
Submitted:
25 April 2023
Posted:
26 April 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first identified in people in the city of Wuhan at the end of December 2019, and it has since spread to all continents. Global interest has been focused on discovering a treatment and developing a successful coronavirus vaccine as a result of the emergence of this new coronavirus. In this research, we sequenced the whole genome of the SARS-COV-2 virus that were isolated from 40 patients in Duhok, Iraq. 95 different mutations were identified in our isolation when such the whole genome sequence of the SARS-COV-2 isolated from the city of Wuhan, China (Accession number: NC 045512.2) was matched to the virus's sequence using Sequencing technology (Illumina , USA ) and Assembly method (iVar 1.3.1). Sequence analysis revealed that 38 mutations were found at spike glycoprotein (S), 30 of which were found in the ORF1b , 11 mutations were found in ORF1b ,7,3,2,1 mutations were found in ( N,M,ORF6 ,ORF9 /E ) genes which had been linked to structural changes at various places. The conclusion that the cases in Iraq were of different origins of infections and had a close relationship with the isolates from different country and state were further confirmed by phylogenetic analysis and transmission. The whole genome sequence of the SARS-CoV-2 , which were identified from the Iraqi Kurdistan region, and reported as a first study in Kurdistan region.
Keywords:
COVID-19 patient
; whole Genome sequence
; Of SARS-COV-2
; phylogenetic analysis and Transmission supported
Introduction
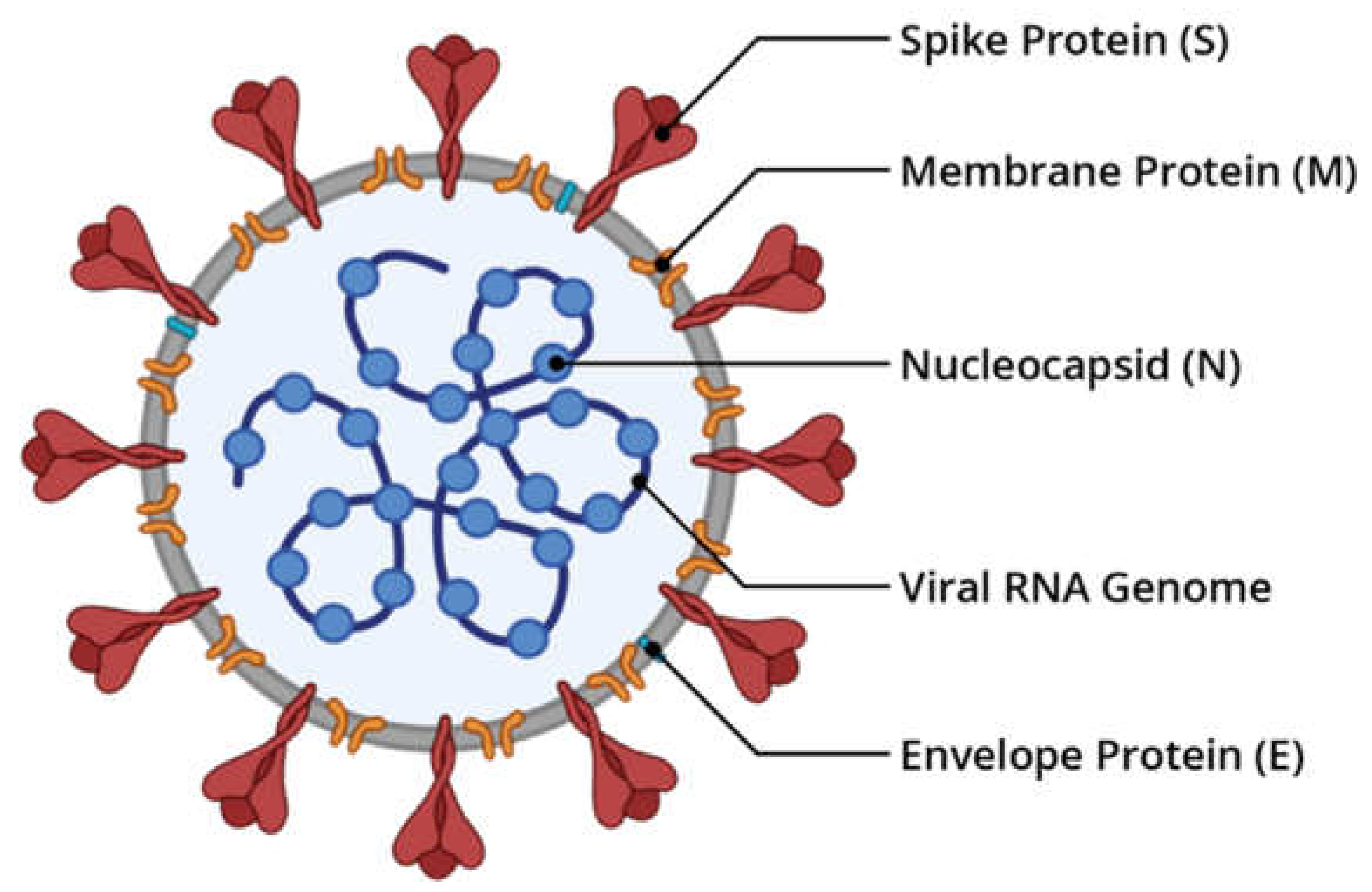
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) caused an unusual global outbreak in Wuhan, China in December 2019 that was quickly identified as a pandemic and known as COVID-19. This outbreak has suddenly gained international awareness and prompted a global campaign to develop a treatment and vaccine (Zhu et al. 2020). Over 5 million deaths and more than 260 million confirmed cases of COVID-19 have been reported by the World Health Organization (WHO) since November 2021. SARS-CoV-2 is a single-stranded -positive-sense RNA virus. The typical SARS-CoV-2 has one of the longest genomes among all RNA virus families, measuring 30 kb and encoding nearly 9860 amino acids. (1,2).
Figure 1.
Illustration of the SARS-CoV-2 structure (figure made in Bio Render).
In consideration of how urgent the epidemic is, different efforts have been made to make effective vaccines. However, just like other infections, viruses are more likely to spread in an open-air environment. From a health standpoint, the rise in visitors to crowded areas seems absurd, but it might be less shocking than anticipated. it is difficult to accurately estimate the risk of self-behavior, and the length of the current situation is challenging the limits of human patience and self-control (3) For the diagnosis of viral infectious illnesses and the design of vaccines, full viral genome sequencing is essential. The virus's virulence and pathogenicity in addition the evolutionary connections between hosts and viruses could also be studied via genome sequencing (4). Rapid country-to-country transmission of SARS-CoV-2 has raised questions regarding whether virus mutations are to reason for the spreading (5,6,7). Random mutations occur because SARS-CoV-2, an RNA virus, lacks a "proofreading" mechanism as it multiplies. These are the main strains of the virus that are spreading in numerous places and have multiplied quite a bit (8). Additionally, it is thought that the virus is modified under all circumstances by nucleotide changes, one of the most significant indications of viral progression (9,10,11,12,13,14).
This study's primary objective was to perform local SARS-CoV2 mutant whole-genome sequencing (WGS) to study more about the virus's mutational diversity when compared to Wuhan first isolated genome sequence of the SARS-COVID-2 which are important determinants of virus detection and vaccine performance.
