
Submitted:
25 April 2023
Posted:
26 April 2023
You are already at the latest version
Abstract
The use of spirocycles in drug discovery and medicinal chemistry has been booming in the last two decades. This has clearly translated in the landscape of approved drugs. Among two dozen clinically used medicines containing a spirocycle, 50% have been approved in the 21st century. The present review focuses on the notable synthetic routes to such drugs invented in industry and academia and is intended to serve as a useful reference source of synthetic as well as general drug information for researchers engaging in the design of new spirocyclic scaffolds for medicinal use or embarking upon analog syntheses inspired by the existing approved drugs.
Keywords:
Spirocycles
; three-dimensional
; high-Fsp3
; spirocyclizations
; cyclodehydration
; ketalization
; intramolecular Claisen condensation
; intramolecular alkylation
; approved drugs
In memory of Prof. Mikhail Krasavin
In commemoration of the 300th anniversary of St Petersburg State University’s founding
1. Introduction
Spirocycles are becoming increasingly popular in drug discovery. Inherently three-dimensional, these high-Fsp3 (Fsp3 = number of sp3 hybridized carbons/total carbon count) motifs are expected to rid drug candidate molecules of many liabilities which would be inevitably countered with the formerly typical scaffolds built on flat aromatic carbo- and heterocycles. Hence it is perhaps unsurprising that the trend (articulated for the first time in the ‘escape from the flatland’ publication by Lovering and colleagues [1]) has now translated itself in an increasing number of approved drugs containing spirocyclic motifs. We and the colleagues recently published a review article on the use of spirocycles in drug discovery [2]. In this review, we partly touched upon the syntheses of approved spirocyclic drugs. However, that overview was far from exhaustive. Moreover, it only episodically touched upon approved spirocyclic drugs, also focusing on advanced, though not approved, drug candidates. Considering the apparent interest to the subject from the research community (at the time of writing this review, publication [2] was cited over 100 times in 1.5 years since its appearance in the literature), we thought it prudent to compile a more comprehensive compendium of synthetic routes to approved drugs containing a spirocyclic motif with a particular emphasis on alternative synthetic approaches to the same drug molecule if such exist. The present review is the result of realizing that intent. The review covers 23 drug molecules approved for medical use over the period of 1959 to 2021.
2. Griseofulvin
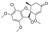
Griseofulvin is an antifungal agent used to treat fungal infections of the fingernails and toes. It is one of the earliest spirocyclic drugs which was approved in 1959. The exact mechanism of its action remains unclear with putative targets including tubulin beta chain and keratin, type I cytoskeletal 12 through which griseofulvin interferes with fungal mitosis.
Two total syntheses of griseofulvin were accomplished in the 1990s. Yamoto and co-workers reported the synthesis of racemic griseofulfin in 1990 [3]. The synthesis commenced with 7-chloro-4,6-dimethoxy-3(2H)-benzofuranone (1) which was condensed with acetaldehyde diethyl acetal in the presence of Ti(IV) chloride to give (Z)-configured ethylidene derivative 2. In order to bring the olefin configuration to the desired (E), compound 2 was subjected to somewhat low-yielding photochemical isomerization. The (E)-configured olefin 3 was now set for a Diels-Alder reaction with diene 4 (10 equiv.) which was accomplished in refluxing toluene to furnish racemic griseofulvin in 46% yield (Scheme 1).
Pirrung and co-workers reported the first asymmetric synthesis of natural enantiomer (+)-griseofulvin in 1991 [4]. Commercially available phenol 5 was regiospecifically chlorinated to provide intermediate 6. The latter underwent a Fries rearrangement in the presence of aluminum chloride and the liberated phenol was involved in the Mitsunobu coupling with non-racemic allylic alcohol 7 to give rise to pentasubstituted benzene building block 8. The acetyl group in the latter was deprotonated and methoxycarbonylated by treatment with Mander’s reagent (9) and the resulting -keto ester was subjected to Regitz diazo transfer to provide diazo compound 10. Decomposition of the latter was accomplished with the use of rhodium pivalate catalyst and the resulting rhodium carbene underwent intramolecular trapping with the nearby oxygen atom followed by sigmatropic rearrangement to give benzofuranone 11. Conversion of the latter to non-racemic methyl ketone 14 was accomplished via ozonolysis, Wittig reaction with phosphonium ylide 12, tert-butyl ester cleavage with TFA and decarboxylation on treatment with diphenylphosphoryl azide (13) followed by exposure to refluxing aqueous hydrochloric acid. Finally, methyl ketone 14 was subjected to treatment with sodium methoxide on which it underwent the Dieckmann cyclization. The resulting cyclohexane-1,3-dione (griseofulvinic acid) was O-methylated with diazomethane to furnish (+)-griseofulvin (Scheme 2).
3. Spironolactone
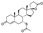
Spironolactone is a relatively old multi-target drug that is primarily used to treat high blood pressure and heart failure. Additional therapeutic applications of this drug include edema, cirrhosis of the liver and primary aldesteronism. The mechanism of action of spironolactone includes binding to intracellular mineralocorticoid receptors (MRs) in kidney epithelial cells which leads to the inhibition of aldosterone binding [5].
The first industrial synthesis of spironolactone was reported in 1957-1959 by Cella and co-workers of G. D. Searle and Co. [6-8]. The synthesis commences with previously reported ethynyl steroid building block 15. It was carbonylated at the alkyne terminus by treatment with methyl magnesium bromide followed by quenching with carbon dioxide. Hydrogenation of alkyne 16 over Lindlar catalyst elaborated the propionic acid side chain in 17 required for the formation of the spirocyclic lactone at the next step. Cyclization on treatment with p-toluenesulfonic acid yielded unsaturated lactone which was hydrogenated over palladium on carbon to yield spirocyclic lactone 18.
Oppenauer oxidation of the alcohol functionality in 18 was accompanied by the migration of the double bond to give α,β-unsaturated ketone 19. The conjugated system in 19 was extended by γ,δ -oxidation with chloranil to give compound 20 and set the scene for the conjugate addition of thioacetic acid at the d-position which proceeded with high diastereoselectivity and furnished spironolactone in 86% yield (Scheme 3).
Another industrial synthesis of spironolactone was patented in 1980 by Ciba Geigy scientists [9]. The synthesis stems from dehydroepiandrosterone (21) which was treated with 3-chloropropionaldehyde ethylene acetal and lithium to furnish tertiary alcohol 22 in diastereoselective fashion. The following sequence of steps (which proceeded in 75% overall yield) included bromination of the double bond, oxidation of the secondary alcohol to ketone and, finally, base-promoted double dehydrobromination, which led to α,β,γ,δ-unsaturated ketone (androstadienone) 23. Interestingly, the conjugate addition of thioacetic acid was in this case performed prior to the elaboration of the spirocyclic lactone in the last step. Thioacetic acid also caused the deprotection of the aldehyde to furnish 24. Oxidation of the aldehyde to the carboxylic acid in the presence of sulfuric acid brought about the closure of the lactone ring and led to the formation of spironolactone in 51% yield from 23 (Scheme 4).
A fundamentally different industrial route to spironolactone was developed by the Chemical Process Research and Development scientists at The Upjohn Co. in the late-1980s [10]. The synthesis relied on the readily available ethynyl steroid building block 25 the ketone group of which was protected as ethylene glycol ketal in the first step. The key transformation which led to the formation of spirocyclic lactol intermediate 27 was rhodium-catalyzed hydroformylation at elevated temperature and pressure. Oxidation of 27 to lactone was achieved with PDC and the ketal protecting group was removed to afford 28. The formation of α,β,γ,δ -unsaturated ketone 20 was achieved with chloranil (vide supra) and conjugate addition of thioacetic acid (catalyzed by TMSOTf) at the -position furnished spironolactone (Scheme 5).
Among many non-industrial syntheses of spironolactone described in the literature [5] the most recent one reported by Nagamitsu, Ohtawa and co-workers is quite notable [11]. The synthesis commences with dehydroepiandrosterone (21) and is reliant on the innovative approach to -lactonization of homopropargyl alcohols which involved terminal borylation of a homopropargyl moiety followed by m-CPBA oxidation. The latter sequence leads to the transformation of the alkyne moiety into a ketene intermediate which is trapped intramolecularly to give a -lactone. Hence, starting material 21 was treated with propargyl magnesium bromide (under Hg(II) catalysis) to give homopropargyl alcohol 29 in nearly quantitative yield. Application of the -lactonization protocol described above led to the formation of spirocyclic lactone 18. Oxidation of the secondary alcohol moiety in the latter with Dess-Martin periodinane (DMP) gave -unsaturated ketone 28. Transformation of the latter into spironolactone was analogous to the Upjohn Co. synthesis described above. Oxidation with chloranil installed the polyunsaturated ketone system (20) which is suitable to the conjugate addition of thioacetic acid (also TMSOTf-catalyzed in this case) resulting in the formation of spironolactone (Scheme 6).
4. Fluspirilene
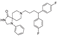
This spirocyclic antipsychotic drug was discovered at Janssen Pharmaceutica in 1963 and by 1970 it was approved for the treatment of schizophrenia [12]. The primary molecular targets of fluspirilene in the central nervous system are dopamine D2 and serotonin 5HT2A receptors as well as 1 subunit of voltage-dependent calcium channel [13].
Fluspirilene is an old drug and reviewing the vast patent literature on its synthesis would be tedious. Instead, we will focus on the most notable cases of fluspirilene assemble from relatively recent literature. For instance, an efficient synthesis of the key spirocyclic building block 30 needed for the synthesis of drug was published in 2018 by a team of Italian and Polish scientists [14]. The synthesis commenced with 1-benzyl piperidin-4-one which was involved in the Strecker reaction with aniline and trimethylsilyl cyanide to give nitrile 31. The latter was hydrated in sulfuric acid and resulting a-anilino carboxamide 32 was condensed with N,N-dimethylformamide dimethyl acetal to form spirocyclic imidazolinone 33. The C-N double bond in 33 was reduced to give imidazolidinone 34 and the benzyl group was removed by hydrogenation to give the target spirocycle 30 (Scheme 7).
Interestingly, it is the synthetic assembly of the 4,4-bis(4-fluorophenyl)butyl halide building block (for the alkylation of key spirocycle 30) that received much of the synthetic route development. For instance, in 2001, an Italian group reported an interesting synthesis of fluspirilene starting from 4,4’-difluorobenzophenone (35). Acetylene bis-lithium salt was added to the keto group of 35 in the presence of ethylene diamine to give allylic alcohol 36. The latter was hydroformylated in the presence of a rhodium catalysts cyclic hemiacetal 37. Sodium borohydride reduction of the latter furnished diol 38 which lost its doubly benzylic tertiary alcohol hydroxy group on hydrogenation. Alcohol 39 was brominated to alkyl halide 40 which then was used in the completion of fluspirilene synthesis (Scheme 8) [15].
A different approach to bromide 40 was reported in 2011 by a Shanghai Institute of Organic Chemistry group [16]. Diol 38 was obtained by reacting -butyrolactone with excess 4-fluorophenyl magnesium bromide. Thereupon, it was dehydrated in refluxing aqueous hydrochloric acid to give olefinic alcohol 41, bromination of which gave unsaturated alkyl bromide 42. Hydrogenation of the latter resulted in double bond saturation and went without a loss of the bromide to give alkylating agent 40 which was used in the preparation of fluspirilene (Scheme 9).
5. Fenspiride
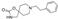
This spirocyclic oxazolidinone, acting as an antagonist of a1-adrenergic receptors, was first approved in selected European countries for medical use in the 1970s for the treatment of otolaryngological or ENT (ear, nose and throat) organ diseases as well as respiratory tract diseases [17]. The structure of fenspiride comprises a well-established feature for binding to the hERG channel (aromatic ring at a certain distance from a basic amine center) [18]. Unfortunately, it does bind to this off-target and causes prolongation of QT interval. For this reason, i. e. the increased risk of torsades de pointes, fenspiride was withdrawn from the pharmaceutical market in most markets in the World [19].
One of the early patents, obtained by Italian Voghera company for the synthesis of fenspiride, describes the assembly of the target molecule starting from 1-phenethyl piperidin-4-one (43) which was involved in the Reformatsky reaction with ethyl bromoacetate. After the conversion of resulting -hydroxy ester 44 to hydrazide 45, the latter was diazotized which triggered a Curtius rearrangement of the intermediate acyl azide and the isocyanate product of the rearrangement was trapped, intramolecularly, by the nearby hydroxy group to give fenspiride (Scheme 10) [20].
In 1994, a Mexican group disclosed an alternative approach to elaborating the spirocyclic oxazolidinone based on 1-phenethyl piperidin-4-one which includes the Strecker-type reaction of the ketone with trimethylsilyl cyanide. Resulting O-silylated a-hydroxy nitrile 46 was subjected to reduction with lithium alumohydride to give -amino alcohol 47. The latter was treated with trichloromethyl carbonate which donated the carbonyl group and resulted in the closure of the oxazolidinone ring of fenspiride (Scheme 11) [21].
In 2019, Indian company Emcure Pharmaceuticals jointly with a University of Pune group published a completely novel route to fenspiride which is based on nitro aldol reaction and is amenable to industrial scale production of the drug substance [22]. In fact, the researchers disclosed two possible approaches to fenspiride for comparison and made a conclusion that the nitro aldol route was preferred from the standpoint of pharmaceutical production. In the first approach, Corey-Chaykovsky epoxidation gave epoxide 48 which was opened up by sodium azide to give -azido alcohol 49. Reduction by hydrazine (presumably, to alcohol 47) and treatment with CDI (donor of the carbonyl) afforded fenspiride. In the second, preferred, approach, ketone 43 was condensed with nitromethane anion and resulting nitro compound 50 was reduced to the respective amine and Boc-protected to afford intermediate 51. Deprotonation of the hydroxy group in the latter with potassium tert-butoxide triggered intramolecular displacement of the tert-butoxy group and ring closure to fenspiride (Scheme 12).
6. Amcinonide
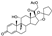
Amcinonide is a topical corticosteroid approved in 1979 for the treatment of various inflammatory dermatological conditions such as atopic dermatitis and allergic contact dermatitis. Amcinonide displayed high affinity to the glucocorticoid receptor which is thought to be the main biological target mediating its anti-inflammatory action. Binding to the receptor and its activation by amcinonide eventually leads to the induction of phospholipase A2 inhibitory proteins, which suppresses production of arachidonic acid and the downstream production of prostaglandins as chemical mediators of inflammation [23].
Unfortunately, synthetic routes to amcinonide are poorly described in the literature. Even the original patent issued to American Cyanamide company [24] features little synthetic detail. An industrial synthesis of amcinonide patented by an Indian company in 2018 [25] is described in quite sufficient detail. It starts with readily available natural steroid 2-((10S,13S,14S)-10,13-dimethyl-3-oxo-6,7,8,10,12,13,14,15-octahydro-3H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl acetate (52). The latter is subjected to epoxidation of the non-conjugated double bond using 1,3-dibromo-5,5-dimethylhydantoin (dibromantin, 53) and aqueous perchloric acid followed by the treatment of the resulting bromohydrin with potassium carbonate. Epoxide 54 is formed with high diastereoselectivity. The next step is dihydroxylation of the cyclopentene ring which is brought about by potassium permanganate and formic acid in acetone. Finally, diol 55, which is formed diastereoselectively, then subjected by the treatment with 70% hydrofluoric acid followed by the addition of cyclopentanone. The net result of the latter step is epoxide opening with the fluoride anion and spirocyclic ketal formation leading to amcinonide (Scheme 13).
7. Guanadrel
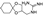
Guanadrel is an anti-hypertension drug containing a guanidine moiety which was approved in 1979 [26]. It exerts its therapeutic action by blocking sodium-dependent noradrenaline transporter and thus is a typical adrenoblocker [27]. The industrial synthesis of guanadrel was patented in 1970 by its inventors [28]. In the first step, cyclohexanone undergoes ketalization with 3-chloro-1,2-propandiol (56), forming 2-chloromethyl-1,4-dioxyspiro[4,5]decane (57). The latter building block is used to alkylate phthalimide sodium salt to give protected amine derivative 58. Removal of the phthalimide protecting group with hydrazine liberated primary amino group in 59 which is reacted with S-methyl thiourea sulfate in aqueous medium on steam bath heating to give guanadrel as a sulfate salt (Scheme 14).
In 1999, Goodman of UC San Diego developed a novel guanidine synthesis using triflyl-diurethane protected guanidines and applied it to the synthesis of guanadrel. In this synthesis, cyclohexanone was ketalized with acetylamino-substituted diol 60. The resulting spirocyclic building block 61 was deacetylated with hydrazine and reacted with N,N′-di-Cbz-N′′-triflylguanidine (62). Upon the displacement of the triflic amide leaving group in 62, bis-Cbz-protected guanidine 63 was hydrogenated over palladium on charcoal to give guanadrel in quantitative yield as a free base (Scheme 15). Interestingly, attempted use of Boc protecting groups in lieu of Cbz failed since the deprotection under acidic conditions led also to the removal of the acid-prone ketal [29].
8. Buspirone
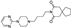
First synthesized in 1966 and approved for medical use in 1986, this spirocyclic serotonin 5-HT1A receptor agonist is primarily used to treat anxiety disorders [30]. Several patented protocols for the preparation of buspirone [31-33], including industry synthesis, have been reported in the literature.
On the plant scale, the following route was initially employed for the production of buspirone. 1-(Pyrimidin-2-yl)piperazine (64) was alkylated with 4-chlorobutyronitrile (65) and the nitrile group was hydrogenated over Raney nickel catalyst to give butylamine 66. The latter was condensed with commercially available 3,3-tetramethylene glutaric anhydride (67) in refluxing pyridine to give, after crystallization, buspirone [32] (Scheme 16).
Over more than three decades following buspirone approval for medical use, several newer synthetic routes appeared in the journal literature, some of which will be discussed below.
In 2008, Wei and co-workers from China University of Mining and Technology reported a new process for the production of buspirone. In the first step, condensation of cyclopentanone with methyl isocyanoacetate catalyzed by ammonia followed by acidic ester hydrolysis afforded cyclopentane-1,1-diacetic acid (68). Heating of the latter with ammonium carbonate at 200 ℃ afforded 3,3-tetramethyleneglutarimide (69). The latter was alkylated with butyl bromide 70 prepared, in turn, from 1-(pyrimidin-2-yl)piperazine (64) by alkylation with 1,4-dibromobutane. The last step afforded buspirone in nearly quantitative yield (Scheme 17) [34].
In 2019, Steven Ley and co-workers reported a continuous flow method to prepare buspirone which involves direct alkylation of 1-(pyrimidin-2-yl)piperazine (64) with alcohol 71 via a Ru(II)-catalyzed hydrogen borrowing approach (Scheme 18) [35].
In 2022, Beller and co-workers reported a remarkable, Ni-catalyzed reductive cross-coupling of nitriles with primary and secondary amines. In order to exemplify the scope of their methodology, a novel synthesis of buspirone was presented. Nitrile 72 prepared by alkylation of 3,3-tetramethylene glutarimide (69) with 4-bromobutyronitrile reacted 1-(pyrimidin-2-yl)piperazine (64) under 40 bar of hydrogen in the presence of nickel triflate and a special triphosphine ligand to give 80% yield of buspirone (Scheme 19) [36].
9. Ivermectin
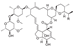
Discovered in 1975, this antiparasitic drug was initially approved for veterinary use in 1981. After extensive clinical research, ivermectin was approved in 1987 for use in humans, for the treatment for a wide range of parasitic infections [37-38]. Ivermectin acts by binding to parasites’ glutamate-gated sodium channels and forcing them to remain open, which causes the overflow of chlorine anions and, consequently, hyperpolarization of the cell membranes. This eventually kills the parasite [39].
Ivermectin is an approximately 80:20 mixture of two individual macrocyclic lactone (macrolide) compounds, ivermectin B1a and B1b. These compounds are produced by hydrogenation of the 22,23 double bond of the mixture of avermectins B1a and B1b which are, in turn, produced by fermentation of Streptomyces avermitilis, the bacteria discovered in 1970 by Satoshi Ōmura, for which discovery he and William Campbell of Merck were awarded the Nobel Prized in Physiology or Medicine [40]. In one of the more recent patents, Bayer described the hydrogenation of avermectins to ivermectins performed over RhCl3∙H2O catalyst in the presence of triphenylphosphine (Scheme 20) [41]. In addition, several successful total syntheses of avermectin and ivermectin by academic research groups have been reported in the literature [42]. However, these syntheses are not economically viable for the production of the drug.
10. Rifabutin
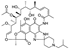
This macrocyclic semisynthetic drug of rifamycin family [43] was developed by Italian drug company Farmitalia Carlo Erba and received FDA approval in 1992 for the treatment of tuberculosis as well as other bacterial infections such as Mycobacterium avium complex [44]. The drug works via the inhibition of DNA-dependent RNA polymerase in bacteria, leading to a suppression of RNA synthesis and cell death [45].
The original synthesis of rifabutin was reported by Marsili and colleagues from Framitalia in 1981 [46]. The synthesis relies on 3-aminorifamycin S (73) prepared via azidation of rifamycin S (74) which was accompanied by a spontaneous loss of nitrogen molecule [47]. Compound 73 was treated with ammonia solution in THF to give 3-amino-4-deoxo-4-iminorifamycin S (75). The latter was isolated by crystallization and further involved in cyclocondensation with N-isobutyl piperidone performed at ambient temperature in the presence of ammonium acetate as a mild acidic catalyst. As the result spirocyclic imidazoline moiety was installed and pure rifabutin was isolated without chromatographic purification (Scheme 21).
11. Spirapril
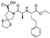
This spirocyclic inhibitor of angiotensin-converting enzyme (ACE) had been originated and developed by Schering-Plough Corporation (now Merck & Co.) and was approved in 1995 for the treatment of hypertension. ACE is a part of the renin-angiotensin system regulating blood pressure. Inhibition of ACE suppresses production of potent vasoconstrictor angiotensin II from its inactive precursor pro-angiotensin and thus prevents the increase in blood pressure [48]. Spirapril is an ethyl ester prodrug. It undergoes biotransformation to the respective carboxylic acid, spiraprilat which is the active ACE inhibitor [49].
Several syntheses of spirapril have been reported in the patent and periodic literature around the time of the drug’s approval. The original Schering-Plough route was patented in 1984 [50] and later disclosed in greater detail and with a few reaction sequence variants in a 1989 research article [51]. The synthesis commenced with Cbz-protected 4-oxoproline methyl ester (76) the carbonyl group of which was protected as spirocyclic thioketal to give methyl ester 77. The Cbz group was removed by 20% hydrobromic acid in glacial acetic acid to give amino acid 78. The ester side chain building block was prepared by reductive amination of keto ester 79. The resulting amino acid 80 was obtained as a mixture of diastereomers. Without separation the mixture was activated as N-hydroxysuccinimide ester (81) and reacted with 78 in the presence of triethylamine to give a good yield of spirapril after diastereomer separation (Scheme 22).
An alternative synthesis reported by the Squibb Corporation (now Bristol-Myers-Squibb) in 1988 also made use of spirocyclic amino acid 78. The latter was Boc-protected and the carboxylic function was esterified with 2-(trimethylsilyl)ethanol in the presence of DCC and DMAP to give good yield of ester 82. The Boc group was then removed by p-toluenesulfonic acid (PTSA) and the liberated free pyrrolidine 83 was acylated with carboxylic acid 80 activated by DCC/HOBt. This two-step sequence gave ester 84 in 70% after diastereomer separation. The removal of the 2-(trimethylsilyl)ethyl group with tetrabutylammonium fluoride trihydrate gave spiropril in quantitative yield (Scheme 23) [52].
12. Irbesartan
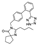
This spirocyclic antihypertensive drug was developed by Sanofi Research (now Sanofi-Aventis) and approved for medical use in 1997. The compound is a typical representative of the sartan family of hypertensives which act by blocking the angiotensin type 1 (AT1) receptor and preventing its activation and downstream events leading the blood pressure increase [53]. Irbesartan possesses four key elements of structure which are typical of sartans and could be traced back to losartan, the first-in-class AT1 receptor blocker: biphenyl substituted with tetrazole in the ortho position of the distal ring, five-membered nitrogen heterocycle (in this case, imidazoline-5-one) and n-butyl side chain [54].
The original synthetic route to irbesartan was described in detail in their 1995 publication [55]. The synthesis relies on commercially available tetramethylene glycine ethyl ester (85) as the starting material. Amino ester 85 was condensed with imidate 86 in the presence of a catalytic amount of acetic acid in refluxing xylene to give imidazolin-5-one 87. The latter was alkylated with 4'-(bromomethyl)-[1,1'-biphenyl]-2-carbonitrile and the resulting derivative 88 was treated with tributyl tin azide in refluxing xylene to give irbesartan (Scheme 24).
Interestingly, the key spirocyclic imidazolin-5-one building block 87 was recently accessed by Wu and DiPoto via a novel annulation of azaoxyallyl cations. Cyclopentanoyl chloride was first transformed into O-benzyl hydroxamic acid 89. The latter was treated with the base to generate azaoxyallyl cation 90 which reacted with pentanenitrile to give benzyloxy-substituted imidazolin-5-one 91. N-Deoxygenation with samarium iodide gave 87 thereby completing the formal synthesis of irbesartan (Scheme 25) [56].
Intermediate 87 was also key in the synthesis of irbesartan patented by a group of Israeli scientists in 2004 [57]. The convergent route started with C-phenyl tetrazole (92) which was protected with a trityl group and borylated in the para position via lithiation to give boronic acid 93. In parallel, intermediate 87 was N-alkylated with p-bromobenzyl bromide and resulting intermediate 94 was involved in Suzuki coupling with boronic acid 93 to give trityl-protected irbesartan 95. Removal of the trityl group furnished the target molecule (Scheme 26).
Among the synthetic routes to irbesartan featured in the patent literature, the following synthesis from Dr. Reddy’s Laboratories is notable. Similarly to the Sanofi route (vide supra), the synthesis starts with tetramethylene glycine (96) which was acylated with pentanoyl chloride under phase transfer conditions to give carboxylic acid 97 which, in turn, was amidated with 4'-(aminomethyl)-[1,1'-biphenyl]-2-carbonitrile to give ‘fully decorated’ precursor 98. The latter was cyclodehydrated in refluxing toluene in the presence of trifluoroacetic acid to give nitrile 88 and, after [3+2] cycloaddition with tributyltin azide, irbesartan (Scheme 27) [58].
An approach rather similar in spirit to the Dr. Reddy’s route was described in 2006 by ArQule scientists [59]. It is based on three-component, microwave-promoted cyclodehydrative synthesis of irbesartan’s key precursor 88 from ester 85, 4'-(aminomethyl)-[1,1'-biphenyl]-2-carbonitrile and pentanoic acid in the presence of triphenoxyphosphine. The conversion of nitrile 88 to irbesartan was achieved via in situ generation of tributyltin azide (Scheme 28).
Finally, a fundamentally different approach to irbesartan was developed in 2022 at Beijing University of Chemical Technology [60]. In this route, the spirocyclic scaffold of the drug was elaborated at a more advanced stage of the synthesis as shown in Scheme 29. Glycine methyl ester was acylated by pentanoyl chloride and ester 99 was amidated with 4'-(aminomethyl)-[1,1'-biphenyl]-2-carbonitrile under forcing, base-promoted conditions to give diamide 100. The latter was cyclodehydrated in refluxing toluene in the presence of methanesulfonic acid to give intermediate 101 which was found to be sufficiently C-H acidic to be doubly alkylated with 1,4-dibromobutane to furnish spirocyclic imidazoline-5-one 88. Notably, the nitrile-to-tetrazole conversion was achieved without the use of the toxic tin reagent (used in all previously described syntheses) which makes this 2022 route quite environmentally friendly and amenable to the plant scale production of the drug.
13. Cevimeline
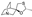
This spirocyclic derivative of quinuclidine acts as an agonist of muscarinic M1 and M3 receptors [61]. Originated from Israel Institute of Biological Research, the drug was developed by Daiichi Sankyo company and gained regulatory approval in 2000 for the treatment of dry mouth and Sjögren's syndrome [62]. Recently, the drug has been investigated on such therapeutic areas as cognitive impairment and neurodegenerative diseases [63].
In the original synthesis of cevimeline patented by the Israeli group [64], commercially available quinuclidin-3-one was converted to epoxide 102 using the Corey-Chaykovsky reaction with trimethyloxosulfonium iodide (Me3S+(O)I-) and sodium hydride. Resulting epoxide 102 was treated with hydrogen sulfide in basic aqueous medium, which led to epoxide opening and the formation of hydroxy thiol 103. The latter was subjected to thio/oxa acetal formation on treatment with acetaldehyde in the presence of boron trifluoride etherate. Cevimeline contained in the diastereomeric mixture of products 104 was separated by fractional crystallization (Scheme 30). The patent [64] presented a rather elaborate scheme for the fractional crystallization.
In 2008, Canadian company Apotex Pharmachem, Inc. filed a patent application on an improved synthesis of cevimeline which is similar in spirit to the original Israeli route. The Corey-Chaykovsky epoxidation of quinuclidine-3-one was performed with potassium tert-butoxide and the opening of epoxide 102 was achieved with thioacetic acid in lieu of hydrogen sulfide gas. Thioacetate 105 was condensed directly with acetaldehyde dimethyl acetal which gave an excellent yield of diastereomeric mixture 104 from which cevimeline was isolated by diastereomer separation (Scheme 31) [65].
In 2013, the Active Pharmaceutical Ingredient R&D Center of Indian company Emcure Pharmaceuticals, Ltd. published a yet further improved route to cevimeline in which safe and odorless thiourea is utilized as a thiolating reagent. Epoxide 102 was first converted to hydroxy bromide 106 which was then refluxed with thiourea in aqueous medium to furnish thiolactone 107. The latter was hydrolyzed with aqueous sodium hydroxide to give key hydroxy thiol 103 which was converted to cevimeline using the same routine as was developed in the original synthesis (vide supra) (Scheme 32) [66].
An intriguing, stand-alone approach to enantiomerically pure hydroxy thiol (S)-103 was published by F. Hoffmann-La Roche, Ltd. six years prior to cevimeline approval for medical use [67]. In this synthesis, the quinuclidine nucleus was elaborated quite late into the synthetic route. The synthesis relied on olefin 108 synthesized from pyridine-4-carboxaldehyde in a few trivial steps. Sharpless epoxidation gave enantiomerically pure (ee 94%) epoxide 109 which was opened with benzyl mercaptan to give diol 110. Mesylation of the primary hydroxy group set the scene for the intramolecular SN2 reaction to form the quinuclidine core. This was achieved by removal of the Boc group from the piperidine nitrogen atom in 111 and triggering the nucleophilic substitution by DIPEA. The bnzyl group was removed from intermediate 112 by dissolving metal (Ca) reduction in ammonia (Scheme 33).
14. Eplerenone
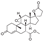
This spirocyclic antagonist of mineralocorticoid receptors developed by Ciba-Geigy was approved in 2002 for the treatment of hypertension and hear failure [68]. It is related to spironolactone but displays better selectivity to its primary target over the affinity to androgen, progestogen, glucocorticoid, or estrogen receptors and thus is devoid of undesired sexual side effects typical for spironolactone which limit its use [69].
Among industrial syntheses of eplerenone, the route patented in 1998 by Searle & Co. is quite notable [70]. The synthesis starts with steroid intermediate 20 (also useful in the synthesis of spironolactone, vide supra) which is subjected to regiospecific and stereoselective hydroxylation by fermentation with Aspergillus ochraceus mold. The hydroxy group in 113 serves as a kind of masked double bond which is unmasked later into the synthesis. Condensation with two equivalents of cyanide (in the form of acetone cyanohydrin) results in the formation of polycyclic enamine 114. The latter is hydrolyzed to ketone 115 which was then put through a sequence of steps including treatment with sodium methoxide (opening the ketone linkage as the methyl ester with simultaneous elimination of the cyanide), mesylation of the hydroxy group and elimination of methane sulfonic acid to give olefinic intermediate 116. The latter was epoxidized on treatment with hydrogen peroxide in phosphate buffered solution to give eplerenone (Scheme 34).
In 1997, Ciba-Geigy chemists published their synthetic route to eplerenone which basically provides another approach to the synthesis of intermediate 116 starting from spirocyclic steroid lactone 117. Conjugate addition of hydrogen cyanide at the -position proceeded stereoselectively and gave intermediate 118. Reduction with DIBAL-H gave aldehyde alcohol 119 which was oxidized into keto carboxylic acid 120. Esterification with diazomethane gave intermediate 116 (at which point the overall yield over four steps was a remarkable 69%). Finally, epoxidation with hydrogen peroxide gave eplerenone (Scheme 35) [71].
An interesting eplerenone synthesis was published in 2006 by Pfizer’s process chemistry team [72]. The synthesis commences with the same spirocyclic lactone 117 as was employed by Ciba-Geigy (vide supra). 2-Methylfuran was employed as a chemical equivalent of the carboxylic group. On Lewis acid catalysis, it underwent arylation of the distal conjugated double bond. The furan moiety in 121 was hydrolyzed to enedione 122 and the latter, without isolation, was ozonolyzed to give hemiacetal 123. The latter was subjected to Beyer-Villiger oxidation to give carboxylic acid 124 which was forced to close to isolable bis-lactone 125. The strained lactone in 125 was opened up via elimination and intermediate carboxylic acid was esterified with methyl sulfate to give ester 116. The latter is only one epoxidation step away from eplerenone (Scheme 36).
Intermediate 125 is clearly key to the large-scale synthesis of eplerenone. In 2011, a Chinese Academy of Sciences group published an interesting approach to it from triene 117 which involves conjugate addition of silyl methyl cuprate, Tamao oxidation and another intramolecular oxa-Michael addition to give cyclic ether 126. Curiously, methyltrifluoromethyl dioxirane was found to selectively oxidize ether 126 to lactol 127. PDC oxidation of the latter gave a nearly quantitative yield of lactone 125 which could now be opened and epoxidized to eplerenone as described by Pfizer (vide supra) (Scheme 37) [73].
15. Drospirenone
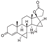
This is another steroid spirocyclic lactone drug structurally related to spironolactone and eplerenone, antagonists of mineralocorticoid receptors. However, pharmacologically drospirenone is different from the latter two drugs. In addition to high affinity to mineralocorticoid receptors, it binds rather tightly to progesterone receptors and less tightly to androgen receptors [74], which underlines its use as a birth control medication approved in 2000 [75].
The earliest synthesis of drospirenone was published by Schering AG in 1982 [76]. The synthesis commenced with compound 128 obtained from 3-hydroxy-5,15-androstadien-17-one by Corey cyclopropanation. Microbial hydroxylation of 128 gave diol 129 which was mono-pivaloylated at the less hindered hydroxy group to give allylic alcohol 130. The olefinic portion of 130 was epoxidized with tert-butyl hydroperoxide under vanadium(IV) oxide-acetylacetonate catalysis. Resulting epoxide 131 which was converted to chloride 132 by treatment with Ph3P/CCl4 in pyridine. Reductive elimination of the chlorine atom on treatment with zinc in acetic acid followed by saponification produced diol 133. The latter was subjected to Simmons-Smith cyclopropanation to establish the basic core of the target compound (134). Now the scene was set to establish the spirocyclic lactone moiety. Propargylic alcohol was doubly deprotonated and added to the ketone portion of 134 to give propargylic tetraol 135. The triple bond in 135 was hydrogenated over calcium carbonate. Resulting compound 136 was treated with PDC which triggered three events: oxidation of the primary alcohol moiety to carboxylic acid and lactonization, oxidation of the secondary alcohol to ketone and dehydration to form -unsaturated system of drospirenone (Scheme 38).
