
Submitted:
26 April 2023
Posted:
27 April 2023
You are already at the latest version
Abstract
Staphylococci sp. have become the primary pathogens implicated in infective endocarditis, especially within high-income nations. This along with the increasing burden of healthcare, aging populations and the protracted course the infections may take, contribute to a significant challenge for healthcare systems. A systematic review was conducted using relevant search criteria from PubMed, Ovid’s version of MEDLINE, and EMBASE, and data were tabulated from randomized controlled trials (RCT), observational cohort studies, meta-analysis, and basic research articles. The review was registered with the OSF register of systematic reviews and followed the PRISMA reporting guidelines. 35 studies met the inclusion criteria and were included in the final systematic review. The role of Staphylococcus aureus and its interaction with the protective shield and host protection functions was identified and highlighted in several studies. The interaction between infective endocarditis pathogens, vascular endothelium, and blood constituents was also explored giving rise to the potential use of antiplatelets as preventative and/or curative agents. Several factors allow Staphylococcus aureus infections to proliferate within the host with numerous promoting and perpetuating agents. The complex interaction with the hosts' innate immunity also potentiates its virulence. Ameliorating these molecular pathways may serve as a therapeutic avenue for the prevention and treatment of these infections in near future.
Keywords:
Staphylococcus Aureus Infection
; Staphylococcus Aureus Immunity
; Staphylococcus Aureus Cytotoxin
; Biofilm resistance. Host innate immunity.
1. Introduction
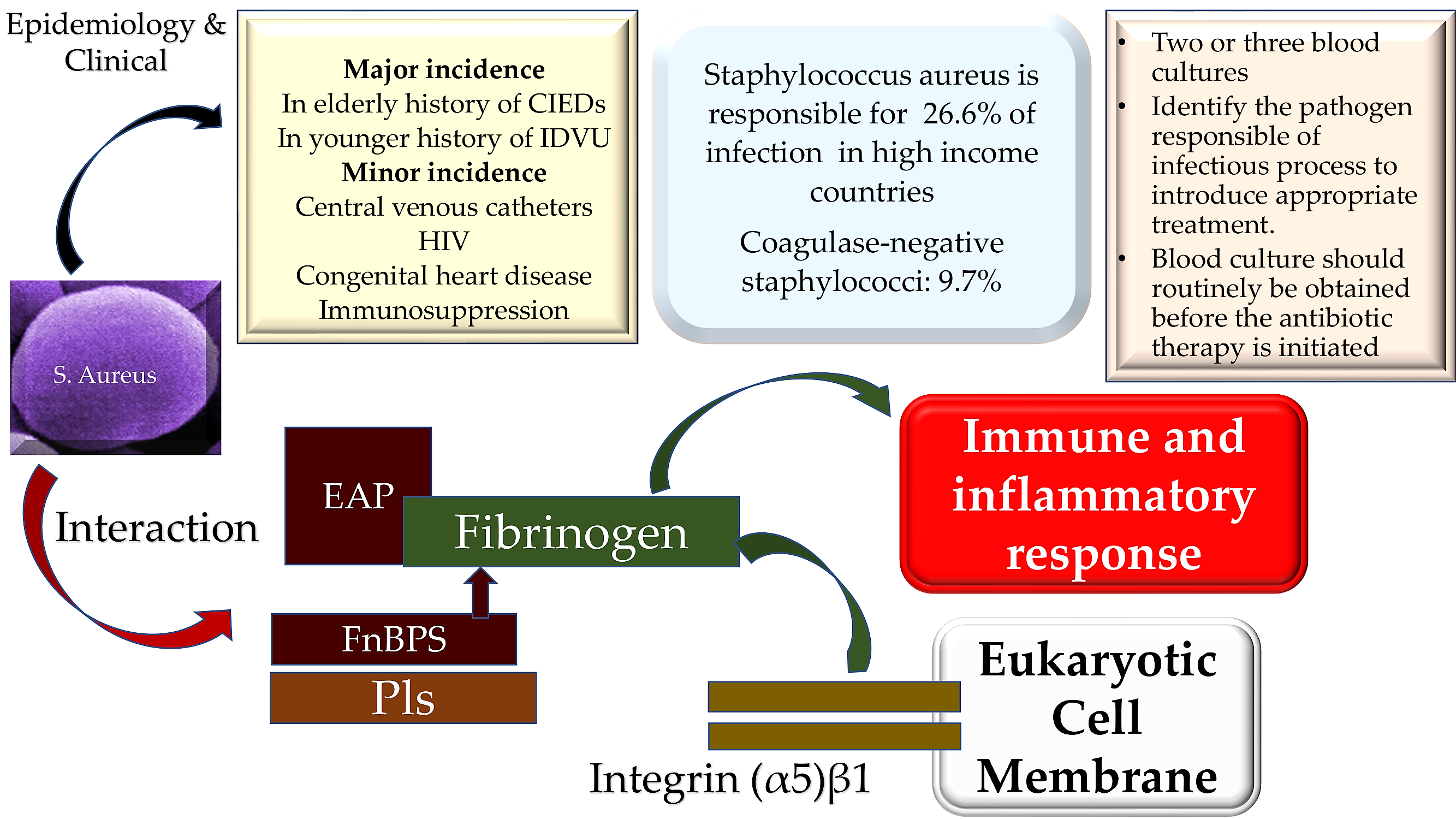
In many high-income countries, virulent staphylococci represent the leading causative pathogens promoters of infective endocarditis (IE) pushing into a corner the penicillin-sensitive streptococci. [1,2,3]. Similarly, the subjects at risk of contracting an IE by staphylococcus bacteremia have increased the burden on healthcare facilities, and tackling this infection represents one of the paramount challenges to infection in the 21st century [4,5,6]. This concern is related to the biomolecular characteristics of Staphylococcus aureus infection, which often has increased resistance to many antibiotics, constituting a major conundrum in modern health care [7,8,9]. Staphylococcus aureus interacts with the host’s innate immunity, playing a pivotal role in sustaining and maintaining the infectious state. The pathogen generates a protective shield that interferes with the host's protective mechanisms by means of two coagulases, von Willebrand factor binding protein (vWFbp) and Coagulase (Coa), leading to its critical virulence [10,11,12,13,14,15] These molecules make up a functionally intricate framework which offers S. aureus a defensive shield due to the assembly of fibrinogen/fibrin complex so as to surround the pathogen and conferring the ability to generate large vegetations. A substantial concern for staphylococcal infection is related to the specific characteristics of these vegetations. These can be large, mobile, and very frequently located in the mitral valve; this phenomenon has been linked to a markedly increased risk of symptomatic embolic incidents [16,17,18,19]. Although in 50% of patients, embolic events occur subtly and asymptomatically, up to 80% of patients’ systematic magnetic resonance imaging (MRI) of the brain may highlight cerebral injuries, [20,21,22]. The former condition may generate mycotic aneurysmal lesions resulting from a septic arterial embolism which is associated with the migration of the pathogen into the intraluminal space or vasa vasorum, followed by the diffusion of the infection through the vascular structure. The mycotic aneurysm was recorded in 5% of IE older patients with weak contrast immunity to S. aureus infection. Recently the detection of lesions is more frequently recorded through the increasing use of advanced imaging methods [24,25,26]. Graphical Abstract
2. Methods
2.1. Search Strategy
In January 2023, PubMed, Ovid’s version of MEDLINE, and EMBASE the systematic review has been designed, and the database was investigated using the terms “Staphylococcus Aureus Infection (9.716 to the present)” “Staphylococcus aureus immunity (1.102 to the present)” “Staphylococcus aureus cytotoxin (300 to the present)” “Staphylococcus aureus coagulation (455 to the present)” and “Staphylococcus aureus biofilm (884 to the present)”. The search was directed to the identification of data from randomized controlled trials (RCT), meta-analysis, observational cohort studies and basic research articles. The review was registered with the OSF register of systematic reviews and followed the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) reporting guidelines. A DOI is available for the project online (https://osf.io/mnu9s)
2.2. Study Selection and Data Extraction
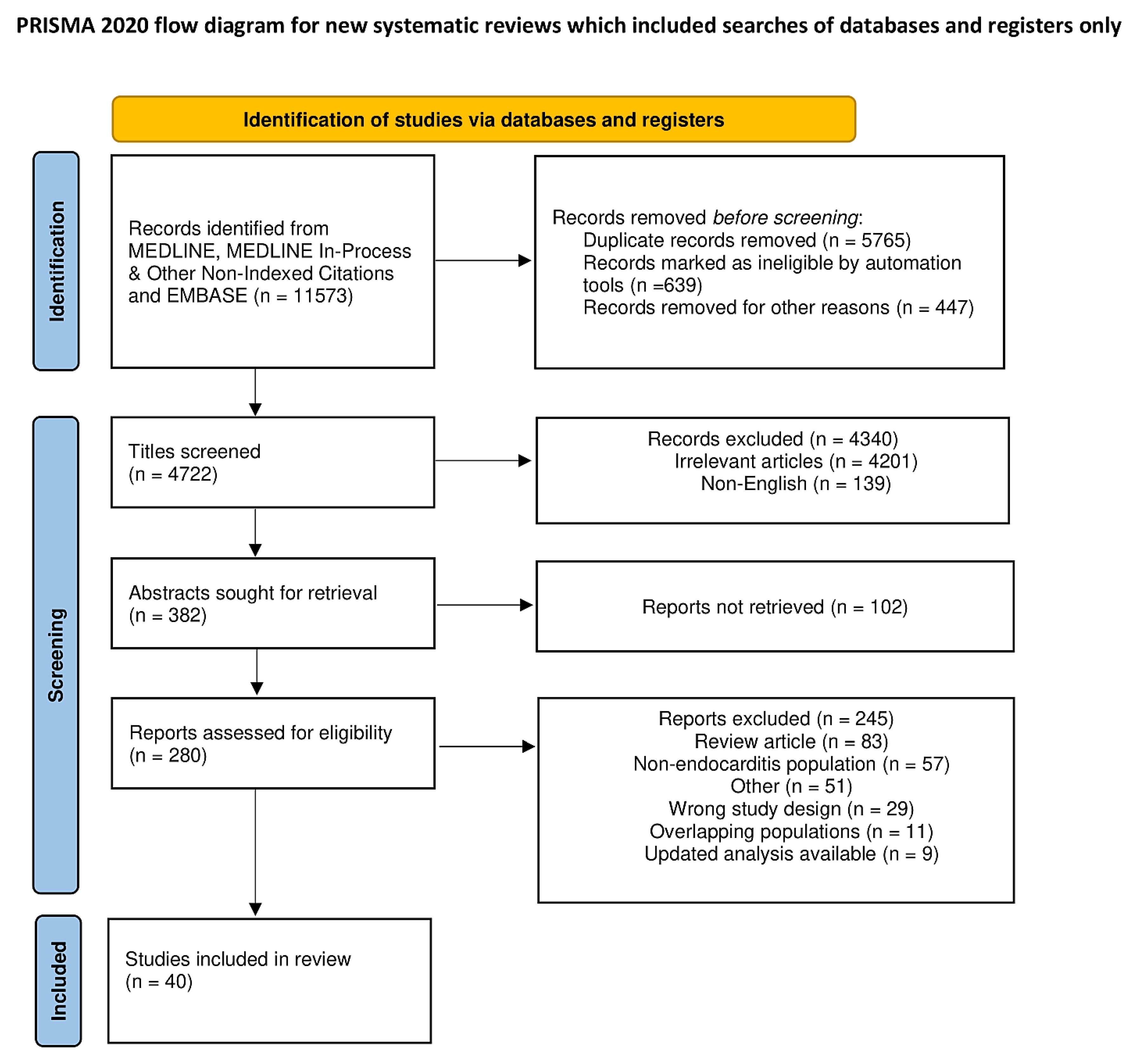
Relevant abstracts were searched (11, 573) and after deduplication 4 722 relevant citations were screened. The predefined inclusion criteria guided the review of titles and abstracts. Very impressive articles in English based on infective endocarditis, S aureus infection, mechanisms related to S aureus immunity, and mechanisms of action of S aureus cytotoxin were included. Furthermore, particular attention has been given to the pathophysiology of the biofilm and to the mechanisms of resistance to S aureus infection. Pertinent animal model studies were selected, as the topics raised improved understanding of the role played by S aureus as a causal pathogen in promoting infection and shed light on the interaction between S. aureus, the immune response and the coagulation process. Case reports, conference presentations, editorials, and expert opinions were excluded. A total of 280 citations were evaluated of which 40 studies met inclusion criteria and were included in the final systematic review. In Figure 1 is reported PRISMA Flowchart. PRISMA 2020 checklist in supplementary material
Figure 1.
PRISMA 2021 Flow diagram for new systematic review which include searches of database and registers only.
Figure 1.
PRISMA 2021 Flow diagram for new systematic review which include searches of database and registers only.
2.3. Endpoints and Effect Summary
The endpoints assessed the effects of the prominent role of Staphylococcus aureus immunity, conferring particular attention to host innate immunity, immune modulation, B-cell vs T-cell cooperation, as well as immune response and vaccine. We also investigated new evidence from the infectious array of Staphylococcus aureus focusing on the involvement of the protective shield and host protection functions, the interaction between infective endocarditis pathogens, vascular endothelium, and blood constituents. Again, the role of biofilm formation was discussed. Included studies that meet inclusion criteria are reported in tables 1-3
3. Result and Discussion
3.1. Staphylococcal manipulation of host immune responses
Gram-positive cocci of the staphylococcus, streptococcus, and enterococcus species are accountable for 80–90% of infective endocarditis. In the context of these causative bacteria, S aureus is the highly usually isolated and related pathogen of IE in high-income countries, accounting for up to 30% of infection events [1,2,3,4,5,6,27,28,29]. The lineage of coagulase-negative staphylococci, including Staphylococcus epidermidis, Staphylococcus lugdunensis and Staphylococcus capitis, stands out as far-reaching skin commensals. Coagulase-negative staphylococci maintain distinct characteristics involving frequent colonization of indwelling lines and CIEDs. Moreover, they are the highly recurrent causative bacteria responsible of infective field in patients suffering from early prosthetic valvular endocarditis [30,31,32,33,34]. These pathogens are often accountable as the cause of hospital-acquired native valvular endocarditis. [35,36,37] Furthermore, coagulase-negative staphylococci express the worrying function of generating biofilms that can advocate high rates of abscess formation and multi-antibiotic resistance [36]. Given the specificity of the immune response to infection and the capacity to develop a marked resistance to antibiotics, IE caused by staphylococcal outbreaks affects a particular population of patients. These include at-risk hemodialysis patients and intravenous drug users, but also those with native valves, prostheses, and cardiac implantable electronic devices (CIEDs) [38,39,40,41,42,43]. Staphylococcus sp have an ingrained tendency to increase antibiotic resistance where methicillin-resistant strains have emerged and remain a serious concern worldwide. [2,44,45]
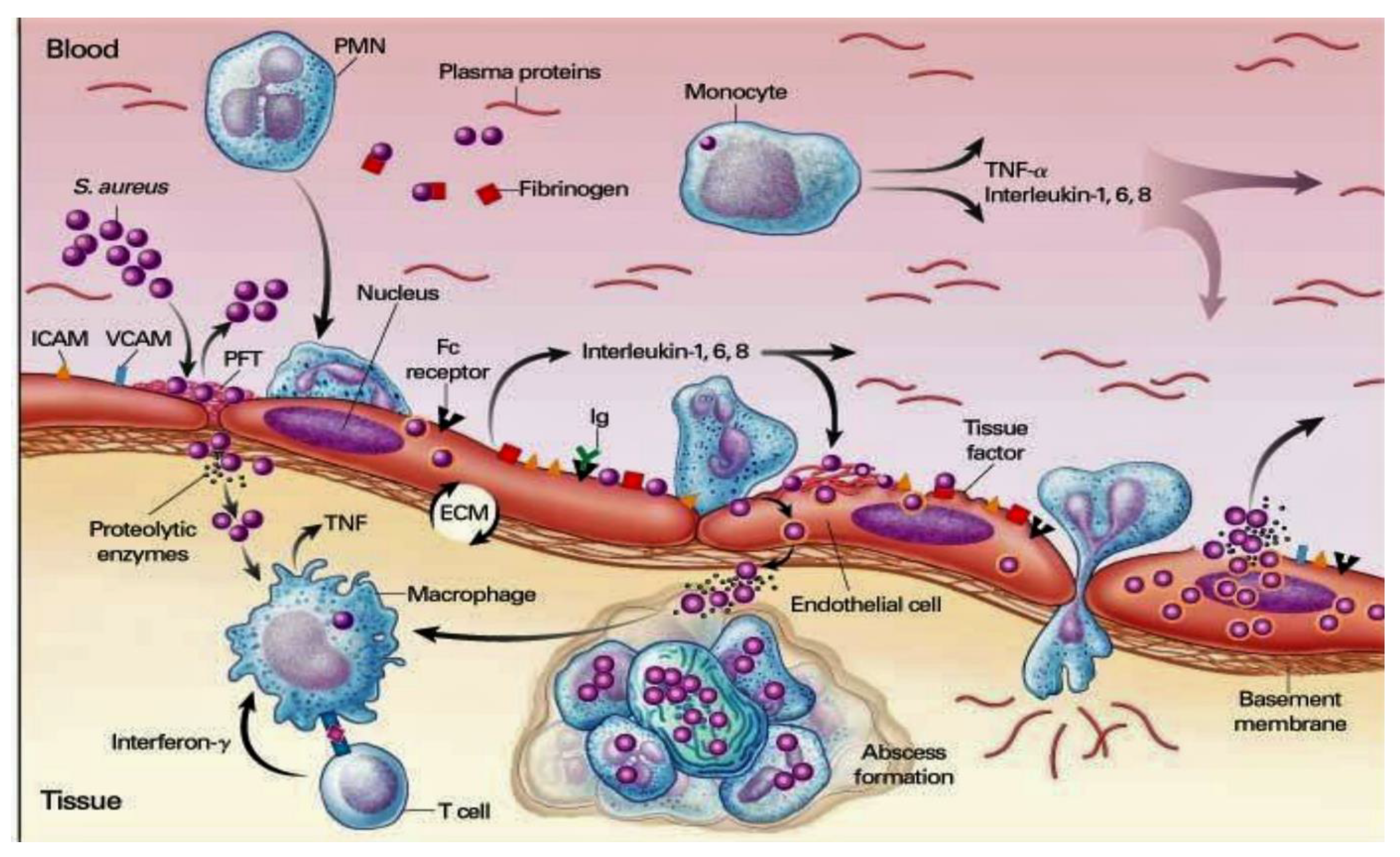
In the immune response advocated by pathogens, in lacking heart disease, the cardiac endothelium is not subject to recurrent bacteremia. However, the latter can be promoted by ordinary quotidian activities, the most routinely depicted by brushing and chewing the teeth. [46] Bacterial fastening to the tissue appoints one of the crucial steps in the pathophysiological process of IE. Once the endothelial injury is initiated, bacterial adhesion is promoted through two main steps. Initially, the release of inflammatory cytokines associated with tissue factors is recorded while in the second time the expression of fibronectin is observed, which advocates the generation of a thrombus constituted by conglomerate of fibrin and platelets [47,48,49]. Common causative pathogens implicated in the development of ED can colonize heart valves either with pre-existing sterile vegetations or in the presence of slightest endothelial injuries. The superimposed inflammatory response induces the assembly of cytokines, integrins, and tissue factors, which in turn attract monocytes and platelets. Due to effect induced by chemokines, the combined production of fibronectin can be observed. The crucial action of the chemokines allows the bacteria to adhere further favoring the activation of the inflammatory cascade which offers, through the incorporation of the bacteria, an anomalous protection mechanism by the host defenses. [48,49]. Figure 2
Figure 2.
