Submitted:
24 April 2023
Posted:
25 April 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
SARS-CoV-2 was detected in China in December 2019. Myocardial injury is a crucial presentation of COVID-19, based on the association of ACE-2 and SARS-CoV-2. Down-regulating ACE-2 decreases the cardioprotective effects of angiotensin, leading to a higher TNF-α activation. TNF-α causes the inflammatory response in the myocardial damage as an apoptotic inducer. Moreover, as an inducer of necroptosis, TNF-α binds to a part of TNF receptor 1, which involves receptor-interacting protein 1 (RIP1) and causes cell death through RIP1 inhibition and NF-κB stimulation, which are also done through Tpl-2. Calcineurin controls the Tpl-2-driven NFAT stimulation. Bcl-2 or Bcl-XL entirely blocks these pathways. Bcl-2 overexpression reduces FasL expression with a mechanism based on Bcl-2 inhibiting the NFAT. Moreover, the Fas/FasL system activates apoptosis in various cells. Bcl-XL stimulates Fas-related cell death. Additionally, TNF-α, as a part of inflammatory cytokine storms, indirectly interacts with NFAT/Bcl-2 through Tpl-2/NF-κB. Diversely, TNF-α and IL-1ẞ, the basis of inflammatory cytokine storms in COVID-19, can stimulate generating NO. Also, IL-2 is highly up-regulated in COVID-19 patients and stimulates NO generation in patients. TNF-α can provoke the generation of superoxides in neutrophils. A well-determined mechanism is the intracellular production of NO via calcium-calmodulin-dependent NO synthase (NOS). NO enhances NFAT’s calcium-dependent activity. Also, Intra/extracellular calcium exchange activates calcineurin and its related molecule, NFAT. Nitration provokes RIP1 necroptosis cascade, with respiratory complex I. Nitrites converse protection against ischemia-reperfusion injuries in the myocardium. Regulating this intrinsic molecular pathway can prevent the necroptosis of cardiomyocytes.
Keywords:
COVID-19
; Necroptosis
; NFAT
; NO
; TNF-α
; NF-κB
1. Background
In December 2019, a local outbreak of a new type of pneumonia was detected in Wuhan, China, which was quickly determined to be caused by a novel coronavirus, called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2 or COVID-19) [1]. Scientists worldwide quickly started to design and perform studies to recognize this new type of coronavirus. Myocardial injury is the main presentation of COVID-19. Besides, myocarditis is another cause of morbidity among COVID-19 patients. The suggested mechanisms of myocardial injury during COVID-19 include direct viral damage to cardiac cells, viral-induced myocardial fibrosis, systemic inflammation, overstated cytokine reaction due to the activation of type 1 and 2 helper T cells, and hypoxia [2].
Direct viral attack on the myocardium may be the other possible fundamental pathway leading to myocardial damage, in which the noticeable affinity of SARS-CoV-2 with the ACE-2 receptor may lead to direct viral infection in the vascular endothelium and myocardium [3,4,5,6]. SARS-CoV infection of the myocardium is based on the ACE-2 receptors, and the disturbance of ACE-2 causes cardiomyopathy, cardiac dysfunction, and heart failure [7,8]. The association of SARS-CoV and ACE-2 in the myocardium can lead to SARS-related myocardial inflammation and direct damage [9]. The harmful effect of ACE-2 down-regulation may lead the cardioprotective effects of angiotensin to decrease, causing higher TNF-α activation, which as a well-known inflammatory cytokine, develops the inflammatory response in the myocardial damage [2,7,10,11]. Furthermore, TNF-α and JAK/STAT molecular pathway, as other accepted inflammatory pathway, is responsible for several inflammatory processes within the cellular zone [12].
Janus kinase (JAK)/signal transducer and activator for transcription (STAT) pathway have an essential effect in coordinating cell-mediated immune system, particularly cytokine receptors, and may control the modification of T-helper cells. STAT family members are expressed in cardiomyocytes and are the fundamental molecules in myocardial ischemia pathogenesis. Although the association between the renin-angiotensin system (RAS) and STAT clarifies the inhibition of RAS activation, it incites many cardiomyocytes to be saved in myocardial ischemia. Besides, COVID-19’s viral attack and inflammatory cascades may decline during JAK/STAT pathway inhibition over ACE-2 [13,14].
We theorize that COVID-19-related molecular pathways are critical in myocardial damage and necroptosis. SARS-CoV-2-derived molecular pathways lead to myocardial necroptosis due to inflammatory cytokine storm, higher TNF-α activation, and induction of the inflammatory response. This study describes the molecular pathways affecting the myocardium during the infliction of this new type of disease.
2. NFAT Molecular Pathway
Calcium signals result in translocating the nuclear factor of activated T cells (NFAT) from the cytoplasm to the nucleus. Nevertheless, the calcium-activated phosphatase calcineurin, which can be co-transported to the nucleus with NFAT to maintain transcriptionally activity during calcium signaling, regulates this translocation process [15]. NFAT is a key regulatory factor in gene transcription. NFATc1–NFATc4 are four different recognized calcium-related members. Different cell types, such as B lymphocytes, NK cells, mast cells, and neurons, modulate these transcription factors. They can be stimulated by different pathways, for instance, signaling through the B cell receptor (BCR) and other receptors [16,17,18,19,20,21]. NFAT is translocated into the cytoplasm while calcium, NFAT, and calcium-mediated phosphatase calcineurin pathway signal shut down. Different NFAT kinases deal with calcineurin actions and control the nuclear transfer of NFAT. P38/MAPK phosphorylates deal with NFATp.
Furthermore, stimulating this pathway by triggering MKK6 and p38 stabilizes the calcium-induced nuclear increase in NFATp. The activation of the JNK or ERK pathway ineffectively adjusts the nuclear transportation of NFATp. Regularly stimulating p38 leads to down-regulating NFATp-mediated transcription. Hence, the p38 signaling pathway seems to have a key role in controlling NFATp and cellular homeostasis [15].
Although the stimulation process is related to calcium through activating the dephosphorylation of NFAT by the calcium-dependent phosphatase calcineurin, the exact downstream molecular pathway of NFAT is unknown. NFAT functions in T- and B-cell activation and differentiation [22,23]. NFATc1 and NFATc2 knocked-out genes show elevated IgG1 and IgE antibody levels [24]. Likewise, in T-cells, the Tpl-2 kinase accelerates IL-2 expression and stimulates NFAT. Mutant signaling molecules, which inhibit the mitogen-activated protein kinase (MAPK) or the calcineurin/NFAT pathways, may inhibit the IL-2 promoter stimulation by Tpl-2. Signaling molecules’ stimulation triggers the pathways discussed above. As the NFAT-mediated promoter, IL-2 promoter stimulation depends on Raf1 signals. Consequently, the stimulating NFAT is related to MAPK, and Tpl-2 would be independent of MEK1 and MEK2. Likewise, Tpl-2 also stimulates NF-κB. A combination of calcineurin-dependent and independent pathways controls the Tpl-2-related NFAT stimulation. These dependent and independent pathways are also entirely blocked by Bcl-2 or Bcl-XL [25]. Nevertheless, Bcl-2 inhibits the apoptosis cascade, which could be induced by several intra/extracellular stimuli. Dissemination of microtubule dynamics kills cells in a Fas/Fas ligand (FasL)-driven situation. The overexpression of Bcl-2 reduces the expression of FasL. This mechanism is based on preventing NFAT by Bcl-2 [26]. Fas/FasL system activates apoptosis in various cell types. Moreover, Bcl-XL stimulates Fas-related cell death [27].