Materials and Methods
This study was approved by the ethical committee (Application number: 16727) at the Directorate General of Health Duhok, Iraq. Once the cases completed routine SARS-CoV-2 diagnostic testing at Covid-19 Center and Duhok Burn Hospital (Covid-19 Hospital), the SARS-CoV-2 positive samples were subjected to next-generation sequencing.
Clinical Sample and Processing
A nasopharyngeal swab collected from 160 patients accompanied by a respiratory tract infection in Duhok received at the COVID-19 Center for research and diagnosis, University of Duhok in December 2021 in the early five waves was applied to this study. According to the instructions provided by the manufacturer, a QI amp RNA extraction kit was used to extract viral nucleic acids. 40 samples were selected to RNA extraction and quantitative detection using the QIAprep and amp Viral RNA UM Kit for real-time PCR-based virus identification. As required by the sequencing laboratory, the Ct value of the selected samples was (< 20). Following RNA extraction from the viral transport medium using a QIAamp Viral RNA Mini Kit, then samples were shipped on dry ice to the USA for whole-genome sequencing.
Genomic Sequencing
After the samples arrived in the USA, They were tested again for identification of targets and RNA integrity. Reverse transcription and cDNA synthesis were performed using Qiagen's (Germany) RT Kit and nanomere primers. Using an Illumina sample preparation kit, the individual samples were indexed and tagged. Using illumina Miseq instruments iVar 1.3.1 an and Coverage: 2423.56x (Illumina, San Diego, CA, USA), next-generation sequencing was carried out following the manufacturer's instructions. BWA-MEM alignment technique was used to assemble and align the short-read sequencing with the reference genome (NC 045512.2). The whole-genome sequence was obtained and submitted to the GISAID database, where it was given the assigned accession numbers. EPI_ISL_12604438 , EPI_ISL_12604442 ,EPI_ISL_12604444 ,EPI_ISL_12604448 ,EPI_ISL_12604451,EPI_ISL_12604457 EPI_ISL_12604460 ,EPI_ISL_12604463, EPI_ISL_12604471,EPI_ISL_12604476 EPI_ISL_12604477, EPI_ISL_12604478 ,EPI_ISL_12604481, EPI_ISL_12604482 ,EPI_ISL_12604483,EPI_ISL_12604487 ,EPI_ISL_12604488,EPI_ISL_12604489, EPI_ISL_12604490 , EPI_ISL_12604495, EPI_ISL_12604496,EPI_ISL_12604501 ,EPI_ISL_12604502,EPI_ISL_12604503 ,EPI_ISL_12604507, EPI_ISL_12604508 EPI_ISL_12604509, EPI_ISL_12604510 ,EPI_ISL_12604514 ,EPI_ISL_12604516 ,EPI_ISL_12604517, EPI_ISL_12604521 , EPI_ISL_12604526, EPI_ISL_12604527 ,EPI_ISL_12604528,EPI_ISL_12604532, EPI_ISL_12604845, EPI_ISL_12604846 ,EPI_ISL_12604847, EPI_ISL_12604848 .
The genome sizes were 29,488, 29,550,29,559,29,562,29,565 bp when compared to the Wuhan sequence (NC_045512.2).
Genome Alignment, and Phylogenetic Analysis
Entire genome sequences were aligned to the SARS-CoV-2 reference genome (NC 045512.2), using the program Nextclade version (Version 2.8.0)
A phylogenetic analysis was done using Molecular Evolutionary Genetics Analysis version 11.0 (MEGA 11.0) to examine similarities and differences between the Duhok genome sequence and other SARS-CoV-2 genome sequences downloaded from GISAID database and GenBank (Figure 2).
The samples from the neighboring countries, the blast search result sequences globally, the whole-genome sequences of this study's (n = 40) sequences, and more have all been obtained from the databases include GISAID and the National Center for Biotechnology Information. Good quality sequences without running (Ns) that were collected from September 2021 to April 2022 and identified as Omicron variations were the selection criteria.
Results and Discussions
Early January 2020 had seen the discovery of a novel coronavirus as the source of many pneumonia cases that had been reported from China in late December 2019 but were of unknown origin (16). The virus was later identified as the cause of Coronavirus Disease 2019 and recognized as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (COVID-19). The virus has spread worldwide despite significant efforts to restrict the disease in China, and The World Health Organization classified COVID-19 as a global pandemic (WHO) in March 2020 (17). All CoV have the characteristic that single-stranded RNA makes up their genomes with positive polarity, which means that the RNA's base sequences are oriented in a manner that is identical to the later messenger RNA (mRNA). The CoV genome is the biggest RNA genome known to exist, measuring 26.4–31.7 kilobases (18).
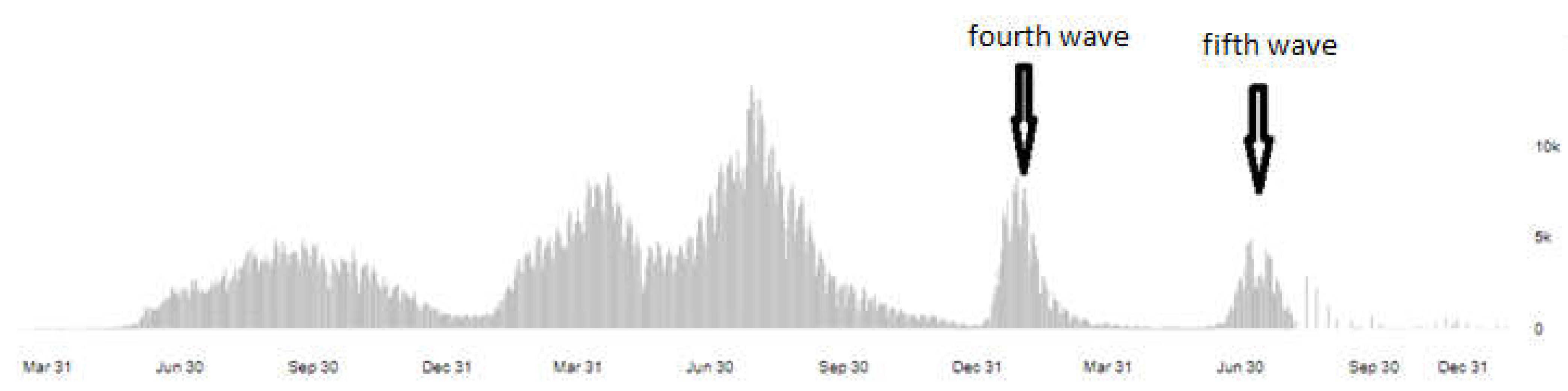
In April 2022, we uploaded our isolated virus strain's genome sequences between four and five waves for genome analysis to the GISAID databases. According to the WHO coronavirus dash-board of the reported cases in Iraq, the samples of the latest fifth waves belonged to the Omicron variant of concern (pango_liniage BA.2) that is only discovered in this study and had never been identified in the previous studies in the country.
Figure 4.
Daily confirmed cases of SARS-COV-2between 2021 and 2022 on the WHO Coronavirus dash-board in Iraq.
Figure 4.
Daily confirmed cases of SARS-COV-2between 2021 and 2022 on the WHO Coronavirus dash-board in Iraq.
