
Submitted:
25 April 2023
Posted:
26 April 2023
You are already at the latest version
Abstract
Understanding molecular mechanisms of neurological disorders is required for development of personalized medicine. When the diagnosis considers not only the disease symptoms, but also their molecular basis, the treatments tailored for individual patients may be suggested. The vitamins-responsive neurological disorders are induced by deficiencies in the vitamin-dependent processes. The deficiencies may occur due to genetic impairments of proteins whose functions are involved with the vitamins. This review considers the enzymes encoded by DHTKD1, PDK3 and PDXK genes, whose mutations are observed in patients with Charcot-Marie-Tooth (CMT) disease. The enzymes bind or produce the coenzyme forms of vitamins B1 (thiamine diphosphate, ThDP) and B6 (pyridoxal-5’-phosphate, PLP). Alleviation of such disorders by administration of the lacking vitamin or its derivative calls upon a better introduction of the mechanistic knowledge to medical diagnostics and therapies. Recent data on lower levels of the vitamin B3 derivative, NAD+, in the blood of patients with CMT disease, compared to the control subjects, are also considered in view of the NAD-dependent mechanisms of pathological axonal degeneration, suggesting therapeutic potential of vitamin B3. Thus, improved diagnostics of the underlying causes of the CMT disease may allow the patients with the vitamin-responsive disease forms to benefit from the administration of the vitamins B1, B3, B6, their natural derivatives or pharmacological forms.
Keywords:
vitamin B1
; vitamin B6
; vitamin B3
; pyridoxal-5’-phosphate
; thiamine diphosphate
; NAD+
; mutation in vitamin-binding protein
; Charcot-Marie-Tooth disease
; DHTKD1
; PDK3
; PDXK
1. Introduction
Pharmacological doses of vitamins may be successfully applied to fight neurological diseases, both acquired and genetically determined [1,2,3,4,5]. The goal of this review is to demonstrate how our understanding of the metabolism and action of vitamins in mammals helps suggesting therapies whose urgent application saves genetically, nutritionally and/or aging-compromised organisms from progressive damage. A relatively advanced understanding of mammalian metabolism of vitamin B1 (thiamine), B3 (nicotinamide, nicotinic acid) and vitamin B6 (pyridoxal, pyridoxine and pyridoxamine), added by the well-known mechanisms of metabolic action of their coenzyme forms, i.e. thiamine diphosphate (ThDP), NAD+ and pyridoxal-5’-phosphate (PLP), underlies the choice of these vitamins to address the review goal. In view of a wide spectrum of neurological disorders induced by impairments in metabolism of these vitamins or their coenzyme action, the focus of this review is on the vitamin B1-, B3- or B6-related disorders manifesting in Charcot-Marie-Tooth (CMT) disease. When diverse cues converge into the similar pathology, exemplified by axonal degeneration in CMT disease, understanding the different molecular mechanisms of the pathology is essential for finding the therapy counteracting the cause of the pathology, not only its symptoms.
Common symptoms due to the compromised metabolic action of each of the vitamins may result from their metabolic interplay. First of all, the coenzyme derivatives of vitamins B1, B3 and B6 function in interconnected metabolic nodes. For instance, de novo production of the major neurotransmitters glutamate and gamma-aminobutyric acid (GABA) from glucose requires participation of ThDP, PLP and NAD+ (Figure 1), exposing their critical and interconnected value for neuronal function.
An amphibolic metabolite 2-oxoglutarate is generated from glucose in mitochondrial tricarboxylic acid cycle. The further ThDP-dependent degradation of 2-oxoglutarate results in energy production. Alternatively, 2-oxoglutarate is required for biosynthesis of glutamate, that occurs through the PLP-dependent transamination or NAD+-dependent reductive amination of 2-oxoglutarate. Glutamate further generates GABA in the PLP-dependent decarboxylation (Figure 1). This example demonstrates that perturbed levels of any of the involved vitamin derivatives may adversely affect the metabolic network of the major neurotransmitters. Therefore, different regulatory mechanisms exist to support stable function of the metabolic network upon perturbations in specific reactions. In particular, the enzyme pyridoxal kinase producing the coenzyme form of vitamin B6, is regulated by phosphorylated forms of vitamin B1 [6]. As a result, the interplay of B1, B3 and B6 vitamins in the metabolic pathways essential for neural functions, provides a good explanation for the notion that different perturbations in the vitamin-dependent processes may converge into axonal degeneration underlying neuropathological symptoms of Charcot-Marie-Tooth (CMT) disease. Moreover, exacerbation of the disease symptoms with aging may be contributed by the known age-associated impairments in the vitamin status and metabolism [7,8,9,10].
The pathologies induced by perturbed vitamin action or status, are often alleviated by administration of the corresponding vitamin, with the timely administration preventing irreversible secondary damage [2,3,11]. Hence, especially at the initial stages of the disease, knowledge on the specific primary impairment is of critical therapeutic significance. This review draws attention to the translational value of the basic knowledge on the disease mechanisms by demonstrating the relationship between CMT disease, perturbed vitamin-dependent processes and the therapeutic potential of the vitamins administration.
2. General Information on Charcot-Marie-Tooth Disease and Associated Mutations
CMT disease is a group of non-fatal neurological disorders affecting app. 1 in 2500 people. Despite clinical and genetic heterogeneity, common manifestations of the disease are impairments of the motor and/or sensory peripheral nerves, resulting in muscle weakness and atrophy, accompanied by sensory loss. Neural cells of patients with this disease cannot support the required electrostimulation due to axonal anomalies in the motor and sensory neurons. Impaired peripheral signaling in CMT disease is generally suggested to be due to de-energization of the axon endings, linked to mitochondrial abnormality. Dependent on the primary cause, the two major forms of CMT are the demyelinating form and axonal form [12]. Although the convergent symptoms point to interdependence of the pathological events in different forms of CMT disease, identification of its specific molecular mechanisms is crucial for the cause-directed therapies. Yet in autosomal recessive CMT disease causal genetic variants are identified only in ¼ of all the cases.
Approximately 1000 mutations in 90 different genes are known to cause CMT disease, with most of the mutations affecting cytoskeleton or axonal trafficking [13,14]. Genetic impairments in the mitochondrial proteins represent the most direct pathway to mitochondrial insufficiency associated with CMT disease. The two genes (DHTKD1, PDK3) encode for the mitochondrial enzymes binding the coenzyme form of vitamin B1 (ThDP), and another gene (PDXK) encodes for the producer of the coenzyme form of vitamin B6 (PLP). Mitochondrial metabolism and energy production strongly depend on both the ThDP- and PLP-catalyzed reactions.
In addition to mutated genes, a balance in biosynthesis and degradation of a major mitochondrial redox substrate NAD+ (vitamin B3 derivative) has a significant role in axonal degeneration [15,16]. The link between the NAD+ levels and CMT disease is supported by reduced levels of NAD+ in the blood of CMT patients vs healthy subjects [17]. Thus, the pathogenic mechanisms of CMT disease arising due to the currently known mutations in the ThDP- or PLP-binding enzymes, are considered below along with the data from animal and cellular models demonstrating molecular mechanisms underlying potential usage of vitamins B1, B3 and B6 as therapeutic agents to overcome the CMT pathology.
3. CMT Disease Upon Mutations of the Vitamin-Dependent Enzymes
3.1. Pyridoxal Kinase
PDXK gene encodes for pyridoxal kinase (PDXK) catalyzing the ATP-dependent phosphorylation of the vitamers of vitamin B6 (pyridoxal, pyridoxin, pyridoxamine) to their phosphorylated forms. The phosphorylated derivative of pyridoxal, PLP, is a coenzyme of a number of enzymes, including those essential for energy production and neurotransmitter metabolism. Those are exemplified in Figure 1 as transaminases and PLP-dependent decarboxylases.