Besides, SRF can control the expression of MCL-1 as a Bcl family member. NFAT and SRF are the key molecules in controlling cytokine production in lymphocyte lines. NFAT stimulation by the BCR happens through the Ca2+/calcineurin pathway. Conversely, SRF responds to low cellular Ca2+ levels and is less dependent on IP3R expression than NFAT. Ca2+-regulated calcineurin is critical in SRF activation combined with diacylglycerol (DAG). DAG is essential for NFAT stimulation. Several DAG effectors, like Ras and Rap1, protein kinase C, and the MEK/ERK pathway, are also needed for SRF and NFAT regulation; however, NFAT is dependent on JNK signals individually [28].
3. Necroptosis
Necroptosis is a programmed necrosis that employs various death receptors such as TNF receptor (TNFR) 1, TNFR2, and Fas. It is validated that death receptors possibly induce necroptosis associated with receptor-interacting protein 1 (RIP1) [29]. Moreover, inducing Toll-like receptor (TLR) agonists are known to control caspase-independent necrosis and necroptosis [30,31]. Various genes involved in the TLR pathway are regularly found in necroptotic signaling cascades and lead to necroptosis. So, the TNF-α/TNFR-mediated pathways lead to the necroptosis process. TNF-α, produced by activated macrophages [32], is usually defined as an apoptotic inducer, so, as the first identified agent, it can induce necrosis in tumoral cells [33,34]. Moreover, TNF-α, as a necroptosis inducer, binds to the extracellular part of TNFR1 [35]. This extracellular part of TNFR1, called cysteine-rich domains (CRDs) or pre-ligand assembly domain (PLAD), is essential to develop pre-assembled TNF-α binder receptors [36]. TNF-α binder molecules such as silencer of death domains (SODD) trigger TNFR1 and TNFR2. Subsequently, the downstream signaling cascade forms complex I with TNF-α receptor-associated death domain (TRADD), TNF-α receptor-associated factor 2/5 (TRAF2/5), Fas-associated death domain (FADD), RIP1, and Inhibitor of apoptosis proteins (IAPs), e.g., cIAP1 and cIAP2 [37].
The ubiquitination state of RIP1, as a part of the RIP family homologous N-terminal kinase domain determinant, promotes cell death. TNFR1 involves RIP1, which leads to poly-ubiquitination of TNFR1 by TRAF2/5, cIAP1, and cIAP2 [38,39]. The ubiquitination process of RIP1 activates the IKK complex and NEMO. Therefore, it may up-regulate the NF-κB molecular pathway. RIP1 deubiquitination may inhibit NF-κB molecular pathway and cause a disposition in the cell death pathways program. At this point, cylindromatosis (CYLD), the inhibitor of NF-κB, plays a crucial role in the cell death program [40]. Tumor cells expressing inactive CYLD decrease the incidence of apoptosis [41,42]. Proteasome-dependent degradation of E3 ligases down-regulates NF-κB pathway [43,44]. The ubiquitination of RIP1 is an essential part of the NF-κB molecular pathway activation [45]. Nevertheless, the RIP1 kinase activity [46,47] as the key element in regulating the TNF-induced NF-κB molecular pathway is the ubiquitination status of RIP1, regardless of whether the RIP1 kinase is activated or not. De-ubiquitination of RIP1 is related to complex I and, consequently, to complex II, also called the death-inducing signaling complex (DISC) [48]. The aggregation of TRADD, FADD, RIP1, and caspase-8 forms complex II. The Knock-down of CYLD inhibits TNF-α-induced necroptosis [31], which shows the role of the RIP1 deubiquitination in TNF-α-induced necroptosis [49].
Also, TRAF2, as the other E3 ligase, is demonstrated to be one of the essential molecules in TNF-induced necrosis [50]. This effect is potentially based on TRAF2’s requirement to form complex I. FADD is a critical molecule for TNF-α-induced necroptosis [37], as well as the necroptotic process in several cellular processes which are not based on this cascade [51] (Figure 1). FADD plays a down-regulator role in the necroptosis process for the proliferative phase of the T-cell line [52]. The principal mechanism is based on various roles of FADD, which are explicitly unknown [53,54]. Therefore, although TRADD is essential for the necroptosis process, it may be based on the stimulation type. Complex II might trigger apoptosis and necroptosis as two downstream signaling pathways. While activated, caspase-8 dispossesses RIP1 of its role by splitting dynamic complex II into a pro-apoptosis complex. RIP1 interaction with caspase-8 leads to down-regulating RIP1 and the NF-κB pathway [55] and also shows an inhibitory effect on the necroptosis process [54]. Furthermore, the receptor-interacting protein 3 (RIP3) interface with caspase-8 suppresses RIP3’s capability to increase the caspase-independent cell death rate [56,57].
4. Myocardial Injuries and the Role of Necroptosis
Myocardial infarction is a cause of morbidity worldwide [58]. Despite the significant improvements, current therapies describe the central issue incompletely, derived from acute ischemia or infarction to the chronic type of heart failure. The considerable loss of cardiomyocytes with subsequent remodeling and contractile dysfunction is the leading cause of clinical presentations. Apoptosis, which is related to TNF-mediated death receptors [59] or fas receptor/CD95 [60], is reflected as a well-known molecular cascade of the described events [61]. TNF-α is up-regulated through myocardial infarction (MI) and increases the apoptosis rate [62]. Nevertheless, the overall rate of apoptosis ratio in the infarcted area is approximately 1% in research studies [63,64]. Necroptosis or “programmed necrosis” is suggested as another chief modulator of cell death in myocardial events [63]. This process is closely controlled by individual molecular pathways [65,66,67,68]. Conversely, several studies indicated that the TNF-α-mediated construction of the RIP1 and RIP3 complex is critical in prompting necroptosis [65,66,69]. RIP3 plays a significant role in this process as the regulatory molecule of the RIP1 phosphorylation process [65,69]. An essential function of RIP3-facilitated necroptosis is validated during hepatic viral infections and tissue damage due to inflammatory bowel disease [70]. In the myocardium, inhibition of RIP1 by the small molecule necrostatin-1 causes a decrease in the infarct size [71,72,73], where the efficient consequence of RIP3 on the heart is indistinctly unidentified [74]. RIP3-facilitated necroptosis also expressed in the myocardium, makes a complex with RIP1 in cardiomyocytes, activated by TNF stimulation. However, the RIP1/RIP3 complex inhibition via caspase 8 molecular cascade is suggested as a potential protective mechanism of necroptosis [74,75,76].
Necroptosis and apoptosis are significant severe cardiac pathological situations. While apoptotic signaling pathways are well-defined, the molecular mechanisms inducing cardiomyocyte necroptosis are not yet defined. RIP3 activates the necroptosis cascade by Ca2+-calmodulin-dependent protein kinase (CaMKII) activation. Thus, RIP3-induced triggering of CaMKII causes myocardial necroptosis through molecular events [77].
