
Submitted:
26 April 2023
Posted:
27 April 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since the first description of COVID-19 infection, among clinical manifestations of the disease including fever, dispnea, cough, fatigue, it was observed a high incidence of thromboembolic events potentially evolving towars ARDS and COVID-associated-coagulopathy (CAC).The hypercoagulation state is based on an interaction between thrombosis and inflammation. The so-called CAC represents a key aspect in the genesis of organ damage from SARS-CoV-2. The prothrombotic status in COVID-19 disease can be explained by the increase of coagulation levels of D-dimer, lymphocytes, fibrinogen, IL-6 and prothrombin time. Several mechanisms have been hypothesized to explain this hypercoagulable process such as inflammatory cytokine storm, platelet activation, endothelial dysfunction and stasis for a long time. The purpose of this narrative review is to provide an overview of the current knowledge on the pathogenic mechanisms of coagulopathy that may characterize COVID-19 infection and inform on new areas of research. New vascular therapeutic strategies are also reviewed.
Keywords:
COVID-19 infection
; coagulopathy
; endothelial dysfunction
; platelet activation
; citokine storm
; anticoagulant therapy
1. Background
At the end of December 2019 a novel coronavirus, denominated SARS-CoV-2 according to the similarity with the previous SARS viral epidemy, was described for the first time in China and, in March 2020 it was declared a global pandemic by the WORLD Health Organization (WHO) due to high morbidity and mortality, Italy was one of the most affected countries at the beginning of the infection spreading1. The disease caused by this virus was denominated Coronavirus disease 2019 (COVID-19) which still represent a critical challenge for the worldwide health community despite the reduction in mortality following the global vaccination campaign. To date, there are more than 762 million confirmed cases of COVID-19 infection, including almost 6,8 million deaths reported to WHO, with a total of 13,340,275,493 vaccine doses been administered2. Among the wide range of SARS-CoV-2 clinical manifestations (cough, fever, pharyngodynia, myo-arthralgia, fatigue) a respiratory tract involvement has been observed, potentially evolving with pneumonia, acute respiratory distress syndrome (ARDS), hyperinflammation, and COVID-associated-coagulopathy3,4,5. Hypercoagulable state is strongly associated with COVID-19 infection and may explain several phenomena observed in clinical practice. Since the beginning of the pandemic a very high incidence of thrombo-embolic events was observed including arterial and venous thrombosis, cerebral and myocardial infarction, limb arterial thrombosis, and venous thrombosis leading to higher incidence of stroke, acute coronary syndrome and myocardial infarction, venous thromboembolism (VTE) and pulmonary thromboembolism (PTE)6,7,8,9,10,11. Pathophysiological characteristic of acute pulmonary thromboembolism12 and abnormal coagulation status13 have been reported in these patients. Additional important laboratory findings such as high levels of D-dimer, fibrinogen (FIB) and its related degradation products (FDP) have been correlated with a poorer outcome13,1415,.In patients who died from COVID-19 infection, the micro thrombosis of alveolar capillary was more prevalent (9 times) than in patients died from influenza and about 15.2% to 79% of patients with severe COVID-19 infection have shown thrombotic events16. The involvement of the coagulation cascade and its abnormalities were previously identified in experimental investigations on mice infected with SARS-CoV-1 and in human autopsies. These findings suggested the hypothesis of a diffuse thrombotic microangiopathic mechanism involved in the pathogenesis of acute pulmonary interstitial disease caused by SARS-CoV-2 infection17,18. The prothrombotic status seems to be caused by the immune cell activation, excessive coagulation, and endothelial dysfunction19. Immuno-thrombosis appears to be involved in the pathological mechanism of SARS-CoV-2 and it is characterized by the interaction between the hemostatic system and the innate immune system, especially between monocytes, macrophages and neutrophils. After the endocytosis of SARS-CoV-2 in the host cells, a vascular damage is induced leading to a proinflammatory form of programmed cell death with cell lysis named “pyroptosis”, and the release of various substances, the so called Damage-Associated Molecular Patterns (DAMPs) such as adenosine triphosphate (ATP), nucleic acids and inflammasomes20 thus intensifying the inflammatory environment. Several mechanisms have been hypothesized to be involved in this hypercoagulable process such as inflammatory cytokine storm, platelet activation, endothelial dysfunction and stasis for a long time11,21,22. The purpose of this narrative review is to provide an overview of the current knowledge on the pathogenic mechanisms of coagulopathy that may characterize COVID-19 infection and inform on new areas of research. New vascular therapeutic strategies are also reviewed.
