as chloramphenicol acetyltransferases (CATs) facilitates the modification of chloramphenicol. The transfer of the acetyl group from acetyl-coenzyme A (AcCoA) to the 3-hydroxyl group of chloramphenicol had caused bacterial resistance towards the antibiotic [26]. In both Gram-negatives and Gram-positives bacteria, multiple cat genes have been found and grouped into two main types; Type A that results in high-level resistance and Type B that results in low-level resistance [27]. These two types of CATs also differs in terms of their structure component where type A CAT monomers range from 207 to 238 amino acids while type B CAT monomers is smaller with 209 to 219 amino acids [28].
2.2. Degradation of antibiotic
The enzyme-catalysed antibiotic degradation is another major antibiotic resistance mechanism that plays a major part in the discovery of β-lactamase since antibiotics were first used in 1940 [29]. Since then, there have been thousands of enzymes that have the ability to degrade antibiotics of different classes including macrolides and β-lactams [30]. β-lactamases are degradation enzymes used to hydrolyse the β-lactam antibiotics such as cephalosporins, monobactams, carbapenems, and penicillins [31]. β-lactam antibiotics are bactericidal and act by interfering with the remodelling and synthesis of the peptidoglycan layer of bacteria [32]. β-lactam inactivates the penicillin-binding proteins (PBPs) by forming covalent bonds to it. PBPs are transpeptidase that cross-linked the amino acids consisting of β-(1-4)-N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) together in order to form the cell wall of bacteria [33]. Bacteria able to develop resistance to β-lactam antibiotics through mutations of the gene that encodes PBPs.
β-lactamases can be acquired via HGT. This can be seen in the penicillin-resistant S. aureus that carries plasmid-encoded β-lactamases which can be readily transmitted between the S. aureus strains [34]. β-lactamase genes are generally called bla, followed by the specific enzyme name, for example; blaKPC is the gene that encodes for K. pneumoniae carbapenemase (KPCs) that confer resistant towards carbapenems [35]. It can be found within a chromosome or as part of the accessory genes located in MGEs [32]. TEM-1 (class A) is another example of β-lactamase capable of hydrolyzing ampicillin and typically found in Gram-negative bacteria such as E. coli and Klebsiella spp. [37]. It is termed as TEM-1 after the name Temoneira, the patient in which it was originally found [36].
2.3. Decrease permeability
Most antibiotics have specific intracellular bacterial targets. The antibiotics must go through the cytoplasmic membrane in order to be effective, however, some bacteria have the ability to avert the antibiotic from reaching its target sites by lowering the uptake of the antibiotic molecules [9]. The Gram-negative bacteria’s outer membrane plays an important role in providing the organism with an additional protective layer without compromising the material exchange needed to sustain life [38]. Therefore, the Gram-negative bacteria are less permeable to many antibiotics intrinsically as compared to Gram-positive bacteria as their outer membrane can act as a barrier [39].
The outer membrane of Gram-negative bacteria consists of proteins called porins. Various porin types have been described and it can be categorized by their selectivity, structure (monomeric vs. trimeric), and their regulation of expression. The Pseudomonas aeruginosa’s OprD also referred to as protein D2 and the three major proteins in E. coli called OmpC, OmpF and PhoE are among the best-characterized porins [40]. These two bacteria are also examples of Gram-negative bacteria that employ porin-mediated antibiotic resistance.
Antibiotics with hydrophilic properties such as tetracyclines, some fluoroquinolones and β-lactam are mostly affected by the permeability changes of outer membranes since they used porins to go through the barrier [41]. Permeability changes occurred by three general processes which are the (i) change in the porin expression level, (ii) shift in the type of porins expressed and (iii) the impairment of porin functions [42].
One of the general processes of porin-mediated antibiotic resistance is the changes in the expression level of the porin. Data shows that reduction in expression of porin contributes significantly to the development of resistance in Enterobacteriaceae family, Acinetobacter spp. and Pseudomonas spp. to newer antibiotics such as cephalosporins and carbapenems to which enzyme degradation usually mediates the resistance [43]. For instance, it has been observed in Enterobacteriaceae family that in the absence of carbapenemase production, resistance towards carbapenem can still be achieved due to the decrease of porin production (as a result of gene mutation) [43,44].