Permanent cardiac injury causes an emergent contest in dealing with ischemic heart diseases. Molecular chaperone heat shock protein 70 (HSP70) reduces heat-mediated autophagy-related apoptosis. Hence, autophagy may induce the necroptosis process [78].
As discussed above, necroptosis is revealed as a novel cell death model and may be crucial in heart disease. Therefore, specific microRNA (miRNA) and ring finger protein 11 (RNF11)-related cascades presume that oxidative stress, inflammation response, and the mitogen-activated protein kinase (MAPK) pathway may cause necroptosis. Stimulatingly, these molecules play a key role in cardiomyocyte necroptosis occurrence [79].
The discordant stimulation of cell death is strictly associated with the pathogenesis of cardiovascular diseases [80]. Necroptosis is one of the classic pathways for regulating necrosis and depends on the activity of RIP3 kinase. RIP1 substantially relates to RIP3, activating death receptors to produce necrosomes and filamentous amyloid protein complexes [81].
Thus, the mixed lineage kinase (MLKL) domain, by phosphorylation of activated RIP3, oligomerizes and translocates to cellular membranes, cooperating with their capability to preserve homeostasis [82]. The activation of necroptosis organizes a pathophysiological incident in myocardial injury [83,84]. However, whether or not cardiac microvascular endothelium injury is related to necroptosis in myocardial injury is not explicitly defined. PI3K-AKT-eNOS pathway activation up-regulates NO production and decreases necroptosis [85].
Necroptosis is a highly planned mechanism to induce cellular death by caspase activation in order to preserve the homeostasis balance [86]. Expressing the death molecular pathways receptors like Fas and TNF-α stimulates the apoptotic cellular pathway. Nevertheless, it is confirmed that death receptor activation can induce cell death as necrosis in several cell types, while the extrinsic apoptotic pathway is inactivated [87], indicating the existence of regulated necrosis. As defined in necroptosis pathways, RIP1’s serine/threonine kinase action has a key role in cell death. Whereas necrostatin-1, a tryptophan-based molecule, also plays a crucial part as the inhabitant [88,89,90]. This molecule-mediate block of TNF-α induces necrotic cell death by inhibiting RIP1 kinase activity [91,92].
5. COVID-19 and Necroptosis
The COVID-19 virus genome leads to the expression of nonstructural proteins, as an essential requirement for replication, and other proteins related to the receptors of the host cell and interacts with the ACE2 to allow the virus to enter. ACE inhibitors display vasoconstrictive, pro-inflammatory, prooxidative, and prothrombotic effects. SARS-CoV-2 probably controls the inactivation of eukaryotic Initiation Factor 2 (eIF2) by two of the three cellular eIF2 kinases, i.e., protein kinase R (PKR) and PKR-like endoplasmic reticulum kinase (PERK). Stimulatingly, PKR up-regulates the autophagy protein p62, related to Nrf2, a regulatory molecule of ACE2, for binding to KEAP1 and further promotes the autophagic degradation of KEAP1, leading to the stimulation of Nrf2 transcriptional activity. Consequently, Nrf2 may reduce the inflammatory process of viral infection. Also, the up-regulation of the Nrf2-transcriptional target heme oxygenase 1 (HO-1, gene name HMOX1) is related to an antiviral process like many viruses like influenza virus and respiratory syncytial virus (RSV). HO-1 can facilitate antiviral reactions by developing a heterodimeric complex. Thus, triggering the Nrf2/HO-1 pathway facilitates SARS-CoV-2 infection [93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141].
Nrf2 is a transcription factor for up-regulating the expressions of many anti-oxidative factors such as SODD and catalase; therefore, it plays a crucial role in maintaining cellular redox homeostasis. However, the level and activity of Nrf2 decrease with age. This statement illustrates the reason for older patients’ morbidity and mortality during COVID-19. Therefore, considering the Nrf2 activators may benefit the elderly who suffer from COVID-19. Well-known nitric oxide (NO), superoxide, and hydroxyl free radicals react with intra/extracellular molecules, cause protein deterioration and cell death, and lead to organ failure. Viral infection induces massive production of free radicals. The pathogenic roles of free radicals in viral infections are profound but overlooked. Free radicals’ damaging role in morbidity and mortality is not declared in several COVID-19 management guidelines. Inflammatory cytokine storms are reported in both COVID-19 and SARS, and the cytokine storm is the most frequently mentioned pathological theory in COVID-19. These inflammatory cytokines are proteins that act as signaling molecules to recruit immune cells to the site of inflammation, induce vascular leakage and exudation, and stimulate the production of free radicals and proteases. For example, IFNɤ, IL-1ẞ, IL-6, and TNF-α can stimulate NO synthesis. IL-2 is highly up-regulated in COVID-19 patients and is known to significantly stimulate producing NO in patients, while NO is the main mediator of IL-2-induced hypotension and vascular leak syndrome. IL-6 is another major inflammatory cytokine up-regulated in COVID-19 patients. IL-6 and TNF-α can provoke generating superoxide in neutrophils, while the production of IL-6 is stimulated by hydrogen peroxide. The inhibition of NO synthesis can decrease the IL-6 generation by more than 50%. The cytotoxicity effect of inflammatory cytokines can be blocked by lipid peroxidation inhibitors [142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157].
Intra/extracellular calcium exchange leads to the activation of calcineurin and its related molecule, NFAT. Intracellular production of NO via calcium-calmodulin-dependent NO synthase (NOS) is a well-determined mechanism in this pathway. Also, NO enhances the calcium-dependent activity of NFAT and promotes the phosphorylation of glycogen synthase kinase-3β (GSK-3β) [158].
Furthermore, vascular endothelial growth factor (VEGF) controls the NOS/NO pathway of endothelial progenitor cells (EPCs) by stimulating the calcineurin/NFAT signaling pathway [159].
NO overproduction acts as RNS, potent oxidants that initiate protein oxidation and peroxidation [160]. Recently, nitration has been displayed to provoke RIP1- and RIP3-related necroptosis cascade, with respiratory complex I [161], which contradicts the well-defined cytoprotective effects of NO, which reduce oxidative stress effects, mitochondrial damage, and tissue infarction [162]. Nitrites also converse protection against ischemia-reperfusion injuries, affecting one of the main intracellular molecules and signal transducers in the myocardium [163].
6. Conclusive Statement
Myocardial injury caused by SARS-CoV-2 infection is a critical event in the pathogenesis of COVID-19. It occurs due to myocardial inflammation caused by ACE-2 and SARS-CoV-2 association, in which ACE-2 down-regulation results in an inflammatory cytokine storm and increases TNF- activation, which results in myocardial damage, necroptosis, and apoptosis. This necroptosis-initiating factor appears to have a role in the pathogenesis of myocardial infarction. TNF-a can cause neutrophils to produce superoxide and induce NO production in the body. The intracellular generation of NO by NOS, dependent on calcium and calmodulin, is a well-established process. NO increases the calcium-dependent activity of the nuclear factor of activated T cells (NFAT).