depicts the mechanism of bacterial adhesion. The first pathophysiological process leading to IE is the development of proinflammatory cell lines such as PMN, monocyte, and macrophage is supported by the production of cytokines (TNF, α, interleukine 1,6 and 8), adhesion molecules (ICAM, VCAM) integrins and tissue factor. These mediators of inflammation draw monocytes and platelets through the intervention of chemokines with the associated production of fibronectin. S Aureus releases cytotoxins that trigger the immune response both innate and mediate (T-cell and B- cell). Abbreviations; ICAM, IE, infective endocarditis; Inter Cellular Adhesion Molecule; S. Aureus, staphylococcus aureus; TNF, tumor necrosis factor; VCAM, vascular cell adhesion molecule.
Figure 2.
depicts the mechanism of bacterial adhesion. The first pathophysiological process leading to IE is the development of proinflammatory cell lines such as PMN, monocyte, and macrophage is supported by the production of cytokines (TNF, α, interleukine 1,6 and 8), adhesion molecules (ICAM, VCAM) integrins and tissue factor. These mediators of inflammation draw monocytes and platelets through the intervention of chemokines with the associated production of fibronectin. S Aureus releases cytotoxins that trigger the immune response both innate and mediate (T-cell and B- cell). Abbreviations; ICAM, IE, infective endocarditis; Inter Cellular Adhesion Molecule; S. Aureus, staphylococcus aureus; TNF, tumor necrosis factor; VCAM, vascular cell adhesion molecule.
The pathoanatomy of IE is characterized by three substantial harmful factors that are addressed towards the endothelium: the direct activity of the bacterial pathogen, valvular sclerosis, and/or rheumatic valvulitis. The former is strongly advocated through the interaction of S. aureus at the site of infection. [50].
The pathophysiological and clinical assessments of IE, involving heterogeneous cohorts of subjects, allowed to range from individuals treated successfully without experiencing adverse events, to subjects who instead showed serious complications up to a high mortality event. As there has been a modification in the temporal trend in the pattern of infective endocarditis in developed countries over the past 5 decades, the study of pathophysiology and clinics has involved increasingly aging subjects. These contract IE with increasing incidence of Staphylococcus aureus as the causative bacterium and often the infection develops in the health care setting. From this, physicians have acquired a greater understanding of the mechanisms that support the formation, growth, and embolization of vegetation that occur on damaged or inflamed heart valves on cardiac devices. Improved knowledge of these mechanisms has led to a greater understanding of how to address the growing problem of antimicrobial resistance.
As concern the pathophysiology, two mechanisms causing IE has been shown to play a substantial role in its treatment: the modulation of the immune response in older patients with IE, the use of new platforms for the treatment of structural pathologies of the heart such as the transcatheter procedure for valve replacement or repair that can trigger septic shock. The latter drive to a substantially increased risk of death in patients with IE [51,52,53,54,55].
3.2. Subversion of innate immune responses
The peculiar virulence of S aureus is due to the presence of specific factors, present both on the surface of the bacterium and in its secretory molecules. Both, once triggered, give the bacterium a greater ability to counteract the host's immune defense procedure [56,57]. S. aureus has a crucial virulence program, the Accessory Gene Regulatory System (AGR), which operates for the quorum detection of pathogens. Our knowledge suggests that AGR acts on the control of the expression of phenol-soluble modulins (PSM) which are effective against immune cells and keratinocytes (KC). However, how and when this mechanism is triggered has not been fully understood [58]. The innate immune response supports a reaction by dead KCs, which generates a physical fence exerted by the deliverance of antimicrobial molecules such as cathelicidins, human β-defensins 2 and 3, and RNase 7 while bacteriostasis against S aureus infection is promoted.
Two independent study [59,60] reported the antibacterial role of KCs that is also mediated by pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD) proteins. Molecular patterns associated with invading pathogens (PAMPs) are integrated into these two surveillance systems thus encouraging timely defense against S. aureus. [59,60] In addition, the innate immune response is sustained by the activity of other cells such as B and T cells, plasma cells, natural killer (NK) cells, dendritic cells, macrophages, mast cells and fibroblasts individualized in the dermis [61,62]. S aureus infection is promoted by number of processes by which the breach of innate immune system triggers is instituted. Two other phases have also been observed that feed the infection of the pathogen, from when it enters the bloodstream and subsequently spreads into the host tissue once it leaves the bloodstream. Both stages are strictly connected to the activity of specific molecules expressed by S aureus, which work alongside the endothelium, the blood, and the extracellular matrix. With a well-defined role, FnBPA and FnBPB bind fibronectin work along-side α5β1 integrin on the surface of the vascular endothelium, causing transmigration and cell invasion. Subsequently, along with wall-wall teichoic acid (WTA) and lipoteichoic acid (LTA), which are expressed as polymers in the outer envelope of S aureus, invasion of host cells is promoted. The second step of S aureus infection is facilitated by the production of fibrin thrombi, across the trigger of the agglutination mechanism, induced by Coa/vWbp and ClfA. Binding to von Willebrand factor (vWF) on endothelial surfaces leads to the formation of polymers such as Ultra Large vWF (ULVWF). The third phase of S. Aureus infection is typically characterized by the secretion of Hla, a toxin that work alongside with the ADAM10 receptor, which leads to disruption of the physiological barrier function exerted by the vascular endothelium. Lastly, due to the activation of a Trojan horse model, neutrophils, containing intracellularly engulfed S. aureus, lose the ability to deliver bacteria into host tissues [10,11,12,13,14,63].
Since S Aureus is devoted to interacting with a large kind of immune cells during infection, the pathogen's delivery of cytotoxins is decisive and includes leukocidins, hemolysins, and PSM. The leukocidin family comprises leukotoxins such as gamma hemolysin with HlgAB, HlgCB, LukED and LukAB as well as Panton-Valentine Leukocidin (PVL). Three independent studies have clearly described the role played by leukotoxins [64,65,66]. Malachova et al [64] suggested that LukAB was worthwhile only on human polymorphonuclear leukocytes (PMNs) and can destroy, monocytes, macrophages and dendritic cells. This evidence was corroborated by Alonzo et al [65,66] who demonstrated that LukED recognizes C-C chemokine receptor 5 expressed on the cell resulting in the elimination of lymphocytes, macrophages, and dendritic cells.
Likewise, at the micromolar level, a noticeable function is offered by the intervention of PSM and alphahemolysin (Hla). The latter operates with a considerable capacity to destroy neutrophils after phagocytosis. [67] Thus, it can modulate the action of disintegrin A and metalloprotease 1 (ADAM1) and promote the eradication of monocytes, macrophages, neutrophils, and T cells [68]. A substantial role is offered by cytotoxins that serve functionally as a Trojan horse to encourage the diffusion of S aureus. Foster and collegues [69] observed that this activity is separate from the role offered by S Aureus in evading the host's immune response. Cytotoxins rule, by notably dampening, both the innate and adaptive immune responses, thus agreeing on the protection of S aureus through its movement in the host.
The pathophysiology by which S aureus circumvent the host immune surveillance is umpired by proteins suppressor of phytochrome A-105 (SpA proteins), which are embedded in the wall structure of the S aureus. These molecules are issued during the growing momentum of the bacterium. The existence of five domains in the SpA, which are implied with the linkage of immunoglobulins was demonstrated. Silverman and collegues [70] observed that the 5 immunoglobulin-binding domains tie to the IgG Fcγ domain and the Fab domain of the VH3 IgG, as well as to the IgM clan. This function is guided by the cross-links of the B-cell receptors which promote the polyclonal proliferation of the B cells thus advocating the undertaking of the superantigen SpA. It is foremost to improve that throughout the different phases of the infection, a differing growing response was noticed, arousing a varying expression of SpA. This event contributes to the delivery of the Hla toxin which trigger the activity of specific B lymphocytes detected in sites far from the S aureus. The described phenomenon is the immunological elucidation for which humans mostly generate antibodies resistant to Hla despite most of the detected SpA strains. Another important point to consider is linked to the fact that the Hla deliverance function is also umpired from the cell wall of the bacterium [70]. The superantigenic activity exercised by SpA proteins can advocate a purpose at a distance from the site of infection providing a decisive matter for vaccine progress. A specific effect of SpA proteins that evade recognition by B cells has been suggested by promoting a state termed "lethargy" – a usual early response to antigen. In this case, the B lymphocytes may not pick up a secondary signal to sustain their activation advocating a state of shock termed "anergy". The latter is a phenomenon that arises in the colonization of S. aureus, in the perseverance of its infective momentum, and in the weakening of the defensive protection of T lymphocytes, caused by an impairment of their recruitment by superantigens and cytotoxins which leads to a reduced affinity for antibodies [71,72].
3.3. Pathogen-Host Interaction in Determining Inflammation
A particular recognition is awarded to the pathogenic role of S. Aureus, which is mediated by adhesion proteins such as the fibronectin-binding protein and staphylococcal aggregation factors A and B. These molecules deploy the role of bacterial mediators of adhesion and are determinants for bacterial pathogenicity [73,74,75,76]. Likewise, the contribution offered by the induced experimental endocarditis in the animal models were of higher importance in demonstrating the pathological role sustained by the expression of Staphylococcus adhesins in Lactococcus lactis. Clumping-factor A(ClfA) and fibronectin binding protein A (FnBPA) have been suggested to play a crucial role in valve colonization. [73]
Que and colleagues [78] studied the development of infective endocarditis in an animal model over 3 days. Successful colonization of damaged valves by ClfA-positive lactococci was observed. Removal of the infection was noted spontaneously within 48 hours. FnBPA-positive lactococci showed titers of pathogens that were progressively enhanced in both vegetations and spleens. The imaging results disclosed that whilst the ClfA-positive lactococci were confined to the vegetations, the FnBPA-positive lactococci had spread to the contiguous endothelium. This explained the ability of FnBPA to trigger cell internalization in vitro. FnBPA conveys either fibrinogen and fibronectin binding domains, so the activity of these two selective functionalities in advocating infection was evaluated by dispossessing FnBPA of the fibrinogen binding domain and incorporating it with the fibrinogen binding domain of ClfA in cis. or trans configurations. Although the withdrawal of the fibrinogen binding domain of FnBPA did not modify fibronectin binding and cellular internalization in vitro, it strongly determined the dismissal of valve infectivity in vivo. Interestingly, the propensity for causing infections was resumed in cis configuration by inserting the fibrinogen binding domain of ClfA into truncated FnBPA, whilst in trans, was reached by co-expressing full-length ClfA and truncated FnBPA, by using two distinct plasmids. Thus, it may be argued that in S. aureus infection the binding of fibrinogen and fibronectin might con-tribute to valve colonization and endothelial encroachment in vivo. [73]
Staphylococcus aureus infection is supported by bacteremia which not only drives complications such as infective endocarditis and osteo-myelitis but promotes the exit of the pathogen from the bloodstream to cause metastatic abscesses. The bacterial interaction with endothelial cells works a considerable role in promoting these complications. At this stage of the infection, several bacterial proteins are implied. A fundamental role is provided by the extracellular adhesion protein (Eap) of S. aureus which has many functions including that of binding numerous host glycoproteins. [77,78,79,80,81]
The Eap complex of S. aureus has also been observed to exert both pro- and anti-inflammatory activity. Difficulties have emerged in robustly evaluating the role of Eap in vivo, due to the difficulties shown in defining its assets in mutant strains. There is evidence of the pro-inflammatory role of Eap and the activity that purified native adhesion protein of S. aureus has in triggering the delivery of TNFα in human whole blood in a dose-dependent mode. TNFα generation advocated S. aureus adhesion to endothelial cells with a 4-fold increase through a mechanism requiring protein A on the bacterial surface and gC1qR / p33 on the surface of endothelial cells. This finding suggested that Eap's contribution to disease during the course of S. aureus bacteremia is decisive. It was genetically engineered for an isogenic set of strains, in which the Eap gene was inactivated and integrated after inserting an intact copy of the gene elsewhere on the bacterial chromosome. Using a mouse bacteremia model, it was displayed that Eap-expressing strains determined a more serious infection, advocating the pivotal role of Eap in invasive disease. [78,80,81]
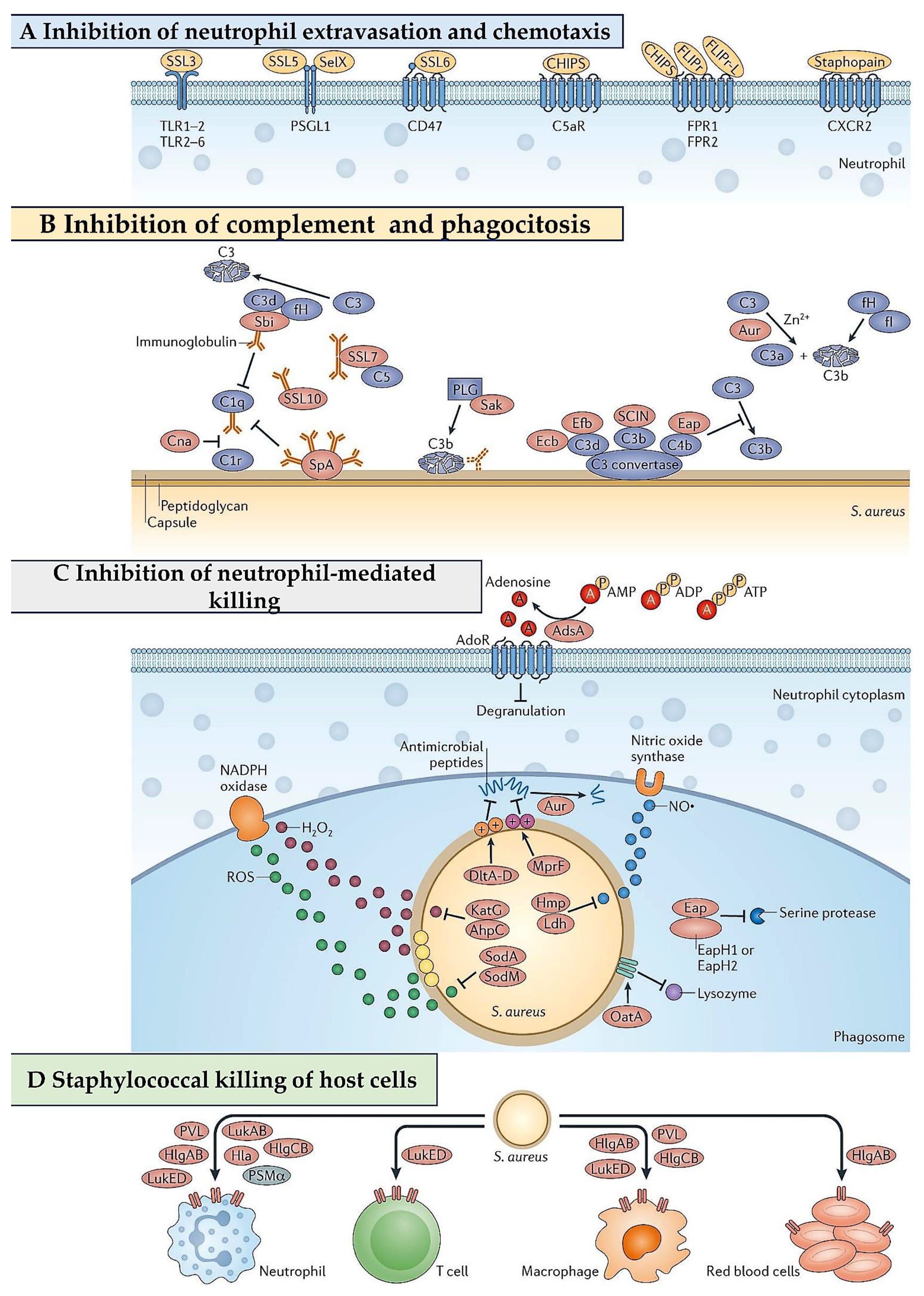
Bacterial colonization provides the trigger for additional cycles of endothelial harm and thrombus deposition resulting in the implantation of infected vegetations. In this stage, the formation of a biofilm which is generated by a multilayer bacterial aggregate containing a polysaccharide combined with a protein matrix assists bacterial persistence and contributes to antibiotic tolerance [82]. In Figure 3 Staphylococcal manipulation of host immune responses is disclosed.
