
Submitted:
25 April 2023
Posted:
26 April 2023
You are already at the latest version
Abstract
Barrett’s esophagus (BE) is a premalignant lesion for esophageal adenocarcinoma (EAC). Development of Barrett’s esophagus is caused by biliary reflux that provokes intensive mutagenesis in stem cells of epithelium in distal esophagus and gastro-esophageal junction. Other possible cell origins of Barrett’s esophagus include stem cells of mucosal esophageal glands and their ducts, of stomach, residual embryonic cells and circulating bone marrow stem cells. Classic conception of healing of caustic lesion was replaced by idea of cytokine storm that forms inflammatory microenvironment for phenotypic shift toward intestinal metaplasia of distal esophagus. The review summarizes contemporary concepts of BE and EAC pathogenesis.
Keywords:
Barrett’s esophagus
; inflammatory signaling pathways
; intestinal metaplasia
; mutational load
; p53
; dysplasia
; carcinogenesis
; esophageal adenocarcinoma
1. Introduction
Barrett’s esophagus (BE) is a premalignant lesion that means a development of intestinal metaplasia in distal esophagus, which is caused by long-term exposure with predominantly bile reflux. Histological evaluation reveals several gland phenotypes across the metaplasia segment intercepted in mosaic fashion [1,2,3,4]. These phenotypes include cardiac, oxynto-cardiac, mature and immature intestinal phenotypes. Complex histological, immunohistochemical approach and sequencing of mitochondrial DNA revealed that cardiac phenotype is the earliest that gives rise to all the other gland phenotypes in metaplasia segment during clonal evolution. High mutational load in BE [5,6] along with marked clonal heterogeneity [4] serves as a premise for development of dysplasia and esophageal adenocarcinoma.
Two main hypothesis of BE source include classical mechanism of healing of caustic injury [2,7,8,9] and phenotype shift in context of so called “cytokine storm” [10,11,12,13,14]. These concepts are not mutually exclusive, but rather complete each other, leading to stem cells (SC) reprogramming with subsequent changes in architectonics of esophageal mucosa, that includes change in epithelial type and attraction of inflammatory microenvironment in lamina propria mucosa and submucosa. Our review analyzes molecular pathways in pathogenesis of BE.
2. Gastro-biliary reflux as inductor of intestinal metaplasia
Huge evidence suggests that gastro-biliary reflux plays the main role in BE development that’s why BE is observed in 2-14% patients with gastro-esophageal reflux disease (GERD) [15,16,17]. Pathogenetic association of acid and bile reflux with BE is shown in studies that used pH-metry in human [18,19,20], and also at laboratory models of BE [10,12,13,21,22], and in cell lines [23,24,25,26,27,28,29,30,31,32,33,34]. Acid reflux provides gradient of pH along the segment of metaplasia to reach optimal solubility of bile salts that allow them enter into epithelial cells [35].
Multiple signaling pathways are involved in BE that are activated by bile reflux inside the epithelial cells and inspire epithelial-stromal interaction. Inside epithelial cells in distal esophagus bile acids cause injury of organelles including mitochondrial membranes that trigger uncontrolled generation of reactive oxygen species (ROS), oxidative stress and DNA damage. Bile acids also drive the release of proinflammatory cytokines, including IL1β, IL6, IL8, TNF-α [10,12,13,36,37,38], PGE2 [24] and COX-2 [25], that activate signaling pathway NF-kB, that prevents apoptosis and enhance proliferation of epithelial cells that favors regeneration of injury. Therefore development of intestinal metaplasia acts as an adaptational mechanism in distal esophagus. At the same time, repeated reflux exposure with DNA injury, including TP53, and accumulation of multiple mutations and genomic instability drives the way to dysplasia and EAC [4,5,6,39,40].
Bile acid exposure of keratinocyte cell lines EPC1 and EPC2 induce changes in expression of numerous genes, including genes of squamous differentiation, oxidative stress, DNA repair, cell cycle and others [29]. The key event of phenotypic shift toward intestinal differentiation is activation of transcriptional factor CDX2. Cholic and dehyrdocholic acid cause the most prominent activation of CDX2 in cellular cultures of keratinocytes. Moreover, keratinocytes transfected with Cdx2 expression vectors start the transcription of MUC2, which is an early sign of cellular reprogramming from squamous to intestinal differentiation [23]. This process is caused by activation of signaling pathway NF-kB [23,33] and inhibition of NOTCH [26-28]. Bile acids exposure of EAC cell cultures (OE19, OE33) and immortalized squamous epithelium (Het-1A) showed decreased expression of NOTCH receptors that leads to inhibition of transcriptional factor Hes-1 and activation of ATOH-1 that directly stimulates the expression of CDX2. Moreover, activation of NOTCH receptor ligand Dll1 also increases the expression of ATOH-1, and high expression of CDX2 inhibits Hes-1, that leads to fixation of intestinal phenotype (Figure 1).
Notch-signalling pathway inhibition in immortalized keratinocytes leads to changes in morphology of basal layer of squamous epithelium with acquisition of columnar features with expression of CDX2, KRT8, KRT18, KRT19, KRT20, MUC2, MUC3B, MUC5B, MUC17, SOX9, villin, Das-1 and reduced expression of squamous markers CK4, Tap63, KRT5, КRT13 и КRT14 [31,32].
Bile acid exposure in acidic pH of cell line Het-1A leads to hedgehog (Hh) signaling pathway activation that is normally seen in esophagus during embyogenesis and is absent in squamous epithelium. Hh-signaling in Barrett’s esophagus is realized in both – epithelium and stromal elements. Transmembrane receptor PTCH is activated after binding of Hh-ligands and inhibits protein SMO that leads to activation of transcriptional factor Gli causing the expression of different genes [41,42], including SOX9 in epithelium and BMP4 in stromal elements. Due to epithelial-stromal interaction BMP4 activates pSMAD1/5/8 [43] in keratinocytes that leads to expression of SOX9 [42], that normally is expressed in colon. Coactivation of transcriptional factor CDX2 is necessary for development of intestinal phenotype KRT20+ and MUC2+, because CDX2 forms complex with pSMAD and binds to promoter of Muc2 to induce its transcription [44].
Besides Wang D.H. et al. 2014 [45] revealed that Hh-dependent transcriptional factor FOXA2 can induce acquisition of intestinal phenotype without CDX2 activation by direct activation of MUC2 expression and via increase of AGR2 expression that controls processing of MUC2 (Figure 2).
Animal models can’t totally elucidate the pathogenesis of BE because after operations reflux exposure on esophagus is far from physiological. At the same time researches on cell lines can explain biological mechanisms of bile exposure but they can’t reliably provide evidence on cellular origins of BE.
Therefore, development of intestinal metaplasia in distal esophagus is complex and multiple stage process that is determined by realization of several complimentary signaling pathways including epithelial-stromal interactions. This process doesn’t imply mature epithelial cells but rather involve SC and progenitor cells that give rise to several cell populations that via clonal evolution leads to development of BE.
3. Possible cellular origin of BE
Several possible origins of BE were proposed that imply different pathogenetic mechanisms. Chronic bile and acid exposure of distal esophagus causes intestinal metaplasia due to cellular reprogramming that involves changes in transcriptional factors expression and shift in cellular phenotype. Proposed pathways of cellular reprogramming include transdifferentiation of squamous epithelium and transcommitment of progenitor cells [40,46,47,48].
Transdifferentiation is a process when one well differentiated cellular type (g.e., squamous epithelium) turns into another cellular type (columnar epithelium) [46,49]. Direct transdifferentiation means that change in cellular phenotype doesn’t imply cell division. Undirect transdifferentiation first involves dedifferentiation of squamous epithelium to progenitor cells and then acquisition of new phenotype – metaplastic columnar epithelium. Therefore undirect transdifferentiation suggests transcommitment of progenitor cells as well. The term transcommitment is more precise, because transdifferentiation means generation of one cell type from one another while in BE several cellular phenotypes (lines of differentiation) arise from multipotent SC [50].
Six potential cellular origins of columnar metaplasia in distal esophagus are proposed:
- SC and progenitor cells of squamous epithelium
- SC and progenitor cells of gastro-esophageal junction
- SC and progenitor cells of submucosal glands and their ducts
- SC and progenitor cells of first oxyntic gland of stomach
- Residual embryonic cells
- Circulating bone marrow-derived multipotent SC
Multilayered epithelium at squamo-columnar junction that consists of several layers of immature squamous epithelium covered with mucous-secreting columnar cells favours transcommitment of squamous progenitor cells. Multilayered epithelium demonstrates ultrastructural and immunohistochemical features of both squamous and columnar epithelium. Scanning electron microscopy (SEM) revealed that multilayered epithelium displayed both intercellular ridges (feature of squamous epithelium) and short, stubby microvilli and bulging mucus (typical for metaplastic columnar epithelium) [51]. Moreover basal cells of multilayered epithelium co-expressed CK19 (feature of columnar epithelium) and CK4 typical for squamous epithelium [52]. Multilayered epithelium was associated with GERD and doesn’t present in normal gastro-esophageal junction [53]. Based on these data and own findings, Chandrasoma P.T. et al. created revolutionary conception of cardia as reflux damaged dilated distal esophagus lined with metaplastic columnar epithelium [54,55,56]. Therefore first oxyntic gland serves histological demarcation between stomach and esophagus.
Origin of BE from progenitor cells of squamous esophagus was also confirmed by Nicholson A.M. et al. 2012 [1], who revealed the same mutations in mitochondrial DNA in metaplastic columnar epithelium and adjoined squamous epithelium.
SC of gastro-esophageal junction were suggested by Jiang M. et al. 2017 [57], who showed in multilayered epithelium in transitional zone in human biopsies and in mice basal progenitor cells with phenotype p63+ KRT5+ KRT7+. In mice upon ectopic expression of CDX2 these transitional basal progenitors differentiated into intestinal-like epithelium including MUC2+ TFF3+ goblet cells. These progenitor cells are likely to be the same squamous progenitor cells as discussed earlier.