An approach similar in spirit to the synthesis of drospirenone by Schering AG was patented in 2010 by Evestra, Inc. [77]. The synthesis starts with bis-cyclopropane 134 to the carbonyl group of which C-anion of ethyl propiolate was added. Hydrogenation of the triple bond in resulting intermediate 137 gave triol 138 in which the secondary alcohol moiety was oxidized by TEMPO. Treatment of ketone 139 with methanolic potassium hydroxide followed by acidification with acetic acid led to the formation of spirocyclic lactone and dehydration of the -hydroxy cyclohexanone moiety which resulted in drospirenone (Scheme 39).
A different approach to the construction of drospirenone’s spirocyclic lactone motif was reported in 2008 by a University of Bologna group [78]. Ketone 134 was treated with excess vinyl magnesium bromide. Resulting tertiary allylic alcohol 140 was involved in olefin metathesis reaction with methyl acrylate catalyzed by 2nd generation Grubbs-Hoveyda catalyst. Resulting intermediate 141 having all requisite carbon atoms necessary for the formation of spirocyclic -lactone was hydrogenated over palladium on charcoal which resulted in the reduction of the double bond and the lactone ring closure to give 142. Final steps toward drospirenone were Dess-Martin oxidation of the secondary alcohol moiety and dehydration of the resulting ketone (Scheme 40).
16. Ledipasvir
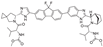
This inhibitor of hepatitis C virus non-structural protein 5A (HCV NS5A) was developed by Gilead Sciences and was approved in 2014 for the treatment of genotype 1 HCV in combination with sofosbuvir [79]. The pertinence of this drug to the present review is determined by the spirocyclic pyrrolidine moiety in its structure.
The complexity of ledipasvir’s structure mandates a lengthy synthetic route to assemble the drug molecule. The original synthesis patented by Gilead in 2013 [80] is presented in Scheme 41. The synthetic scheme includes the assembly of requisite building blocks from four different directions, thereby being remarkably convergent. From one direction, 2-bromofluorene (143) was iodinated in position 7 to give 144. The methylene group in the latter was bis-fluorinated with N-fluorobenzenesulfonimide (NFSI) to give compound 145 which was converted to an aryl magnesium species at the more reactive, iodo side and coupled with chloroacetic acid Weireb amide to give building block 146. From another direction, Boc-protected 4-methylene L-proline (147) was converted to spiro dibromo cyclopropane 148 which was dibrominated and converted to potassium carboxylate 149. The latter was involved in a nucleophilic substitution reaction with chloroacetyl compound 146 to give keto ester 150 which was reacted with ammonium acetate to give imidazole 151. Assembly of bicyclic pyrrolidine portion of ledipasvir started with enantiopure -methyl benzylamine (152) which was used to form a Schiff base with methyl glyoxylate hemiacetal. The Schiff base was involved in a Lewis acid-catalyzed Diels-Alder reaction with cyclopentadiene and the desired bicyclic structure was established (compound 153). Three trivial transformations (debenzylation with double bond reduction, Boc-protection and ester hydrolysis) gave carboxylic acid 154 which was employed in benzimidazole synthesis to give compound 155. Now the two halves of ledipasvir molecule (151 and 155) were brought together via a Suzuki coupling involving in situ formation of aryl boronic acid intermediate. Resulting intermediate 156 was deprotected and acylated at both liberated nitrogen atoms with N-methoxycarbonyl valine to give lidepasvir.
The Gilead Sciences route to ledipasvir may as well be quite optimal, considering that there have not been any attempts to improve it as judged by the literature on the subject. However, in 2021, a Suzuki-Miyaura coupling of the two halves of ledipasvir molecule was selected as a drug synthesis example to illustrate the power of novel aryl halide to aryl boronic acid coupling protocol where no palladium catalyst is employed. The two research groups from China reported the use of organocatalyst 157 to bring about the coupling of aryl bromide 151 with boronic acid pinacol ester 158. Considering the high yield of advanced ledipasvir precursor 156 reported for this coupling, this palladium-free approach appears much more amenable to large-scale pharmaceutical production than the original Gilead Sciences route (Scheme 42) [81].
17. Rolapitant
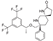
This spirocyclic neurokinin NK1 receptor antagonist comprising three stereogenic centers originated in the laboratories of Schering-Plough and was approved for the treatment of chemotherapy-induced nausea and vomiting in 2015 [82]. The drug’s syntheses are primarily featured in the patent literature.
The original synthesis of rolapitant patented by Schering-Plough [83] relies of the supply of key building block 159. Synthetically, the latter can be traced back to (S)-phenylglycine over 8 synthetic operations, including the preparation of oxazolidinone 160, as detailed further below. Cyclic enamine 159 was subjected to standard hydroboration-oxidation conditions to afford alcohol 161 which was oxidized to ketone 162 under the Swern conditions. Ketone 162 was involved in a standard hydantoin synthesis with potassium cyanide and ammonium carbonate to afford spirocyclic compound 163. The latter was Boc-protected, hydrolyzed and Boc-protected again to give -amino acid 164. The carboxylic group in 164 was reduced via mixed anhydride to give amino alcohol 165 which was oxidized (again under the Swern conditions) and involved, without protection of the amino group, in a Horner-Wadsworth-Emmons olefination to give acrylate ester 166. The latter turned out to be a direct precursor of rolapitant and was converted to the drug by hydrogenation (accompanied by lactam formation) and protecting group removal (Scheme 43).
As to the synthesis of key building block 159 from (S)-phenylglycine ((S)-Phg) it was accomplished as follows. (S)-Phg was converted to diastereomerically pure oxazolidinone 160 on condensation with benzaldehyde and Cbz-protection [84]. Oxazolidinone 160 was C-alkylated is diastereoselective fashion with chiral bromide 167. The carbonyl group of resulting intermediate 168 was reduced by lithium alumohydride and lactol 169 was obtained. The aldehyde form of the latter reacted with phosphonium ylide generated from alkyl phosphonium bromide 170 and elaborate olefinic building block 171 was obtained. It was hydrogenated over platinum dioxide and resulting acetal 172 was treated with p-toluene sulfonic acid in refluxing ethanol. This led to the removal of the acetal protecting group and the cyclization of the Cbz-protected amino group onto the liberated aldehyde carbonyl group to give target key intermediate 159 (Scheme 44) [83].
In 2013, Opko Health, Inc. patented an alternative route to rolapitant which is also based on key building block 159 [85]. This intermediate was nitrated and reduced by lithium borohydride to give nitro alkane 173. Removal of the Cbz group (to give 174) and subsequent alkylation of the nitro alkane portion with methyl acrylate over basic alumina gave compound 175 which was converted to methanesulfonic acid salt 176 for easy isolation. Reduction of 176 with zinc metal in acetic acid triggered lactamization and gave rolapitant (Scheme 45). Thus, the Opko Health, Inc. synthesis appears to be shorter than the original Schering-Plough route and more amenable to large scale API production.
In 2016, Opko Health, Inc. patented a fundamentally different route to rolapitant which relies of chiral building block 177 (for the sake of brevity, we do not discuss its synthesis) [86]. It was reductively alkylated with masked aldehyde building block 178 to give bis-olefinic compound 179. In the latter, a scene was set for intramolecular olefin metathesis which was successfully brought about using Hoveyda-Grubbs 2nd generation ruthenium catalysts (HGII) and resulting spirocycle 180 was isolated as hydrochloride salt. The latter advanced intermediate was free-based by aqueous sodium hydroxide and hydrogenated over palladium on charcoal to give rolapitant (Scheme 46).
18. Apalutamide
This spirocyclic androgen receptor antagonist discovered by the Sawyers/Jung team at UCLA was approved for the treatment of prostate cancer in the US in 2018 and in Europe in 2019 [87]. The original synthesis was patented in 2007 by the discovery team [88]. It started with benzoyl chloride 181 which was reacted with methylamine and resulting amide 182 was involved in aromatic nucleophilic substitution reaction with p-methoxybenzylamine to give 183. Upon removal of the PMB group, aniline 184 was involved in a Strecker reaction with cyclobutanone to give nitrile 185. The monothiohydantoin ring of apalutamide was formed on microwave-promoted condensation with thioisocyanate 186 followed by acidic hydrolysis on the initially formed imine (Scheme 47).
Later, a Sloan-Kettering team patented an apalutamide synthesis in which thiohydantoin formation was performed via in situ generation of thioisocyanate 186 from aniline 187 by reacting the latter with Strecker nitrile 185 in the presence of thiophosgene (Scheme 48) [89].
A number of different approaches to apalutamide, mostly focused on the synthesis of pyridine-3-amine 187 were disclosed [87]. A somewhat different approach to apalutamide assembly which involves two copper(I)-catalysed N-arylation steps was patented in 2018 by Hangzhou Cheminspire Technologies Co., Ltd. (China) [90]. The synthesis avoided the use of thiophosgene or thiophosgene equivalents by generating the thiohydantoin using potassium thiocyanate. Cyclobutane -amino acid 188 was used as a partner in copper(I)-catalyzed reaction with aryl bromide 189 to give carboxylic acid 190. Following esterification, compound 191 was reacted with potassium thiocyanate to give thiohydantoin portion of apalutamide (192) from which the target drug molecule was crafted via copper(I)-catalyzed arylation with 3-bromopyridine 193 (Scheme 49).
19. Moxidectin
This macrolide antiparasitic drug containing an (S)-1,7-dioxaspiro[5.5]undecane motif was derived from Streptomyces bacteria discovered in the late 1980s in an Australian soil sample [91]. It was initially employed for the treatment of helminths in house animals. However, in 2018, moxidectin received FDA approval to treat onchocerciasis (river blindness) in patients aged 12 and older [92]. Moxidectin exerts its antiparasitic action by selectively binding to the parasite's GABA-A and glutamate-gated chloride ion channels [93].
Chemically, moxidectin belongs to milbemycin class of macrocyclic lactones. It is obtained by semisynthetic modification of nemadectin (F-29249α) which, in turn, is isolated from the fermentation broth of bacterium Streptomyces cyanogriseus [94]. The following four-step synthesis of moxidectin from nemadectin was disclosed in a 1991 patent [95]. The secondary alcohol hydroxy group on the hexahydrobenzofuran moiety was chemoselectively protected a p-nitrobenzoate (PNB) ester (194). Then the other secondary alcohol hydroxy group was subjected to Albright–Goldman oxidation (DMSO/acetic anhydride) to give ketone 195 which was converted to O-methyl oxime 196 and the PNB group was removed by alkaline hydrolysis (without any damage to the macrolide ester linkage) to give moxidectin (Scheme 50).
20. Ubrogepant
Migraine is a debilitating disease which was traditionally treated only symptomatically by serotonin receptor modulators or non-steroidal anti-inflammatory drugs. However, more recent in-depth investigation of the mechanisms underlying the onset of migraine established calcitonin gene related peptide (CGRP) as a primary player in this pathological process. Produced in peripheral and central neurons, CGRP is a potent vasodilator and mediator of nociception. Thus, antagonizing the binding of CGRP to its receptor was established as a novel approach to the treatment of acute migraine at the fundamental level [96]. Ubrogepant is the first-in-class drug developed by Merck and Co. and approved in 2020 [97].
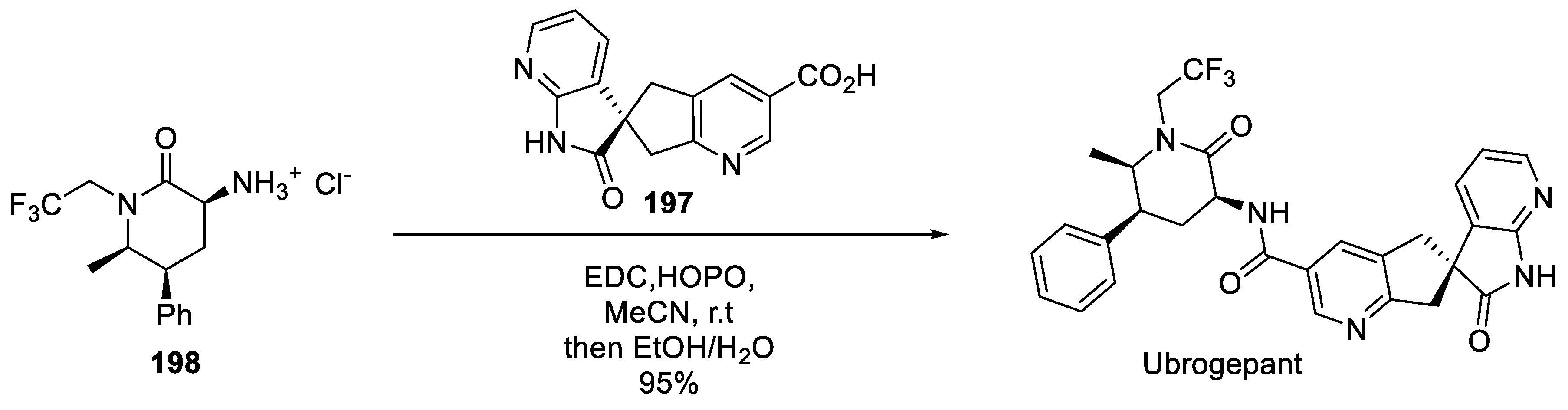
The detailed process chemistry route towards ubrogepant was published by the Merck team in 2017 [98]. The final step in the assembly of the target molecule was amidation of enantiopure spirocyclic carboxylic acid 197 with enantiopure amine 198 hydrochloride salt.
Scheme 51.
Final amidation in the assembly of ubrogepant.
Therefore, it is the assembly of enantiopure building blocks 197 and 198 which required most of the innovation. The synthesis of amine 198 started with a diastereomeric mixture of keto esters 199. The latter was subjected to dynamic kinetic transaminase-mediated cyclization (using an evolved enzyme strain) to give piperidone 200 as a 3:2 mixture of epimers. N-Alkylation of the latter with 3,3,3-trifluoroethyl triflate afforded predominantly compound 201 (separated in 87% yield from the unreacted starting material and bis-alkylation product). Removal of the Boc group gave epimeric amine 202 which was subjected to crystallization induced diastereoselective transformation (CIDT) with p-toluic acid and 3,5-dichlorosalicylic aldehyde to give building block 203 which was converted to hydrochloride salt 198 (Scheme 52).
Spirocyclic carboxylic acid 197 was assembled as follows. 2,3-Dibromo-5-chloropyridine (204) was transformed in four steps (involving two additions of pyridine magnesium species to DMF) to tetrahydropyran-protected aldehyde 205. The latter was condensed with 7-azaindolin-2-one 206 to give olefin 207 which was reduced by sodium borohydride and the THP group was removed to give alcohol 208. Conversion of 208 into alkyl chloride 209 set the scene for spirocyclization. On the action of aqueous sodium hydroxide, in the presence of cinchona alkaloid-derived chiral catalyst 210, chloride 209 underwent an efficient and enantioselective intramolecular C-alkylation to form spirocycle 211. Palladium-catalyzed carbonylation of the chloropyridine portion in the latter followed by removal of the tert-butyl group from the lactam nitrogen atom afforded key spirocyclic carboxylic acid 197 (Scheme 53).
An alternative approach to spirocyclic carboxylic acid 197 was reported in 2022 by Hu and co-workers [99]. It involves direct asymmetric spiroannulation of Boc-protected 7-azaindolin-2-one (213) with doubly benzylic bis-bromide 212. Thus, from cheap and readily available starting materials, spirocycle 214 was obtained in good yield and enantioselectivity. Removal of the Boc group followed by nitrile hydrolysis gave requisite spiro acid 197 thus completing a formal synthesis of ubrogepant (Scheme 54).
21. Oliceridine
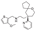
This spirocyclic biased [100] agonist of -opioid receptors was developed by Trevena, Inc. and was approved in 2020 for the intravenous treatment of acute moderate-to-severe post-operative pain [101].
The only full detailed route to the oliceridine drug substance is the original Trevena, Inc. approach patented in 2012 [102]. The source of the spirocyclic motif was ketone 216 obtained by oxidation of perhydropyran-4-ol 215 synthesized, in turn, by the Prins cyclization between cyclopentanone and homoallylic alcohol [103]. Base-promoted condensation with methyl cyanoacetate afforded olefin 217 to which pyrid-2-yl organometallic reagent (magnesium or lithium) was added in conjugate fashion to give cyano ester 218. The latter was subjected to basic hydrolysis and decarboxylation at elevated temperature to afford racemic product which was resolved by preparative chiral supercritical fluid chromatography to give enantiopure nitrile 219. This product was reduced using LAH and resulting amine 220 was reductively alkylated with 3-methoxythiophene-2-carboxaldehyde to give oliceridine (Scheme 55).
22. Risdiplam
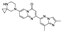
This spirocyclic drug is the first oral medication to treat spinal muscular atrophy, an orphan disease [104]. The drug was developed by PTC Therapeutics and Spinal Muscular Atrophy Foundation and received FDA approval in 2020 for the treatment of the disease in patients aged 2 months or older [105]. The drug acts by modifying mRNA splicing by binding to survival of motor neuron 2 (SMN2) [106]. It is currently marketed by Genentech, a subsidiary of Roche.
The synthetic routes to risdiplam disclosed in the original process chemistry patents by Roche rely on the commercial supply of the spirocylic piperazine building block and thus do not involve the formation of the spirocyclic moiety in the course of the synthesis.
In their 2015 patented route, PTC Therapeutics and Roche scientists started with 3,6-dichloro-5-methylpyridazine (221) in which the less accessible chlorine atom was substituted with ammonia and the imidazo ring was formed on condensation with bromoacetone dimethyl ketal (222). The overall yield of resulting imidazo pyridazine 223 was obtained in relatively low yield (unsurprisingly, considering the unfavorable course of the initial nucleophilic aromatic substitution). Compound 223 was coupled, under palladium(II) catalysis, with bis(pinacolato)diboron to give boronic acid 224. The latter was coupled again, in Suzuki fashion, with pyrido pyrimidone building block 225 (obtained, in turn, from 2-amino-5-fluoropyridine on condensation with dimethyl malonate). The Susuki coupling 224 + 225 resulted in core risdiplam scaffold 226 which was decorated with spirocyclic piperazine 227 under thermal conditions to give risdiplam (Scheme 56) [107].
The more recent route from their 2019 patent application, Roche scientists started by involving 5-bromo-2-chloropyridine (228) in Buchwald-Hartwig amination with Boc-protected spirocyclic piperazine (229). The unreacted, under Pd(II)-catalyzed conditions, chlorine atom in 230 was displaced by ammonia under Pd(0) catalysis and the amino portion of resulting intermediate 231 was converted in two steps into tosylate 232. The latter was coupled with boronic acid 224 (prepared as described in the previous Roche patent) in Suzuki fashion and the Boc group in resulting coupling products 233 was removed to give risdiplam (Scheme 57) [108].
23. Atogepant
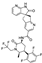
This next-in-class antagonist of calcitonin gene related peptide (CGRP) is a direct analog of ubrogepant (the first-in-class CGRP antagonist developed by Merck, vide supra) which is different from ubrogepant since it contains 2,3,6-trifluorophenyl ring. Discovered by Merck and developed by AbbVie, atogepant was approved in 2021 for the preventive treatment of episodic migraine in adults [109].
The synthesis of atogepant was described in detail in 2016 patent application by Merck [110] and it relies on essentially the same routine as was used in the preparation of ubrogepant. Specifically, the same enantiopure spiro acid 197 was in use. The enantiopure 3-aminopyrid-2-one building block was also assembled in a fashion similar to the amine half of ubrogepant (202). The synthesis commenced with phenylacetic acid 234 which was converted to the respective acyl chloride and then Weinreb amide 235. The latter was treated with methylmagnesium chloride to give methyl ketone 236. The latter was alkylated at the most acidic, benzylic position with mesylate 237 to give keto ester 238. The latter is a direct analog of compound 199 as precursor to 202 - and the exact same routine, involving the use of dynamic kinetic transaminase, was used to convert compound 238 to enantiopure 3-aminopyrid-2-one 239. Amidation of spiro acid 197 with amine 239 gave atogepant (Scheme 58).
24. Trilaciclib
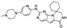
This spirocyclic inhibitor of cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) was developed by G1 Therapeutics. The drug was indicated for cancer patients receiving certain types of chemotherapy to protect the bone marrow from chemotherapy-induced damage. As trilaciclib was the first-in-class drug developed for this important indication, it received a priority review and a breakthrough therapy designation from the FDA. It was approved for medical use in 2021 [111].
The drug’s originator patented two synthetic routes to the compound both involving an intramolecular N-acylation as the key step in the construction of the spirocyclic moiety [112-113]. Of these two approaches, the later one [113] is described in much greater detail and is most likely the API production route which we chose to discuss herein.
The synthesis commences with an efficient and high-yielding Strecker reaction of cyclohexanone with glycine methyl ester and trimethylsilyl cyanide to afford amino nitrile 240. The latter was hydrogenated over platinum(IV) oxide. This caused reduction of the nitrile to aminomethyl compound which underwent lactamization to afford key spirocyclic building block 241. Without protection, sterically hindered secondary amine 241 was used to displace the labile chlorine atom in pyrimidine template 242. The nucleophilic aromatic substitution reaction proceeded under forcing conditions and afforded spirocycle 243 which was now set for the formation of the pyrrolo portion of the target molecule. As this step would require C-H deprotonation of the piperazinone moiety, the lactam nitrogen of intermediate 243 was Boc-protected to give compound 244. Treatment of the latter with DBU at 0 ℃ in THF triggered the desired deprotonation and intramolecular cyclization and gave 3-hydroxypyrrolo compound 245 in quantitative yield over the last two steps (including the preceding Boc-protection). Clearly, the hydroxy group in 245 had to be removed, which was achieved by O-triflation of the compound with subsequent palladium(0)-catalyzed reductive coupling with triethylsilane. The two steps proceeded in 70% combined yield and afforded spirocyclic pyrrolo pyrimidine 246 which was deprotected with trifluoroacetic acid to give advanced trilaciclib precursor 247. To complete the synthesis, the methylthio group in 247 was oxidized to the better leaving, methylsulfonyl group with oxone in aqueous acetonitrile to give 248 in excellent yield. The final nucleophilic aromatic substitution step between spirocyclic template 248 and pyridine amine 249 (prepared, in turn via nucleophilic aromatic substitution with 5-halopyridine precursor and N-methylpiperazine) required the use of a strong base (LiHMDS) and gave trilaciclib (Scheme 59).
25. Conclusions and perspectives
As noted previously, the use of spirocycles in drug discovery and medicinal chemistry has been on an explosive rise [2]. This trend is clearly reflected in the number of drugs containing a spirocyclic motif which received approval for medical use over the year. Indeed, over 50% of such drug molecules have been approved in this century. A range of synthetic methods to form a spirocyclic scaffold have been employed in the production routes towards these drugs though at times the source of the spirocyclic moiety is the naturally occurring, biosynthesized compound or a commercially available spirocyclic building block (Table 1).
The trend is likely to continue resulting in new advanced drug candidates and approved drug molecules which mandates that the arsenal of synthetic methods applicable to spirocycle formation is expanded beyond the most popular key steps such as lactonization and lactamization, cyclocondensation (including ketalization and acetalization) and well as intramolecular C-alkylation and acylation.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the Russian Foundation for Basic Research (grant # 21-53-12001).
References
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Dar'in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [Google Scholar] [CrossRef] [PubMed]
- Tomozane, H.; Takeuchi, Y.; Choshi, T.; Kishida, S.; Yamato, M. Syntheses and Antifungal Activities of dl-Griseofulvin and Its Congeners. I. Chem. Pharm. Bull. 1990, 38, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Brown, W.L.; Rege, S.; Laughton, P. Total Synthesis of (+)-Griseofulvin. J. Am. Chem. Soc. 1991, 113, 8561–8562. [Google Scholar] [CrossRef]
- Larik, F.A.; Saeed, A.; Shahzad, D.; Faisal, M.; El-Seedi, H.; Mehfooz, H.; Channar, P.A. Synthetic approaches towards the multi target drug spironolactone and its potent analogues/derivatives. Steroids 2017, 118, 76–92. [Google Scholar] [CrossRef]
- Cella, J.A.; Kagawa, C.M. Steroidal Lactones. J. Am. Chem. Soc. 1957, 79, 4808–4809. [Google Scholar] [CrossRef]
- Cella, J.A.; Brown, E.A.; Burtner, R.R. Steroidal Aldosterone Blockers. I. J. Org. Chem. 1959, 24, 743–748. [Google Scholar] [CrossRef]
- Cella, J.A.; Tweit, R.C. Steroidal Aldosterone Blockers. II. J. Org. Chem. 1959, 24, 1109–1110. [Google Scholar] [CrossRef]
- Anner, G.; Wehrli, H. Process for the preparation of steroid carbolactones. Swiss Patent CH617443A5 (publication 30.05.1980).
- Wuts, P.G.M.; Ritter, A.R. A Novel Synthesis of Spironolactone. An Application of the Hydroformylation Reaction. J. Org. Chem. 1989, 54, 5180–5182. [Google Scholar] [CrossRef]
- Yamane, D.; Tanaka, H.; Hirata, A.; Tamura, Y.; Takahashi, D.; Takahashi, Y.; Nagamitsu, T.; Ohtawa, M. One-Pot γ-Lactonization of Homopropargyl Alcohols via Intramolecular Ketene Trapping. Org. Lett. 2021, 23, 2831–2835. [Google Scholar] [CrossRef] [PubMed]
- Stepnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Janssen, P.A.; Niemegeers, C.J.; Schellekens, K.H.; Lenaerts, F.M.; Verbruggen, F.J.; van Nueten, J.M.; Marsboom, R.H.; Hérin, V.V.; Schaper, W. K. The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug. Arzneimittelroschung 1970, 20, 1689–1698. [Google Scholar]
- Morciano, G.; Preti, D.; Pedriali, G.; Aquila, G.; Missiroli, S.; Fantinati, A.; Caroccia, N.; Pacifico, S.; Bonora, M.; Talarico, A.; et al. Discovery of Novel 1,3,8-Triazaspiro[4.5]decane Derivatives That Target the c Subunit of F1/FO-Adenosine Triphosphate (ATP) Synthase for the Treatment of Reperfusion Damage in Myocardial Infarction. J. Med. Chem. 2018, 61, 7131–7143. [Google Scholar] [CrossRef]
- Botteghi, C.; Marchetti, M.; Paganelli, S.; Persi-Paoli, F. Rhodium catalyzed hydroformylation of 1,1-bis(p-fluorophenyl)allyl or propargyl alcohol: A key step in the synthesis of Fluspirilen and Penfluridol. Tetrahedron 2001, 57, 1631–1637. [Google Scholar] [CrossRef]
- Chen, G.; Xia, H.; Cai, Y.; Ma, D.; Yuan, J.; Yuan, C. Synthesis and SAR study of diphenylbutylpiperidines as cell autophagy inducers. Bioorg. Med. Chem. Lett. 2011, 21, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Stewart, A.G. Value of Fenspiride in the Treatment of Inflammation Diseases in Otorhinolaryngology. Eur. Respir. Rev. 1991, 1, 105–110. [Google Scholar]
- Vandenberg, J.I.; Perozo, E.; Allen, T.W. Towards a structural view of drug binding to hERG K+ channels. Trends Pharmacol. Sci. 2017, 38, 899–907. [Google Scholar] [CrossRef]
- Wiśniowska, B.; Holbrook, M.; Pollard, C.; Polak, S. Utilization of mechanistic modelling and simulation to analyse fenspiride proarrhythmic potency - Role of physiological and other non-drug related parameters. J. Clin. Pharm. Ther. 2022, 47, 2152–2161. [Google Scholar] [CrossRef]
- Taccone, I. Method for the preparation of derivatives of spiro[4,5]decane and derivatives thus obtained. US Patent 4,028, 351.
- Somanathan, R.; Rivero, I.A.; Nunez, G.I.; Hellberg, L.H. Convenient synthesis of 1-oxa-3,8-diazaspiro[4,5]decan-2-ones. Synth. Commun. 1994, 24, 1483–1387. [Google Scholar] [CrossRef]
- Pramanik, C.; Bapat, K.; Patil, P.; Kotharkar, S.; More, Y.; Gotrane, D.; Chaskar, Mahajan, U. ; Tripathy, N.K. Nitro-Aldol Approach for Commercial Manufacturing of Fenspiride Hydrochloride. Org. Proc. Res. Dev. 2019, 23, 1252–1256. [Google Scholar] [CrossRef]
- Woodford, R.; Barry, B.W.; Frosch, P.J. Activity and Bioavailability of Amcinonide and Triamcinolone Acetonide in Experimental and Proprietary Topical Preparations. Curr. Ther. Res. 1977, 21, 877–886. [Google Scholar]
- Shultz, W.; Sieger, G.M.; Krieger, C. Novel triamcinolone derivative. Brittish patent GB1442925 (published 14.07.1976).
- Trimathi, V.; Kumar, R.; Bhuwania, R.; Bhuwania, B.K. Novel process for preparation of corticosteroids. PCT Int. Appl. WO2018037423 (published 01.03.2018).
- Finnerty, F.A.; Brogden, R.N. Guanadrel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in hypertension. Drugs 1985, 30, 22–31. [Google Scholar] [CrossRef]
- Vardanyan, R.S.; Hruby, V.J. 12 - Adrenoblocking Drugs, In Synthesis of Essential Drugs; R.S. Vardanyan, V.J. Hruby, Eds.; Elsevier, 2006, pp. 161–177.
- Hardie, W.R.; Aaron, J.E. 1,3-Dioxolan-4-yl-alkyl guanidines. US Patent 3,547, 951.
- Baker, T.J.; Goodman, M. Synthesis of Biologically Important Guanidine-Containing Molecules Using Triflyl-Diurethane Protected Guanidines. Synthesis 1999, 1423–1426. [Google Scholar] [CrossRef]
- Taylor, D.P.; Moon, S. L. Buspirone and related compounds as alternative anxiolytics. Neuropeptides 1991, 19, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rayburn, J. N-(Heteroarcyclic)piperazinylalkylazaspiroalkanediones. US Patent 3,717, 634.
- Wu, Y.H.; Rayburn, J.W. N-[(4-Pyridyl-piperazino)alkyl]azaspiroalkanediones. US Patent 3,907, 801.
- Wu, Y.H.; Rayburn, J.W. Tranquilizer process employing N-(heteroarcyclic)piperazinylalkylazaspiroalkanediones. US Patent 3,976, 776.
- Mou, J.; Zong, Z.-M.; Wei, X.-Y. Facile synthesis of anxyolytic buspirone. Org. Prep. Proc. Int. 2008, 40, 391–394. [Google Scholar] [CrossRef]
- Labes, R.; Mateos, C.; Battilocchio, C.; Chen, Y.; Dingwall, P.; Cumming, G.R.; Rincon, J.A.; Nieves-Remacha, M.J.; Ley, S.V. Fast continous alcohol amination employing a hydrogen borrowing protocol. Green Chem. 2019, 21, 59–63. [Google Scholar] [CrossRef]
- Chandrashekhar, V.G.; Baumann, W.; Beller, M.; Jagadeesh, R.V. Nickel-catalyzed hydrogenative coupling of nitriles and amines for general amine synthesis. Science 2022, 376, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.C. Ivermectin as an antiparasitic agent for use in humans. Ann. Rev. Microbiol. 1991, 45, 445–474. [Google Scholar] [CrossRef] [PubMed]
- Crump, A. Ivermectin: Enigmatic multifaceted 'wonder' drug continues to surprise and exceed expectations. J. Antibiotics 2017, 70, 495–505. [Google Scholar] [CrossRef]
- Martin, R.J.; Robertson, A.P.; Choudhary, S. Ivermectin: An Anthelmintic, an Insecticide, and Much More. Trends Parasitol. 2021, 37, 48–64. [Google Scholar] [CrossRef]
- Molyneux, D.H.; Ward, S.A. Reflections on the Nobel Prize for Medicine 2015 - The Public Health Legacy and Impact of Avermectin and Artemisinin. Trends Parasitol. 2015, 31, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Arlt, D.; Bonse, G.; Reisewitz, F. Method for the preparation of Ivermectin. Eur. Patent EP072997 (published 04.09.1996).
- Yamashita, S.; Hayashi, D.; Nakano, A.; Hayashi, Y.; Hirama, M. Total synthesis of avermectin B1a revisited. J. Antibiotics 2016, 69, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, D.M. Rifamycins, Alone and in Combination. Cold Spring Harb. Perpect. Med. 2016, 6, a027011. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Ebrahimi, G.; Arefzadeh, S.; Zamani, S.; Nikpor, Z.; Mirsaeidi, M. Antibiotic therapy success rate in pulmonary Mycobacterium avium complex: A systematic review and meta-analysis. Expert Rev. Anti. Infect. Ther. 2020, 18, 263–273. [Google Scholar] [CrossRef]
- Pinheiro, M.; Nunes, C.; Caio, J.M.; Lucio, M.; Brezesinski, Reis, S. The Influence of Rifabutin on Human and Bacterial Membrane Models: Implications for Its Mechanism of Action. J. Phys. Chem. B 2013, 117, 6187–6193. [Google Scholar] [CrossRef]
- Marsili, L.; Pasqualucci, C.; Vigevani, A.; Gioia, B.; Schioppacassi, G.; Oronzo, G. New rifamycins modified at positions 3 and 4. Synthesis, structure and biological evaluation. J. Antiobiot. 1981, 34, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Marsili, L.; Rosetti, V.; Pasqualucci, C. Method of preparing derivatives of rifamycin S. US Patent 4,007, 169.
- Noble, S.; Sorkin, E.M. Spirapril: A Preliminary Review of its Pharmacology and Therapeutic Efficacy in the Treatment of Hypertension. Drugs 1995, 49, 750–766. [Google Scholar] [CrossRef]
- Hossein-Nia, M.; Surve, A.H.; Weglein, R.; Gerbeau, C.; Holt, D.W. Radioimmunoassays for spirapril and its active metabolite spiraprilate: Performance and application. Ther. Drug Monit. 1992, 14, 234–242. [Google Scholar] [CrossRef]
- Gold, E.H.; Neustadt, B.R.; Smith, E.M. 7-Carboxyalkylaminoacyl-1,4-dithia-7-azaspiro[4.4]-nonane-8-carboxylic acids. US Patent 4,470, 972.
- Smith, E.M.; Swiss, G.F.; Neustadt, B.R.; McNamara, P.; Gold, E.H.; Sybertz, E.J.; Baum, T. Angiotensin Converting Enzyme Inhibitors: Spirapril and Related Compounds. J. Med. Chem. 1989, 32, 1600–1606. [Google Scholar] [CrossRef]
- Krapcho, J.; Turk, C.; Cushman, D.W.; Powell, J.R.; DeForrest, J.M.; Spitzmiller, E.R.; Karanewsky, D.S.; Duggan, M.; Rovnyak, G.; Schwartz, J.; et al. Angiotensin-Converting Enzyme Inhibitors. Mercaptan, Carboxyalkyl Dipeptide, and Phosphinic Acid Inhibitors Incorporating 4-Substituted Prolines. J. Med. Chem. 1988, 31, 1148–1160. [Google Scholar] [CrossRef]
- Muszalska, I.; Sobczak, Dolhan, A. ; Jelinska, A. Analysis of Sartans: A review. J. Pharm. Sci. 2014, 103, 2–28. [Google Scholar] [CrossRef] [PubMed]
- Siragy, H. Angiotensin II receptor blockers: Review of the binding characteristics. Am. J. Cardiol. 1999, 84, 3S–8S. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, C.A.; Perreaut, P.M.; Ferrari, B.P.; Muneaux, Y.A.; Assens, J.-L. A.; Clement, J.; Haudricourt, F.; Muneaux, C.F.; Taillades, J.E.; Vignal, M.-A.; et al. A New Series of Imidazolones: Highly Specific and Potent Nonpeptide AT1Angiotensin II Receptor Antagonists. J. Med. Chem. 1993, 36, 3371–3380. [Google Scholar] [CrossRef] [PubMed]
- DiPoto, M.C.; Wu, J. Synthesis of 2-Aminoimidazolones and Imidazolones by (3 + 2) Annulation of Azaoxyallyl Cations. Org. Lett. 2018, 20, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Nisnevich, G.; Rukhman, I.; Pertsikov, B.; Kaftanov, J.; Dolitzky, B.-Z. Novel synthesis of irbesartan. US Patent Appl. US2004192713 (published 30.09.2004).