Two biallel variants of the PDXK gene (c.682G>A and c.659G>A with the protein substitutions Ala228Thr and Arg220Gln, correspondingly) are identified in five individuals from two unrelated families (OMIM 618511). The variants cause the autosomal recessive CMT disease of 6C type (CMT6C) with primary axonal polyneuropathy and optic atrophy, also called as hereditary motor and sensory neuropathy type VIC with optic atrophy (HMSN6C) [3]. Distal muscle weakness and atrophy predominantly affecting the lower limbs, have an onset in the first life decade. Optic atrophy and vision loss occur during adulthood.
The substituted amino acids Ala228 and Arg220 are evolutionary conserved residues participating in the PDXK interaction with its substrate ATP. Hence, the substitutions of these conserved residues impair the substrate binding, resulting in the reduced PDXK activity and low PLP levels, observed in the patients. Another homozygous missens mutation in PDXK gene (c.225T>A with the protein substitution Asn75Lys) is revealed in two children from one family, exhibiting symptoms of CMT disease [18]. This substitution decreases stability of the PDXK dimer and increases the protein degradation. As a result, the patients’ levels of the PDXK activity and PLP are very low.
Oral PLP administration at a dose of 50 mg/kg, known to have no neurotoxic effects that are possible at high doses of vitamin B6, increases the levels of PLP in the blood of patients with pathogenic substitutions in PDXK, characterized by impaired production of PLP [3,18]. In the CMT disease with the Ala228Thr substitution in PDXK, the PLP replacement scheme is shown to stably increase the PLP levels in the patients up to 24 months [3]. Moreover, in the long-term follow-up, the PLP increase is associated with alleviation of the symptoms and slowing down the progress of CMT disease, induced by mutations of PDXK gene. The data obtained [3,18] stress the need of screening the potential PDXK mutations in patients with autosomal recessive polyneuropathy of early onset, as prompt PLP supplementation may cause long-term improvement in clinical outcomes.
3.2. Vitamin B1-Dependent Enzymes
Thiamine (vitamin B1), its natural derivative ThDP (cocarboxylase) and pharmacological forms (benfotiamin, sulbutiamin etc. [19]) are known to exhibit cardio- and neuroprotective actions under variety of conditions associated with dysfunctional mitochondria [1,20,21,22]. Potential link between thiamine and the CMT disease symptoms is supported by an outbreak of a peripheral neuropathy similar to CMT disease in 88 male prisoners who exhibited hyporeflexia/areflexia of the lower extremities, sensory deficit and motor weakness. The pathology has been diagnosed along with the observation of thiamine deficiency in 80% of the prisoners [23].
Regarding benefits of thiamine or its known pharmacological forms to treat the CMT disease, the cases of the disease induced by pathogenic mutations in the genes encoding for mitochondrial enzymes regulated by ThDP, are of special interest. These are mutations in the DHTKD1 and PDK3 genes encoding for the mitochondrial enzymes 2-oxoadipate dehydrogenase (OADH) and the neural-tissue-specific isoenzyme 3 of pyruvate dehydrogenase kinase (PDK3), correspondingly. ThDP is a coenzyme of OADH and an inhibitor of PDK3 [24,25,26]. Remarkably, ThDP is also a coenzyme of the PDK3 target pyruvate dehydrogenase, that is inactivated by the PDK3-catalyzed phosphorylation. As the first component of the key mitochondrial pyruvate dehydrogenase complex (PDC) coupling glycolysis to the mitochondrial metabolism, pyruvate dehydrogenase is indispensable for oxidation of the glucose-derived pyruvate in mitochondria.
While the PDK3-dependent PDC phosphorylation is a well-known regulatory mechanism [27], the role of the OADH-dependent PDC glutarylation is only emerging [17]. Nevertheless, the finding that OADH is involved with post-translational modifications of PDC in addition to PDK3, is of pathophysiological significance, as excessive protein glutarylation is observed in a number of pathological states [28].
Molecular mechanisms of mitochondrial dysfunction upon the CMT-disease-associated mutations in the mitochondrial ThDP-regulated OADH and PDK3, are considered below in view of the therapeutic potential of vitamin B1.
3.2.1. Isoenzyme 3 of Kinase of the ThDP-Dependent Pyruvate Dehydrogenase
Mutation in the PDK3 gene causes the substitution R158H in PDK3, representing the genetic cause of an X-linked dominant form of axonal CMT disease (CMTX6) (OMIM 300905) [29,30]. Males are more severely affected by this mutation than females. The symptoms onset occurrs within the first 13 years, when the muscle weakness and atrophy, predominantly in lower limbs, and sensory deficits are detected. Some of the affected men have moderate sensorineural hearing loss and perturbed auditory brainstem response.
The substitution R158H leads to a hyper-active PDK3 exhibiting an increased affinity to PDC, compared to the wild-type enzyme. As a result, higher levels of phosphorylation and inactivation of cellular PDC are observed in patients carrying this mutation [30]. Metabolic characteristics of fibroblasts and iPSC-derived motor neurons of the patients reveal failures in energy production and mitochondrial function. In vivo models where the R158H-substituted PDK3 ortholog, known as PDHK2 in C. elegans, is knocked-in, or the wild-type and mutated PDK3 are overexpressed in the GABA-ergic motor neurons, recapitulate deficits in mitochondrial function and synaptic neurotransmission, observed in the cellular studies [13]. Besides, the models reveal such characteristics of CMT disease as the locomotion defects and signs of progressive neurodegeneration.
ThDP is known to inhibit kinases of pyruvate dehydrogenase [24,25,26]. Hence, patients with CMT disease induced by hyperactive PDK3, may benefit from administration of the PDK3 inhibitor ThDP, that is well-known in medicine and pharmacology as cocarboxylase. Pharmacological lipophilic forms of thiamine (vitamin B1), such as sulbutiamin (enerion) and benfotiamin, are also widely available. Remarkably, ThDP inhibition of PDK3 would increase the activity of pyruvate dehydrogenase by decreasing the enzyme phosphorylation level. Simultaneously, as a coenzyme of pyruvate dehydrogenase, ThDP may activate PDC-catalyzed oxidation of glycolytic product pyruvate by an independent mechanism, such as the saturation of the pyruvate dehydrogenase with its coenzyme. As a result, the thiamine administration may counteract the hyperactivity of the R158H-substituted PDK3 by inhibiting PDK3 and activating PDC, potentially relieving the CMT disease caused by this mutation.
3.2.2. Molecular Mechanisms of CMT Disease Caused by Mutations in the DHTKD1-Encoded ThDP-Dependent 2-Oxoadipate Dehydrogenase
Axonal type of CMT disease type 2Q (OMIM 615025) is described upon heterozygous loss-of-function mutation of the DHTKD1 gene encoding for the mitochondrial ThDP-dependent OADH [31]. The mutation c.1455T>G in exon 8 of the DHTKD1 gene causes preterm termination of the transcription at Tyr485 codon and a 2-fold decrease in the OADH mRNA. The resulting autosomal dominant form of CMT disease is characterized by muscle atrophy, predominant weakness of the lower limbs, decreased or absent deep tendon reflex, and mild to moderate sensory impairment. The symptom onset is detected from 13 to 25 years.
Mice models of homozygous knockout of the DHTKD1 gene mimic CMT disease phenotype, demonstrating anatomic and functional features of peripheral neuropathy with characteristic perturbations of the motor and sensory functions, axonal degeneration and muscle atrophy. The phenotype is accompanied by serious metabolic abnormalities, with significant increases in the urine levels of 2-oxoadipate and its transamination product 2-aminoadipate [32].
