
Submitted:
23 April 2023
Posted:
24 April 2023
You are already at the latest version
Abstract
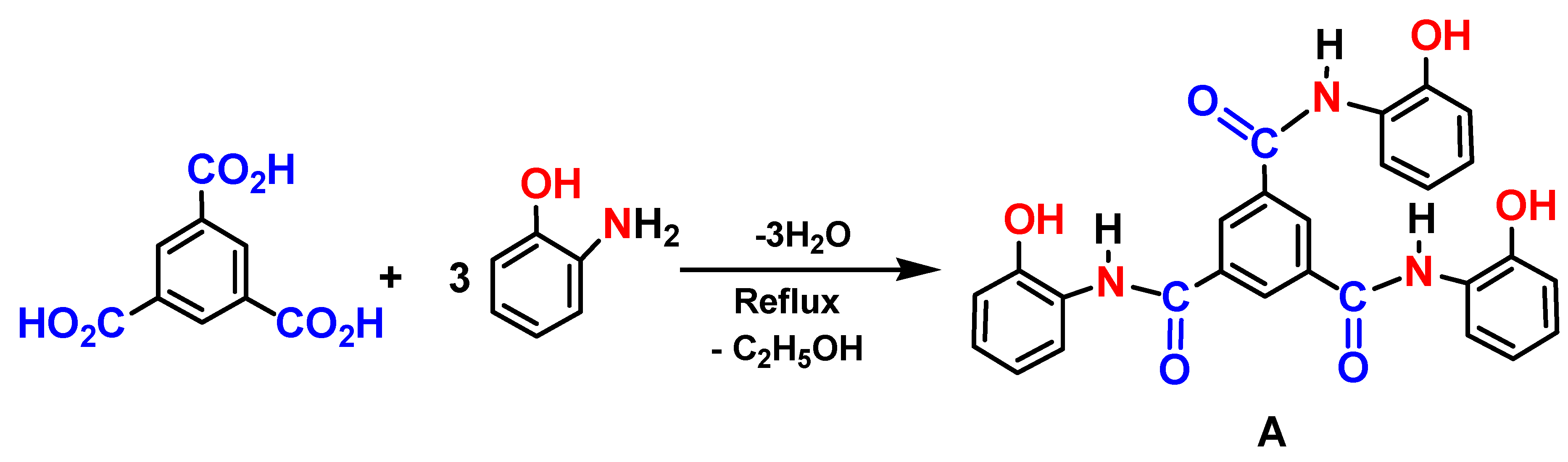
The substrate N1, N3, N5 -tris(2-hydroxyphenyl)benzene-1,3,5-tricarboxamide (Sensor A) was prepared in the reaction of 1,3,5-benzenetricarboxylic acid (trimesic acid) and o-aminophenol in ethanol. The prepared organic sensor fulfills the chemiluminescent requirements including luminophore, spacer, and suitable binding receptor that distress the probe's luminescent features, providing selective and sensitive detection of mercury and iron ions in aqueous solutions. The sensor selectively detects mercury and iron ions in a water matrix containing various metal ions including sodium, calcium, magnesium, zinc, and nickel. Strong and immediate binding was observed between mercury ions and the substrate at pH 7.0 with a binding affinity toward Hg (II) enhanced by nine folds higher than that observed for iron sensor binding affinity, which makes the substrate a distinctive luminescence sensor for mercury detection at ambient conditions. The sensor shows a linear response toward Hg (II) in the concentration range from 4.2 x 10-5 to 2.0 x 10-8 M with a limit of detection of 1.0 x 10-8 M. Further, Sensor A provides linear detection for iron ions in the range from 1.5 x 10-3 to 1.5 x 10-8 M. The measured adsorption capacity of Sensor A toward mercury ions ranged from 1.25 to 1.97 mg/g and the removal efficiency from water samples reached 98.8% at pH 7.0. The data demonstrate that Sensor A is an excellent probe for detecting and removing mercury ions from water bodies.
Keywords:
luminescent sensors
; mercury ions
; iron ions
; binding affinity
; selective detection
; adsorption capacity
1. Introduction
Pollutants deposited in the ecological systems especially heavy metals are a major concern due to their toxicity. Heavy metals are of great interest due to the role they play in environmental and biological systems. Compared to other organic pollutants, heavy metals are very stable substrates at ambient conditions so they accumulate in soil and water bodies and thus are transported to humans and mammals through the food chain [1,2,3,4]. Most of the reported heavy metals receive great attention from environmental sectors and health agencies due to their harmful effect on humans and living environments even in very small concentrations. In specific, studies showed that the presence of lead, cadmium, and mercury contaminants may damage many organs and tissues including the brain, bone, liver, and calcium metabolism disorders, leading to certain types of cancers [5,6,7,8,9,10]. Despite the harm they cause to the environment and human health, they are widely used in our daily life since they play a key role in many industries. For instance, mercury is deposited into the environment through various industrial actions including mining, coal combustion, metal smelter exhaust, and paper mills [11,12,13]. Combustion and industrial sources are significant for mercury emission into the environment where most of the anthropogenic mercury emissions is associated with the burning of fossil fuel, coal and oil. In addition, being part of major industrial products of daily use including batteries, paper, latex paints, and electronics, mercury deposition to the environment is significant especially when no effective and direct recycling processes are applied. Mercury accumulated in the environment in three forms with Hg(II) is the most widely spread in the environment.
In contrast to mercury, iron is one of the major elements that is widely used in various industrial applications [14,15]. As an essential element, iron is a very important element to humans and other living organisms. Despite its importance in human health, the iron content should be optimized where cases with deficient or saturated levels of iron are reported to cause function disorders, weak immunity, and lack of sleep [16,17,18]. As a result, high levels of iron contamination in drinking water poses health concerns, thus the World Health Organization recommended that the iron (III) levels in drinking water to be very low [19].
Given the high stability of heavy metal along with the direct and indirect effect they pose to human health, there is a demand to selectively and sensitively detect their presence in both environmental and biological samples. Several instruments and methods were modified to detect and quantify mercury and other toxic metal ions in water bodies. The common techniques in use include atomic absorption spectrometry (AAS) [20,21], inductively coupled plasma techniques [22,23,24], and electrochemistry [25]. In some cases, these methods were modified and used along with various separation chromatographic methods including ion chromatography, gas chromatography, and high-performance liquid chromatography [26,27]. While these methods are very sensitive with a wide range of linearity, still the high cost and long operation, and complicated sampling processes of such techniques increase the need of finding simpler and faster methods that provide similar results in a shorter time. Recently, chemiluminescent sensors were modified to investigate organic and inorganic analytes in biological and environmental systems [28,29,30]. This provides a simple method to detect target molecules/ions with low detection limits over a short time. Importantly, luminescence-based sensors can be further designed to attain a wide range of signaling “antenna” when the analyte covalently binds to the luminescent probe providing opportunities to monitor the concentrations of the target within a short time. This antenna can be observed through changes in the signal’s intensities, wavelength, lifetime, and chirality [28,29,30,31,32].
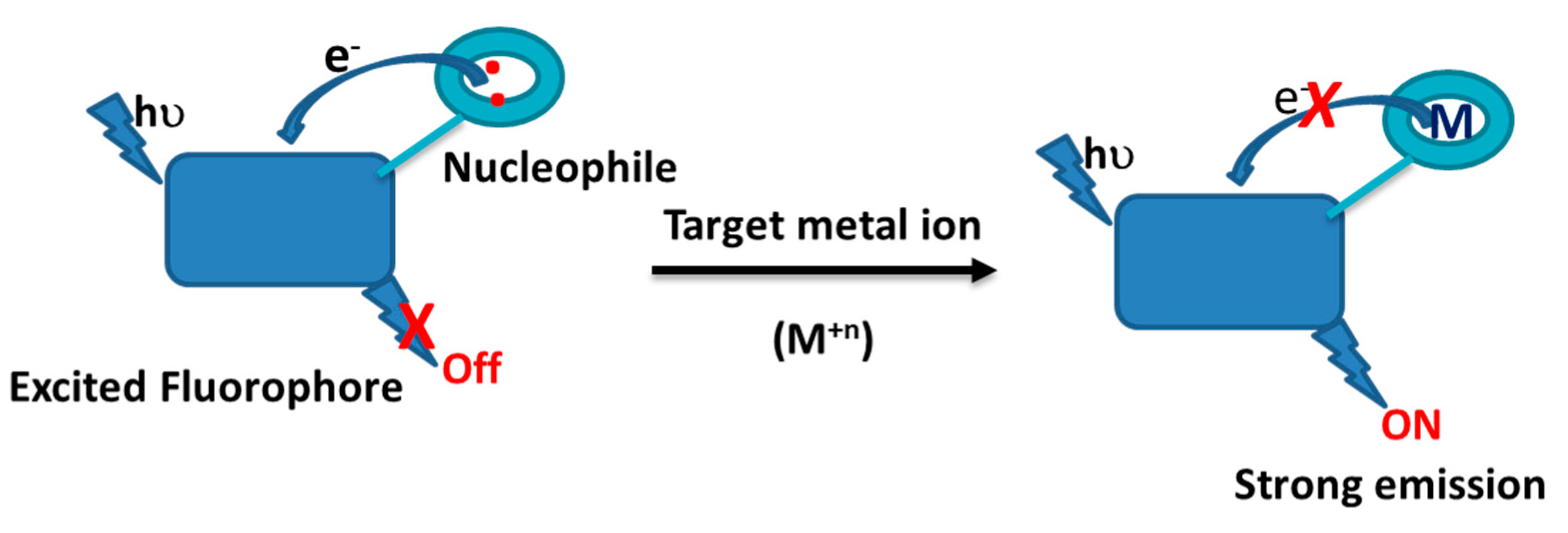
The photophysical properties of a fluorophore can be designed by establishing proton-, energy-, or electron-transfer processes that are sensitive toward the target molecules including heavy metal ions. One of the most common strategies used to build luminescent devices is to facilitate the photoinduced electron transfer (PET) process that turns the emission on or off upon target analyte binding/interaction (see Scheme 1). Three major features must be present for designing a luminescent sensor: the luminophore, spacer, and receptor. When the target analyte binds to the receptor, an immediate change to the emission occur where emission intensity may be enhanced or quenched making ‘off-on’ or ‘on-off’ sensors as illustrated in Scheme 1. Variations to the emission spectra may occurs through various mechanisms including PET, Ligand-to-metal charge transfer (LMCT), or metal-to-ligand charge transfer (MLCT) [30,31,32,33,34]. Several studies reported that the LMCT involves electronic transitions from the localized orbitals in the conjugated linker to a metal-centered orbital and is highly sensitive to the ionic size and the coordination geometry of the linker [30,31,32,33,34,35,36,37].