1.1. Role of platelets and complement as prothrombotic factors in COVID-19 infection
Platelets have a pivotal role in the innate immune system by activating the complement, thus playing a key role in COVID-19 “immune-thrombosis”23. The aggregation of PLT activated by endothelium damage and its interaction with other cells, increase their potential for pathologic thrombosis; their activation is essential to structural remodeling of pulmonary vasculature, inflammation and cardiovascular disease24,25. The mechanism of platelet activation may include different and multiple pathways even more complex in Covid- 19 infection, as the virus is able to infect cells using several entry mechanisms such as TLRs and/or ACE2-AngII axis26. The activated endothelial cells express P-selectin and other adhesion molecules with the recruitment of platelets and leukocytes. Bioactive molecules (e.g. adenosine diphosphate [ADP], polyphosphates, coagulation factors) and immunological mediators (e.g. complement factors) are released from activated platelets, activating the immune system through a positive feedback23. P-selectin, a platelet activation marker, is increased in patients with COVID-19 and can lead to a procoagulant phenotype by inducing tissue factor (TF) expression in monocytes. Moreover, Von Willebrand factor (VWF) is a glycoprotein derived from activated endothelial cells, platelets or sub-endothelial cells mediating the adhesion and aggregation of platelets. In patients affected by COVID-19, VWF is significantly increased and may suggest a tendency to thrombosis27. The activation of complement system was documented in COVID-19 with formation of the terminal membrane attack complex (MAC) that, in turn, can activate platelets with subsequent endothelial damage and secretion of VWF28. For this reason, the complement activation is associated with an amplification of the prothrombotic phenotype in COVID-19 disease. In fact, C5a can stimulate the release of TF and plasminogen activator inhibitor-1 (PAI-1) expression and activate neutrophils, which are responsible for the increased release of cytokines and the formation of neutrophil extracellular traps (NETs)29.
1.2. Role of hypoxia, blood viscosity and vasoconstriction as prothrombotic factors in COVID-19 infection
Hypoxia may represent itself a factor inducing a prothrombotic status in patient with SARS-CoV-2 infection with the production of a hypoxia inducible transcription factor (HIF-1α), which promotes the secretion of PAI-1 (plasminogen activator inhibitor) and macrophages by the endothelium. On the other hand, the mechanisms of altered coagulation are responsible of hypoxia that favours in turn the thrombo-inflammatory loop. Furthermore, hypoxia causes a release of cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6)30, critical inflammatory cytokines with prothrombotic effects. Positive correlations have been found between IL-6 and D-Dimer, especially during exacerbation of the disease31. Consequently, an increased blood viscosity and the release of procoagulant antibodies develop32. Recent studies showed that the appearance of antiphospholipid antibodies and lupus anticoagulant immunoglobulins may also play a role in the pathogenesis of coagulopathy. Indeed, the presence of IgA anti-cardiolipin antibodies and IgA and IgG anti-2-glycoprotein I antibodies have been found in association with coagulopathy, thrombocytopenia, and the development of peripheral and cerebral ischemic events. Harzallah and coworkers33 investigated 56 patients with confirmed or suspected SARS-CoV-2 infection. Among these, 25 were found to be lupus anticoagulant immunoglobulin, while 5 were found positive for IgM or IgG anti-cardiolipin or anti-2-glycoprotein I antibodies. Endothelial dysfunction is characterized by the loss of characteristics of endothelial native cells like the ability to regulate vascular tonus which may conduce to vasoconstriction and, subsequently, a prothrombotic status. Moreover, the down-regulation of the endothelial ACE2 receptor as a consequence of SARS-CoV-2 infection gives a pro-inflammatory, pro-coagulant and pro-apoptotic phenotype to endothelial cells34.
1.3. Interlink between coagulation and inflammation in COVID-19 disease
The so-called COVID-associated coagulopathy (CAC) represents a key aspect in the genesis of organ damage from SARS-CoV-2 and the hypercoagulation state is based on an interaction between thrombosis and inflammation. A close relationship between inflammation and coagulation had been widely demonstrated in previous research35,36. The coagulation system consists in a finely regulated balance between procoagulant and anticoagulant mechanisms and inflammation can compromise this equilibrium, leading to impaired coagulation. As a result, the final clinical consequence in inflammatory conditions may consist in bleeding, thrombosis or both of them37. Pathogens, inflammatory mediators such as IL-6, IL-8, TNF-α as well as DAMPs from injured host tissue can activate monocytes and induce the expression of tissue factor on monocytes and endothelial cell surfaces38 (Figure 1).