To find mutations, Sequences from SARS-CoV-2 isolates were compared to a Wuhan reference sequence (NC 045512.2). The position of the mutations was then predicted using the Nextclade (version 2.7.1) software. This study's analysis of the Omicron VOC of SARS-CoV-2 sequences revealed that the S-gene had the most changes, proceeded by the ORF1ab, N, M, ORF6, ORF3a, ORF9b, and E genes. Among the genes, ORF3a, ORF6, E-gene, and ORF9b had the least mutations.
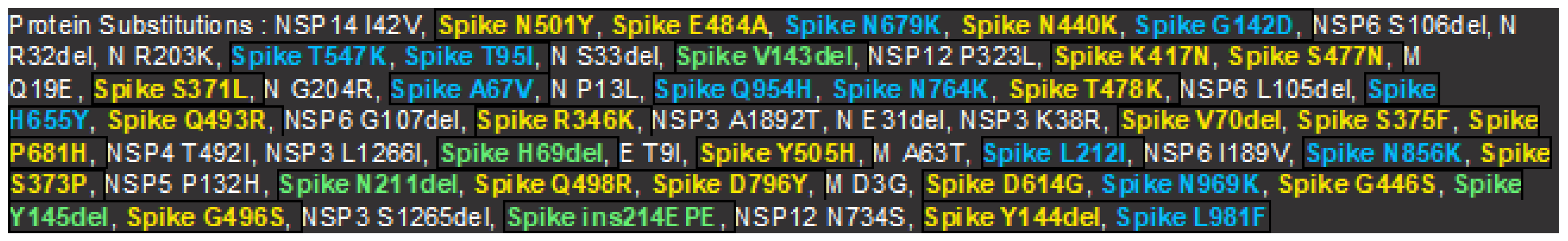
Since spike glycoprotein is the primary target for both therapy and diagnosis, it is also the essential protein that defines viral host affinity and pathogenesis. The structural protein that is encoded by the S-gene with the greatest mutations acts as a protein that binds to viruses for host cell receptors and recognizes the host range (19). Over 90% of the neutralizing antibodies in COVID-19 convalescent plasma, which is subunit S1, are anti-RBD, the S1 viral protein is the most immunodominant one (20). There are a total of 38 non-synonymous mutations in this gene. including, T 76 I, T95I, G142D, Y 145 D, L 212 I, V 213 G, G339D, R346K, S371L, S373P, S375F, T376A, R408S, K417N, N440K, G446S, S477N, T478K, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969Kand L981Fdel211/211 del3674/3676 del69/70 del142/144 del22/23 del68 /70, del142 /145, and del211 /212 deletions are also present. According to reports, this gene's mutation potential is higher than that of other genomic locations (21). The global prevalence of the S protein D614G variant has progressively increased over time and is currently present in about 74% of all known variants, according to the GISAID SARS-CoV-2 database as of June 25, 2020. One of the most common SARS-CoV-2 mutations, On January 24, 2020, a mutation called D614G, which changes the amino acid glycine (G) with a nonpolar side chain from the amino acid aspartate (D) with a polar negative charged side chain, was first identified in China. (22). It was discovered that D614G enhances the infectivity, transmission rate, and effectiveness of cellular entry for the SARS-CoV-2 virus across a wide spectrum of human cell types as a result of positive natural selection (23,24,25). One of the earliest and most common mutations is D614G, where glycine is substituted for aspartic acid at position 614 (G614). The RBM domain mutation D614G has been demonstrated to increase the density of S-proteins on the viral surface, increasing infectivity (26). The D614G mutation is present in the majority of circulating VOCs, such as Alpha (B.1.1.7), Beta (B.1.351), Delta (B.1.617.2), Gamma (P.1), and the more modern delta plus (AY lineage) and Omicron (B.1.1.529) variants (27). This is associated with elevated rates of transmission, infection, and viral evasion from neutralizing antibodies (28) .in this study It was discovered that some of the other frequently occurring RBD changes improved ACE2 binding including N501Y, S477N and E484K and same substitutions seen in the majority of VOCs, which are associated with greater transmissibility (29 ).in the existing change of VOC, the T478K, Q498R and Q493K mutations have demonstrated to increase the electrostatic potential, boosting the RBD-ACE2 binding affinity (29). Immune escape has also been connected to the existing change E484K (30). A variety of VOCs in the previous study, including B.1.617.2 (E484K/E484Q B.1.351 (E484K), P.1, and B.1.1.529, have been shown to have E484 replacements (E484A) (31).
It's interesting to note that during the evolution of SARS-CoV-2, the S1-NTD in the present study has additionally organized many mutations, including deletions idel69/70, and del 142/144. Similarly, the frequently deleted regions in the NTD are those at positions 69–70, 141–144, 146, 210, and 243-244 reported in the previous study, most NTD mutations were found to change antigenicity or eliminate epitopes, enabling immune escape. (32).
Most mutations are likely to have an impact on virus entry because the S2 remains stable across CoVs and has a low mutation rate. Additionally, it is less antigenic than S1, perhaps as a result of the substantial N-linked glycosylation, and is consequently not subject to as much selective pressure (33). The BA.1.BA.1.1 and BA.1.18 (Omicron variant) in the present study unexpectedly display several S2 changes, including D796Y, N856K, Q954H, N969K, and L981F. Similarly, the previous study reported that the Omicron variant B.1.1.529 has the following S2 substitutions: D796Y, N856K, Q954H, N969K, and L981F. (34). The global sustainability dissemination of B.1.1.529 shows that these mutations have a benefit. However,

Table 1.
The Entire genome sequences (40 Sequences) were aligned to the SARS-CoV-2 reference genome (NC 045512.2), using the program Nextclade version (Version 2.8.0).
Table 1.
The Entire genome sequences (40 Sequences) were aligned to the SARS-CoV-2 reference genome (NC 045512.2), using the program Nextclade version (Version 2.8.0).