Another process involved is the shift in the type of porin expressed. When a particular porin is replaced with a different type that forms more selective channels, reduced permeability of the external membrane can be achieved consequently limiting the entry of antibiotic into the cell of bacteria [39,45]. Example of the shift in the porin expression can be seen in K. pneumoniae strains which is a multiple drug-resistant bacteria. K. pneumoniae strains become less susceptible towards β-lactam such as cephalosporins and carbapenems due to the shift of porin expression from OmpK35 to OmpK36 which possessed a smaller size of channel [46]. OmpK36 porin caused a four to eight fold decrease of susceptibility for a wide range of β-lactam antibiotics [47].
3. Antibiotic Efflux
Another common resistant mechanism is antibiotic efflux. This mechanism often occurs in conjunction with other mechanisms such as modification of a target binding site or the antibiotic. Efflux pump consists of protein within the channel that works as pumps to extrude the antibiotics out of the cell. It acts on various classes of antibioticss, including β-lactams, aminoglycoside fluoroquinolones, polymyxins and carbapenems [48]. The genes that encode efflux pumps can be found in bacterial chromosomes or MGEs.
ABC (ATP Binding Cassette), MATE (Multidrug and Toxin Extrusion), MFS (Major Facilitator Superfamily), SMR (Small Multidrug Resistance) and RND (Resistance-Nodulation-Division) are the five major families of efflux pump [49]. The number of components (multiple or single), sequence, substrate specificity, the number of transmembrane spanning region and energy sources have been determined for each of the families [50]. The ABC family utilize hydrolysis from ATP in the export of substrates while other families used proton motive force as the energy source. The ABC, MATE, MFS, and SMR families, are distributed widely among the Gram-positive and -negative bacteria, while the RND superfamily is specifically found in Gram-negative bacteria.
The tetracycline-specific efflux pump of MFS is one example of efflux-mediated antibiotic resistance that mediates tetracycline resistance [51]. This pump is used to extrude the tetracycline molecules from within the cells at the expense of a proton [52]. Another efflux-mediated antibiotic resistance that can be found is in the resistance of macrolides where erythromycin can be extruded by the efflux pumps that is encoded by the mef genes (mef E and mefA) [53,54].
4. Changes in Target Sites
Inhibiting antibiotic action by interfering with its target site is a common strategy of resistance in bacteria. Few different tactics has been evolved by bacteria to decrease the potency and affinity of antibiotics towards its target sites and this includes modification of the target sites and target protection (prevents the antibiotic from reaching the site of binding).
4.1. Target protection
Proteins that are physically associated with the antibiotic's target site can defend the bacteria against the antibiotic's inhibitory effects [55]. The genes involved in this mechanism of resistance encodes target or ribosomal protection proteins (RPPs) and are usually carried by the MGEs. Generally, this mechanism works by dislodging and preventing the rebinding of the antibiotics [55]. Tetracycline, fusidic acid and fluoroquinolones are the examples of antibiotics that are affected by this resistance mechanism [56].
Examples of best characterised RPPs are Tet(M) found in Streptococcus spp. and Tet[O] in Campylobacter jejuni, which confer resistance to tetracycline [57]. Tet(M) bind to the ribosome specifically at domain IV of the 16S rRNA which is the binding site for the tetracycline. Through this interaction, Tet(M) dislodges and releases tetracycline from the ribosome. Consequently, the rebinding of the antibiotics is prevented due to conformational changes caused by the interaction of Tet[M] within the drug-binding site [58].
Other than Tet(M), quinolone resistance protein; Qnr is another example of target protection protein that reduces susceptibility towards quinolones [59]. Qnr protects DNA gyrase and topoisomerase IV from quinolones inhibition. This plasmid-mediated fluoroquinolone resistance is frequently found in K. pneumoniae making it clinically significant [60]. The target sites for quinolones are the topoisomerase IV and DNA gyrase; bacterial enzymes that involve in DNA replication [61]. Therefore, by acting as DNA homologue, Qnr competes for the binding site, preventing the binding of quinolones to topoisomerase IV and DNA gyrase [62].
4.2. Modification of target site
Modification of the target sites is the resistance mechanisms affecting almost all families of antibiotics. It consists of mutations of genes encoding target site proteins, enzymatic alterations and also complete replacement or bypass of target sites. However, no matter what type of modification that takes place, the final impact of the mechanism is always the same, which is the decreased in antibiotic's affinity for the target site [63]. This mechanism acts as a mechanism of resistance against few antibiotic classes such as aminoglycosides, glycopeptides, β-lactams, macrolides, lincosamide and streptogramins (MLS) [64]. The examples of those three strategies will be presented below.