- Reddy, R.B.; Suhakar, S.; Sridhar, C.; Rao, S.N.; Venkataraman, S.; Reddy, P.P. Satyanarayana, B. Process for preparing irbesartan. PCT Int. Appl. WO2005113518 (published 01.12.2005).
- Ye, P.; Sargent, K.; Stewart, E.; Liu, J.-F.; Yohannes, D.; Yu, L. Novel and Expeditious Microwave-Assisted Three-Component Reactions for the Synthesis of Spiroimidazolin-4-ones. J. Org. Chem. 2006, 71, 3137–3140. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Guo, Y.; Niu, H.; Wang, J.; Li, J.; Li, C.; Qiao, R. Development of a New Synthetic Route of the Key Intermediate of Irbesartan. Org. Proc. Res. Dev. 2022, 26, 2438–2446. [Google Scholar] [CrossRef]
- Yasuda, H.; Niki, H. Review of the Pharmacological Properties and Clinical Usefulness of Muscarinic Agonists for Xerostomia in Patients with Sjögren's Syndrome. Clin. Drug Investig. 2002, 22, 67–73. [Google Scholar] [CrossRef]
- Ono, M.; Takamura, E.; Shinozaki, K.; Tsumura, T.; Hamano, T.; Yagi, Y.; Tsubota, K. Therapeutic effect of cevimeline on dry eye in patients with Sjögren's syndrome: A randomized, double-blind clinical study. Am. J. Ophthalmol. 2004, 138, 6–17. [Google Scholar] [CrossRef]
- Oleksak, P.; Novotny, M.; Patocka, J.; Nepovimova, E.; Hort, J.; Pavlik, J.; Klimova, B.; Valis, M.; Kuca, K. Neuropharmacology of Cevimeline and Muscarinic Drugs—Focus on Cognition and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 8908. [Google Scholar] [CrossRef]
- Fisher, A.; Karton, I.; Heldman, E.; Levy, A.; Grunfeld, Y. Derivatives of quinuclidine. US Patent 4,855, 290.
- Bratovanov, S.; Bejan, E.; Wang, Z.-X.; Horne, S. Process for the preparation of 2-methylspiro(1,3-oxathiolane-5,3')quiniclidine. US Patent Appl. US2008249312 (published 09.10.2008).
- Pramanik, C.; Patil, P.; Kotharkar, S.; Tripathy, N.K.; Gurjar, M.K. A new route to cevimeline. Tetrahedron Lett. 2013, 54, 3043–3045. [Google Scholar] [CrossRef]
- Bös, M.; Canesso, R. EPC-synthesis of (S)-3-hydroxy-3-mercaptomethylquinucudine, a chiral building block for the synthesis of the muscarinic agonist AF1028. Heterocycles 1994, 38, 1889–1896. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Menard, J. The 45-year story of the development of an anti-aldosterone more specific than spironolactone. Mol. Cell. Endocrinol. 2004, 217, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.S.; Liu, C.; Anderson, D.K.; Lawson, J.P.; Wieczorek, J.; Kunda, S.A.; Letendre, L.J.; Pozzo, M.J.; Sing, Y.-L. L.; Wang, P.T.; et al. Processes for preparation of 9,11-epoxy steroids and intermediates useful therein. PCT Int. Appl. WO9825948 (published 18.06.1998).
- Grob, J.; Boillaz, M.; Schmidlin, J.; Wehrli, H.; Wieland, P.; Fuhrer, H.; Rihs, G.; Joss, U.; de Gasparo, M.; Haenni, H.; et al. Steroidal, Aldosterone Antagonists: Increased Selectivity of 9,11-Epoxy Derivatives. Helv. Chim. Acta 1997, 80, 566–585. [Google Scholar] [CrossRef]
- Pearlman, B.A.; Padilla, A.G.; Hach, J.T.; Havens, J.L.; Pillai, M.D. A New Approach to the Furan Degradation Problem Involving Ozonolysis of the trans-Enedione and Its Use in a Cost-Effective Synthesis of Eplerenone. Org. Lett. 2006, 8, 2111–2113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, H.; Feng, H.; Li, Y. Stereoselective construction of a steroid 5a,7a-oxymethylene derivative and its use in the synthesis of eplerenone. Steroids 2011, 76, 56–59. [Google Scholar] [CrossRef]
- Krattenmacher R: Drospirenone: Pharmacology and pharmacokinetics of a unique progestogen. Contraception 2000, 62, 29–38. [CrossRef] [PubMed]
- Muhn P, Fuhrmann U, Fritzemeier KH, Krattenmacher R, Schillinger E: Drospirenone: A novel progestogen with antimineralocorticoid and antiandrogenic activity. Ann. N. Y. Acad. Sci. 1995, 761, 311–335. [CrossRef]
- Bittler, D.; Hofmeister, H.; Laurent, H.; Nickisch, K.; Nickolson, R.; Petzoldt, K.; Wiechert, R. Synthesis of Spirorenone- A Novel Highly Active Aldosterone Antagonist. Angew. Chem. Int. Ed. Engl. 1982, 21, 696–697. [Google Scholar] [CrossRef]
- Nickisch, K.; Acosta, K.; Santhamma, B. Methods for the preparation of drospirenone. US Patent Appl. US2010261896 (published 14.10.2010).
- Bandini, M.; Contento, M.; Garelli, A.; Monari, M.; Tolomelli, A.; Umani-Ronchi, A.; Andriolo, E.; Montorsi, M. A Nonclassical Stereoselective Semi-Synthesis of Drospirenone via Cross-Metathesis Reaction. Synthesis 2008, 3801–3904. [Google Scholar] [CrossRef]
- Scott, L.J. Ledipasvir/Sofosbuvir: A Review in Chronic Hepatitis C. Drugs 2018, 78, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.W.; Vitale, J.P.; Matthews, K.S.; Teresk, M.G.; Formella, A.; Evans, J.W. Synthesis of antiviral compound. US Patent Appl. US20130324740 (published 05.12.2013).
- Xu, L.; Liu, F.-Y.; Zhang, Q.; Chang, W.-J.; Liu, Z.-L.; Lv, Y.; Yu, H.-Z.; Xu, J.; Dai, J.-J.; Xu, H.-J. The amine-catalysed Suzuki–Miyaura-type coupling of aryl halides and arylboronic acids. Nat. Catal. 2021, 4, 71–78. [Google Scholar] [CrossRef]
- Heo, Y.-A.; Deeks, E.D. Rolapitant: A Review in Chemotherapy-Induced Nausea and Vomiting. Drugs 2017, 77, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.; Reichard, G.A.; Wang, C.; Xiao, D.; Tsui, H.-C.; Shih, N.-Y.; Arredondo, J.D.; Wrobleski, M.L.; Palani, A. US Patent 7,049,320 (published 23. 05. 2006.
- O'Donnell, M.; Fang, Z.; Ma, X.; Huffman, J.C. New methodology for the synthesis of -dialkylamino acids using the "self-regeneration of stereocenters" method: -ethyl--phenylglycine. Heterocycles 1997, 46, 617–630. [Google Scholar] [CrossRef]
- Mergelsberg, I.; Scherer, D.H.; Huttenloch, M.E.; Tsui, H.-C.; Paliwal, S.; Shih, N.-Y. Process and intermediates for the synthesis of 8-[{1-(3,5-bis-(thrifluoromethyl)phenyl)ethoxy}methyl]-8-phenyl-1,7-diazadpiro[4,5]decan-2-one compounds. US Patent 8,552, 191.
- Wu, G.G.; Werne, G.; Fu, X.; Orr, R.K.; Chen, F.X.; Cui, J.; Sprague, V.M.; Zhang, F.; Xie, J.; Zeng, L.; et al. Process and intermediates for the synthesis of 8-[{1-(3,5-bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1,7-diaza-spiro[4.5]decan-2-one compounds. US Patent 9,249, 144.
- Hughes, D.L. Review of Synthetic Routes and Crystalline Forms of the Antiandrogen Oncology Drugs Enzalutamide, Apalutamide, and Darolutamide. Org. Proc. Res. Dev. 2020, 24, 347–362. [Google Scholar] [CrossRef]
- Jung, M.E.; Sawyers, C.L.; Ouk, S.; Tran, C.; Wongvipat, J. Androgen Receptor Modulator for the Treatment of Prostate Cancer and Androgen Receptor-Associated Diseases. PCT Int. Patent Appl. WO 2007126765 (published 08.11.2007).
- Ouerfelli, O.; Dilhas, A.; Yang, G.; Zhao, H. Synthesis of Thiohydantoins. PCT Int. Patent Appl. WO 2008119015 (published 02.10.2008).
- Zheng, X.; Zhang, Y. Synthesis method of apalutamide and its intermediates. Chinese Patent CN108383749 (published 10.08.2018).
- Takiguchi, Y.; Mishima, H.; Okuda, M.; Terao, M. Milbemycins, a new family of macrolide antibiotics: Fermentation, isolation and physico-chemical properties. J. Antibiot. 1980, 23, 1120–1127. [Google Scholar] [CrossRef]
- Prichard, R.K.; Geary, T.G. Perspectives on the utility of moxidectin for the control of parasitic nematodes in the face of developing anthelmintic resistance. Int. J. Parasitol. Drugs Drug Resist. 2019, 10, 69–83. [Google Scholar] [CrossRef]
- Milton, P.; Hamley, J.I.D.; Walker, M.; Basáñez, M.G. Moxidectin: An oral treatment for human onchocerciasis. Exp. Rev. Anti-infect. Ther. 2020, 18, 1067–1081. [Google Scholar] [CrossRef]
- Takiguchi, Y.; Ono, M.; Muramatsu, S.; Ide, J.; Mishima, H.; Terao, M. Milbemycins, a new family of macrolide antibiotics. Fermentation, isolation and physico-chemical properties of milbemycins D, E, F, G, and H. J. Antibiot. 1983, 36, 502–508. [Google Scholar] [CrossRef]
- Maulding, D.R.; Kumar, A. Process for the preparation of 23-(C1-C6 alkyloxime)-LL-F28249 compounds. US Patent 4,988, 824.
- Russell, F.A.; King, R.; Smillie, S.J.; Kodji, X.; Brain, S. D. Calcitonin gene-related peptide: Physiology and pathophysiology". Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef]
- Moore, E.; Fraley, M.E.; Bell, I.M.; Burgey, C.S.; White, R.B.; Li, C.C.; Rega, C.P.; Danziger, A.; Michener, M.S.; Hostetler, E.; et al. Characterization of Ubrogepant: A Potent and Selective Antagonist of the Human Calcitonin Gene‒Related Peptide Receptor. J. Pharmacol. Exp. Ther. 2020, 373, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, N.; Cleator, E.; Kosjek, B.; Yin, J.; Xiang, B.; Chen, F.; Kuo, S.-C.; Belyk, K.; Mullens, P.R.; Goodyear, A.; et al. Practical Asymmetric Synthesis of a Calcitonin Gene-Related Peptide (CGRP) Receptor Antagonist Ubrogepant. Org. Proc. Res. Dev. 2017, 21, 1851–1858. [Google Scholar] [CrossRef]
- Gao, M.; Li, Y.; Li, X.; Hu, L. Phase-Transfer-Catalyzed Asymmetric Annulations of Alkyl Dihalides with Oxindoles: Unified Access to Chiral Spirocarbocyclic Oxindoles. Org. Lett. 2022, 24, 875–880. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Oliceridine: First Approval. Drugs 2020, 80, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, D.; Gotchev, D.; Pitis, P.; Chen, X.-T.; Liu, G.; Yuan, C.C.K. Opioid receptor ligands and methods of using and making same. PCT Int. Appl. WO2012129495 (published 27.09.2012).
- Hanschke, E. Zur Kenntnis der Prinsschen Reaktion, III. Mitteil.1): Über die Reaktion von Allylcarbinol mit Aldehyden und Ketonen. Chem. Ber. 1955, 88, 1053–1061. [Google Scholar] [CrossRef]
- D'Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Ratni, H.; Scalco, R.S.; Stephan, A.H. Risdiplam, the First Approved Small Molecule Splicing Modifier Drug as a Blueprint for Future Transformative Medicines. ACS Med. Chem. Lett. 2021, 12, 874–877. [Google Scholar] [CrossRef]
- Ratni, H.; Green, L.; Naryshkin, N.A.; Weetall, M.L. Compounds for treating spinal muscular atrophy. PCT Int. Appl. WO2015173181 (published 19.11.2015).
- Adam, J.-M.; Fantasia, S.M.; Fishlock, D.V.; Hoffmann-Emery, F.; Moine, G.; Pfleger, C.; Moessner, C. Process for the prepration of 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one derivatives. PCT Int. Appl. 2019057740 (published 28.03.2019).
- Ailani, J.; Lipton, R.B.; Goadsby, P.J.; Guo, H.; Miceli, R.; Severt, L.; Finnegan, M.; Trugman, J.M.; ADVANCE Study Group. Atogepant for the Preventive Treatment of Migraine. N. Engl. J. Med. 2021, 385, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Molinaro, C.; Wuelfing, W.P.; Yasuda, N.; Yin, J.; Zhong, Y.-L.; Lynch, J.; Andreani, T. Process for Making CGRP Receptor Antagonists. US Patent Appl. US2016130273 (published 12.05.2016).
- Dhillon, S. Trilaciclib: First Approval. Drugs 2021, 81, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Strum, J.C.; Bisi, J.E.; Roberts, P.J.; Tavares, F.X. Highly Active Anti-Neoplastic and Anti-Proliferative Agents. WO2014144740A2 (published 18.09.2014).
- Smith, A.; White, H.S.; Tavares, F.X.; Krasutsky, S.; Chen, J.-X.; Dorrow, R.L.; Zhong, H. Synthesis of N-(Heteroaryl)Pyrrolo[3,2-d]Pyrimidin-2-Amines. WO201800 5865A1, 2018.
Scheme 1.
Synthesis of dl-griseofulvin by Yamoto and co-workers.
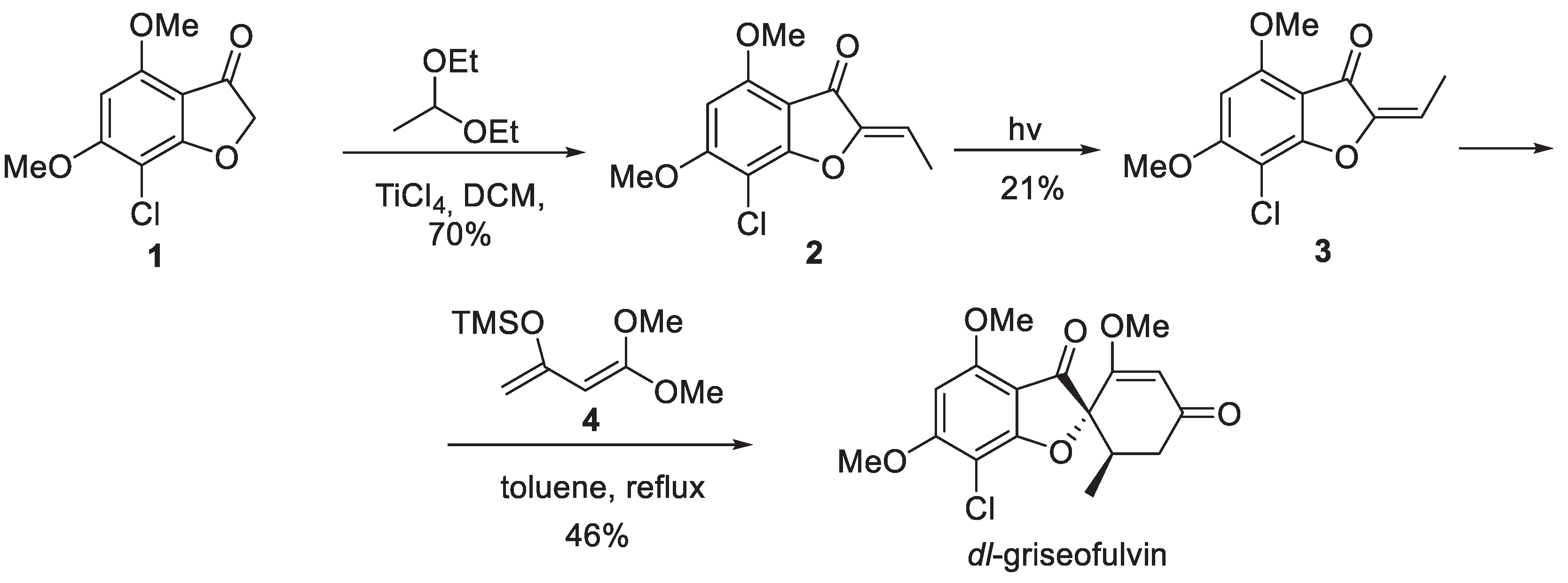
Scheme 2.
Synthesis of (+)-griseofulvin by Pirrung and co-workers.
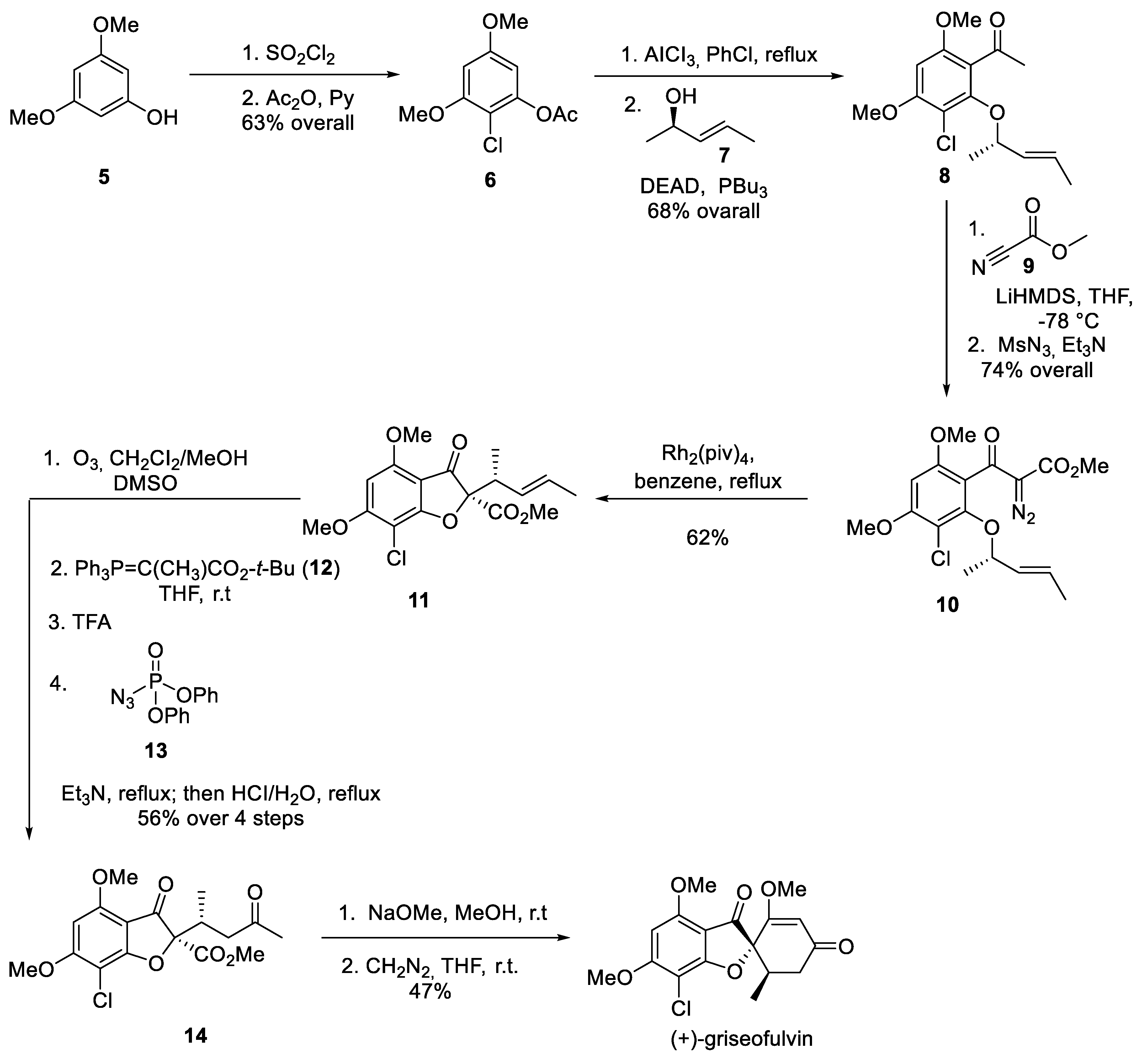
Scheme 3.
Spironolactone synthesis by G. D. Searle and Co.
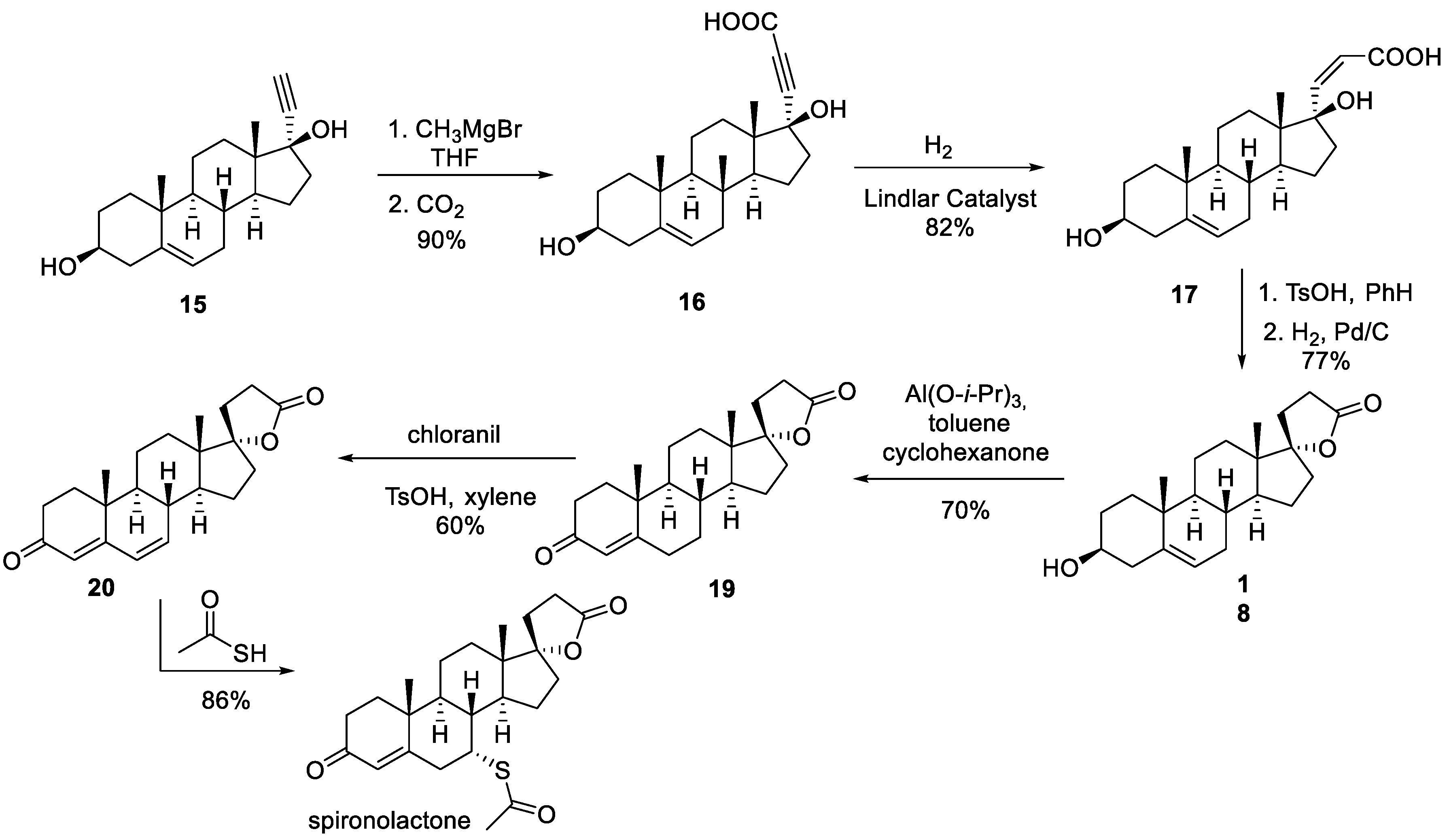
Scheme 4.
Ciba Geigy industrial synthesis of spironolactone.
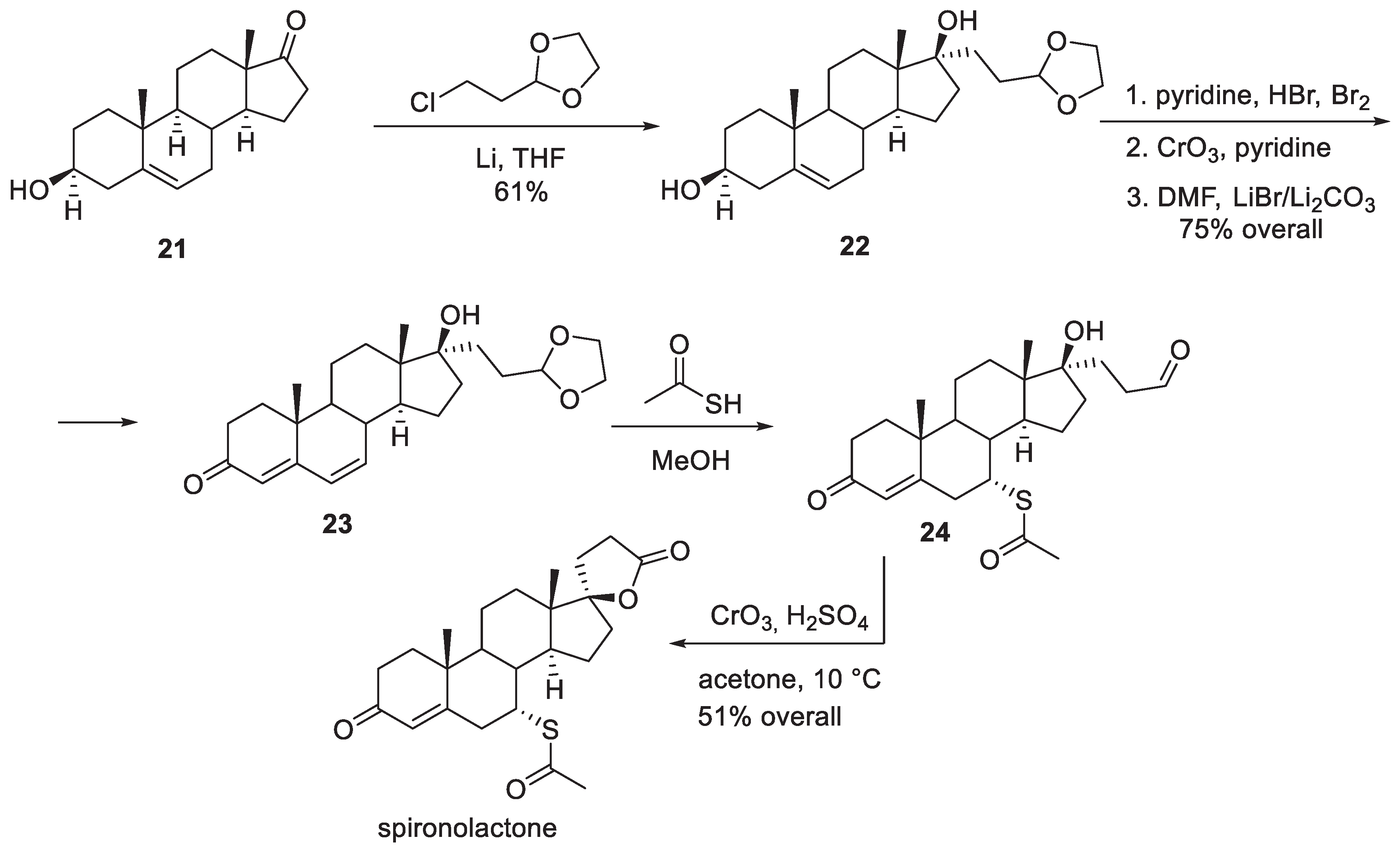
Scheme 5.
The Upjohn Co. synthesis of spironolactone.
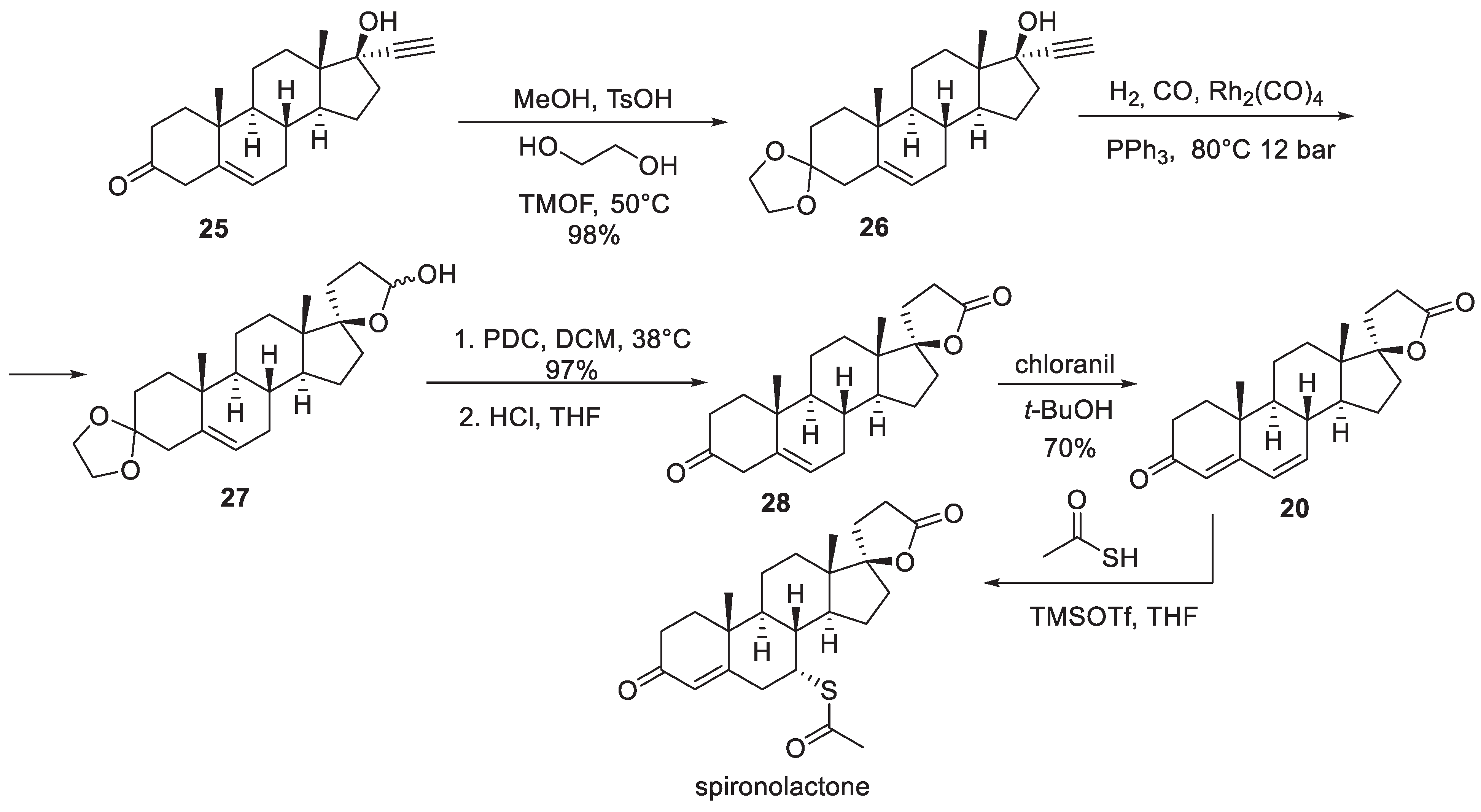
Scheme 6.
Synthesis of spironolactone via -lactonization of homopropargyl alcohol.
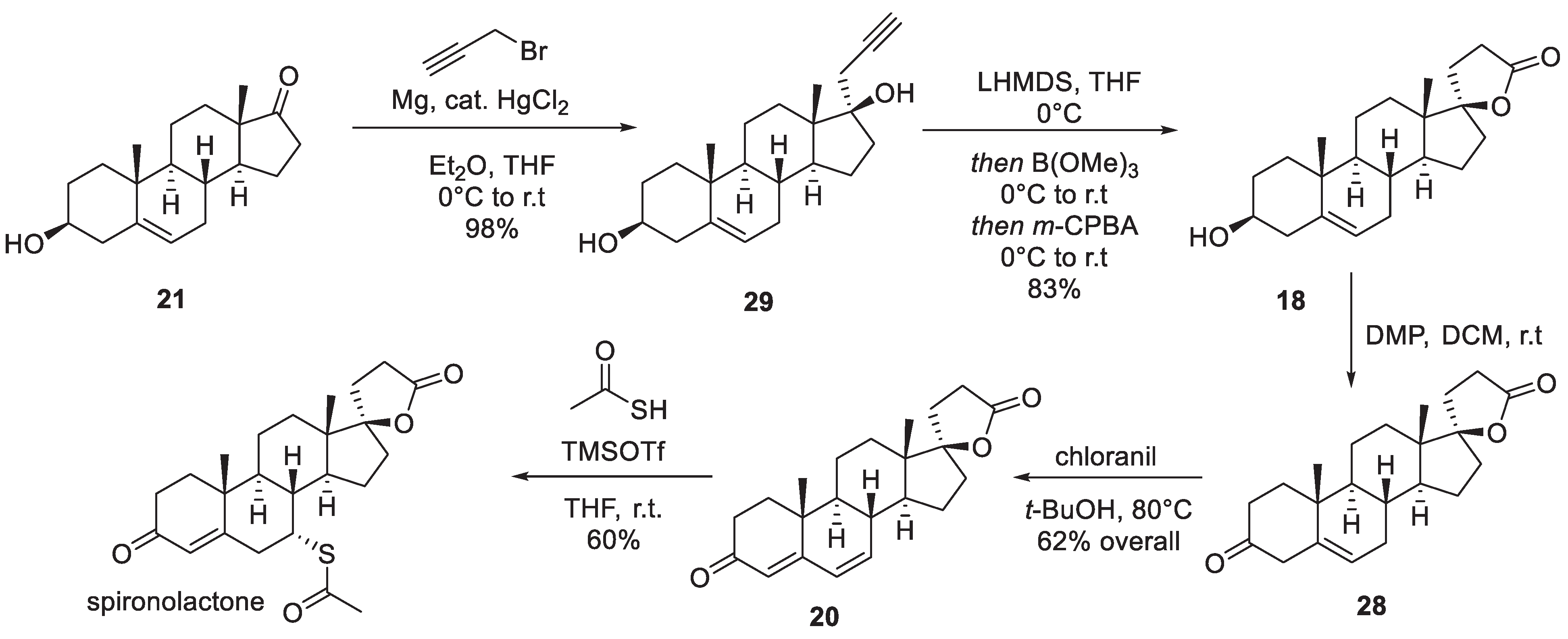
Scheme 7.