SC and progenitor cells of submucosal glands of esophagus are other possible source of columnar metaplasia. Thorough histological examination showed that multilayered epithelium was a continuation of submucosal esophageal gland ducts [58,59]. Glickman J.N. et al. 2001 [58] revealed the same expressional profile of CK7, 8/18, 19 и 20 in columnar epithelium, squamous epithelium, submucosal glands and their ducts. Moreover, the proliferative index of multilayered epithelium of ducts was high in 88% of cases. Therefore multilayered epithelium at the surface that is thought to be the earliest sign of reflux injury may arise from SC of esophageal submucosal gland ducts.
In pigs multipotent SC of submucosal glands are involved in regeneration after injury of squamous epithelium. Moreover these cells express columnar epithelial markers SOX9, CK7 and CK8 [60]. Owen R.P. et al. 2018 [61] found similar expression of different markers (including LEFTY1 and OLFM4) in SC of submucosal glands and SC in metaplastic columnar epithelium. These findings favors SC of submucosal glands to be the source of BE. But the most crucial evidence is that DNA sequencing revealed similar mutations in CDKN2A and TP53 (including LOH) in metaplastic epithelium and ducts of submucosal glands [62].
At the same time there is evidence that metaplastic epithelium arises from LGR5+ SC and progenitor cells of first oxyntic gland in the stomach. Quante M. et al. 2012 [63] developed mice model with permanent overexpression of IL-1β in esophagus (L2-IL-1β mice). At the age of 12-15 months these mice demonstrated MUC5AC+ TFF2+ Notch1+ columnar metaplasia of distal esophagus without goblet cells. After treatment of deoxycholic acid these mice developed Barrett’s-like metaplasia with expression of мРНК Tff2, Cckbr, Muc5ac, Cdx2, Krt19, Bmp4 and Shh. Inhibition of Notch-signalling pathway led to acquisition of intestinal phenotype enriched with goblet cells. These data dive evidence that metaplastic epithelium evolves from cardiac to intestinal phenotype. And the reason for cardiac phenotype are LGR5+ SC and Dclk1+ progenitor cells of first oxyntic gland that proliferate and spread to distal esophagus. The expression of mRNA LGR5 and Dclk1 also was seen in human biopsy specimens by real-time PCR. Further research showed the role of Notch-signaling pathway and its association with NF-κB activation in LGR5+ SC in development of metaplasia, dysplasia and EAC [64].
LGR5 expression increases in high-grade dysplasia and EAC meaning that LGR5+ SC are involved in carcinogenesisе [65,66]. Overexpression of LGR5 is associated with poor survival in EAC patients independently from stage of the disease, age and neoadjuvant or adjuvant therapy [66,67].
Lavery D.L. et al. 2014 [68] demonstrated that metaplastic glands of BE on structural level are similar to pyloric glands of the stomach with proliferative zone at the middle third of mucosa with expression of LGR at the bottom and bidirectional migration of IdU-traced cells. Epithelial cells in upper compartment express MUC5AC and TFF1, and in lower compartment – TFF2 and MUC6. Goblet cells are localized at the upper third of mucosa and express MUC2 and TFF3. Jang B.G. et al. 2015 [69] suggest that LGR5+ in areas of intestinal metaplasia in stomach and esophagus with expression of intestinal SC markers ASCL2, OLFM4 and EPHB2 are the distinct population of SC that replaces pre-existing SC.
Recidual embryonic SC [70,71] and bone marrow-derived multipotent SC [72,73] were also suggested as BE cellular origin, but gained little evidence.
The most established hypothesis of BE origin is transcommitment of multipotent SC and progenitor cells. Although seems reasonable that different progenitor cells may be involved in pathogenesis of BE (Figure 3), that are responsible for all the cellular phenotypes of columnar metaplasia. It explains heterogeneity, polyclonal and mosaic spread of metaplastic glands.
4. Mechanism of injury repair in distal esophagus: experimental findings
Injury of squamous epithelium of esophagus by reflux causes development of erosive esophagitis. Injury repair in distal esophagus is stereotypic [2]: at the first stage granulation tissue forms under a cover of fibrinous exudate for protection of deep tissues, then granulations are covered with reparative epithelium without functional properties including secretion of mucous. That the defect is covered with lateral growth of adjacent glands/crypts (ulcer-associated mucosal lineage – UACL) till functional epithelium is restored [2,9].
According to this model repair of local injury leads to proliferation of progenitor cells in squamous epithelium and first oxyntic gland in the margins of ulceration. Repeated reflux exposure drives a selection of clones resistant in these conditions that are mucous secreting cardiac-type epithelium. Consecutively columnar epithelium spreads more and more in distal esophagus and goes through clonal evolution to form all the gland phenotypes of BE. Therefore similar to pyloric gland compartmentalization pattern [50,68] may be explained by typical process of healing injury with UACL that gives phenotype of pyloric metaplasia all through gastrointestinal tract [8,9,74].
Surgical model of esophagojejunostomy in rat prevents migration of SC from stomach to esophagus. Two weeks after operation there was ulceration in distal esophagus near the anastomosis that was epithelized by distal margin with immature crypts of jejunum [7]. Metaplastic crypts had phenotype similar to epithelium of small intestine with expression of CDX2, villin, CD10, MUC2 and negative expression of gastric markers (MUC5AC and MUC6), intestinal marker Das-1 and squamous marker p63. Reepithelization of ulcer caused epithelial to mesenchymal transition (positive expression of Е-cadherin in epithelial cells of newly developed crypts and co-expression of E-cadherin and TWIST in spindle-like cells in stroma at margin of ulcer) and migration of cells from jejunum to distal esophagus. At proximal margin of ulcer there was proliferation of immature squamous epithelium.
In cell cultures of BE without dysplasia and dysplastic BE bile salts also activate epithelial to mesenchymal transition (decreased expression of cadherin 1, increased expression of fibronectin 1, vimentin and matrix metalloprotease 2, and increase in cell mobility), that was associated with VEGF signaling [75]. Phipps S.M. et al. 2020 [76] found genes GPS1 and RRM2, that were suppressed by low pH that caused epithelial to mesenchymal transition in BE with high-grade dysplasia and EAC. Therefore bile salts at low level of pH induce epithelial to mesenchymal transition and modulate both reparation of injury at distal esophagus and invasion in EAC.
5. «Cytokine storm» provides microenvironment for BE development
Conception of reflux induced and cytokine mediated injury of mucosa in distal esophagus was proposed by Souza R.F. et al. 2009, 2010, 2016, 2017 [10,11,12,13,77]. Souza R.F. et al. 2009 [77] found that in rats after esophagoduodenostomy ulcers appeared weeks after operation that’s why they can’t be initiated directly by acid and bile reflux in manner of caustic injury. Rather authors observed morphological features of reflux esophagitis with lymphocytic infiltration of submucosa at the 3rd day after operation. Then lymphocytic infiltration spread into lamina propria mucosa and in squamous epithelium. Infiltration of lamina propria significantly increased at the end of first week and in epithelium – 3 weeks after operation. Intensity of infiltration was stable from 3rd to 8th week after operation. On the 3rd day after operation infiltrate was composed only from CD3+CD20- T-lymphocytes, but since the 7th day few neutrophils presented as well. Basal cell hyperplasia was observed 1 week after operation and papillary hyperplasia developed 2 weeks and reached the peak 4 weeks after operation. And only from the 4th week there were found erosions in distal esophagus. The dynamics of morphological changes were associated with levels of IL-8 in different compartments of mucosa and submucosa. Other studies demonstrated the role of other pro-inflammatory cytokines IL-1β [37,38] and TNFα [78,79] in reflux-induced injury of esophageal mucosa. These cytokines cause chemoattraction of immune cells and activate NF-κB signaling that leads to persistent inflammation. Therefore the pathogenesis of erosive esophagitis is associated with immune cells mediated injury caused by release of proinflammatory cytokines by keratinocytes with chemoattraction of T-lymphocytes and other immune cells.
This model was validated on biopsies of patients with severe reflux esophagitis 1-2 weeks after cessation of protone pump inhibitors (PPIs). Histological findings included huge infiltration of mucosa with predominantly CD3+ T-lymphocytes with few or absent neutrophils and eosinophils, with aggravation of basal cells and papillary hyperplasia and increased spongiosis [78]. Immunohistochemical evaluation showed overexpression of HIF-2α and phosphorylated p65, that was associated with increased levels of mPNA of proinflammatory cytokines IL-8, IL-1β, TNF-α, COX-2 and ICAM-1 [79,80].
Bile acid exposure in acidic pH leads to increase of ROS in keratinocytes and metaplastic epithelium in BE [81] that leads to HIF-2α stabilization [82], translocation in nucleus and binding with HIF-responsive elements (HRE), that triggers synthesis and release of proinflammatory cytokines. Therefore, HIF-2α regulates inflammatory response to reflux injury that is associated with NF-κB signaling via p65 hosphorylation (Figure 4).
NF-κB activation in distal esophagus not only leads to persistence of inflammation but induces development of intestinal metaplasia via activation of CDX2, which is crucial for intestinal differentiation. CDX2 harbors binding site for NF-κB and may act as a downstream target for NF-κB [12,13,23,33]. Moreover after exposure of deoxycholic acid NF-κB directly activates MUC2 expression [83].
Levels of IL-8 and IL-1β rise in line erosive esophagitic – BE – EAC that is accompanied with increase in level of NF-κB [37] that leads to epithelial cells proliferation and prevention of apoptosis that promotes carcinogenesis [36].
Other important cytokine in BE pathogenesis is IL-6. L2-IL-1β/IL-6−/− deficient mice didn’t develop metaplasia in distal esophagus [63]. IL-6 is produced in metaplastic epithelium and causes activation and translocation of STAT3 in nucleus with concecutive synthesis of antiapoptotic proteins Bcl-xL and Mcl-1 [84,85,86]. This signaling pathway favors epithelial cells survival in reflux aggressive environment. Moreover, autocrine IL-6 signaling in EAC induces cell proliferation and angiogenesis that promotes cancer progression (Figure 5). Interaction and reciprocal activation of IL-6/STAT3 and NF-κB pathways forms persistent inflammation and drives carcinogenesis.