Additionally, the exchange of intracellular calcium for external calcium activates calcineurin and its related protein, NFAT. Nitration activates the RIP1 necroptosis pathway associated with respiratory complex I. The protective effects of nitrates in the myocardial and activation of the PI3K-AKT-eNOS pathway enhances NO generation and necroptosis decrease in the myocardium.
NO also activates the necrosis molecular pathway by attaching to a portion of the TNFR1 molecule, which activates RIP1, which is a significant participant in cell death during necroptosis. Additionally, NO activates the NF-B, which Tpl-2 also activates. Aside from that, calcineurin regulates Tpl-2-mediated NFAT and is blocked by Bcl-2 or Bcl-XL, respectively. In contrast, the NFAT pathway is dependent on JNK signals; Bcl-2 overexpression produces a decrease in FasL, which triggers apoptosis and inhibits the NFAT pathway, whereas RIP3 activation is responsible for the activation of apoptotic signaling pathways in cardiomyocytes, which are also responsible for the molecular processes of cardiomyocyte necroptosis. Thus, in combination with DAG, which is required for NFAT stimulation, calcium-regulated calcineurin contributes to SRF activation to a limited extent. NFAT depends on signals from DAG effectors such as protein kinase C, Ras, and Rap1 and signals from downstream. Finally, it should be emphasized that inhibiting this intrinsic molecular process can prevent the necroptosis of cardiomyocytes from occurring in some cases.
Author Contributions
Mohammadjavad Sotoudeheian: Conceptualization, Supervision, Writing – review & editing, Writing – original draft Reviewing the literature, Methodology, Investigation, Writing, review & editing, Designing the figure; Mohammad Soleimani: Writing, Methodology, Investigation; Navid Farahmandian and Seyed-Mohamad-Sadegh Mirahmadi: Methodology, Investigation, Data curation, Editing, Designing the figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.
Acknowledgments
Thanks to Dr. Soroush Nematollahi, Dr. Mohsen Hassanzadeh and Dr. Mohammadamin Mofleh for their helpful comments. Thanks also to the researchers from different areas of science for all their work undertaken as the basis for constructing this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dong, Ensheng, Hongru Du, and Lauren Gardner. “An interactive web-based dashboard to track COVID-19 in real time” The Lancet infectious diseases 20, no. 5 (2020): 533-534.
- Babapoor-Farrokhran, Savalan, Deanna Gill, Jackson Walker, Roozbeh Tarighati Rasekhi, Behnam Bozorgnia, and Aman Amanullah. “Myocardial injury and COVID-19: Possible mechanisms” Life Sciences (2020): 117723.
- Bonow, Robert O., Gregg C. Fonarow, Patrick T. O’Gara, and Clyde W. Yancy. “Association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality”JAMA cardiology (2020).
- Shi S, Qin M, Shen B, et al. association of cardiac injury with mortality in hospitalized patients with COVID-19 inWuhan, China. JAMA Cardiol. Published online March 25, 2020.
- Guo T, Fan Y, Chen M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. Published online March 27, 2020. [CrossRef]
- Yang C, Jin Z. An acute respiratory infection runs into the most common noncommunicable epidemic—COVID-19 and cardiovascular diseases. JAMA Cardiol. Published online March 25, 2020. [CrossRef]
- M.A. Crackower, R. Sarao, G.Y. Oudit, C. Yagil, I. Kozieradzki, S.E. Scanga, A.J. Oliveira-dos-Santos, J. da Costa, L. Zhang, Y. Pei, J. Scholey, C.M. Ferrario, A.S. Manoukian, M.C. Chappell, P.H. Backx, Y. Yagil, J.M. Penninger, Angiotensinconverting enzyme 2 is an essential regulator of heart function, Nature 417 (2002) 822–828. [CrossRef]
- K. Yamamoto, M. Ohishi, T. Katsuya, N. Ito, M. Ikushima, M. Kaibe, Y. Tatara, A. Shiota, S. Sugano, S. Takeda, H. Rakugi, T. Ogihara, Deletion of angiotensinconverting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II, Hypertension 47 (2006) 718–726. [CrossRef]
- G.Y. Oudit, Z. Kassiri, C. Jiang, P.P. Liu, S.M. Poutanen, J.M. Penninger, J. Butany, SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS, Eur. J. Clin. Investig. 39 (2009) 618–625. [CrossRef]
- G.Y. Oudit, Z. Kassiri, M.P. Patel, M. Chappell, J. Butany, P.H. Backx, R.G. Tsushima, J.W. Scholey, R. Khokha, J.M. Penninger, Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice, Cardiovasc. Res. 75 (2007) 29–39. [CrossRef]
- Zheng, Ying-Ying, Yi-Tong Ma, Jin-Ying Zhang, and Xiang Xie. “COVID-19 and the cardiovascular system” Nature Reviews Cardiology 17, no. 5 (2020): 259-260.
- Ahmad, Sheikh Fayaz, Mushtaq Ahmad Ansari, Khairy MA Zoheir, Saleh A. Bakheet, Hesham M. Korashy, Ahmed Nadeem, Abdelkader E. Ashour, and Sabry M. Attia. “Regulation of TNF-α and NF-κB activation through the JAK/STAT signaling pathway downstream of histamine 4 receptor in a rat model of LPS-induced joint inflammation” Immunobiology 220, no. 7 (2015): 889-898.
- AbdelMassiha, Antoine Fakhry, David Ramzyc, Lauren Nathanc, Silvia Azizc, Mirette Ashrafc, Nourhan Hatem Youssefc, Nouran Hafezc, Rana Saeedc, and Hala Aghaa. “Possible molecular and paracrine involvement underlying the pathogenesis of COVID-19 cardiovascular complications” pathogenesis 2, no. 8 (2020): 10-15. [CrossRef]
- Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/ reperfusion dysfunction, and heart failure. J Mol Cell Cardiol 2002; 34:1443–1453. [CrossRef]
- del Arco, Pablo Gómez, Sara Martı́nez-Martı́nez, Janet Lynn Maldonado, Inmaculada Ortega-Pérez, and Juan Miguel Redondo. “A role for the p38 MAP kinase pathway in the nuclear shuttling of NFATp.” Journal of Biological Chemistry 275, no. 18 (2000): 13872-13878.
- Haylett, Romney S., Norbert Koch, and Lothar Rink. “MHC class II molecules activate NFAT and the ERK group of MAPK through distinct signaling pathways in B cells” European journal of immunology 39, no. 7 (2009): 1947-1955.
- 10 Ho, A. M., Jain, J., Rao, A. and Hogan, P. G., Expression of the transcription factor NFATp in a neuronal cell line and in the murine nervous system. J. Biol. Chem. 1994. 269: 28181–28186. [CrossRef]
- Yaseen, N. R., Maizel, A. L., Wang, F. and Sharma, S., Comparative analysis of NFAT (Nuclear factor of activated T cells) complex in human T and B lymphocytes. J. Biol. Chem. 1993. 268: 14285–14293. [CrossRef]
- Aramburu, J., Azzoni, L., Rao, A. and Perussia, B., Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: regulation upon CD16 ligand binding. J. Exp. Med. 1995. 182: 801–810. [CrossRef]
- Hutchinson, L. E. and McCloskey, M. A., Fc epsilon RI-mediated induction of nuclear factor of activated T-cells. J. Biol. Chem. 1995. 270: 16333–16338.