Subsequently, activated monocytes release inflammatory cytokines and chemokines that enlarge the inflammatory response and stimulate neutrophils, lymphocytes, platelets, and vascular endothelial cells. Healthy endothelial cells have an anti-thrombogenic attitude due to the expression of glycocalyx and its binding protein, antithrombin. When endothelial cells go through injury, the glycocalyx is disrupted, the anticoagulant factors are lost and consequently these cells change their properties to procoagulant39. Furthermore, also neutrophils are involved in an important defense mechanism that may lead to a procoagulant status by means of NETs. NETs are structures of DNA, histones and neutrophil antimicrobial proteins that binds and kill pathogens, an excessive production of NETs can facilitate microthrombosis by creating a scaffold for platelets aggregation40 .When an infection occurs, the first leukocytes recruited are neutrophils that, producing and releasing NETs, stimulate the formation and deposition of fibrin to trap and destroy invading microorganisms. It has been previously demonstrated a NETs increase in sepsis and other inflammatory conditions41. NETs also cause platelet adhesion and, in some experimental models, their connection with deep vein thrombosis has been demonstrated40 They stimulate both the extrinsic and intrinsic coagulation pathway playing a major role towards a coagulative pattern during infection-mediated inflammation. Patients with severe COVID-19 have been shown to present elevated levels of circulating histones and myeloperoxidase DNA (MPO-DNA) which are two specific markers of NETs42. As a consequence of the described mechanisms, an extreme inflammatory response may also occur, causing disseminated intravascular coagulation (DIC), which leads to multiple organs failure. This life-threatening acquired syndrome is characterized by disseminated and often uncontrolled activation of coagulation and is associated with a high risk of macro-and microvascular thrombosis. In this setting, also natural coagulation inhibitors become inefficient to downregulate thrombin generation. Moreover, it can be observed a progressive consumption coagulopathy, which leads to an increased bleeding risk43. Other clinical manifestations of the altered coagulation system are hemolytic uremic syndrome, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura44 and hemophagocytic lymphohistiocytosis (sHLH). Globally, all these evidences suggest that the hypercoagulative state described in patients with COVID-19, is likely to be caused by a deep and complex inflammatory response to the virus, based on an interaction between thrombosis and inflammation as resumed in Figure 2. Another important interlink between inflammation and pro-thrombotic status is represented by underlying clinical conditions such as chronic comorbidities that are linked to the mortality in COVID-19 infection. In particular, obesity has been shown to increase the risk of hospitalization and COVID-19 complications45 suggesting an interplay between obesity and inflammation. The adipose tissue, in fact, express higher ACE2 levels than lung tissue, being a powerful inflammatory reservoir for the replication of SARS-Cov-246. In addition, obese people are characterized by a low-grade inflammation, associated with the over-expression of pro-inflammatory cytokines and chemokines such as TNF-α, IL-6, and MCP-1, high leptin levels with known pro-inflammatory effects, low adiponectin levels with anti-inflammatory effects and consequently a procoagulant status. It has been calculated that one third of total circulating concentrations of IL-6 originate from adipose tissue47. In addition, obese patients show higher blood IL-6 and TNF- α levels and a polarization of natural killer (NK) cells to non-cytotoxic NK cells. As both obesity and COVID-19 seem to share common metabolic and inflammatory pathways, it has been recommended by many authors to consider and classify obese and severely obese patients as high-risk patients for COVID-19 disease. Also, sleep disturbances during pandemics have been suggested to be related to a major risk of infection linked to an increased inflammatory status and a reduction in efficiency of immune system48, an interesting linkage was found between sleep deprivation, inflammation and immune response to SARS-CoV-2 that may have a role in predisposing to the infection49.
1.4. Interlink between coagulopathy in viral infections and in COVID-19 disease
Since the beginning of the pandemic a very high incidence of thrombo-embolic events (VTE) was observed (Table 3). The hypercoagulative state, described in patients with COVID-19 derives from a complex inflammatory response to the virus in which hemostasis and immune system collaborate together to limit the spread of viral infection. Physiological immune-thrombosis can evolve to an excessive, dysregulated formation of immunologically mediated thrombi and spread, especially in the microcirculation. Several viral infections may share abnormal coagulation processes such as bleeding, thrombosis, or both.