Pathogenic homozygous DHTKD1 mutations are associated with strongly increased levels of the OADH substrate 2-oxoadipate and its derivatives (2-aminoadipate, 2-hydroxyadipate), causing severe neurological manifestations, delayed development, chronic diseases of respiratory pathways and early muscle atrophy [33,34,35]. Nevertheless, in some mutants clinical manifestations are absent even when biochemical parameters are perturbed [34,35].
Pathogenicity of heterozygous mutations in the DHTKD1 gene varies. The mutations may be associated with neuropathies, such as CMT disease and similar states, with autoimmune disease, such as eosinophilic esophagitis, or with epilepsy, but often the same mutations may be present in the asymptomatic persons [31,36,37,38]. Most of the nucleotide substitutions in the DHTKD1 gene are asympthomatic [39]. Thus, understanding molecular basis of pathophysiological impact of the DHTKD1-encoded OADH is not straightforward, with the impact potentially involving interactions with other pathways which are considered below.
Current level of characterization of the structure-function relationship in the OADH molecule allows one to predict pathogenicity of the amino acids substitutions in the enzyme active site and protein-protein interfaces. Structures of recombinant OADH [39,40] and homology modelling of the enzyme complexes with its ligands [41,42] reveal the impact of OADH mutations in the active site and dimeric interface of the enzyme on the enzyme-catalyzed reaction. Besides, some information on the protein residues involved in heterologous interactions upon formation of the OADH multienzyme complex, where OADH catalyzes oxidative decarboxylation of 2-oxoadipate with generation of glutarylCoA and NADH [43,44,45], enables predictions of functional impairments due to mutations affecting the complex formation. Nevertheless, several considerations are important to note, regarding the varied pathophysiological impact of the same DHTKD1 mutations in different carriers of a mutation. First, the catalytic function of OADH is redundant and could well be substituted by an ubiquitously expressed OADH isoenzyme, 2-oxoglutarate dehydrogenase (OGDH). In vitro, OGDH catalyzes oxidative decarboxylation of 2-oxoadipate at a rate up to 50% of that of oxidative decarboxylation of 2-oxoglutarate, with OGDH expression usually exceeding that of OADH [46]. Second, in animal tissues OADH undergoes post-translational modifications strongly affecting the enzyme structure, compared to that of the characterized recombinant enzyme, that may correspond to the existence of a yet unidentified enzyme function [47]. Third, biochemical effects of the OADH mutations, i.e. increases in the 2-oxo/aminoadipate levels, are not necessarily accompanied by clinical symptoms [34]. These considerations suggest that the (patho)physiological impact of OADH relies on the OADH-specific interplay with other proteins/pathways rather than solely OADH catalytic function.
The assumption is supported by the data on the most characterized associations of OADH with insulin resistance, obesity and type II diabetes. The data not only reveal a significance of genetic background for the OADH biological impact, but also suggest perturbed glucose metabolism as molecular mechanism underlying the OADH-associated neurological disorders. Indeed, increased 2-oxoadipate and/or 2-aminoadipate levels in the mice knockouts of DHTKD1 [32,48,49] cause insulin resistance phenotype [32]. Liver expression of DHTKD1 is the major regulator of the level of 2-aminoadipate, the transamination sibling of the OADH substrate 2-oxoglutarate, in serum [50]. Dependent on external (diet) and genetic (different alleles) factors, the DHTKD1 gene expression also correlates with the levels of glucose, cholesterol and the diabetic status [50]. In the blood plasma of humans and mice, glucose levels are negatively correlated with those of 2-aminoadipate, while the levels of 2-aminoadipate and insulin correlate positively [51]. When β-cells of pancreas are treated with 2-aminoadipate, more insulin is secreted, corresponding to increased insulin secretion upon the accumulation of 2-aminoadipate in pancreas, as observed in mice on a high-fat diet [51]. At the same time, the mice knockouts of Csf2 gene (controlling the function of granulocytes and macrophages) are obese without diabetic symptoms. Remarkably, these mice exhibit increased expression of DHTKD1 and decreased 2-aminoadipate level, compared to the wild-type mice [52]. DHTKD1 expression is also increased by a natural compound mangiferin preventing obesity [53]. Vitamin B1 (thiamine), which is a precursor of the OADH coenzyme ThDP, upregulates OADH activity in the rat cerebellum [54].
Thus, relatively low expression of DHTKD1 may be an early indicator of obesity and type II diabetes, while increased expression of DHTKD1 is associated with clinical improvement of such states [55]. Pharmacological regulation (e.g., mangiferin, vitamin B1) or genetic factors (exemplified by knockout of Csf2 gene) may increase DHTKD expression. Compensating for functional impairments in the OADH mutants, this conditional increase in the DHTKD expression may contribute to heterogeneity of the mutant DHTKD1 phenotypes. The available data thus imply that pharmacological up-regulation of the OADH expression and/or activity by administration of vitamin B1 may be of therapeutic value.
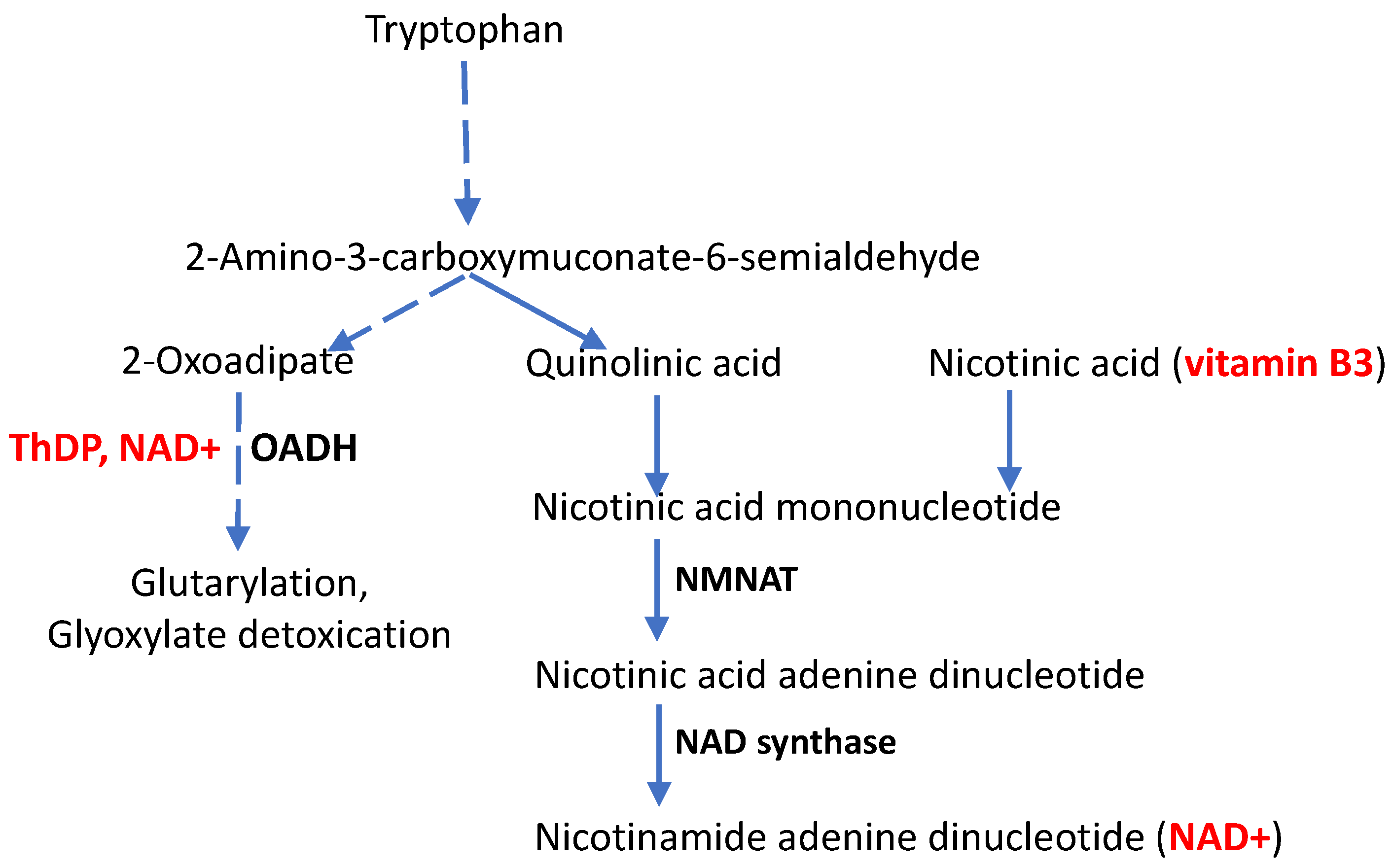
If phenotypic manifestations of the DHTKD1 mutations, particularly in CMT disease, depend on the function of other genes, what could be these other genes? To answer this question, a closer look on metabolic pathways potentially affected by OADH function, is required (Figure 2).