While several conjugated organic molecules were reported for metal ion detection at low concentration levels [30,31,32,33,34,35,36,37], the available fluorescent sensors that selectively detect mercury at ambient environmental conditions are limited. Much attention has been focused on porphyrin derivatives with strong fluorescent properties that are quenched upon the addition of mercury ions as on-off type sensors [38,39,40]. Additionally, most of the reported luminescent chemo sensors lack selectivity and water solubility which mimic their use in real environmental samples. In the present investigation we synthesized N1, N3, N5-tris(2-hydroxyphenyl)benzene-1,3,5-tricarboxamide (Sensor A) that is tailored to three different suitable sites to capture metal ions forming five-membered ring associated between the linker and the target metal ion of use. Both the orientation of the donor sites and the associated space are designed to covalently bind to Hg (II). For the potential use of Sensor A for real water samples, we tested several water samples of environmental, tap, and deionized water. Since our goal is to find a selective and sensitive sensor for the detection of specific metal ions in water bodies, the photophysical properties of Sensor A toward various concentrations of metal ions including Hg(II), Mg2+, Na+, Zn2+, Ni2+, Pb2+, Fe3+, and Ca2+ ions were investigated at various aqueous solutions adjusted at three different pH values (pH 5, 7, and 10).
2. Materials and Methods
All chemicals and reagents including 1,3,5-benzenetricarboxylic acid (trimesic acid), o-aminophenol, iron (III) nitrate, mercury (II) nitrate, zinc nitrate, magnesium nitrate, sodium nitrate, nickel (II) nitrate, ethanol, and phosphate buffers were purchased from Sigma-Aldrich and used without further purifications. Also, 1000 ppm standard solutions were purchased from Sigma-Aldrich and used for quality control in this study. All solvents used are of analytical reagent grade and the water was doubly distilled. The metal ion solutions were prepared from their nitrate salts with specific metal concentrations calculated to represent the quencher concentrations.
UV-visible measurements were recorded on a single-beam UV-visible spectrophotometer (carry 50 conc) equipped with a xenon arc lamp. Fluorescence spectra were recorded from a carry eclipse Varian spectrophotometer equipped with 150 W continuous xenon lamp along with a sensitive grating and PMT detector. The machine has a feature to survey the excitation and emission profile of the fluorophore. Excitation and emission curves were recorded upon exposure to the selected emission and excitation wavelengths, respectively. Infrared spectra were recorded using the ABB Bomem MB3000 series combined with the intuitive Horizon MBTM FTIR software. NMR spectra were recorded on Bruker 400 MHz.
2.1. Synthesis of Sensor A.
Compound A was synthesized by a solvothermal process where trimesic acid (0.693 g, 3.3 mmol) and o-aminophenol (1.06 g, 9.9 mmol) were dissolved in ethanol (20 mL each). The trimesic acid solution was connected to reflux at 80°C. When all the solids dissolved, the o-aminophenol solution was gradually added and continued under reflux for 6 h with continuous stirring where no starting materials remains as depicted from thin layer chromatography. The red precipitate formed after slow cooling to room temperature and was filtered and crystallized from ethanol. The collected precipitate was dried in an oven at 80°C for 2 h and then air-dried for 1 h before further use in this study. The material was characterized using UV-visible, Luminescence, FTIR, and NMR spectroscopic techniques. The reaction yield obtained was 93% with a high melting point of m.p. > 300℃. FTIR (KBr): ν (cm−1) 3373, 1691, 1604, 1495, 1460, 1381, 1280, 1110, 928 cm-1. Proton NMR (1H NMR, 400 MHz, d6-DMSO) showed δ (ppm) 8.90 (s, 3H), 8.0 (s, 3H), 7.47 (d, 3H), 6.7-6.84 (d, 9H). 5.0 (s, 3H).
2.2. Binding Studies
We rely on the SSLS to monitor the changes associated to the fluorimetric and colorimetric features for detecting different metal ionswater samples. 30-ppm solutions of Sensor A were prepared in methanol: water, 10:90 v:v buffered solutions at pH 5, 7, and 10 using phosphate buffers. Several concentrations of the tested analytes including Zn (II), Hg (II), Mg2+, Na+, Ca2+, Ni (II), and Fe (III) ions were prepared from their nitrate salts with concentrations ranging from 0.5 to 1000 ppm. The initial luminescence intensity was recorded for a solution containing 2.75 mL of Sensor A diluted with water to a 3.0 mL total volume. The fluorescence intensity was monitored to reach stable emission thus maximum binding and quenching affinity was reached. Using 100 ppm solution of the metal ion of interest, it was noticed that immediate responses occurred with iron and mercury ions with no change to the emission profile occurred when other metal ions are used even after being monitored for 30 min. Even though the binding affinity occurred immediately, we followed the binding affinity for all tested various metal ion concentrations after 2-min intervals for a complete binding to be established. To ensure that the observed inhibition to the luminescence intensity is directed to the binding affinity between the sensor and the target metal ions, equal volumes of water (0.25 mL) were added to the organic sensor for an initial emission intensity that will be used as F0.
2.3. Equilibration adsorption measurements.
To evaluate the adsorption of mercury ions on Sensor A, a 2 mL solution containing 6.0 mg of Sensor A was monitored upon adding various volumes of 50 ppm mercury ions solution at pH 7 for 10 min. The initial and the final equilibrium concentrations were determined from the emission intensity at 515 nm. The equilibrium adsorption capacity of mercury ion on Sensor A was then determined using equation 1.
where Qeqm is the equilibrium adsorption capacity, Co is the initial concentration of the sensor and Ceqm is the equilibrium concentration of the adsorbent with V and W represent the solution's volume and the mass of adsorbent, respectively. Finally, the the removal efficiency from the initial concentrations, η %, is determined using equation 2:
Qeqm = (Co−Ceqm) V/W
η % = (Co−Ceqm)/Co) ×100
3. Results and Discussion
The luminescent Sensor A was prepared in a one-pot reaction of trimesic acid and o-aminophenol in a 1:3 mole ratio, Scheme 2. The product was crystallized from ethanol and dried at 100℃ for 1 h.
Figure 1 shows the UV-Vis absorption spectrum along with the excitation and emission spectra of Sensor A depicted at the indicated wavelengths. The UV-Vis spectrum of A showed absorption peaks with maximum absorbance appearing at 275 and 436 nm along with a shoulder peak at 515 nm. The compound also possesses strong excitation and emission profiles in the UV and visible regions. The emission profile depends on the selected excitation wavelength. For instance, a strong emission appears at 571 nm upon excitation at 515 nm, whereas strong emissions were observed at 375 and 515 nm upon excitation at 325 nm. The excitation profile monitored at 550 nm also indicates bands at 512 and 325 nm aligned to the absorption bands depicted from the UV-Vis absorption spectrum. The obtained absorption and emission profiles are due to the high conjugation chain along with plausible n-π* transitions. Figure 1B shows a full-range 2D excitation-emission profile where the maximum luminescence features appear in the visible range from 500-700 nm. The appearance of the emission bands in the visible region corresponds to the presence of the extended conjugation of the designed fluorophore with the two aminophenol substrates via the possible formation of amide-imido tautomers in the solution that extends the conjugation to all fused rings, Scheme 3.
The selection of Sensor A for metal ion detection stems from the strong chain of conjugation that provides reasonable clear and strong fluorescence and absorption properties with a special molecular skeleton, with efficient binding sites for metal ions that causes a direct and sudden change to its luminescence features. Finally, the material is highly soluble in polar solvents thus the testing protocols can be applied to real environmental samples as illustrated therein. Given the broad features of the UV-visible absorption bands and the lack of well-defined emission bands due to the selected excitation wavelengths, Figure 1A, we rely on the Synchronous Scan Luminescence Spectroscopy (SSLS) rather than ordinary emission or excitation spectra for monitoring the binding interaction with the target metal ions. This method involves recording the luminescence intensity while both the excitation and emission monochromators are varied at a constant wavelength difference (Δλ) [41,42]. Using this technique, one can monitor luminescence profiles for materials with weak luminophores or low concentrations with well-defined bands especially when both excitation and emission intensities will be gathered as previously reported [41,42,43].