| Mutation | Position | Nucleotide change | Code | Amino acid Change | Type of Mutation | ||
|---|---|---|---|---|---|---|---|
| ORF1a (266...13468) | |||||||
| 444 | GTT > GCT | V 60 A | Valin>Alanine | Non-synonymous SNV | |||
| 593 | CAT > TAT | H 110 Y | Histidine>Tyrosine | Non-synonymous SNV | |||
| 670 | AGT > AGG | S 135 R | Serine>Arginine | Non-synonymous SNV | |||
| 1415 | CTT > TTT | L 384 F | Leucine>Phenylalanine | Non-synonymous SNV | |||
| 2790 | ACT > ATT | T 842 I | Threonine>Isoleucine | Non-synonymous SNV | |||
| 2832 | AAG > AGG | K 856 R | Lysine>Arginine | Non-synonymous SNV | |||
| 2883 | TGT > TAT | C 873 Y | Cisteine>Tyrosine | Non-synonymous SNV | |||
| 3896 | GTT > TTT | V 1211 F | Valine>Phenylalanine | Non-synonymous SNV | |||
| 4184 | GGT > AGT | G 1307 S | Glycine>Serine | Non-synonymous SNV | |||
| 4893 | ACA > ATA | T 1543 I | Threonin>Isoleucine | Non-synonymous SNV | |||
| 5007 | ACG > ATG | T 1581 M | Threonin>Methionine | Non-synonymous SNV | |||
| 510 - 518 | ATG > -TG | del82/84 | del82/84 | Non-frame shift deletion | |||
| 519 | ATG > -TG | M 85 V | Methionine>Valine | Non-synonymous SNV | |||
| 6176 | GAT > AAT | D 1971 N | Aspartic acid>Asparagine | Non-synonymous SNV | |||
| 6513 - 6515 | del2083/2083 | del2083/2083 | Non-synonymous deletion | ||||
| 6516 | TTA > -TA | L 2084 I | Leucine>Isoleucine | Non-synonymous SNV | |||
| 7036 | TTA > TTT | L 2257 F | Leucine>Phenylalanine | Non-synonymous SNV | |||
| 7488 | ACT > ATT | T 2408 I | Threonine>Isoleucine | Non-synonymous SNV | |||
| 8393 | GCT > ACT | A 2710 T | Alanine>Threonin | Non-synonymous SNV | |||
| 9344 | CTT > TTT | L 3027 F | Leucine>Phenylalanine | Non-synonymous SNV | |||
| 9474 | GCT > GTT | A 3070 V | Alanine>Valine | Non-synonymous SNV | |||
| 9534 | ACT > ATT | T 3090 I | Threonine>Isoleucine | Non-synonymous SNV | |||
| 9866 | CTT > TTT | L 32201 I | Leucine>Isoleucine | Non-synonymous SNV | |||
| 10029 | ACC > ATC | T 3255 I | Threonin>Isoleucine | Non-synonymous SNV | |||
| 10323 | AAG > AGG | K 3353 R | Lysine>Arginine | Non-synonymous SNV | |||
| 10449 | CCC > CAC | P 3395 H | Proline>Histidine | Non-synonymous SNV | |||
| 11405 | GTC > TTC | V 3714 F | Valine>Phenylalanine | Non-synonymous SNV | |||
| 11285-11293 | del3674/3676 | del3674/3676 | Non-frame shift deletion | ||||
| 11537 | ATT > GTT | I 3758 V | Isoleucine>Valine | Non-synonymous SNV | |||
| 12534 | ACT > ATT | T 409 I | Threonine>Isoleucine | Non-synonymous SNV | |||
| ORF1b (13468...21555) | |||||||
| 13756 | ATA > GTA | I 97 V | Isoleucine>Valine | Non-synonymous SNV | |||
| 14408 | CCT > CTT | P 314 L | Proline>Leucine | Non-synonymous SNV | |||
| 14821 | CCA > TCA | P 452 S | Proline>Serine | Non-synonymous SNV | |||
| 15641 | AAT > AGT | N 725 S | Asparagine>Serine | Non-synonymous SNV | |||
| 15982 | GTA > ATA | V 839 I | Valine>Isoleucine | Non-synonymous SNV | |||
| 16744 | GGT > AGT | G 1093 S | Glycine>Serine | Non-synonymous SNV | |||
| 17410 | GGT > TGT | R 1315 C | Arginine>Cisteine | Non-synonymous SNV | |||
| 18163 | ATA > GTA | I 1566 V | Isoleucine>Valine | Non-synonymous SNV | |||
| 18433 | GAT > CAT | D 165 H | Aspartic acid>Histidine | Non-synonymous SNV | |||
| 19999 | GTT > TTT | V 2178 F | Valine>Phenylalanine | Non-synonymous SNV | |||
| 20003 | GAT > GGT | P 2179 G | Proline>Glycine | Non-synonymous SNV | |||
| S (21563...25384) | |||||||
| 21765 - 21770 | TACATG > - - - | del69/70 | del69/70 | Non-synonymous deletion | |||
| 21789 | ACT > ATT | T 76 I | Threonine>Isoleucine | Non-synonymous SNV | |||
| 21846 | ACT > ATT | T95I | Threonine>Isoleucine | Non-frame shift deletion | |||
| 21987 | GGT > GAT | G142D | Glycine>Aspartic acid | Non-synonymous SNV | |||
| 21987 - 21995 | del142/144 | del142/144 | Non-frame shift deletion | ||||
| 21996 | TAC > -AC | Y 145 D | Tyrosine>Aspartic acid | Non-synonymous SNV | |||
| 22194 - 22196 | AAT > A-- | del211/211 | del211/211 | Non-synonymous deletion | |||
| 22197 | TTA > -TA | L 212 I | Leucine>Isoleucine | Non-synonymous SNV | |||
| 222000 | GTG > GGG | V 213 G | Valine>Glycine | Non-synonymous SNV | |||
| 22578 | GCT > GAT | G339D | Glycine>Aspartic acid | Non-synonymous SNV | |||
| 22599 | AGA > AAA | R346K | Arginine>Lysine | Non-synonymous SNV | |||
| 22673 | T > C | S371L | Serine>Leucine | Non-synonymous SNV | |||
| 22674 | C > T | S 373 P | Serine>Proline | Non-synonymous SNV | |||
| 22686 | TCC > TTC | S 375 F | Serine>Phenylalanine | Non-synonymous SNV | |||
| 22688 | ACT > GCT | T 376 A | Threonine>Isoleucine | Non-synonymous SNV | |||
| 22786 | AGA > AGC | R408S | Arginine>Serine | Non-synonymous SNV | |||
| 22813 | AAG > AAT | K 417 N | Lysine>Asparagine | Non-synonymous SNV | |||
| 22882 | AAT > AAG | N440K | Asparagine>Lysine | Non-synonymous SNV | |||
| 22898 | GGT > AGT | G446S | Glycine>Serine | Non-synonymous SNV | |||
| 23013 | GAA > GCA | E 484 A | Glutamic acid > isoleucine | Non-synonymous SNV | |||
| 22992 | AGC > AAC | S477N | Serine>Asparagine | Non-synonymous SNV | |||
| 22995 | ACA > AAA | T478K | Threonine>Lysine | Non-synonymous SNV | |||
| 23040 | CAA > CGA | Q493R | Glutamine>Arginine | Non-synonymous SNV | |||
| 23048 | G > A | G496S | Glycine>Serine | Non-synonymous SNV | |||
| 23055 | A > G | Q498R | Glutamine>Arginine | Non-synonymous SNV | |||
| 23063 | AAT > TAT | N501Y | Asparagine>Tyrosine | Non-synonymous SNV | |||
| 23075 | TAC > CAC | Y505H | Tyrosine>Histidine | Non-synonymous SNV | |||