Participating in the production of glutaryl-CoA for protein glutarylation, OADH is involved with metabolic regulation by post-translational modifications through the acylation of protein lysine residues. Recent discovery of glutarylation of the pyruvate dehydrogenase complex, a key player in the oxidative glucose metabolism, provides a mechanistic link between the OADH function and oxidative glucose metabolism [17], characterized in the association studies discussed above. Depending on pathophysiological state, a short-term inhibition of OADH in the rat brain may cause a long-term increase in the enzyme expression, associated with increased glutarylation of the brain proteins and elevated expression of deglutarylase sirtuin 5 [17].
Furthermore, existence in mammalian tissues of an N-terminus-truncated isoform of OADH, lacking the protein part involved into the formation of the multienzyme complex, but comprising the active site [47], agrees with our earlier suggestion [43], that OADH may also have some functions outside of the multienzyme complex. An example of such functions is glyoxylate detoxication that involves non-oxidative decarboxylation of 2-oxoacids by their ThDP-dependent dehydrogenases.
Finally, functioning in the tryptophan degradation pathway, OADH is linked to de novo biosynthesis of NAD+ and its signaling derivatives from tryptophan (Figure 2). The interplay is supported by perturbed nicotinamide (vitamin B3) homeostasis upon the OADH inhibition [56]. Perturbed NAD+ metabolism is also shown in independent animal and cellular models of OADH impairment [32,57], further exposing the role of NAD+ in CMT disease arising upon the DHTKD1 mutations. In fact, mutations and knockouts of the DHTKD1 gene decrease NAD pool and energy metabolism [32,57]. Combined with the data on perturbed pool of vitamin B3 in the OADH-inhibited cells [56] and neurological manifestations of impaired tryptophan catabolism [58], the available data argue for significance of the NAD+-dependent pathways in the pathophysiological contribution of OADH function. Hence, administration of vitamin B3, especially in the form of its derivative nicotinamide riboside which helps overcoming the NAD+ biosynthetic hurdles [59,60], may be promising to alleviate the neurological symptoms in CMT disease caused by the DHTKD1 mutations. The underlying molecular mechanisms are considered in more details below.
4. CMT Disease, Aging and the Vitamin B3 Derivative NAD+
The symptoms of CMT disease may appear at an earlier or later age. Accordingly, the disease progress is very different. Many patients remain active and have normal life expectancy, but severe cases are also known, when respiratory difficulties may be lethal [61]. Increased manifestations of CMT disease with age coincide with the age-dependent decrease in cellular NAD+ levels [62,63,64,65,66]. The link deserves more attention, especially regarding CMT disease developed in people with mutated DHTKD1 gene, encoding the OADH protein that is linked to the metabolic and signaling pathways involving NAD+ (Figure 2).
Decreasing NAD+ levels in cells and tissues upon normal aging is considered to be due to the aging-induced drop in NAD+ biosynthesis and/or increase in NAD+ degradation. Apart from the well-known function of being a substrate of redox reactions, NAD+ is involved in the post-translational regulation of proteins and DNA damage response as a substrate of sirtuins and poly-(ADP-ribose) polymerase (PARP), correspondingly [67]. NAD+ is also used in biosynthesis of signaling molecules by nucleosidases CD38, CD157, SARM1, regulating calcium homeostasis and NAD+ metabolism [64,65]. In particular, production of adenosine diphosphate ribose and its cyclic form in the SARM1-catalyzed reaction of NAD+ degradation (NADase reaction) is a signal for axonal degeneration. The NADase activity of SARM1 is greatly stimulated by axonal damage. The loss of intracellular NAD+ upon such a damage may be a signal for the SARM1 activation, as high concentrations of NAD+ inhibit SARM1 [68]. Associated with energy failure, Ca2+ overload and proteolysis activation, the loss of NAD+ due to axonal damage activates SARM1, thus initiating axonal self-destruction. In contrast, increasing NAD+ levels may not only attenuate the activation of SARM1 by neurodegenerative insults, but also improve the energy status, supporting calcium homeostasis and associated events [68]. The role of increased NAD+ biosynthesis upon pathological self-destruction of damaged axons, known as Wallerian degeneration, is demonstrated by studies of the mouse mutation in the WLD gene. When the gene is mutated to WLDS (Wallerian degeneration slow), the pathological axon degeneration under a number of neurodegenerative conditions, including CMT disease, is suppressed. Remarkably, WLD gene is mapped to the distal region of the mouse chromosome 4, with an axonal form of CMT disease mapped to the homologous location in humans, 1p36. This region includes the gene encoding an enzyme for NAD+ biosynthesis, nicotinamide/nicotinic acid mononucleotide adenylyltransferase 1 (NMNAT, Figure 2), whose mutation causes the axonal form of CMT disease (OMIM 608700). In the WLDS mice, axonal NAD+ biosynthesis is activated, as the WLDS mutation enables expression of a chimeric protein (WLDS), comprising NMNAT1 [69]. In the same work, SIRT1-encoded sirtuin 1 is shown as a downstream activator for increased activity of NMNAT1. Thus, fighting the NAD+ depletion in the axonal degeneration is suggested as a therapeutic approach for CMT disease [70].
In this regard, the observation of decreased NAD+ levels in the blood of patients with CMT disease, compared to healthy subjects [17], is remarkable. This NAD+ decrease supports independent studies on the role of NAD+ in pathological axon degeneration, that is observed in CMT disease [16,68]. Positive action of the administration of different NAD+ precursors is known in a number of pathologies with neurological manifestations [64,65]. In particular, NAD+ precursor, nicotinamide riboside, is shown to protect from excitotoxicity-induced neurodegeneration [15]. Thus, the available data suggest vitamin B3 and its derivatives to be of therapeutic potential in CMT disease.
4. Spatial Specificity of the Impact of Gene Mutations Perturbing the Vitamin-Dependent Processes
This review deals with the perturbed processes dependent on vitamins B1 or B6, which result in non-fatal peripheral polyneuropathy, such as CMT disease. The considered mutations cause peripheral perturbations affecting axonal function and morphology. Remarkably, when the status of the same vitamins is perturbed in the brain, the much more severe neurological disorders develop. For example, mutations in the PNPO- or PROSC-encoded proteins of vitamin B6 metabolism cause severe epilepsies in newborns [71]. Moreover, epileptic seizures are also known in animal models with dysregulated expression of PDXK in the brain [72]. However, with the PDXK mutations known in CMT disease, the B6-dependent impairments are limited to peripheral signaling. It may therefore be suggested that this peripheral pathological manifestation is due to axon-specific features of metabolism and heterological interaction of the mutated protein. It is also known from independent studies that the regulation of PDXK expression differs in the peripheral tissues, compared to the brain [3].