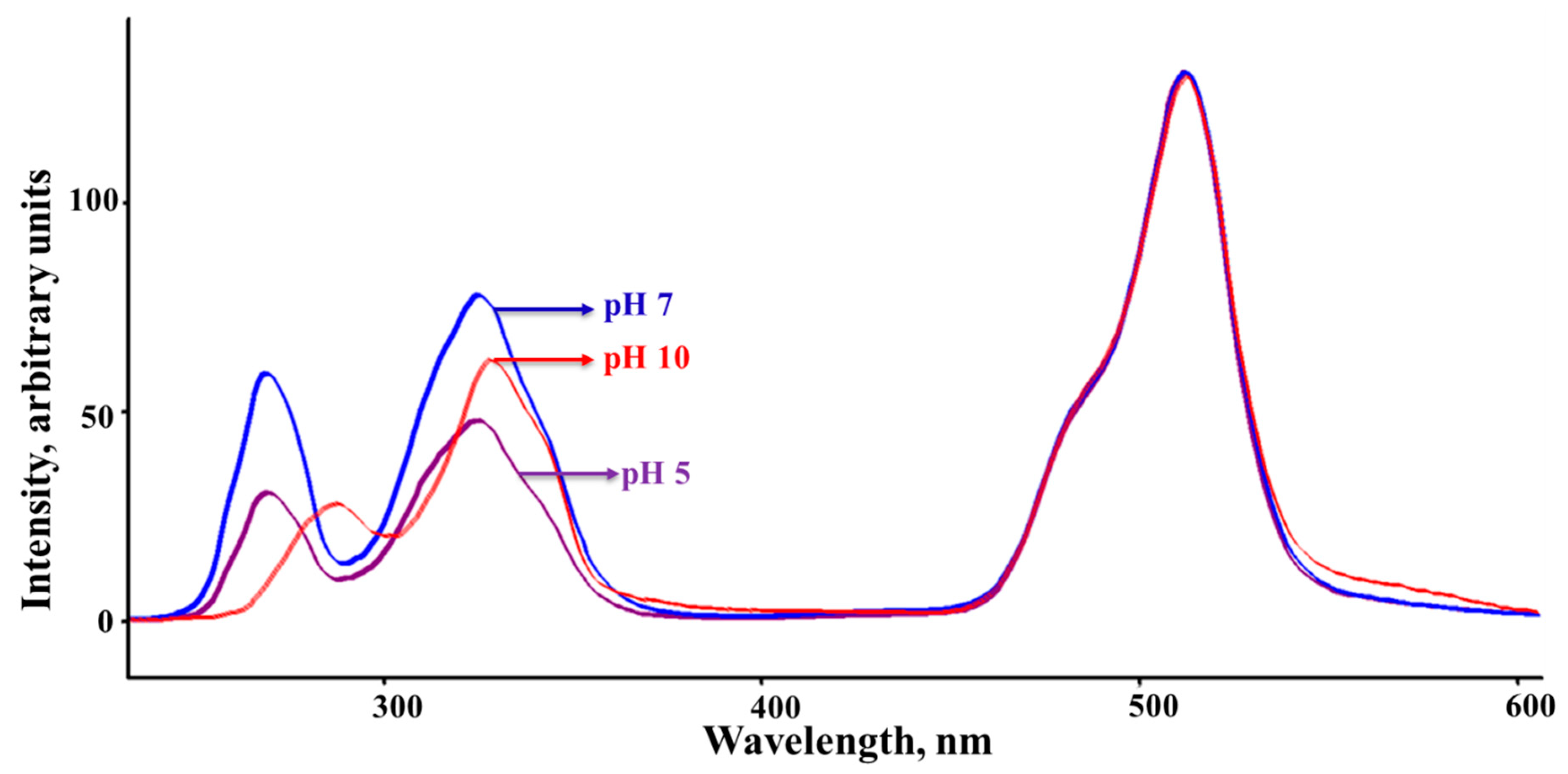
The degree of covalency between the metal ions and the organic sensor depends on the pH and the linkage sites require tailoring the sensor under optimized conditions. For example, a study illustrated the solvent effect on the structural relaxation that follows the displacement of the electron cloud in the dipolar merocyanine dyes where variations were observed in the fluorescence quantum yield that is associated with the dielectric constant of the solvent. In specific, using a series of solvents ordered by increasing dielectric constant the optical gap was found to decrease as the polarity of the solvent increases which results in the positive solvatochromic shift observed in the absorption and emission spectrum [44]. Therefore, all the SSLS recorded in this study represent analysis for solutions using similar aqueous compositions. Besides the high sensitivity and the low detection limit, SSLS also helps to study changes that may occur during the process when new luminophores are formed during the binding processes under various pH buffer solutions and regular ambient water samples. Figure 2 shows the SSLS recorded at Δλ = 50 nm for Sensor A prepared in water solution as well as adjusted pH buffer solutions. As shown in Figure 2, Sensor A has a strong luminescence band in the visible range along with two bands in the UV region at 275 and 325 nm indicating the presence of several luminophores. As shown in Figure 2, the band that appears in the visible range is solvent and pH-independent while the high energy bands are pH as well as solvent dependent. This supports the fact that the low energy band (visible region) is associated with the long chain of conjugation, while the high-energy modes are related to n-π* transitions that may be affected by varying solutions pH in polar solvents. This provides both luminescent and colorimetric detection of the metal ions. The SSLS recorded under basic conditions, showed a redshift in the high energy bands compared to the analogue spectra recorded at neutral and acidic conditions. This shift occurs because of the formation of a highly conjugated system obtained via the phenolate ions formation.
Since our goal is to find a selective and sensitive sensor for the detection of specific metal ions in water bodies, the photophysical properties of Sensor A toward various concentrations of metal ions including Hg(II), Mg2+, Na+, Zn2+, Ni2+, Pb2+, Fe3+, and Ca2+ ions were investigated at various aqueous solutions adjusted at three different pH values (pH 5, 7, and 10). To test the ability of Sensor A as a luminescent sensor for the given target metal ion, its SSLS was monitored as the metal ion concentration increased in the solution. In brief, SSLS were first recorded for 2.75 mL of 30 ppm solution of Sensor A mixed with 0.25 mL of water and the obtained intensity is taken as a reference (F0). Then, 2.75 mL of the sensor solution was mixed with 0.25 mL of various concentrations of the target metal ion and the SSLS were recorded (referred to as F). To test the time required for complete binding (if any) between Sensor A and the target metal ion, we monitored the emission intensity of Sensor A combined with each metal ion for 15 min. Interestingly, an immediate response has occurred with mercury and iron ions (within 30 seconds) with no response observed for the other metal ions even at higher exposure time. Repeating the measurements for various concentrations, it was noticed that the sensor provides responses in a short time where the luminescence intensity is stabilized immediately upon mixing. However, in the testing protocol followed herein, all solutions were mixed for 2 min after which the SSLS were recorded to get stable and consistent results as presented thereafter.
Sensor A shows an immediate and strong response to both mercury and iron ions with no changes occurring for all other tested metal ions including Na+, Mg2+, Ca2+, Ni2+, Pb2+, and Zn (II). This is evidenced by the clear significant changes to the luminescence bands of Sensor A before and after the addition of various concentrations of mercury and iron ions. Figure 3 shows the SSLS of Sensor A before and after adding various concentrations of mercury ions as monitored at pH 5.0. As shown in Figure 3, the luminescence band intensities are gradually reduced when higher concentrations of mercury ions are added with the quenching magnitude found to be directly proportional to the concentration of the quencher Hg (II). As presented in Figure 3, a complete quenching for the band at 515 nm occurred at a 100 ppm Hg (II) concentration level. Similarly, the emission intensity is quenched when different concentrations of Fe(III) were adequately mixed with sensor A. As presented in Figure 4, the emission intensity observed at 510 nm tends to decline when various levels of ferric ions were added with complete quenching occurred at 1000ppm metal ion concentration. The conjugated probe tend to react with the target metal ion through the suitable donor sites affecting the electron cloud distribution of the sensor, thereby changing the output of the emission intensity of the starting material. Therefore, both results, presented in Figure 3 and Figure 4, indicate the high affinity for Sensor A to capture mercury and iron ions from solutions forming five-membered chelating rings through complexation with the probe via the imidazole nitrogen atom, pyridine nitrogen atom, and phenolic oxygen atom, Scheme 4.
We used FT-ATR spectroscopy to monitor the changes that occurred between the starting material, sensor, and the sensor binding to metal ions. Figure 5 shows the FT-ATR spectra for trimesic acid, and Sensor A before and after binding to 50 ppm mercury ions solution (after removing the solvent). Trimeric acid showed bands in the range of 1105, 1250, and 1272 cm-1 assigned for the aromatic C-H in-plane bending vibrations. The bands that appear at 690, 740, and 990 cm-1 are assigned to the C-H out-of-plane bending modes. The characteristic infrared absorption bands of C=O appear at 1691 and 1713 cm-1. The ring C=C stretching modes are observed at 1453 and 1604 cm−1. Compared to the bands observed for trimesic acid, Sensor A shows stretching bands at 3303 and 3373 cm−1 assigned to the N-H stretching mode. The presence of the C=O mode at 1690 cm−1 along with the C=C and C=N modes appear at 1460 and 1498 cm-1 supporting the formation of the amide moieties. Upon binding to mercury ions, bands were observed at 1627, and 1468, along with a broad band centered at 1313 cm-1. The C=N and C=O modes were shifted to appear at 1627 cm−1 supporting their binding to mercury ions that weakened the C=O and C-N modes, Scheme 4.