4.3. Mutations of genes encoding the target site proteins
There are two examples of mutational resistance which are the rifampin (RIF) resistance and the fluoroquinolones (FQ) resistance. Rifamycin inhibits the DNA-dependent RNA polymerase and blocks the transcription in bacteria. RIF is an effective antibiotic treatment for tuberculosis [65]. RIF acts by inhibiting bacterial RNA polymerase activity and considered as one of the powerful broad spectrums antibiotics against bacterial pathogens. In M. tuberculosis, RIF prevents the elongation of mRNA by binding to the β-subunit of RNA polymerase. The binding of RIF molecule at this highly conserved pocket will directly block the path of the nascent RNA, subsequently interrupts the DNA transcription [65,66]. The β subunit within the RNA polymerase is encoded by rpoB gene and mutations within this gene has been shown to confer RIF-resistant phenotype. It has been observed among the RIF-resistant isolates that mutations occurred within 81 bp RIF-resistance determining region (RRDR) of rpoB gene [67]. The mutation caused an alteration within the binding pocket (β subunit) that decrease the binding affinity of RIF to the RNA polymerase.
FQ resistance mechanism is another example of mutational resistance. Fluoroquinolones inhibit the topoisomerase IV and DNA gyrase’s supercoiling activity within cells, resulting in cell death (at lethal concentrations) and impaired DNA replication (at lower concentrations) [68]. However, alterations of those two crucial enzymes (DNA gyrase and topoisomerase IV) caused resistance towards fluoroquinolones action [61]. This can be achieved by chromosomal mutations in the genes encoding the DNA gyrase subunits (gyrA and gyrB gene) or enzyme topoisomerase IV (parC and parE gene) [69]. In these genes, the region where mutation arises is known as the quinolone resistance-determining region (QRDR) [70]. The point mutation within QRDR sequence will results in the substitutions of amino acid that alters the structure of the target protein followed by the decrease of fluoroquinolone-binding affinity, leading to resistance towards the antibiotic [71].
4.4. Enzymatic alteration of target sites
Macrolide resistance through ribosome methylation catalysed by an enzyme encoded by the erm (erythromycin ribosomal methylation) is one of the best examples for this type of resistance [72]. Adenine residue at the A2058 position of domain V of 23S rRNA within 50S ribosomal subunit can be monomethylated or dimethylated by rRNA methyltransferase. These biochemical changes impair the binding of the macrolide to its target [73].
Other than macrolide resistance, another relevant example is the Cfr-mediated linezolid resistance [74]. This transferable multidrug resistance genes: cfr that encodes for cfr-methyltransferase, confers resistance towards oxazolidinones, lincosamides, phenicols, streptogramin A and plueromutilins [75]. Adenine residue at the A2503 position of the 23S rRNA can be modified by the cfr-methyltransferase that results in impaired binding of linezolid with their target sites [76]. Subsequently this will inhibit the protein synthesis by preventing the binding of amino-acyl tRNA to the A site of ribosome [77]. However, a newer generation of oxazolidinone namely as tedizolid was generated to overcome the resistance problem by the substitution of acetamide group with a smaller hydroxymethyl group. This substitution allows the antibiotic to bind to its target site in the presence of cfr-methyltransferase [78].
4.5. Complete replacement or bypass of target site
Bacteria are capable of preventing the inhibitions of the antibiotic molecule by evolving new targets that fulfilled the biochemical function of the original target. By producing an additional low-affinity target, bypass of the original target is accomplished. Vancomycin resistant-enterococci (VRE) and methicillin resistant-S. aureus (MRSA) have been observed to employ the replacement and bypass strategy to achieve antibiotic resistance [9].
Other than producing blaZ encoded-β-lactamase (that hydrolyses the β-lactam ring, rendering it ineffective), MRSA also carries mecA gene that encodes PBP2a (Penicillin-Binding Protein 2a) [79]. Due to its low affinity for β-lactam, PBP2a provides transpeptidase activity that enables the synthesis of the cell wall by the bacteria to be continued at a concentration that inhibit the β-lactam-sensitive PBPs normally produced by S. aureus [80]. PBP2a consist of an active-site serine (S403) at N-terminus of α2 helix in the sequence motif SXXK. Through the binding of β-lactam antibiotic with the active-site serine (S403), a rapidly reversible Michaelis complex (EI) was formed and was converted to stable covalent adduct by nucleophilic attack by S403 on the β-lactam ring [81]. Even though the binding occurs, the binding of β-lactam to the active site of PBP2a does not inhibit its transpeptidase activity, thus cross-linking of the peptidoglycan chains to form rigid cell walls is not inhibited.