Similarly, a well-known consequence of the perturbed function of the ThDP-dependent enzymes in the brain is Wernicke's encephalopathy. Its unusual presentation may include epileptic seizures, as observed in pregnancy [73,74]. An outbreak of the life-threatening Wernicke's encephalopathy is also known in infants on a thiamine-deficient soy-based formula [11]. In the long-term follow-up of the 8 initial survivors, 6 developed severe epilepsy [75]. These cases suggest that untreated deficiency of vitamin B1 in the brain may progress from Wernicke’s encephalopathy to epilepsy, probably interacting with such factors as increased nutritional and metabolic demand in pregnant or developing organisms. Severe epileptic seizures have also been reported in some patients with the DHTKD1 mutations, although the interaction with non-identified co-morbidities cannot be excluded in these cases [35].
A characteristic feature of seizures arising upon impaired metabolism of vitamins B1 or B6 in the brain, is no response to conventional anticonvulsant treatments. Instead, the seizures disappear upon administration of the corresponding vitamins, similarly to the less severe symptoms of CMT disease caused by axonal impairments of the vitamin-dependent processes.
5. Conclusions
CMT disease may develop due to a great number of gene mutations, some of which affect the vitamin-dependent processes. Ensuing perturbations in the mutations-affected action of vitamins B1 and B6 may be counteracted by administration of the lacking vitamin or its derivative.
Vitamin B3 is a precursor or NAD+. In aging, the increased NAD+ degradation in the DNA damage response and sirtuin-dependent pathways is not properly compensated by the NAD+ biosynthesis. This generally observed disbalance in the metabolism of NAD+ due to aging should be taken into account regarding increased manifestations of CMT disease with age and the NAD-dependent regulation of axonal degeneration. The decreased NAD+ levels in the blood of CMT patients, compared to healthy subjects, suggest that the patients may benefit from the administration of NAD+ precursors. The NAD+ biosynthetic intermediates, such as nicotinamide riboside, may decelerate the progression of CMT disease with age.
Funding
This research was funded by Russian Foundation of Basic Research, grant number 20-54-7804.
Acknowledgments
Technical support of Ms. Irina Karlina in formatting this manuscript is acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
References
- Gibson, G.E.; Feldman, H.H.; Zhang, S.; Flowers, S.A.; Luchsinger, J.A. Pharmacological thiamine levels as a therapeutic approach in alzheimer's disease. Front Med (Lausanne) 2022, 9, 1033272. [Google Scholar] [CrossRef]
- Fukui, K.O.; Kubota, M.; Terashima, H.; Ishiguro, A.; Kashii, H. Early administration of vitamins b1 and b6 and l-carnitine prevents a second attack of acute encephalopathy with biphasic seizures and late reduced diffusion: A case control study. Brain Dev 2019, 41, 618–624. [Google Scholar] [CrossRef]
- Chelban, V.; Wilson, M.P.; Warman Chardon, J.; Vandrovcova, J.; Zanetti, M.N.; Zamba-Papanicolaou, E.; Efthymiou, S.; Pope, S.; Conte, M.R.; Abis, G.; et al. Pdxk mutations cause polyneuropathy responsive to pyridoxal 5'-phosphate supplementation. Ann Neurol 2019, 86, 225–240. [Google Scholar] [CrossRef]
- Rusch, C.T.; Wortmann, S.B.; Kovacs-Nagy, R.; Grehten, P.; Haberle, J.; Latal, B.; Stettner, G.M. Thiamine pyrophosphokinase deficiency due to mutations in the tpk1 gene: A rare, treatable neurodegenerative disorder. Neuropediatrics 2021, 52, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Qin, J.; Liu, D.; Wang, Y.; Shen, X.; Yang, N.; Zhou, H.; Cai, X.T.; Wang, Z.L.; Yu, D.; et al. Reduced thiamine binding is a novel mechanism for tpk deficiency disorder. Mol Genet Genomics 2019, 294, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.; Aleshin, V.; Nogues, I.; Kahne, T.; Parroni, A.; Contestabile, R.; Salvo, M.L.; Graf, A.; Tramonti, A. Thiamine-dependent regulation of mammalian brain pyridoxal kinase in vitro and in vivo. J Neurochem 2022, 161, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo nad assay reveals the intracellular nad contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo nad(+) synthesis specifies immune function in aging and inflammation. Nat Immunol 2019, 20, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fei, G.; Lu, J.; Jin, L.; Pan, S.; Chen, Z.; Wang, C.; Sang, S.; Liu, H.; Hu, W.; et al. Measurement of blood thiamine metabolites for alzheimer's disease diagnosis. EBioMedicine 2016, 3, 155–162. [Google Scholar] [CrossRef]
- Pan, X.; Sang, S.; Fei, G.; Jin, L.; Liu, H.; Wang, Z.; Wang, H.; Zhong, C. Enhanced activities of blood thiamine diphosphatase and monophosphatase in alzheimer's disease. PLoS One 2017, 12, e0167273. [Google Scholar] [CrossRef]
- Fattal-Valevski, A.; Kesler, A.; Sela, B.A.; Nitzan-Kaluski, D.; Rotstein, M.; Mesterman, R.; Toledano-Alhadef, H.; Stolovitch, C.; Hoffmann, C.; Globus, O.; et al. Outbreak of life-threatening thiamine deficiency in infants in israel caused by a defective soy-based formula. Pediatrics 2005, 115, e233–e238. [Google Scholar] [CrossRef]
- Luan, C.J.; Guo, W.; Chen, L.; Wei, X.W.; He, Y.; Chen, Y.; Dang, S.Y.; Prior, R.; Li, X.; Kuang, Y.; et al. Cmt2q-causing mutation in the dhtkd1 gene lead to sensory defects, mitochondrial accumulation and altered metabolism in a knock-in mouse model. Acta Neuropathol Commun 2020, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.K.; Brewer, M.H.; Perez-Siles, G.; Ellis, M.; Ly, C.; Burgess, A.; Neumann, B.; Nicholson, G.A.; Vucic, S.; Kennerson, M.L. Charcot-marie-tooth disease causing mutation (p.R158h) in pyruvate dehydrogenase kinase 3 (pdk3) affects synaptic transmission, atp production and causes neurodegeneration in a cmtx6 c. Elegans model. Hum Mol Genet 2021, 31, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Dohrn, M.F.; Glockle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hortnagel, K.; et al. Frequent genes in rare diseases: Panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J Neurochem 2017, 143, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Vaur, P.; Brugg, B.; Mericskay, M.; Li, Z.; Schmidt, M.S.; Vivien, D.; Orset, C.; Jacotot, E.; Brenner, C.; Duplus, E. Nicotinamide riboside, a form of vitamin b(3), protects against excitotoxicity-induced axonal degeneration. FASEB J 2017, 31, 5440–5452. [Google Scholar] [CrossRef]