Figure 3.
Four steps of pathophysiology of S. Aureus in interfering with chemotaxis, complement and killing by phagocytes are depicted. A) S. aureus promotes the inhibition of neutrophil extravasation and chemotaxis by mean of the secretion of staphylococcal superantigen-like (SSL) molecules. SSL3 lead to inhibition of Toll-like receptor (TLR) heterodimers, SSL5, SSL1. In addition, SelX hampers PSGL1 signaling and SSL6 impedes the interaction between G protein-coupled receptor CD47. Among the other active secreted proteins, we recognize the S. aureus chemotaxis inhibitory protein (CHIPS), which hinders the interaction with the complement receptor C5aR. We still find Formyl-peptide 1 (FPR1) and FPR2 receptor, Formyl peptide receptor-like 1 inhibitor (FLIPr) and FLIPr-like (FLIPrL), which hinder the action of FPR1 and FPR2. Staphopain instead work by inhibiting signaling from the C-X-C chemokine receptor chemokine receptor (CXCR2). B) The interference with the opsonization process is mediated by the secretion of inhibitory factors, which interfere with the activation of the complement factors C1q and C1r, compromising the phagocytosis of staphylococci. Specifically, collagen adhesin (Cna) blocks the association of the immunoglobulin-bound complement factor C1q with the complement receptor C1r. Staphylococcal protein A (SpA) and staphylococcal immunoglobulin ligand (Sbi) that bind to the immunoglobulin block its association with C1q. Sbi, SpA, SSL7 and SSL10 sequester immunoglobulins to block their ability to promote complement activation. Sbi (when associated with host factors C3d and factor H (fH)) and SSL7 also inactivate complement factors C3 and C5, respectively. Sak associates with plasminogen (PLG) and activates the zymogen to cleave complement factor C3b and immunoglobulin. Extracellular complement binding protein (Ecb), extracellular fibrinogen binding protein (Efb), staphylococcal complement inhibitor (SCIN), and extracellular adherence protein (Eap) inhibit C3 convertases and aureolysin (Aur) cleave complement factor C3, which impairs opsonization because the C3b cleavage product is degraded by a complex of host proteins fI and fH. C) S. aureus prevents neutrophil-mediated killing of phagocytosed bacteria through the expression of several enzymes and inhibitors. The adenosine synthesis enzyme AdsA helps block granulation via adenosine receptor (AdoR) signaling. Staphyloxanthin, superoxide dismutase A (SodA) and SodM, catalase KatG and alkyl hydroperoxide reductase (AhpC) are antioxidants that induce oxidative stress promoted by phagosomal reactive oxygen species (ROS) and H2O2 generation. Aureolysin (Aur) cleaves antimicrobial peptides and DltA-DltD lead to d-alanyl esterification of teichoic acids to protect staphylococci from antimicrobial peptides. MprF alters phosphatidylglycerol with alanine or lysine, another mechanism to protect staphylococci from antimicrobial peptides. l-lactate dehydrogenase (Ldh) and flavohemoglobin (Hmp) inhibit nitrosative stress, Eap and its homologues EapH1 and EapH2 inhibit neutrophil serine proteases and OatA O-acetylated peptidoglycan, which prevents its lysozymal degradation. D) Secreted β-barrel pore forming toxins (β-PFTs) bind specific receptors on immune cells to impair immune cell functions or advocate cell lysis. These β-PFTs include leukocidin ED (LukED) that ties to neutrophils, T cells and macrophages, γ-haemolysin AB (HlgAB) that ties to neutrophils, macrophages and red blood cells), HlgCB and Panton–Valentine leukocidin (PVL) that tie to neutrophils and macrophages, and LukAB and α-haemolysin (Hla) that tie to neutrophils. Phenol-soluble modulin-α (PSMα), which is another factor secreted by S. aureus but not a β-PFT, can also lyse leukocytes.
Figure 3.
Four steps of pathophysiology of S. Aureus in interfering with chemotaxis, complement and killing by phagocytes are depicted. A) S. aureus promotes the inhibition of neutrophil extravasation and chemotaxis by mean of the secretion of staphylococcal superantigen-like (SSL) molecules. SSL3 lead to inhibition of Toll-like receptor (TLR) heterodimers, SSL5, SSL1. In addition, SelX hampers PSGL1 signaling and SSL6 impedes the interaction between G protein-coupled receptor CD47. Among the other active secreted proteins, we recognize the S. aureus chemotaxis inhibitory protein (CHIPS), which hinders the interaction with the complement receptor C5aR. We still find Formyl-peptide 1 (FPR1) and FPR2 receptor, Formyl peptide receptor-like 1 inhibitor (FLIPr) and FLIPr-like (FLIPrL), which hinder the action of FPR1 and FPR2. Staphopain instead work by inhibiting signaling from the C-X-C chemokine receptor chemokine receptor (CXCR2). B) The interference with the opsonization process is mediated by the secretion of inhibitory factors, which interfere with the activation of the complement factors C1q and C1r, compromising the phagocytosis of staphylococci. Specifically, collagen adhesin (Cna) blocks the association of the immunoglobulin-bound complement factor C1q with the complement receptor C1r. Staphylococcal protein A (SpA) and staphylococcal immunoglobulin ligand (Sbi) that bind to the immunoglobulin block its association with C1q. Sbi, SpA, SSL7 and SSL10 sequester immunoglobulins to block their ability to promote complement activation. Sbi (when associated with host factors C3d and factor H (fH)) and SSL7 also inactivate complement factors C3 and C5, respectively. Sak associates with plasminogen (PLG) and activates the zymogen to cleave complement factor C3b and immunoglobulin. Extracellular complement binding protein (Ecb), extracellular fibrinogen binding protein (Efb), staphylococcal complement inhibitor (SCIN), and extracellular adherence protein (Eap) inhibit C3 convertases and aureolysin (Aur) cleave complement factor C3, which impairs opsonization because the C3b cleavage product is degraded by a complex of host proteins fI and fH. C) S. aureus prevents neutrophil-mediated killing of phagocytosed bacteria through the expression of several enzymes and inhibitors. The adenosine synthesis enzyme AdsA helps block granulation via adenosine receptor (AdoR) signaling. Staphyloxanthin, superoxide dismutase A (SodA) and SodM, catalase KatG and alkyl hydroperoxide reductase (AhpC) are antioxidants that induce oxidative stress promoted by phagosomal reactive oxygen species (ROS) and H2O2 generation. Aureolysin (Aur) cleaves antimicrobial peptides and DltA-DltD lead to d-alanyl esterification of teichoic acids to protect staphylococci from antimicrobial peptides. MprF alters phosphatidylglycerol with alanine or lysine, another mechanism to protect staphylococci from antimicrobial peptides. l-lactate dehydrogenase (Ldh) and flavohemoglobin (Hmp) inhibit nitrosative stress, Eap and its homologues EapH1 and EapH2 inhibit neutrophil serine proteases and OatA O-acetylated peptidoglycan, which prevents its lysozymal degradation. D) Secreted β-barrel pore forming toxins (β-PFTs) bind specific receptors on immune cells to impair immune cell functions or advocate cell lysis. These β-PFTs include leukocidin ED (LukED) that ties to neutrophils, T cells and macrophages, γ-haemolysin AB (HlgAB) that ties to neutrophils, macrophages and red blood cells), HlgCB and Panton–Valentine leukocidin (PVL) that tie to neutrophils and macrophages, and LukAB and α-haemolysin (Hla) that tie to neutrophils. Phenol-soluble modulin-α (PSMα), which is another factor secreted by S. aureus but not a β-PFT, can also lyse leukocytes.
3.4. Immuno-Response and Vaccine
The spread of an antagonistic vaccine towards S. Aureus is a crucial challenge that would allow the emergence of antibiotic-resistant strains to be addressed. Resistance to antibiotic therapy has made it possible to direct research toward alternative treatments, such as the use of immunotherapeutic drugs. However, better knowledge of the mechanisms driving the immune response during S aureus infection and the manufacturing of an active vaccine are two parallel paths. In several published reports, based on infected mouse models, the ability of a S. aureus vaccine antigen has been evaluated to elicit an immune response that can be scaled up to safeguard multiple mouse models infected with various strains of the bacterium. This procedure allowed scientists to evaluate cross-immune protection across diverse models and with the appearance of unlike strains of S aureus [83,84,85,86].
Considerations related to the progress achieved by successful immuno-humoral response may be mitigated by converging immune-evasion mechanisms of S aureus. Given the experiences accumulated to date regarding the immune response to staphylococcal infections, there is no doubt that the progress needed to obtain a promising vaccine in terms of effectiveness and safety averse to S aureus apparatus relies on an even better understanding of the immunity, both innate and adaptive. We learned that the immune response to S aureus is articulated on the effectiveness of the humoral response, T-cell function, stopping complement proteins function, and slaying immune mediators by its toxins. The main contrasting mechanism exerted by S. aureus to the host concerns the ability of the pathogen to hinder the immune action. Precisely, this peculiar characteristic epitomizes the main factor responsible for the lack of success in the progress of targeted vaccines. Thus, the core of the problem can be related to the evolution of immunological interventions that are capable of fruitfully hampering the mechanisms by which S. aureus restrains immunity. This procedure could guarantee promising upcoming achievement in vaccine spread [83,84,85,86].
A line of investigative speculation has been the role of ESAT-6-like proteins secreted by S aureus, designed as S aureus EsxA (SaEsxA) and SaEsxB, which have been studied as possible targets for a vaccine. Although tall titers of anti-SaEsxA and anti-SaEsxB antibodies were generated in mouse models vaccinated with the administration of purified proteins, a finding revealing an antibody-mediated immune response, S aureus infection was not prevented. However, mice processed with the usage of recombinant SaEsxA (rSaEsxA) and rSaEsxB recorded sustained immunity to Th1 and Th17. Additionally, this cohort was observed to have considerably improved survival rates when subjected to S. aureus with respect to the control cohort. This evidence elucidated the functioning of SaEsxA and SaEsxB as two hopeful Th1 and Th17 antigen candidates, with the likelihood of future expansion towards the development of multivalent and serotype-independent vaccines hostile to S. aureus induced bacteremia [84].
Brady et colleagues [85] focused on the genetically inactivated mutant HlaH35L of toxin alpha and analyzed the protection provided by these antigenic molecules in three infection models using the same vaccine quantity, regimen, immunization route, challenge strain and adjuvant. The use of a systemic infection model challenged by HlaH35L immunized mice revealed a small but statistically remarkable reduction in bacterial colonization juxtaposed to that noted in control mice. In contrast, using a prosthetic implant model of chronic biofilm infection, no notable discrepancies in bacterial standards compared to checks were observed. These results suggest that although vaccines may protect from one form of S. aureus disease, they seem to be inactive in providing an effective defence versus various manifestations of the disease, thus underscoring the significant challenge that exists in vaccine development against S. aureus. [85]
Epidemiological studies have revealed the high colonization potential that characterizes S. aureus, reaching 20 to 80% in humans. This implies the potential to generate a variety of diseases that constitute a nightmare for healthcare-associated and community-associated bacterial infections [83,86]. It is evident that in such a context the development of the vaccine against S aureus has been burdened by abortion, producing failures every time its enforcement has been endeavored to date. However, it is possible that the reason for this failure is due to incomplete knowledge of the tools that support the immune defense resistant to this bacterium. In humans, S aureus advocates bacteremia with the potential to progress to sepsis. The genesis of infectious fields can promote endocarditis, osteomyelitis, pneumonia and meningitis, as well as skin and soft tissue infections. People who are vectors of S aureus are at an increased risk of infection and conveyance of bacteria to others. The diffusion of multidrug-resistant strains of S aureus restricts first line medical treatment through the administration of effective antibiotics. [83,86]
Zhang et colleagues suggested a multipronged B-cell, Th1-, and Th17-mediated response averse to S aureus antigens. Similarly, this precise immune response provides increased and extensive protection versus S aureus by anticipating the stage of invasive infection, mucosal colonization as well as skin and soft tissue infection. [86] Today, the impact of immunotherapy is continuously cultivated and sustained, and can also be indefinitely conferred by the administration of the vaccine hostile to S aureus bacteremia. A decisive part is offered by S aureus manganese transport protein C (MntC). This protein is a highly conserved cell surface molecule that may evoke safeguarding immunity versus S. aureus and Staphylococcus epidermidis. Wei et al evaluated the humoral immune response and CD4+ T cell-mediated immune responses disclosing vital defense for mice to decrease the incursion of S aureus that was supported by MntC-specific antibodies. The findings firmly underpinned the definite role of MntC-induced immunity response disclosing that Th17 works substantially in counteracting S aureus infection. Again, the evidence noted that MntC-specific antibodies and MntC-specific Th17 cells work side by side in forestalling S aureus infection. Rather, Yu and colleagues [87] observed that MntC-promoted protective immunity was declined following the neutralization of IL-17 by the antibody in vivo. Thus, adoptive Th17 shifted from mice may not be fully refractory to the S. aureus challenge. Table 1

Table 1.