6. Genomic alterations in BE carcinogenesis
Repeated bile acid exposure in acidic pH causes oxidative stress in squamous and metaplastic epithelium in BE, that leads to DNA damage [24] and particularly double-strand breaks [87,88]. Moreover, hydrochloric acid in the lumen of esophagus reacts with nitrites of saline that causes the release of nitric oxide (NO), that per se may lead to double-strand breaks of DNA [87,89,90,91]. In turn, double-strand breaks are the most hazardous as they repair with loss of origin DNA sequence that contributes to frameshift and truncating mutations, and to loss of large DNA regions including several genes that may lead to loss of heterozygotici (LOH).
Mutataional load in BE without dysplasia widely vary an is estimated as 0,42-1,28 mutations per 1 Mbase and higher [6,92,93,94]. Expectedly, Mutataional load increases in line: BE without dysplasia – low grade dysplasia – high grade dysplasia – EAC [95], reaching 7,33-9,9 mutations per 1 Mbase in EAC [94,96,97]. The higher mutational load is associated with higher risk of neoplastic progression [6,92,98]. However, small series of samples was observed in most studies. On a large sample of patients Eluri S. et al. 2018 [93] showed no differences in mutational load in progressors and non-progressors in large sample series.
The most important for neoplastic progression in metaplastic segment are clones with mutations in TP53 that provokes exponential increase of genetic abnormalities. TP53 mutations are demonstrated in 72% of EAC [96]. New generation sequencing showed that these mutations may arise years before histological diagnosis of dysplasia in progressors and are seen only in 2,5-5% of non-progressors [5,6,99].
TP53 mutations facilitate realization of further genetic mechanisms of carcinogenesis in BE. Thus, loss of function mutations of TP53 provoke exponential growth in number of mutations due to impaired mechanisms of DNA repair and apoptosis. Moreover mutated p53 acquires non-canonical functions, such as induction of epithelial to mesenchymal transition, activation of NF-kβ and others [100].
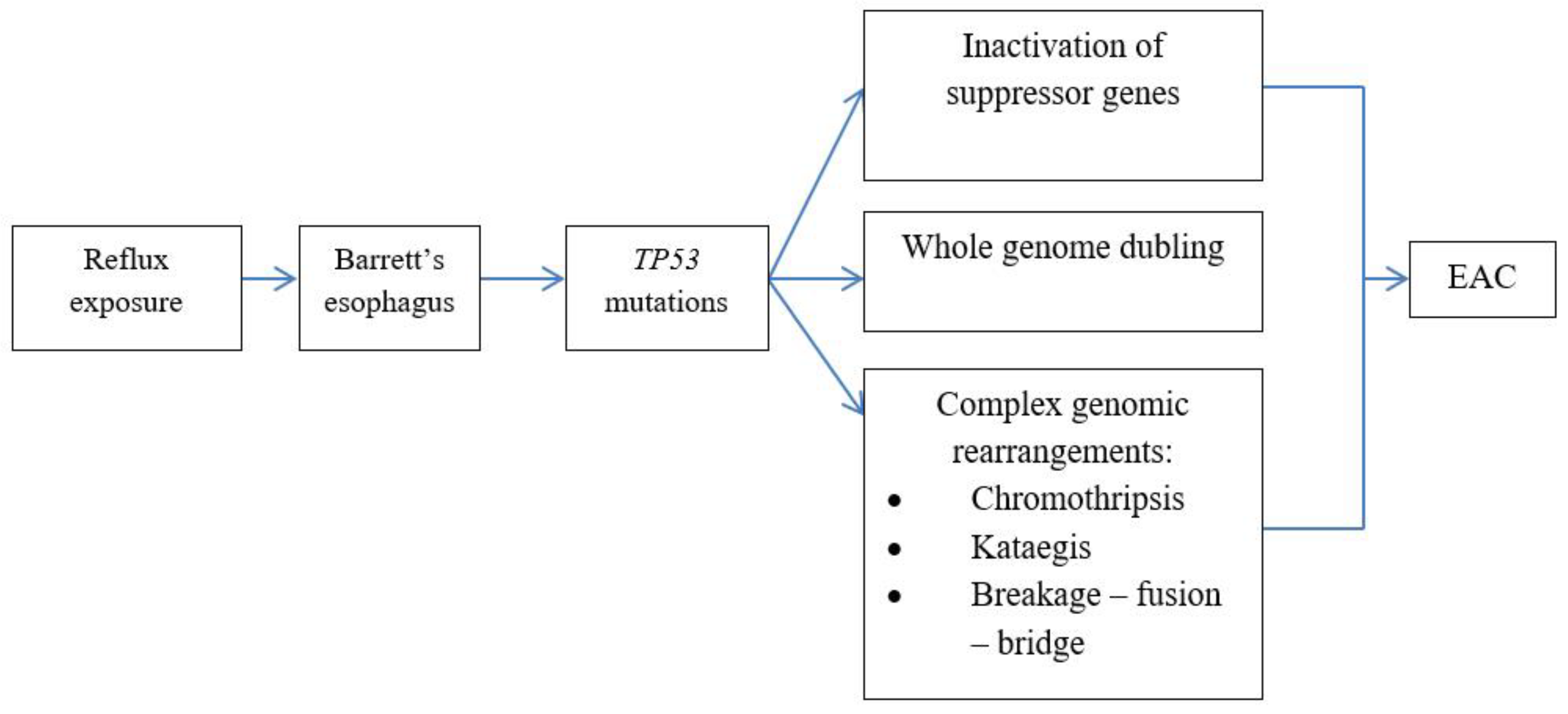
Mutations of TP53 serve as bifurcation in realization of different genetic mechanisms (Figure 6). Classical pathway with gradual increase in number of mutations with consecutive inactivation of suppressor genes (CDKN2A, SMAD4 и TP53) is rarely observed. More often (62,5% of EAC) mutations of TP53 induce rapid whole genome doubling and chromosome instability with amplification of oncogenes in cancer cells [5]. Third pathway includes catastrophic genetic events such as chromothripsis, kataegic and breakage-fusion-bridge due to impaired DNA reparation [39,97,101].
Chromothripsis is a catastrophic genomic event with simultaneous large genomic rearrangement including chromosomal shattering, gains and losses that involve regions with several genes and may cause rapid activation of oncogenes and inactivation of tumor suppressor genes [102]. Particularly, chromothripsis may lead to amplification of MYC and MDM2 oncogenes [97].
Genetic catastrophes are also associated with TP53 mutations. Rausch T. et al. 2012 [103] showed such association in children with Sonic-Hedgehog medulloblastoma as a part of Li-Fraumeni syndrome. Authors elucidated 3 mechanisms of chromothripsis linked with TP53 mutations: 1) critical telomere shortening followed by chromosome end-to-end fusions, 2) premature chromosome compaction due to cell cycle impairment (G2/M transition checkpoint), 3) impaired DNA repair and apoptosis induction mechanisms. High frequency of TP53 mutations and telomere shortening can explain high frequency of chromothripsis in EAC – 30-32,5% of cases [97,101].
Chromothripsis often coexists with kataegis. Kataegis is a region of hypermutation with cluster changes C>T and C>G in TpC dinucleotides, that was first described in breast cancer [104,105]. Kataegis is associated with activity of apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC) protein members. APOBEC protein family is a group of cytosine deaminases that target nucleic acids to induce C > U changes that may induce mutations [106]. In cytoplasm APOBECs prevent replication of DNA-containing viruses (first of all human immunodeficiency virus) and serve as a component of innate retroviral defense [107]. APOBEC target single stranded DNA and produce cluster of strand-coordinated mutations. Kataegis is observed at points of DNA fragmentation during chromothripsis after telomere crisis [108]. The frequency of kataegis in EAC varies from 31 to 86,4% of cases [97,101].
Breakage-fusion-bridge begins with loss of telomeres followed by fusion of chromosome ends or sister chromatid fusion with formation of double-minute chromosomes that break during anaphase [109]. This process repeats through several cell cycles leading to inverted duplications with amplification of certain chromosome regions. Malignant transformation is induced when such amplified regions include oncogenes. Breakage-fusion-bridge cycle is seen in 27% cases of EAC. It triggers amplification of potent oncogenes RCF3, MDM2, VEGFA, BCAT1 and KRAS [97,101].
These data give evidence that genomic catastrophes are important in malignant transformation of BE and represent an alternative mechanism of carcinogenesis. Genomic catastrophes that are frequently revealed in high grade dysplasia and EAC may probably be a reason for rapid neoplastic progression in BE [39,94,97,101]. Moreover, catastrophic events are a point of no return when malignant transformation becomes indispensable [110].