To find the affinity of the binding association (Ksv) between Sensor A (fluorophore F) and the metal ion of interest (quencher) we use the Stern-Volmer equation shown below providing that static quenching is the dominant quenching process, equation 4 [42,43].
Where [Q] is the metal ion concentration (quencher), F0 and F represent the fluorescence intensity of the sensor before and after adding the quencher.
F0/F = 1 + Ksv [Q]
Figure 6 shows the Stern-Volmer plot that describes the affinity of the quencher to reduce the initial emission intensity of the sensor upon mixing reported at pH = 5. As shown in Figure 6, the plot provides a good correlation between the normalized emission intensity before and after adding various concentrations of Hg(II) and Fe(III) where the R2=0.9908 and 0.9923, respectively. This is expected given that more covalency occured between the iron or mercury ions that are classified as soft acids with the intermediate basic sites represented via oxygen and imine nitrogen linkage . As presented in Figure 6, the binding affinity of Sensor A to Hg (II) is four times higher than that observed for Fe (III), Table 1. The binding affinity also tends to vary when the pH of the solution is varied using buffers at pH 7 and 10. The binding constants obtained for Sensor A at various pH conditions are summarized in Table 1. As depicted in Table 1, Sensor A has a strong binding affinity for both metal ions detected in acidic and neutral conditions.
The response toward Hg (II) was linear in the concentration range of 4.2 x 10-5 to 2.0 x 10-8 M with a limit of detection of 1.0 x 10-8 M. In addition, Sensor A showed a wide linear response in the range of 1.5 x 10-3 to 1.5 x 10-8 M toward iron ions. This is very important to detect low levels of Fe (III) in an aqueous medium at the nM scale since Fe (III) is considered among the most important metal ions in biological systems and exerts an incomparable role in many biological processes including RNA and DNA synthesis, and metabolism. While the sensor showed a good response toward both mercury and iron ions, the binding affinity was found to vary at a given pH condition. For example, the relative binding affinity of Sensor A with mercury ions to that of iron ions is 1.5 at pH 10. This affinity enhanced to around 4 and 9 folds under acidic and neutral conditions, respectively. The stong binding with mercury is expected due to its preference to bind to N-based donor ligands especially the one associated to aromatic and conjugated systems as presented herein. The sensor also provides special skeletal arrangement that allows good size fitting to capture large metal ions under all pH conditions employed, Scheme 4.
For the potential use of Sensor A for real water samples, we tested several environmental, tap, and deionized water samples. Further, tap water samples contaminated with 50 ppm iron ions and mercury ions in isolated and mixture forms were also evaluated. The SSLS of Sensor A was not affected by adding deionized water. The tap water showed a slight reduction in the synchronous scan luminescence bands estimating the iron level to a few ppm levels which were confirmed by ICP analysis. The spiked water samples were added in a different sequence. When 0.25 mL of the spiked sample with 50 ppm iron ions was mixed with Sensor A, the emission intensity was reduced by 65% with this reduction reaching 97% when the same volume of 50 ppm of mercury ions spiked water sample was added. Similarly, when the spiked sample with mixed ions or with mercury ions alone was used, a direct quenching occurred with above 95% reduction observed to the initial emission intensity. This supports the results obtained in Table 1 where the sensor provides a high binding affinity toward mercury ions compared to iron ions.
In summary, a highly conjugated organic molecule was synthesized in one step reaction using low cost and safe starting materials. Besides its high solubility in polar solvents, the unique electronic and skeletal structures depicted by the orientation of the N-, and O-, provides multiple active sites that are suitable to attract mercuric and ferric ions (scehme 4) with low detection limits and wide linear detection range (mM to nM) thus providing a wide range of applications to biological and environmental samples. Further, sensor A, is a highly conjugated organic framework that provides both colorimetric and fluorometric signatures when it is covalently binds to the target metal ions. It is shown that both the emission intensity and the color of the dye have been immediately changed upon mixing with mercuric ions (less than 30 seconds). Finally, the material was tested for detection of mercuric and ferric ions in real water samples with high affinity to adsorb mercuric ions from water bodies with the measured adsorption capacity of 1.2-2.0 mg/g and the removal efficiency from water samples reached 98.8% at pH 7.0.
4. Conclusions
The prepared substrate, N1, N3, N5-tris(2-hydroxyphenyl)benzene-1,3,5-tricarboxamide, was found to be a good substrate to capture mercury(II) and iron(III) contaminants in water samples. Sensor A showed strong luminescent profiles that are sensitive toward trace concentrations of mercury and iron ions under neutral, acidic, and basic conditions. The binding affinity toward mercury ions was always higher than that observed for iron ions under similar conditions. Given the sensor–Hg(II) interaction was found best at pH value range of 5.0-7.0, the sensor fabricated is good substrate for mercuric ions detection at ambient environmental conditions. The affinity of Sensor A to bind to Hg (II) at pH 7, is almost nine times higher than its binding affinity toward iron ions. No effect is demonstrated in when other ions that are common in water bodies was observed in this study.
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by all authors. The first draft of the manuscript was written by Sofian Kanan and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by the Research Office at the American University of Sharjah (FRG22-C-S77).
Availability of data and materials
Not applicable
Acknowledgments
This work was funded by the Research Office at the American University of Sharjah Grant Number: FRG22-C-S77.
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Consent to Participate
Not applicable, no human subjects are involved
Consent to Publish
The participant has consented to the submission of this work to the journal.
References
- Renzoni, A.; Zino, F.; Franchi, E. Mercury levels along the food chain and risk for exposed populations. Environmental Research 1998, 77, 68–72. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, S.; Liu, M.; Chen, Z.; Xu, J.; Dong, Y. Transport and transformation of atmospheric metals in ecosystems: A review. Journal of Hazardous Materials Advances 2023, 9, 100218. [Google Scholar] [CrossRef]
- Mng'ong'o, M.; Munishi, L.K.; Ndakidemi, P.A.; Blake, W.; Comber, S.; Hutchinson, T.H. Toxic metals in East African agro-ecosystems: Key risks for sustainable food production. Journal of Environmental Management 2021, 294, 112973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, Y.; Wang, Z.; Liu, Y. Distribution characteristics, bioaccumulation and trophic transfer of heavy metals in the food web of grassland ecosystems. Chemosphere 2021, 278, 130407. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.F.; Hu, Y.A. Lead (Pb) Isotopic Fingerprinting and Its Applications in Lead Pollution Studies in China: A Review. Environ. Pollut. 2009, 58, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, C.; Calagna, A.; et al. Risk assessment of cadmium, lead, and mercury on human health in relation to the consumption of farmed sea bass in italy: a meta-analytical approach. Front. Mar. Sci. 2021, 8. [Google Scholar] [CrossRef]
- Panhwar, A.H.; Kazi, T.G.; Afridi, H.I.; Arain, S.A.; Arain, M.S.; Brahaman, K.D.; Naeemullah, A.S. Correlation of Cadmium and Aluminum in Blood Samples of Kidney Disorder Patients with Drinking Water and Tobacco Smoking: Related Health Risk. Geochem. Health. 2016, 38, 265–274. [Google Scholar] [CrossRef]
- Tong, S.Y.; Meija, J.; Zhou, L.; Methven, B.; Mester, Z.; Yang, L. High-precision Measurements of the Isotopic Composition of Common Lead Using MC-ICP MS: Comparison of Calibration Strategies Based on Full Gravimetric Isotope Mixture and Regression Models. Anal. Chem. 2019, 91, 4164–4171. [Google Scholar] [CrossRef]
- Luo, X.; Liu, L.; Deng, F.; Luo, S. Novel Ion-imprinted Polymer Using Crown Ether as a Functional Monomer for Selective Removal of Pb(II) Ions in Real Environmental Water Samples. J. Mater. Chem. A. 2013, 1, 8280–8286. [Google Scholar] [CrossRef]
- Wani, A.B.; Ara, A.; Usmani, J.A. Lead Toxicity: A Review. Interdiscip. Toxicol. 2015, 8, 55–64. [Google Scholar] [CrossRef]
- Charvát, P.; Klimeš, K.; Pospíšil, J.; Klemeš, J.; Varbanov, P. An overview of mercury emissions in the energy industry - A step to mercury footprint assessment. Journal of Cleaner Production 2020, 267, 122087. [Google Scholar] [CrossRef]
- Dworak, S.; Rechberger, H. Mercury throughput of the Austrian manufacturing industry – Discussion of data and data gaps. Resources, Conservation and Recycling 2021, 166, 105344. [Google Scholar] [CrossRef]
- Dziok, T.; Bury, M.; Burmistrz, P. Mercury release from municipal solid waste in the thermal treatment process. Fuel 2022, 329, 125528. [Google Scholar] [CrossRef]
- Sun, W.; Wang, Q.; Zhou, Y.; Wu, J. Material and Energy Flows of the Iron and Steel Industry: Status Quo, Challenges and Perspectives. Appl. Energy. 2020, 268, 114946. [Google Scholar] [CrossRef]
- Xin, H.; Wang, S.; Chun, T.; Xue, X.; Long, W.; Xue, R.; Zhang, R. Effective pathways for energy conservation and emission reduction in iron and steel industry towards peaking carbon emissions in China: Case study of Henan. Journal of Cleaner Production 2023, 399, 136637. [Google Scholar] [CrossRef]
- Zhang, X.; Xue, C.; Wang, R.; Shen, R.; Lan, P. Physiological and proteomic dissection of the rice roots in response to iron deficiency and excess. Journal of Proteomics 2022, 267, 2022. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wu, P.; Jiang, W.; Liu, Y.; Kuang, S.; Jiang, J.; Tang, L.; Tang, W.; Zhang, Y.; Zhou, Q.; Feng, L. The impaired immune function and structural integrity by dietary iron deficiency or excess in gill of fish after infection with Flavobacterium columnare: Regulation of NF-κB, TOR, JNK, p38MAPK, Nrf2 and MLCK signaling. Fish & Shellfish Immunology 2018, 74, 593–608. [Google Scholar] [CrossRef]
- Perng, V.; Li, C.; Navazesh, S.; Klocke, C.; Pinneles, D.; Lein, P.; Ji, P. Iron Deficiency and Iron Excess Alter Dendritic Architecture of Pyramidal Neurons in the Hippocampus of Neonatal Pigs. Current Developments in Nutrition 2020, 4, 4141232. [Google Scholar] [CrossRef]
- Guidelines for Drinking-Water Quality: Recommendations; World Health Organization, 2004.