Characteristics of the Included Studies.
| First author / Year/ Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Lockhart et al (2008) Circulation [46] |
Human RCT Single Center (USA) |
290 pts Brushing Gro 98 Vs Extraction-Amoxicillin 96 Vs Extraction-Placebo 96 |
To compare the incidence, duration, nature, and magnitude of IE related bacteremia from single-tooth extraction and toothbrushing. To determine the impact of amoxicillin prophylaxis on single-tooth extraction. |
Amoxicillin has a significant impact on bacteremia resulting from a single-tooth extraction. Toothbrushing may be a greater threat for individuals at risk for infective endocarditis. |
| Mancini et al (2018) Virulence [49] |
Animal (Switzerland pilot) |
Rat with catheter-induced aortic vegetations | To investigate the role of Coa and vWbp in IE initiation | Coa does not support the initial colonization of IE (in L. lactis). vWbp contributes to initiation of IE (in L. lactis) however is marginal in the present of ClfA. |
| Reguiero et al (2019) Circ Cardiovasc Interv [51] |
Human Comparative Multicenter (Canada pilot) |
245 pts SEV 115 Vs BEV 130 |
To determine the incidence, clinical characteristics, and outcomes of patients with IE post-TAVR | IE post-TAVR did not reveal early or late mortality |
| Rodríguez-Vidigal et al (2019) Enferm Infecc Microbiol Clin [52] |
Human Observational Retrospective (Spain) |
200 pts with TAVI | To evaluate single-centre experience of incidence, mortality and associated factors of IE after TAVI. | Incidence of IE post TAVI greater than other series. |
| Di Carluccio et al (2021) RSC Chem Biol. [55] |
Human Multicenter (Italy pilot) |
Collected anatomical specimen | To evaluate the mechanism of interaction of SLBR-B and SLBR-H from S. gordonii in causing IE | Streptococcal Siglec-like adhesins sparks the development of tailored synthetic inhibitors and therapeutics specific for Streptococcal adhesins to counteract IE. No impairment the interplay between Siglecs and glycans. |
| Manukumar et al (2017) Sci Rep [56] |
Human Single Center (India) |
Collected blood draws | To characterize MRSA strain using MALDI-Biotyper multiplex PCR to distinguish between MRSA and MSSA. To screen PCR-SSCP | PCR-SSCP technique for rapid detection of MSSA and MRSA strains was developed |
| Mempel et al (2002) Br J Dermatol. [57] |
Human Single Center (Germany) |
†S. aureus DU 5720 Vs S. aureus DU 8325-4 Vs S. aureus DU 5883 |
To investigate haemolysin-independent virulence to human keratinocytes. | Staphylococcal invasion of human keratinocytes independently of alpha- and beta-hemolysins, leads to necrotic and apoptotic cell damage. |
| Nakagawa et al (2017) Cell Host Microbe J [58] |
Animal Multicenter Center (Japan pilot) |
Murine epicutaneous infection model | To evaluate how S. aureus trigger inflammation | Increased production of IL-1α, IL-36α and Il 17 via IL-1R and IL-36R. Increased γδ T cells, ILC3 and neutrophil. Keratinocyte* Myd88 signaling in response to S. aureus PSMα drives an IL-17-mediated skin inflammatory response to epicutaneous S. aureus infection. |
| Schwarz et al (2021) Virulence [63] |
Human in vitro and in vivo Multicenter (Germany) |
34 S. aureus Pts with S. aureus endocarditis Vs healthy individuals |
To evaluate pathomechanisms in the induction of IE | in vitro assays did not correlate with the severity of IE. i S. aureus isolates differed in the activation and inhibition of pathways connected to the extracellular matrix and inflammatory response |
| Malachowa et al (2011) PLoS One [64] |
Human/Animal Single center (USA) |
S. aureus LAC Vs S. aureus LACΔhlgABC |
To study the S. aureus USA300 transcriptome | Limited contribution of any single two-component leukotoxin lukS-PV and lukF-PV to USA300 immune evasion and virulence. |
| Alonso et al (2013) Nature [65] |
Animal Single center (USA) |
CCR5-deficient mice | To study activity of S. aureus leukotoxin ED (LukED) | CCR5-deficient mice are resistant to lethal S. aureus infection |
| Kim et al (2010) J Exp Med. [71] |
Animal Single center (USA) |
ℷ Mice with SpA (KKAA) | To study S. aureus protective immunity. | SpA (KKAA) immunization enabled MRSA-challenged mice to organize antibody responses to many different staphylococcal antigens. |
| Becker et al (2014) Proc Natl Acad Sci U S A. [72] |
In vitro Single center (USA) |
S. aureus Newman cultures | To demonstrate that SpA is released with murein tetrapeptide-tetraglycyl [L-Ala-D-iGln-(SpA-Gly5) L-Lys-D-Ala-Gly4] linked to its C-terminal threonyl | SpA, a B cell superantigen, is released with peptidoglycan linked to its C terminus. Murein hydrolases cleave the anchor structure of released SpA to modify host immune responses. |
| Zhang et al (2015) Infect Immun. [84] |
Animal Single center (China) |
Mice SaEsxA and SaEsxB Vs Mice rSaEsxA and rSaEsxB |
To investigate SaEsxA and SaEsxB, as possible targets for a vaccine. | SaEsxA and SaEsxB are effective toward Th1 and Th17 candidate antigens. |
| Brady et al (2013) PLoS One [85] |
Animal Single center (USA) |
Mice HlaH35L Vs Control Vs Prosthetic implant model of chronic biofilm |
To evaluate the ability of one S. aureus vaccine antigen to protect in three mouse models of infection | Vaccines may confer protection against one form of S. aureus disease without conferring protection against other disease presentations |
| Zhang et al (2018) mBio [86] |
Animal Multicenter (USA pilot) |
C57BL/6 mice | To study the role of adaptive immunity induced by an S. aureus vaccine in protection against S. aureus bacteremia | Multipronged humoral and cellular (B-cell, Th1, Th17) responses to S. aureus antigens may be critical to achieve effective and comprehensive immune defense |
| Yu et al (2018) Sci Rep [87] |
Animal Single center (China) |
Mouse peritonitis model | To evaluate the humoral immune response and CD4+ T cell-mediated immune responses | The MntC-specific antibodies and MntC-specific Th17 cells play cooperative roles in the prevention of S. aureus infection. |
Abbreviations; BEV, balloon-expandable valve; C57BL/6 , C57 black 6; CCR5; C-C chemokine receptor type 5; ClfA, clumping factor A; Coa, plasma-clotting factors staphylocagulase; DU, S. aureus mutant; IE, infective endocarditis; γδ T cells, Gamma delta T cells; IL, interleukine; ILC3, group 3 innate lymphoid cells; HaCaT, aneuploid immortal keratinocyte cell; LAC, wild-type USA300 strain; LACΔhlgABC, hlgABC-deletion strain ; L. Lactis; Lactococcus lactis; lukS/F-PV, leukotoxin S/F -Panton-Valentine ; LukED, S. aureus leukotoxin ED; MntC, S. aureus manganese transport protein C; MRSA, methicillin resistant Staphylococcus aureus; MSSA, methicillin susceptible S. aureus; PCR, protein chain reaction; PCR-SSCP, PCR-coupled single strand conformation polymorphism; PSM, phenol-soluble modulin α; Pt, patient; PVL, Panton-Valentine Leukocidin; rSaEsx, recombinant; SaEsx, S. aureus Esx; SEV, self-expanding valve; SLBR, Siglec-like binding region; SpA, staphylococcal protein A; TAVI, transcatheter aortic valve implantation; Th17, T helper 17 cells, TSB, trypticase soy broth. †S. aureus mutant DU 5720 alpha-haemolysin, beta-haemolysin double-negative ; S. aureus mutant virulent strain DU 8325-4 ; S. aureus variant DU 5883 isogenic fibronectin-binding protein A/B-negative. .* Myd88, keratinocyte-specific deletion of the IL-1R and IL-36R ; ℷ variant KKAA staphylococcal protein A
3.5. Biofilm formation
Biofilms allow pathogens to live by conforming to the functions and metabolism of the self-produced matrix which is composed of hydrated extracellular polymeric substances (EPS). Therefore, biofilms behave as an immediate functional environment constituted directly by the bacteria. The primary constituents that organize EPS are molecules of polysaccharides, proteins, nucleic acids, and lipids. EPS performs varied functions involving the conferral of mechanical stability of biofilms. Furthermore, EPS mediates the adhesion of bacteria to surfaces by forming a cohesive and three-dimensional polymer network that interconnects and transiently immobilizes the biofilm cells. The external digestive system by the biofilm matrix keeps extracellular enzymes close to the cells which can be metabolized and dissolved into colloidal and solid biopolymers [82,88,89].
In course of infective endocarditis, the production of bacterial biofilms is a basic phase for the fatal evolution of the disease. IE manifests itself as a lesion of the cardiac structure and causes a healing reaction, which advocates the recruitment of fibrin and immune cells. In the first cicatricial stage the vegetations are sterile but potentially at risk of causing colonization over temporary bacteremia, thus promoting well-established IE. In vitro, experimental models using a simulated IE vegetation model, produced from venous whole blood, have been demonstrated to be of great utility for assessing biofilms generation in course of infective endocarditis. Similarly, these models allowed the establishment of stable bacterial colonization after 24 hours. Once organized in biofilm aggregates, the pathogens revealed higher tolerance to antibiotics [88,89].
Swartz and colleagues recently studied the momentum required to produce biofilms and how these affect the maturing of antibiotic tolerance. Evidence advocated that reference strains of Staphylococcus aureus, as well as three clinical secludes of IE, produced biofilms modeled on IE vegetation 6 hours later than the onset of infection. Thus, the earlier the antibiotic was administered, the more marked its pharmacological action in containing biofilm maturation, advocating that early treatment was more effective in restraining the spread of the disease. The investigators followed the biofilm development under the microscope by observing the bacterial aggregates growing on the IE vegetation model and the interaction with the antibiotic. The generation of mature, antibiotic-resistant biofilms was recorded 6 hours later, thus precipitating the screening for optimal treatment strategies for IE [90].
Biofilm formations raise concerns in patients requiring treatment of heart valve endocarditis (HVE) [91,92,93,94]. In this context, the aggressiveness of Gram-positive bacteria becomes crucial due to the lack of an external membrane that is replaced by surrounding peptidoglycan, less sensitive to serum-induced killing. Subsequently to bacteria colonization and adhesion, the pathophysiology of HVE is characterized by bacterial proliferation cycles. In this phase local thrombotic processes, recruitment of monocytes, and inflammation, leading to the formation of mature vegetations occur [50]. Regarding HVE, the production of biofilm is representative of numerous causative pathogens, including staphylococci, streptococci, and enterococci with other rarer germs, such as Pseudomonas aeruginosa and Candida species that promote bacterial incorporation into polysaccharide extracellular slime-like matrix. In patients who developed staphylococcal prosthetic valve endocarditis (PVE), undergoing valve replacement with the use of a homograft or autograft [91,92,93,94], the specificity of biofilms induces a cell-to-cell communication and synchronized gene expression that promotes the assembly and maturation of pathogens. In this population of patients, once the biofilm arises, it protects the bacteria from the host’s immune system, and reduces antimicrobial efficacy, while shielding the organisms [50].
The characteristics of the generating biofilm are now recognized as a virulent trait in the development of PVE especially related to Staphylococcus aureus, for which the use of allogeneic or autologous tissue as an ideal valve substitute is recommended. Cryopreserved Aortic Homograft (CAH) is widely used in prosthetic valve endocarditis (58.1% vs 28.8%, P =.002) and methicillin-resistant Staphylococcus infection (25.6% vs 12.1%, P = .002), compared to those whom surgical correction was performed using conventional prostheses [95]. In another report, 64% of patients with PVE involving the aortic valve received an aortic homograft in 56 (64%) patients while mechanical prosthesis was used in 23% of cases and a bioprosthetic in 13%, respectively. Surgical correction using aortic homograft was independently associated with a reduced risk of infection relapse (P = 0.006) compared to conventional valves [96]. Active endocarditis supported by causative pathogens generating biofilm is often responsible for recurrence [97,98,99,100,101] and is a statistically significant univariable risk factor for increased early and late mortality as revealed by studies with short [95,100] and long-term follow-up (over 20 years) [102,103,104,105,106,107]. As far as PVE is concerned, the use of CAH appears indisputable, unlike native valve endocarditis whereby the preference for conventional prosthesis and synthetic material is still prevailing [96].
We used cryopreserved aortic homograft as a substitute to replace aortic and mitral valve diseases in 56,2% and 21% of patients, in which abscess formation occurred. The process was sustained by causative pathogen-generating biofilm and resistance to antibiotic treatment [18,98,101,102,104,107]. Sometimes in the presence of aggressive IE with extension to the aorto-mitral junction and mitral valve, we used a double homograft valve implant [18,102,107,108,109,110,111,112]. During the cryopreservation process, the homograft was processed in combination with the application of antibiotics (gentamicin, vancomycin, metronidazole, piperacillin, flucloxacillin, tobramycin, meropenem, colistin, and antifungal amphotericin B) which promoted a significant influence on the resistance of the allogeneic tissue to infections. To note that ascending aortic homograft tissue revealed significantly improved resistance against S epidermidis and S aureus with a lower propensity for bacterial contamination than homograft aortic valves. For the latter, the highest risk of bacterial biofilm formation persists, especially induced by staphylococcus aureus, which appears to be difficult to perforate. Along the same lines, more effective resistance was observed against P aeruginosa using flucloxacillin and E coli using meropenem and colistin [113]. Table 2
Table 2.
Characteristics of the Included Studies.
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Schwartz et al (2021) APMIS [88] |
In vitro patch enriched with platelet and Leucocyte-rich fibrin Multicenter (Danemark) |
IE organoid-like model by colonization with IE-associated bacterial isolates S. aureus, S. mitis and Enterococcus faecalis (IE vegetation (IEV) | To establish an in vitro vegetation simulation IE model for fast screening of novel treatment strategies | The surface-associated bacteria displayed increased tolerance to antibiotics compared to planktonic bacteria. IE simulation model with the relevant pathogens S. aureus, S. mitis group, and E. faecalis was established and IE model mirrors the natural IE process. |
| Di Domenico et al (2019) BMC Microbiol [89] |
Human Multicenter (IT) |
Samples of infected heart tissue. S. s aureus 50%, Enterococcus faecalis 25% and Streptococcus gallolyticus 25% | To assess a rapid biofilm identification assay and a targeted antimicrobial susceptibility profile of biofilm-growing bacteria in patients with IE, which were unresponsive to antibiotic therapy. | Biofilm-producing bacteria, from surgically treated IE, display a high tolerance to antibiotics, which is undetected by conventional antibiograms. |
| Schwartz et al (2012) APMIS [90] |
Animal model Multicenter (Danemark) |
IE organoid-like model by colonization with IE-associated bacterial isolates S. aureus, S. mitis and Enterococcus faecalis (IEV) | To evaluate the time course of biofilm formation and the impact on antibiotic tolerance development. | The antibiotic effect was significantly higher than when treatment was started after the biofilm was allowed to mature. |
| Kim et al (2016) JTCVS [95] |
Human Single Center (USA) |
86 pts Homografts Vs 139 pts Xenograft prostheses Vs 79 pts Mechanical prostheses |
To evaluate resistance to infection | Homografts were more used in PVE (P = .002) and methicillin-resistant Staphylococcus (P = .002), compared with conventional prostheses. No significant benefit to use of homografts was demonstrable with regard to resistance to reinfection in the setting of IE. |
| Nappi et al (2018) JTCVS [102] |
Human Single center (France) |
210 pts | To evaluate long-term results of aortic allografts and to identify factors influencing long-term durability. | The use of allograft is a valid option in complex infective endocarditis and in women of childbearing age |
| Steffen et al (2016) JTCVS. [113] |
In vitro Single center (Germany) |
10 cryopreserved human allografts | To evaluate the in vitro antimicrobial activity of 3 antibiotic regimens | Allograft antibacterial activity despite long-term storage over 5 years. Antibiotic combinations applied during CHA processing have a significant influence on their infection resistance. Ascending aortic tissue shows a significantly enhanced bacterial resistance against staphylococcal bacteria compared with aortic valves. |
Abbreviations ; IEV, infective endocarditis vegetation ; pts, patients ; PVE, prosthetic valve endocarditis ; S. aureus, staphylococcus aureus ; S. mitis, Streptococcus mitis
3.6. Interaction of Staphylococcus aureus with coagulation mechanisms.
Staphylococcus aureus infections have been extensively studied with the use of different animal models, specially adapted to invasive infections of this pathogen, suggesting the fundamental role of two coagulases, von Willebrand factor binding protein (vWbp) and coagulase (Coa), which lead to critical virulence. These molecules form a functionally intricate architecture that S. aureus uses to generate a protective fibrinogen/fibrin shield that surrounds it. The emergence of this armor yields the pathogen the potential to circumvent the main defense immunity system implemented by the host's phagocytic cells. One of the pivotal functions of coagulases promotes the non-proteolytic activation of the zymogen pro-thrombin to transform fibrinogen into fibrin, thus contributing to the emergence of the fibrinogen/fibrin safeguarding shield.
There are many essential functions of coagulases. One of these influences the non-proteolytic activation of the prothrombin zymogen to con-vert fibrinogen to fibrin, thus leading to the genesis of the protective fibrinogen/fibrin shield. Another function promoted by coagulases is to serve as a linkage with fibrinogen, whose interactions greatly sustain infection. The mechanism or mechanisms that enable the binding between vWbp and Coa and fibrinogen entail well-defined interactions of the two proteins with the molecule, although they show a similar structure. Coa binding to soluble fibrinogen has a significantly higher affinity than fibrinogen coated on a plastic surface. The vWbp, on the other hand, did not show any preference between the two forms of fibrinogen [10,11,12,13,14]. Figure 4
Figure 4.
Virulent factors of S. aureus are reported. MSCRAMMs drive with a substantial key role in the initiation of endovascular, bone, and joint, alongside prosthetic-device infections. These structures can bind to molecules such as collagen (mostly via Cna), fibronectin (via FnbAB), and fibrinogen (with ClfAB and Fib) and thus evade the immune system. The development of infective process is induced by Coa and von Willebrand factor binding protein that led to critical virulence. Coa binds preferentially soluble fibrinogen while vWbp did not disclose any preference across the two forms of fibrinogen. Abbreviations; Coa, coagulase; MSCRAMM, microbial surface components recognizing adhesive matrix molecules; vWbp, von Willebrand factor binding protein.
Figure 4.