| 23202 | ACA > AAA | T547K | Threonine>Lysine | Non-synonymous SNV | |||
| 23403 | GAT > GGT | D614G | Aspartic acid>Glycine | Non-synonymous SNV | |||
| 23525 | CAT > TAT | H655Y | Histidine>Tyrosine | Non-synonymous SNV | |||
| 23599 | T > G | N679K | Asparagine>Lysine | Non-synonymous SNV | |||
| 23604 | CCT > CAT | P681H | Proline>Histidine | Non-synonymous SNV | |||
| 23854 | AAC > AAA | N764K | Asparagine>Lysine | Non-synonymous SNV | |||
| 23948 | GAT > TAT | D796Y | Aspartic acid>Tyrosine | Non-synonymous SNV | |||
| 24130 | ACC > AAA | N856K | Asparagine>Lysine | Non-synonymous SNV | |||
| 24424 | CAA > CAT | Q954H | Glutamine>Histidine | Non-synonymous SNV | |||
| 24469 | AAT > AAA | N969K | Asparagine>Lysine | Non-synonymous SNV | |||
| 24503 | CCT > TTT | L981F | Leucine>Phenylalanine | Non-synonymous SNV | |||
| ORF3a (25393…26220) | |||||||
| 25471 | GAT > TAT | D 27 Y | Aspartic acid>Tyrosine | Non-synonymous SNV | |||
| 26060 | ACT > ATT | T 223 I | Threonine>Isoleucine | Non-synonymous SNV | |||
| M (26523... 27191) | 26530 | GAT > GGT | D 3 G | Aspartic acid>Glycine | Non-synonymous SNV | ||
| 26577 | CAA > GAA | Q 19 E | Glutamine>Glutamic acid | Non-synonymous SNV | |||
| 26709 | GCT > ACT | A 63 T | Alanine>Threonin | Non-synonymous SNV | |||
| ORF6 (27202…27387) | 27269 | AAA > -AA | K 23 * | K23* | Non-synonymous SNV | ||
| 27266 - 27268 | TTA > - - - | del22/23 | del22/23 | Non-frame shift deletion | |||
| ORF9b (28284…28577) | 28311 | CCC > TCC | P 10 S | Proline>Serine | Non-synonymous SNV | ||
| N (28274…29533) | 28881 | AGG > AAA | R 203 K | Arginine>Lysine | Non-synonymous SNV | ||
| 28882 | AGG > AAA | R203 K | Arginine>Lysine | Non-synonymous SNV | |||
| 28883 | GGA > ACG | G 204 R | Glycine>Arginine | Non-synonymous SNV | |||
| 28311 | CCC > CTC | P 13 L | Proline>Leucine | Non-synonymous SNV | |||
| 28725 | CCT > CTT | P 151 L | Proline>Leucine | Non-synonymous SNV | |||
| 29000 | GGC > AGC | G 243 S | Glycine>Serine | Non-synonymous SNV | |||
| 29005 | CAA > CAC | Q 244 H | Glutamine>Histidine | Non-synonymous SNV | |||
| 29510 | AGT > CGT | S 413 R | Serine > Arginine | Non-synonymous SNV | |||
It is yet unknown how these alterations may affect the pathogenicity of the virus and the polyclonal mAb response.
The ORF1a/b gene is a key target for SARS-CoV-2 nucleic acid assays. It presents the non-structural proteins (nsp1–16) required for the reproduction, maintenance, and repair of the viral DNA (35).
RdRp (nsp12), 3-chymotrypsin-like protease (3CLpro), nsp5, also known as major protease or Mpro, and papain-like proteinase protein are a few of these proteins that antiviral medications used to treat COVID-19 use as targets. (PLpro, nsp3) (36)
The ORF1ab gene produces an RNA-dependent RNA polymerase enzyme and a helicase protein that are both necessary for viral replication (37). The most frequently altered non-structural protein of ORF1ab was nsp3, which also had a deletion at the amino acid sites del2083/2083, A 2710 T, and K 856 R variations. Variant T 3255 I and I 3758 V with non-frame shift deletion (del3674/3676) were found on nsp4 and nsp6, respectively. Transmembrane proteins NSP3, NSP4, and NSP6 have functions in modulating host immunity and enhancing the functionality of host cell organelles for virus reproduction (38). The largest NSP and a crucial part of the virus's transcription and replication are NSP3, which is present at this stage. The host cell membrane is where the transcription and replication of the virus genome take place (39).
The N (nucleocapsid) is a viral protein or gene of significance for diagnostics (nucleic acid and antigen detection) and unique vaccine formulation (40). It is essential for viral assembly, budding, and the recipient cell's reaction to viral infection. Its function is to maintain the genome's structure inside the membrane (41). R203K and G204R mutations are the most frequent types seen in N-protein in the present study (42). The previous study reported that the SARS-CoV-2 variants have improved virulence and transmission due to R203K and G204R mutations. In addition to spike protein changes, nucleocapsid protein mutations are crucial for the pandemic virus' ability to transmit (43). The N-gene in the omicron contains a significant number of deletions, which have been observed to affect diagnostics, primarily the primer binding of a few commercially available kits. It is yet unclear how these alterations affect the pathogenicity of viruses. However, the accessory proteins ORF6 and ORF9b function to inhibit innate immunity, signaling pathways, and interferon (IFN) expression by concentrating on the MAVS adapter associated with mitochondria (44). The previous study results reported that the existing discovered ORF9b gene mutation was>85% prevalent across all Omicron (n = 70) sequences (45).
In comparison to other VOCs, the SARS-CoV-2 genome's high mutation rate, particularly on the spike protein, may promote viral transmission and immunological evasion. Additionally, the development of novel vaccines that incorporate the Omicron variety as a potential reference strain became necessary due to an assortment of these large mutations on the immunogenic epitopes of Spike protein. In the meanwhile, more research is needed to determine the infectivity and efficacy of the current vaccines against Omicron.
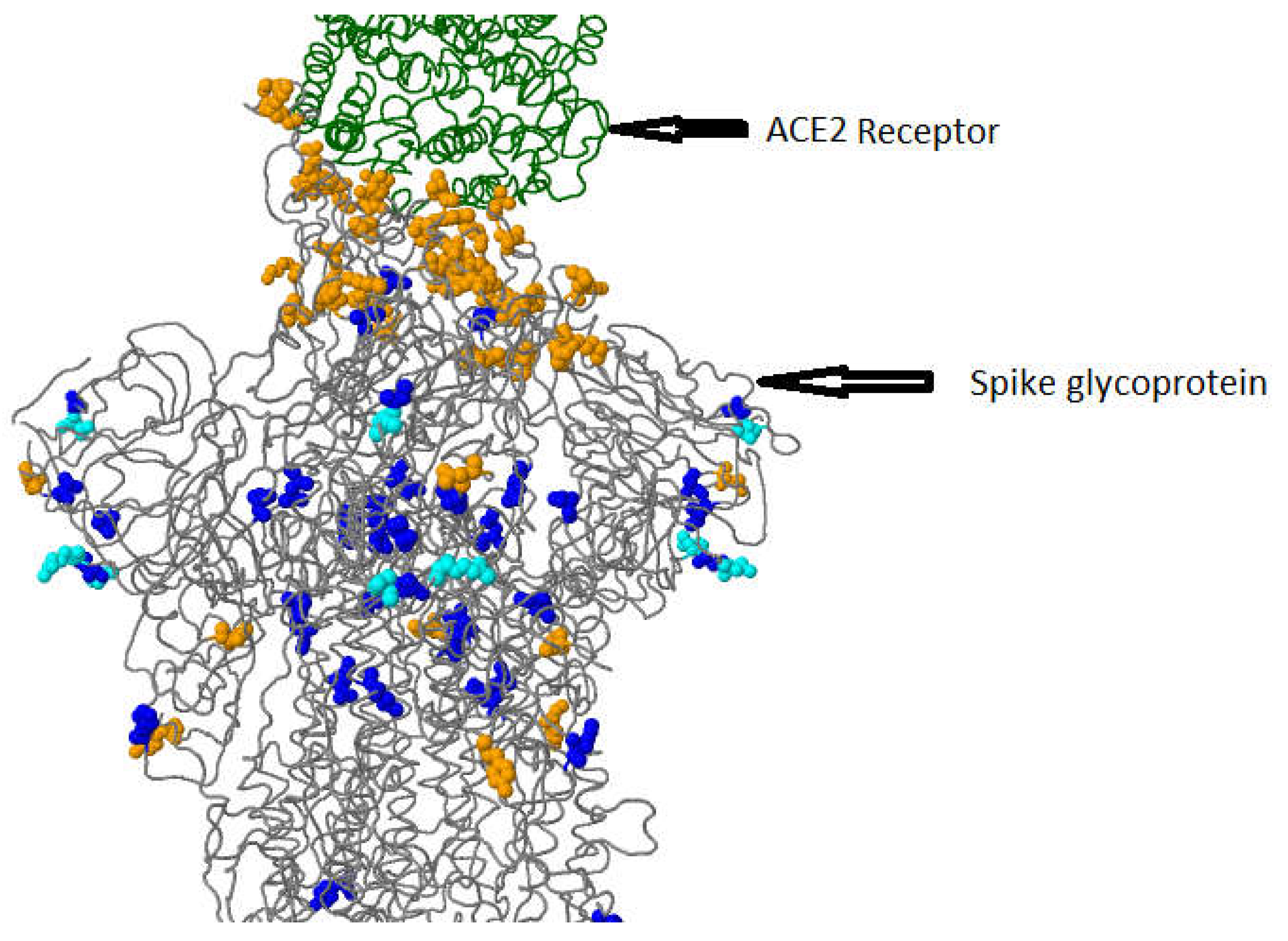
Figure 5.