2. Thrombosis
The increased incidence of VTE in COVID-19 patients was similarly also in patients with other viral infections, i.e., the severe acute respiratory syndrome (SARS) and Middle East Respiratory Syndrome (MERS-CoV)50,51. H1N1 influenza infection is associated with an 18-fold increased risk of developing VTE when compared to critically ill patients with ARDS with no H1N1 influenza infection6. A previous study by Avnon et al. found that VTE occurred in 25% of patients with severe H1N1 influenza admitted to the intensive care unit (ICU)52. Particular evidence for thromboembolic events was also reported during Cytomegalovirus (CMV) infection in which two arterial thrombotic events were described in nine Israelitic immunocompetent CMV infected patients (spleen and liver)53. The pathophysiological mechanism is yet unknown but it seems to be related to higher levels of VWF in plasma of CMV infected people54. It is likely that SARS-CoV-2 virus does not have intrinsic procoagulant effects, while the coagulopathy appears as a consequence of the intense COVID-19 inflammatory response and endothelial activation/damage55. Two possible mechanisms implicated in the pathogenesis of coagulation dysfunction during SARS-CoV2 infection have been proposed: the cytokine storm which seems to play a pivotal role, and virus-specific mechanisms related to the virus interaction with renin angiotensin system and the fibrinolytic pathway56.
2.1. Cytokine storm
Pro-inflammatory cytokines are involved in a so-called “cytokine release syndrome” responsible of the innate immunity system activation and severe clinical manifestation of the disease57. The immune system dysfunction is a candidate risk factor for adverse outcomes in COVID-19 and the most important cause of morbidity and mortality in patients suffering from COVID19 infection seems to be the cytokine storm causing an immune dysregulation in the peripheral tissues and in the lungs58(p2),57,59,60,61,62. More specifically, IL-6 plays an important role in cytokine release syndrome and contributes, together with TNF-α and interleukin-1 (IL-1), to a blood hyper-coagulability and to a severe inflammation, sometimes evolving in disseminated intravascular coagulation (DIC)63. Figure 3.
Current evidence from clinical studies shows that IL-6 seems to play a prominent role in cytokine-induced activation of coagulation. Also, IL-6 promotes the proliferation of megakaryocytes64 and the release of TF, the latter detected in inflamed tissues and in particular in the lungs of patients affected by COVID1965. A postulated mechanism consider that SARS-CoV-2-infected megakaryocytes may interfere with platelet’s function and count itself, as already described in previous studies that reported thrombocytopenia during SARS-CoV infection. The virus induces release of cytokines like IL-6 conducting to megakaryocytic proliferation and differentiation, although the mechanism remains not completely clarified66,67.
Furthermore, vascular permeability is mediated by IL-6 through stimulation of vascular endothelial growth factor (VEGF) secretion and the release of other coagulation factors such as FIB and factor VIII68. A great effort during pandemic has been done to find inflammatory markers reflecting disease severity and eventually predicting disease prognosis. Among the most studied, the increased levels of a pivotal serum cytokine, IL-1, which is a principal source of tissue damage interacting in both innate and acquired immunity, have been detected in patients suffering from severe COVID-19 infection. IL-1 stimulates the secretion of mediators stored in the granules of mast cells and macrophages, such as TNF-α, IL-6, and the release of arachidonic acid products such as prostaglandins and thromboxane A269,70,71,72. Another important marker in the cytokine network of COVID-infection is IL-18. The catastrophic clinical course in COVID-19 shares similar features with macrophage activation syndrome (MAS) encountered also in other conditions with a potentially rapidly fatal course without treatment. IL-1, IL-6, IL-8, IL-10, IL-18, interferon (IFN)-γ and TNF-α are the most important responsible for MAS development. IL-18 is produced by macrophages at very early stages of viral infections and induces production of IL-6 and IFN-γ which are considered critical for optimal viral host defense. A study by Satis and coworkers observed a four-fold levels of IL-18 in 58 people suffering from a severe form of COVID-19. These findings contrasted with the mildly affected patients and led to the conclusion of a correlation between IL-18 and the severity of the disease73. An additional role is determined by TNF-α, responsible of the activation of glucuronidases, which degrades the endothelial glycocalyx, and the upregulation of hyaluronic acid synthase 2, that leads to hyaluronic acid deposition and fluid retention74. Due to the systemic hypoxia induced by ARDS COVID-19 related, a reduction in endothelial nitric oxide synthase activity and nitric oxide levels have been indicated as a possible pathogenic process typical of endothelial dysfunction75.