- Volynkin, S.S.; Demakov, P.A.; Shuvaeva, O.V.; Kovalenko, K.A. Metal-organic framework application for mercury speciation using solid phase extraction followed by direct thermal release–electrothermal atomization atomic absorption spectrophotometric detection (ETA AAS). Analytica Chimica Acta 2021, 1177, 338795. [Google Scholar] [CrossRef]
- dos Santos, N.; dos Santos, L.; Damin, I.; Vale, M.; Dessuy, M. Multielement determination of metals in edible seeds by HR-CS GF AAS and direct analysis. Journal of Food Composition and Analysis 2022, 111, 104625. [Google Scholar] [CrossRef]
- Lores-Padín, A.; Fernández, B.; García, M.; González-Iglesias, H.; Pereiro, R. Real matrix-matched standards for quantitative bioimaging of cytosolic proteins in individual cells using metal nanoclusters as immunoprobes-label: A case study using laser ablation ICP-MS detection. Analytica Chimica Acta 2022, 1221, 340128. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Su, Z.; Mao, Y.; Liu, C.; Qi, B.; Fang, G.; Wang, S. Carboxyl-functionalized hollow polymer microspheres for detection of trace metal elements in complex food matrixes by ICP-MS assisted with solid-phase extraction. Ecotoxicology and Environmental Safety 2021, 208, 111729. [Google Scholar] [CrossRef] [PubMed]
- Dimpe, K.; Ngila, J.; Mabuba, N.; Nomngongo, P. Evaluation of sample preparation methods for the detection of total metal content using inductively coupled plasma optical emission spectrometry (ICP-OES) in wastewater and sludge. Physics and Chemistry of the Earth, Parts A/B/C 2014, 76–78, 42–48. [Google Scholar] [CrossRef]
- Pal, H.; Majumder, S. Ultra-low-level detection of mercury (Hg2+) heavy metal carcinogens in aqueous medium using electrochemistry. Materials Today: Proceedings 2020, 29, 1129–1131. [Google Scholar] [CrossRef]
- Tong, Y.; Wu, Y.; Bai, H.; Li, S.; Jiang, L.; Zhou, Q.; Chen, C. Highly efficient and simultaneous magnetic solid phase extraction of heavy metal ions from water samples with l-Cysteine modified magnetic polyamidoamine dendrimers prior to high performance liquid chromatography. Chemosphere 2023, 313, 137340. [Google Scholar] [CrossRef] [PubMed]
- Dehdashtian, S.; Pourreza, N.; Rostamnia, S. Electrochemical sensing of indole in plasma using Pd nanoparticles modified metal-organic framework Cr-MIL-101/ionic liquid sensor. Microchemical Journal 2022, 181, 107839. [Google Scholar] [CrossRef]
- Darwish, N.; Kurdi, A.; Alshihri, S.; Tabbakh, T. Organic heterocyclic-based colorimetric and fluorimetric chemosensors for the detection of different analytes: a review (from 2015 to 2022). Materials Today Chemistry 2023, 27, 101347. [Google Scholar] [CrossRef]
- Li, A.; Liu, Y.; Chen, Z.; Li, S.; Zhong, R.; Cheng, D.; Chen, L.; He, L. Development of a Golgi-targeted fluorescent chemosensor for detecting ferrous ions overload under Golgi stress. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2023, 294, 122560. [Google Scholar] [CrossRef]
- Pandey, H.P.; Aggarwal, S.; Vats, M.; Rawat, V.; Pathak, S. Coumarin-based Chemosensors for Metal Ions Detection. Asian Journal of Organic Chemistry 2022, 11, 23–75. [Google Scholar] [CrossRef]
- Kanan, S.; Abu-Yousef, I.; Hassouneh, N.; Malkawi, A.; Abdo, N.; Kanan, M. A Highly Selective Luminescent Sensor for Detecting Mercuric Ions in Water. Australian Journal of Chemistry 2019, 62, 1593–1599. [Google Scholar] [CrossRef]
- Rasin, P.; Haribabu, J.; Malappuram, K.; Manakkadan, V.; Palakkeezhillam, V.N.; Cesar Echeverria, C.; Sreekanth, A. A “turn-on” fluorescent chemosensor for the meticulous detection of gallium (III) ion and its use in live cell imaging, logic gates and keypad locks. Journal of Photochemistry and Photobiology A: Chemistry 2023, 437, 114493. [Google Scholar] [CrossRef]
- Xie, H.; Hu, Q.; Qin, X.; Zhang, Y.; Li, L.; Li, J. Naked-eye chemosensor with high absolute fluorescence quantum yield for selective detection of Cu(II) and cell imaging. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2022, 283, 121740. [Google Scholar] [CrossRef] [PubMed]
- Kanan, S.; Malkawi, A. Recent Advances in Nanocomposite Luminescent Metal-Organic Framework Sensors for Detecting Metal Ions. Comments on Inorganic Chemistry 2021, 41, 1–66. [Google Scholar] [CrossRef]
- Jiang, J.; Lu, Y.; Zhou, Y.; Zhao, D.; Li, C. An Acid-base Resistant Zn-based Metal-organic Framework as a Luminescent Sensor for mercury(II), J. Solid State Chem. 2020, 283, 121–153. [Google Scholar] [CrossRef]
- Kang, W.; Han, C.; Liu, D.; Cui, G. A Bifunctional Benzimidazole-based Luminescent Zn(II) Coordination Polymer for Detection of Hg2+ and Photocatalytic Degrading of Methylene Blue. Inorg. Chem. Commun. 2019, 106, 81–85. [Google Scholar] [CrossRef]
- Wan, Y.; Zou, D.; Cui, Y.; Yang, Y.; Qian, G. A Zn Based Anionic Metal-organic Framework for Trace Hg2+ Ion Detection. J. Solid State Chem. 2018, 266, 70–73. [Google Scholar] [CrossRef]
- Latt, K.K.; Takahashi, Y. Fabrication and characterization of a α,β,γ,δ-Tetrakis(1-methylpyridinium-4-yl)porphine/silica nanocomposite thin-layer membrane for detection of ppb-level heavy metal ions. Analytica Chimica Acta 2011, 689, 103–109. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, J.; Shen, G.; Yu, R. An optical sensor for mercury ion based on the fluorescence quenching of tetra(p-dimethylaminophenyl)porphyrin. Analytica Chimica Acta 2009, 636, 83–88. [Google Scholar] [CrossRef]
- Jian, Y.; Zhe, W.; Li, Y.; Wenru, Q.; Jinlou", G. Porphyrinic MOFs for reversible fluorescent and colorimetric sensing of mercury(ii) ions in aqueous phase. RSC Adv. 2016, 6, 69807–69814. [Google Scholar]
- Kanan, S.; Kanan, M.; Patterson, H. Silver nanoclusters doped in X and mordenite zeolites as heterogeneous catalysts for the decomposition of carbamate pesticides in solution. Res. Chem. Intermed. 2006, 32, 871. [Google Scholar] [CrossRef]
- Kanan, S.; Kanan, M.; Patterson, H. Photoluminescence spectroscopy as a probe of silver doped zeolites as photocatalysts. Current Opinion in Solid State & materials Science 2003, 7, 443. [Google Scholar] [CrossRef]
- Fan, J.; Peng, Y.; Wu, E.; Lu, J.; Hou, J.; Zhang, H.; Zhang, R.; Fu, X. A new PET fluorescent sensor for Zn2+. J. Luminescence 2005, 114, 125. [Google Scholar] [CrossRef]
- Hoche, J.; Schulz, A.; Fietrich, L. The origin of the solvent dependence of fluorescence quantum yields in dipolar merocyanine dyes. Chem. Sci., 2019, 10, 11013. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Schematic illustration of luminescent sensors based on the fluorophore-receptor photoinduced electron transfer process.