- Shaw, J. P. , Utz, P. J., Durand, D. B., Toole, J. J., Emmel, E. A. and Crabtree, G. R., Identification of a putative regulator of early T cell activation genes. Science 1988. 241: 202–205. [CrossRef]
- Winslow, M. M. , Gallo, E. M., Neilson, J. R. and Crabtree, G. R., The calcineurin phosphatase complex modulates immunogenic B cell responses. Immunity 2006. 24: 141–152. [CrossRef]
- Barrington, R. A. , Borde, M. Rao, A., Caroll, M. C., Involvement of NFAT1 in B cell self-tolerance. J. Immunol. 2006. 177: 1510–1515.
- Peng, S. L. , Gerth, A. J., Ranger, A. M. and Glimcher, L. H., NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 2001. 14: 13–20. [CrossRef]
- Tsatsanis, Christos, Christos Patriotis, and Philip N. Tsichlis. “Tpl-2 induces IL-2 expression in T-cell lines by triggering multiple signaling pathways that activate NFAT and NF-κB” Oncogene 17, no. 20 (1998): 2609-2618.
- Srivastava, Rakesh K., Carl Y. Sasaki, J. Marie Hardwick, and Dan L. Longo. “Bcl-2–mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription” The Journal of experimental medicine 190, no. 2 (1999): 253-266.
- Biswas, Rajat S., Hyuk J. Cha, J. Marie Hardwick, and Rakesh K. Srivastava. “Inhibition of drug-induced Fas ligand transcription and apoptosis by Bcl-XL” Molecular and cellular biochemistry 225, no. 1-2 (2001): 7-20.
- Hao, Shengli, Tomohiro Kurosaki, and Avery August. “Differential regulation of NFAT and SRF by the B cell receptor via a PLCγ–Ca2+-dependent pathway” The EMBO Journal22, no. 16 (2003): 4166-4177.
- Fiers W, Beyaert R, Boone E, et al. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J Inflamm 1995;47:67–75.
- Kalai M, Van Loo G, Vanden Berghe T, et al. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsDNA. Cell Death Differ 2002;9:981–94.
- Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008;135:1311–23. [CrossRef]
- Chen G, GoeddelDV. TNF-R1 signaling: a beautiful pathway. Science2002;296:1634–5. [CrossRef]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 1975;72:3666–70. [CrossRef]
- Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptotic and necrotic forms of cell lysis. J Immunol 1988;141:2629–34.
- Andera, L. Signaling activated by the death receptors of the TNFR family. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2009;153:173–80.
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000;288:2351–4. [CrossRef]
- Wertz IE, Dixit VM. Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev 2008;19:313–24. [CrossRef]
- Mahoney DJ, Cheung HH, Mrad RL, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA 2008;105:11778–83.
- Varfolomeev E, Goncharov T, Fedorova AV, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha(TNFalpha)- induced NF-kappaB activation. J Biol Chem 2008;283:24295–9.
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D,Courtois G. The tumor suppressor CYLD negatively regulates NF-κB signaling pathway by deubiquitination. Nature 2003;424:801–5.
- Alameda JP, Moreno-Maldonado R, Navarro M, et al. An inactivating CYLD mutation promotes skin tumor progression by conferring enhanced proliferative, survival and angiogenic properties to epidermal cancer cells. Oncogene 2010;29:6522–32. [CrossRef]
- Urbanik T, Kohler BC, Boger RJ, et al. Down-regulation of CYLD as a trigger for NF-k activation and a mechanism of apoptotic resistance in hepatocellular carcinoma cells. Int J Oncol 2011;38:121–31.
- Wertz IE, O’Rourke KM, Zhou H, et al. deubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004;430:694–9.
- Shembade N, Ma A, Harhaj EW. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010;327:1135–9.
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNF requires site-specific ubiquitination of RIP1 and polyubiquitin by NEMO. Mol Cell 2006;22:245–57.
- Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-κB. Cell Death Differ 2010;17:482–7.
- Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor--induced IkB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem 2004;279:33185–91.
- Schneider-BrachertW, Tchikov V, Merkel O, et al. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J Clin Invest 2006;116:2901–13.
- Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promotes RIP1 ubiquitination. Mol Cell 2008;30:689–700. [CrossRef]
- Lin Y, Choksi S, Shen HM, et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem 2004;279:10822–8. [CrossRef]
- Chan FK, Shisler J, Bixby JG, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 2003;278:51613–21. [CrossRef]
- Osborn SL, Diehl G, Han SJ, et al. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci USA 2010;107:13034–9. [CrossRef]
- Ermolaeva MA, Michallet MC, Papadopoulou N, et al. Function of TRADD in tumor necrosis factor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol 2008;9:1037–46. [CrossRef]
- Wang L, Du F, Wang X. TNF-alpha induced two distinct caspase-8 activation pathways. Cell 2008;133:693–703. [CrossRef]
- Kim JW, Choi EJ, Joe CO. Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene 2000;19:4491–9. [CrossRef]
- Feng S, Yang Y, Mei Y, et al. Cleavage of RIP3 inactivates its caspaseindependent apoptosis pathway by removal of kinase domain. Cell Signal 2007;19:2056–67. [CrossRef]
- Wu, Wei, Peng Liu, and Jianyong Li. “Necroptosis: an emerging form of programmed cell death” Critical reviews in oncology/hematology 82, no. 3 (2012): 249-258.
- World Health Organization. The top 10 causes of death. Fact sheet N8 310, 2014.
- Sugano M, Koyanagi M, Tsuchida K, Hata T, Makino N. In vivo gene transfer of soluble TNF-alpha receptor 1 alleviates myocardial infarction. FASEB J 2002;16:1421–1422. [CrossRef]
- Wollert KC, Heineke J, Westermann J, Ludde M, Fiedler B, Zierhut W, Laurent D, Bauer MK, Schulze-Osthoff K, Drexler H. The cardiac Fas (APO-1/CD95) Receptor/ Fas ligand system: relation to diastolic wall stress in volume-overload hypertrophy in vivo and activation of the transcription factor AP-1 in cardiac myocytes. Circulation 2000;101:1172–1178.
- Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest 1997;100:1363–1372. [CrossRef]
- Sun M, DawoodF,WenWH,Chen M, Dixon I, KirshenbaumLA, Liu PP. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation 2004;110:3221–3228. [CrossRef]
- Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart. Circ Res 2011;108:1017–1036.
- Dorn GW, II. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res 2009;81:465–473.
- Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell 2009;138:229–232. [CrossRef]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137: 1100–1111. [CrossRef]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009;325:332–336. [CrossRef]
- Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 2010;22:263–268. [CrossRef]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylationdriven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009;137:1112–1123. [CrossRef]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011;477:330–334. [CrossRef]
- Lim SY, Davidson SM, Mocanu MM, YellonDM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther 2007;21:467–469. [CrossRef]
- Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, Sluijter JP. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol 2012;107:270. [CrossRef]
- Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent. Cardiovasc Drugs Ther 2007;21:227–233. [CrossRef]
- Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovascular research. 2014;103(2):206-16. [CrossRef]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011;471:368–372. [CrossRef]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011;471:363–367.