- McGuinness, H.Y.; Gu, W.; Shi, Y.; Kobe, B.; Ve, T. Sarm1-dependent axon degeneration: Nucleotide signaling, neurodegenerative disorders, toxicity, and therapeutic opportunities. Neuroscientist 2023, 10738584231162508. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.I.; Karlina, I.S.; Zavileyskiy, L.G.; Aleshin, V.A.; Artiukhov, A.V.; Kaehne, T.; Ksenofontov, A.L.; Ryabov, S.I.; Graf, A.V.; Tramonti, A.; et al. Delayed impact of 2-oxoadipate dehydrogenase inhibition on the rat brain metabolism is linked to protein glutarylation. Front Med (Lausanne) 2022, 9, 896263. [Google Scholar] [CrossRef]
- Keller, N.; Mendoza-Ferreira, N.; Maroofian, R.; Chelban, V.; Khalil, Y.; Mills, P.B.; Boostani, R.; Torbati, P.N.; Karimiani, E.G.; Thiele, H.; et al. Hereditary polyneuropathy with optic atrophy due to pdxk variant leading to impaired vitamin b6 metabolism. Neuromuscul Disord 2020, 30, 583–589. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of non-coenzyme action of thiamine: Protein targets and medical significance. Biochemistry (Mosc) 2019, 84, 829–850. [Google Scholar] [CrossRef]
- Boyko, A.; Tsepkova, P.; Aleshin, V.; Artiukhov, A.; Mkrtchyan, G.; Ksenofontov, A.; Baratova, L.; Ryabov, S.; Graf, A.; Bunik, V. Severe spinal cord injury in rats induces chronic changes in the spinal cord and cerebral cortex metabolism, adjusted by thiamine that improves locomotor performance. Front Mol Neurosci 2021, 14, 620593. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Ucal, M.; Mullebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim Biophys Acta Bioenerg 2018, 1859, 925–931. [Google Scholar] [CrossRef]
- Weidinger, A.; Milivojev, N.; Hosmann, A.; Duvigneau, J.C.; Szabo, C.; Toro, G.; Rauter, L.; Vaglio-Garro, A.; Mkrtchyan, G.V.; Trofimova, L.; et al. Oxoglutarate dehydrogenase complex controls glutamate-mediated neuronal death. Redox Biol 2023, 62, 102669. [Google Scholar] [CrossRef]
- Tiamkao, S.; Boonsong, A.; Saepeung, K.; Kasemsap, N.; Apiwattanakul, M.; Suanprasert, N.; Hemachudha, T.; Pithak, P.; Juntee, K.; Waisaen, C.; et al. An outbreak of peripheral neuropathy in a prison. Case Rep Neurol 2019, 11, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V. Vitamin-dependent complexes of 2-oxo acid dehydrogenases: Structure, function, regulation and medical implications. Nova Science Publishers: New York, 2017. [Google Scholar]
- Hanberry, B.S.; Berger, R.; Zastre, J.A. High-dose vitamin b1 reduces proliferation in cancer cell lines analogous to dichloroacetate. Cancer Chemother Pharmacol 2014, 73, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Jonus, H.C.; Byrnes, C.C.; Kim, J.; Valle, M.L.; Bartlett, M.G.; Said, H.M.; Zastre, J.A. Thiamine mimetics sulbutiamine and benfotiamine as a nutraceutical approach to anticancer therapy. Biomed Pharmacother 2020, 121, 109648. [Google Scholar] [CrossRef] [PubMed]
- Klyuyeva, A.; Tuganova, A.; Kedishvili, N.; Popov, K.M. Tissue-specific kinase expression and activity regulate flux through the pyruvate dehydrogenase complex. J Biol Chem 2019, 294, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xiao, Y.; Meng, F.; Li, Y.; Shi, Z.; Qian, K. Functions and mechanisms of lysine glutarylation in eukaryotes. Front Cell Dev Biol 2021, 9, 667684. [Google Scholar] [CrossRef] [PubMed]
- Kennerson, M.L.; Yiu, E.M.; Chuang, D.T.; Kidambi, A.; Tso, S.C.; Ly, C.; Chaudhry, R.; Drew, A.P.; Rance, G.; Delatycki, M.B.; et al. A new locus for x-linked dominant charcot-marie-tooth disease (cmtx6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (pdk3) gene. Hum Mol Genet 2013, 22, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Perez-Siles, G.; Cutrupi, A.; Ellis, M.; Screnci, R.; Mao, D.; Uesugi, M.; Yiu, E.M.; Ryan, M.M.; Choi, B.O.; Nicholson, G.; et al. Energy metabolism and mitochondrial defects in x-linked charcot-marie-tooth (cmtx6) ipsc-derived motor neurons with the p.R158h pdk3 mutation. Sci Rep 2020, 10, 9262. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Y.; Gu, M.M.; Sun, L.H.; Guo, W.T.; Zhu, H.B.; Ma, J.F.; Yuan, W.T.; Kuang, Y.; Ji, B.J.; Wu, X.L.; et al. A nonsense mutation in dhtkd1 causes charcot-marie-tooth disease type 2 in a large chinese pedigree. Am J Hum Genet 2012, 91, 1088–1094. [Google Scholar] [CrossRef]
- Xu, W.Y.; Zhu, H.; Shen, Y.; Wan, Y.H.; Tu, X.D.; Wu, W.T.; Tang, L.; Zhang, H.X.; Lu, S.Y.; Jin, X.L.; et al. Dhtkd1 deficiency causes charcot-marie-tooth disease in mice. Mol Cell Biol 2018, 38. [Google Scholar] [CrossRef]
- Danhauser, K.; Sauer, S.W.; Haack, T.B.; Wieland, T.; Staufner, C.; Graf, E.; Zschocke, J.; Strom, T.M.; Traub, T.; Okun, J.G.; et al. Dhtkd1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria. Am J Hum Genet 2012, 91, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J.; te Brinke, H.; Wanders, R.J.; Knegt, A.C.; Oussoren, E.; Hoogeboom, A.J.; Ruijter, G.J.; Becker, D.; Schwab, K.O.; Franke, I.; et al. Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria. J Inherit Metab Dis 2015, 38, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; Venturoni, L.; Mucci, G.; Elbalalesy, N.; Woontner, M.; Goodman, S.; Abdenur, J.E. New cases of dhtkd1 mutations in patients with 2-ketoadipic aciduria. JIMD Rep 2016, 25, 15–19. [Google Scholar] [PubMed]