3D visualization of Spike glycoprotein (PDB: 6acj, EM 4.2 Angstrom) in complex with host cell receptor ACE2in this study.
Figure 5.
3D visualization of Spike glycoprotein (PDB: 6acj, EM 4.2 Angstrom) in complex with host cell receptor ACE2in this study.
Conclusions
This study demonstrates several previously unknown mutations, including mismatches and deletions in translated and untranslated SARS-CoV-2 genomic regions from a clinical sample collected from Duhok, Iraq. In the nucleotide sequences, we discovered position-specific mutations, such as single-nucleotide variations. To establish potential prophylaxis and mitigation for dealing with the pandemic COVID-19 issue, further research should concentrate on structural validations, the functional stability of the proteins, and the effects of the deletions and mismatches in transmission dynamics of the current epidemics. Our study discovered that the coding areas of the SARS-CoV-2 proteins contain 96 distinct variants. Three structural proteins are next in order of most mutated protein, S (ORF1ab, ORF3a, N). Compared to S proteins and ORF1ab, ORF3a, and N proteins have fewer mutations. Both the second and third codon positions are where a greater percentage of single point mutations, C > T, are found. Interestingly the present study reported the new Pango lineage (BA.2) that was previously not described in other studies in Kurdistan, Iraq. The phylogenetic analysis of the viral genome showed several similarities with isolates from USA, UK, Germany, Austria, Canada, Australia, Denmark, Poland, and Colorado, suggesting that several introductions to the state may have taken place. A thorough investigation would be helpful to get additional details about the potential role in terms of infectivity virulence, and virus release. However, creating a new vaccine, particularly one that contains numerous VOCs and is multivalent, could be a significant step in the right direction for limiting current illnesses.
Funding
No funding has been received.
Data Availability Statement
The Global Initiative on Sharing all Personal Data (GISAID) has received the SARS-CoV-2 sequences from this investigation.
Acknowledgments
The authors acknowledge the Covid-19 Center for research and diagnosis for the valuable support of this study.
Conflicts of Interest
The authors declare no conflicts of interes.
Ethics Approval
The ethical Committee of the Directorate General of Health in Duhok province has approved this study (Application number: 16727). The next-generation sequencing of the SARS-CoV-2 positive samples has been done after the cases underwent regular SARS-CoV-2 diagnostic tests at Covid-19 Center and Duhok Burn Hospital (Covid-19 Hospital).
References
- Kim D, Lee J-Y, Yang J-S, Kim JW, Kim VN, Chang H, et al. The architecture of SARS-CoV-2 transcriptome. Cell. 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Sidiqi KR, Sabir DK, Ali SM, Kodzius R, et al. Does early childhood vaccination protect against COVID-19? Frontiers in molecular biosciences 2020, 7, 120. [Google Scholar]
- Huremović, D. Psychiatry of pandemics: a mental health response to infection outbreak: Springer; 2019.
- Shaibu JO, Onwuamah CK, James AB, Okwuraiwe AP, Amoo OS, Salu OB, et al. Full length genomic sanger sequencing and phylogenetic analysis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Nigeria. PloS one. 2021, 16, e0243271. [Google Scholar]
- Aldiabat K, Kwekha Rashid A, Talafha H, Karajeh A,et al. The extent of smartphones users to adopt the use of cloud storage. J Comput Sci. 2018, 14, 1588–1598. [Google Scholar] [CrossRef]
- Alhayani B, Abbas ST, Mohammed HJ, Mahajan HB,et al. Intelligent secured two-way image transmission using corvus corone module over WSN. Wireless Personal Communications. 2021, 120, 665–700. [Google Scholar] [CrossRef]
- Hasan HS, Abdallah AA, Khan I, Alosman HS, Kolemen A, Alhayani B,et al. Novel unilateral dental expander appliance (udex): a compound innovative materials. Computers, Materials and Continua. 2021, 3499–3511.
- Mostafaei S, Sayad B, Azar MEF, Doroudian M, Hadifar S, Behrouzi A, et al. The role of viral and bacterial infections in the pathogenesis of IPF: a systematic review and meta-analysis. Respiratory research. 2021, 22, 1–14. [Google Scholar]
- Hui EK-W. Reasons for the increase in emerging and re-emerging viral infectious diseases. Microbes and infection. 2006, 8, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Viruses at the edge of adaptation. Virology. 2000, 270, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Taheri M, Rad LM, Hussen BM, Nicknafs F, Sayad A, Ghafouri-Fard S,et al. Evaluation of expression of VDR-associated lncRNAs in COVID-19 patients. BMC Infectious Diseases. 2021, 21, 1–10. [Google Scholar]
- Ibrahim FM, Alkaim A, Kadhom M, Sabir DK, Salih N, Yousif E,et al. Chemistry of selected drugs for SARS-CoV-2 inhibition: tested in vitro and approved by the FDA. Chem Int. 2021, 7, 212–216. [Google Scholar]
- Khan S, Hussain A, Vahdani Y, Kooshki H, Hussen BM, Haghighat S, et al. Exploring the interaction of quercetin-3-O-sophoroside with SARS-CoV-2 main proteins by theoretical studies: A probable prelude to control some variants of coronavirus including Delta. Arabian Journal of Chemistry. 2021, 14, 103353. [Google Scholar] [CrossRef] [PubMed]
- Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Global challenges. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. others.(2020). A novel coronavirus from patients with pneumonia in China, 2019. New England Journal of Medicine.