7. Conclusions
Development of intestinal metaplasia in distal esophagus is a multiple step process that takes place under exposure of bile reflux in acidic pH that acts as a trigger for creation of proinflammatory microenvironment. Bile acids exposure and cytokine storm causes reprogramming of SC and progenitor cells that leads to development of cardiac metaplasia and then via clonal evolution it gives rise to different cell populations along the metaplasia segment. Increase in clonal diversity and active mutagenesis with massive genome rearrangements underlie neoplastic progression to EAC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
All authors were actively involved in with the work presented on in this manuscript. KM, LM and MT designed the study; MN, RV and MG searched available literature on the signaling pathways in pathogenesis of Barrett’s esophagus and esophageal adenocarcinoma, K.M., LM and DA wrote the manuscript; IB and MG performed the figures, LM and MT supervised the study.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nicholson, A.M.; Graham, T.A.; Simpson, A.; Humphries, A.; Burch, N.; Rodriguez-Justo, M.; Novelli, M.; Harrison, R.; Wright, N.A.; McDonald, S.A.; et al. Barrett's metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 2012, 61, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Quante, M.; Leedham, S.; Jansen, M. The metaplastic mosaic of Barrett's oesophagus. Virchows Arch 2018, 472, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.A.; McDonald, S.A. The Complex, Clonal, and Controversial Nature of Barrett's Esophagus. Adv Exp Med Biol 2016, 908, 27–40. [Google Scholar] [CrossRef]
- Evans, J.A.; Carlotti, E.; Lin, M.L.; Hackett, R.J.; Haughey, M.J.; Passman, A.M.; Dunn, L.; Elia, G.; Porter, R.J.; McLean, M.H.; et al. Clonal Transitions and Phenotypic Evolution in Barrett's Esophagus. Gastroenterology 2022, 162, 1197–1209 e1113. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett's esophagus and adenocarcinoma. Nat Genet 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; et al. Detection of Mutations in Barrett's Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Agoston, A.T.; Pham, T.H.; Odze, R.D.; Wang, D.H.; Das, K.M.; Spechler, S.J.; Souza, R.F. Columnar-Lined Esophagus Develops via Wound Repair in a Surgical Model of Reflux Esophagitis. Cell Mol Gastroenterol Hepatol 2018, 6, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J Pathol 2018, 245, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.A. Aspects of the biology of regeneration and repair in the human gastrointestinal tract. Philos Trans R Soc Lond B Biol Sci 1998, 353, 925–933. [Google Scholar] [CrossRef]
- Souza, R.F. The role of acid and bile reflux in oesophagitis and Barrett's metaplasia. Biochem Soc Trans 2010, 38, 348–352. [Google Scholar] [CrossRef]
- Souza, R.F. From Reflux Esophagitis to Esophageal Adenocarcinoma. Dig Dis 2016, 34, 483–490. [Google Scholar] [CrossRef]
- Souza, R.F. Reflux esophagitis and its role in the pathogenesis of Barrett's metaplasia. J Gastroenterol 2017, 52, 767–776. [Google Scholar] [CrossRef]
- Souza, R.F.; Bayeh, L.; Spechler, S.J.; Tambar, U.K.; Bruick, R.K. A new paradigm for GERD pathogenesis. Not acid injury, but cytokine-mediated inflammation driven by HIF-2alpha: a potential role for targeting HIF-2alpha to prevent and treat reflux esophagitis. Curr Opin Pharmacol 2017, 37, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Spechler, S.J. Oesophagus: A new candidate for the progenitor cell of Barrett metaplasia. Nat Rev Gastroenterol Hepatol 2018, 15, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P. Barrett Esophagus: A Review. JAMA 2022, 328, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, L.H.; Cirota, G.G.; Zagari, R.M.; Ford, A.C. Global prevalence of Barrett's oesophagus and oesophageal cancer in individuals with gastro-oesophageal reflux: a systematic review and meta-analysis. Gut 2021, 70, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, L.H.; Telese, A.; Cirota, G.G.; Haidry, R.; Zagari, R.M.; Bazzoli, F.; Ford, A.C. Effect of gastro-esophageal reflux symptoms on the risk of Barrett's esophagus: A systematic review and meta-analysis. J Gastroenterol Hepatol 2022, 37, 1507–1516. [Google Scholar] [CrossRef]
- Gutschow, C.A.; Bludau, M.; Vallbohmer, D.; Schroder, W.; Bollschweiler, E.; Holscher, A.H. NERD, GERD, and Barrett's esophagus: role of acid and non-acid reflux revisited with combined pH-impedance monitoring. Dig Dis Sci 2008, 53, 3076–3081. [Google Scholar] [CrossRef]
- Hak, N.G.; Mostafa, M.; Salah, T.; El-Hemaly, M.; Haleem, M.; Abd El-Raouf, A.; Hamdy, E. Acid and bile reflux in erosive reflux disease, non-erosive reflux disease and Barrett's esophagus. Hepatogastroenterology 2008, 55, 442–447. [Google Scholar]
- Koek, G.H.; Sifrim, D.; Lerut, T.; Janssens, J.; Tack, J. Multivariate analysis of the association of acid and duodeno-gastro-oesophageal reflux exposure with the presence of oesophagitis, the severity of oesophagitis and Barrett's oesophagus. Gut 2008, 57, 1056–1064. [Google Scholar] [CrossRef]
- Terabe, F.; Aikou, S.; Aida, J.; Yamamichi, N.; Kaminishi, M.; Takubo, K.; Seto, Y.; Nomura, S. Columnar Metaplasia in Three Types of Surgical Mouse Models of Esophageal Reflux. Cell Mol Gastroenterol Hepatol 2017, 4, 115–123. [Google Scholar] [CrossRef]
- Matsui, D.; Omstead, A.N.; Kosovec, J.E.; Komatsu, Y.; Lloyd, E.J.; Raphael, H.; Kelly, R.J.; Zaidi, A.H.; Jobe, B.A. High yield reproducible rat model recapitulating human Barrett's carcinogenesis. World J Gastroenterol 2017, 23, 6077–6087. [Google Scholar] [CrossRef] [PubMed]
- Kazumori, H.; Ishihara, S.; Rumi, M.A.; Kadowaki, Y.; Kinoshita, Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett's epithelium. Gut 2006, 55, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, K.; Payne, C.M.; Chavarria, M.; Ramsey, L.; Dvorakova, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; Guy, N.; Condon, A.; et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett's oesophagus. Gut 2007, 56, 763–771. [Google Scholar] [CrossRef]
- Song, S.; Guha, S.; Liu, K.; Buttar, N.S.; Bresalier, R.S. COX-2 induction by unconjugated bile acids involves reactive oxygen species-mediated signalling pathways in Barrett's oesophagus and oesophageal adenocarcinoma. Gut 2007, 56, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.J.; Avissar, N.E.; Toia, L.; Redmond, E.M.; Watson, T.J.; Jones, C.; Raymond, D.P.; Litle, V.; Peters, J.H. Pathogenesis of Barrett's esophagus: bile acids inhibit the Notch signaling pathway with induction of CDX2 gene expression in human esophageal cells. Surgery 2009, 146, 714-721; discussion 721-712. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, Y.; Ishimura, N.; Uno, G.; Yuki, T.; Kazumori, H.; Ishihara, S.; Amano, Y.; Kinoshita, Y. Notch signaling pathway and Cdx2 expression in the development of Barrett's esophagus. Lab Invest 2012, 92, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, Y.; Ishimura, N.; Uno, G.; Aimi, M.; Oshima, N.; Yuki, T.; Sato, S.; Ishihara, S.; Kinoshita, Y. Bile acids induce Delta-like 1 expression via Cdx2-dependent pathway in the development of Barrett's esophagus. Lab Invest 2016, 96, 325–337. [Google Scholar] [CrossRef]
- Reveiller, M.; Ghatak, S.; Toia, L.; Kalatskaya, I.; Stein, L.; D'Souza, M.; Zhou, Z.; Bandla, S.; Gooding, W.E.; Godfrey, T.E.; et al. Bile exposure inhibits expression of squamous differentiation genes in human esophageal epithelial cells. Ann Surg 2012, 255, 1113–1120. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, H.; Wang, P.; Feng, J.; Li, J.; Xu, Y.; Zhang, A.; Shao, S.; Yu, X.; Yan, W.; et al. Deoxycholic acid (DCA) confers an intestinal phenotype on esophageal squamous epithelium via induction of the stemness-associated reprogramming factors OCT4 and SOX2. Cell Cycle 2016, 15, 1439–1449. [Google Scholar] [CrossRef]
- Vega, M.E.; Giroux, V.; Natsuizaka, M.; Liu, M.; Klein-Szanto, A.J.; Stairs, D.B.; Nakagawa, H.; Wang, K.K.; Wang, T.C.; Lynch, J.P.; et al. Inhibition of Notch signaling enhances transdifferentiation of the esophageal squamous epithelium towards a Barrett's-like metaplasia via KLF4. Cell Cycle 2014, 13, 3857–3866. [Google Scholar] [CrossRef]