Another example of replacement and bypass strategy is observed in VRE. Enterococci, particularly Enterococcus faecium is closely related to the resistance of vancomycin [82]. The vancomycin resistance is conferred by van gene clusters involving biochemical machinery that remodels the synthesis of peptidoglycan. There are two biochemical types of machinery designated in the remodelling of peptidoglycan synthesis; (i) by preventing the binding of vancomycin to the cell wall precursors through destroying the D-Ala-D-Ala ending precursors and (ii) by changing the last D-Ala for either D-serine or D-lactate [83].
5. Biofilm Formation
Biofilms are complex systems that are composed of many different types of cells, including bacteria, fungi, and algae. The extracellular matrix that surrounds these cells is also heterogeneous, with varying compositions of proteins, lipids, polysaccharides, and other molecules [84,85,86]. Biofilms are common in natural and human-made environments and are well known to be one of the major contributors to antibiotic resistance [87,88,89].
Biofilm formation can contribute to antibiotic resistance through several mechanisms: i) physical barrier: biofilms provide a physical barrier between the antibiotics and the bacteria, which makes it difficult for the agent to penetrate and reach the bacterial cells [90]; ii) slow growth rate: biofilms have a slower growth rate and metabolism than planktonic bacteria [91], which reduces their susceptibility to antibiotics, as these antibiotics typically target rapidly growing bacterial cells; iii) quorum sensing: bacteria in biofilms can communicate with each other through quorum sensing, allowing them to coordinate their behaviour and respond to environmental cues [92]. This can lead to the activation of genes that confer resistance to antibiotics; iv) phenotypic resistance: bacteria in biofilms can exhibit phenotypic resistance, where their gene expression and metabolic activity change in response to their environment [93]. This can make them less susceptible to antibiotics. Overall, biofilm formation can protect bacteria from antibiotics, making them more resistant and difficult to eliminate, leading to persistent infections. Continuous screening for antibiofilm activity of natural products may shed new light on the control of biofilm formation and antibiotic resistance [94,95,96].
6. Conclusions
The emergence of resistance bacteria has increased due to misuse and overuse of antibiotics, patients not finishing the entire antibiotic course, and non-laboratory oriented antibiotic therapy. A thorough understanding of the molecular mechanisms of antibiotic resistance is vital to provide the key to devise new strategies to deal with the threat. Designing better drugs that will not be affected by the bacteria’s defense mechanisms or preventing the spread and dissemination of antibiotic resistance genes need to be further explored.
Acknowledgments
The author acknowledges with much appreciation to the Faculty of Applied Sciences, Universiti Teknologi MARA, Shah Alam, Selangor, Malaysia.
References
- Nathan, C. (2004). Antibiotics at the crossroads. Nature, 431(7011), 899-902.
- Conly, J., & Johnston, B. (2005). Where are all the new antibiotics? The new antibiotic paradox. Canadian Journal of Infectious Diseases and Medical Microbiology, 16(3), 159-160.
- Sabtu, N., Enoch, D. A., & Brown, N. M. (2015). Antibiotic resistance: what, why, where, when and how? British Medical Bulletin, ldv041.
- Livermore, D. (2004). Can better prescribing turn the tide of resistance? Nature Reviews Microbiology, 2(1), 73-78.
- Manson, J. M., Hancock, L. E., & Gilmore, M. S. (2010). Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proceedings of the National Academy of Sciences, 107(27), 12269-12274.
- Thomas, C. M., & Nielsen, K. M. (2005). Mechanisms of, and Barriers to, Horizontal Gene Transfer between Bacteria. Nature Reviews Microbiology, 3(9), 711-721.
- Colavecchio, A., Cadieux, B., Lo, A., & Goodridge, L. D. (2017). Bacteriophages Contribute to the Spread of Antibiotic Resistance Genes among Foodborne Pathogens of the Enterobacteriaceae Family – A Review. Frontiers in Microbiology, 8.
- Biggest Threats and Data. (2020, June 18). Retrieved July 03, 2020, from https://www.cdc.gov/drugresistance/biggest-threats.htm.
- Munita, J. M., & Arias, C. A. (2016). Mechanisms of Antibiotic Resistance. Virulence Mechanisms of Bacterial Pathogens, 481-511.
- Cox, G., & Wright, G. D. (2013). Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. International Journal of Medical Microbiology, 303(6-7), 287-292.
- Reygaert, W. C. (2018). An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiology, 4(3), 482-501.
- Gullberg, E. Selection of Resistance at very low Antibiotic Concentrations. PhD thesis. Uppsala University. 2014. ISBN 978-91-554-9101-7.
- Wilson, D. N. (2013). Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nature Reviews Microbiology, 12(1), 35-48.