A 2018 synthesis of the core piperidine spirocycle needed for fluspirilene assembly.
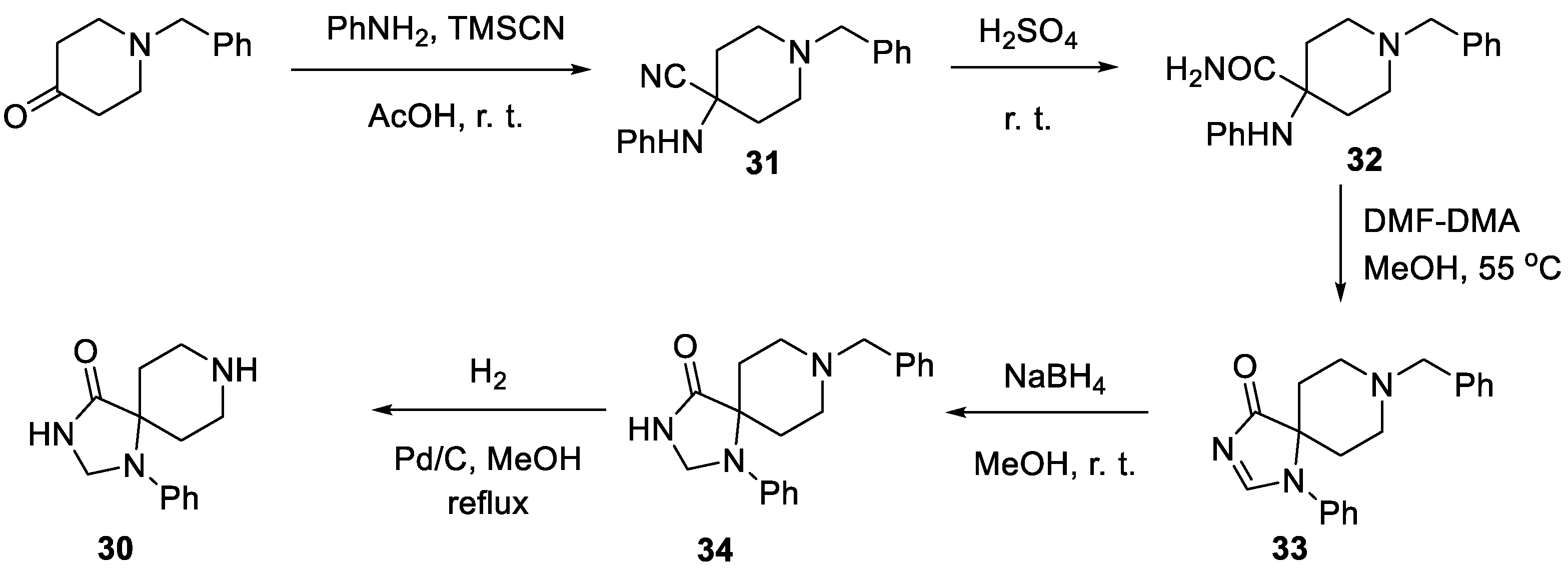
Scheme 8.
A 2001 synthesis of fluspirilene from an Italian group.
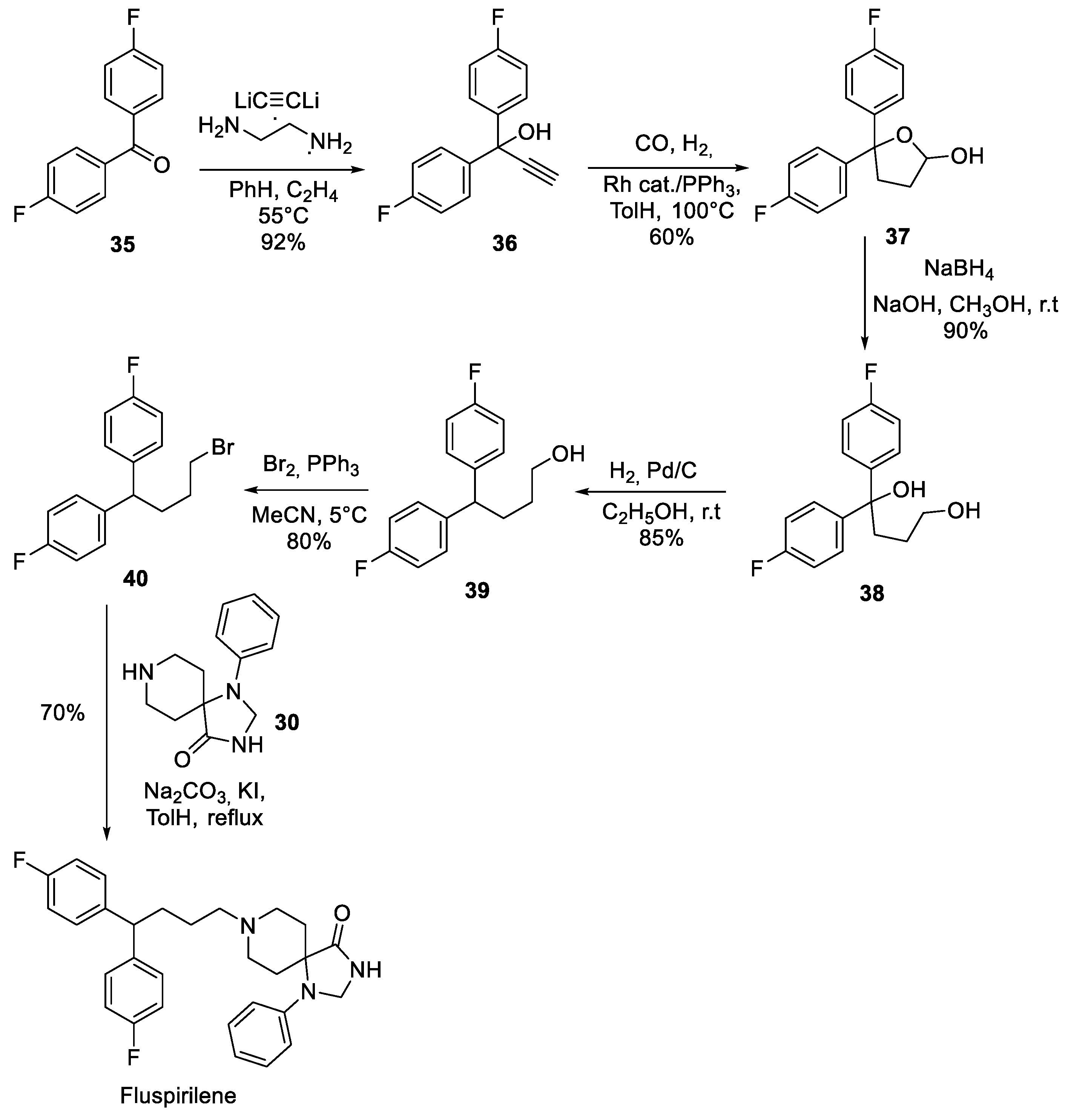
Scheme 9.
Fluspirilene synthesis by Shanghai Institute of Organic Chemistry.
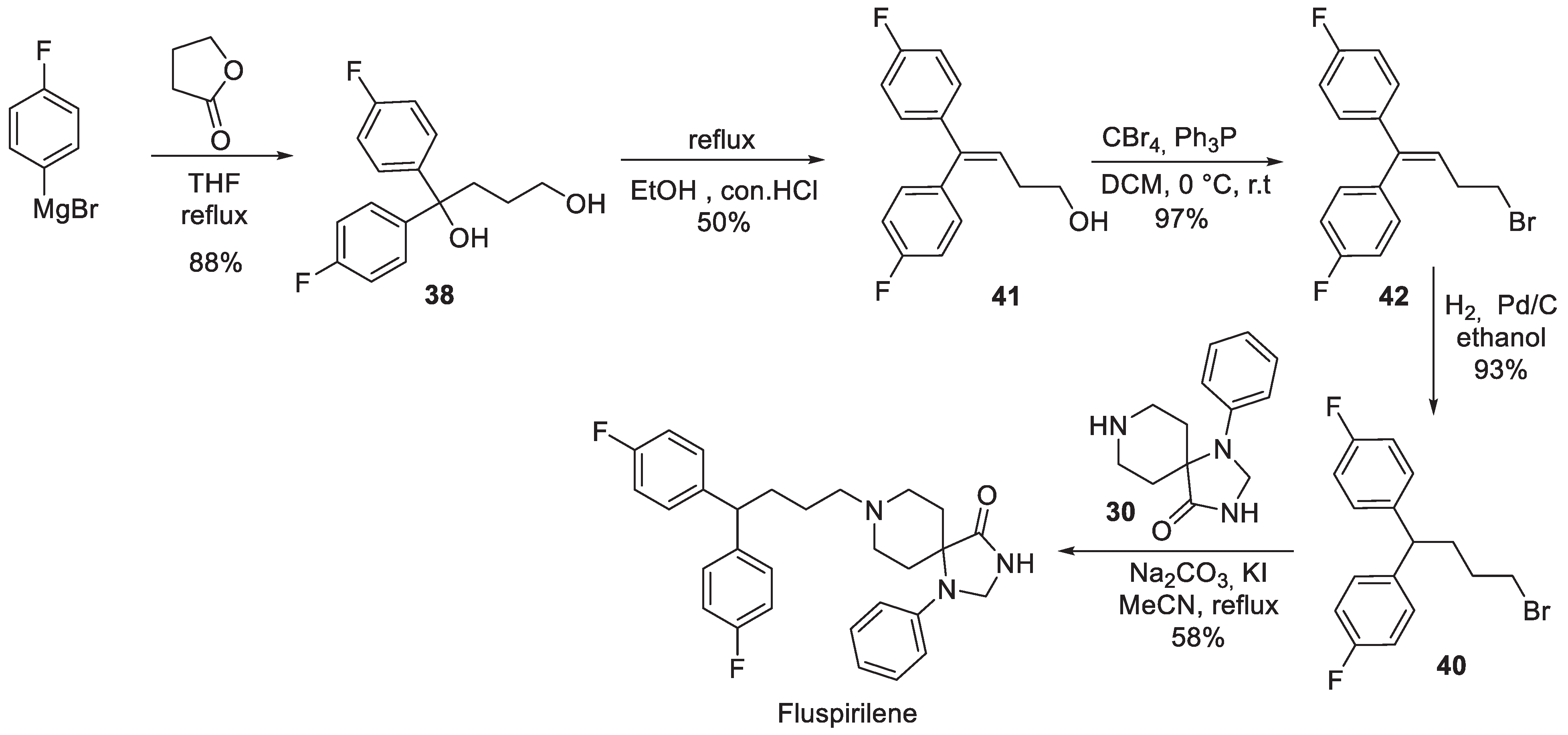
Scheme 10.
Synthesis of fenspiride by Voghera company.
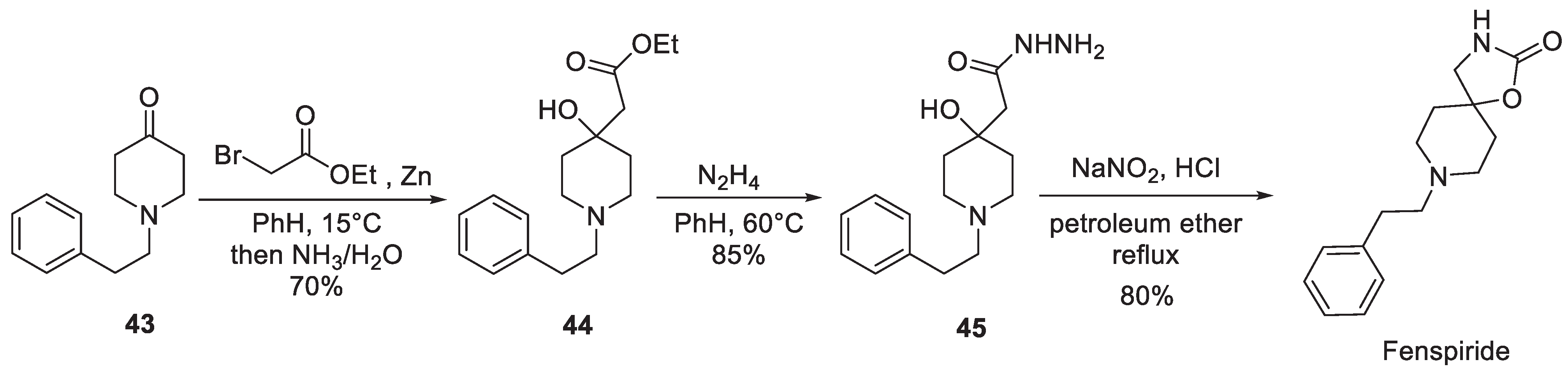
Scheme 11.
Synthesis of fenspiride by a Mexican group.
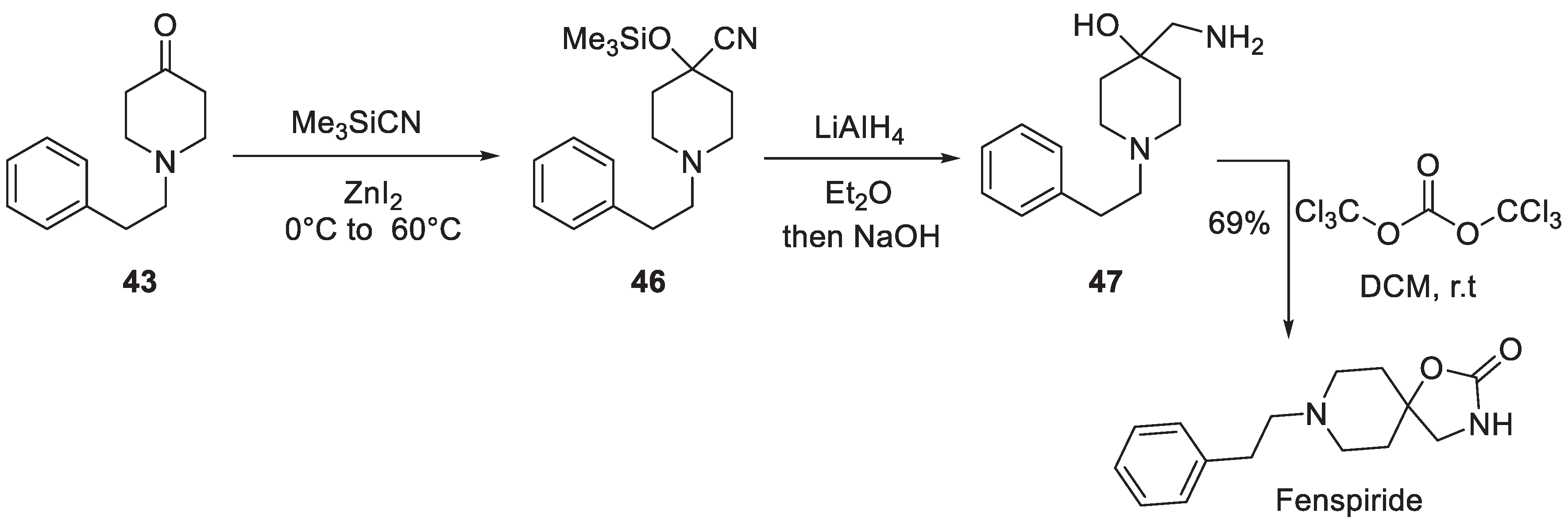
Scheme 12.
Synthesis of fenspiride by Emcure Pharmaceuticals.
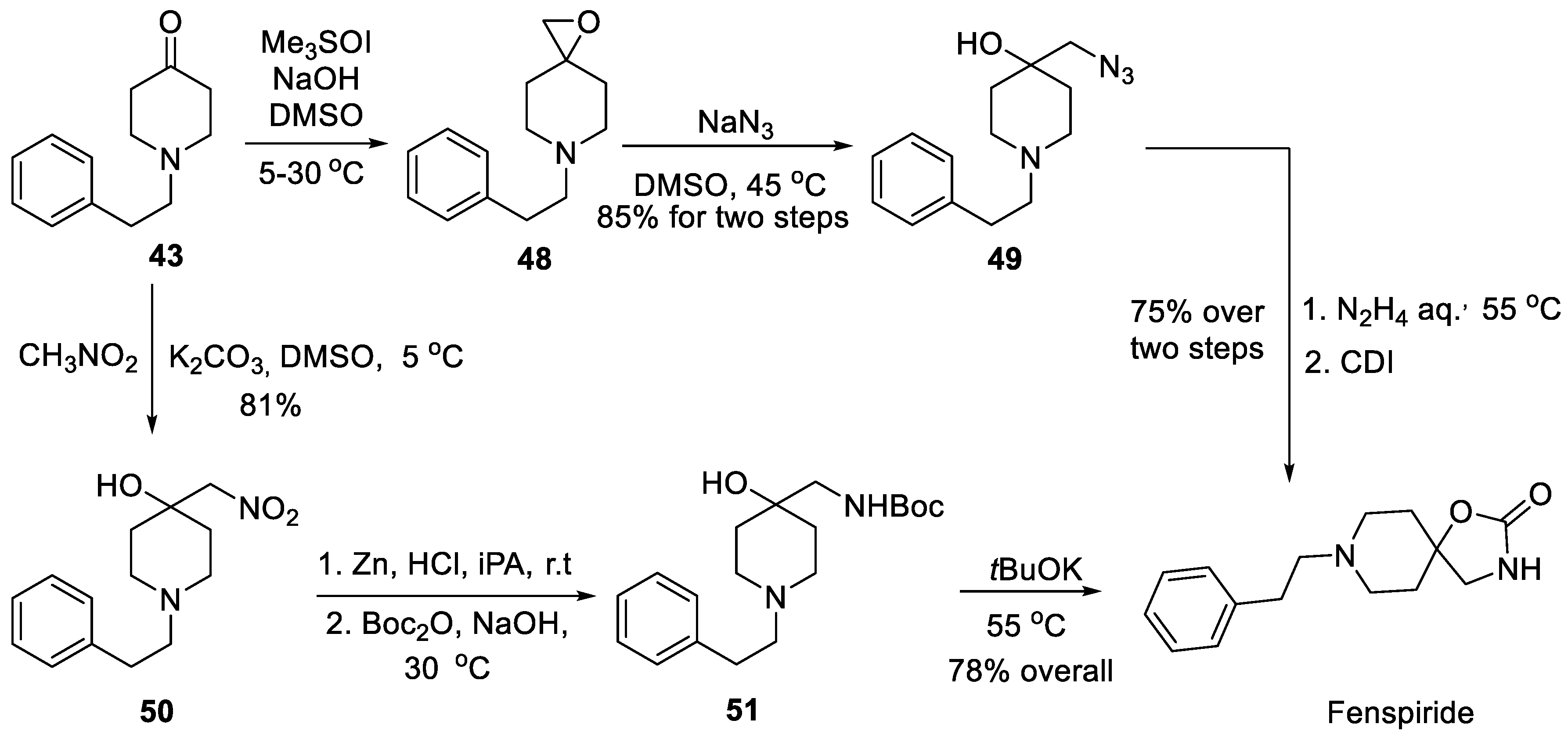
Scheme 13.
Synthesis of amcinonide.
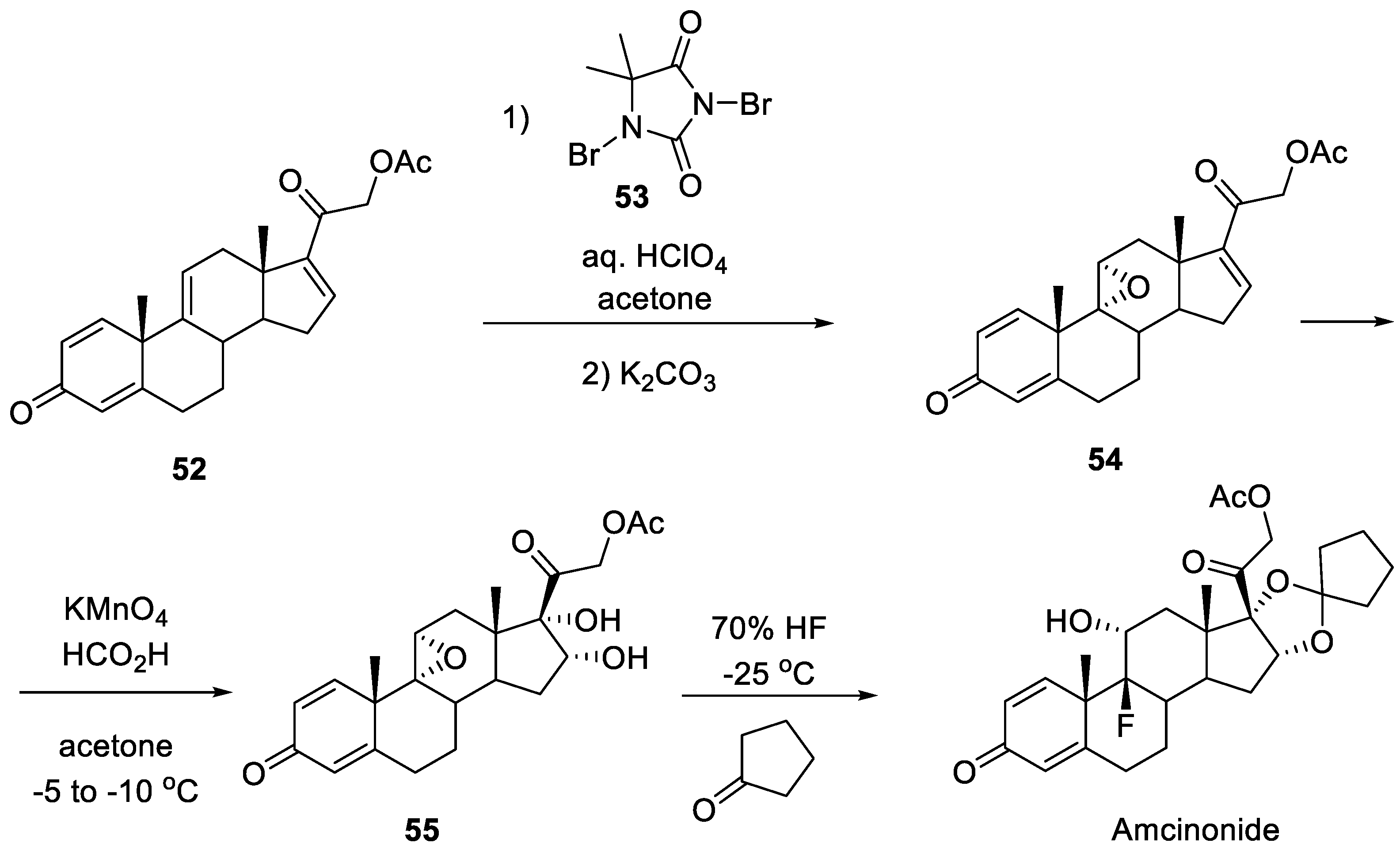
Scheme 14.
Industrial synthesis of guanadrel.
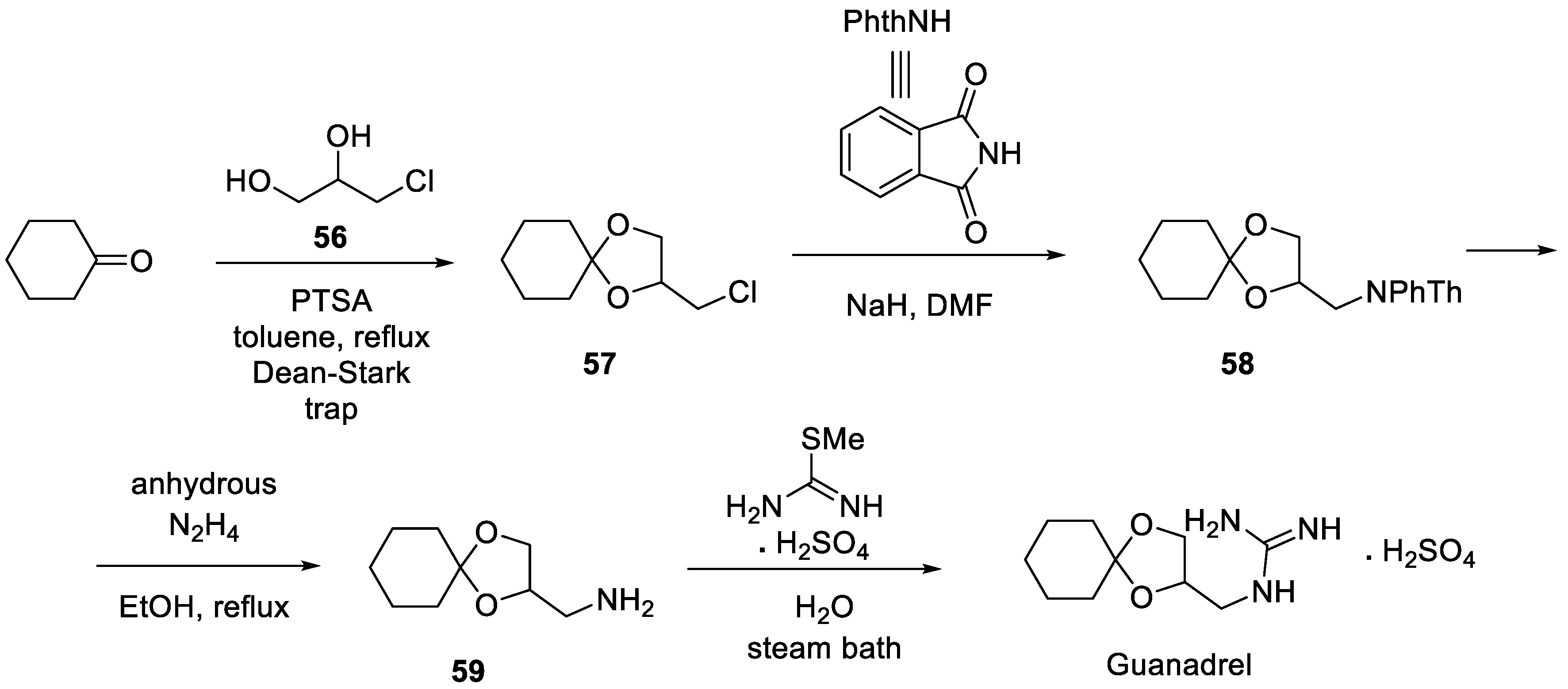
Scheme 15.
Synthesis of guanadrel by Goodman.
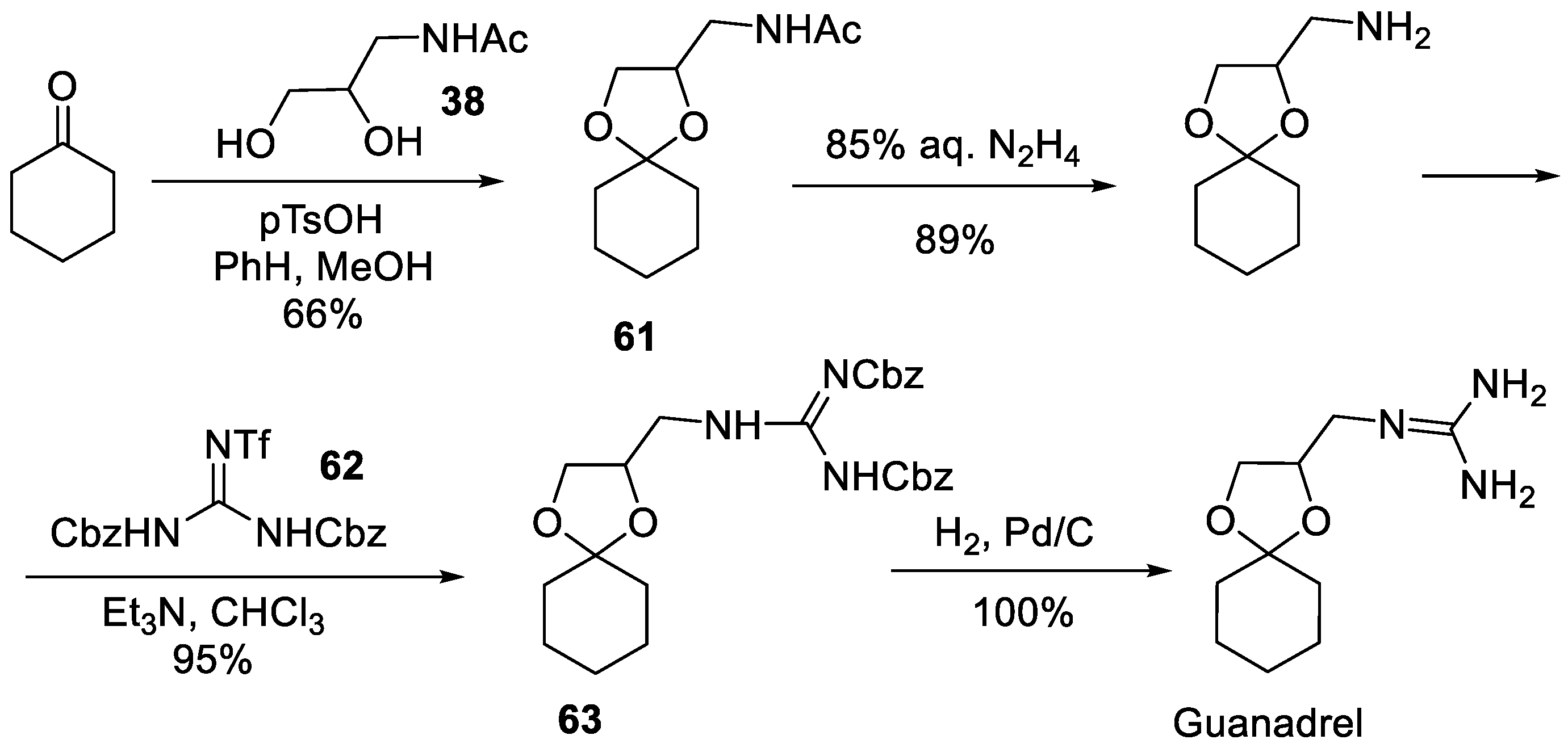
Scheme 16.
Industry synthesis of buspirone.
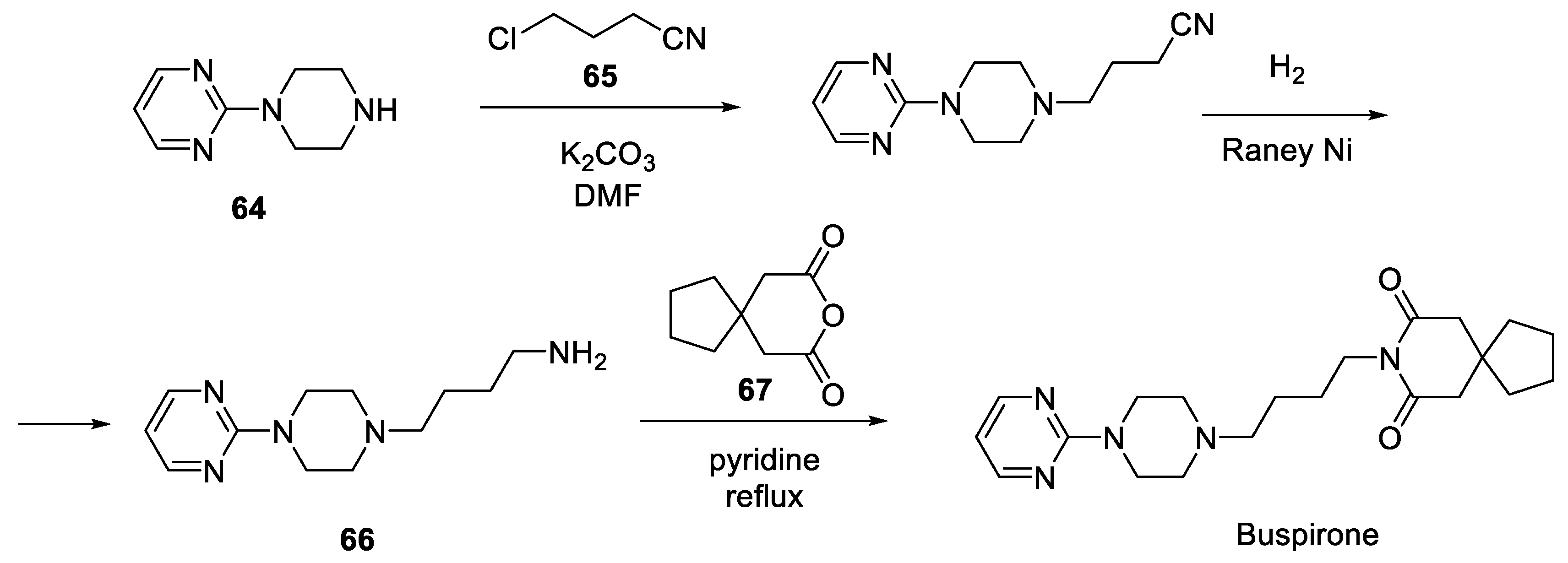
Scheme 17.
Synthesis of buspirone by Wei and co-workers (2008).
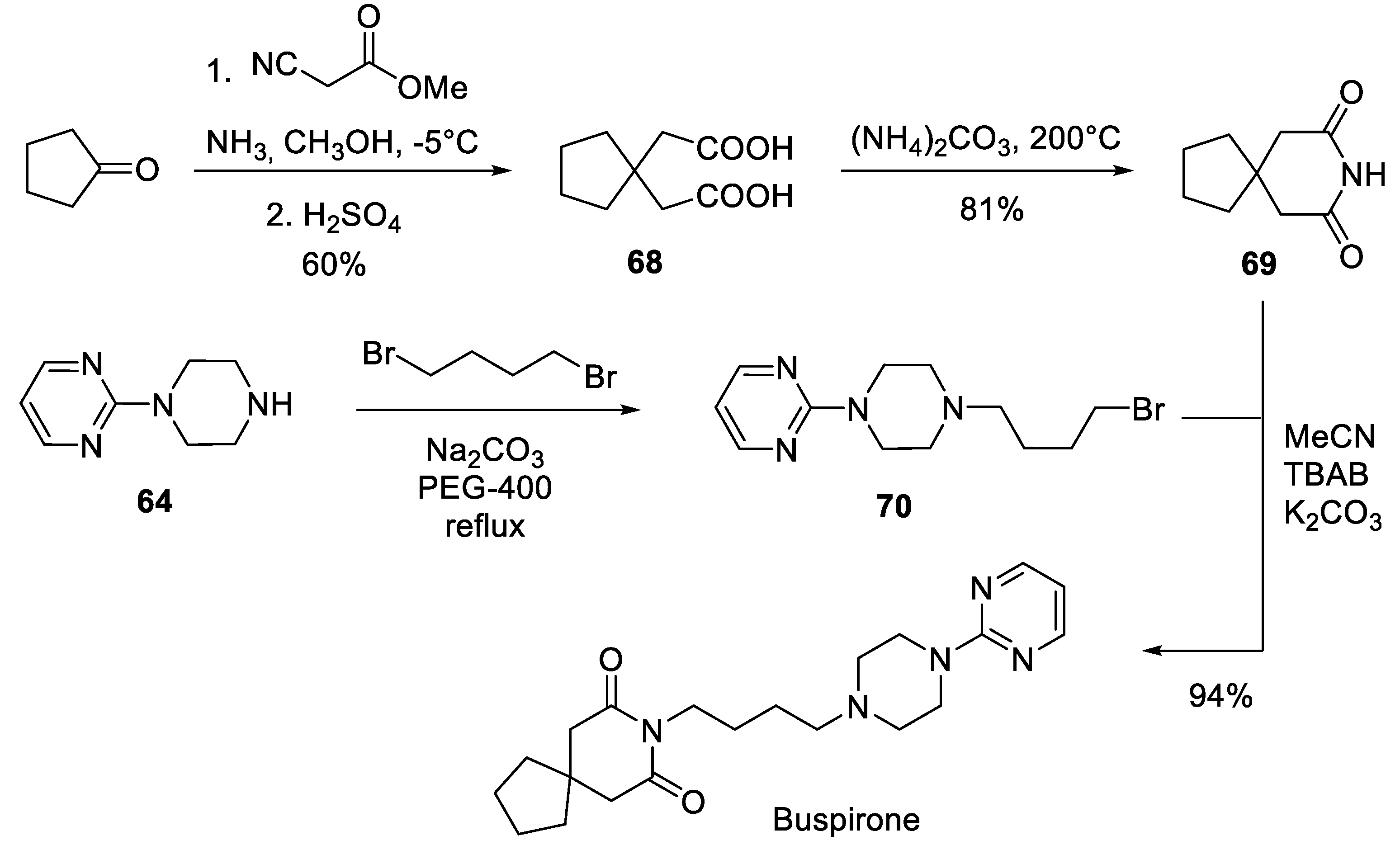
Scheme 18.