- Minacapelli, C.D.; Bajpai, M.; Geng, X.; Cheng, C.L.; Chouthai, A.A.; Souza, R.; Spechler, S.J.; Das, K.M. Barrett's metaplasia develops from cellular reprograming of esophageal squamous epithelium due to gastroesophageal reflux. Am J Physiol Gastrointest Liver Physiol 2017, 312, G615–G622. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, H.Y.; Zhang, X.I.; Lynch, J.P.; Strauch, E.D.; Wang, J.Y.; Melton, S.D.; Genta, R.M.; Wang, D.H.; Spechler, S.J.; et al. Acid and bile salt-induced CDX2 expression differs in esophageal squamous cells from patients with and without Barrett's esophagus. Gastroenterology 2010, 139, 194–203 e191. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Dunbar, K.B.; Zhang, X.; Zhang, Q.; Spechler, S.J.; Souza, R.F. In Barrett's epithelial cells, weakly acidic bile salt solutions cause oxidative DNA damage with response and repair mediated by p38. Am J Physiol Gastrointest Liver Physiol 2020, 318, G464–G478. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, D.; Ayazi, S.; DeMeester, S.R.; Zehetner, J.; Peyre, C.G.; Grant, K.S.; Augustin, F.; Oh, D.S.; Lipham, J.C.; Chandrasoma, P.T.; et al. Intraluminal pH and goblet cell density in Barrett's esophagus. J Gastrointest Surg 2012, 16, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Tambunting, L.; Kelleher, D.; Duggan, S.P. The Immune Underpinnings of Barrett's-Associated Adenocarcinogenesis: a Retrial of Nefarious Immunologic Co-Conspirators. Cell Mol Gastroenterol Hepatol 2022, 13, 1297–1315. [Google Scholar] [CrossRef] [PubMed]
- O'Riordan, J.M.; Abdel-latif, M.M.; Ravi, N.; McNamara, D.; Byrne, P.J.; McDonald, G.S.; Keeling, P.W.; Kelleher, D.; Reynolds, J.V. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol 2005, 100, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.C.; Abdalla, S.; Onwuegbusi, B.A.; Sirieix, P.; Saeed, I.T.; Burnham, W.R.; Farthing, M.J. Inflammatory gradient in Barrett's oesophagus: implications for disease complications. Gut 2002, 51, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Contino, G.; Vaughan, T.L.; Whiteman, D.; Fitzgerald, R.C. The Evolving Genomic Landscape of Barrett's Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2017, 153, 657–673 e651. [Google Scholar] [CrossRef]
- Peters, Y.; Al-Kaabi, A.; Shaheen, N.J.; Chak, A.; Blum, A.; Souza, R.F.; Di Pietro, M.; Iyer, P.G.; Pech, O.; Fitzgerald, R.C.; et al. Barrett oesophagus. Nat Rev Dis Primers 2019, 5, 35. [Google Scholar] [CrossRef]
- Caspa Gokulan, R.; Garcia-Buitrago, M.T.; Zaika, A.I. From genetics to signaling pathways: molecular pathogenesis of esophageal adenocarcinoma. Biochim Biophys Acta Rev Cancer 2019, 1872, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Clemons, N.J.; Miyashita, T.; Dupuy, A.J.; Zhang, W.; Szczepny, A.; Corcoran-Schwartz, I.M.; Wilburn, D.L.; Montgomery, E.A.; Wang, J.S.; et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett's metaplasia. Gastroenterology 2010, 138, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Milano, F.; van Baal, J.W.; Buttar, N.S.; Rygiel, A.M.; de Kort, F.; DeMars, C.J.; Rosmolen, W.D.; Bergman, J.J.; J, V.A.M.; Wang, K.K.; et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology 2007, 132, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Mari, L.; Milano, F.; Parikh, K.; Straub, D.; Everts, V.; Hoeben, K.K.; Fockens, P.; Buttar, N.S.; Krishnadath, K.K. A pSMAD/CDX2 complex is essential for the intestinalization of epithelial metaplasia. Cell Rep 2014, 7, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Tiwari, A.; Kim, M.E.; Clemons, N.J.; Regmi, N.L.; Hodges, W.A.; Berman, D.M.; Montgomery, E.A.; Watkins, D.N.; Zhang, X.; et al. Hedgehog signaling regulates FOXA2 in esophageal embryogenesis and Barrett's metaplasia. J Clin Invest 2014, 124, 3767–3780. [Google Scholar] [CrossRef]
- Que, J.; Garman, K.S.; Souza, R.F.; Spechler, S.J. Pathogenesis and Cells of Origin of Barrett's Esophagus. Gastroenterology 2019, 157, 349–364 e341. [Google Scholar] [CrossRef]
- Wang, D.H.; Souza, R.F. Transcommitment: Paving the Way to Barrett's Metaplasia. Adv Exp Med Biol 2016, 908, 183–212. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, D.H. Origins of Metaplasia in Barrett's Esophagus: Is this an Esophageal Stem or Progenitor Cell Disease? Dig Dis Sci 2018, 63, 2005–2012. [Google Scholar] [CrossRef]
- Wang, D.H. The Esophageal Squamous Epithelial Cell-Still a Reasonable Candidate for the Barrett's Esophagus Cell of Origin? Cell Mol Gastroenterol Hepatol 2017, 4, 157–160. [Google Scholar] [CrossRef]
- McDonald, S.A.; Lavery, D.; Wright, N.A.; Jansen, M. Barrett oesophagus: lessons on its origins from the lesion itself. Nat Rev Gastroenterol Hepatol 2015, 12, 50–60. [Google Scholar] [CrossRef]
- Sawhney, R.A.; Shields, H.M.; Allan, C.H.; Boch, J.A.; Trier, J.S.; Antonioli, D.A. Morphological characterization of the squamocolumnar junction of the esophagus in patients with and without Barrett's epithelium. Dig Dis Sci 1996, 41, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Shields, H.M.; Zwas, F.; Antonioli, D.A.; Doos, W.G.; Kim, S.; Spechler, S.J. Detection by scanning electron microscopy of a distinctive esophageal surface cell at the junction of squamous and Barrett's epithelium. Dig Dis Sci 1993, 38, 97–108. [Google Scholar] [CrossRef]
- Boch, J.A.; Shields, H.M.; Antonioli, D.A.; Zwas, F.; Sawhney, R.A.; Trier, J.S. Distribution of cytokeratin markers in Barrett's specialized columnar epithelium. Gastroenterology 1997, 112, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Chandrasoma, P.T.; Lokuhetty, D.M.; Demeester, T.R.; Bremmer, C.G.; Peters, J.H.; Oberg, S.; Groshen, S. Definition of histopathologic changes in gastroesophageal reflux disease. Am J Surg Pathol 2000, 24, 344–351. [Google Scholar] [CrossRef]
- Chandrasoma, P. Controversies of the cardiac mucosa and Barrett's oesophagus. Histopathology 2005, 46, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Chandrasoma, P.; Wijetunge, S.; Ma, Y.; Demeester, S.; Hagen, J.; Demeester, T. The dilated distal esophagus: a new entity that is the pathologic basis of early gastroesophageal reflux disease. Am J Surg Pathol 2011, 35, 1873–1881. [Google Scholar] [CrossRef]
- Jiang, M.; Li, H.; Zhang, Y.; Yang, Y.; Lu, R.; Liu, K.; Lin, S.; Lan, X.; Wang, H.; Wu, H.; et al. Transitional basal cells at the squamous-columnar junction generate Barrett's oesophagus. Nature 2017, 550, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Glickman, J.N.; Chen, Y.Y.; Wang, H.H.; Antonioli, D.A.; Odze, R.D. Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett's esophagus. Am J Surg Pathol 2001, 25, 569–578. [Google Scholar] [CrossRef]
- Coad, R.A.; Woodman, A.C.; Warner, P.J.; Barr, H.; Wright, N.A.; Shepherd, N.A. On the histogenesis of Barrett's oesophagus and its associated squamous islands: a three-dimensional study of their morphological relationship with native oesophageal gland ducts. J Pathol 2005, 206, 388–394. [Google Scholar] [CrossRef]
- Kruger, L.; Gonzalez, L.M.; Pridgen, T.A.; McCall, S.J.; von Furstenberg, R.J.; Harnden, I.; Carnighan, G.E.; Cox, A.M.; Blikslager, A.T.; Garman, K.S. Ductular and proliferative response of esophageal submucosal glands in a porcine model of esophageal injury and repair. Am J Physiol Gastrointest Liver Physiol 2017, 313, G180–G191. [Google Scholar] [CrossRef]
- Owen, R.P.; White, M.J.; Severson, D.T.; Braden, B.; Bailey, A.; Goldin, R.; Wang, L.M.; Ruiz-Puig, C.; Maynard, N.D.; Green, A.; et al. Single cell RNA-seq reveals profound transcriptional similarity between Barrett's oesophagus and oesophageal submucosal glands. Nat Commun 2018, 9, 4261. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.J.; Preston, S.L.; McDonald, S.A.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett's oesophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Wein, F.; Fang, H.Y.; Anand, A.; Baumeister, T.; Strangmann, J.; Gerland, S.; Ingermann, J.; Munch, N.S.; Wiethaler, M.; et al. Notch Signaling Mediates Differentiation in Barrett's Esophagus and Promotes Progression to Adenocarcinoma. Gastroenterology 2020, 159, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Schellnegger, R.; Quante, A.; Rospleszcz, S.; Schernhammer, M.; Hohl, B.; Tobiasch, M.; Pastula, A.; Brandtner, A.; Abrams, J.A.; Strauch, K.; et al. Goblet Cell Ratio in Combination with Differentiation and Stem Cell Markers in Barrett Esophagus Allow Distinction of Patients with and without Esophageal Adenocarcinoma. Cancer Prev Res (Phila) 2017, 10, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.; Huang, Q.; Mashimo, H. Lgr5, an intestinal stem cell marker, is abnormally expressed in Barrett's esophagus and esophageal adenocarcinoma. Dis Esophagus 2010, 23, 168–174. [Google Scholar] [CrossRef] [PubMed]
- von Rahden, B.H.; Kircher, S.; Lazariotou, M.; Reiber, C.; Stuermer, L.; Otto, C.; Germer, C.T.; Grimm, M. LgR5 expression and cancer stem cell hypothesis: clue to define the true origin of esophageal adenocarcinomas with and without Barrett's esophagus? J Exp Clin Cancer Res 2011, 30, 23. [Google Scholar] [CrossRef]
- Lavery, D.L.; Nicholson, A.M.; Poulsom, R.; Jeffery, R.; Hussain, A.; Gay, L.J.; Jankowski, J.A.; Zeki, S.S.; Barr, H.; Harrison, R.; et al. The stem cell organisation, and the proliferative and gene expression profile of Barrett's epithelium, replicates pyloric-type gastric glands. Gut 2014, 63, 1854–1863. [Google Scholar] [CrossRef]