Scheme 1.
Schematic illustration of luminescent sensors based on the fluorophore-receptor photoinduced electron transfer process.
Scheme 2.
The synthetic scheme for the preparation of Sensor A.
Scheme 3.
Plausible amide-imido tautomers of A in aqueous solution.
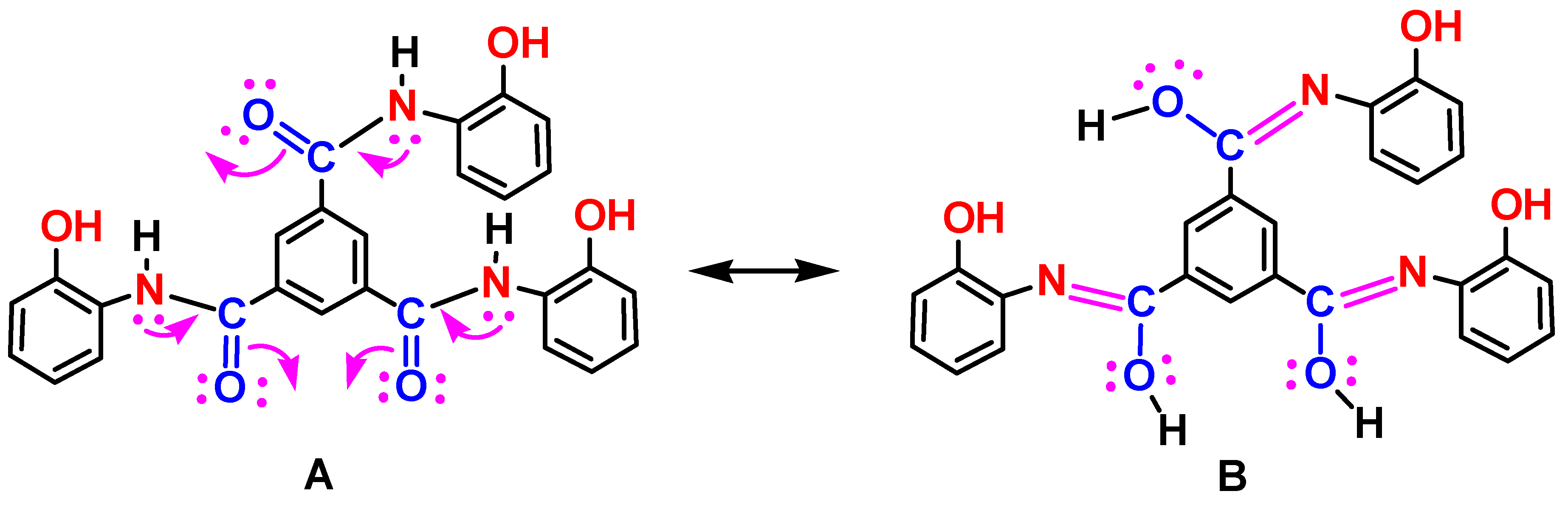
Figure 1.
A) absorption spectrum of Sensor A along with excitation and emission profiles monitored at specific λem and λexc as labeled, and B) 2D full range excitation and emission profile for Sensor A.
Figure 1.
A) absorption spectrum of Sensor A along with excitation and emission profiles monitored at specific λem and λexc as labeled, and B) 2D full range excitation and emission profile for Sensor A.
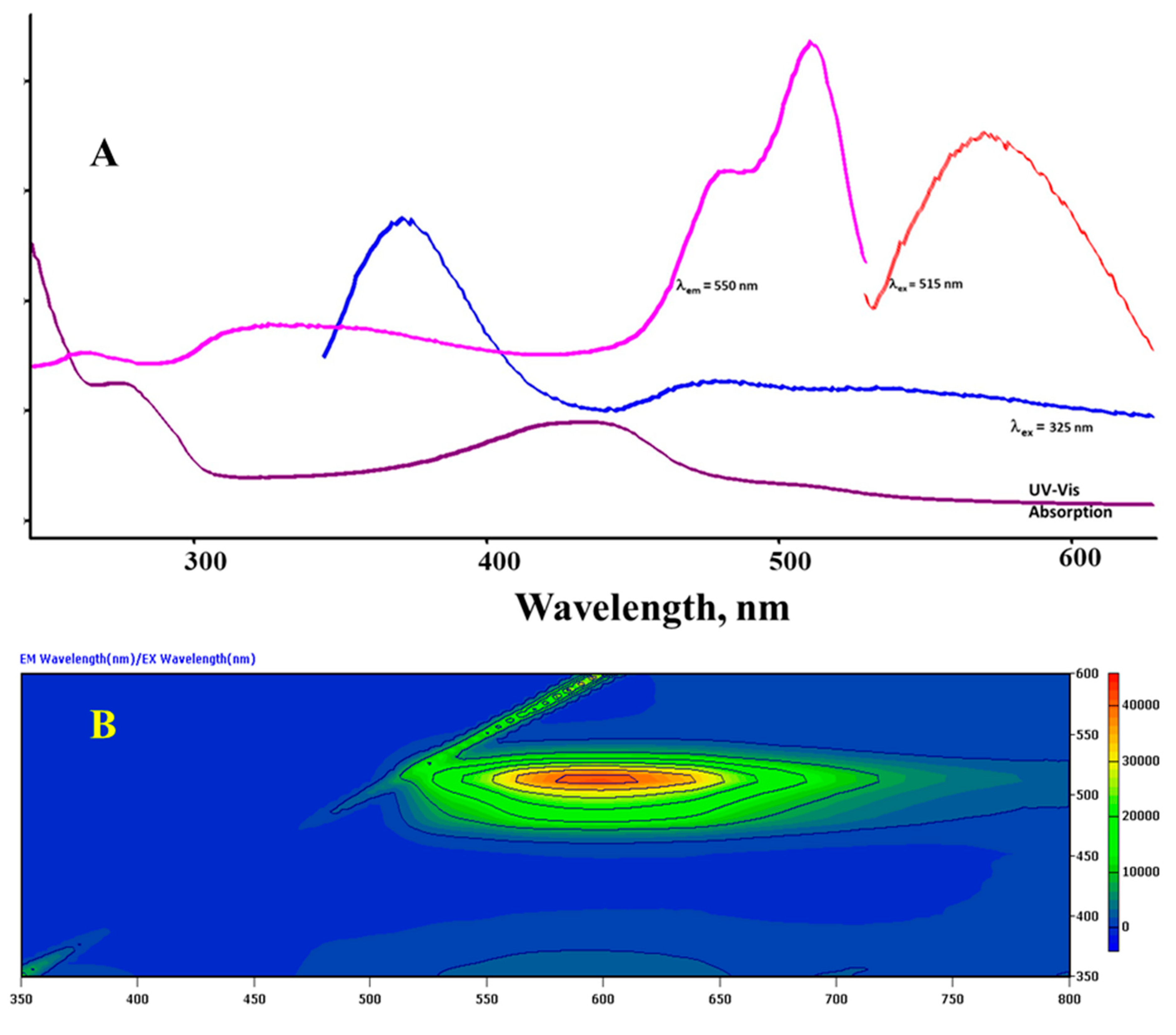
Figure 2.
Synchronous Scan Luminescence Spectra recorded for Sensor A solutions at the indicated pH solutions as monitored at Δλ = 50 nm.
Figure 2.
Synchronous Scan Luminescence Spectra recorded for Sensor A solutions at the indicated pH solutions as monitored at Δλ = 50 nm.
Scheme 4.
Binding model between Sensor A and the depicted metal ions.
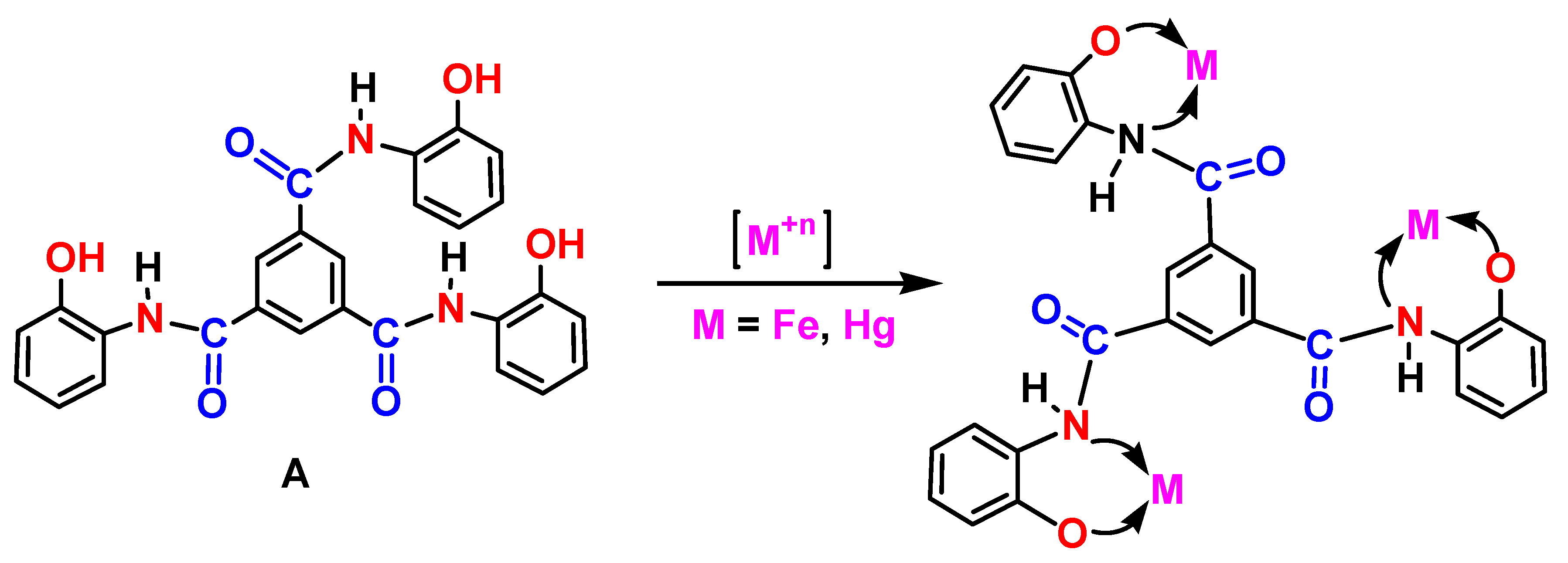
Figure 3.