Virulent factors of S. aureus are reported. MSCRAMMs drive with a substantial key role in the initiation of endovascular, bone, and joint, alongside prosthetic-device infections. These structures can bind to molecules such as collagen (mostly via Cna), fibronectin (via FnbAB), and fibrinogen (with ClfAB and Fib) and thus evade the immune system. The development of infective process is induced by Coa and von Willebrand factor binding protein that led to critical virulence. Coa binds preferentially soluble fibrinogen while vWbp did not disclose any preference across the two forms of fibrinogen. Abbreviations; Coa, coagulase; MSCRAMM, microbial surface components recognizing adhesive matrix molecules; vWbp, von Willebrand factor binding protein.
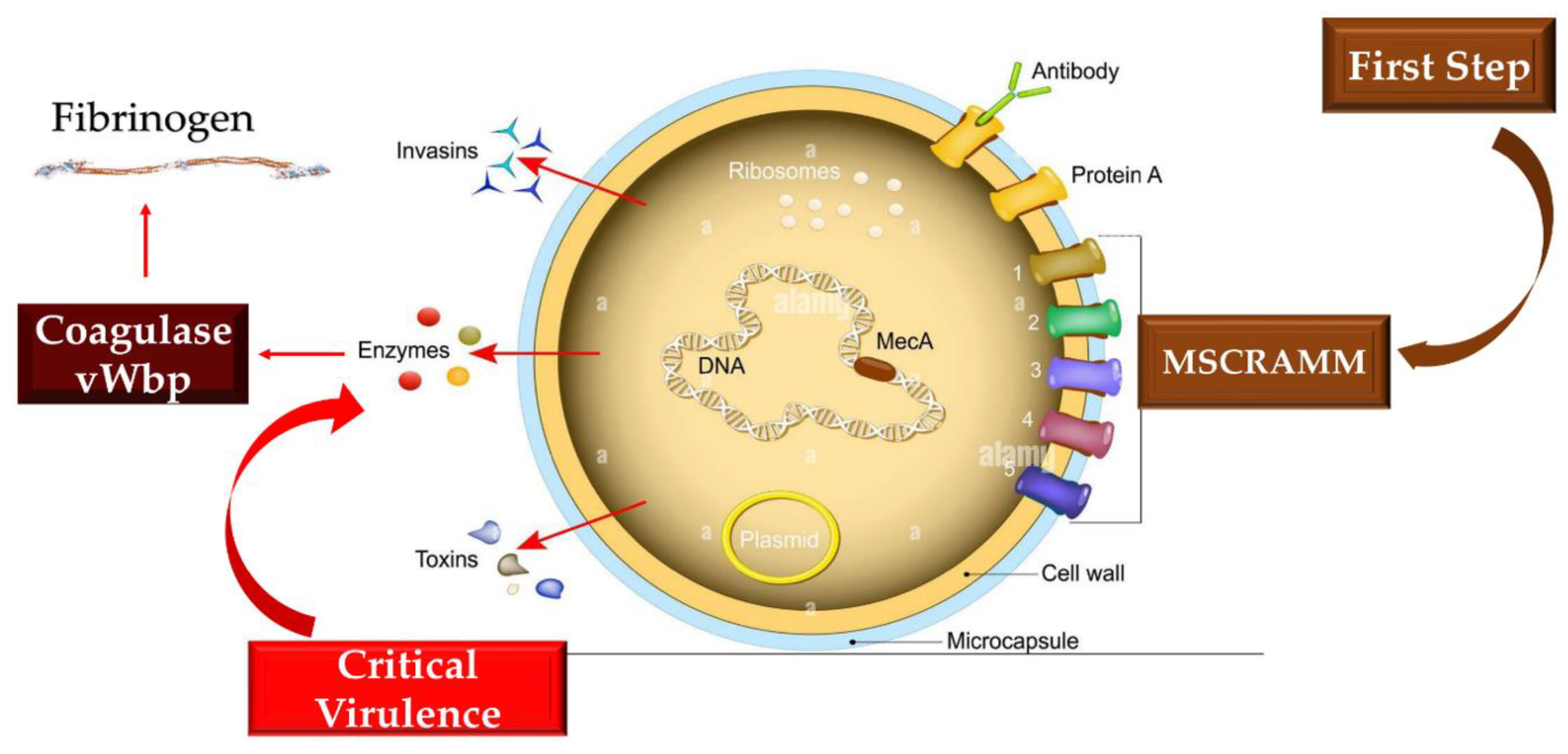
Thomas and colleagues investigated the complex interactions between fibrinogen and S. aureus suggesting a different action exerted by vWbp and Coa targeting different sites on fibrinogen, demonstrating an absence of conflict among the two molecules in fibrinogen binding. Both Coa and vWbp have N- and C-terminal halves that drive fibrinogen binding activity [13,14]. These vWbp coagulases have higher fibrinogen binding affinity in the vWbp-N region, in divergence to Coa in which the major bias towards the fibrinogen binding site has been related to the C-terminal region. It has been observed that the peptides constituting the formerly recognized Fibrinogen Coa/Efb1 binding motif do not impede the vWbp-C constituent from attaching to fibrinogen. Therefore, non-attendance of a functional homolog to this motif has been suggested for vWbp-C. It was also observed that although the N-terminal prothrombin-binding domains of both coagulases recognize the β-chain of fibrinogen, they nevertheless seem to interrelate with several sequence motifs in the host protein. It is therefore possible to speculate that the interplay of the two coagulases seems to be exhibited with divergent sequence motifs in the host protein. The findings reported by Thomas et al give new awareness to the intricate interlinkage among Fg and S. aureus coagulases [14].
Multidrug-resistant S aureus strains are accountable for life-threatening diseases deploying a worldwide public health concern. The restrictions for dealing with S aureus infection rely on both the treatment and the absence of a fruitful vaccine. As formerly indicated S aureus develops complex and errorless mechanisms that preserve it through the protection of a shield by fibrinogen/fibrin. This coating serves two objectives: 1) it permits the bacterium to survive in the blood rendering it invisible to the host's immune protection and 2) it provides the likelihood of spreading and giving rise to invasive diseases. Modifying this process depicts an encouraging aim for new antistaphylococcal treatment strategies, however, the mechanisms that adjust the phenomena are not yet entirely inquired. S. aureus expresses many proteins that tie to fibrinogen. A redundant action exerted by some of these molecules with vWbp can limit its function. Sharing between proteins expressing similar functions in the structural or functional motif has often been suggested.
Thomas and colleagues [14] argued the expression of a protein homolog vhp corresponding to the C-terminus of the von Willebrand factor binding protein (vWbp) contributes to shield assembly and fibrinogen binding. They recognized a common Fg binding motif between vhp and vWbp.
Recently Schwartz and colleagues [63] illustrated the potential pathomechanisms using both in vitro and in vivo models of 34 isolates of Staphylococcus aureus which were evaluated by gathering causative pathogens from patients with S. aureus endocarditis and healthy subjects. The strains of S. aureus isolated were assessed in vitro to analyze cytotoxicity and the invasion and interrelation with platelets typically revealed by these bacteria. In order to correlate the faculty of S. aureus to advocate the development of vegetations on the aortic valves in vivo, the virulence factor expression profiles and cellular response were also assessed using an animal model. The existence of IE involving valves was evaluated in vivo with the use of magnetic resonance imaging at 9.4 T. Histological assessment with enrichment gene expression analysis was also fulfilled. S. aureus isolated and investigated in vivo revealed the potential of causing IE by reliably inducing inflammatory responses associated with the aortic valve’s injuries. However, the differentiation and classification of IE as well as the characterization of inflammation based on the measurement of in vitro virulence profiles and cytotoxicity were not established [63].
Schwartz and colleagues [63] observed that in vitro test results did not correlate with IE severity. However, the researchers noted that the Staphylococcus isolates differed considerably in the degree of activation and inhibition of pathoanatomical processes related to the extracellular matrix and in the features of the inflammatory reaction. It was therefore suggested that the pathogenic ability of pathogen did not bestow a constant response and that more comprehensive approaches to host-pathogen interactions were required for its assessment. Furthermore, this approach promoted new insights into the corresponding immune pathways to highlight differences in host/pathogen interaction [63].
With regards to the etiology of S. aureus-promoted infective endocarditis, Schwarz and colleagues [63] permitted better comprehension on the interaction between virulence factors and immune response in S. aureus-borne infective endocarditis, thus promoting the spread of innovative therapeutic strategies and specific diagnostic imaging markers.
3.7. Involvement of vascular endothelium and blood constituents in S. aureus induced endocarditis.
Staphylococcus aureus surface molecules operate crucially to favor colonization of the vascular endothelium which is a pivotal primary event in the pathogenesis of IE. The faculty of these molecules to elicit associated endothelial procoagulant and proinflammatory responses, promoting the progress of infective endocarditis, has been well stated [73,114,115,116,117]. Heying and colleagues [114] assessed the peculiar role of three fundamental molecules expressed on the surface of S aureus. Fibronectin-binding protein A (FnBPA) and B (FnBPB) as well as clumping factor A (ClfA) act to advocate bacterial adherence that identifies the cultured human endothelial cells (ECs) inter-acting with S. aureus. Likewise, these molecules encourage phenotypic and functional modifications in ECs. The investigators used a non-invasive surrogate bacterium Lactococcus lactis. Lactococcus lactis, by gene transfer, ex-pressed staphylococcal molecules FnBPA, FnBPB, or ClfA. In this way, the recombinant Lactococci positive for FnBPA or FnBPB revealed an increase in the incidence of infection at the EC level by up to 50-100 times the base-line threshold. Other evidence highlighted provocation of an inflammatory response with activation of the EC characterized by an increased ex-pression of ICAM-1 and VCAM-1 on the surface, and the production of interleukin-8 associated with the concomitant adhesion of monocytes. On the contrary, infection determined by ClfA-positive lactococci did not activate EC. The leading action of FnBPA-positive L. lactis promoted a notable inflammatory response that was enhanced by cell-bound monocytes and mediated by tissue factor-dependent endothelial coagulation. Evidence suggested that S. aureus FnBPs, but not ClfA, promoted the invasiveness and pathogenicity of nonpathogenic L. lactis microorganisms, pointing out that bacterium-EC interactions mediated by these adhesins were strongly inclined to promote coagulation and inflammation in infected endovascular sites [114].
Studies carried out in experimental endocarditis induced by Staphylococcus aureus have highlighted two important phases of the infection. The purpose of sequential fibrinogen binding in charge of valve colonization and the pivotal role of fibronectin-binding promoting endothelial invasion was demonstrated. These biological phenomena were supported by peptidoglycan-linked adhesins. The function played by fibronectin-binding protein A (FnBPA) promoted a combination of these two determined properties, merged with the binding of elastin, in favoring experimental endocarditis.
One study reported the substantial role played by the minimal sub-domain of FnBPA accountable for fibrinogen and fibronectin binding in promoting cell invasion in endocarditis in vivo. FnBPA was expressed in Lactococcus lactis and was assessed in vitro and in animal models [115]. The subdomain needed to induce IE comprised of 127 amino acids that depicted the hub of the fibrinogen- and fibronectin-binding regions of FnBPA and were adequate to bestow the charge to these assets. Although in animals evidence noted the crucial role of fibrinogen binding to deter-mine endocarditis induction; however, the role exerted by fibronectin binding was not significantly coupled with endocarditis development. In-stead, as for disease acuteness, both fibrinogen binding and fibronectin binding were of substantial importance. Besides, the synergistic merger of fibrinogen binding and fibronectin binding suggested a considerable enhancement in the infectious foray of cultured cell lines, emphasizing a decisive feature to be linked with the severity of endocarditis. Accordingly, the concept based on sequential action offered by fibrinogen binding and fibronectin binding in fostering colonization and invasion could be used for the development of anti-adhesin strategies [115]. Figure 5
Figure 5.
Depict the mechanism of experimental endocarditis caused by S. aureus. The process is marked the crucial role of sequential fibrinogen binding responsible for valve colonization and the critical action of fibronectin-binding that promotes endothelial invasion. FnBPA responsible for fibrinogen and fibronectin binding may advocate cell invasion in vivo endocarditis. Abbreviations; Akt or PKB, protein kinase B, FAK, focal adhesion kinase; P13-k, Phosphoinositide 3-kinase; Src, proto-oncogene tyrosine-protein kinase.
Figure 5.
Depict the mechanism of experimental endocarditis caused by S. aureus. The process is marked the crucial role of sequential fibrinogen binding responsible for valve colonization and the critical action of fibronectin-binding that promotes endothelial invasion. FnBPA responsible for fibrinogen and fibronectin binding may advocate cell invasion in vivo endocarditis. Abbreviations; Akt or PKB, protein kinase B, FAK, focal adhesion kinase; P13-k, Phosphoinositide 3-kinase; Src, proto-oncogene tyrosine-protein kinase.
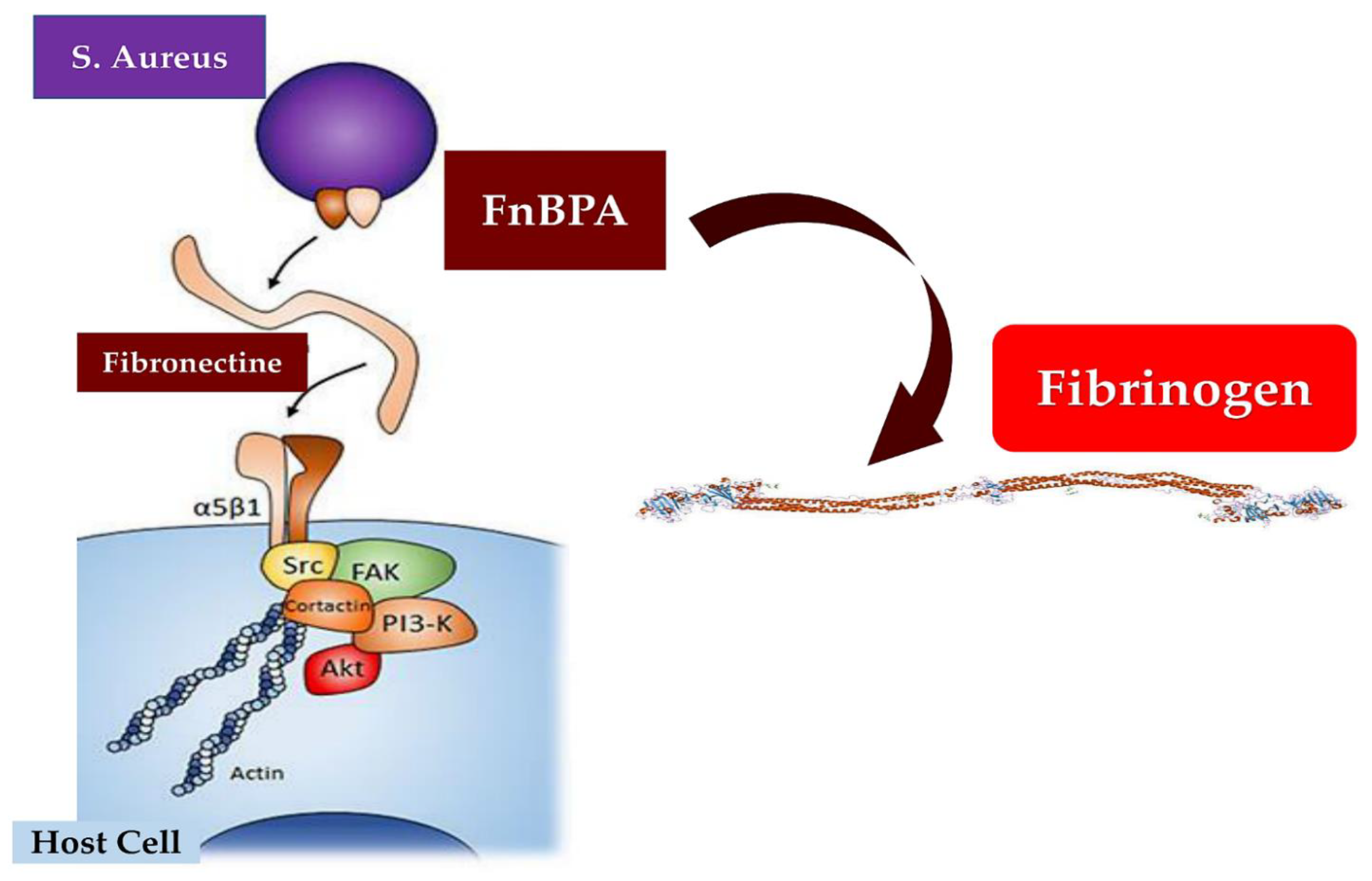
Bacterial proteins such ClfA and FnBPA intercede for the adhesion of S aureus to EC surface molecules. This purpose is shared with subendothelial matrix proteins involving fibrinogen, fibrin, fibronectin, and von Willebrand factor (vWF) [116]. It is important to underline the work of Pappelbaum et al [117] who suggested ultra-large von Willebrand factor (ULVWF) substantially concurred with the inceptive pathogenic step of S aureus-induced endocarditis in subjects with healthy untouched endothelium. The synergistic role of ClfA, FnBPA, and von Willebrand factor (vWF) in determining the adhesion of Staphylococcus aureus to endothelial cells (ECs) has been investigated in three recent reports that markedly endorse the fundamental importance of these molecules in IE [118,119,120]. Evidence pointed out that ultra-large von Willebrand factor (ULVWF) substantially promoted the initial pathogenic phase of S aureus-induced endocarditis in patients with undamaged endothelium. The use of heparin and ADAMTS13 reduced ULVWF formation and may serve as a novel therapeutic choice to avoid IE [117].