- Liu J, Dai S, Wang M, Hu Z, Wang H, Deng F,et al. Virus like particle-based vaccines against emerging infectious disease viruses. Virologica Sinica. 2016, 31, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Woo PC, Lau SK, Lam CS, Lai KK, Huang Y, Lee P, et al. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. Journal of virology. 2009, 83, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Domains and functions of spike protein in Sars-Cov-2 in the context of vaccine design. Viruses. 2021, 13, 109. [Google Scholar] [CrossRef]
- Gupta D, Sharma P, Singh M, Kumar M, Ethayathulla A, Kaur P,et al. Structural and functional insights into the spike protein mutations of emerging SARS-CoV-2 variants. Cellular and Molecular Life Sciences. 2021, 78, 7967–7989. [Google Scholar] [CrossRef]
- Mahajan S, Kode V, Bhojak K, Karunakaran C, Lee K, Manoharan M, et al. Immunodominant T-cell epitopes from the SARS-CoV-2 spike antigen reveal robust pre-existing T-cell immunity in unexposed individuals. Sci Rep. 2021, 11, 13164. [Google Scholar] [CrossRef]
- Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. Journal of medical virology. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nature communications. 2020, 11, 19. [Google Scholar]
- Daniloski Z, Jordan TX, Ilmain JK, Guo X, Bhabha G, TenOever BR, et al. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. Elife. 2021, 10, e65365. [Google Scholar] [CrossRef]
- Zhang J, Cai Y, Xiao T, Lu J, Peng H, Sterling SM, et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science. 2021, 372, 525–530. [Google Scholar] [CrossRef]
- Ahmad, L. Implication of SARS-CoV-2 immune escape spike variants on secondary and vaccine breakthrough infections. Frontiers in immunology. 2021, 2021, 4563. [Google Scholar] [CrossRef] [PubMed]
- https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.
- Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. 2020, 182, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Shah M, Woo HG. Omicron: a heavily mutated SARS-CoV-2 variant exhibits stronger binding to ACE2 and potently escapes approved COVID-19 therapeutic antibodies. Frontiers in immunology. 2022, 12, 6031. [Google Scholar]
- Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, Van Der Merwe PA,et al. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife. 2021, 10, e70658. [Google Scholar] [CrossRef] [PubMed]
- (https://covariants.org/variants/S.E484).
- Khateeb J, Li Y, Zhang H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Critical Care. 2021, 25, 1–8. [Google Scholar]
- Barrett CT, Neal HE, Edmonds K, Moncman CL, Thompson R, Branttie JM, et al. Effect of mutations in the SARS-CoV-2 spike protein on protein stability, cleavage, and cell-cell fusion function. bioRxiv. 2021.
- (https://covariants.org/variants/21K.Omicron).
- Mishra SK, Tripathi T. One year update on the COVID-19 pandemic: Where are we now? Acta tropica. 2021, 214, 105778.
- Guruprasad, K. Geographical distribution of amino acid mutations in human SARS-CoV-2 orf1ab poly-proteins compared to the equivalent reference proteins from China. ChemRxiv. 2021. [Google Scholar] [CrossRef]
- Graham RL, Sparks JS, Eckerle LD, Sims AC, Denison MR,et al. SARS coronavirus replicase proteins in pathogenesis. Virus research. 2008, 133, 88–100. [Google Scholar] [CrossRef]
- Thomas, S. Mapping the nonstructural transmembrane proteins of severe acute respiratory syndrome coronavirus 2. Journal of Computational Biology. 2021, 28, 909–921. [Google Scholar] [CrossRef]
- Wolff G, Melia CE, Snijder EJ, Bárcena M,et al. Double-membrane vesicles as platforms for viral replication. Trends in microbiology. 2020, 28, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Diao B, Wen K, Zhang J, Chen J, Han C, Chen Y, et al. Accuracy of a nucleocapsid protein antigen rapid test in the diagnosis of SARS-CoV-2 infection. Clinical Microbiology and Infection. 2021, 27, 289–e1. [Google Scholar]
- Gao T, Gao Y, Liu X, Nie Z, Sun H, Lin K, et al. Identification and functional analysis of the SARS-COV-2 nucleocapsid protein. BMC microbiology. 2021, 21, 1–10. [Google Scholar]
- Ramesh S, Govindarajulu M, Parise RS, Neel L, Shankar T, Patel S, et al. Emerging SARS-CoV-2 variants: a review of its mutations, its implications and vaccine efficacy. Vaccines. 2021, 9, 1195. [Google Scholar] [CrossRef] [PubMed]
- Washington NL, Gangavarapu K, Zeller M, Bolze A, Cirulli ET, Barrett KMS, et al. Emergence and rapid transmission of SARS-CoV-2 B. 1.1. 7 in the United States. Cell. 2021, 184, 2587–2594. [Google Scholar] [CrossRef]
- Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS,et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. Journal of virology. 2007, 81, 9812–9824. [Google Scholar] [CrossRef]
- Kannan SR, Spratt AN, Cohen AR, Naqvi SH, Chand HS, Quinn TP, et al. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. Journal of autoimmunity. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Kannan, S.R., Spratt A.N., Quinn T.P., Heng X., Lorson C.L., Sonnerborg A., et al. Infectivity of SARS-CoV2: there is something more than D614G? J. Neuroimmune Pharmacol. 2020, 15, 574–577. [CrossRef]
Figure 2.
Phylogenetic investigation using SARS-COV-2 isolates from various regions and their almost complete gene sequences. the sequences marked Blue dots were isolated from Duhok province, Kurdistan region Iraq the GenBank and GISAID databases’ websites were used to download the sequence. The MEGA 11 tree-building program employed the neighbor joining approach, bootstrapped with 1,000 repeat runs, to analyze the viral sequences.
Figure 2.
Phylogenetic investigation using SARS-COV-2 isolates from various regions and their almost complete gene sequences. the sequences marked Blue dots were isolated from Duhok province, Kurdistan region Iraq the GenBank and GISAID databases’ websites were used to download the sequence. The MEGA 11 tree-building program employed the neighbor joining approach, bootstrapped with 1,000 repeat runs, to analyze the viral sequences.