Synchronous Scan Luminescence Spectra of Sensor A before and after adding various concentrations of mercury ions recorded at Δλ of 50 nm and at pH = 5. The color represents Sensor A (top) and Sensor A after adding 10 ppm Hg (II) solution (bottom).
Figure 3.
Synchronous Scan Luminescence Spectra of Sensor A before and after adding various concentrations of mercury ions recorded at Δλ of 50 nm and at pH = 5. The color represents Sensor A (top) and Sensor A after adding 10 ppm Hg (II) solution (bottom).
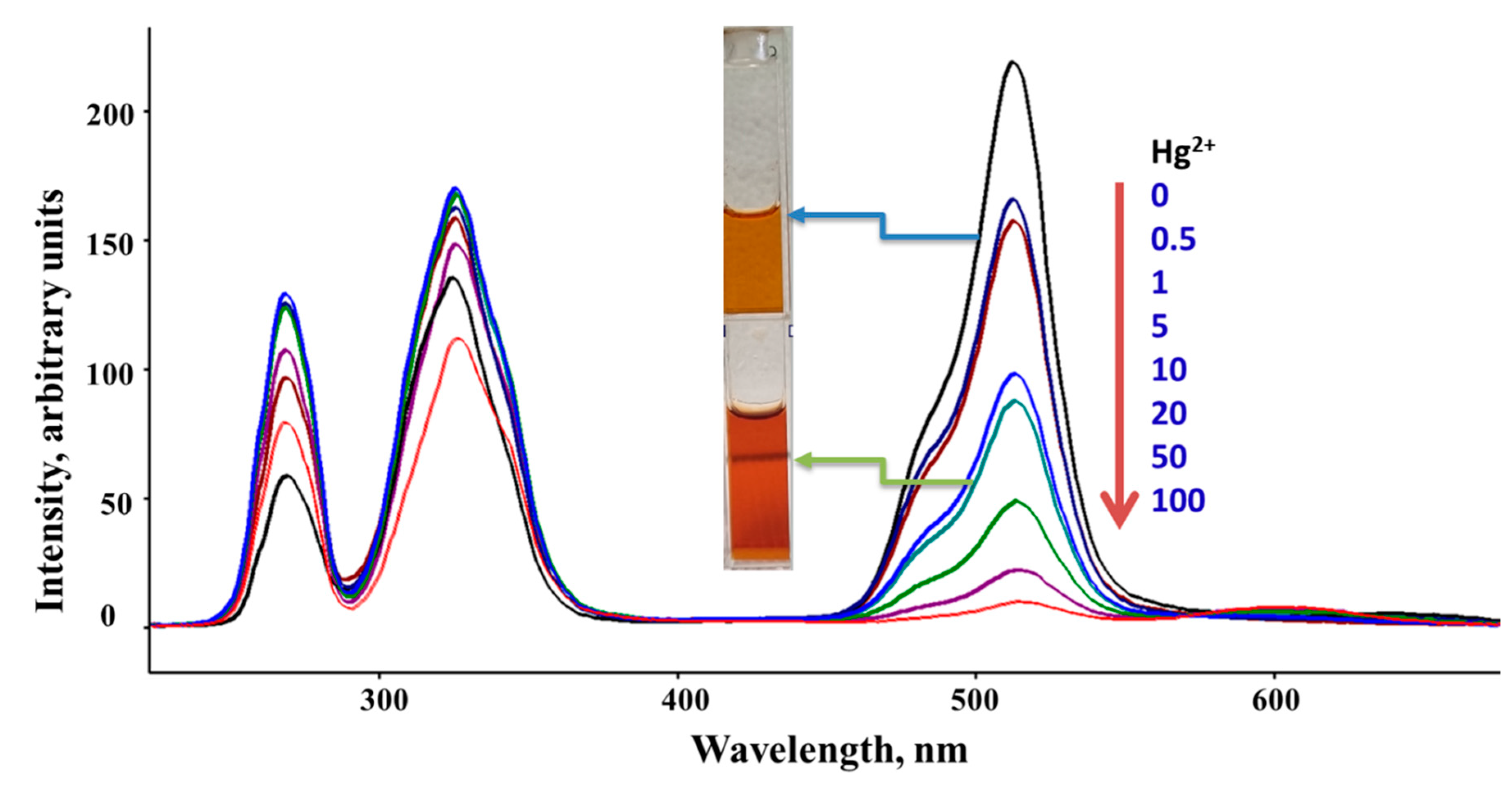
Figure 4.
Synchronous Scan Luminescence Spectra of Sensor A before and after adding various concentrations of Fe (III) ions recorded at Δλ of 50 nm and at pH = 5. .
Figure 4.
Synchronous Scan Luminescence Spectra of Sensor A before and after adding various concentrations of Fe (III) ions recorded at Δλ of 50 nm and at pH = 5. .
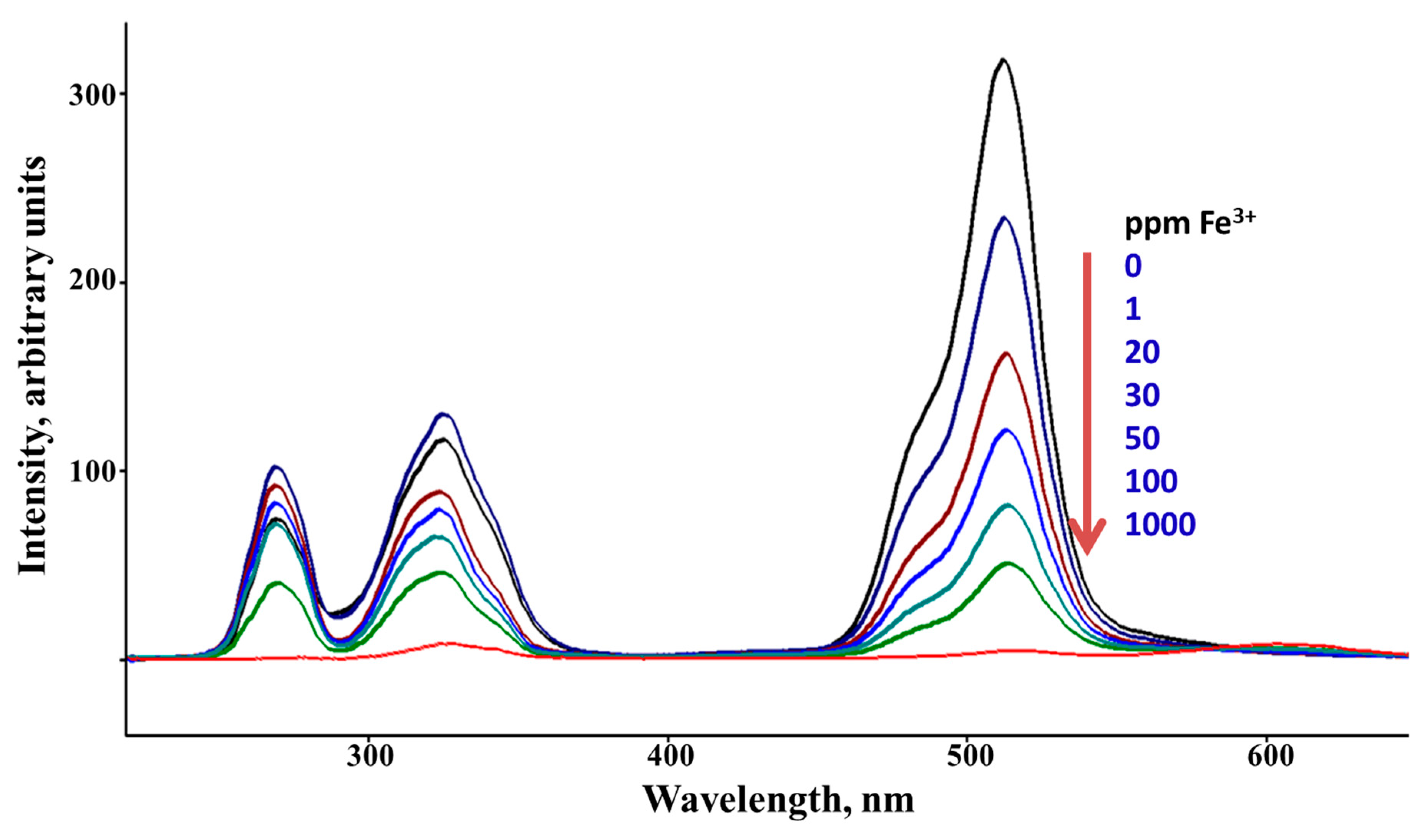
Figure 5.