2.2. Virus-specific mechanisms
Experiments in vitro demonstrated that SARS-CoV2 can infect primary endothelial cells76 and there is some evidence of the infection of endothelial cells in severe cases of COVID-1911. Moreover, the replication within endothelial cells is able to induce cell death causing the activation of procoagulant reactions77. The membrane glycoprotein (Spike) of the SARS-CoV-2 virus interacts with Angiotensin Converting Enzyme 2 (ACE-2), an integral membrane receptor expressed in lung but also heart, kidney and intestine by reducing its activity. Normally, ACE-2 reduces the availability of angiotensin II through the counter-regulated activity of ACE78. As a result, the virus-mediated engagement of ACE2 decreases its expression and activates the renin-angiotensin system (RAS), promoting activation of epithelial cells, monocytes, neutrophils and procoagulant factors with platelet adhesion and aggregation, with consequently vasoconstriction and release of inflammatory cytokines79, also a reduction of fibrinolytic activity mediated by RAS can be observed80 as represented in Figure 4.
Among virus-related mechanisms high levels of PAI-1, the principal inhibitor of fibrinolysis interfering with tissue plasminogen activator (tPA) and urokinase, have been related with increased risk of thromboembolic events80. Interestingly, previous studies reported high blood levels of PAI-1 in patients with SARS-CoV infection suggesting a possible direct effect of infection on the production of anti-coagulant factors81. One study described an important increase of another mediator of platelet adhesion, the platelet derived vitronectin (VN), in SARS-CoV pneumonia, however it was not possible to discriminate its origin from increased expression by the liver or from lung damage82. Another possible virus-specific effect could be related to the induction of autoimmunity, also described in SARS patients37. Recent studies showed that the appearance of antiphospholipid antibodies and lupus anticoagulant immunoglobulins may have a role in the pathogenesis of coagulopathy. Indeed, the presence of IgA anti-cardiolipin antibodies and IgA and IgG anti-2-glycoprotein I antibodies have been found in association with coagulopathy, thrombocytopenia, and the development of peripheral and cerebral ischemic events. Antiphospholipid antibodies (aPL), recognized as risk factors for arterial and venous thrombosis, have been associated with different viral infections, such as parvovirus B19, herpes viruses, hepatitis viruses and human immunodeficiency virus. A first case-report of COVID-19 patient with aPL and arterial ischemia was described by Chinese Authors83 although, subsequently, a larger multicentric cohort demonstrated a low rate of aPL positivity, as defined by classification criteria, suggesting that aPL found in COVID-19 patients are different from aPL found in antiphospholipid syndrome84. It is likely that the mechanisms of altered coagulation due to SARS-CoV-2 infection, also responsible of hypoxia, may favors in turn the thrombo-inflammatory loop and consequently an increased blood viscosity and the release of procoagulant antibodies32. These observations were confirmed by a study of Harzallah and coworkers investigating 56 patients with confirmed or suspected SARS-CoV-2 infection. Among these, 25 were found to lupus anticoagulant immunoglobulin, while 5 were found positive for IgM or IgG anti-cardiolipin or anti-2-glycoprotein I antibodies33. Further studies though, are needed to address this issue.
3. Thrombocytopenia
COVID19-related coagulopathy firstly determines elevated D-dimer levels that combine in turn with mildly prolonged PT, APTT and mild thrombocytopenia. At late stages this process evolves toward a classical DIC85. These findings were identified in the clinical setting in a meta-analysis where 7.613 patients suffering from COVID-19 infection were examined. In these cohort thrombocytopenia was worse in the critically ill group than in those with non-severe disease86. Also, platelets count was lower in elderlies, in males and in patients with higher APACHE II score at admission87. This study highlights an association between low platelets count and an increased risk of severity of the disease and mortality. As per the SARS-CoV-2 infection related thrombocytopenia, it appears that the platelets can be more rapidly removed or sequestrated by the reticuloendothelial system after activation of antigen-antibody complexes88,89. Also, megakaryocyte’s function and the consequent platelet production can be reduced by the virus activity90. A possible mechanism of thrombocytopenia has been described after COVID-19 vaccination. It has been observed in rare cases an immune thrombotic thrombocytopenia (VITT) syndrome vaccine induced especially related to ChAdOx1 nCoV-19 vaccine. The main pathogenetic hypothesis supporting this evidences is the possible promotion of antibodies synthesis against PF4 by some anti-covid vaccines promoting the synthesis of antibodies against PF4 that provoke platelets’ massive activation, inducing immune thrombotic thrombocytopenia91. As anti-PF4 antibodies were detected in patients with VITT, current guidelines recommends PF4-heparin ELISA blood test before performing vaccine when VITT is clinically suspected92. Clot’s risk in general population is estimated around 1:250.000 while it is higher in young people (20-29 years old) at 1.1: 100.00093.