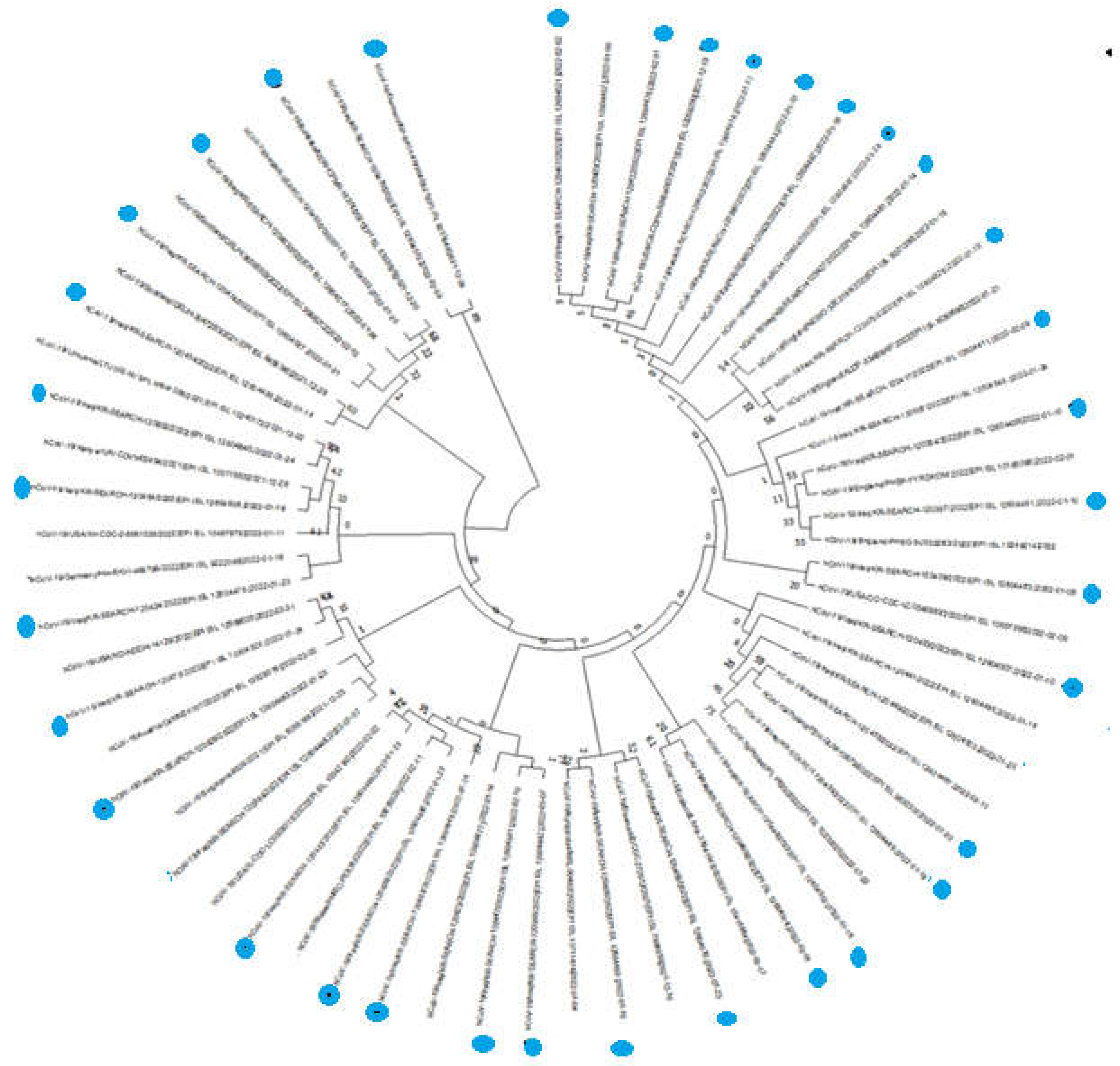
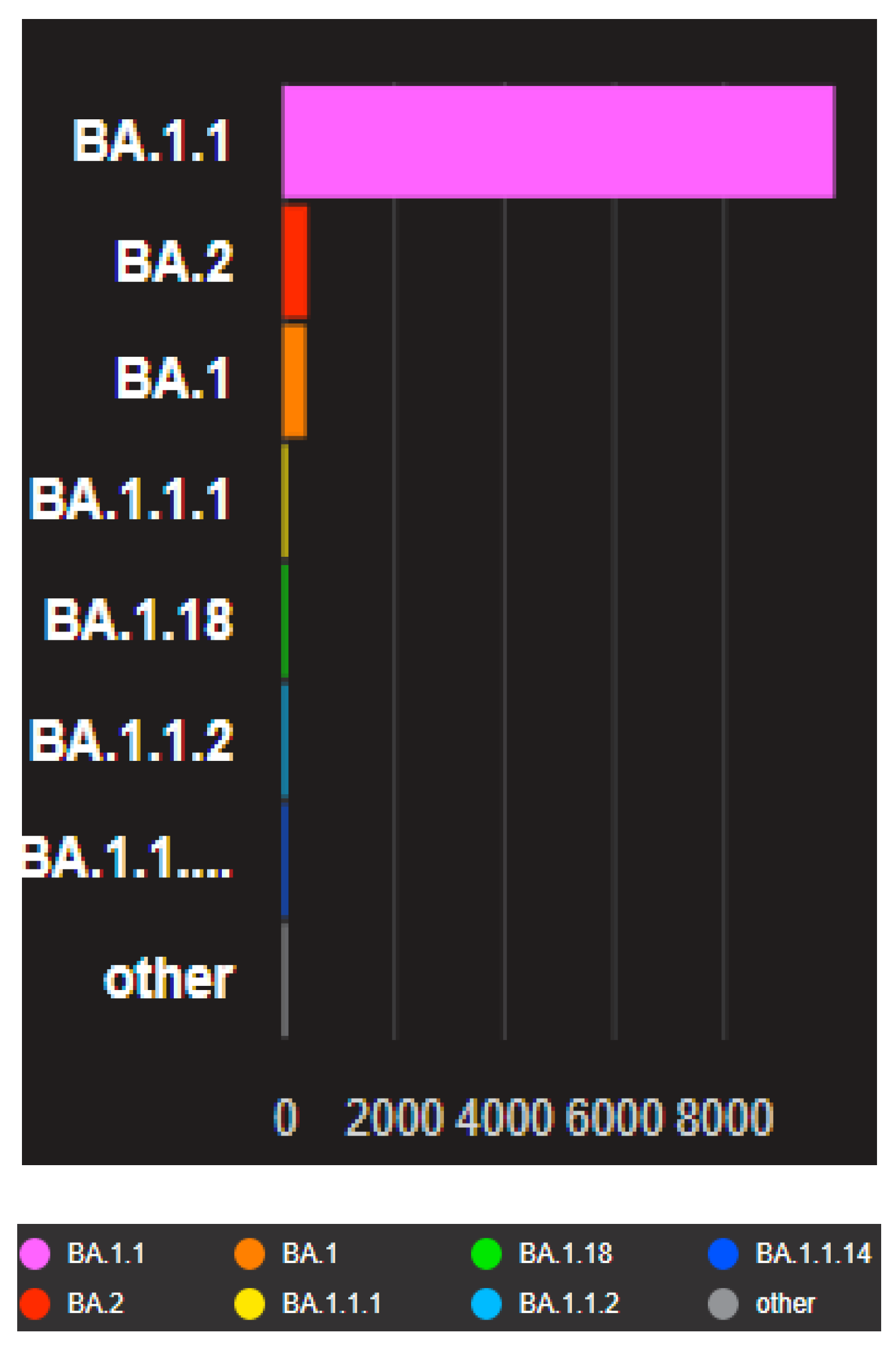
Figure 3.
The Barographs show the distribution of the closely related genomes to the uploaded genomes according to the Pango lineage in the present study. The analyzed data were downloaded from GISAID databases, and the uploaded data in this study were compared with other neighboring countries downloaded from GISAID databases and Gen bank.
Figure 3.
The Barographs show the distribution of the closely related genomes to the uploaded genomes according to the Pango lineage in the present study. The analyzed data were downloaded from GISAID databases, and the uploaded data in this study were compared with other neighboring countries downloaded from GISAID databases and Gen bank.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