Recently Claes et colleagues [118] revealed the interaction between vWbp and proteins expressed on the surface of S aureus that moderated the bacterium adhesion to VWF and to vascular endothelium under shear stress. Mutants deficient in Sortase A (SrtA) and SrtA-dependent surface proteins, as well as Lactococcus lactis transmitting single staphylococcal surface proteins have been used. In detail, S. aureus firstly tied to the endothelium via vWF, raising levels of VWF-binding protein (vWbp) that finalized the adhesion of S. aureus to VWF under shear stress and lastly, vWbp interconnected with vWF and the Sortase, a ClfA dependent surface protein. So, it is possible to affirm that vWF-vWbp-ClfA anchored S. aureus to the vascular endothelium under shear stress [92]. In another report, the same investigators studied the effect of shear flow and plasma on the binding of ClfA and FnBPA, comprising its sub-domains A, A16+, ABC, CD, vWF, fibrinogen/fibrin, fibronectin or confluent ECs. With the use of a genetically engineered Lactococcus lactis that exhibited these adhesins heterologously, Claes et al [119] found that comprehensive adherence profiles were almost alike in static and flow conditions. The level of adhesion of L. lactis- FnBPA to EC-bound fibronectin and of L. lactis- ClfA to EC-bound fibrinogen was like to that of L. lactis- ClfA to coated vWF domain A1, in the presence of vWF-binding protein (vWbp). Thus, in plasma, the adhesion of L. lactis-ClfA to activated EC-vWF/vWbp was reduced by 80% within the time limit of 10 min and this event was associated with the paramount role of disintegrin-mediated and metalloproteinase-mediated vWF hydrolysis with thrombospondin motif type 1, member 13. Equally, in lacking plasma components the adhesion of L. lactis, FnBPA was decreased by > 70%. Instead, plasma fibrinogen noted high binding affinity of L. lactis-ClfA to resting and activated ECs. These findings suggested that in plasma S aureus adhesion to active endothelium was dependent mostly on two supportive pathways: a rapid but short-lived vWF/vWbp pathway and a stable integrin-coupled - fibrinogen pathway. Observations derived from these findings suggest that pharmacological inhibition of ClfA-fibrinogen interactions may play a role in the adjunctive treatment of infective endocarditis. [119]
Staphylococcus aureus actively invades the endothelium promoting detrimental action that causes apoptosis and endothelial damage. The literature supports the knowledge of the crucial role of Staphylococcus in causing IE by means of the pivotal function mediated by protein clumping factor A (ClfA), which interconnects to the cell wall of S. aureus. Several reports have recently elucidated mechanisms of secreted plasma coagulation factors staphylocagulase (Coa) and protein binding von Willebrand factor (vWbp). Mancini et colleagues [49] assessed rat models with catheter-induced aortic vegetations. They studied the function of staphylococcal secreted coagulase (Coa-positive staphylococci) and Staphylococcus aureus encoding von Willebrand factor binding protein (vWbp) in the development of IE. As previously reported, a model based on Lactococcus lactis mutants expressing coa, vWbp, ClfA or vWbp / clfA and S. aureus Newman Δcoa, ΔvWbp, ΔclfA or Δcoa / ΔvWbp / ΔclfA was used. The investigators noted that vWbp expression statistically raised L. lactis-induced valve infection in contrast to strains expressing coa. Likewise, ClfA expression revealed an increase in the infectiousness produced by L. lactis, which was not further affected by vWbp co-expression. Of note the finding that effacement of the Coa or vWbp genes in S aureus did not reduce infectivity whilst annulment of ClfA function dramatically diminished valve infection. A decisive observation advocated that the function of clfA was not influenced by the triple deletion of Δcoa / ΔvWbp / ΔclfA. This result allowed hypothesizing that Coa did not promote colonization of inceptive IE using L. lactis as a pathogen in the absence of other key virulence factors. The presence of vWbp concurred with the onset of IE induced by L. lactis, but its role was borderline in the attendance of ClfA [49].
Although evidence has shown the pathogenic extracellular role of Staphylococcus aureus, , this causative pathogen also can be integrated by host cells, including nonprofessional phagocytes. Therefore, it can be a deterrent inside endothelial cells, epithelial cells, or osteoblasts. The intracellular S aureus location concurs with the establishment of infection. The entry gate of the bacterium is umpired by the binding of integrin α5β1 expressed on the membrane of the host cell which recognizes fibronectin. This bridge encourages the recognition between pathogen and host cell promoting subsequent cell integration. [121,122,123,124]. Although the osteoblasts revealed tall expression of α5β1-integrin and fibronectin with demonstrable adherence to osteoblasts, Niemann et colleagues [125] suggested, using internalization tests and immunofluorescence microscopy, that S aureus was less engulfed by osteoblasts compared to epithelial cells. The authors noted that throughout cell infection, adding exogenous fibronectin in the presence of S aureus increased uptake of the pathogen in epithelial cells that was not disclosed in osteoblasts. This evidence offered understandable contrast to prior claims concerning the pathogen uptake mechanism, which yielded integrin and fibronectin expression, a pivotal action in causing bacterial uptake in host cells. Importantly, the arrangement of extracellular fibronectin surrounding osteoblasts and epithelial cells was dissimilar, revealing a typically structured frame of a fibrillar network in the former. The uptake enhancement of S aureus was significant, arising from the inhibition of fibril production, brief lowering of RNA-mediated fibronectin expression, and disruption of the fibronectin-fibril network. The study of Nieman et colleagues [125] demonstrated that the fibronectin fibril network reduced the uptake of S aureus into a given host cell, suggesting that the supramolecular structure of fibronectin may govern the dissimilar ability of peculiar host cells to internalize the bacterium. The evidence reported by Niemann et al [125] advocated the non-determining function deployed by the crude amount of fibronectin but rather the unfavorable function denoted by the supramolecular structure of the fibronectin molecules.
Once stored on the eukaryotic cell surface, they exert a fundamental action in bacterial uptake by host cells. This evidence may describe the remarkable inconsistency expressed in the efficiency of S aureus uptake by various host cell types. In addition, in vivo discrepancies between bacterial infection courses and bacterial localization have been demonstrated in different clinical settings [125].
From a molecular point of view, the pathogenicity of S aureus is linked to the expression of virulence factors, comprising of proteins that moderate the process of adhesion to host plasma molecules and extracellular matrix proteins. Among these, numerous shreds of findings have demonstrated a marked ability of IsdB-expressing bacteria to tie to both soluble and immobilized vWF [126]. A recent study by Alfeo et al [127] highlighted that iron-regulated surface determinant B (IsdB) protein, besides iron transport and vitronectin binding, interacted with von Willebrand Factor (vWF). The binding between IsdN and recombinant vWF was disrupted by heparin and was also reduced due to the high ionic strength. Thus, the use of administered ristocetin, an allosteric agent that induced the exhibition of the A1 domain of vWF, elicited the considerable effect of rising the binding between IsdB and vWF. It was permissible to speculate that IsdB-binding and S. aureus adhesion were markedly impeded by a monoclonal antibody averse to the A1 domain as well as IsdB reactive IgG isolated from subjects with staphylococcal endocarditis. This evidence suggested two obvious conclusions: the importance of IsdB in promoting S. aureus adhesion and its role in colonization of the endothelium. Again, the potential role of IsdB as a therapeutic target could be offered [127].
3.8. Infective Endocarditis and Platelets
In patients at an increased risk of infective endocarditis, the use of antibiotic prophylaxis is currently recommended, given the difficulty in treating IE and its inherent mortality. It should be underlined that the concerns correlated with the administration of antibiotics are faced with their unquestionable low efficacy for certain strains of Staphylococcus aureus alongside the perpetuation of increasing multidrug-resistant strains of infection. Given this worrying clinical scenario, the need to discover new therapeutic options remains a priority against IE. The role played by platelets is decisive in the early phase of infective endocarditis, making them first-line immune responders. [75,76,128]
Important results have been observed in mechanistic in vitro studies which have highlighted the early action the infection supported by platelets during the first phase of the S. aureus infection involving cardiac structures. The first front of the platelet-dependent immune response can be configured in directing an initial antimicrobial contrast action mediated by the interaction of platelets with the pathogen. This is the proposed case for the therapeutic use of acetylsalicylic acid.
Several experimental and clinical reports have suggested that the purpose of aspirin may restrict bacterial-platelet interactions promoting the prevention of vegetation spread and have revealed hopeful results. However, the data from clinical trials reporting outcomes in patients with IE who received additional aspirin to background therapy has not produced conclusive results. Therefore, conflicting evidence emerged shedding a veil of uncertainty about the advantage of antiplatelet drugs in the prevention of IE sustained by S. aureus. In addition to aspirin, other drugs with anti-platelet action have also been tested for which a therapeutic effect has been observed. For example, the P2Y12 platelet receptor antagonist ticagrelor could couple its potent and well-known antiplatelet role with noticeable antibacterial properties. Furthermore, a recent study based on a mouse animal model reported a pronounced capacity of ticagrelor to eradicate S. aureus bacteremia [129,130,131]. Table 3
Table 3.
Characteristics of the Included Studies.
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Que et al (2005) J Exp Med [73] |
Animal model Single Center (Switzerland) |
Rat model of IE induced | To study valve colonization with experimental endocarditis. To evaluate the role of ClfA and FnBPA positive lactococci | Fibrinogen and fibronectin binding could cooperate for S. aureus valve colonization and endothelial invasion in vivo |
| Edwards et al (2012) PLoS One [74] |
Human Single Center (UK) |
Blood sample | To study in vivo role of Eap to interact with host glyco-proteins | Eap expressing strains cause a more severe infection, demonstrating its role in invasive disease. Increased level of TNFα and gC1qR/p33 expression |
| Veloso et al (2013) Infect Immun [76] |
Animal model Single Center (Switzerland) |
Rat model of induced IE 10(6) CFU L. lactis pIL253 Vs Recombinant L. lactis (ClfA, FnbpA, BCD, or SdrE) |
To explore the contributions of S. aureus virulence factors to the initiation of IE. | Fibrinogen binding in the initiation of S. aureus IE. Activation of platelet aggregation or an inflammatory response may contribute to or promote the development of EI |
| Thomas et al (2021) mBio [77] |
Animal model Single Center (USA) |
Rat model of IE induced | To identify proteins with significant amino acid identities to vWbp | Protein homologous to the C-terminal of vWbp was identified. Its role in Fg shield assembly and binds. |
| Hussain et al (2002) Infect Immun [78] |
In vitro Single center (Germany) |
S. aureus Newman cultures Vs Control mutant |
To investigate the role of Eap by constructing a stable eap::ermB deletion | Eap may contribute to pathogenicity by promoting adhesion of whole staphylococcal cells to complex eukaryotic substrates |
| Palankar et al (2018) Int J Med Microbiol. [79] |
In vitro Single center (Germany |
S. aureus Mu50 | To investigate Eap subdomain and interaction with platelet | Eap subdomain Eap D3D4 specifically interacts and rapidly activates human platelets |
| Hussain et al (2008) Infect Immun [80] |
In vitro Single center (Germany |
S. aureus Newman cultures Vs S. aureus Wood 46 |
To investigate the interactions of full-length Eap and five recombinant tandem repeat domains with host proteins. | More than one Eap tandem repeat domain is required for S. aureus agglutination, adherence, and cellular invasion but not for the stimulation of PBMC proliferation. |
| Heying et al (2007) Thromb Haemost. [114] |
Human Single Center (Germany) |
S. aureus L. lactis culture cultured human EC |
To investigate the role of FnBPA, FnBPB ClfA to promote bacterial adherence to cultured human ECs. | S. aureus FnBPs, but not ClfA, lead pathogenicity to non-pathogenic L. lactis. Adhesins (ICAM-1 and VCAM-1) evokes inflammation (interleukin-8) as well as procoagulant activity. |
| Piroth et al (2008) Infect Immun [115] |
Animal model Single Center (Switzerland) |
S. aureus L. lactis culture In vitro and in vivo |
To study subdomain of FnBPA responsible for fibrinogen and fibronectin binding, cell invasion, and in vivo endocarditis | Fb binding combined with fibronectin binding to synergize the invasion of cultured cell lines is correlate with IE severity |
| Pappelbaum et al (2013) Circulation. [117] |
Human/Animal Single center (Germany) |
6 WT mice with VWF vs 5 knockout mice vs Cultured human EC |
Whether ULVWF mediates bacterial adherence. | ULVWF contributes to the initial pathogenic step of S aureus-induced endocarditis in patients with an apparently intact endothelium. Heparin or ADAMTS13 intervenes in decreasing ULVWF adherence |
| Claes et al (2018) Thromb Haemost [119] |
Human/Animal Multicenter (Belgium pilot) |
L. lactis-clfA Vs L. lactis-fnbpA Vs Cultured human EC |
To study the influence of shear flow and plasma on the binding of ClfA and FnbpA | Pharmacological inhibition of ClfA-Fg interactions may constitute a valuable additive treatment in infective endocarditis. |
| Ko et al (2016) mBio [120] |
Animal model Single Center (USA) |
Rat model of IE induced | To identify variants of a linear Fg binding motif, present in Coa and Efb which are responsible for the Fg binding activities of these proteins | S. aureus coagulase can induce the formation of a fibrinogen shield in experimental abscess models which surrounds and protects bacteria in the microcolony from clearance. |
| Niemann et al (2021) mBio. [125] |
Animal Multicenter (Germany) |
Rat model of IE induced in osteoblasts vs epithelial cells |
To demonstrate that S. aureus was less engulfed in osteoblasts than in epithelial cells. | Large differences of S. aureus uptake efficacy in different host cell types. In vivo differences between courses of bacterial infections and the localization of bacteria in different clinical settings mediated by α5β1-integrin |
| Pietrocola et al (2020) J Biol Chem. [126] |
Animal Multicenter center (Italy pilot) |
Rat model of IE induced | To evaluate a variety of virulence factors that promote infection by S. aureus | Adherence to and invasion of epithelial and ECs by IsdB-expressing S. aureus cells was promoted by Vn, and an αvβ3 integrin-blocking mAb |
| Alfeo et al (2021) Sci Rep [127] |
Animal Multicenter center (Italy pilot |
Rat model of IE induced | To study IsdB protein and Vn binding Interacts with vWF. | Importance of IsdB in adherence of S. aureus to the endothelium colonization and as potential therapeutic target. |
| Ditkowski et al (2021) J Thorac Cardiovasc Surg [129] |
Human Multicenter (Belgium pilot) |
5 graft tissues | To investigate contributions by platelets and plasma fibrinogen to IE initiation on various grafts used for valve replacement | Binding of plasma Fg to especially BJV grafts enables adhesion of single platelets via αIIbβ3. S aureus attaches from blood to activated bound platelet αIIbβ3 via plasma fibrinogen. |
Abbreviations; ADAMTS13, a disintegrin and metalloproteinase with thrombospondin motifs 13; BJV, bovine jugular vein ; ClfA, clumping factor A; Eap, S. aureus extracellular adhesion protein ; EC, endothelial; cell; Fc, fibrinogen; FnBPA, fibronectin-binding protein A; IsdB, iron-regulated surface determinant B protein; IL, interleukine; L. lactis, Lactococcus lactis; SdrE; mAb, monoclonal antibody; PBMC, peripheral blood mononuclear cells; TNF, tumor necrosis factor; ULVWF, ultra large von Willebrand factor; Vn, extracellular matrix protein vitronectin ; vWbp, von Willebrand factor-binding protein ; vhp, vWbp homologous protein
4. Conclusion
Several factors allow Staphylococcus aureus infections to proliferate within the host with numerous promoting and perpetuating agents with direct bacterial pathogenic activity predominating other factors such as rheumatic heart disease. This is further supported by the roles of teichoic and lipoteichoic acids within teichoic acid which favour host cell invasion. The complex interaction with the hosts' innate immunity also potentiates its virulence. The role of vaccines has not been successfully translated to the clinical setting thus far. Ameliorating these molecular pathways may serve as a therapeutic avenue for the prevention and treatment of these infections in the near future with antiplatelet agents showing promising results.