- Jang, B.G.; Lee, B.L.; Kim, W.H. Intestinal Stem Cell Markers in the Intestinal Metaplasia of Stomach and Barrett's Esophagus. PLoS One 2015, 10, e0127300. [Google Scholar] [CrossRef]
- Wang, X.; Ouyang, H.; Yamamoto, Y.; Kumar, P.A.; Wei, T.S.; Dagher, R.; Vincent, M.; Lu, X.; Bellizzi, A.M.; Ho, K.Y.; et al. Residual embryonic cells as precursors of a Barrett's-like metaplasia. Cell 2011, 145, 1023–1035. [Google Scholar] [CrossRef]
- Xian, W.; Duleba, M.; Zhang, Y.; Yamamoto, Y.; Ho, K.Y.; Crum, C.; McKeon, F. The Cellular Origin of Barrett's Esophagus and Its Stem Cells. Adv Exp Med Biol 2019, 1123, 55–69. [Google Scholar] [CrossRef]
- Sarosi, G.; Brown, G.; Jaiswal, K.; Feagins, L.A.; Lee, E.; Crook, T.W.; Souza, R.F.; Zou, Y.S.; Shay, J.W.; Spechler, S.J. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett's esophagus. Dis Esophagus 2008, 21, 43–50. [Google Scholar] [CrossRef]
- Hutchinson, L.; Stenstrom, B.; Chen, D.; Piperdi, B.; Levey, S.; Lyle, S.; Wang, T.C.; Houghton, J. Human Barrett's adenocarcinoma of the esophagus, associated myofibroblasts, and endothelium can arise from bone marrow-derived cells after allogeneic stem cell transplant. Stem Cells Dev 2011, 20, 11–17. [Google Scholar] [CrossRef]
- Nomura, S.; Goldenring, J.R. Mind the Gap: Crossing Boundaries to Establish Reparative Metaplasia. Cell Mol Gastroenterol Hepatol 2018, 6, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Agoston, A.T.; Pham, T.H.; Zhang, W.; Zhang, X.; Huo, X.; Peng, S.; Bajpai, M.; Das, K.; Odze, R.D.; et al. Acidic Bile Salts Induce Epithelial to Mesenchymal Transition via VEGF Signaling in Non-Neoplastic Barrett's Cells. Gastroenterology 2019, 156, 130–144 e110. [Google Scholar] [CrossRef] [PubMed]
- Phipps, S.M.; Garry, C.E.; Kamal, S.; Johnson, J.D.; Gilmer, J.; Long, A.; Kelleher, D.; Duggan, S.P. High Content Imaging of Barrett's-Associated High-Grade Dysplasia Cells After siRNA Library Screening Reveals Acid-Responsive Regulators of Cellular Transitions. Cell Mol Gastroenterol Hepatol 2020, 10, 601–622. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Huo, X.; Mittal, V.; Schuler, C.M.; Carmack, S.W.; Zhang, H.Y.; Zhang, X.; Yu, C.; Hormi-Carver, K.; Genta, R.M.; et al. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 2009, 137, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, K.B.; Agoston, A.T.; Odze, R.D.; Huo, X.; Pham, T.H.; Cipher, D.J.; Castell, D.O.; Genta, R.M.; Souza, R.F.; Spechler, S.J. Association of Acute Gastroesophageal Reflux Disease With Esophageal Histologic Changes. JAMA 2016, 315, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Agoston, A.T.; Dunbar, K.B.; Cipher, D.J.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Pham, T.H.; Tambar, U.K.; et al. Hypoxia-inducible factor-2alpha plays a role in mediating oesophagitis in GORD. Gut 2017, 66, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J.; Merchant, J.L.; Wang, T.C.; Chandrasoma, P.; Fox, J.G.; Genta, R.M.; Goldenring, J.R.; Hayakawa, Y.; Kuipers, E.J.; Lund, P.K.; et al. A Summary of the 2016 James W. Freston Conference of the American Gastroenterological Association: Intestinal Metaplasia in the Esophagus and Stomach: Origins, Differences, Similarities and Significance. Gastroenterology 2017, 153, e6–e13. [Google Scholar] [CrossRef]
- Feagins, L.A.; Zhang, H.Y.; Zhang, X.; Hormi-Carver, K.; Thomas, T.; Terada, L.S.; Spechler, S.J.; Souza, R.F. Mechanisms of oxidant production in esophageal squamous cell and Barrett's cell lines. Am J Physiol Gastrointest Liver Physiol 2008, 294, G411–417. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J.; Harb, H.L. Cytokines and the regulation of hypoxia-inducible factor (HIF)-1alpha. Int Immunopharmacol 2005, 5, 461–483. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gong, J.; Geng, J.; Song, Y. Deoxycholic acid induces the overexpression of intestinal mucin, MUC2, via NF-kB signaling pathway in human esophageal adenocarcinoma cells. BMC Cancer 2008, 8, 333. [Google Scholar] [CrossRef]
- Dvorakova, K.; Payne, C.M.; Ramsey, L.; Holubec, H.; Sampliner, R.; Dominguez, J.; Dvorak, B.; Bernstein, H.; Bernstein, C.; Prasad, A.; et al. Increased expression and secretion of interleukin-6 in patients with Barrett's esophagus. Clin Cancer Res 2004, 10, 2020–2028. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Zhang, Q.; Zhang, X.; Yu, C.; Huo, X.; Cheng, E.; Wang, D.H.; Spechler, S.J.; Souza, R.F. Cancer-related inflammation and Barrett's carcinogenesis: interleukin-6 and STAT3 mediate apoptotic resistance in transformed Barrett's cells. Am J Physiol Gastrointest Liver Physiol 2011, 300, G454–460. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, K.; Dvorak, B. Role of interleukin-6 in Barrett's esophagus pathogenesis. World J Gastroenterol 2013, 19, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Clemons, N.J.; McColl, K.E.; Fitzgerald, R.C. Nitric oxide and acid induce double-strand DNA breaks in Barrett's esophagus carcinogenesis via distinct mechanisms. Gastroenterology 2007, 133, 1198–1209. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Hormi-Carver, K.; Zhang, X.; Spechler, S.J.; Souza, R.F. In benign Barrett's epithelial cells, acid exposure generates reactive oxygen species that cause DNA double-strand breaks. Cancer Res 2009, 69, 9083–9089. [Google Scholar] [CrossRef]
- Suzuki, H.; Iijima, K.; Scobie, G.; Fyfe, V.; McColl, K.E. Nitrate and nitrosative chemistry within Barrett's oesophagus during acid reflux. Gut 2005, 54, 1527–1535. [Google Scholar] [CrossRef]
- Ishiyama, F.; Iijima, K.; Asanuma, K.; Ara, N.; Yoshitake, J.; Abe, Y.; Koike, T.; Imatani, A.; Ohara, S.; Shimosegawa, T. Exogenous luminal nitric oxide exacerbates esophagus tissue damage in a reflux esophagitis model of rats. Scand J Gastroenterol 2009, 44, 527–537. [Google Scholar] [CrossRef]
- Jolly, A.J.; Wild, C.P.; Hardie, L.J. Sodium deoxycholate causes nitric oxide mediated DNA damage in oesophageal cells. Free Radic Res 2009, 43, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Eluri, S.; Brugge, W.R.; Daglilar, E.S.; Jackson, S.A.; Styn, M.A.; Callenberg, K.M.; Welch, D.C.; Barr, T.M.; Duits, L.C.; Bergman, J.J.; et al. The Presence of Genetic Mutations at Key Loci Predicts Progression to Esophageal Adenocarcinoma in Barrett's Esophagus. Am J Gastroenterol 2015, 110, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Eluri, S.; Klaver, E.; Duits, L.C.; Jackson, S.A.; Bergman, J.J.; Shaheen, N.J. Validation of a biomarker panel in Barrett's esophagus to predict progression to esophageal adenocarcinoma. Dis Esophagus 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Patel, K.; Gartside, M.; Krause, L.; Brosda, S.; Aoude, L.G.; Loffler, K.A.; Bonazzi, V.F.; Patch, A.M.; Kazakoff, S.H.; et al. Complex structural rearrangements are present in high-grade dysplastic Barrett's oesophagus samples. BMC Med Genomics 2019, 12, 31. [Google Scholar] [CrossRef]
- Ellsworth, E.; Jackson, S.A.; Thakkar, S.J.; Smith, D.M., Jr.; Finkelstein, S. Correlation of the presence and extent of loss of heterozygosity mutations with histological classifications of Barrett's esophagus. BMC Gastroenterol 2012, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 2013, 45, 478–486. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun 2014, 5, 5224. [Google Scholar] [CrossRef]
- Trindade, A.J.; McKinley, M.J.; Alshelleh, M.; Levi, G.; Stewart, M.; Quinn, K.J.; Thomas, R.M. Mutational load may predict risk of progression in patients with Barrett's oesophagus and indefinite for dysplasia: a pilot study. BMJ Open Gastroenterol 2019, 6, e000268. [Google Scholar] [CrossRef]
- Weaver, J.M.J.; Ross-Innes, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.J.; Lao-Sirieix, P.; Dunning, M.J.; et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet 2014, 46, 837–843. [Google Scholar] [CrossRef]
- Tanaka, T.; Watanabe, M.; Yamashita, K. Potential therapeutic targets of TP53 gene in the context of its classically canonical functions and its latest non-canonical functions in human cancer. Oncotarget 2018, 9, 16234–16247. [Google Scholar] [CrossRef]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O'Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat Genet 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479-480, 131–145. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomeres and chromosome instability. DNA Repair (Amst) 2006, 5, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.; Shin, M.R.; Cheng, C.; Bajpai, M. Barrett's Epithelium to Esophageal Adenocarcinoma: Is There a "Point of No Return"? Front Genet 2021, 12, 706706. [Google Scholar] [CrossRef]
Figure 1.
Notch-signalling pathway inhibition that leads to intestinal differentiation.