- Sherrill, J.D.; Kc, K.; Wang, X.; Wen, T.; Chamberlin, A.; Stucke, E.M.; Collins, M.H.; Abonia, J.P.; Peng, Y.; Wu, Q.; et al. Whole-exome sequencing uncovers oxidoreductases dhtkd1 and ogdhl as linkers between mitochondrial dysfunction and eosinophilic esophagitis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.H.; Chen, Z.T.; Zhou, R.L.; Wang, Y.Z. A chinese pedigree with a novel mutation in gjb1 gene and a rare variation in dhtkd1 gene for diverse charcot-marie-tooth diseases. Mol Med Rep 2019, 19, 4484–4490. [Google Scholar] [CrossRef] [PubMed]
- Fabrizi, G.M.; Hoyer, H.; Taioli, F.; Cavallaro, T.; Hilmarsen, H.T.; Squintani, G.M.; Zanette, G.; Braathen, G.J. Inherited motor-sensory neuropathy with upper limb predominance associated with the tropomyosin-receptor kinase fused gene. Neuromuscul Disord 2020, 30, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Leandro, J.; Khamrui, S.; Wang, H.; Suebsuwong, C.; Nemeria, N.S.; Huynh, K.; Moustakim, M.; Secor, C.; Wang, M.; Dodatko, T.; et al. Inhibition and crystal structure of the human dhtkd1-thiamin diphosphate complex. ACS Chem Biol 2020, 15, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, G.A.; Foster, W.R.; Bailey, H.J.; Hicks, K.G.; Sauer, S.W.; Dimitrov, B.; McCorvie, T.J.; Okun, J.G.; Rutter, J.; Kolker, S.; et al. Crystal structure and interaction studies of human dhtkd1 provide insight into a mitochondrial megacomplex in lysine catabolism. IUCrJ 2020, 7, 693–706. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Kazantsev, A.V.; Lukashev, N.V.; Bellinzoni, M.; Bunik, V.I. Selective inhibition of 2-oxoglutarate and 2-oxoadipate dehydrogenases by the phosphonate analogs of their 2-oxo acid substrates. Front Chem 2020, 8, 596187. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Nagy, B.; Sanchez, R.; Zhang, X.; Leandro, J.; Ambrus, A.; Houten, S.M.; Jordan, F. Functional versatility of the human 2-oxoadipate dehydrogenase in the l-lysine degradation pathway toward its non-cognate substrate 2-oxopimelic acid. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Bunik, V.I.; Degtyarev, D. Structure-function relationships in the 2-oxo acid dehydrogenase family: Substrate-specific signatures and functional predictions for the 2-oxoglutarate dehydrogenase-like proteins. Proteins 2008, 71, 874–890. [Google Scholar] [CrossRef] [PubMed]
- Ozohanics, O.; Zhang, X.; Nemeria, N.S.; Ambrus, A.; Jordan, F. Probing the e1o-e2o and e1a-e2o interactions in binary subcomplexes of the human 2-oxoglutarate dehydrogenase and 2-oxoadipate dehydrogenase complexes by chemical cross-linking mass spectrometry and molecular dynamics simulation. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.S.; Zhang, X.; Leandro, J.; Zhou, J.; Yang, L.; Houten, S.M.; Jordan, F. Toward an understanding of the structural and mechanistic aspects of protein-protein interactions in 2-oxoacid dehydrogenase complexes. Life (Basel) 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.S.; Gerfen, G.; Nareddy, P.R.; Yang, L.; Zhang, X.; Szostak, M.; Jordan, F. The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their e2 and e3 components for their function and both generate reactive oxygen species. Free Radic Biol Med 2018, 115, 136–145. [Google Scholar] [CrossRef]
- Boyko, A.I.; Artiukhov, A.V.; Kaehne, T.; di Salvo, M.L.; Bonaccorsi di Patti, M.C.; Contestabile, R.; Tramonti, A.; Bunik, V.I. Isoforms of the dhtkd1-encoded 2-oxoadipate dehydrogenase, identified in animal tissues, are not observed upon the human dhtkd1 expression in bacterial or yeast systems. Biochemistry (Mosc) 2020, 85, 920–929. [Google Scholar] [CrossRef]
- Biagosch, C.; Ediga, R.D.; Hensler, S.V.; Faerberboeck, M.; Kuehn, R.; Wurst, W.; Meitinger, T.; Kolker, S.; Sauer, S.; Prokisch, H. Elevated glutaric acid levels in dhtkd1-/gcdh- double knockout mice challenge our current understanding of lysine metabolism. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 2220–2228. [Google Scholar] [CrossRef]
- Leandro, J.; Dodatko, T.; Aten, J.; Nemeria, N.S.; Zhang, X.; Jordan, F.; Hendrickson, R.C.; Sanchez, R.; Yu, C.; DeVita, R.J.; et al. Dhtkd1 and ogdh display substrate overlap in cultured cells and form a hybrid 2-oxo acid dehydrogenase complex in vivo. Hum Mol Genet 2020, 29, 1168–1179. [Google Scholar] [CrossRef]
- Wu, Y.; Williams, E.G.; Dubuis, S.; Mottis, A.; Jovaisaite, V.; Houten, S.M.; Argmann, C.A.; Faridi, P.; Wolski, W.; Kutalik, Z.; et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014, 158, 1415–1430. [Google Scholar] [CrossRef]
- Wang, T.J.; Ngo, D.; Psychogios, N.; Dejam, A.; Larson, M.G.; Vasan, R.S.; Ghorbani, A.; O'Sullivan, J.; Cheng, S.; Rhee, E.P.; et al. 2-aminoadipic acid is a biomarker for diabetes risk. J Clin Invest 2013, 123, 4309–4317. [Google Scholar] [CrossRef]
- Plubell, D.L.; Fenton, A.M.; Wilmarth, P.A.; Bergstrom, P.; Zhao, Y.; Minnier, J.; Heinecke, J.W.; Yang, X.; Pamir, N. Gm-csf driven myeloid cells in adipose tissue link weight gain and insulin resistance via formation of 2-aminoadipate. Sci Rep 2018, 8, 11485. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Liu, Z.; Apontes, P.; Feng, D.; Pessin, J.E.; Sauve, A.A.; Angeletti, R.H.; Chi, Y. Dual mode action of mangiferin in mouse liver under high fat diet. PLoS One 2014, 9, e90137. [Google Scholar] [CrossRef] [PubMed]
- Tsepkova, P.M.; Artiukhov, A.V.; Boyko, A.I.; Aleshin, V.A.; Mkrtchyan, G.V.; Zvyagintseva, M.A.; Ryabov, S.I.; Ksenofontov, A.L.; Baratova, L.A.; Graf, A.V.; et al. Thiamine induces long-term changes in amino acid profiles and activities of 2-oxoglutarate and 2-oxoadipate dehydrogenases in rat brain. Biochemistry (Mosc) 2017, 82, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Atherton, P.J.; Larsson, O.; Sood, S.; Blokhin, I.O.; Brogan, R.J.; Volmar, C.H.; Josse, A.R.; Slentz, C.; Wahlestedt, C.; et al. A coding and non-coding transcriptomic perspective on the genomics of human metabolic disease. Nucleic Acids Res 2018, 46, 7772–7792. [Google Scholar] [CrossRef] [PubMed]