- Labby, K. J., & Garneau-Tsodikova, S. (2013). Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Medicinal Chemistry, 5(11), 1285-1309.
- Landman, D., Babu, E., Shah, N., Kelly, P., Backer, M., Bratu, S., & Quale, J. (2010). Activity of a novel aminoglycoside, ACHN-490, against clinical isolates of Escherichia coli and Klebsiella pneumoniae from New York City. Journal of Antimicrobial Chemotherapy, 65(10), 2123-2127.
- Kotra, L. P., Haddad, J., & Mobashery, S. (2000). Aminoglycosides: Perspectives on Mechanisms of Action and Resistance and Strategies to Counter Resistance. Antimicrobial Agents and Chemotherapy, 44(12), 3249-3256.
- Ramirez, M. S., & Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resistance Updates, 13(6), 151-171.
- Germovsek, E., Barker, C. I., & Sharland, M. (2016). What do I need to know about aminoglycoside antibiotics? Archives of Disease in Childhood - Education & Practice Edition, 102(2), 89-93.
- Krause, K. M., Serio, A. W., Kane, T. R., & Connolly, L. E. (2016). Aminoglycosides: An Overview. Cold Spring Harbor Perspectives in Medicine, 6(6).
- Llano-Sotelo, B., Azucena, E. F., Kotra, L. P., Mobashery, S., & Chow, C. S. (2002). Aminoglycosides Modified by Resistance Enzymes Display Diminished Binding to the Bacterial Ribosomal Aminoacyl-tRNA Site. Chemistry & Biology, 9(4), 455-463.
- Kim, C., & Mobashery, S. (2005). Phosphoryl transfer by aminoglycoside 3′-phosphotransferases and manifestation of antibiotic resistance. Bioorganic Chemistry, 33(3), 149-158.
- Wright, G. D. (1999). Aminoglycoside-modifying enzymes. Current Opinion in Microbiology, 2(5), 499-503.
- Morar, M., Bhullar, K., Hughes, D. W., Junop, M., & Wright, G. D. (2009). Structure and Mechanism of the Lincosamide Antibiotic Adenylyltransferase LinB. Structure, 17(12), 1649-1659.
- Sagar, S., Kaistha, S., Das, A. J., & Kumar, R. (2019). Antibiotic Resistant Bacteria: A Challenge to Modern Medicine.
- Shaw, K. J., Rather, P. N., Hare, R. S., & Miller, G. H. (1993). Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiological Reviews, 57(1), 138-163.
- Biswas, T., Houghton, J. L., Garneau-Tsodikova, S., & Tsodikov, O. V. (2012). The structural basis for substrate versatility of chloramphenicol acetyltransferase CATI. Protein Science, 21(4), 520-530.
- Schwarz, S., Kehrenberg, C., Doublet, B., & Cloeckaert, A. (2004). Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiology Reviews, 28(5), 519-542.
- Roberts, M. C., & Schwarz, S. (2017). Tetracycline and Chloramphenicol Resistance Mechanisms. Antimicrobial Drug Resistance, 231-243. (.
- Abraham, E. P., & Chain, E. (1940). An Enzyme from Bacteria able to Destroy Penicillin. Nature, 146(3713), 837-837.
- Egorov, A. M., Ulyashova, M. M., & Rubtsova, M. Y. (2018). Bacterial Enzymes and Antibiotic Resistance. Acta Naturae, 10(4), 33-48.
- Nordmann, P., Poirel, L., Walsh, T. R., & Livermore, D. M. (2011). The emerging NDM carbapenemases. Trends in Microbiology, 19(12), 588-595.
- Tang, S. S., Apisarnthanarak, A., & Hsu, L. Y. (2014). Mechanisms of β-lactam antimicrobial resistance and epidemiology of major community- and healthcare-associated multidrug-resistant bacteria. Advanced Drug Delivery Reviews, 78, 3-13.
- Bourhis, L. L., & Werts, C. (2007). Role of Nods in bacterial infection. Microbes and Infection, 9(5), 629-636.
- Bush, K. (2013). Proliferation and significance of clinically relevant β-lactamases. Annals of the New York Academy of Sciences, 1277(1), 84-90.
- Meenakshisundaram, J. (2013). Bla KPC gene Detection in Clinical Isolates of Carbapenem Resistant Enterobacteriaceae in a Tertiary Care Hospital. Journal Of Clinical And Diagnostic Research.
- Rawat, D., & Nair, D. (2010). Extended-spectrum ß-lactamases in gram negative bacteria. Journal of Global Infectious Diseases, 2(3), 263.