- Zhang, Ting, Yan Zhang, Mingyao Cui, Li Jin, Yimei Wang, Fengxiang Lv, Yuli Liu et al.””” ““CaMKII is a RIP3 substrate mediating ischemia-and oxidative stress–induced myocardial necroptosis” Nature medicine 22, no. 2 (2016): 175-182.
- Liu, Xiaojuan, Chao Zhang, Chi Zhang, Jingjing Li, Wanwan Guo, Daliang Yan, Chen Yang et al.””” ““Heat shock protein 70 inhibits cardiomyocyte necroptosis through repressing autophagy in myocardial ischemia/reperfusion injury” In Vitro Cellular & Developmental Biology-Animal 52, no. 6 (2016): 690-698.
- Yang, Tianshu, Changyu Cao, Jie Yang, Tianqi Liu, Xin Gen Lei, Ziwei Zhang, and Shiwen Xu. “miR-200a-5p regulates myocardial necroptosis induced by Se deficiency via targeting RNF11” Redox biology 15 (2018): 159-169.
- Zhou, Z. , Zhang, Y., Lin, L., Zhou, J., 2018a. Apigenin suppresses the apoptosis of H9C2 rat cardiomyocytes subjected to myocardial ischemia-reperfusion injury via upregulation of the PI3K/Akt pathway. Mol. Med. Rep. 18 (2), 1560–1570. [CrossRef]
- Zhu, P. , Hu, S., Jin, Q., Li, D., Tian, F., Toan, S., Li, Y., Zhou, H., Chen, Y., 2018. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox. Biol. 16, 157–168. [CrossRef]
- Dovey, C.M. , Diep, J., Clarke, B.P., Hale, A.T., McNamara, D.E., Guo, H., Brown Jr., N.W., Cao, J.Y., Grace, C.R., Gough, P.J., Bertin, J., Dixon, S.J., Fiedler, D., Mocarski, E.S.,Kaiser, W.J., Moldoveanu, T., York, J.D., Carette, J.E., 2018. MLKL requires the inositol phosphate code to execute necroptosis. Mol. Cell. 70 (5), 936–948.
- Dmitriev, Y. ,.V., Minasian, S.M., Demchenko, E.A., Galagudza, M.M., 2013. Study of cardioprotective effects of necroptosis inhibitors on isolated rat heart subjected to global ischemia-reperfusion. Bull. Exp. Biol. Med. 155 (2), 245–248.
- Koshinuma, S. , Miyamae, M., Kaneda, K., Kotani, J., Figueredo, V.M., 2014. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 28 (2), 235–241. [CrossRef]
- Bai, Jiannan, Qingchao Wang, Jiaxin Qi, Hongqiang Yu, Cong Wang, Xiaowei Wang, Yanru Ren, and Fude Yang. “Promoting effect of baicalin on nitric oxide production in CMECs via activating the PI3K-AKT-eNOS pathway attenuates myocardial ischemia–reperfusion injury” Phytomedicine 63 (2019): 153035.
- Smith CA, Williams GT, Kingston R, Jenkinson EJ, Owen JJ. Apoptosis. Nature. 1989;338:10.
- Kitanaka C, Kuchino Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999;6:508-15. [CrossRef]
- Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229-32. [CrossRef]
- Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012;3:e320. [CrossRef]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112-9.
- Degterev A, Hitomi J, Germscheid M, ”” Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313-21.
- Koshinuma, Shizuka, Masami Miyamae, Kazuhiro Kaneda, Junichiro Kotani, and Vincent M. Figueredo. “Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia–reperfusion injury” Journal of anesthesia 28, no. 2 (2014): 235-241.
- Cuadrado, Antonio, Marta Pajares, Cristina Benito, José Jiménez-Villegas, Maribel Escoll, Raquel Fernández-Ginés, Angel J. Garcia Yagüe et al. “Can activation of NRF2 be a strategy against COVID-19?.” Trends in Pharmacological Sciences (2020). [CrossRef]
- Venugopal, R. and Jaiswal, AK (1998) Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 17 (24), 3145-56.
- Cuadrado, A. et al. (2019)Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18 (4), 295-317. [CrossRef]
- Cuadrado, A. et al. (2018) Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol Rev 70 (2), 348-383. [CrossRef]
- Hayes, J.D. and Dinkova-Kostova, A.T. (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39 (4), 199-218. [CrossRef]
- Dinkova-Kostova, A.T. et al. (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 99 (18), 11908-13. [CrossRef]
- Rabbani, P.S. et al. (2019) Dysregulation of Nrf2/Keap1 Redox Pathway in Diabetes Affects Multipotency of Stromal Cells. Diabetes 68 (1), 141-155.
- Schmidlin, C.J. et al. (2019) Redox regulation by NRF2 in aging and disease. Free Radic Biol Med 134, 702-707. [CrossRef]
- Wu, C. et al. (2020) Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. [CrossRef]
- Liu, Q. et al. (2019) Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid Med Cell Longev 2019, 7090534. [CrossRef]
- Acosta-Herrera, M. et al. (2015) Common variants of NFE2L2 gene predisposes to acute respiratory distress syndrome in patients with severe sepsis. Crit Care 19, 256. [CrossRef]
- Hua, C.C. et al. (2010) Functional haplotypes in the promoter region of transcription factor Nrf2 in chronic obstructive pulmonary disease. Dis Markers 28 (3), 185-93.
- Kobayashi, E.H. et al. (2016) Nrf2 suppresses macrophage inflammatory response by blocking pro-inflammatory cytokine transcription. Nat Commun 7, 11624.
- Mills, E.L. et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556 (7699), 113-117.
- Ishii, T. and Mann, G.E. (2014) Redox status in mammalian cells and stem cells during culture in vitro: critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox Biol 2, 786-94. [CrossRef]
- Harvey, C.J. et al. (2011) Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med 3 (78), 78ra32. [CrossRef]
- Seelige, R. et al. (2017) The ancient cytokine IL-17D is regulated by Nrf2 and mediates tumor and virus surveillance. Cytokine 91, 10-12. [CrossRef]
- Saddawi-Konefka, R. et al. (2016) Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep 16 (9), 2348-58. [CrossRef]
- Brune, B. et al. (2013) Redox control of inflammation in macrophages. Antioxid Redox Signal 19 (6), 595-637. [CrossRef]
- Zhu, N. et al. (2020) A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 382 (8), 727-733.
- Chen, Y. et al. (2020) Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. [CrossRef]
- Lopes, R.D. et al. (2020) Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARSCoV- 2)--The BRACE CORONA Trial. Am Heart J 226, 49-59. [CrossRef]
- Mehta, P.K. and Griendling, K.K. (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292 (1), C82-97. [CrossRef]
- Magrone, T. et al. (2020) Focus on Receptors for Coronaviruses with Special Reference to Angiotensin-converting Enzyme 2 as a Potential Drug Target – A Perspective. Endocr Metab Immune Disord Drug Targets.
- Zhao, S. et al. (2018) Nrf2 Deficiency Up-regulates Intrarenal Angiotensin- Converting Enzyme-2 and Angiotensin 1-7 Receptor Expression and Attenuates Hypertension and Nephropathy in Diabetic Mice. Endocrinology 159 (2), 836-852.