- Artiukhov, A.V.; Grabarska, A.; Gumbarewicz, E.; Aleshin, V.A.; Kahne, T.; Obata, T.; Kazantsev, A.V.; Lukashev, N.V.; Stepulak, A.; Fernie, A.R.; et al. Synthetic analogues of 2-oxo acids discriminate metabolic contribution of the 2-oxoglutarate and 2-oxoadipate dehydrogenases in mammalian cells and tissues. Sci Rep 2020, 10, 1886. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhu, H.; Gu, M.; Luo, Q.; Ding, J.; Yao, Y.; Chen, F.; Wang, Z. Dhtkd1 is essential for mitochondrial biogenesis and function maintenance. FEBS Lett 2013, 587, 3587–3592. [Google Scholar] [CrossRef] [PubMed]
- Dehhaghi, M.; Panahi, H.K.S.; Kavyani, B.; Heng, B.; Tan, V.; Braidy, N.; Guillemin, G.J. The role of kynurenine pathway and nad(+) metabolism in myalgic encephalomyelitis/chronic fatigue syndrome. Aging Dis 2022, 13, 698–711. [Google Scholar] [CrossRef]
- Birkisdottir, M.B.; van Galen, I.; Brandt, R.M.C.; Barnhoorn, S.; van Vliet, N.; van Dijk, C.; Nagarajah, B.; Imholz, S.; van Oostrom, C.T.; Reiling, E.; et al. The use of progeroid DNA repair-deficient mice for assessing anti-aging compounds, illustrating the benefits of nicotinamide riboside. Front Aging 2022, 3, 1005322. [Google Scholar] [CrossRef]
- Vreones, M.; Mustapic, M.; Moaddel, R.; Pucha, K.A.; Lovett, J.; Seals, D.R.; Kapogiannis, D.; Martens, C.R. Oral nicotinamide riboside raises nad+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell 2023, 22, e13754. [Google Scholar] [CrossRef]
- Kazamel, M.; Boes, C.J. Charcot marie tooth disease (cmt): Historical perspectives and evolution. J Neurol 2015, 262, 801–805. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. Nad+ and sirtuins in aging and disease. Trends Cell Biol 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Imai, S.I.; Guarente, L. It takes two to tango: Nad(+) and sirtuins in aging/longevity control. NPJ Aging Mech Dis 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Imai, S.I. Nad (+) biosynthesis, aging, and disease. F1000Res 2018, 7, 132. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Nad(+) in aging: Molecular mechanisms and translational implications. Trends Mol Med 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.I. Nad(+) intermediates: The biology and therapeutic potential of nmn and nr. Cell Metab 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, V.A.; Gromyko, D.V.; Nikiforov, A.A. The regulatory role of nad in human and animal cells. Biochemistry (Mosc) 2018, 83, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Ying Cao, Y.W. , Jing Yang. Nad+-dependent mechanism of pathological axon degeneration. Cell Insight 2022, 1. [Google Scholar]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear nad biosynthesis and sirt1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.R.; Hoke, A. Targeting the programmed axon degeneration pathway as a potential therapeutic for charcot-marie-tooth disease. Brain Res 2020, 1727, 146539. [Google Scholar] [CrossRef]
- Barile, A.; Nogues, I.; di Salvo, M.L.; Bunik, V.; Contestabile, R.; Tramonti, A. Molecular characterization of pyridoxine 5'-phosphate oxidase and its pathogenic forms associated with neonatal epileptic encephalopathy. Sci Rep 2020, 10, 13621. [Google Scholar] [CrossRef]
- Gachon, F.; Fonjallaz, P.; Damiola, F.; Gos, P.; Kodama, T.; Zakany, J.; Duboule, D.; Petit, B.; Tafti, M.; Schibler, U. The loss of circadian par bzip transcription factors results in epilepsy. Genes Dev 2004, 18, 1397–1412. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Mandal, A.; Roy, D.; Chatterjee, S.; Ghosh, M.K.; Dubey, S.; Lahiri, D.; Finsterer, J.; Ray, B.K. Seizure as a presenting manifestation of wernicke's encephalopathy induced by hyperemesis gravidarum. J Family Med Prim Care 2021, 10, 567–571. [Google Scholar] [PubMed]
- Mengi, T.; Beckmann, Y. Wernicke encephalopathy with epileptic seizures during pregnancy. Neurocase 2022, 28, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Mimouni-Bloch, A.; Goldberg-Stern, H.; Strausberg, R.; Brezner, A.; Heyman, E.; Inbar, D.; Kivity, S.; Zvulunov, A.; Sztarkier, I.; Fogelman, R.; et al. Thiamine deficiency in infancy: Long-term follow-up. Pediatr Neurol 2014, 51, 311–316. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Interplay of natural derivatives of vitamin B1, B3, B6, i.e. thiamine diphosphate (ThDP), oxidized nicotinamide adenine dinucleotide (NAD+) and pyridoxal-5’-phosphate (PLP), correspondingly, in the de novo production of major neurotransmitters glutamate and gamma-aminobutyrate from glucose. The vitamin-dependent enzymes are mentioned in parenthesis by the type of the catalyzed reactions.
Figure 1.
Interplay of natural derivatives of vitamin B1, B3, B6, i.e. thiamine diphosphate (ThDP), oxidized nicotinamide adenine dinucleotide (NAD+) and pyridoxal-5’-phosphate (PLP), correspondingly, in the de novo production of major neurotransmitters glutamate and gamma-aminobutyrate from glucose. The vitamin-dependent enzymes are mentioned in parenthesis by the type of the catalyzed reactions.
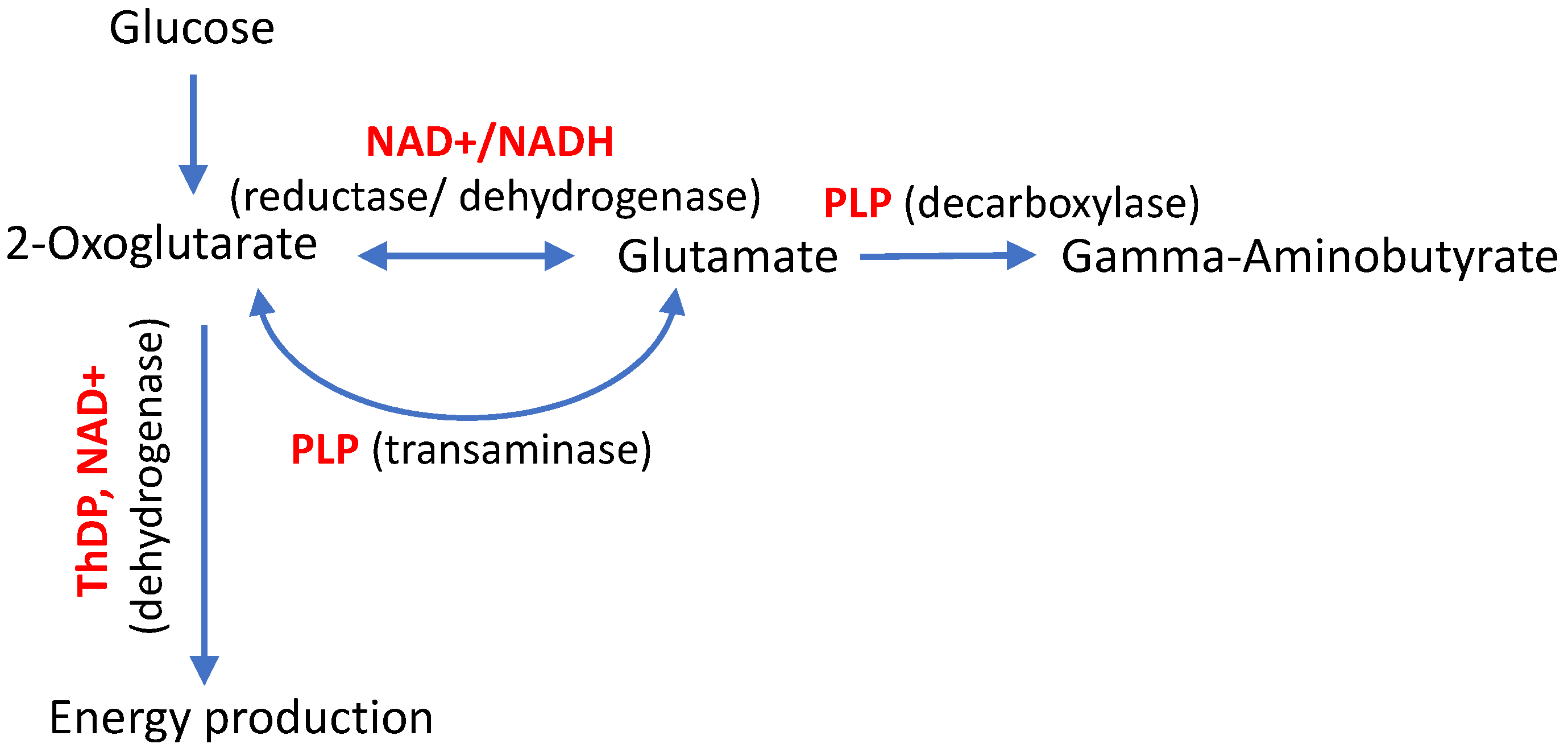
Figure 2.
Participation of the DHTKD1-encoded 2-oxoadipate dehydrogenase (OADH) in the tryptophan catabolism pathway, alternative to de novo NAD+ biosynthesis from tryptophan. Dashed lines represent multistep processes, the vitamins and their natural derivatives are shown in bold red, the enzymes mentioned in the text are shown in bold black. Nicotinamide/nicotinic acid mononucleotide adenylyl transferase (NMNAT) and NAD synthase catalyze the indicated steps of NAD+ biosynthesis.
Figure 2.
Participation of the DHTKD1-encoded 2-oxoadipate dehydrogenase (OADH) in the tryptophan catabolism pathway, alternative to de novo NAD+ biosynthesis from tryptophan. Dashed lines represent multistep processes, the vitamins and their natural derivatives are shown in bold red, the enzymes mentioned in the text are shown in bold black. Nicotinamide/nicotinic acid mononucleotide adenylyl transferase (NMNAT) and NAD synthase catalyze the indicated steps of NAD+ biosynthesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