- Paterson, D. L., & Bonomo, R. A. (2005). Extended-Spectrum β-Lactamases: A Clinical Update. Clinical Microbiology Reviews, 18(4), 657-686.
- Delcour, A. H. (2009). Outer membrane permeability and antibiotic resistance. Biochimica Et Biophysica Acta (BBA) - Proteins and Proteomics, 1794(5), 808-816.
- Vargiu, A. V., & Nikaido, H. (2012). Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proceedings of the National Academy of Sciences, 109(50), 20637-20642.
- Welte, W., Nestel, U., Wacker, T., & Diederichs, K. (1995). Structure and function of the porin channel. Kidney International, 48(4), 930-940.
- Pagès, J., James, C. E., & Winterhalter, M. (2008). The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nature Reviews Microbiology, 6(12), 893-903.
- Nikaido, H. (2003). Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiology and Molecular Biology Reviews, 67(4), 593-656.
- Lavigne, J., Sotto, A., Nicolas-Chanoine, M., Bouziges, N., Pagès, J., & Davin-Regli, A. (2013). An adaptive response of Enterobacter aerogenes to imipenem: Regulation of porin balance in clinical isolates. International Journal of Antimicrobial Agents, 41(2), 130-136.
- Baroud, M., Dandache, I., Araj, G., Wakim, R., Kanj, S., Kanafani, Z., . . . Matar, G. (2013). Underlying mechanisms of carbapenem resistance in extended-spectrum β-lactamase-producing Klebsiella pneumoniae and Escherichia coli isolates at a tertiary care centre in Lebanon: Role of OXA-48 and NDM-1 carbapenemases. International Journal of Antimicrobial Agents, 41(1), 75-79.
- Kojima, S., & Nikaido, H. (2013). Permeation rates of penicillins indicate that Escherichia coli porins function principally as nonspecific channels. Proceedings of the National Academy of Sciences, 110(28).
- Tsai, Y., Fung, C., Lin, J., Chen, J., Chang, F., Chen, T., & Siu, L. K. (2011). Klebsiella pneumoniaeOuter Membrane Porins OmpK35 and OmpK36 Play Roles in both Antimicrobial Resistance and Virulence. Antimicrobial Agents and Chemotherapy, 55(4), 1485-1493.
- Doménech-Sánchez, A., Martínez-Martínez, L., Hernández-Allés, S., Conejo, M. D., Pascual, A., Tomás, J. M., . . . Benedí, V. J. (2003). Role of Klebsiella pneumoniae OmpK35 Porin in Antimicrobial Resistance. Antimicrobial Agents and Chemotherapy, 47(10), 3332-3335.
- Poole, K. (2005). Efflux-mediated antimicrobial resistance. Journal of Antimicrobial Chemotherapy, 56(1), 20-51.
- Sun, J., Deng, Z., & Yan, A. (2014). Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochemical and Biophysical Research Communications, 453(2), 254-267.
- Nikaido, H. (2011). Structure and Mechanism of RND-Type Multidrug Efflux Pumps. Advances in Enzymology - and Related Areas of Molecular Biology Advances in Enzymology and Related Areas of Molecular Biology, 1-60.
- Chopra, I., & Roberts, M. (2001). Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiology and Molecular Biology Reviews, 65(2), 232-260.
- Thaker, M., Spanogiannopoulos, P., & Wright, G. D. (2009). The tetracycline resistome. Cellular and Molecular Life Sciences, 67(3), 419-431.
- Moore, S. D., & Sauer, R. T. (2008). Revisiting the mechanism of macrolide-antibiotic resistance mediated by ribosomal protein L22. Proceedings of the National Academy of Sciences, 105(47), 18261-18266.
- Li, X., & Nikaido, H. (2009). Efflux-Mediated Drug Resistance in Bacteria. Drugs, 69(12), 1555-1623.
- Tomlinson, J. H., Thompson, G. S., Kalverda, A. P., Zhuravleva, A., & O’Neill, A. J. (2016). A target-protection mechanism of antibiotic resistance at atomic resolution: Insights into FusB-type fusidic acid resistance. Scientific Reports, 6(1).
- Tran, J. H., Jacoby, G. A., & Hooper, D. C. (2005). Interaction of the Plasmid-Encoded Quinolone Resistance Protein Qnr with Escherichia coli DNA Gyrase. Antimicrobial Agents and Chemotherapy, 49(1), 118-125.