- Nakagawa, K. et al. (2016) Viral and Cellular mRNA Translation in Coronavirus- Infected Cells. Adv Virus Res 96, 165-192.
- Krahling, V. et al. (2009) Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J Virol 83 (5), 2298-309. [CrossRef]
- Komatsu, M. et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12 (3), 213-23. [CrossRef]
- Taguchi, K. et al. (2012) Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A 109 (34), 13561-6. [CrossRef]
- Chan, C.P. et al. (2006) Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol 80 (18), 9279-87. [CrossRef]
- Cullinan, S.B. et al. (2003) Nrf2 is a direct PERK substrate and effector of PERKdependent cell survival. Mol Cell Biol 23 (20), 7198-209. [CrossRef]
- Loo, Y.M. et al. (2008) Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82 (1), 335-45. [CrossRef]
- Burdette, D.L. et al. (2011) STING is a direct innate immune sensor of cyclic di- GMP. Nature 478 (7370), 515-8. [CrossRef]
- Sun, L. et al. (2012) Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7 (2), e30802. [CrossRef]
- Chen, X. et al. (2014) SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 5 (5), 369-81. [CrossRef]
- Acharya, D. et al. (2020) Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol. [CrossRef]
- Olagnier, D. et al. (2017) Activation of Nrf2 Signaling Augments Vesicular Stomatitis Virus Oncolysis via Autophagy-Driven Suppression of Antiviral Immunity. Mol Ther 25 (8), 1900-1916. [CrossRef]
- Olagnier, D. et al. (2018) Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun 9 (1), 3506. [CrossRef]
- Espinoza, J.A. et al. (2017) Modulation of Antiviral Immunity by Heme Oxygenase- 1. Am J Pathol 187 (3), 487-493. [CrossRef]
- Koliaraki, V. and Kollias, G. (2011) A new role for myeloid HO-1 in the innate to adaptive crosstalk and immune homeostasis. Adv Exp Med Biol 780, 101-11.
- Bafna, K. et al. (2020) Structural Similarity of SARS-CoV2 M(pro) and HCV NS3/4A Proteases Suggests New Approaches for Identifying Existing Drugs Useful as COVID-19 Therapeutics. ChemRxiv.
- Baez-Santos, Y.M. et al. (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115, 21-38.
- Zhu, Z. et al. (2010) Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology 52 (6), 1897-905.
- Fillebeen, C. et al. (2005) Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C Virus. J Biol Chem 280 (10), 9049-57. [CrossRef]
- Fillebeen, C. and Pantopoulos, K. (2010) Iron inhibits replication of infectious hepatitis C virus in permissive Huh7.5.1 cells. J Hepatol 53 (6), 995-9.
- Gao, Y. et al. (2020) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. [CrossRef]
- Tung, W.H. et al. (2011) Enterovirus 71 induces integrin beta1/EGFR-Rac1- dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. J Cell Physiol 226 (12), 3316-29.
- Ma, Z. et al. (2017) Carbon monoxide and biliverdin suppress bovine viral diarrhea virus replication. J Gen Virol 98 (12), 2982-2992.
- Kalyanaraman, H. et al. (2018) Protein Kinase G Activation Reverses Oxidative Stress and Restores Osteoblast Function and Bone Formation in Male Mice With Type 1 Diabetes. Diabetes 67 (4), 607-623. [CrossRef]
- Wu, Jun. “Tackle the free radicals damage in COVID-19” Nitric Oxide (2020).
- T. Akaike, M. Suga, H. Maeda, Free radicals in viral pathogenesis: molecular mechanisms involving superoxide and NO, Proc. Soc. Exp. Biol. Med. 217 (1) (1998) 64–73.
- D. Sun, et al., Clinical Features of Severe Pediatric Patients with Coronavirus Disease 2019 in Wuhan: a Single ”” Center’s Observational Study, World J Pediatr, 2020. [CrossRef]
- E. Prompetchara, C. Ketloy, T. Palaga, Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic, Asian Pac. J. Allergy Immunol. 38 (1) (2020) 1–9. [CrossRef]
- Y. Li, et al., Retrospective analysis of laboratory testing in 54 patients with severeor critical-type 2019 novel coronavirus pneumonia, Lab Invest. 100 (6) (2020) 794–800.
- K.J. Huang, et al., An interferon-gamma-related cytokine storm in SARS patients, J. Med. Virol. 75 (2) (2005) 185–194.
- A.R. Vaz, et al., Pro-inflammatory cytokines intensify the activation of NO/NOS, JNK1/2 and caspase cascades in immature neurons exposed to elevated levels of unconjugated bilirubin, Exp. Neurol. 229 (2) (2011) 381–390.
- J.B. Hibbs Jr.et al., Evidence for cytokine-inducible nitric oxide synthesis from Larginine in patients receiving interleukin-2 therapy, J. Clin. Invest. 89 (3) (1992) 867–877. [CrossRef]
- W.E. Samlowski, et al., Endothelial nitric oxide synthase is a key mediator of interleukin- 2-induced hypotension and vascular leak syndrome, J. Immunother. 34 (5) (2011) 419–427. [CrossRef]
- M. Tsujimoto, et al., Tumor necrosis factor provokes superoxide anion generation from neutrophils, Biochem. Biophys. Res. Commun. 137 (3) (1986) 1094–1100. [CrossRef]
- A. Kharazmi, et al., Interleukin 6 primes human neutrophil and monocyte oxidative burst response, Immunol. Lett. 21 (2) (1989) 177–184. [CrossRef]
- J.T. Colston, B. Chandrasekar, G.L. Freeman, A novel peroxide-induced calcium transient regulates interleukin-6 expression in cardiac-derived fibroblasts, J. Biol. Chem. 277 (26) (2002) 23477–23483. [CrossRef]
- R.A. Willis, et al., induction of nitric oxide synthase in subsets of murine pulmonary fibroblasts: effect on fibroblast interleukin-6 production, Clin. Immunol. Immunopathol. 71 (2) (1994) 231–239. [CrossRef]
- A. Rabinovitch, et al., Cytotoxic effects of cytokines on rat islets: evidence for involvement of free radicals and lipid peroxidation, Diabetologia 35 (5) (1992) 409–413. [CrossRef]
- N. Robledinos-Anton, et al., Activators and inhibitors of NRF2: a review of their potential for clinical development, 2019 Oxid Med Cell Longev, 2019, p. 9372182. [CrossRef]
- L. Zhou, et al., Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells, Redox. Biol. 14 (2018) 35–40. [CrossRef]
- Drenning, Jason A., Vitor A. Lira, Catherine G. Simmons, Quinlyn A. Soltow, Jeff E. Sellman, and David S. Criswell. “Nitric oxide facilitates NFAT-dependent transcription in mouse myotubes” American Journal of Physiology-Cell Physiology294, no. 4 (2008): C1088-C1095.
- Yang, Long, Hongshan Guan, Jionghong He, Liqun Zeng, Zhengqiang Yuan, Min Xu, Wenhui Zhang, Xiaole Wu, and Jing Guan. “VEGF increases the proliferative capacity and eNOS/NO levels of endothelial progenitor cells through the calcineurin/NFAT signalling pathway” Cell biology international 36, no. 1 (2012): 21-27.