Supplementary Materials
(The following supporting information can be downloaded at the website of this paper posted on Preprints.org.). Supplementary Table S1: Prisma checklist 2020, reference [From: Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372: n71. doi: 10.1136/bmj. n71 http://www.prisma-statement.org/] is cited in supplementary materials
Author Contributions
Conceptualization, F.N.; methodology, F. N; software, F.N; formal analysis, F.N.; investigation, F.N.; data curation, F.N.; writing—original draft preparation, F.N.; writing—review and editing, F.N.; visualization, F.N..; supervision, F.N.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations in table and figures
References
- Selton-Suty, C.; Célard, M.; Le Moing, V.; Doco-Lecompte, T.; Chirouze, C.; Iung, B.; Strady, C.; Revest, M.; Vandenesch, F.; Bouvet, A.; et al. Preeminence of Staphylococcus aureus in Infective Endocarditis: A 1-Year Population-Based Survey. Clin. Infect. Dis. 2012, 54, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhan, Y.; Zhang, K.; Gao, Y.; Chen, L.; Zhan, J.; Chen, Z.; Zeng, Z. The Global, Regional, and National Burden and Trends of Infective Endocarditis From 1990 to 2019: Results From the Global Burden of Disease Study 2019. Front. Med. 2022, 9, 774224. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.; Fortes, C.Q.; Nascimento, E.M.D.; Sousa, C.; Fortes, N.R.Q.; Thomaz, D.C.; Pereira, B.d.B.; Pinto, F.J.; de Oliveira, G.M.M. In-hospital Outcomes of Infective Endocarditis from 1978 to 2015: Analysis Through Machine-Learning Techniques. CJC Open 2021, 4, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Allegranzi, B.; Nejad, S.B.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Nejad, S.B.; Allegranzi, B.; Syed, S.; Ellis, B.; Pittet, D. Health-care-associated infection in Africa: a systematic review. 2011, 89, 757–765. [CrossRef]
- Joubert, D.; Cullati, S.; Briot, P.; Righi, L.; Grauser, D.; Ourahmoune, A.; Chopard, P. How to improve hospital admission screening for patients at risk of multidrug-resistant organism carriage: a before-and-after interventional study and cost-effectiveness analysis. BMJ Open Qual. 2022, 11, e001699. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Martínez JL Antibiotics and antibiotic resistance genes in natural environments. Science. 2008 Jul 18 ;321(5887) :365-7.
- Yang M, Zhang J, Wei Y, Zhang J, Tao C Recent advances in metal-organic framework-based materials for anti-staphylococcus aureus infection. Nano Res. 1: 2022, 11 May 2022.
- McAdow, M.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus Secretes Coagulase and von Willebrand Factor Binding Protein to Modify the Coagulation Cascade and Establish Host Infections. J. Innate Immun. 2012, 4, 141–148. [Google Scholar] [CrossRef]
- Thomer, L.; Schneewind, O.; Missiakas, D. Multiple Ligands of von Willebrand Factor-binding Protein (vWbp) Promote Staphylococcus aureus Clot Formation in Human Plasma. J. Biol. Chem. 2013, 288, 28283–28292. [Google Scholar] [CrossRef]
- Nappi, F.; Martuscelli, G.; Bellomo, F.; Singh, S.S.A.; Moon, M.R. Infective Endocarditis in High-Income Countries. Metabolites 2022, 12, 682. [Google Scholar] [CrossRef]
- Thomas, S.; Liu, W.; Arora, S.; Ganesh, V.; Ko, Y.-P.; Höök, M. The Complex Fibrinogen Interactions of the Staphylococcus aureus Coagulases. Front. Cell. Infect. Microbiol. 2019, 9, 106. [Google Scholar] [CrossRef]
- Herrmann, M.; Sinha, B. Mechanism and consequences of invasion of endothelial cells by Staphylococcus aureus. Arthritis Res. Ther. 2005, 94, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Thuny, F.; DiSalvo, G.; Belliard, O. Risk of Embolism and Death in Infective Endocarditis: Prognostic Value of Echocardiography. A Prospective Multicenter Study. ACC Curr. J. Rev. 2005, 14, 31. [Google Scholar] [CrossRef]
- Di Salvo G, Habib G, Pergola V, et al. Echocardiography predicts embolic events in infective endocarditis. J Am Coll Cardiol 2001 ;37 :1069-76.
- Vilacosta I, Graupner C, San Román JA, et al. Risk of embolization after institution of antibiotic therapy for infective endocarditis. J Am Coll Cardiol. 2002 ;39(9):1489-95. 1 May.
- Nappi, F.; Spadaccio, C.; Dreyfus, J.; Attias, D.; Acar, C.; Bando, K. Mitral endocarditis: A new management framework. J. Thorac. Cardiovasc. Surg. 2018, 156, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.A.; Costantino, M.F.; D’addeo, G.; Cardinale, D.; Fiorilli, R.; Nappi, F. A narrative review of diagnosis of infective endocarditis—imaging methods and comparison. Ann. Transl. Med. 2020, 8, 1621–1621. [Google Scholar] [CrossRef] [PubMed]
- Duval X, Iung B, Klein I, et al. Effect of early cerebral magnetic resonance imaging on clinical decisions in infective endocarditis: a prospective study. Ann Intern Med 2010 ;152 :497-504.
- Béraud G, Tubiana S, Erpelding ML, et al; AEPEI study group; COMBAT study group. Combined Bacterial Meningitis and Infective Endocarditis: When Should We Search for the Other When Either One is Diagnosed? Infect Dis Ther. 26 May 2022.
- Vitali P, Savoldi F, Segati F, Melazzini L, et al. MRI versus CT in the detection of brain lesions in patients with infective endocarditis before or after cardiac surgery. Neuroradiology. 2022 May ;64(5) :905-913.
- Corr, P.; Wright, M.; Handler, L.C. Endocarditis-related cerebral aneurysms: radiologic changes with treatment. . 1995, 16, 745–748. [Google Scholar] [PubMed]
- Champey J, Pavese P, Bouvaist H, Maillet M, Kastler A, Boussat B, Francois P; and the investigator groups. Is brain angio-MRI useful in infective endocarditis management ? Eur J Clin Microbiol Infect Dis 2016, 35, 2053–2058.
- Peters, P.J.; Harrison, T.; Lennox, J.L. A dangerous dilemma: management of infectious intracranial aneurysms complicating endocarditis. Lancet Infect. Dis. 2006, 6, 742–748. [Google Scholar] [CrossRef]
- Serrano, F.; Guédon, A.; Saint-Maurice, J.-P.; Labeyrie, M.-A.; Civelli, V.; Eliezer, M.; Houdart, E. Endovascular treatment of infectious intracranial aneurysms complicating infective endocarditis: a series of 31 patients with 55 aneurysms. Neuroradiology 2021, 64, 353–360. [Google Scholar] [CrossRef]
- Murdoch DR, Corey GR, Hoen B, et al, and the International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) Investigators. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med 2009, 169, 463–473.
- Liaqat W, Palaiodimos L, Li W, et al. Epidemiologic and clinical characteristics of infective endocarditis: a single-center retrospective study in the Bronx, New York. Infection. 25 May 2022.
- Paul G, Ochs L, Hohmann C, et al. Surgical Procedure Time and Mortality in Patients with Infective Endocarditis Caused by Staphylococcus aureus or Streptococcus Species Clin Med. 2022 Apr 30 ;11(9):2538.
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin Microbiol Rev 2014; 27: 870–926.
- López, J.; Revilla, A.; Vilacosta, I.; Villacorta, E.; González-Juanatey, C.; Gómez, I.; Rollán, M.J.; Román, J.A.S. Definition, clinical profile, microbiological spectrum, and prognostic factors of early-onset prosthetic valve endocarditis. Eur. Hear. J. 2007, 28, 760–765. [Google Scholar] [CrossRef]
- Alonso-Valle, H.; Fariñas-Álvarez, C.; García-Palomo, J.D.; Bernal, J.M.; Martín-Durán, R.; Díez, J.F.G.; Revuelta, J.M.; Fariñas, M.C. Clinical course and predictors of death in prosthetic valve endocarditis over a 20-year period. J. Thorac. Cardiovasc. Surg. 2010, 139, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Chen, X.; Tang, A.; Huang, D.; Pan, Q.; Fang, Z. Prevalence and Molecular Characterization of Methicillin-Resistant Staphylococci Recovered from Public Shared Bicycles in China. Int. J. Environ. Res. Public Heal. 2022, 19, 4492. [Google Scholar] [CrossRef] [PubMed]
- Argemi, X.; Hansmann, Y.; Prola, K.; Prévost, G. Coagulase-Negative Staphylococci Pathogenomics. Int. J. Mol. Sci. 2019, 20, 1215. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.H.; Woods, C.W.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Pappas, P.A.; Federspiel, J.; Athan, E.; Stryjewski, M.E.; Nacinovich, F.; et al. Emergence of Coagulase-Negative Staphylococci as a Cause of Native Valve Endocarditis. Clin. Infect. Dis. 2008, 46, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.H.; Miro, J.M.; Hoen, B.; Cabell, C.H.; A Pappas, P.; Jones, P.; E Stryjewski, M.; Anguera, I.; Braun, S.; Munoz, P.; et al. Coagulase-negative staphylococcal prosthetic valve endocarditis--a contemporary update based on the International Collaboration on Endocarditis: prospective cohort study. Hear. 2008, 95, 570–576. [Google Scholar] [CrossRef]
- Alawad, M.J.; Ali, G.A.; Goravey, W. Underrecognized pathogen; Staphylococcus warneri -associated native mitral valve endocarditis in an immunocompetent host: A case report and literature review. Clin. Case Rep. 2022, 10, e05591. [Google Scholar] [CrossRef]
- Voigt, A.; Shalaby, A.; Saba, S. Rising Rates of Cardiac Rhythm Management Device Infections in the United States: 1996 through 2003. J. Am. Coll. Cardiol. 2006, 48, 590–591. [Google Scholar] [CrossRef]
- Traykov, V.; Blomström-Lundqvist, C. Antibiotic-Eluting Envelopes for the Prevention of Cardiac Implantable Electronic Device Infections: Rationale, Efficacy, and Cost-Effectiveness. Front Cardiovasc Med. 2022 Mar 28 ;9 :855233.
- Elad, B.; Perl, L.; Hamdan, A.; Yahav, D.; Atamna, A.; Shaked, H.; Rubchevsky, V.; Sharony, R.; Bernstine, H.; Shapira, Y.; et al. The clinical value of the endocarditis team: insights from before and after guidelines implementation strategy. Infection 2021, 50, 57–64. [Google Scholar] [CrossRef]
- Han, H.-C.; Hawkins, N.M.; Pearman, C.M.; Birnie, D.H.; Krahn, A.D. Epidemiology of cardiac implantable electronic device infections: incidence and risk factors. Eur. 2021, 23, iv3–iv10. [Google Scholar] [CrossRef]
- Durante-Mangoni E, Bradley S, Selton-Suty C, et al, and the International Collaboration on Endocarditis Prospective Cohort Study Group. Current features of infective endocarditis in elderly patients: results of the International Collaboration on Endocarditis Prospective Cohort Study. 2: Arch Intern Med 2008; 168, 2008; 168, 2095–2103.
- Zampino, R.; Iossa, D.; Ursi, M.P.; Bertolino, L.; Karruli, A.; Molaro, R.; Esposito, G.; Vitrone, M.; D’amico, F.; Albisinni, R.; et al. Clinical Significance and Prognostic Value of Hemostasis Parameters in 337 Patients with Acute Infective Endocarditis. J. Clin. Med. 2021, 10, 5386. [Google Scholar] [CrossRef]
- Molton, J.S.; Tambyah, P.A.; Ang, B.S.P.; Ling, M.L.; Fisher, D.A. The Global Spread of Healthcare-Associated Multidrug-Resistant Bacteria: A Perspective From Asia. Clin. Infect. Dis. 2013, 56, 1310–1318. [Google Scholar] [CrossRef]
- Çaǧlayan. ; Barnes, S.L.; Pineles, L.L.; Harris, A.D.; Klein, E.Y. A Data-Driven Framework for Identifying Intensive Care Unit Admissions Colonized With Multidrug-Resistant Organisms. Front. Public Heal. 2022, 10, 853757. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008; 117: 3118–25.
- Widmer, E.; Que, Y.-A.; Entenza, J.M.; Moreillon, P. New concepts in the pathophysiology of infective endocarditis. Curr. Infect. Dis. Rep. 2006, 8, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Moreillon, P.; A Que, Y.; Bayer, A.S. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect. Dis. Clin. North Am. 2002, 16, 297–318. [Google Scholar] [CrossRef]
- Mancini, S.; Oechslin, F.; Menzi, C.; Que, Y.A.; Claes, J.; Heying, R.; Veloso, T.R.; Vanassche, T.; Missiakas, D.; Schneewind, O.; et al. Marginal role of von Willebrand factor-binding protein and coagulase in the initiation of endocarditis in rats with catheter-induced aortic vegetations. Virulence 2018, 9, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Werdan, K.; Dietz, S.; Löffler, B.; Niemann, S.; Bushnaq, H.; Silber, R.-E.; Peters, G.; Müller-Werdan, U. Mechanisms of infective endocarditis: pathogen–host interaction and risk states. Nat. Rev. Cardiol. 2013, 11, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Regueiro A, Linke A, Latib A, et al. Infective Endocarditis Following Transcatheter Aortic Valve Replacement: Comparison of Balloon- Versus Self-Expandable Valves. Circ Cardiovasc Interv. e, 2019; 12, e007938.
- Rodríguez-Vidigal, F.F.; Nogales-Asensio, J.M.; Calvo-Cano, A.; González-Fernández, R.; Martínez-Carapeto, A.; Gómez-Sanchez, I.; Bengla-Limpo, B.; Merchán-Herrera, A.; Nogales-Muñoz, N.; Vera-Tomé, A.; et al. Infective endocarditis after transcatheter aortic valve implantation: Contributions of a single-centre experience on incidence and associated factors. Enfermedades Infecc. y Microbiol. Clin. (English ed.) 2019, 37, 428–434. [Google Scholar] [CrossRef]
- Ciofu O, Moser C, Jensen PØ, Høiby N Tolerance and resistance of microbial biofilms. Nat Rev Microbiol. 2022.
- Annappah, D.; Saling, M.; Prodafikas, J.; Badie, A.N. Device-associated aortic valve endocarditis due to a complicated Enterobacter cloacae urinary tract infection. IDCases 2021, 27, e01365. [Google Scholar] [CrossRef]
- Di Carluccio, C.; Forgione, R.E.R.; Bosso, A.; Yokoyama, S.; Manabe, Y.; Pizzo, E.; Molinaro, A.; Fukase, K.; Fragai, M.; Bensing, B.A.; et al. Molecular recognition of sialoglycans by streptococcal Siglec-like adhesins: toward the shape of specific inhibitors. RSC Chem. Biol. 2021, 2, 1618–1630. [Google Scholar] [CrossRef]
- Manukumar, H.M.; Umesha, S. MALDI-TOF-MS based identification and molecular characterization of food associated methicillin-resistant Staphylococcus aureus. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Mempel, M.; Schnopp, C.; Hojka, M.; Fesq, H.; Weidinger, S.; Schaller, M.; Korting, H.; Ring, J.; Abeck, D. Invasion of human keratinocytes by Staphylococcus aureus and intracellular bacterial persistence represent haemolysin-independent virulence mechanisms that are followed by features of necrotic and apoptotic keratinocyte cell death. Br. J. Dermatol. 2002, 146, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Matsumoto, M.; Katayama, Y.; Oguma, R.; Wakabayashi, S.; Nygaard, T.; Saijo, S.; Inohara, N.; Otto, M.; Matsue, H.; et al. Staphylococcus aureus Virulent PSMα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe 2017, 22, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005 Jul ;18(3) :521-40.
- Kawa, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat. Immunol. 2010 May ;11(5) :373-84.
- Kupper, T.S.; Fuhlbrigge, R.C. Immune surveillance in the skin: mechanisms and clinical consequences. Nat. Rev. Immunol. 2004, 4, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Di Meglio, P.; Qin, J.-Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Schwarz C, Töre Y, Hoesker V, et al. Host-pathogen interactions of clinical S. aureus isolates to induce infective endocarditis. Virulence. 2021; 12, 2073–2087.
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global Changes in Staphylococcus aureus Gene Expression in Human Blood. PLOS ONE 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed]
- Alonzo 3rd, F.; Kozhaya, L.; Rawlings, S.A.; et al. CCR5 is a receptor for Staphylococcus aureus leukotoxin, E.D. Nature 2013 Jan 3 ;493(7430):51-5.
- Alonzo 3rd, F.; Torres, V.J. Bacterial survival amidst an immune onslaught: the contribution of the Staphylococcus aureus leukotoxins. PLoS Pathog. 2013 Feb ;9(2) : e1003143.