- Connell, S. R., Tracz, D. M., Nierhaus, K. H., & Taylor, D. E. (2003). Ribosomal Protection Proteins and Their Mechanism ofTetracyclineResistance. Antimicrobial Agents and Chemotherapy, 47(12), 3675-3681.
- Doenhoefer, A., Franckenberg, S., Wickles, S., Berninghausen, O., Beckmann, R., & Wilson, D. (2012). Structural basis for TetM-mediated tetracycline resistance.
- Hooper, D. C., & Jacoby, G. A. (2015). Mechanisms of drug resistance: Quinolone resistance. Annals of the New York Academy of Sciences, 1354(1), 12-31.
- Martínez-Martínez, L., Pascual, A., & Jacoby, G. A. (1998). Quinolone resistance from a transferable plasmid. The Lancet, 351(9105), 797-799.
- Jacoby, G. A. (2005). Mechanisms of Resistance to Quinolones. Clinical Infectious Diseases, 41(Supplement_2).
- Jacoby, G. A. (2017). Plasmid-Mediated Quinolone Resistance. Antimicrobial Drug Resistance, 265-268.
- Lambert, P. (2005). Bacterial resistance to antibiotics: Modified target sites. Advanced Drug Delivery Reviews, 57(10), 1471-1485.
- Peterson, E., & Kaur, P. (2018). Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Frontiers in Microbiology, 9.
- Campbell, E. A., Korzheva, N., Mustaev, A., Murakami, K., Nair, S., Goldfarb, A., & Darst, S. A. (2001). Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell, 104(6), 901-912.
- Palomino, J., & Martin, A. (2014). Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics, 3(3), 317-340.
- Zaw, M. T., Emran, N. A., & Lin, Z. (2018). Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis. Journal of Infection and Public Health, 11(5), 605-610.
- Drlica, K., Hiasa, H., Kerns, R., Malik, M., Mustaev, A., & Zhao, X. (2009). Quinolones: Action and Resistance Updated. Current Topics in Medicinal Chemistry, 9(11), 981-998.
- Piddock, L. J. (1995). Mechanisms of Resistance to Fluoroquinolones. Drugs, 49(Supplement 2), 29-35.
- Yoshida, H., Bogaki, M., Nakamura, M., Yamanaka, L. M., & Nakamura, S. (1991). Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrobial Agents and Chemotherapy, 35(8), 1647-1650.
- Hooper, D. C. (2000). Mechanisms of Action and Resistance of Older and Newer Fluoroquinolones. Clinical Infectious Diseases, 31(Supplement_2).
- Golkar, T., Zieliński, M., & Berghuis, A. M. (2018). Look and Outlook on Enzyme-Mediated Macrolide Resistance. Frontiers in Microbiology, 9.
- Fyfe, C., Grossman, T. H., Kerstein, K., & Sutcliffe, J. (2016). Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harbor Perspectives in Medicine, 6(10).
- Schaenzer, A. J., & Wright, G. D. (2020). Antibiotic Resistance by Enzymatic Modification of Antibiotic Targets. Trends in Molecular Medicine.
- Morales, G., Picazo, J., Baos, E., Candel, F., Arribi, A., Peláez, B., . . . Sánchez-García, M. (2010). Resistance to Linezolid Is Mediated by thecfrGene in the First Report of an Outbreak of Linezolid-ResistantStaphylococcus aureus. Clinical Infectious Diseases, 50(6), 821-825.
- Locke, J. B., Zurenko, G. E., Shaw, K. J., & Bartizal, K. (2014). Tedizolid for the Management of Human Infections: In Vitro Characteristics. Clinical Infectious Diseases, 58(Suppl_1).
- Song, Y., Lv, Y., Cui, L., Li, Y., Ke, Q., & Zhao, Y. (2017). Cfr -mediated linezolid-resistant clinical isolates of methicillin-resistant coagulase-negative staphylococci from China. Journal of Global Antimicrobial Resistance, 8, 1-5.
- Shaw, K. J., Poppe, S., Schaadt, R., Brown-Driver, V., Finn, J., Pillar, C. M., . . . Zurenko, G. (2008). In Vitro Activity of TR-700, the Antibacterial Moiety of the Prodrug TR-701, against Linezolid-Resistant Strains. Antimicrobial Agents and Chemotherapy, 52(12), 4442-4447.
- Blázquez, B., Llarrull, L. I., Luque-Ortega, J. R., Alfonso, C., Boggess, B., & Mobashery, S. (2014). Regulation of the Expression of the β-Lactam Antibiotic-Resistance Determinants in Methicillin-Resistant Staphylococcus aureus (MRSA). Biochemistry, 53(10), 1548-1550.