FT-ATR spectra for trimesic acid, Sensor A, Hg (II) binds to Sensor A.
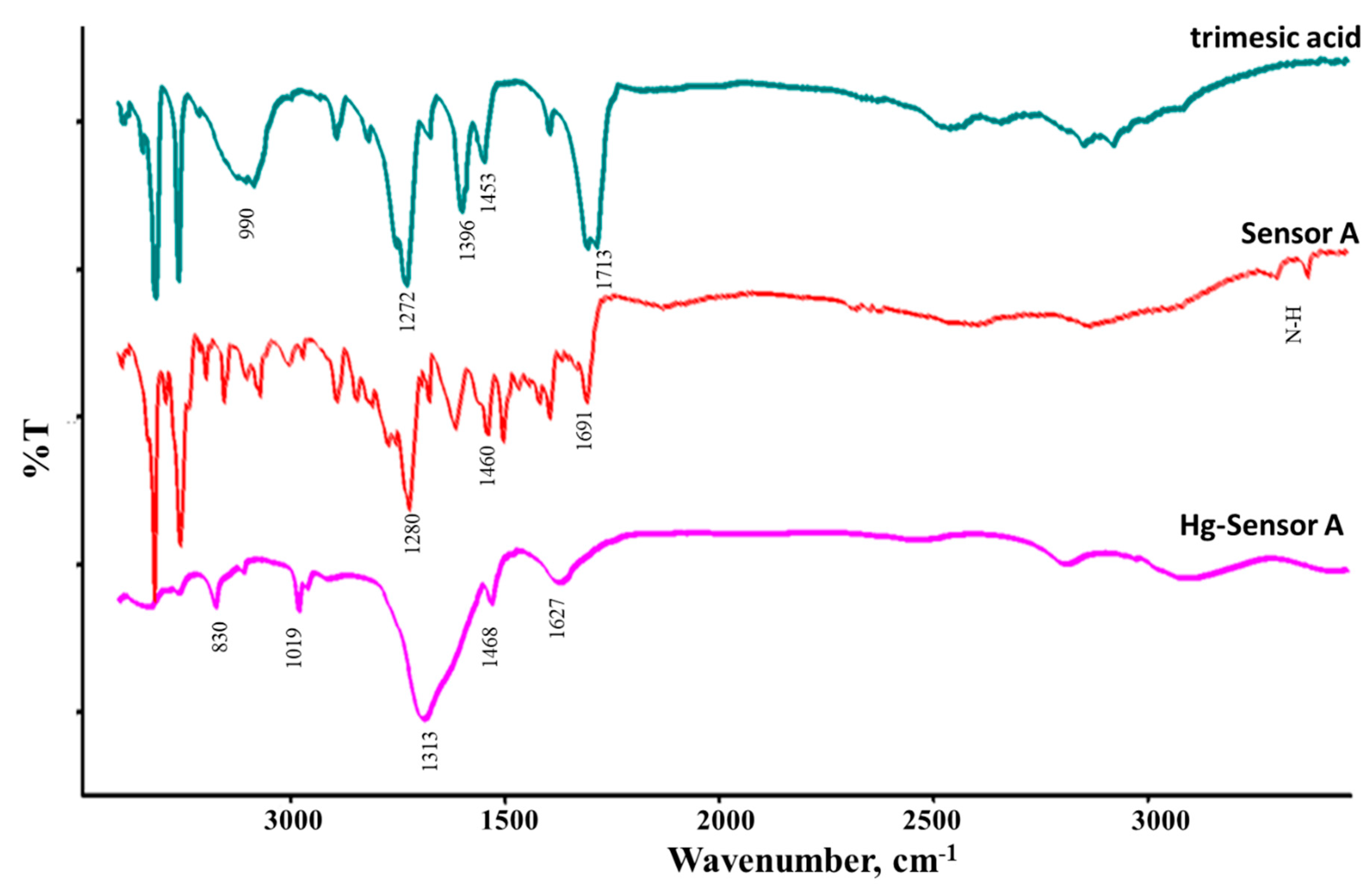
Figure 6.
The Stern-Volmer plots of Sensor A binding to various Hg (II) and Fe (III) concentrations under pH = 5.
Figure 6.
The Stern-Volmer plots of Sensor A binding to various Hg (II) and Fe (III) concentrations under pH = 5.
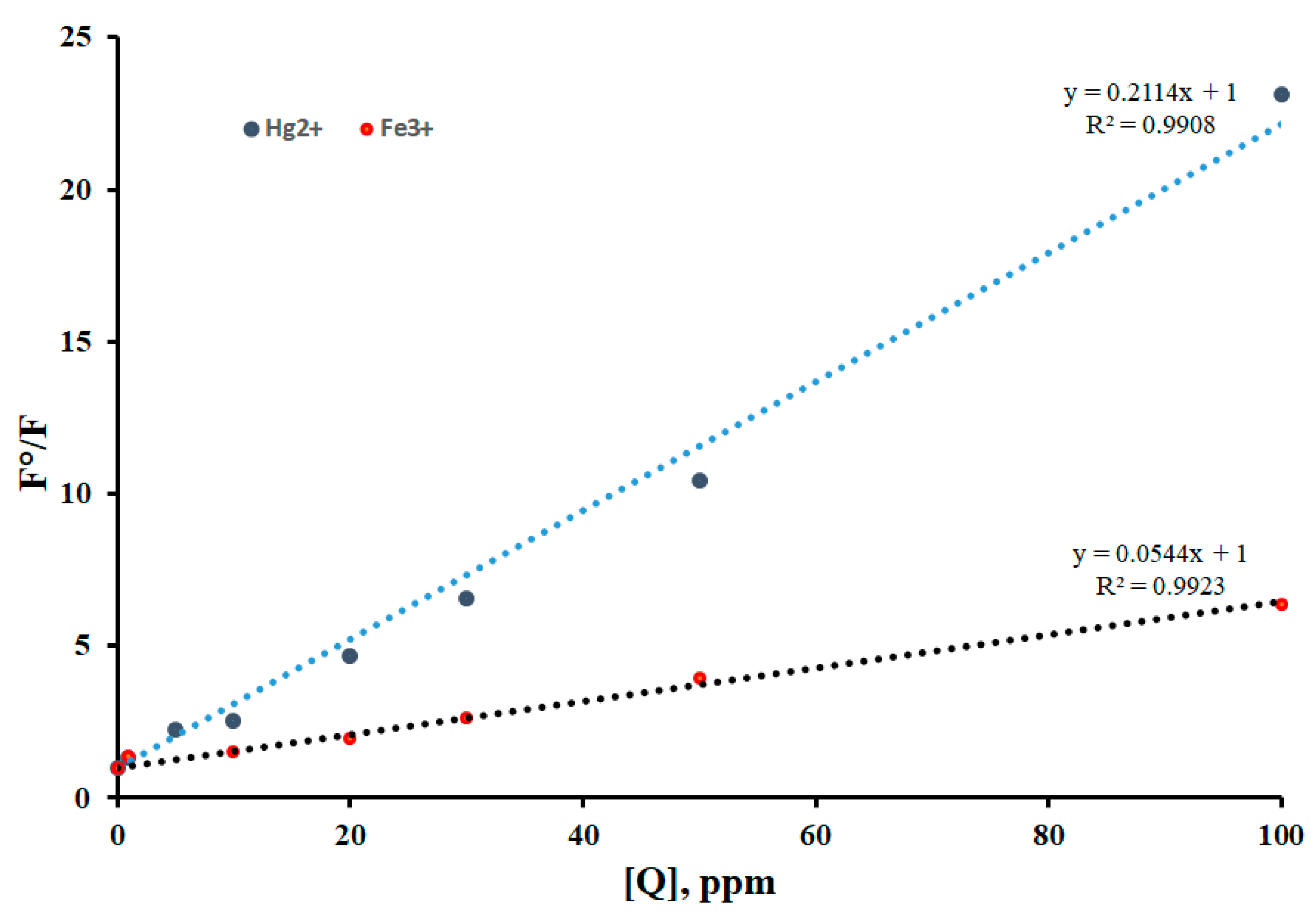

Table 1.
A summary of the calculated binding affinities (Ksv) between Sensor A and mercury and iron ions at various pH conditions.
Table 1.
A summary of the calculated binding affinities (Ksv) between Sensor A and mercury and iron ions at various pH conditions.
| pH 5 | pH 7 | pH 10 | |
|---|---|---|---|
| Ksv Hg (II)/Sensor A | 0.2114 | 0.1650 | 0.0631 |
| Ksv Fe (III)/Sensor A | 0.0544 | 0.0184 | 0.0428 |
| Ksv Hg (II)/Ksv Fe (III) | 3.89 | 8.97 | 1.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