Preparation of buspirone involving piperazine alkylation via hydrogen borrowing.
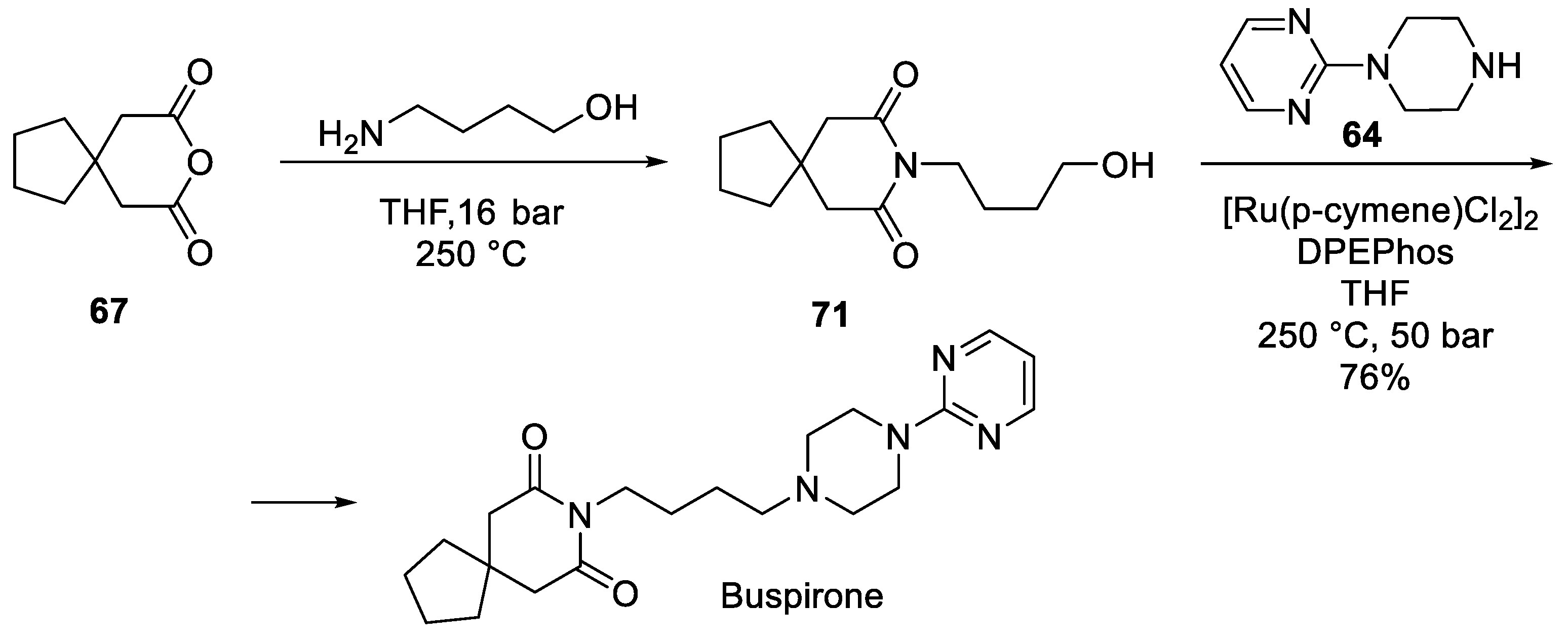
Scheme 19.
Preparation of buspirone involving reductive cross-coupling of 64 with nitrile 72.
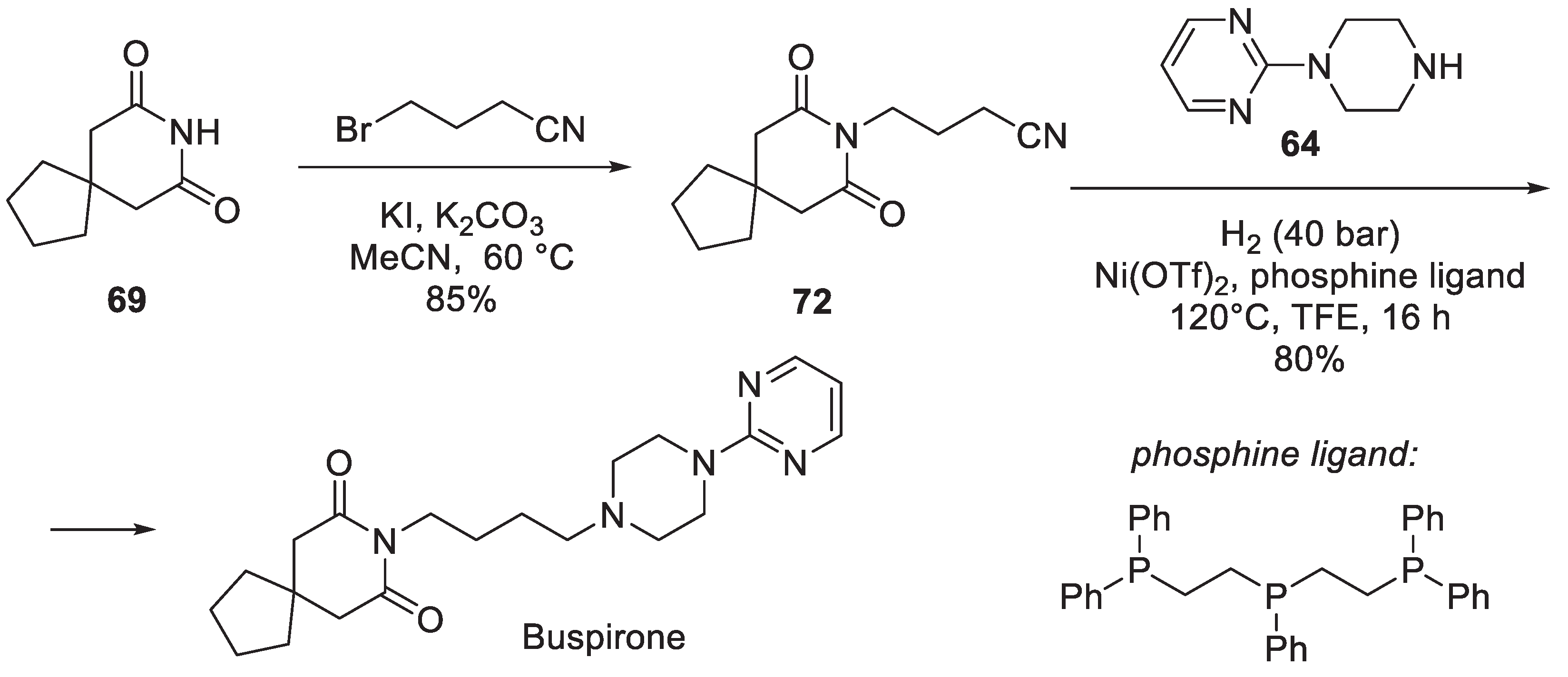
Scheme 20.
Hydrogenation of avermectins B1a and B1b.
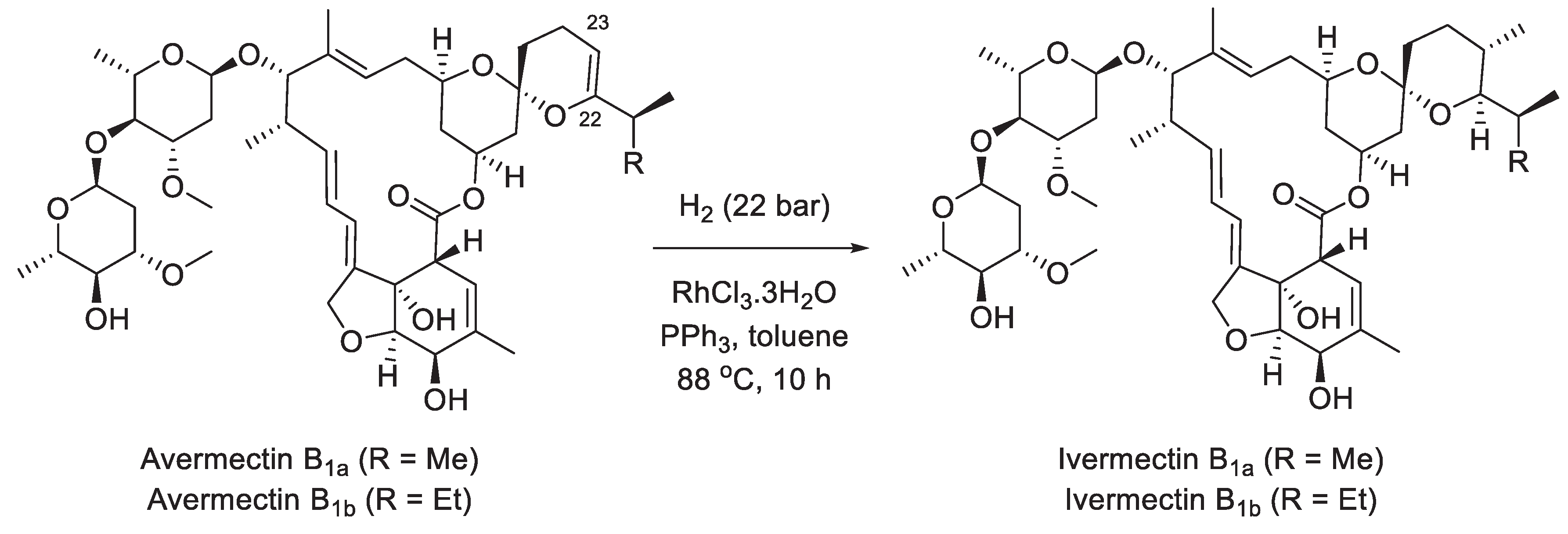
Scheme 21.
Farmitalia synthesis of rifabutin.
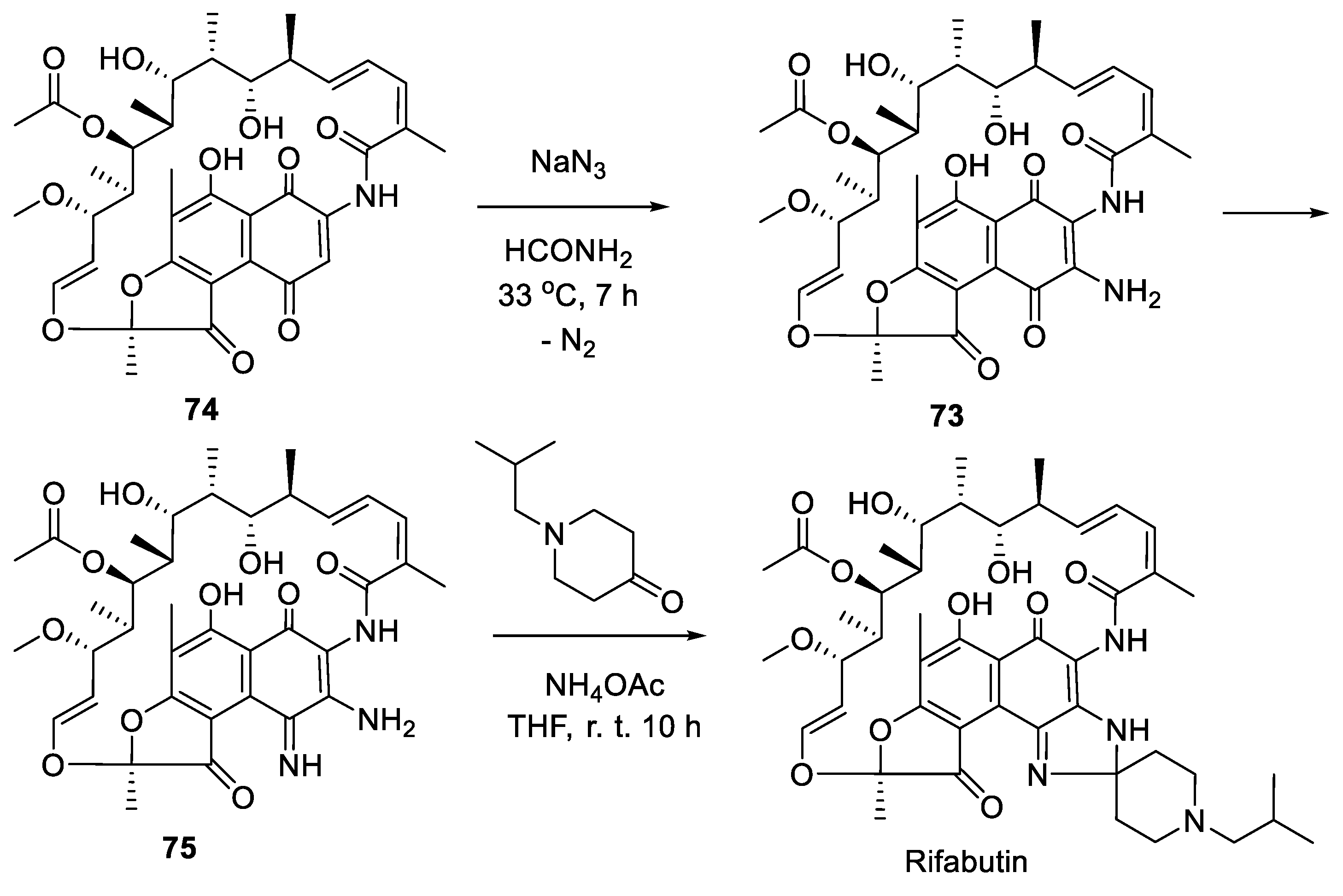
Scheme 22.
Synthesis of spirapril by Schering-Plough Corporation.
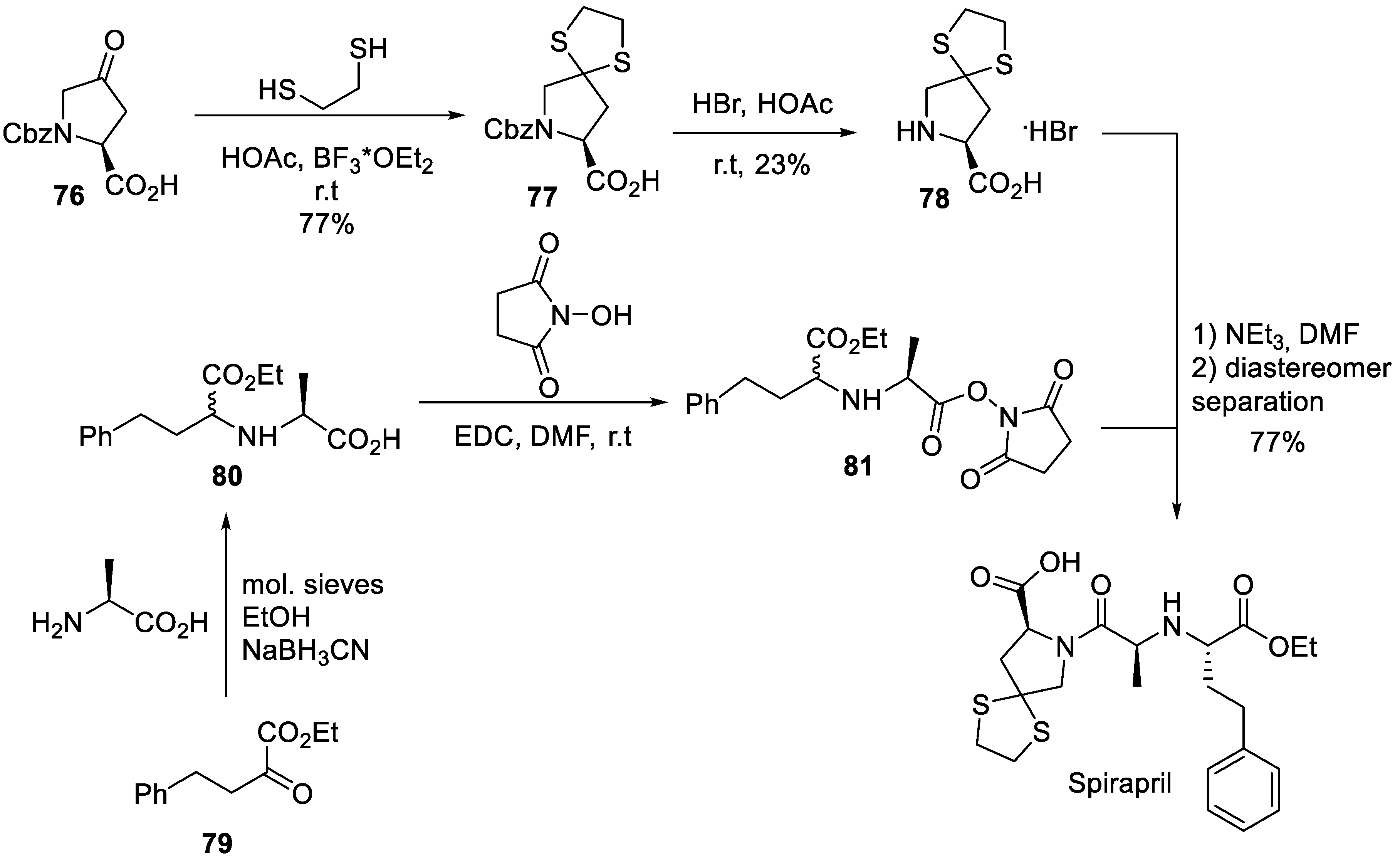
Scheme 23.
Synthesis of spirapril by Squibb Corporation.
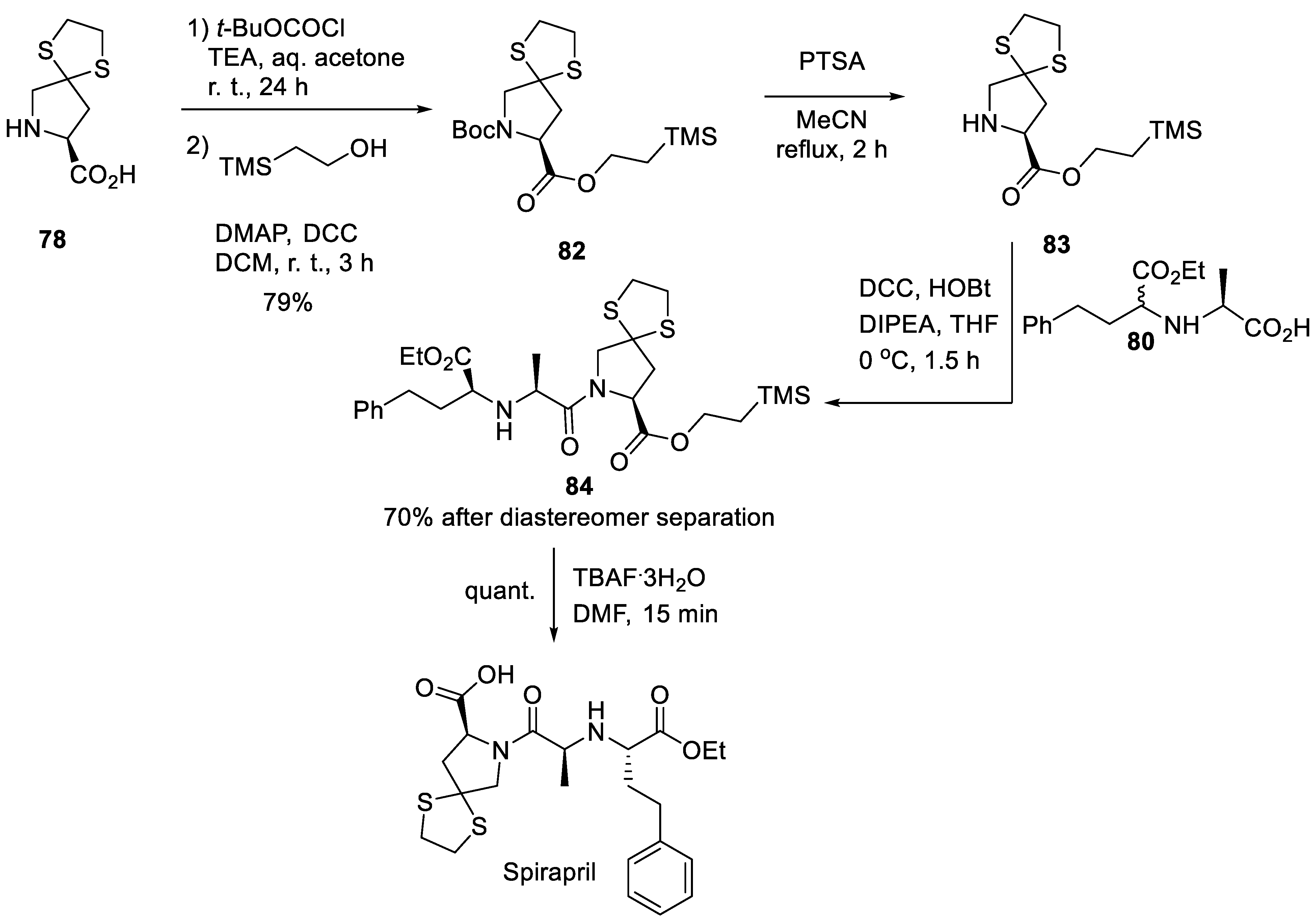
Scheme 24.
Original route to irbesartan by Sanofi Research. .
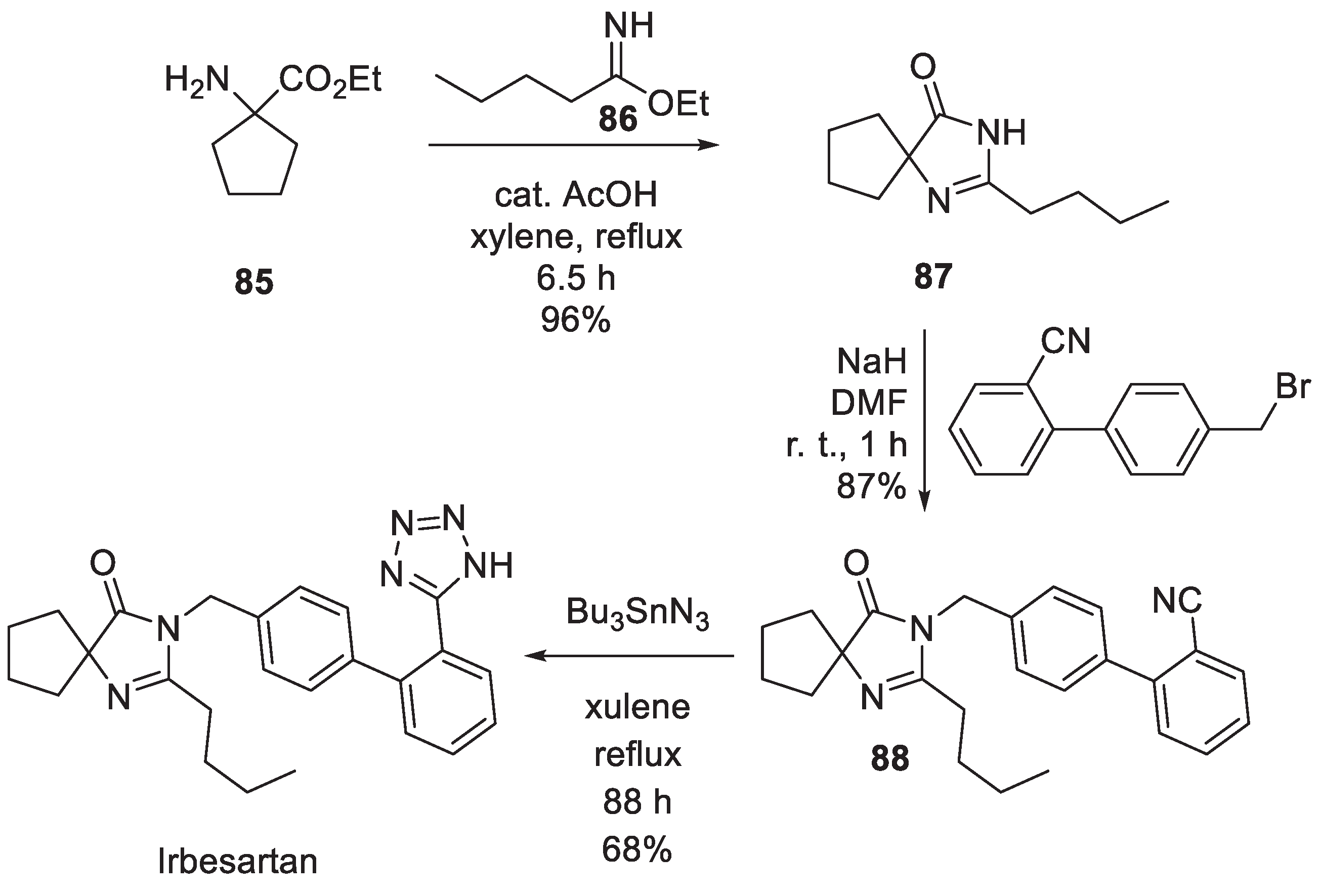
Scheme 25.
Synthesis of key building block 87.
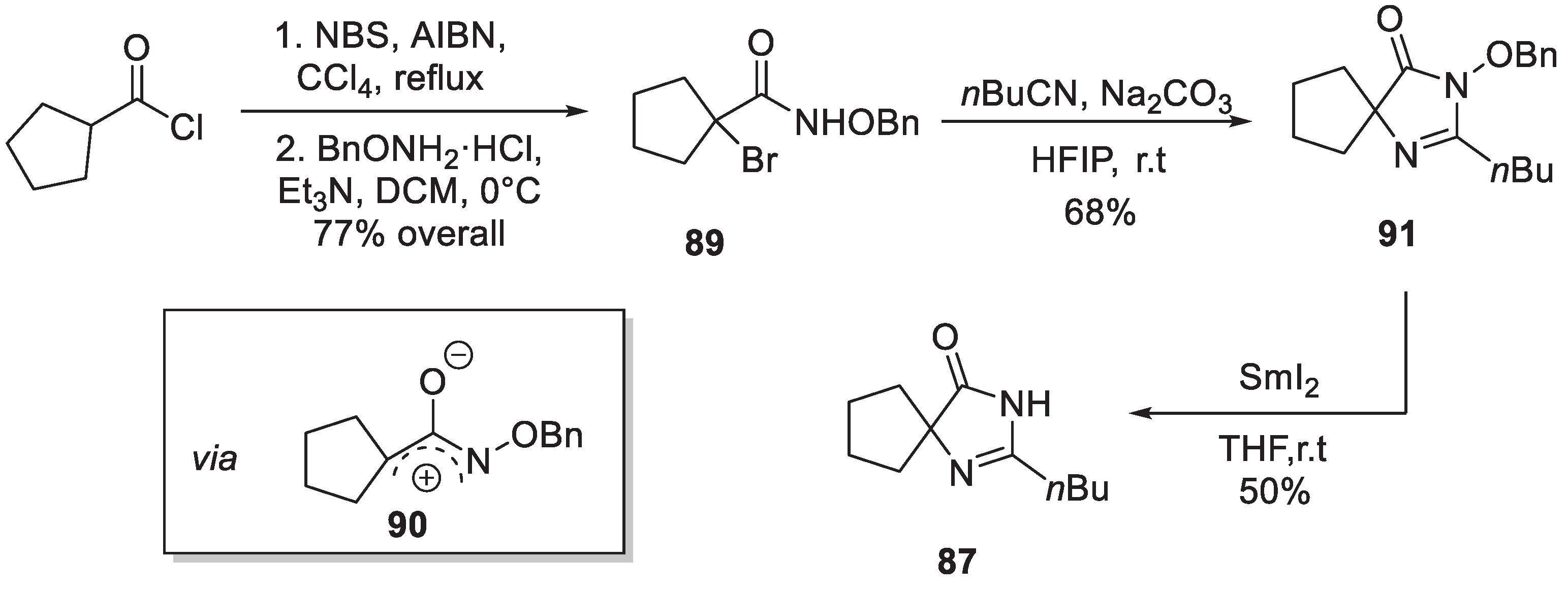
Scheme 26.
Synthesis of irbesartan by Israeli group.
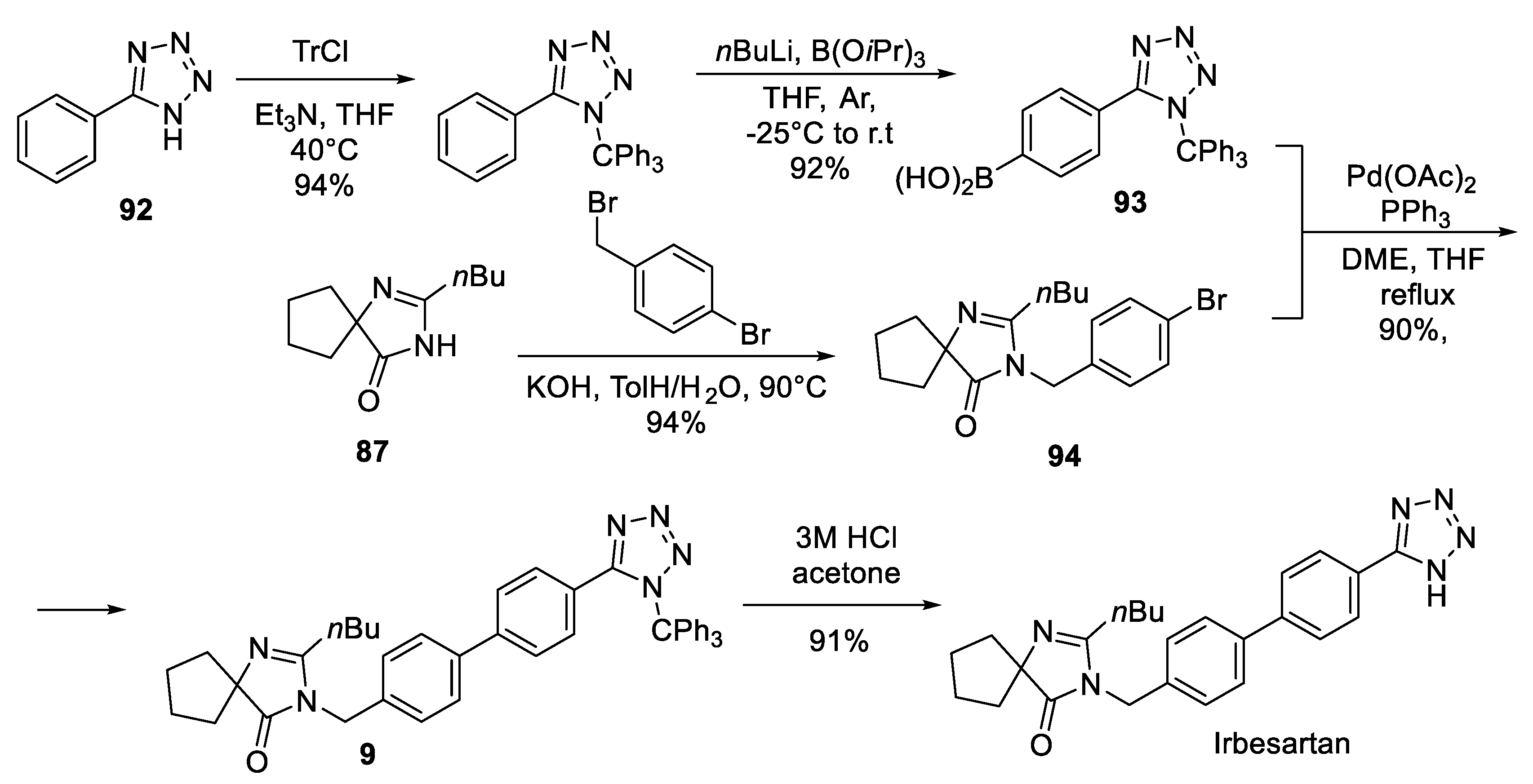
Scheme 27.
Dr. Reddy’s Laboratories synthesis of irbesartan.
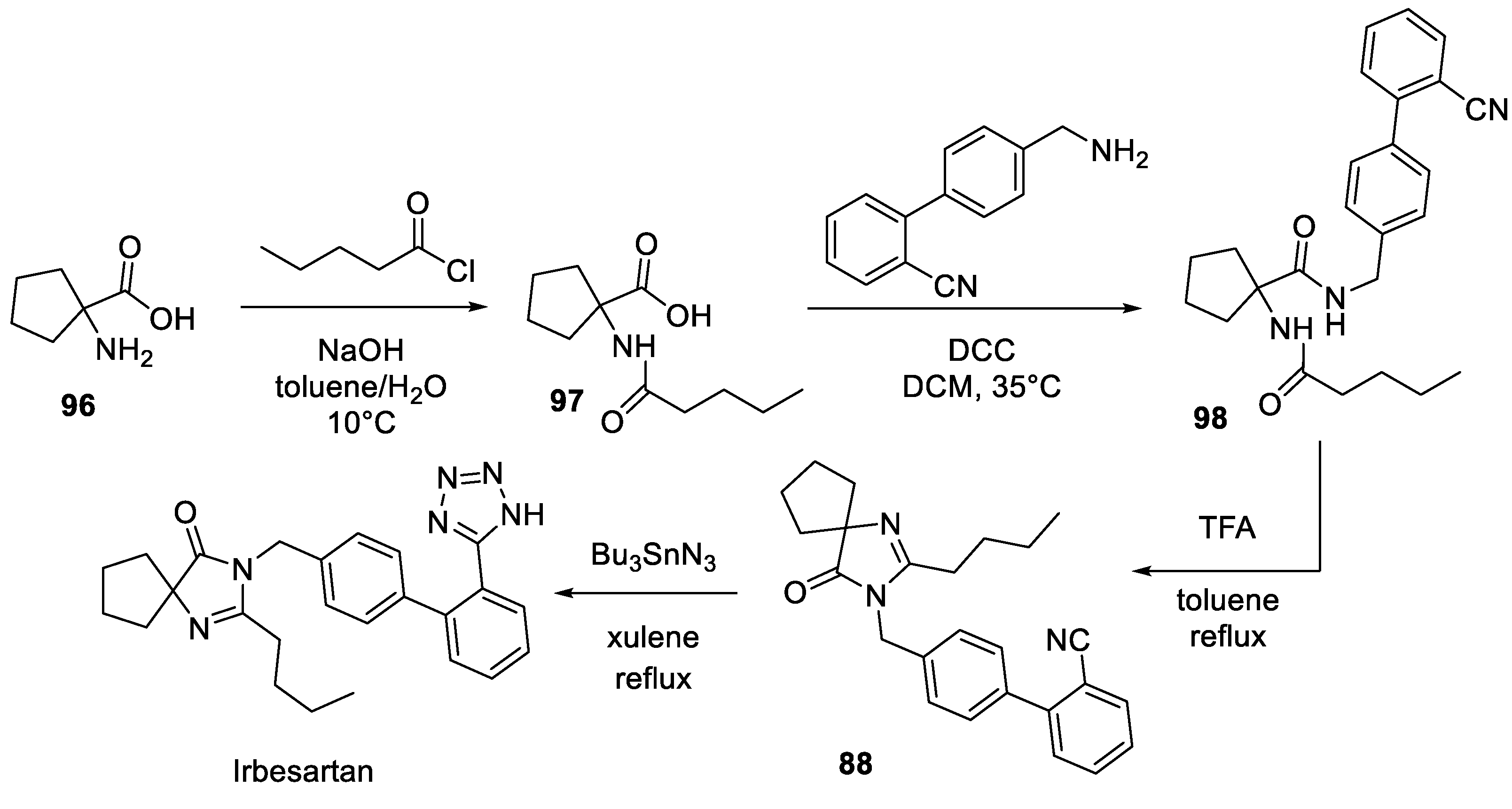
Scheme 28.