- Fishovitz, J., Hermoso, J. A., Chang, M., & Mobashery, S. (2014). Penicillin-binding protein 2a of methicillin-resistan tStaphylococcus aureus. IUBMB Life, 66(8), 572-577.
- Fuda, C., Suvorov, M., Vakulenko, S. B., & Mobashery, S. (2004). The Basis for Resistance to β-Lactam Antibiotics by Penicillin-binding Protein 2a of Methicillin-resistantStaphylococcus aureus. Journal of Biological Chemistry, 279(39), 40802-40806.
- Arias, C. A., & Murray, B. E. (2012). The rise of the Enterococcus: Beyond vancomycin resistance. Nature Reviews Microbiology, 10(4), 266-278.
- Miller, W. R., Munita, J. M., & Arias, C. A. (2014). Mechanisms of antibiotic resistance in enterococci. Expert Review of Anti-infective Therapy, 12(10), 1221-1236.
- Yahya, M. F. Z. R., Alias, Z., & Karsani, S. A. (2018). Antibiofilm activity and mode of action of DMSO alone and its combination with afatinib against gram-negative pathogens. Folia Microbiologica, 63(1), 23-30.
- Yaacob, M. F., Murata, A., Nor, N. H. M., Jesse, F. F. A., & Yahya, M. F. Z. R. (2021). Biochemical composition, morphology and antimicrobial susceptibility pattern of Corynebacterium pseudotuberculosis biofilm. Journal of King Saud University-Science, 33(1), 101225.
- Kamaruzzaman, A. N. A., Mulok, T. E. T. Z., Nor, N. H. M., & Yahya, M. F. Z. R. (2022). FTIR spectral changes in Candida albicans biofilm following exposure to antifungals. Malaysian Applied Biology, 51(4), 57-66.
- Fan, Q., Zuo, J., Wang, H., Grenier, D., Yi, L., & Wang, Y. (2022). Contribution of quorum sensing to virulence and antibiotic resistance in zoonotic bacteria. Biotechnology Advances, 107965. 107965.
- Michaelis, C., & Grohmann, E. (2023). Horizontal gene transfer of antibiotic resistance genes in biofilms. Antibiotics, 12(2), 328.
- Hu, X., Zhang, Y., Chen, Z., Gao, Y., Teppen, B., Boyd, S. A., ... & Li, H. (2023). Tetracycline accumulation in biofilms enhances the selection pressure on Escherichia coli for expression of antibiotic resistance. Science of The Total Environment, 857, 159441.
- Dincer, S., Uslu, F. M., & Delik, A. (2020). Antibiotic resistance in biofilm. In Bacterial biofilms. IntechOpen.
- Miao, L., Yu, Y., Adyel, T. M., Wang, C., Liu, Z., Liu, S., ... & Hou, J. (2021). Distinct microbial metabolic activities of biofilms colonizing microplastics in three freshwater ecosystems. Journal of Hazardous Materials, 403, 123577.
- Narla, A. V., Borenstein, D. B., & Wingreen, N. S. (2021). A biophysical limit for quorum sensing in biofilms. Proceedings of the National Academy of Sciences, 118(21), e2022818118.
- Yinsai, O., Deeudom, M., & Duangsonk, K. (2023). Genotypic Diversity, Antibiotic Resistance, and Virulence Phenotypes of Stenotrophomonas maltophilia Clinical Isolates from a Thai University Hospital Setting. Antibiotics, 12(2), 410.
- Johari, N.A., Amran, S.S.D., Kamaruzzaman, A.N.A., Man, C.A.I.C. & Yahya, M.F.Z.R. 2020. Anti-biofilm potential and mode of action of Malaysian plant species: a review. Science Letters, 14, 34–46.
- Zawawi, W.M.A.W.M., Ibrahim, M.S.A., Rahmad, N., Hamid, U.M.A. & Yahya, M.F.Z.R. 2020. Proteomic analysis of Pseudomonas aeruginosa treated with Chromolaena odorata extracts. Malaysian Journal of Microbiology, 16(2), 124-133.
- Man, C. A. I. C., Razak, W. R. W. A., & Yahya, M. F. Z. R. (2022). Antibacterial and antibiofilm activities of Swietenia macrophylla King ethanolic extract against foodborne pathogens. Malaysian Applied Biology, 51(4), 45-56.
Figure 1.
Antibiotic resistance strategies in bacteria. Redrawn and adapted from [12].
Figure 1.
Antibiotic resistance strategies in bacteria. Redrawn and adapted from [12].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