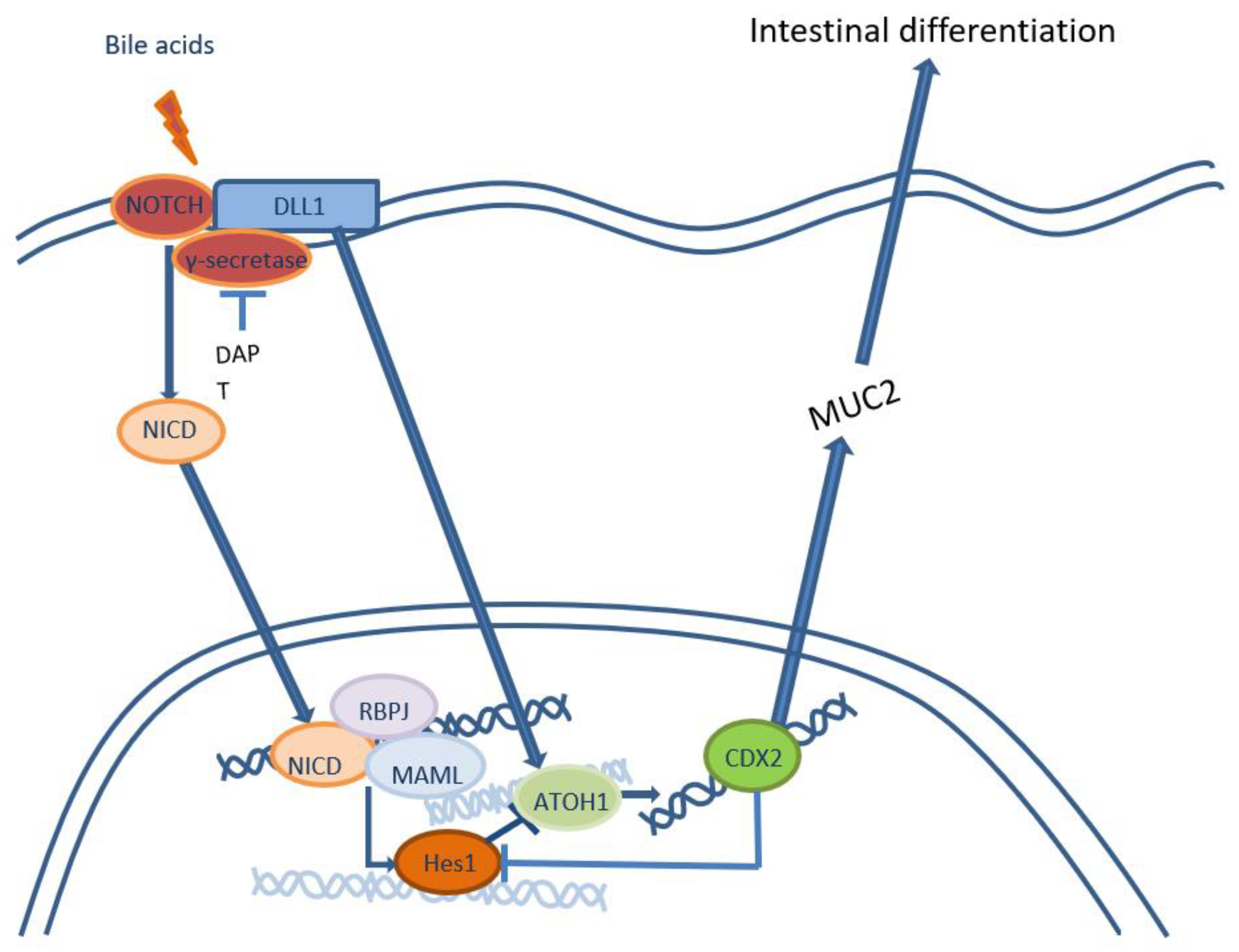
Figure 2.
Hedgehog-signaling pathway in development of intestinal metaplasia in distal esophagus.
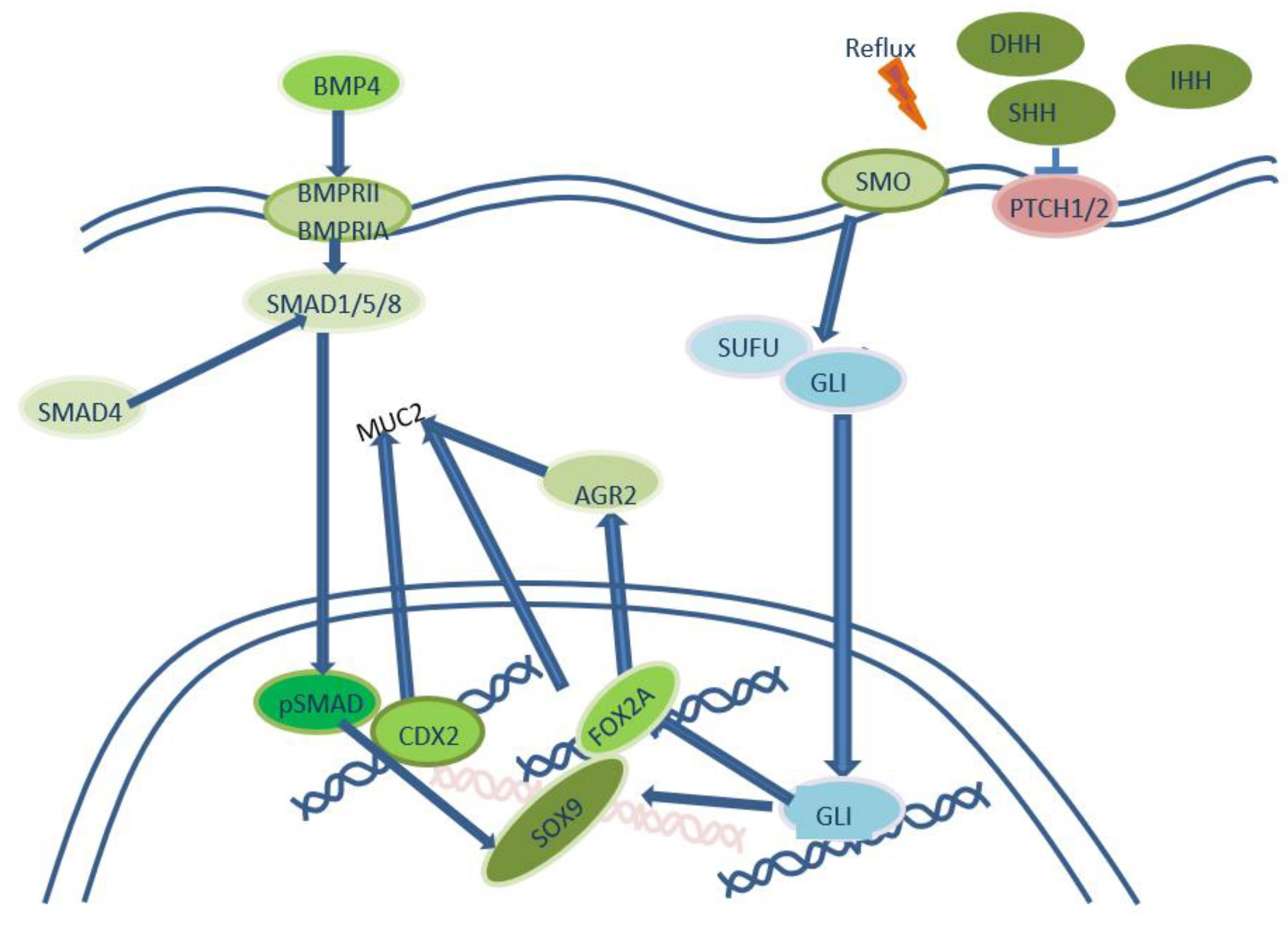
Figure 3.
Cellular origins of metaplasia and pathogenesis of BE and EAC. Abbreviations: SE – squamous epithelium, MSCSE – multipotent stem cells of squamous epithelium, REC – residual embryonic cells, ME – multilayered epithelium, MSCME – multipotent stem cells of multilayered epithelium, SEG – submucosal esophageal glands, MSCSEG – multipotent stem cells of submucosal esophageal glands, FOG – first oxyntic gland of the stomach, MSCFOG – multipotent stem cells of first oxyntic gland of the stomach, BMDCSC – bone marrow-derived circulating stem cells, CM – cardiac-type metaplasia, BE – Barrett’s esophagus, SCBE – stem cells associated with Barrett’s esophagus, EAC – esophageal adenocarcinoma, CSC – cancer-associated stem cells.
Figure 3.
Cellular origins of metaplasia and pathogenesis of BE and EAC. Abbreviations: SE – squamous epithelium, MSCSE – multipotent stem cells of squamous epithelium, REC – residual embryonic cells, ME – multilayered epithelium, MSCME – multipotent stem cells of multilayered epithelium, SEG – submucosal esophageal glands, MSCSEG – multipotent stem cells of submucosal esophageal glands, FOG – first oxyntic gland of the stomach, MSCFOG – multipotent stem cells of first oxyntic gland of the stomach, BMDCSC – bone marrow-derived circulating stem cells, CM – cardiac-type metaplasia, BE – Barrett’s esophagus, SCBE – stem cells associated with Barrett’s esophagus, EAC – esophageal adenocarcinoma, CSC – cancer-associated stem cells.