ArQule synthesis of irbesartan.
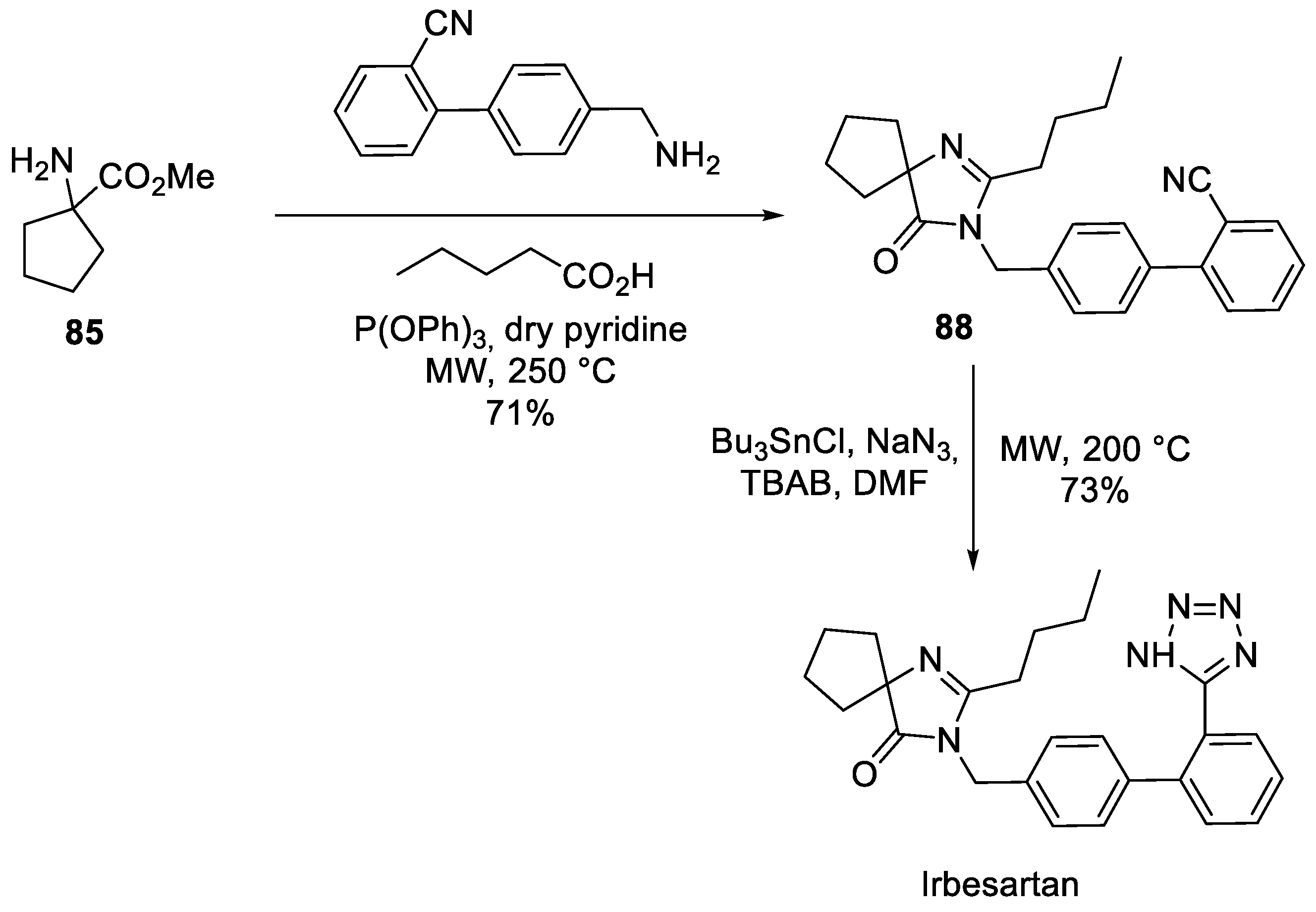
Scheme 29.
Beijing University of Chemical Technology route to irbesartan.
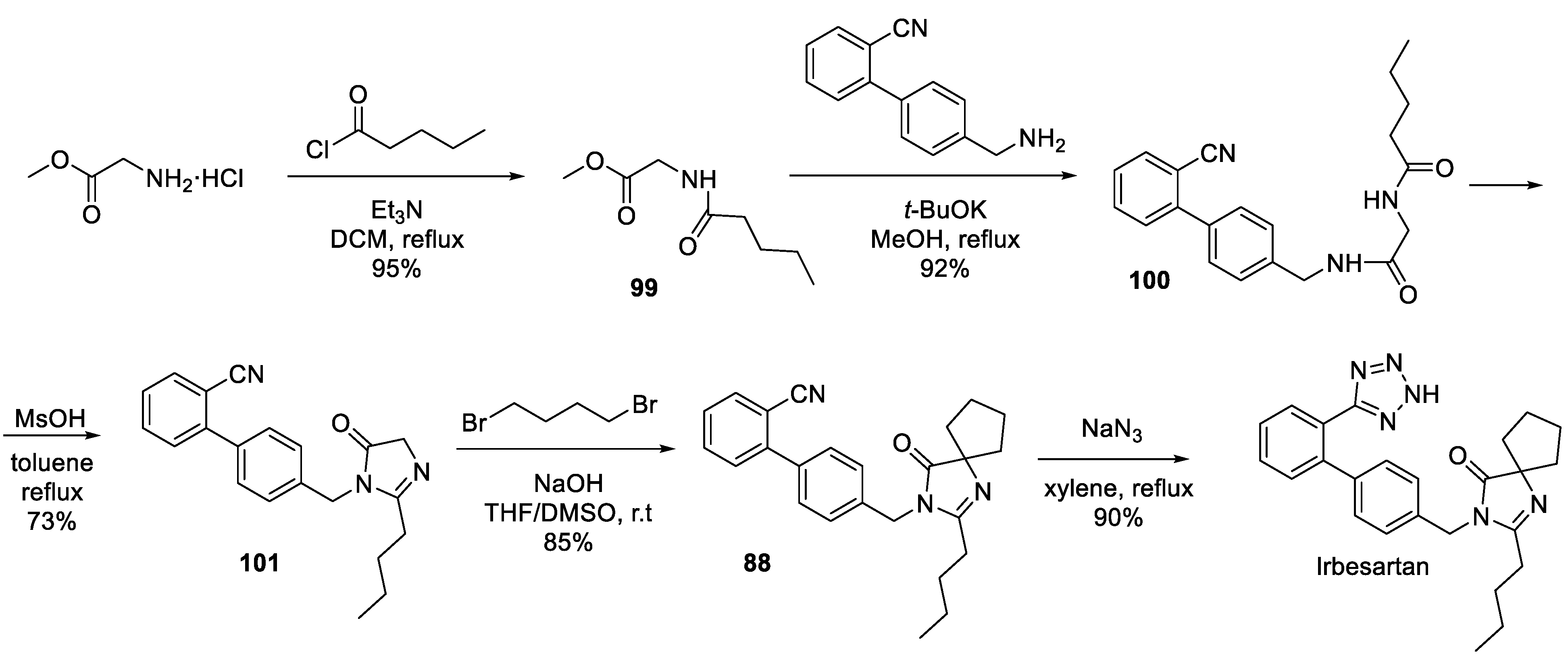
Scheme 30.
Original synthesis of cevimeline.
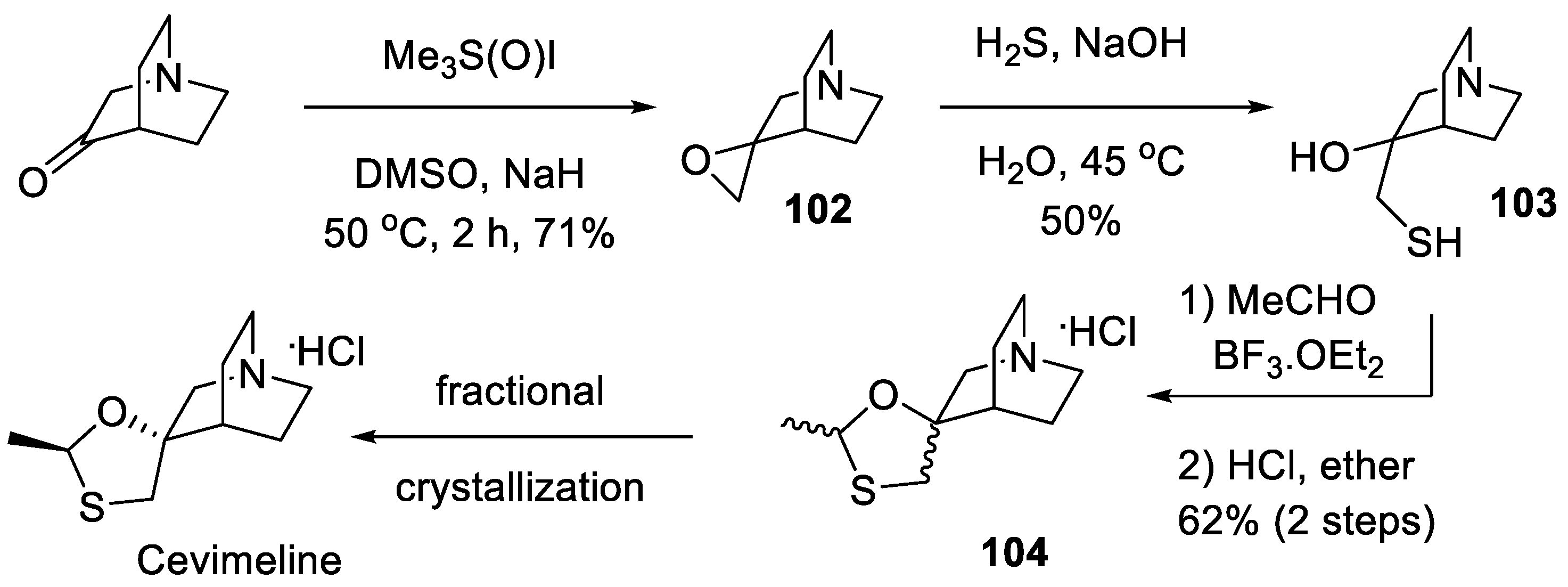
Scheme 31.
Cevimeline synthesis by Apotex Pharmachem, Inc.
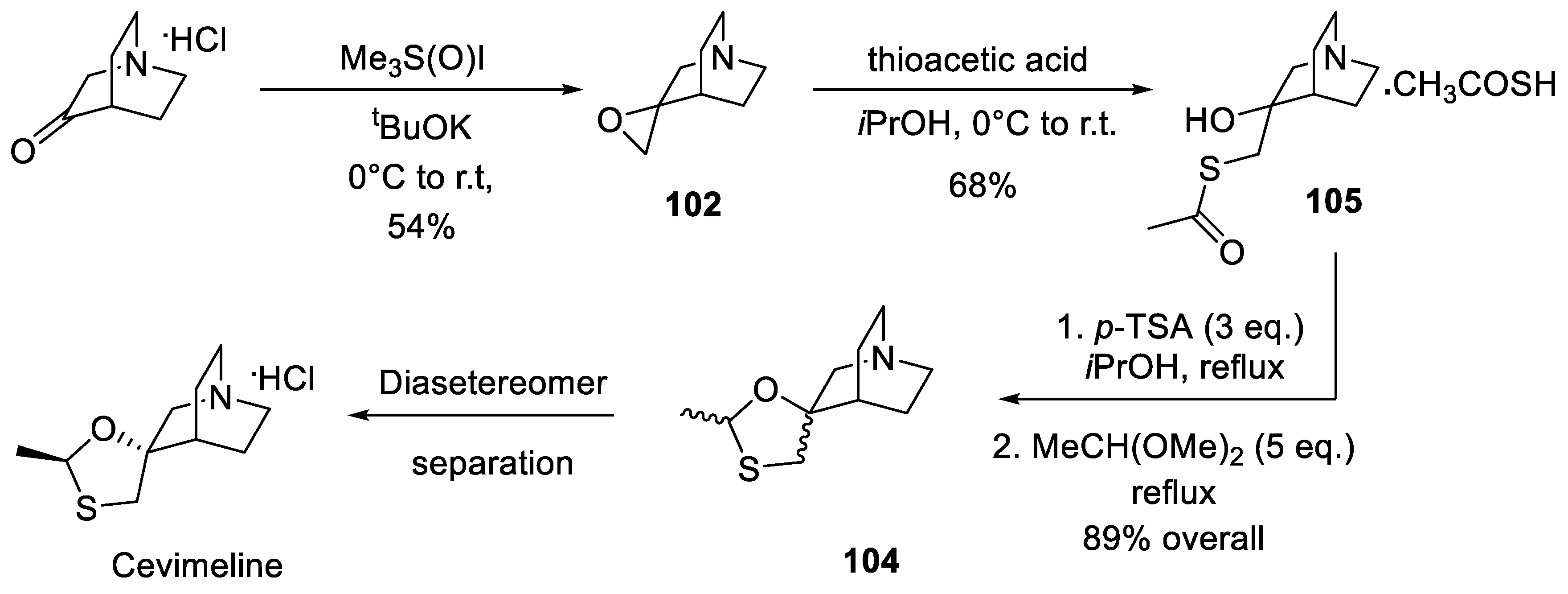
Scheme 32.
Cevimeline synthesis by Emcure Pharmaceuticals, Ltd.
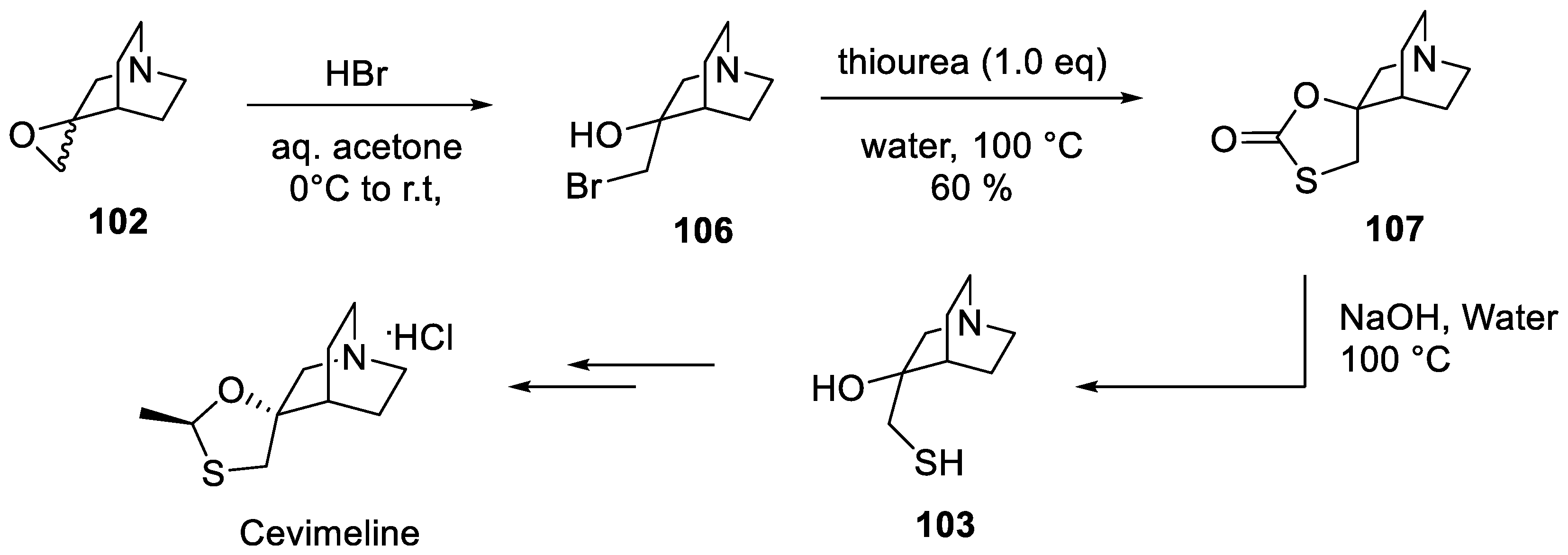
Scheme 33.
Synthesis of key intermediate (S)-103 by F. Hoffmann-La Roche, Ltd.
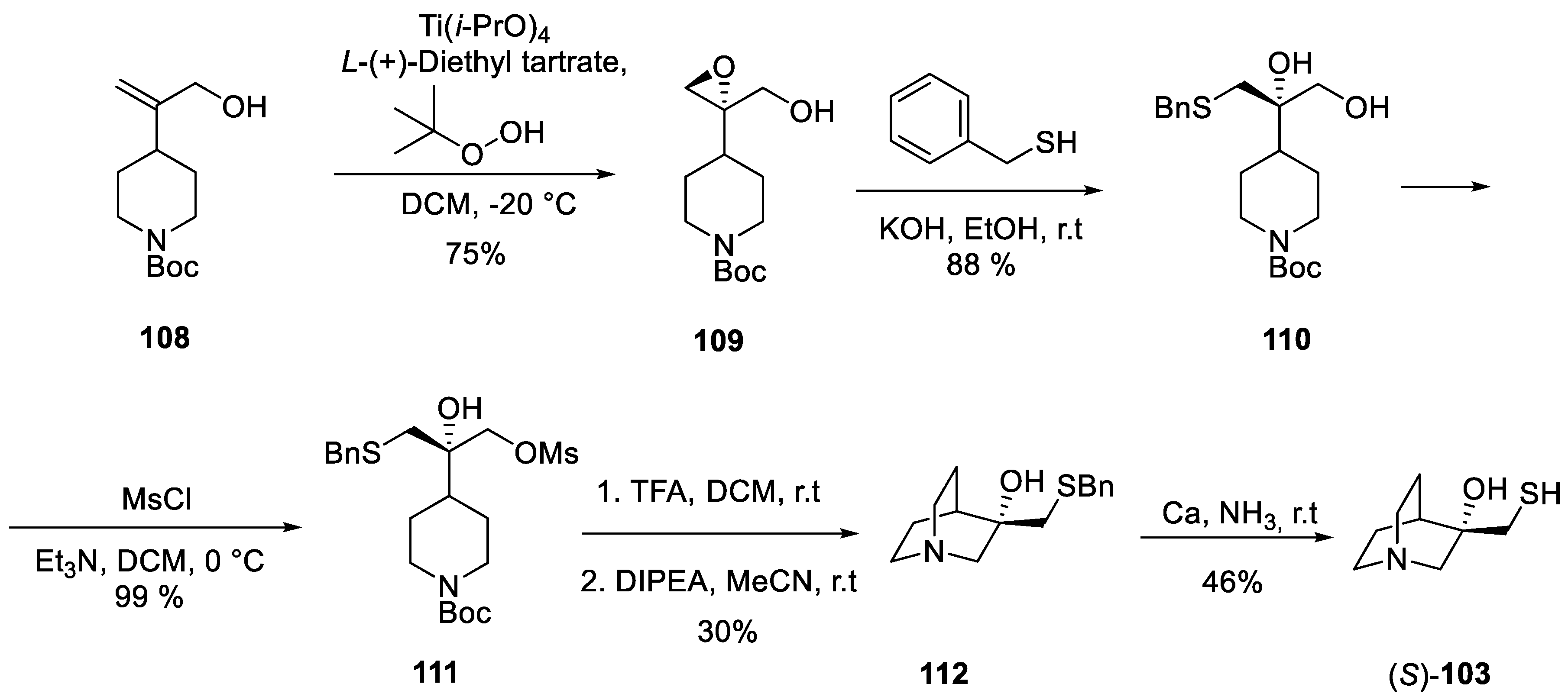
Scheme 34.
Synthesis of eplerenone by Searle and Co.
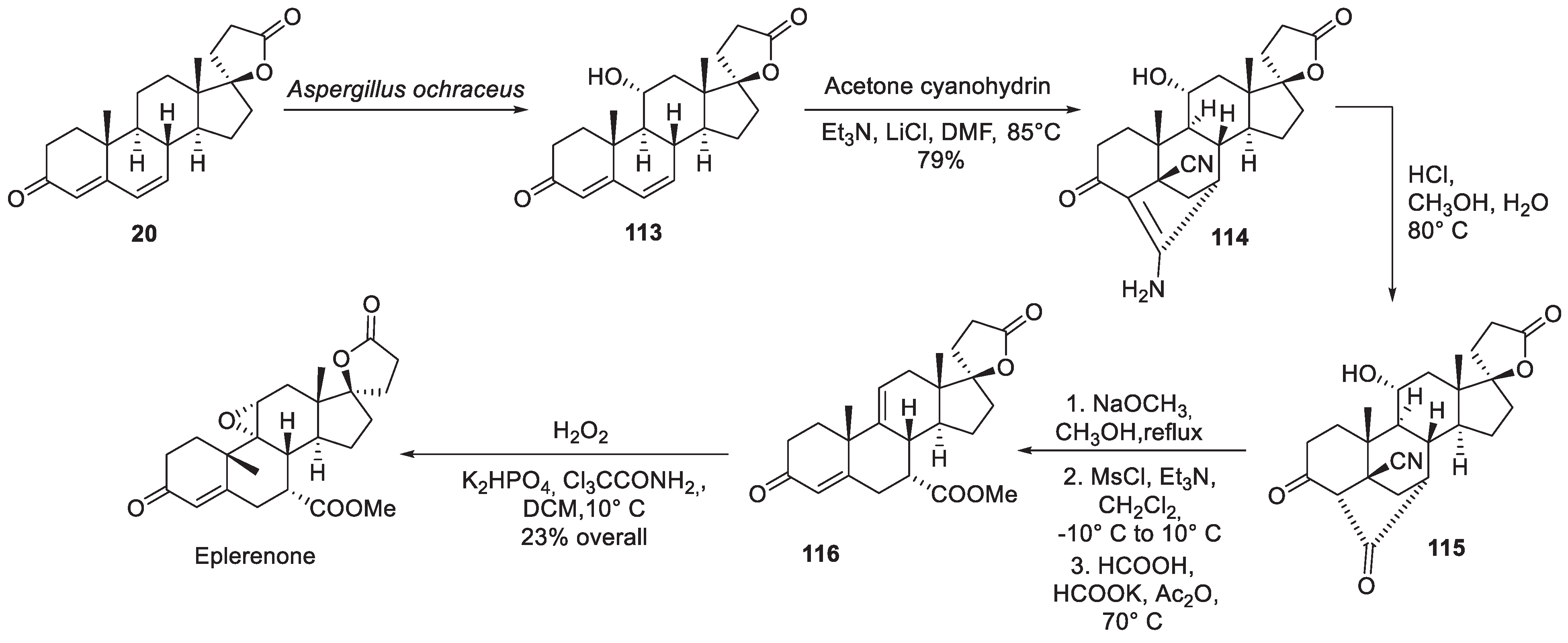
Scheme 35.
Synthesis of eplerenone by Ciba-Geigy.
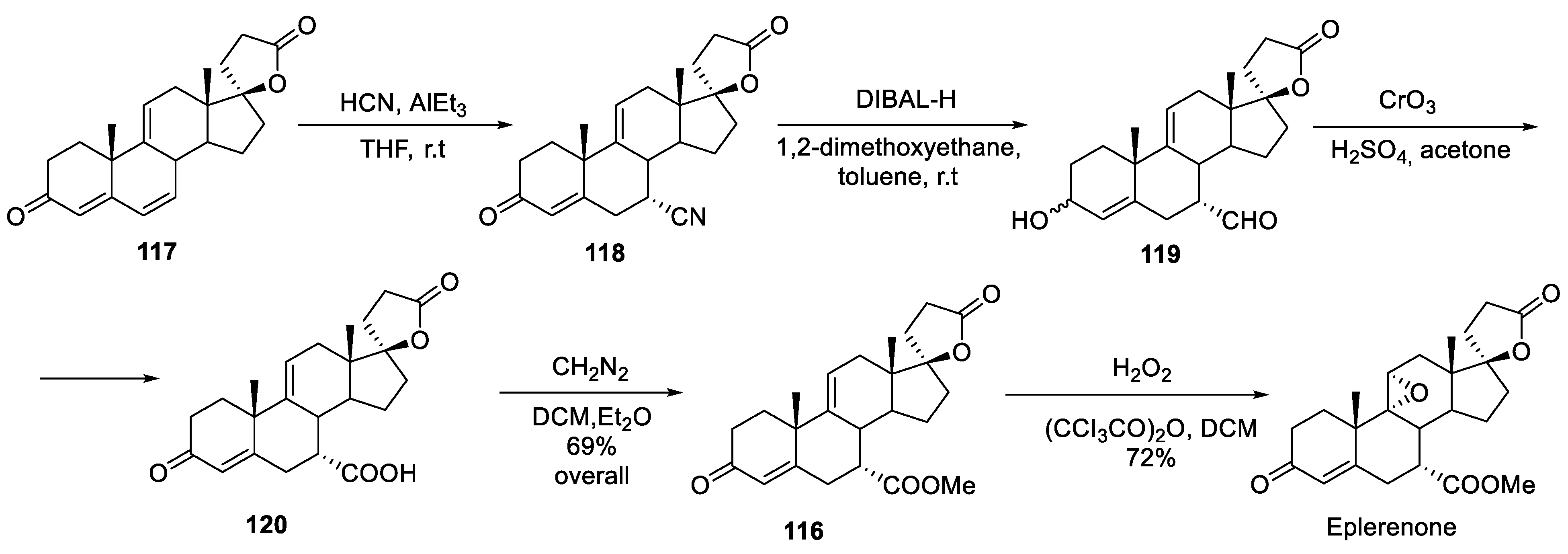
Scheme 36.
Synthesis of eplerenone by Pfizer.
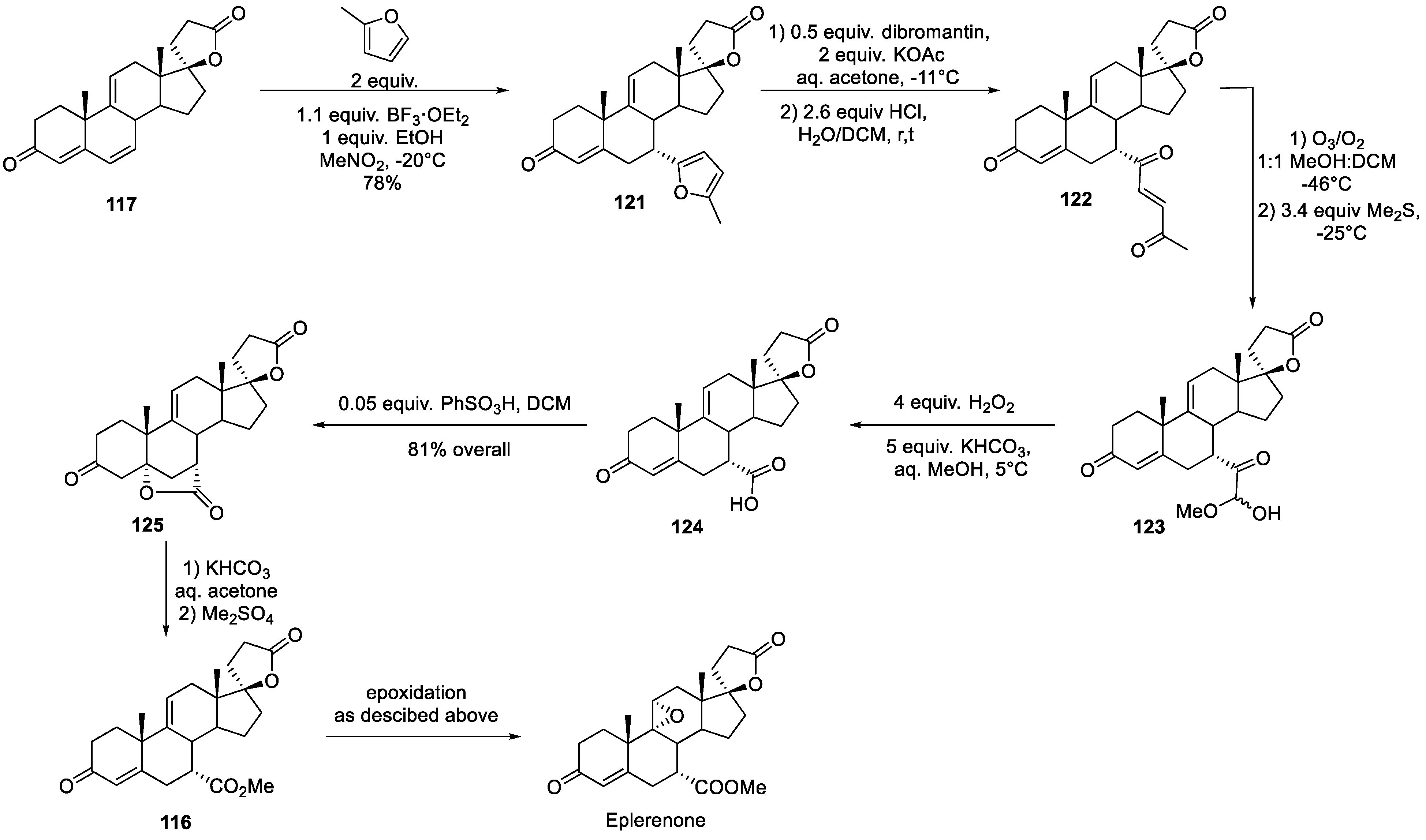
Scheme 37.
Synthesis of eplerenone by Chinese Academy of Sciences.
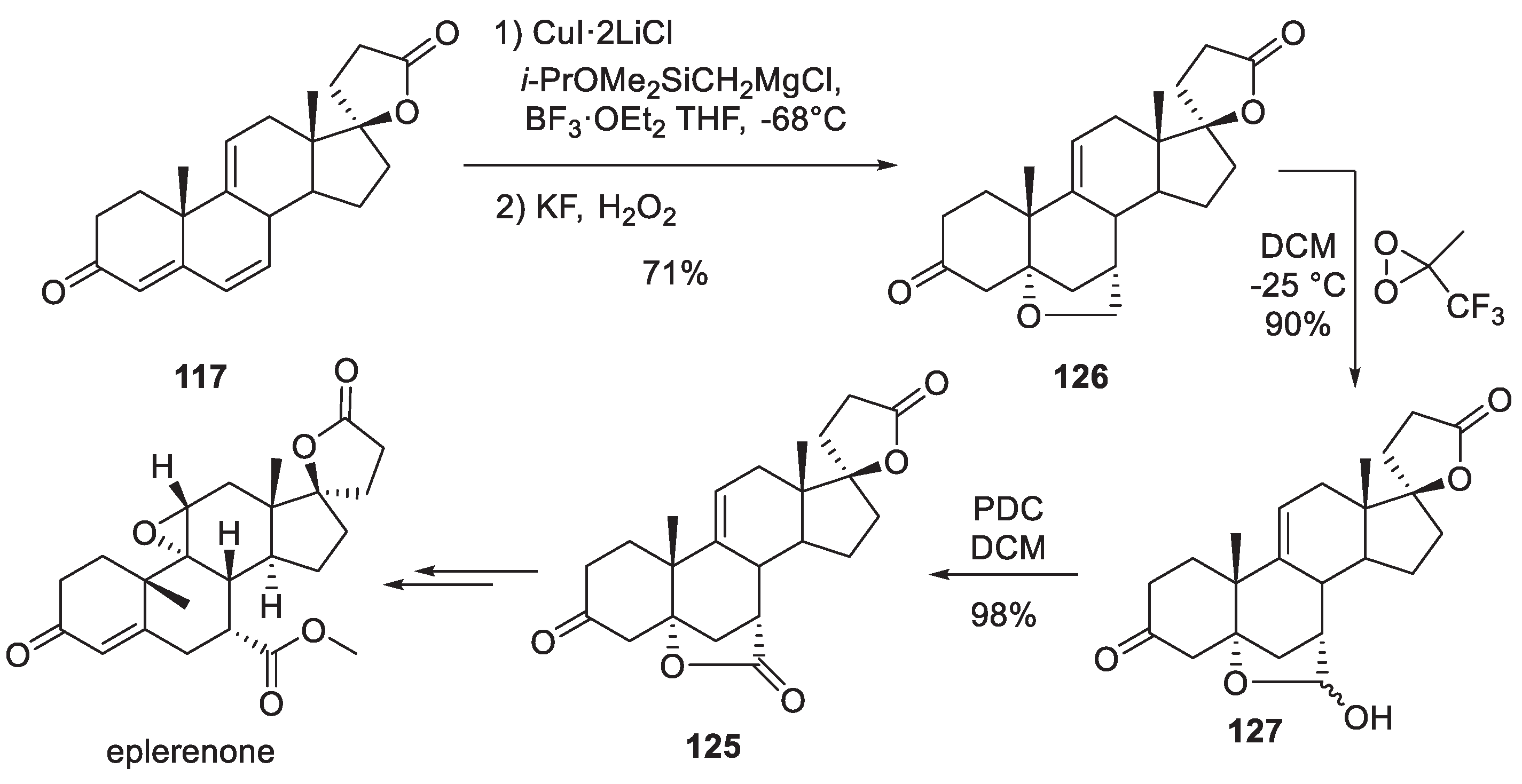
Scheme 38.
Synthesis of drospirenone by Schering AG.
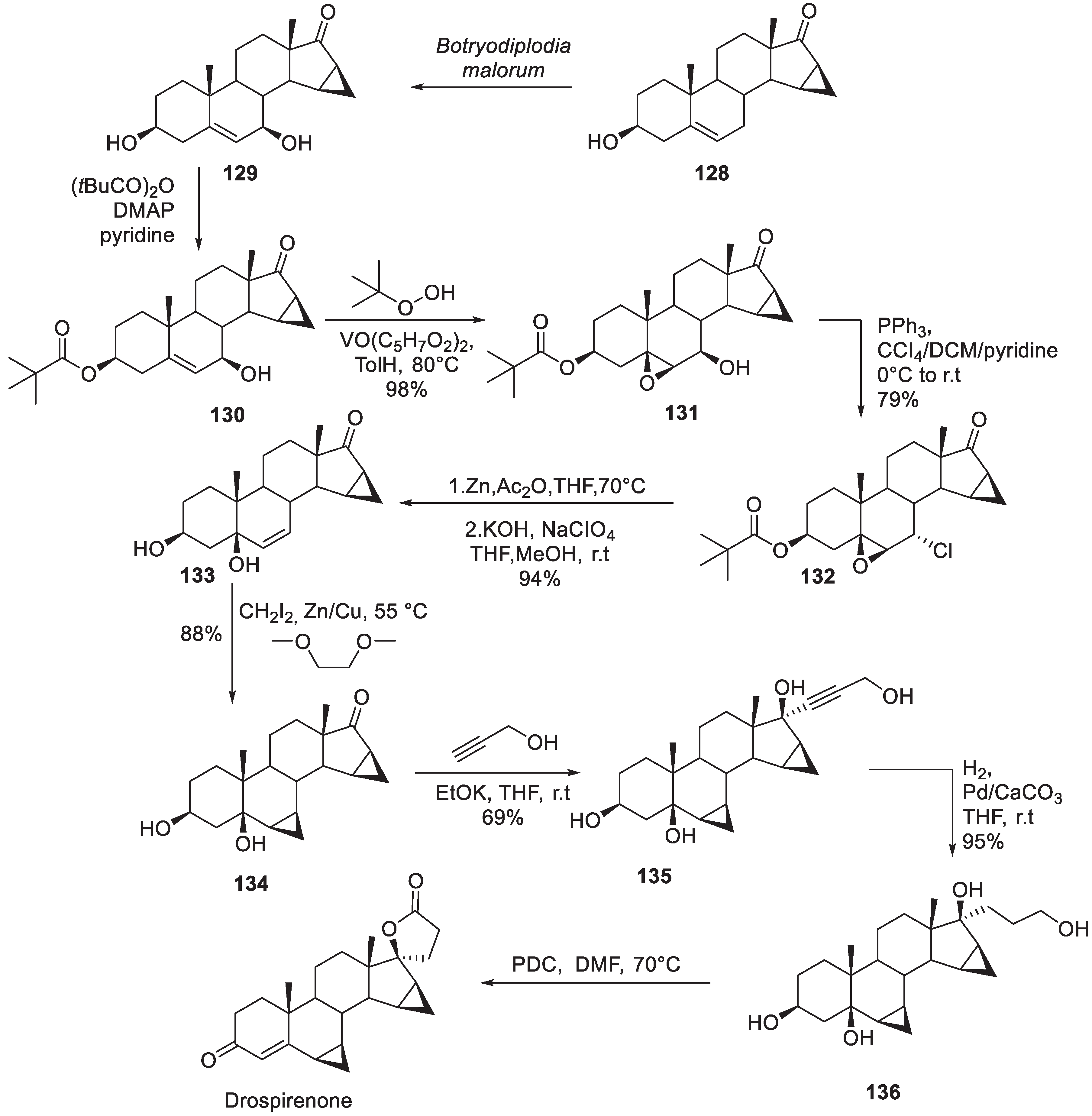
Scheme 39.