- Cheung, G.Y.; Joo, H.-S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins – critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Wardenburg, J.B. Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef]
- Silverman, G.J.; Goodyear, C.S. Confounding B-cell defences: lessons from a staphylococcal superantigen. Nat. Rev. Immunol. 2006, 6, 465–475. [Google Scholar] [CrossRef]
- Kim, H.K.; Cheng, A.G.; Kim, H.-Y.; Missiakas, D.M.; Schneewind, O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 2010, 207, 1863–1870. [Google Scholar] [CrossRef]
- Becker, S.; Frankel, M.B.; Schneewind, O.; Missiakas, D. Release of protein A from the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. 2014, 111, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.-A.; Haefliger, J.-A.; Piroth, L.; François, P.; Widmer, E.; Entenza, J.M.; Sinha, B.; Herrmann, M.; Francioli, P.; Vaudaux, P.; et al. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J. Exp. Med. 2005, 201, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.M.; Bowden, M.G.; Brown, E.L.; Laabei, M.; Massey, R.C. Staphylococcus aureus extracellular adherence protein triggers TNFα release, promoting attachment to endothelial cells via protein A. PLoS One 2012; 7: e43046.
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol 2006; 4: 445–57.
- Veloso, T.R.; Chaouch, A.; Roger, T.; Giddey, M.; Vouillamoz, J.; Majcherczyk, P.; Que, Y.-A.; Rousson, V.; Moreillon, P.; Entenza, J.M. Use of a Human-Like Low-Grade Bacteremia Model of Experimental Endocarditis To Study the Role of Staphylococcus aureus Adhesins and Platelet Aggregation in Early Endocarditis. Infect. Immun. 2013, 81, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Arora, S.; Liu, W.; Churion, K.; Wu, Y.; Höök, M. vhp Is a Fibrinogen-Binding Protein Related to vWbp in Staphylococcus aureus. Mbio 2021, 12, e0116721. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Haggar, A.; Heilmann, C.; Peters, G.; Flock, J.-I.; Herrmann, M. Insertional Inactivation of eap in Staphylococcus aureus Strain Newman Confers Reduced Staphylococcal Binding to Fibroblasts. Infect. Immun. 2002, 70, 2933–2940. [Google Scholar] [CrossRef] [PubMed]
- Palankar, R.; Binsker, U.; Haracska, B.; Wesche, J.; Greinacher, A.; Hammerschmidt, S. Interaction between the Staphylococcus aureus extracellular adherence protein Eap and its subdomains with platelets. Int. J. Med Microbiol. 2018, 308, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Hussain M, Haggar A, Peters G, et al. More than one tandem repeat domain of the extracellular adherence protein of Staphylococcus aureus is required for aggregation, adherence, and host cell invasion but not for leukocyte activation. Infect Immun. 2008 Dec ;76(12) :5615-23.
- Harraghy, N.; Hussain, M.; Haggar, A.; Chavakis, T.; Sinha, B.; Herrmann, M.; Flock, J.-I. The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap. Microbiology 2003, 149, 2701–2707. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat Rev Microbiol 2010; 8: 623–33.
- Zhang, X.; Marichannegowda, M.H.; Rakesh, K.P.; Qin, H.L. Master mechanisms of Staphylococcus aureus: consider its excellent protective mechanisms hindering vaccine development! Microbiol Res. 2018 Jul-Aug ;212-213 :59-66.
- Zhang, B.Z.; Hua, Y.H.; Yu, B.; Lau, C.C.Y.; Cai, J.P.; Zheng, S.Y.; Yam, W.C.; Kao, R.Y.T.; Sze, K.H.; Zheng, B.J.; et al. Recombinant ESAT-6-Like Proteins Provoke Protective Immune Responses against Invasive Staphylococcus aureus Disease in a Murine Model. Infect. Immun. 2015, 83, 339–345. [Google Scholar] [CrossRef]
- Brady, R.A.; Mocca, C.P.; Prabhakara, R.; Plaut, R.D.; Shirtliff, M.E.; Merkel, T.J.; Burns, D.L. Evaluation of Genetically Inactivated Alpha Toxin for Protection in Multiple Mouse Models of Staphylococcus aureus Infection. PLOS ONE 2013, 8, e63040. [Google Scholar] [CrossRef]
- Zhang, F.; Ledue, O.; Jun, M.; Goulart, C.; Malley, R.; Lu, Y.-J. Protection against Staphylococcus aureus Colonization and Infection by B- and T-Cell-Mediated Mechanisms. Mbio 2018, 9, e01949–18. [Google Scholar] [CrossRef]
- Yu, W.; Yao, D.; Yu, S.; Wang, X.; Li, X.; Wang, M.; Liu, S.; Feng, Z.; Chen, X.; Li, W.; et al. Protective humoral and CD4+ T cellular immune responses of Staphylococcus aureus vaccine MntC in a murine peritonitis model. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, F.A.; Christophersen, L.; Laulund, A.S.; Lundquist, R.; Lerche, C.; Nielsen, P.R.; Bundgaard, H.; Høiby, N.; Moser, C. Novel human in vitro vegetation simulation model for infective endocarditis. APMIS 2021, 129, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, E.G.; Rimoldi, S.G.; Cavallo, I.; D’agosto, G.; Trento, E.; Cagnoni, G.; Palazzin, A.; Pagani, C.; Romeri, F.; De Vecchi, E.; et al. Microbial biofilm correlates with an increased antibiotic tolerance and poor therapeutic outcome in infective endocarditis. BMC Microbiol. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Schwartz, F.A.; Nielsen, L.; Andersen, J.S.; Bock, M.; Christophersen, L.; Sunnerhagen, T.; Lerche, C.J.; Bay, L.; Bundgaard, H.; Høiby, N.; et al. Dynamics of a Staphylococcus aureus infective endocarditis simulation model. APMIS 2022, 130, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Singh, S.S.A.; Timofeeva, I. Learning From Controversy: Contemporary Surgical Management of Aortic Valve Endocarditis. Clin. Med. Insights: Cardiol. 2020, 14. [Google Scholar] [CrossRef]
- Nappi, F.; Singh, S.S.A.; Nappi, P.; Spadaccio, C.; Nenna, A.; Gentile, F.; Chello, M. Heart Valve Endocarditis. 2020, 37. 37.
- Nappi, F.; Singh, S.S.A.; Spadaccio, C.; Acar, C. Revisiting the guidelines and choice the ideal substitute for aortic valve endocarditis. Ann. Transl. Med. 2020, 8, 952–952. [Google Scholar] [CrossRef]
- Nappi, F.; Iervolino, A.; Singh, S.S.A. The New Challenge for Heart Endocarditis: From Conventional Prosthesis to New Devices and Platforms for the Treatment of Structural Heart Disease. BioMed Res. Int. 2021, 2021, 1–17. [Google Scholar] [CrossRef]
- Kim JB, Ejiofor JI, Yammine M, Camuso JM, Walsh CW, Ando M, et al. The Journal of Thoracic and Cardiovascular Surgery. 2016;151(5):1239-48. e2.
- Perrotta, S.; Jeppsson, A.; Fröjd, V.; Svensson, G. Surgical Treatment of Aortic Prosthetic Valve Endocarditis: A 20-Year Single-Center Experience. Ann. Thorac. Surg. 2015, 101, 1426–1432. [Google Scholar] [CrossRef]
- David, T.E.; Gavra, G.; Feindel, C.M.; Regesta, T.; Armstrong, S.; Maganti, M.D. Surgical treatment of active infective endocarditis: A continued challenge. J. Thorac. Cardiovasc. Surg. 2007, 133, 144–149. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Acar, C. Use of allogeneic tissue to treat infective valvular disease: Has everything been said? J Thorac Cardiovasc Surg. 2017;153(4):824-8.
- Brouqui, P.; Raoult, D. Endocarditis Due to Rare and Fastidious Bacteria. Clin. Microbiol. Rev. 2001, 14, 177–207. [Google Scholar] [CrossRef]
- Kim, J.B.; Ejiofor, J.I.; Yammine, M.; Ando, M.; Camuso, J.M.; Youngster, I.; Nelson, S.B.; Kim, A.Y.; Melnitchouk, S.I.; Rawn, J.D.; et al. Surgical outcomes of infective endocarditis among intravenous drug users. J. Thorac. Cardiovasc. Surg. 2016, 152, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Spadaccio, C. Simplest solutions are not always the cleverest: Can we stitch in an infected annulus? Should we rethink the current guidelines? J Thorac Cardiovasc Surg. 2017;154(6):1899-900.
- Nappi, F.; Nenna, A.; Petitti, T.; Spadaccio, C.; Gambardella, I.; Lusini, M.; Chello, M.; Acar, C. Long-term outcome of cryopreserved allograft for aortic valve replacement. J. Thorac. Cardiovasc. Surg. 2018, 156, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Arabkhani, B.; Bekkers, J.A.; Andrinopoulou, E.-R.; Roos-Hesselink, J.W.; Takkenberg, J.J.; Bogers, A.J. Allografts in aortic position: Insights from a 27-year, single-center prospective study. J. Thorac. Cardiovasc. Surg. 2016, 152, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F. CRT-721 The Cryopreserved Mitral Homograft Valve: 19 Years Experience. JACC: Cardiovasc. Interv. 2014, 7, S58. [Google Scholar] [CrossRef]
- Fukushima, S.; Tesar, P.J.; Pearse, B.; Jalali, H.; Sparks, L.; Fraser, J.F.; Pohlner, P.G. Long-term clinical outcomes after aortic valve replacement using cryopreserved aortic allograft. J. Thorac. Cardiovasc. Surg. 2014, 148, 65–72. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, M.F.; Harrocks, S.; Stafford, E.G.; A Gardner, M.; Pohlner, P.G.; Tesar, P.J.; Stephens, F. The homograft aortic valve: a 29-year, 99. 3% follow up of 1,022 valve replacements.. 2001, 10, 334. [Google Scholar]
- Olivito, S.; Lalande, S.; Nappi, F.; Hammoudi, N.; D’alessandro, C.; Fouret, P.; Acar, C. Structural deterioration of the cryopreserved mitral homograft valve. J. Thorac. Cardiovasc. Surg. 2012, 144, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Singh, S.S.A.; Lusini, M.; Nenna, A.; Gambardella, I.; Chello, M. The use of allogenic and autologous tissue to treat aortic valve endocarditis. Ann. Transl. Med. 2019, 7, 491–491. [Google Scholar] [CrossRef]
- Nappi, F.; Acar, C. Monobloc or Separate Aortic and Mitral Homografts for Endocarditis of the Intervalvular Fibrosa? Ann. Thorac. Surg. 2021, 112, 1382–1383. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Moon, M.R. A management framework for left sided endocarditis: a narrative review. Ann. Transl. Med. 2020, 8, 1627–1627. [Google Scholar] [CrossRef]
- Benedetto, U.; Spadaccio, C.; Gentile, F.; Moon, M.R.; Nappi, F. A narrative review of early surgery versus conventional treatment for infective endocarditis: do we have an answer? Ann. Transl. Med. 2020, 8, 1626–1626. [Google Scholar] [CrossRef] [PubMed]
- Pollari, F.; Spadaccio, C.; Cuomo, M.; Chello, M.; Nenna, A.; Fischlein, T.; Nappi, F. Sharing of decision-making for infective endocarditis surgery: a narrative review of clinical and ethical implications. Ann. Transl. Med. 2020, 8, 1624–1624. [Google Scholar] [CrossRef] [PubMed]
- Steffen, V.; Marsch, G.; Burgwitz, K.; Kuehn, C.; Teebken, O.E. Resistance to infection of long-term cryopreserved human aortic valve allografts. J. Thorac. Cardiovasc. Surg. 2015, 151, 1251–1259. [Google Scholar] [CrossRef]
- Heying R, van de Gevel J, Que YA, Moreillon P, Beekhuizen H Fibronectin-binding proteins and clumping factor A in Staphylococcus aureus experimental endocarditis: FnBPA is sufficient to activate human endothelial cells. Thromb Haemost. 2007; 97, 617–626.
- Piroth, L.; Que, Y.-A.; Widmer, E.; Panchaud, A.; Piu, S.; Entenza, J.M.; Moreillon, P. The Fibrinogen- and Fibronectin-Binding Domains of Staphylococcus aureus Fibronectin-Binding Protein A Synergistically Promote Endothelial Invasion and Experimental Endocarditis. Infect. Immun. 2008, 76, 3824–3831. [Google Scholar] [CrossRef]
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor–binding protein. Blood 2014, 124, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Pappelbaum, K.I.; Gorzelanny, C.; Grässle, S.; Suckau, J.; Laschke, M.W.; Bischoff, M.; Bauer, C.; Schorpp-Kistner, M.; Weidenmaier, C.; Schneppenheim, R.; et al. Ultralarge von Willebrand Factor Fibers Mediate Luminal Staphylococcus aureus Adhesion to an Intact Endothelial Cell Layer Under Shear Stress. Circulation 2013, 128, 50–59. [Google Scholar] [CrossRef]
- Claes J, Liesenborghs L, Peetermans M, et al. Clumping factor A, von Willebrand factor-binding protein and von Willebrand factor anchor Staphylococcus aureus to the vessel wall. J Thromb Haemost. 2017 May ;15(5) :1009-1019.
- Claes, J.; Ditkowski, B.; Liesenborghs, L.; Veloso, T.R.; Entenza, J.M.; Moreillon, P.; Vanassche, T.; Verhamme, P.; Hoylaerts, M.F.; Heying, R. Assessment of the Dual Role of Clumping Factor A in S. Aureus Adhesion to Endothelium in Absence and Presence of Plasma. Arthritis Res. Ther. 2018, 118, 1230–1241. [Google Scholar] [CrossRef]
- Ko, Y.-P.; Kang, M.; Ganesh, V.K.; Ravirajan, D.; Li, B.; Höök, M. Coagulase and Efb of Staphylococcus aureus Have a Common Fibrinogen Binding Motif. Mbio 2016, 7, e01885–15. [Google Scholar] [CrossRef]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef]
- Ahmed, S.; Meghji, S.; Williams, R.J.; Henderson, B.; Brock, J.H.; Nair, S.P. Staphylococcus aureusFibronectin Binding Proteins Are Essential for Internalization by Osteoblasts but Do Not Account for Differences in Intracellular Levels of Bacteria. Infect. Immun. 2001, 69, 2872–2877. [Google Scholar] [CrossRef]
- Massey, R.C.; Kantzanou, M.N.; Fowler, T.; Day, N.P.J.; Schofield, K.; Wann, E.R.; Berendt, A.R.; Hook, M.; Peacock, S.J. Fibronectin-binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to fibronectin and invasion of endothelial cells. Cell. Microbiol. 2001, 3, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.A.; Douglas, I.; Whawell, S.A. Differential adhesion and invasion by Staphylococcus aureus of epithelial cells derived from different anatomical sites. J. Med Microbiol. 2012, 61, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Nguyen, M.-T.; Eble, J.A.; Chasan, A.I.; Mrakovcic, M.; Böttcher, R.T.; Preissner, K.T.; Roßlenbroich, S.; Peters, G.; Herrmann, M. More Is Not Always Better—the Double-Headed Role of Fibronectin in Staphylococcus aureus Host Cell Invasion. mBio 2021, 12, e0106221. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, G.; Pellegrini, A.; Alfeo, M.J.; Marchese, L.; Foster, T.J.; Speziale, P. The iron-regulated surface determinant B (IsdB) protein from Staphylococcus aureus acts as a receptor for the host protein vitronectin. J. Biol. Chem. 2020, 295, 10008–10022. [Google Scholar] [CrossRef]
- Alfeo, M.J.; Pagotto, A.; Barbieri, G.; Foster, T.J.; Vanhoorelbeke, K.; De Filippis, V.; Speziale, P.; Pietrocola, G. Staphylococcus aureus iron-regulated surface determinant B (IsdB) protein interacts with von Willebrand factor and promotes adherence to endothelial cells. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Leeten K, Jacques N, Lancellotti P, Oury C Aspirin or Ticagrelor in Staphylococcus aureus Infective Endocarditis: Where Do We Stand? Front Cell Dev Biol. 2021; 9, 716302.
- Ditkowski B, Bezulska-Ditkowska M, Jashari R, et al Antiplatelet therapy abrogates platelet-assisted Staphylococcus aureus infectivity of biological heart valve conduits. Congenital Cardiology and Cardiac Surgery Group. J Thorac Cardiovasc Surg. 2021 Jun ;161(6): e457-e472.
- Hannachi N, Habib G, Camoin-Jau L Aspirin Effect on Staphylococcus aureus Platelet Interactions During Infectious Endocarditis. Front Med (Lausanne). 2019 Oct 15 ;6 :217.
- Park, E.; Na, H.S.; Song, Y.-R.; Shin, S.Y.; Kim, Y.-M.; Chung, J. Activation of NLRP3 and AIM2 Inflammasomes by Porphyromonas gingivalis Infection. Infect. Immun. 2014, 82, 112–123. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