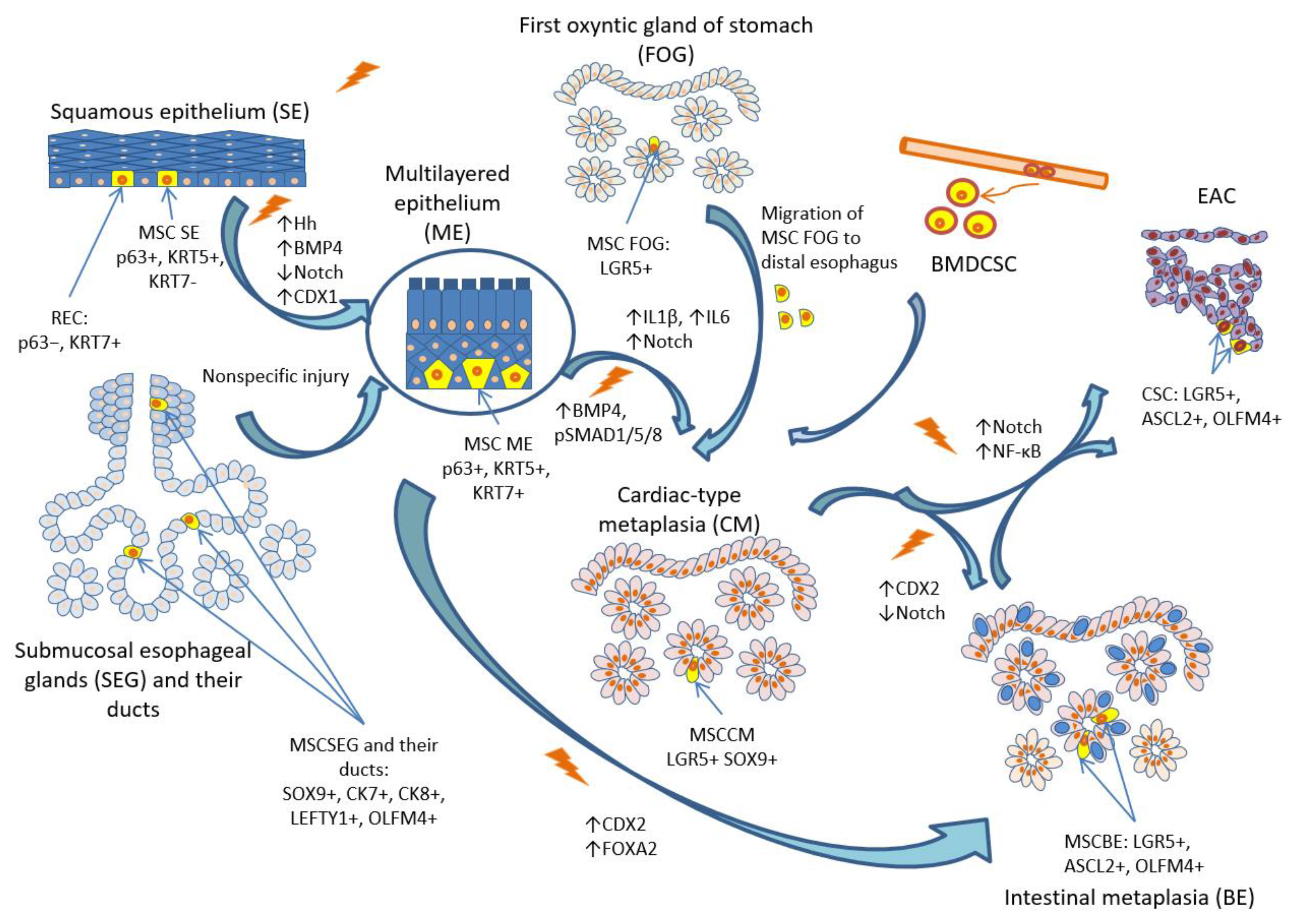
Figure 4.
NFkB signaling pathway in pathogenesis of reflux esophagitis and BE.
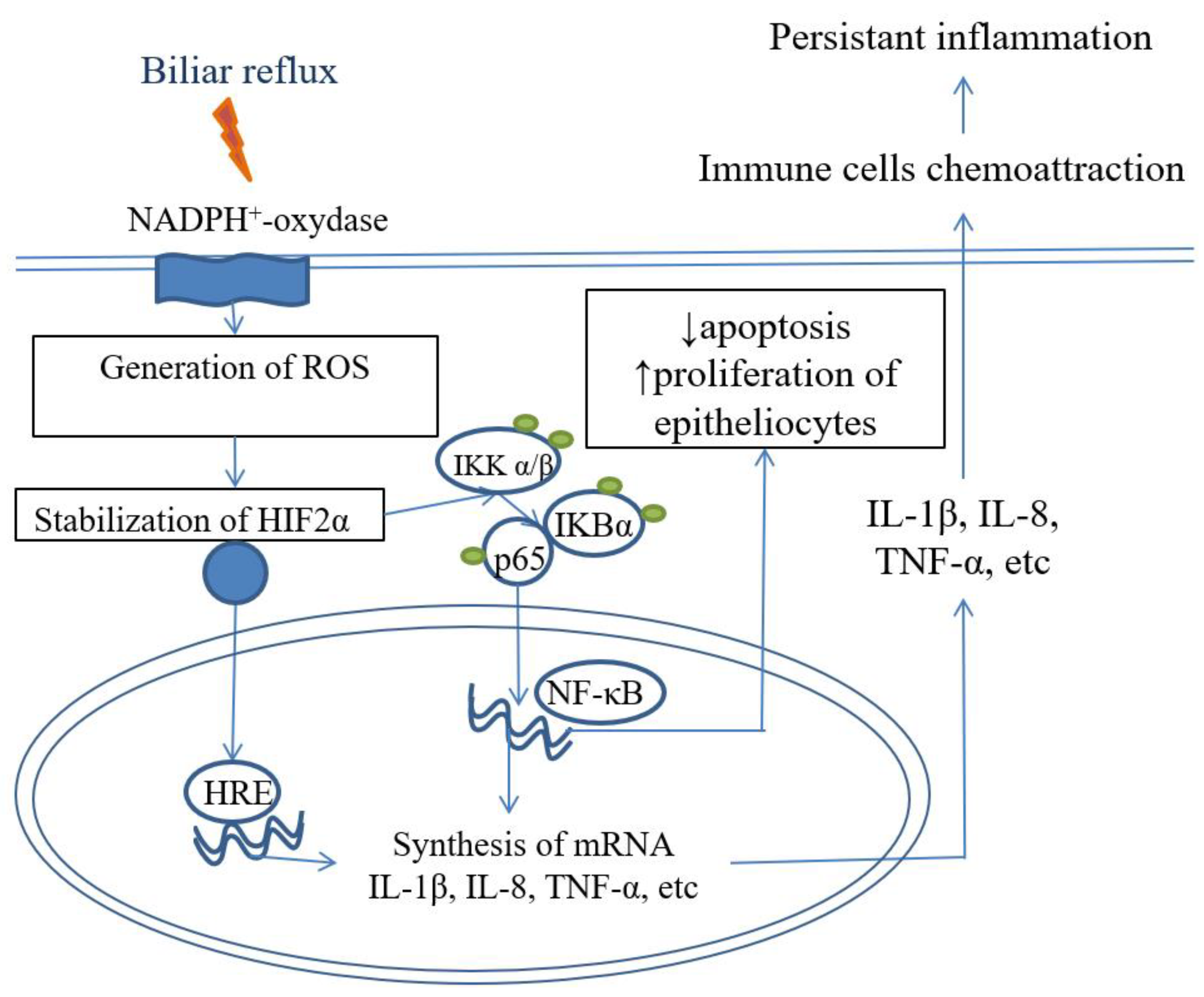
Figure 5.
IL-6/STAT3-signaling pathway in BE.
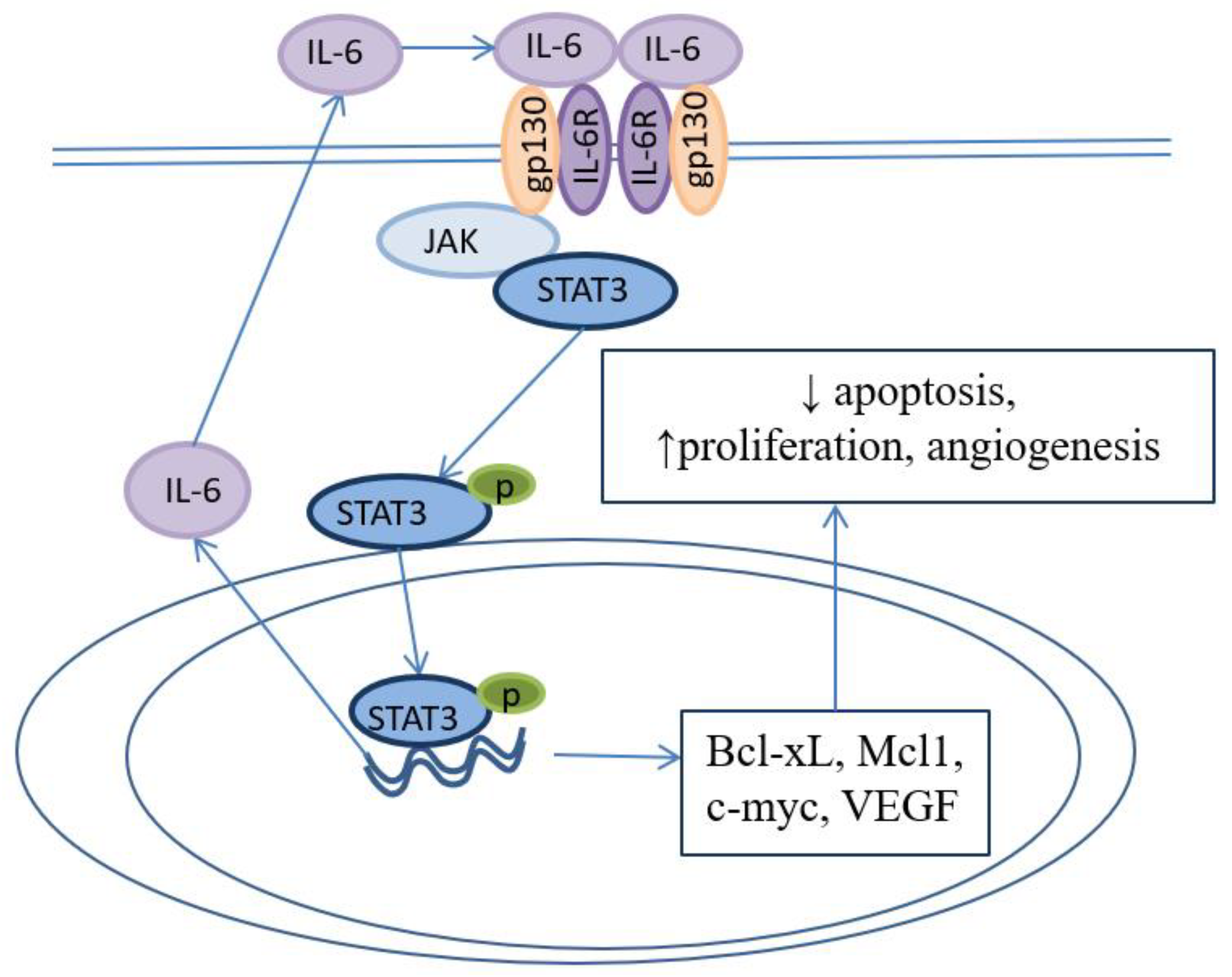
Figure 6.
Genetic mechanisms of neoplastic progression in BE.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