Evestra, Inc. synthesis of drospirenone.
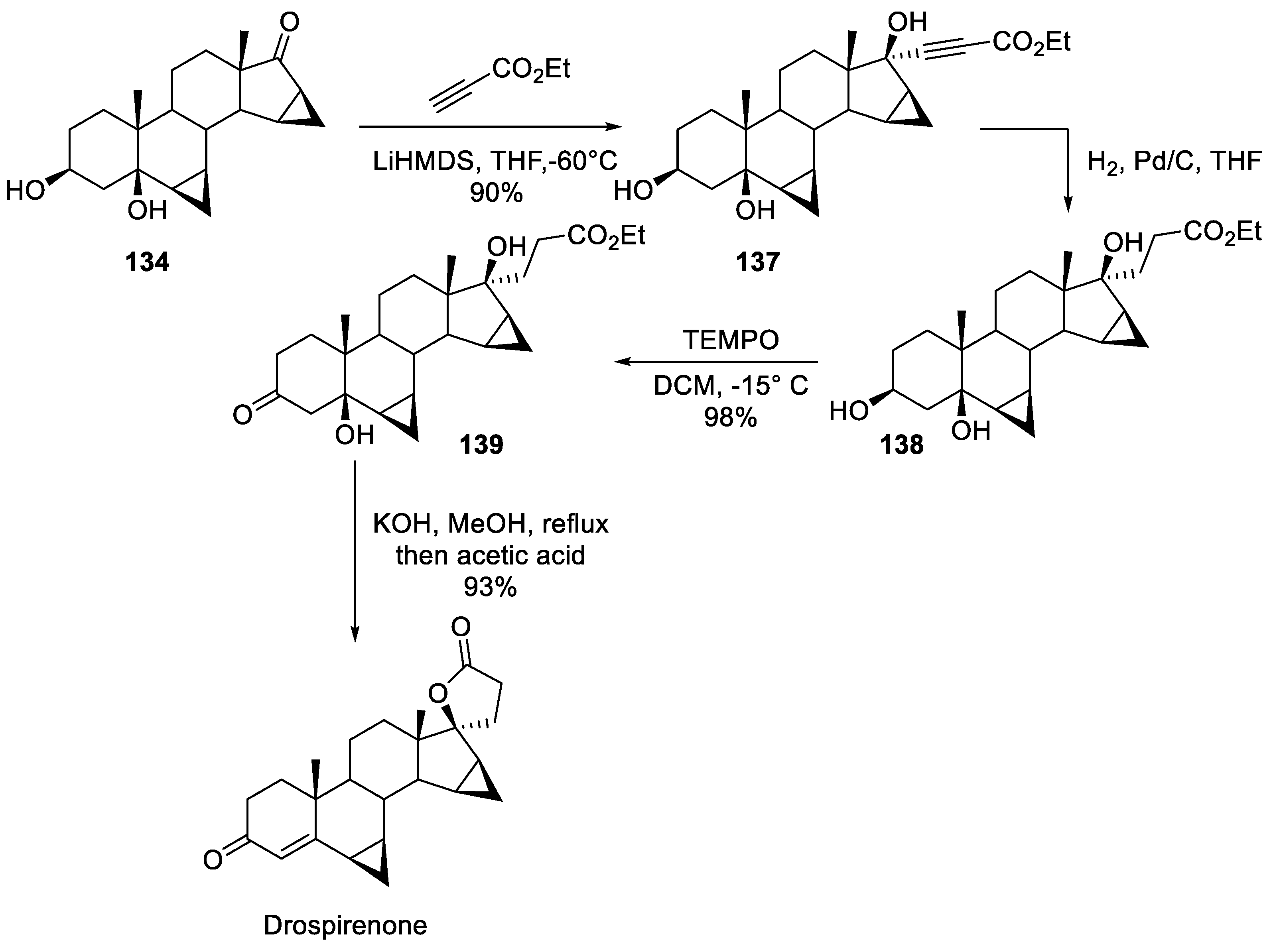
Scheme 40.
University of Bologna synthesis of drospirenone.
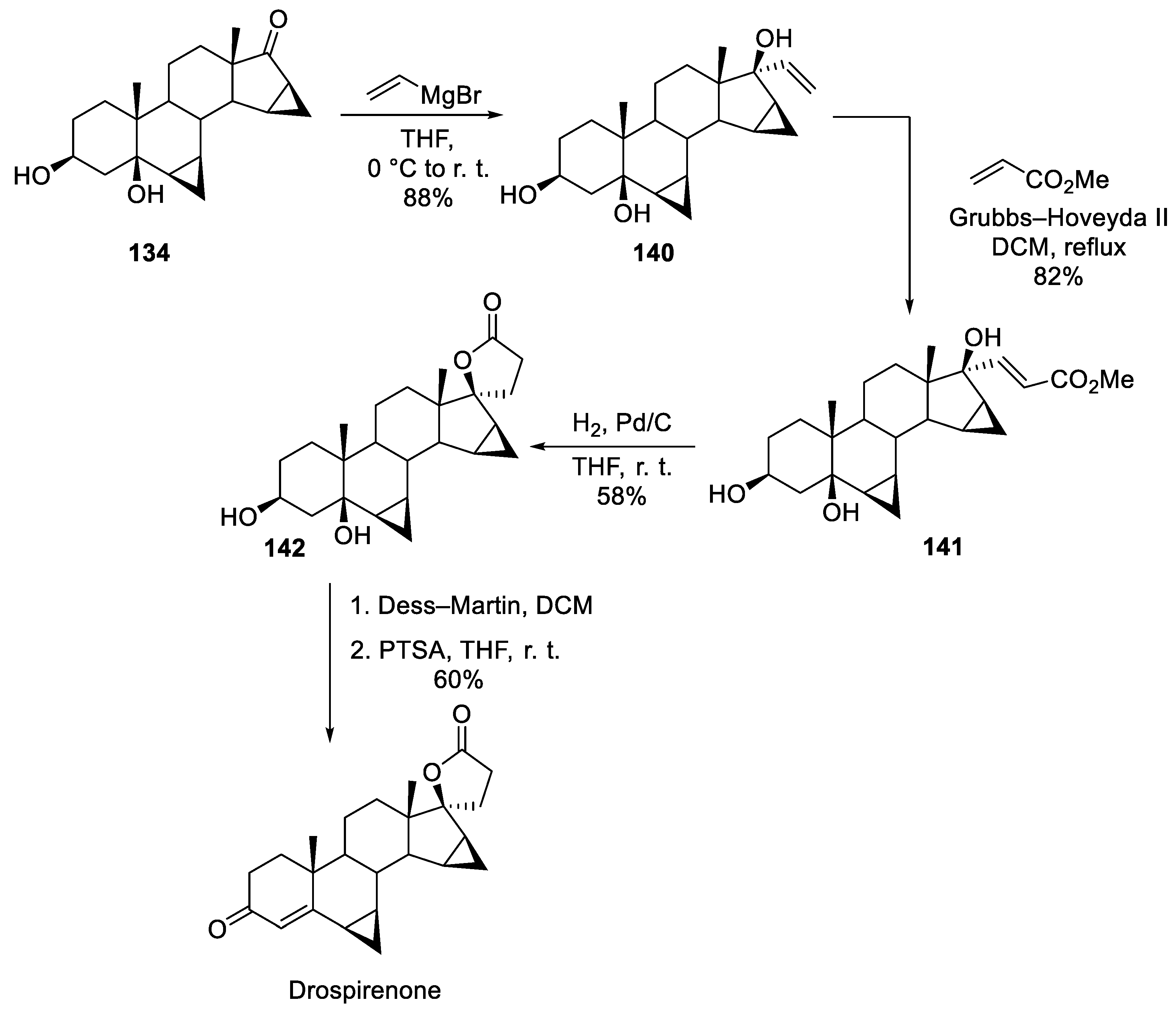
Scheme 41.
Gilead Sciences synthesis of ledipasvir.
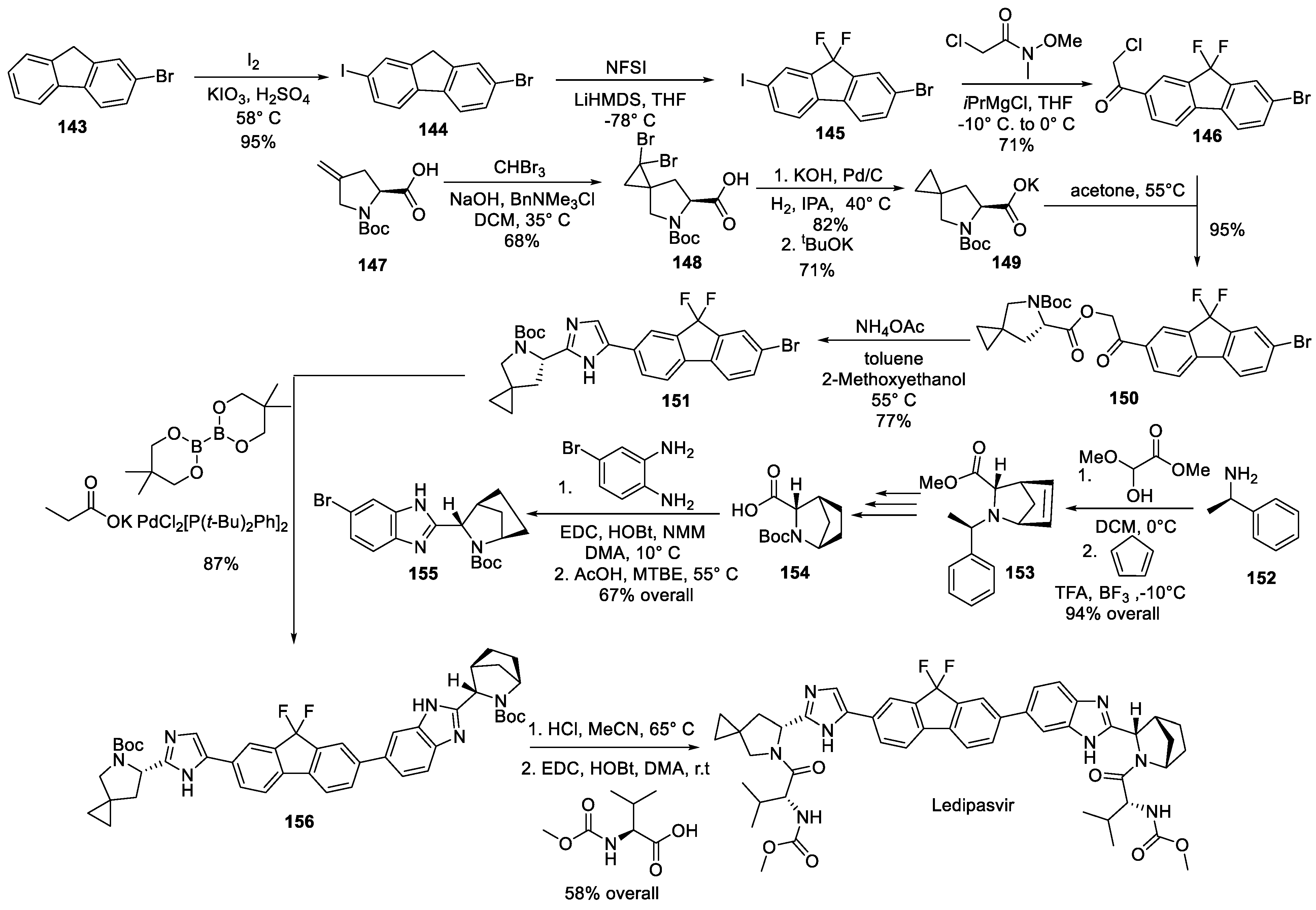
Scheme 42.
Palladium-free Suzuki-Miyaura-type coupling towards ledipasvir.
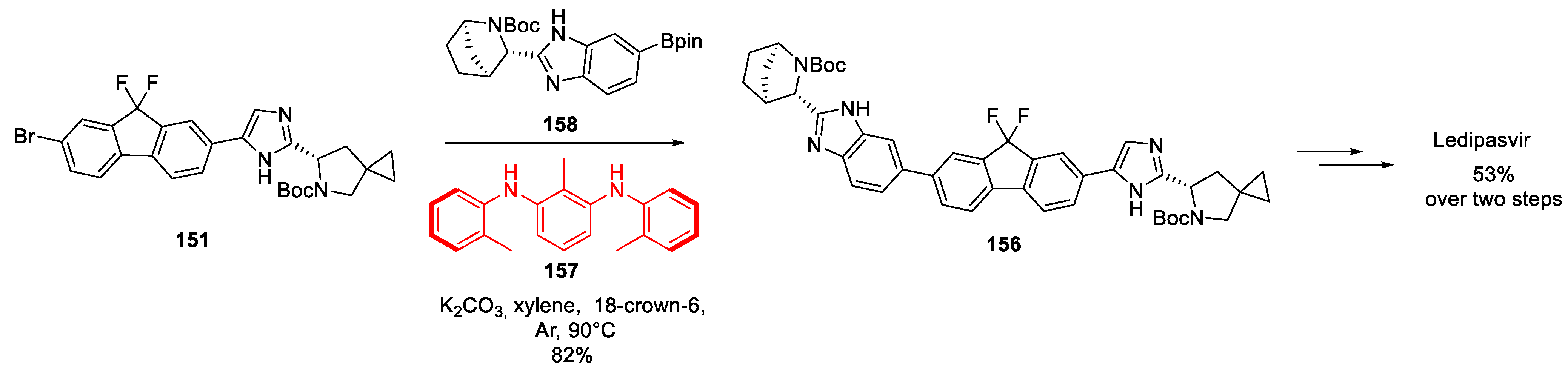
Scheme 43.
Schering-Plough synthesis of rolapitant.
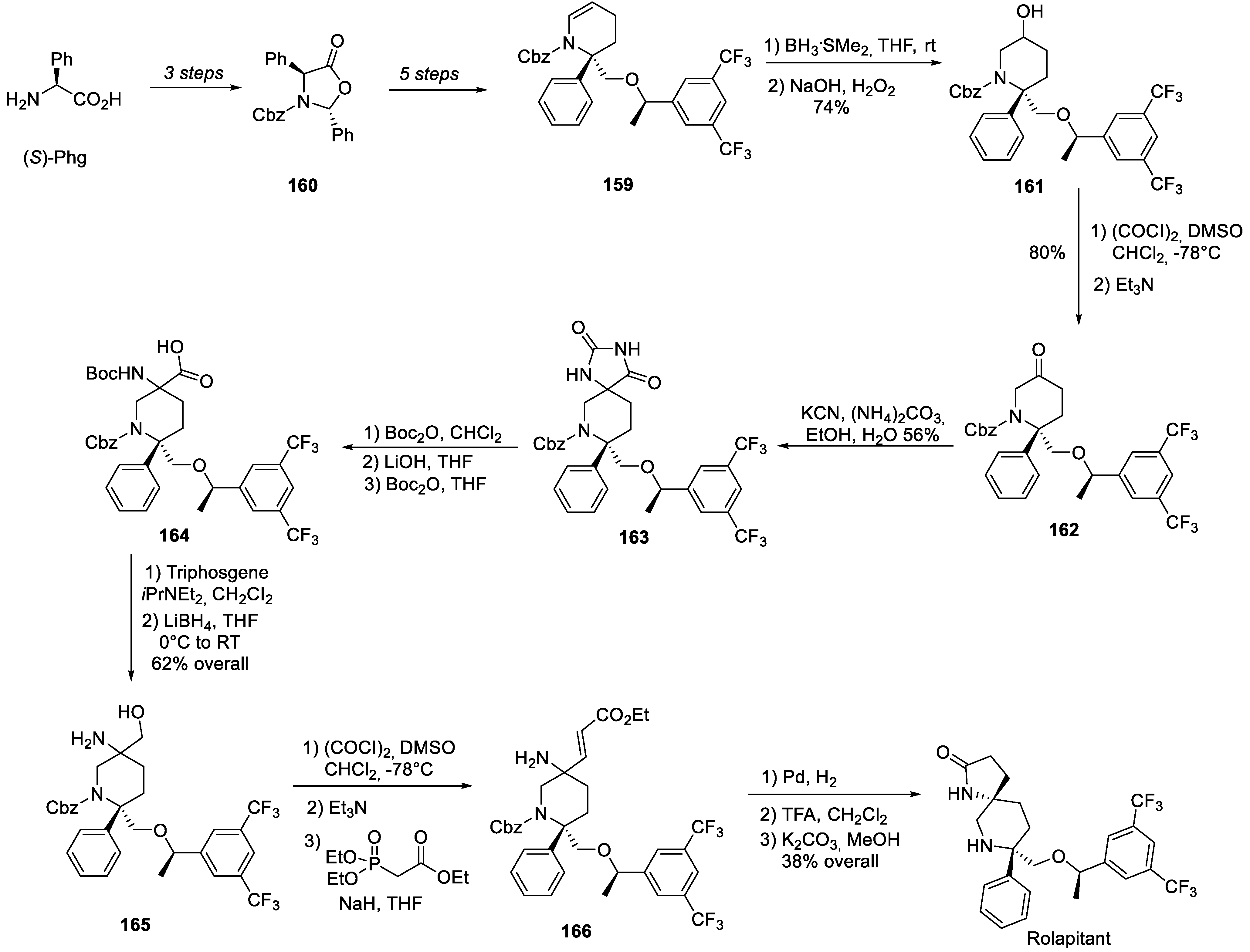
Scheme 44.
Preparation of key building block 159 for the synthesis of rolapitant.
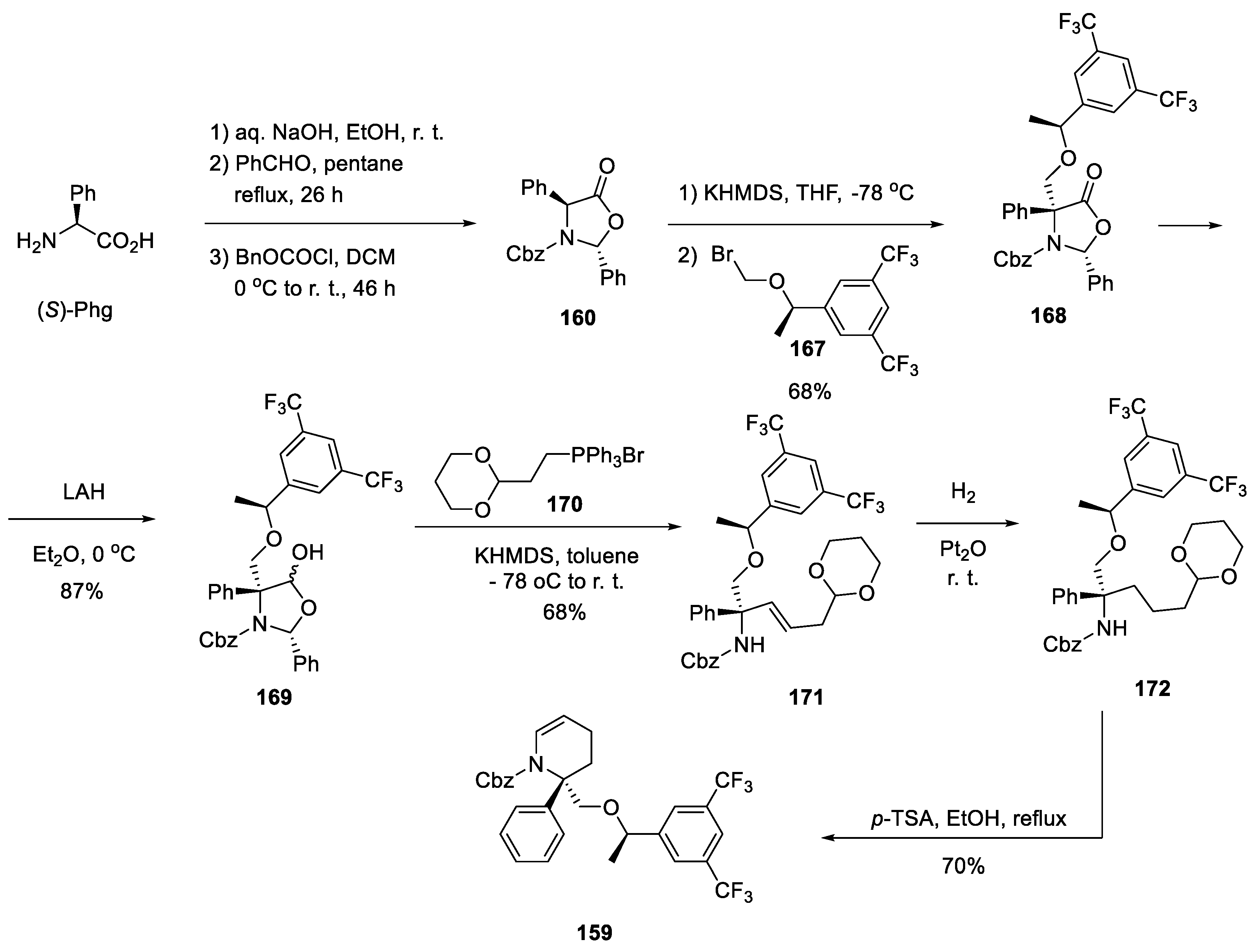
Scheme 45.
A 2013 Opko Health, Inc. synthesis of rolapitant.
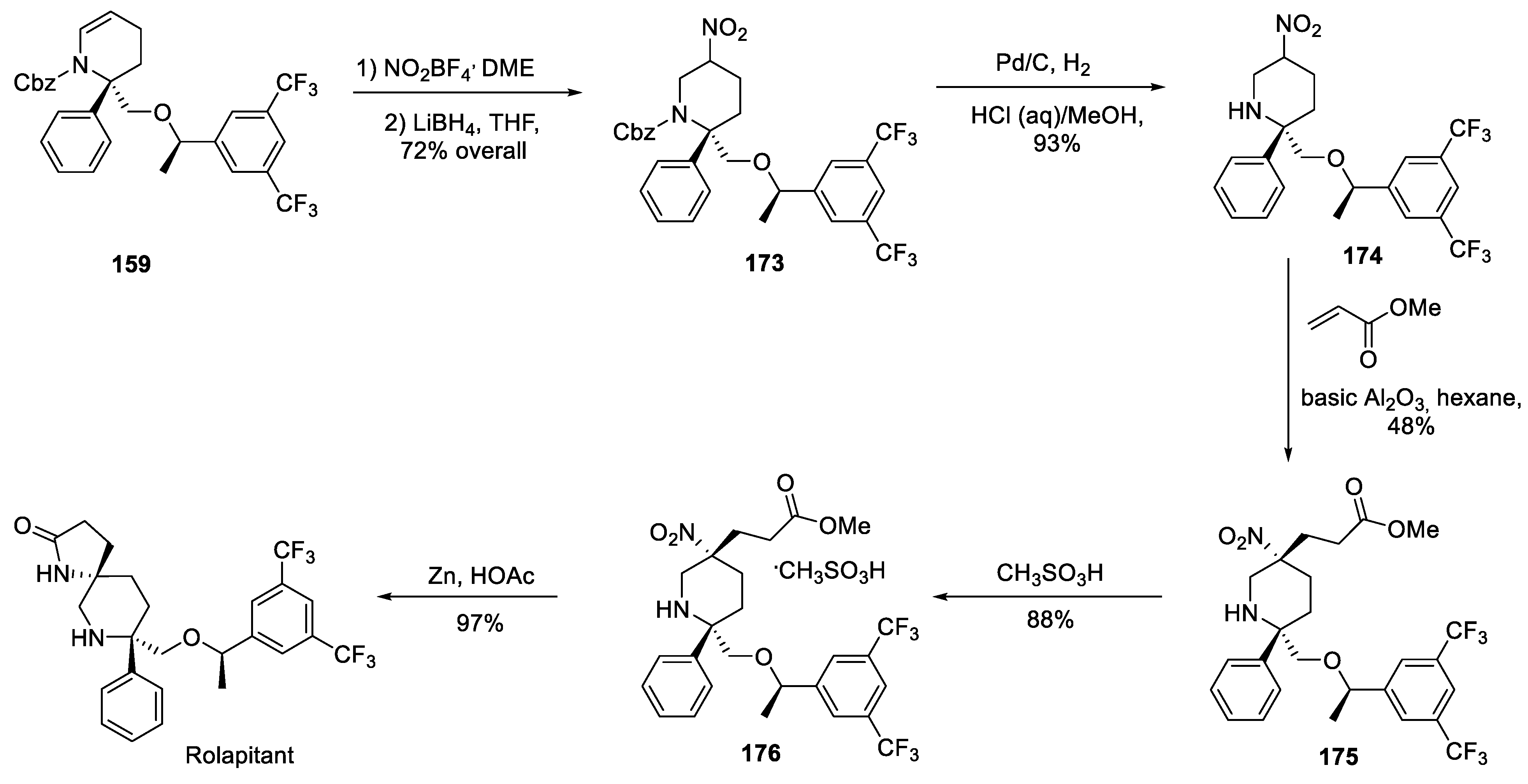
Scheme 46.
A 2016 Opko Health, Inc. synthesis of rolapitant.
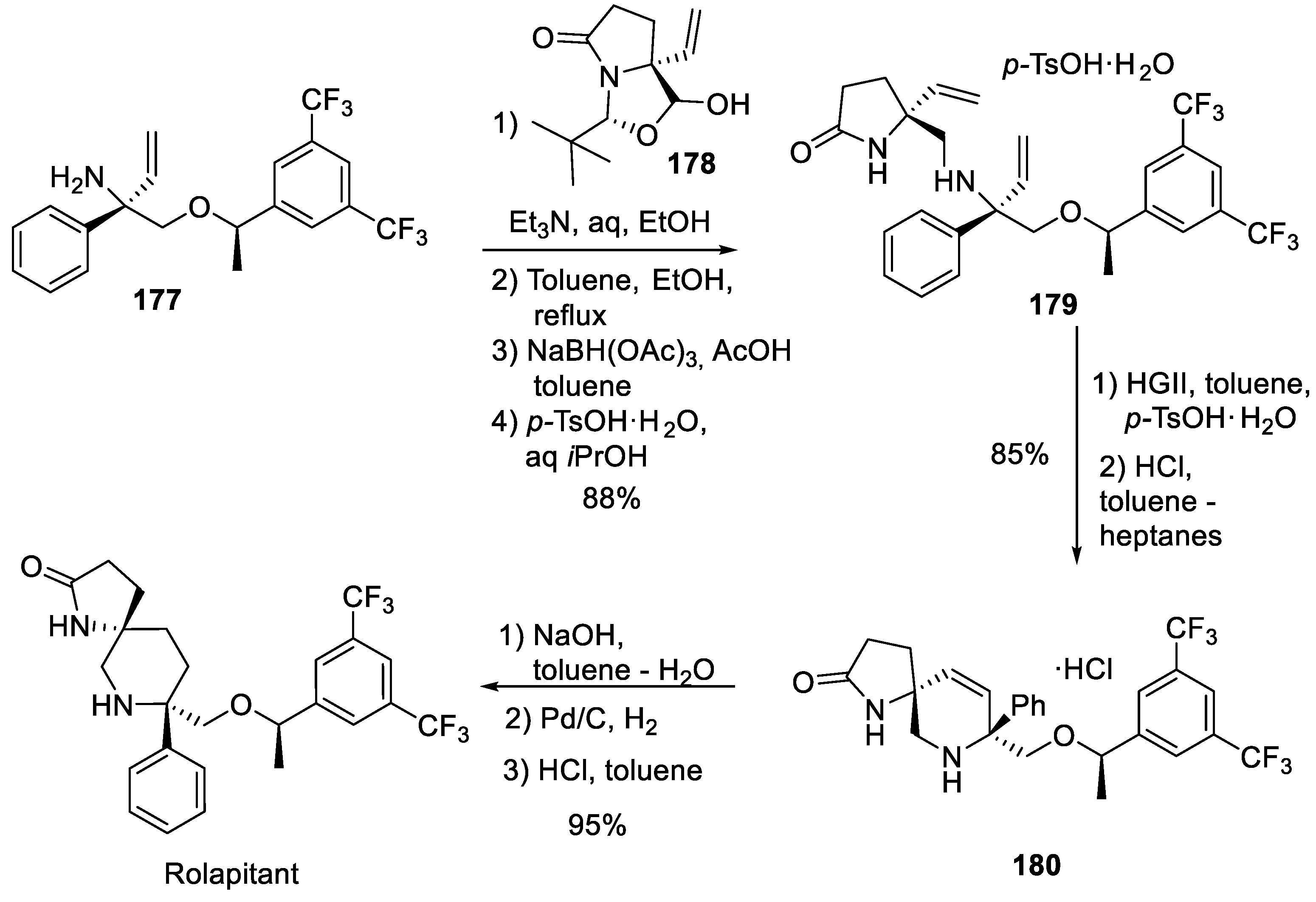
Scheme 47.
University of California original synthesis of apalutamide.
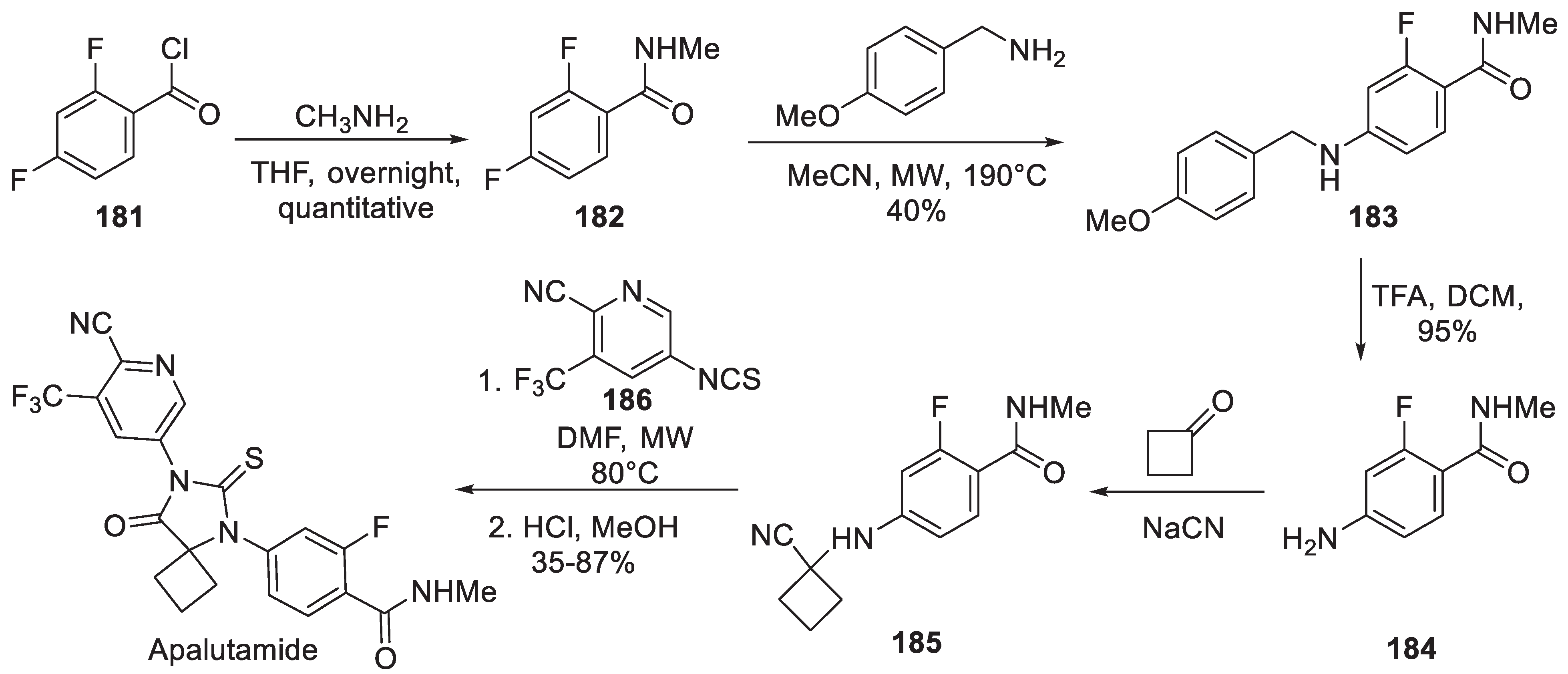
Scheme 48.
Apalutamide synthesis with thiophosgene from Sloan-Kettering.
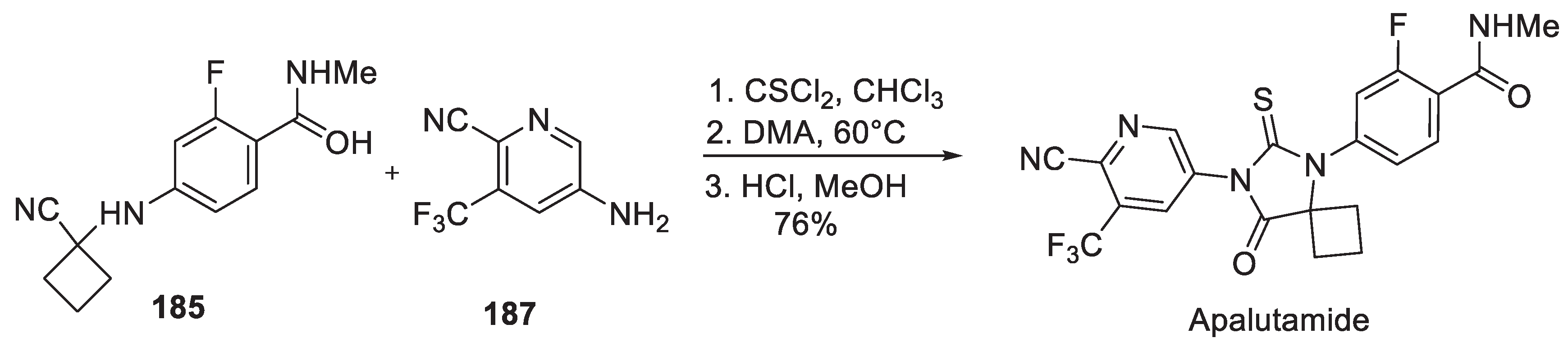
Scheme 49.