- Tien, M. , Berlett, B. S., Levine, R. L., Chock, P. B. & Stadtman, E. R. Peroxynitrite-mediated modification of proteins at physiological carbon dioxide concentration: pH dependence of carbonyl formation, tyrosine nitration, and methionine oxidation. Proc. Natl Acad. Sci. USA 96, 7809–7814 (1999). [CrossRef]
- Davis, C. W. et al. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic. Biol. Med. 48, 306–317 (2010). [CrossRef]
- Cauwels, A. et al. Nitrite protects against morbidity and mortality associated with TNF- or LPS-induced shock in a soluble guanylate cyclase-dependent manner. J. Exp. Med. 206, 2915–2924 (2009). [CrossRef]
- Vandenabeele, Peter, Lorenzo Galluzzi, Tom Vanden Berghe, and Guido Kroemer. “Molecular mechanisms of necroptosis: an ordered cellular explosion” Nature reviews Molecular cell biology 11, no. 10 (2010): 700-714.
Figure 1.
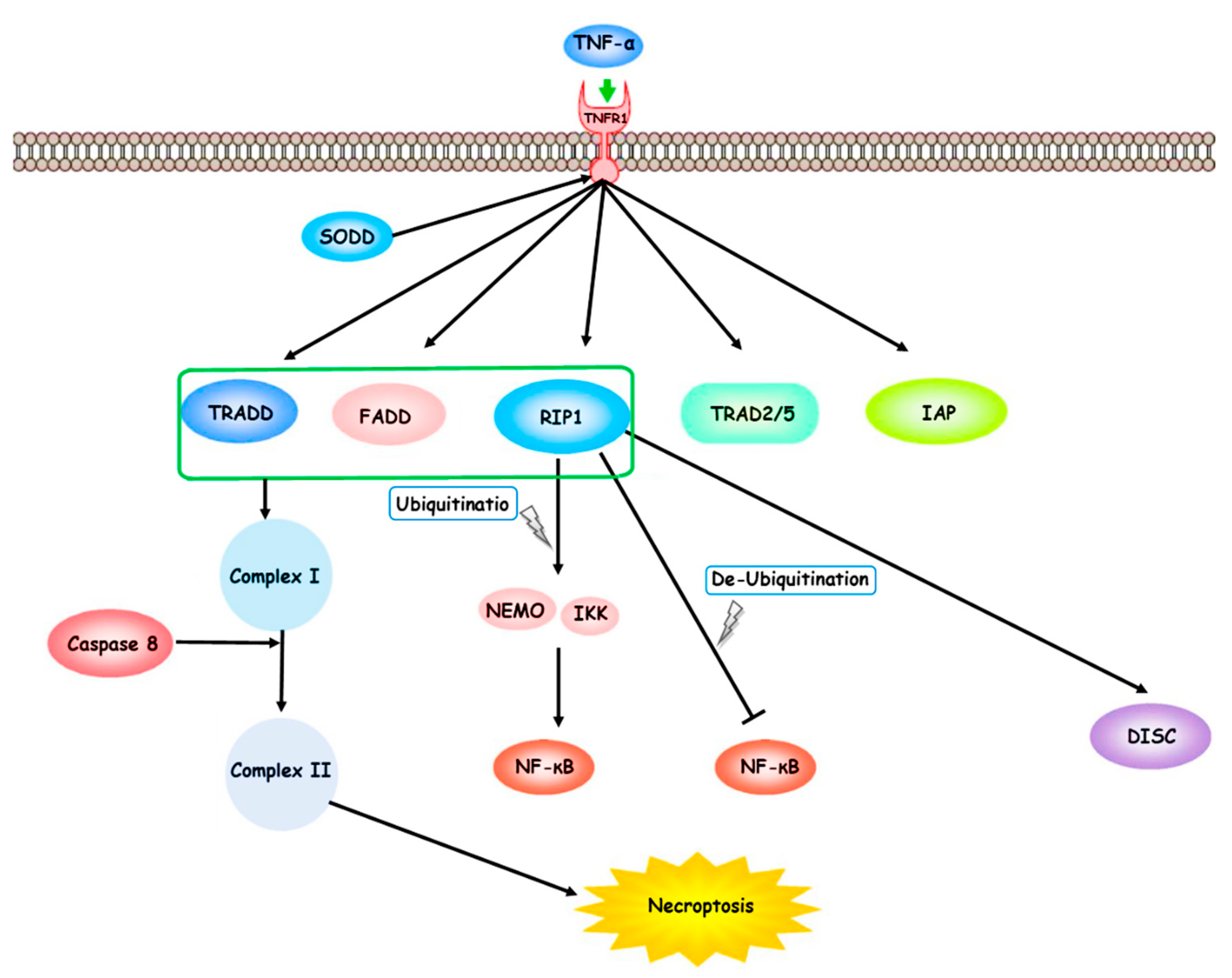
TNF-α, produced by activated macrophages and binds to the extracellular part of TNFR1. TNF-α binder molecules such as SODD trigger TNFR1 and TNFR2. The TNF-α/TNFR-mediated pathways lead to the necroptosis process. Consequently, the downstream signaling cascade forms complex I with TRADD, TRAF2/5, FADD, RIP1, and IAPs. The ubiquitination state of RIP1 promotes cell death. The ubiquitination process of RIP1 activates the IKK complex and NEMO. Therefore, it may up-regulate the NF-κB molecular pathway. RIP1 deubiquitination may inhibit NF-κB molecular pathway and cause a disposition in the cell death pathways program. The aggregation of TRADD, FADD, RIP1, and caspase-8 forms complex II. TNF receptor (TNFR) 1, receptor-interacting protein 1 (RIP1), silencer of death domains (SODD), TNF-α receptor-associated death domain (TRADD), TNF-α receptor-associated factor 2/5 (TRAF2/5), Fas-associated death domain (FADD), receptor-interacting protein 1 (RIP1), and inhibitor of apoptosis proteins (IAPs), death-inducing signaling complex (DISC).
Figure 1.
TNF-α, produced by activated macrophages and binds to the extracellular part of TNFR1. TNF-α binder molecules such as SODD trigger TNFR1 and TNFR2. The TNF-α/TNFR-mediated pathways lead to the necroptosis process. Consequently, the downstream signaling cascade forms complex I with TRADD, TRAF2/5, FADD, RIP1, and IAPs. The ubiquitination state of RIP1 promotes cell death. The ubiquitination process of RIP1 activates the IKK complex and NEMO. Therefore, it may up-regulate the NF-κB molecular pathway. RIP1 deubiquitination may inhibit NF-κB molecular pathway and cause a disposition in the cell death pathways program. The aggregation of TRADD, FADD, RIP1, and caspase-8 forms complex II. TNF receptor (TNFR) 1, receptor-interacting protein 1 (RIP1), silencer of death domains (SODD), TNF-α receptor-associated death domain (TRADD), TNF-α receptor-associated factor 2/5 (TRAF2/5), Fas-associated death domain (FADD), receptor-interacting protein 1 (RIP1), and inhibitor of apoptosis proteins (IAPs), death-inducing signaling complex (DISC).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