Apalutamide synthesis by Hangzhou Cheminspire Technologies Co., Ltd.
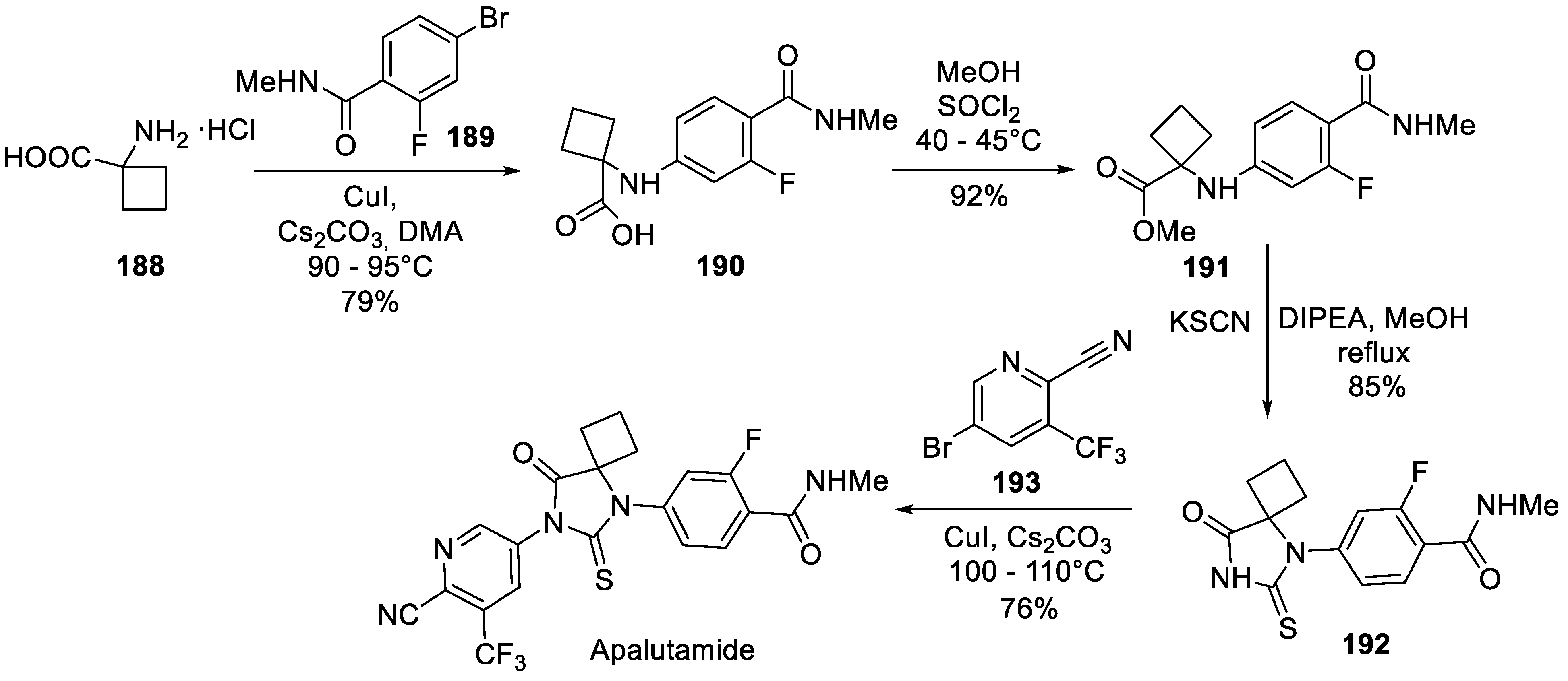
Scheme 50.
Semisynthesis of moxidectin from nemadectin.
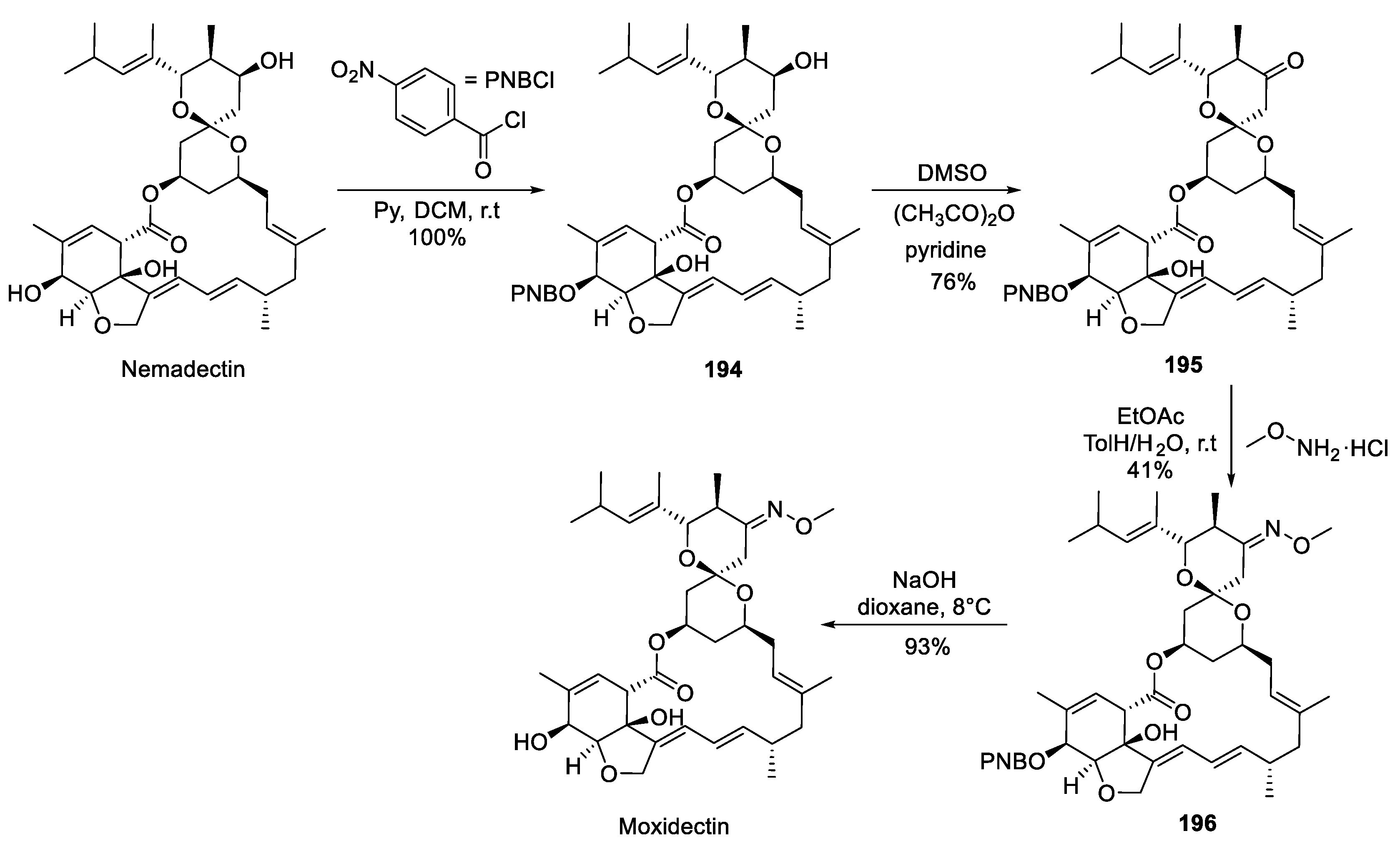
Scheme 52.
Merck synthesis of enantiopure 3-amino piperidone 198.
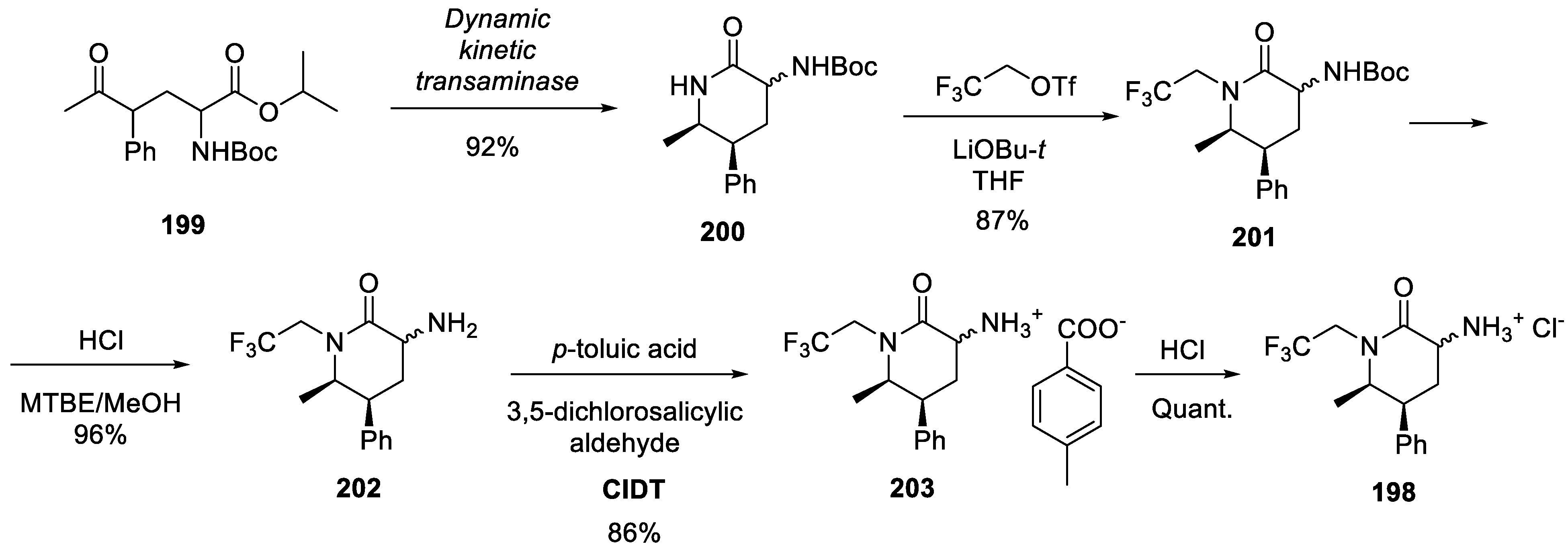
Scheme 53.
Merck synthesis of enantiopure spiro acid 197.
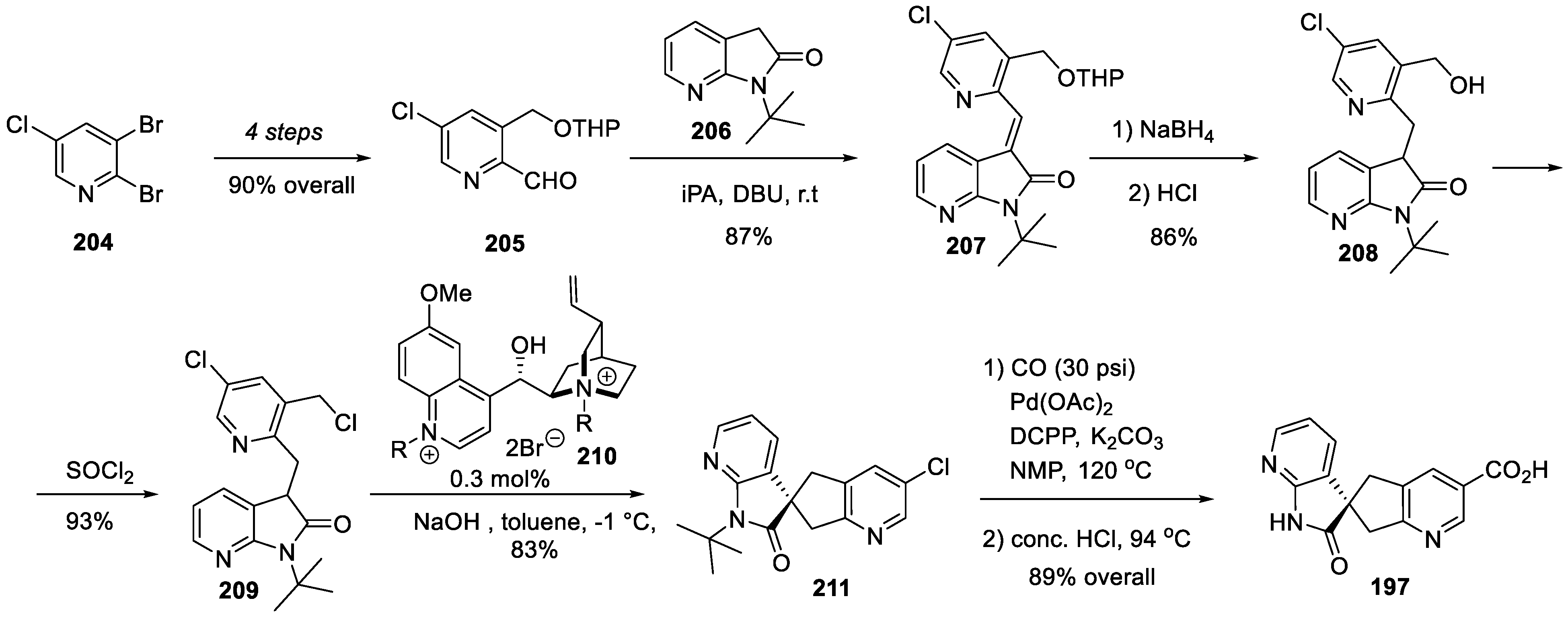
Scheme 54.
Phase-transfer-catalyzed asymmetric synthesis of spiro acid 197.
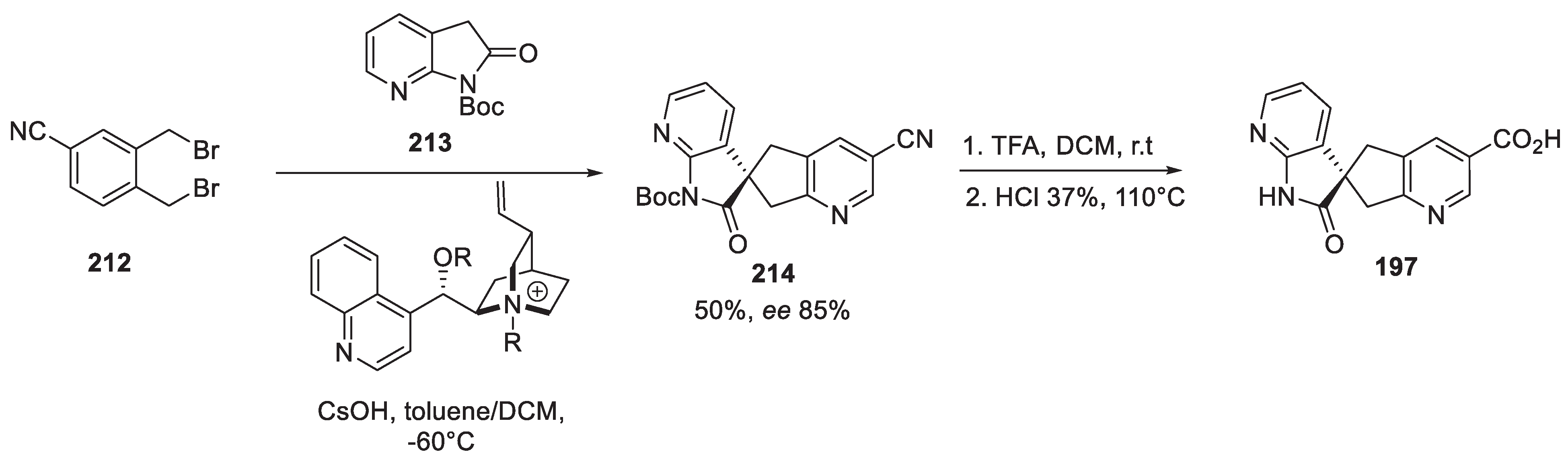
Scheme 55.
Synthesis of Oliceridine by Trevena, Inc.
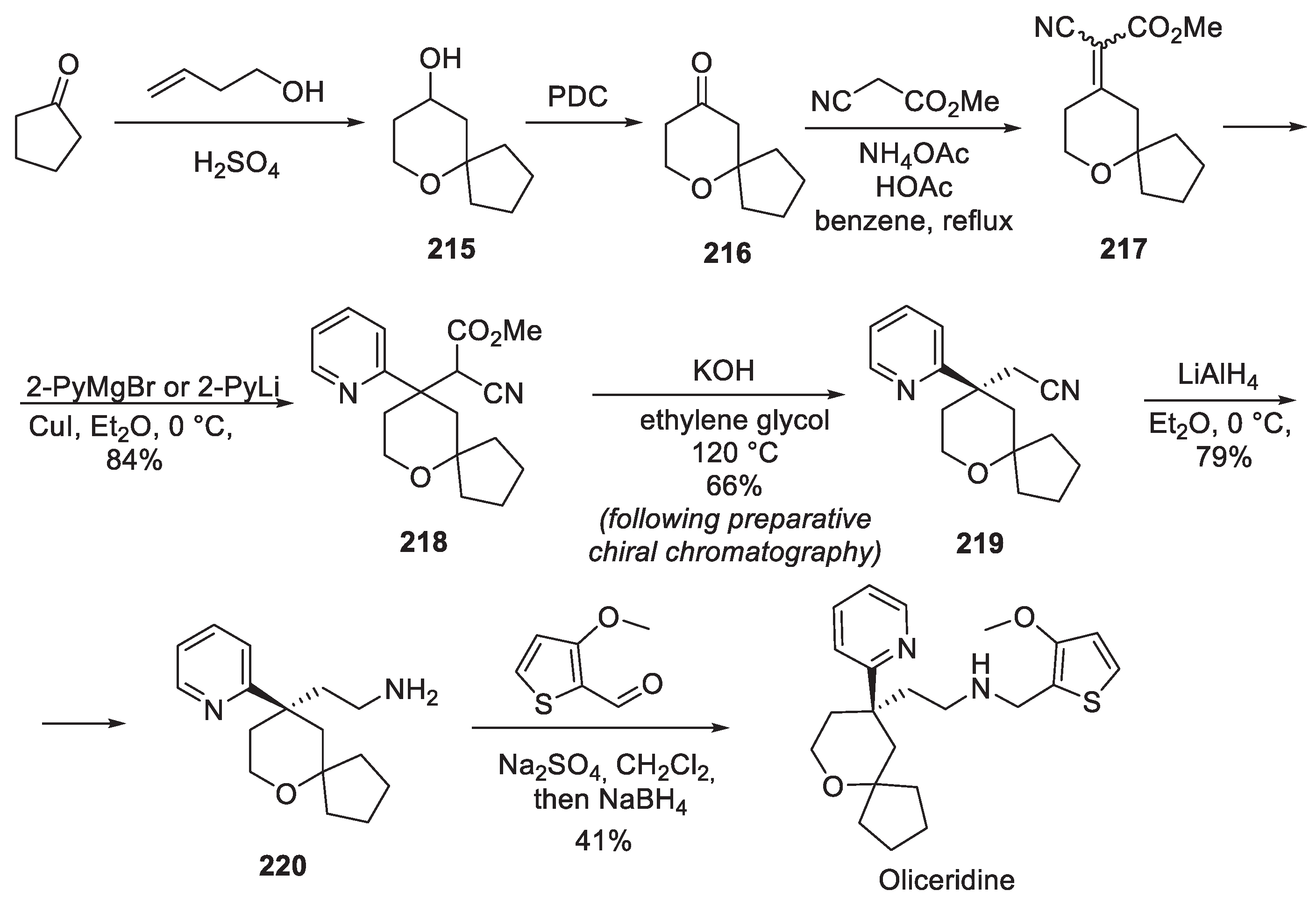
Scheme 56.
A 2015 synthesis of risdiplam by Roche.
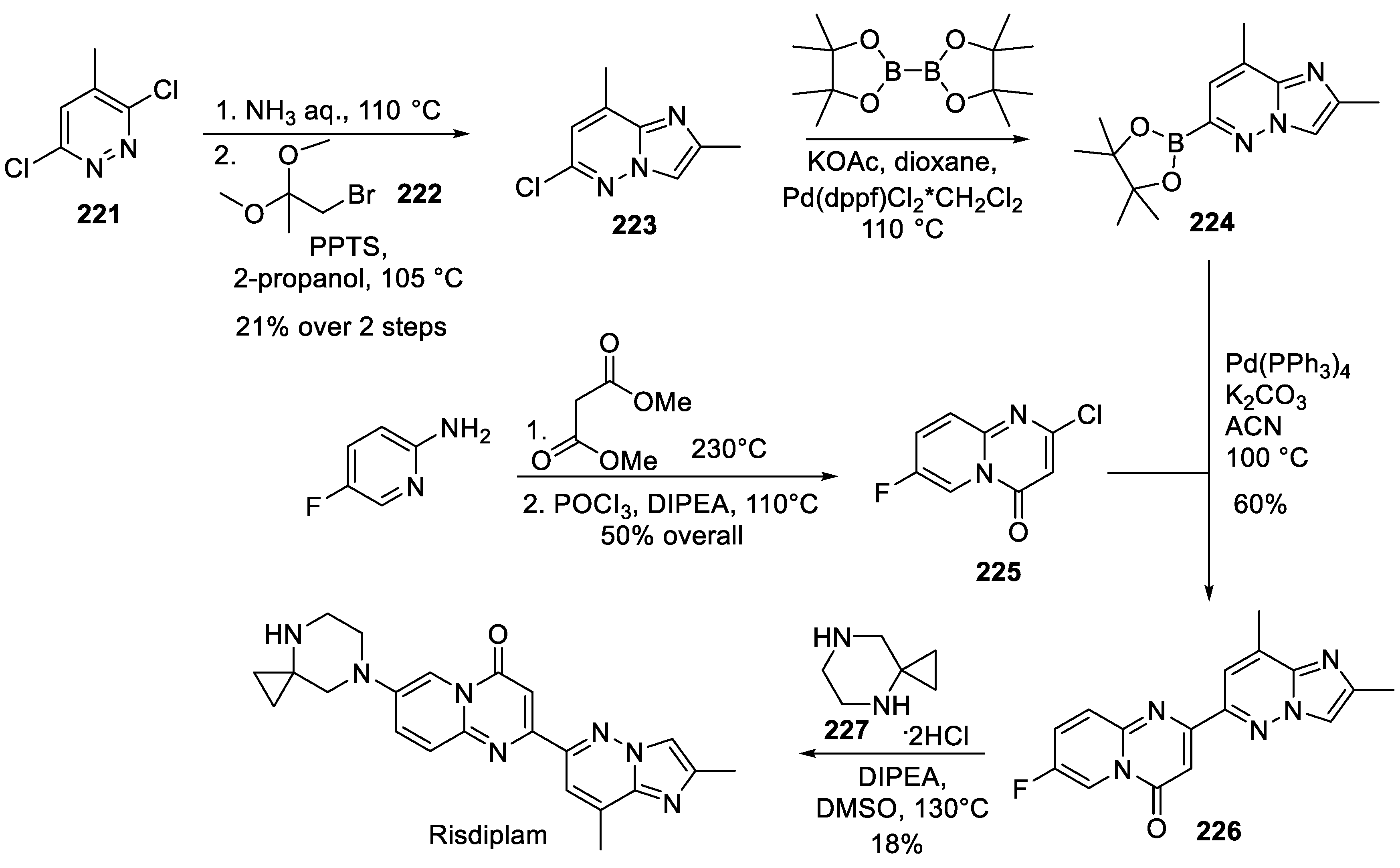
Scheme 57.
A 2019 synthesis of risdiplam by Roche.
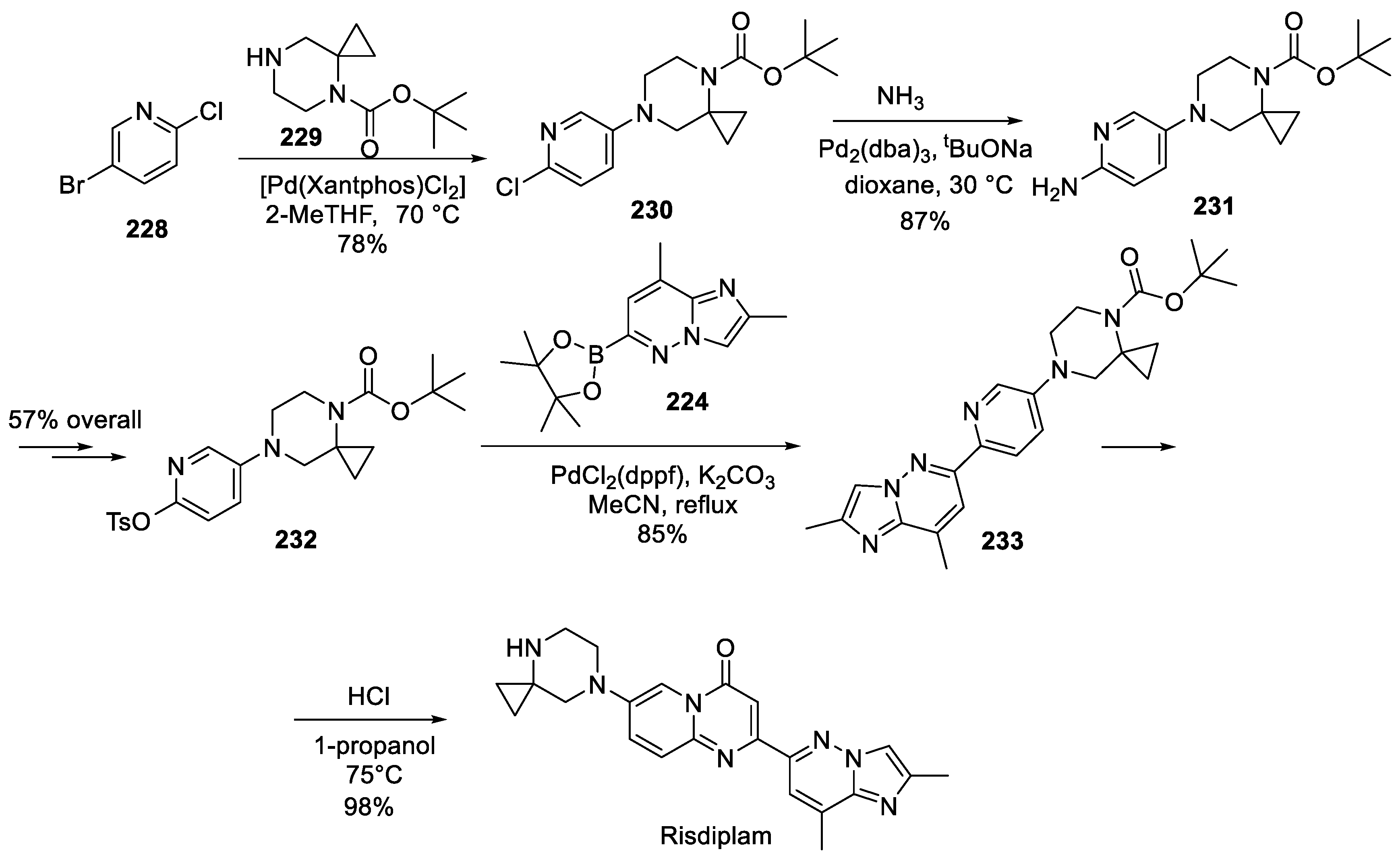
Scheme 58.
Synthesis of atogepant by Merck.
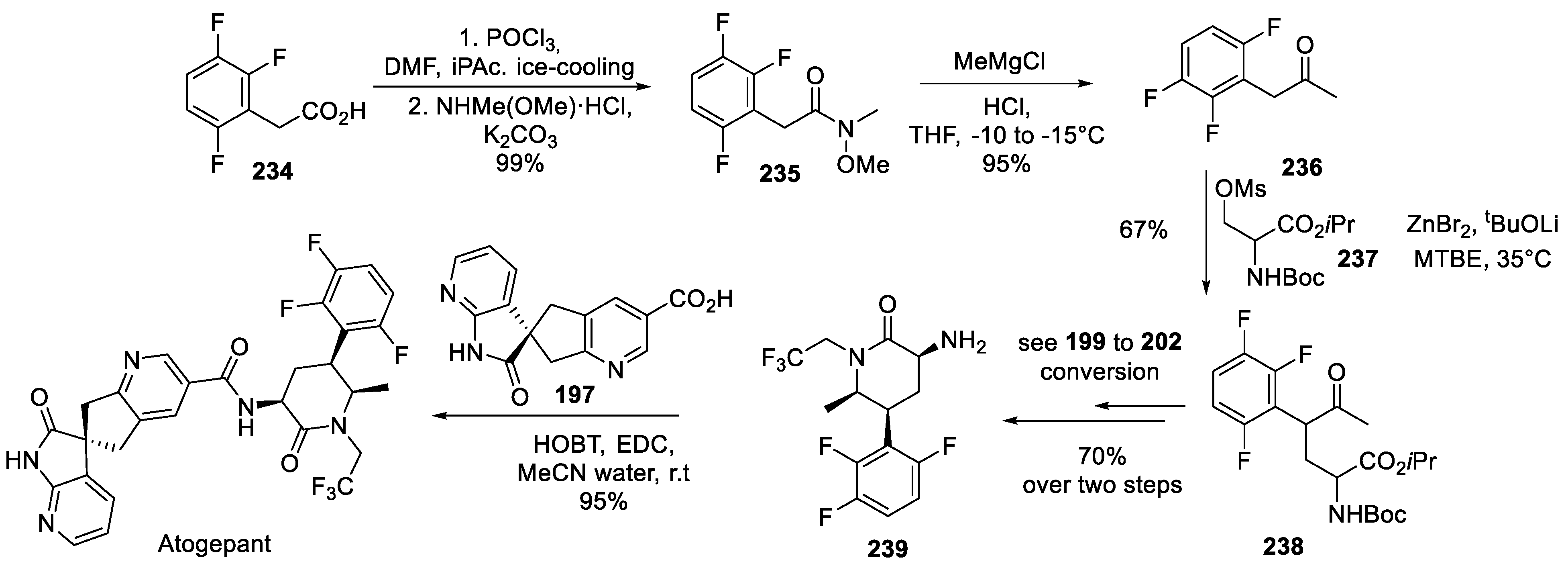
Scheme 59.
G1 Therapeutics industrial synthesis of trilaciclib.
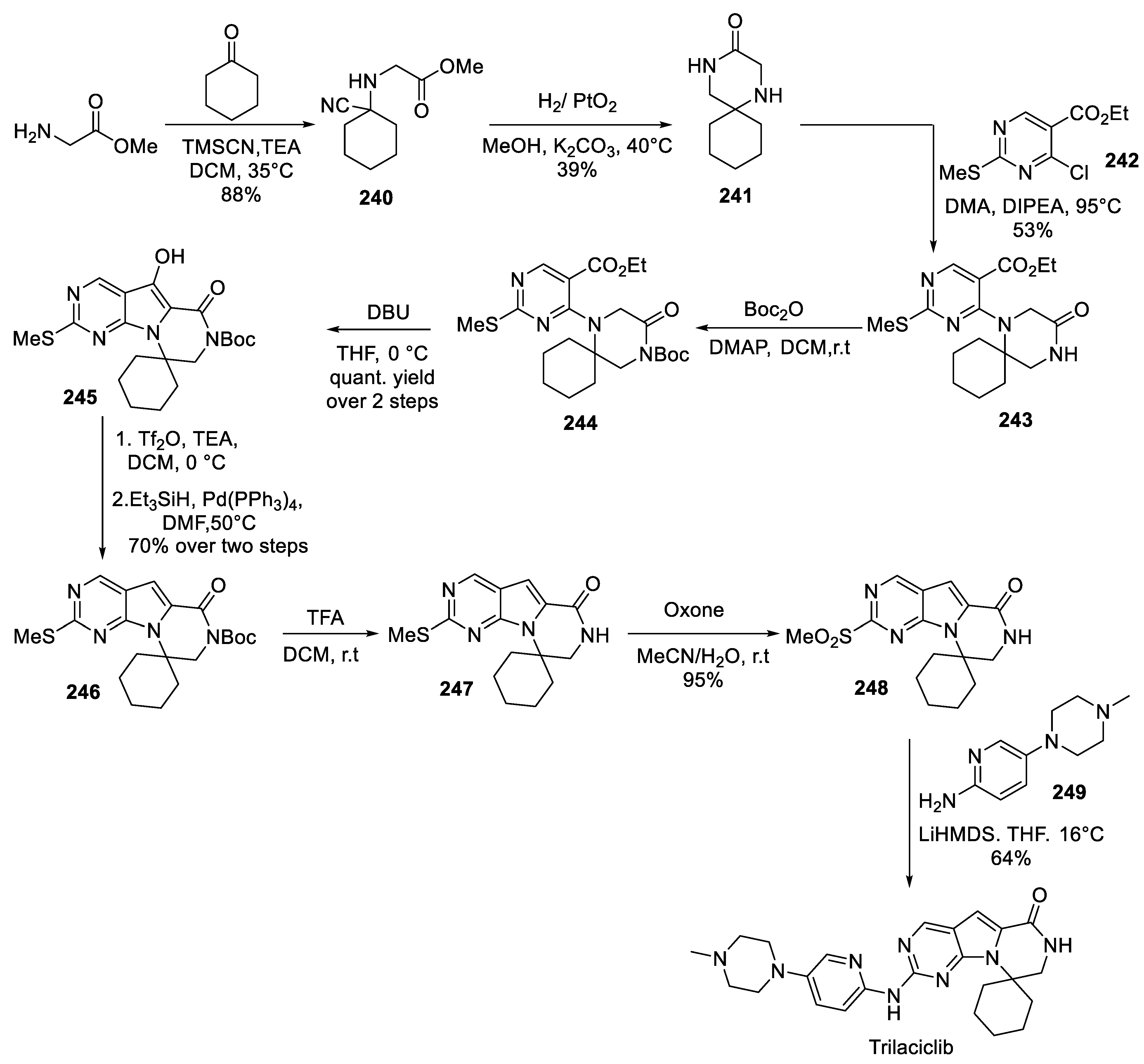

Table 1.
Summary of approved spirocyclic drugs: year of approval and origin of spirocyclic moiety.
| Entry | Drug name | Year of approval | Origin of spirocycle |
|---|---|---|---|
| 1 | Griseofulvin | 1959 | Cycloaddition, intramolecular Claisen condensation |
| 2 | Spironolactone | 1960 | Lactonization |
| 3 | Fluspirilene | 1970 | Cyclocondensation |
| 4 | Fenspiride | 1970 | Lactamization |
| 5 | Amcinonide | 1979 | Ketalization |
| 6 | Guanadrel | 1979 | Ketalization |
| 7 | Buspirone | 1986 | Commercially available spiro building block |
| 8 | Ivermectin | 1986 | Biosynthesis |
| 9 | Rifabutin | 1992 | Cyclocondensation |
| 10 | Spirapril | 1995 | Thioketalization |
| 11 | Irbesartan | 1997 | Cyclocondensation |
| 12 | Cevimeline | 2000 | Acetalization |
| 13 | Eplerenone | 2002 | Biosynthesis |
| 14 | Drospirenone | 2000 | Lactonization |
| 15 | Ledipasvir | 2014 | Cyclopropanation |
| 16 | Rolapitant | 2015 | Lactamization, ring-closing metathesis |
| 17 | Apalutamide | 2018 | Lactonization |
| 18 | Moxidectin | 2018 | Biosynthesis |
| 19 | Ubrogepant | 2020 | Intramolecular C-alkylation |
| 20 | Oliceridine | 2020 | Prins reaction |
| 21 | Risdiplam | 2020 | Commercially available spiro building block |
| 22 | Atogepant | 2021 | Intramolecular C-alkylation |
| 23 | Trilaciclib | 2021 | Intramolecular Claisen condensation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