3.1. Contribution of sepsis in coagulopathy during COVID-19 infection
Sepsis is a life-threatening condition as a response of a primary infection in which the body responds with extreme inflammatory reactions that determine injuries to own tissues and organs. On the other hand, severe COVID-19 infection is commonly complicated with coagulopathy, and, in the latter stages may evolve towards a classical DIC. These manifestations have been object of a major concern during the COVID-19 pandemic. The International Society of Thrombosis and Hemostasis (ISTH) has proposed a new category to identify an early stage of DIC associated with sepsis called sepsis-induced coagulopathy (SIC). Many patients suffering from severe COVID-19 disease meet the Third International Consensus Definitions for Sepsis (Sepsis-3)94 manifesting respiratory dysfunction during a viral infection. The diagnostic criteria of SIC are summarized in Table 1. A score ≥ 4 is diagnostic for SIC. This score can also be applied to COVID-19-affected patients to identify a coagulopathy risk induced by the virus, although it is less reliable than in other pathogens-induced infections since in this case, especially during initial stages of the disease, thrombocytopenia cannot be present. One study by Tang et al, studied the effects of anticoagulant treatment to validate the usefulness95 96 of SIC score finding that patients who met the criteria reported in Table 1 benefit from anticoagulant therapy.12
3.2. Coagulation biomarkers in SARS-CoV-2 infection: a predictive method
In the setting of the altered coagulation state, the measurements of the coagulative parameters may orient the clinicians toward an early identification of a coagulative derangement. Besides the D-Dimer, as above mentioned, other parameters are of bedside interest (Table 2). Increased levels of thrombin-antithrombin complexes, plasmin-alpha-2-antiplasmin, and thrombomodulin complexes have been reported in respiratory tract infections. Increased PAI-1 serum levels were identified, suggesting an impaired fibrinolysis. A study 14 highlighted an alteration of the laboratory parameters deponent for DIC (according to the diagnostic criteria of the ISTH) in 15 subjects (71.4%) who died of COVID-19-related pneumopathy. In the final stage of the disease, elevated levels of D-dimer and FIB degradation products were found. Recent contributions have reported that COVID-19 severity could be associated with some coagulopathy biomarkers, including prothrombin time (PT), activated partial thromboplastin time (APTT), and D-dimer. Nevertheless, the association between coagulopathy and COVID-19 severity still remains undefined.
The severity of the condition is mostly associated with clinical evidence. In particular, one study97 has demonstrated that the majority of patients developed a mild infection, and about 15% of them experienced a severe manifestation with dyspnoea and hypoxia. Another 5% developed respiratory failure in conjunction with ARDS, shock, and multi-organ dysfunction. Many studies have focused on the evaluation of D-dimer, PLT, PT, APTT, and FIB. It was reported that D-dimer and PT values have been shown to be higher in patients with a more severe disease98, moreover, several studies have shown that elevated D-dimer levels are associated with in-hospital mortality. Recent researches have hypothesized that genetic profiles may partly explain individual differences in developing thrombotic complications during COVID-19 infection. An interesting study evaluated the genotypic distribution of targeted DNA polymorphisms in COVID-19 complicated by pulmonary embolism during hospitalization finding significant associations between higher D-dimer levels and ACE I/D and APOE T158C polymorphism in patients with and without pulmonary embolism, suggesting a potential useful marker of poor clinical outcome99. In another meta-analysis, it was demonstrated that the platelet count decreased progressively with the degree of disease severity100. However, a previous meta-analysis analysis101 demonstrated that there were no differences in PLT and APTT levels between wild and severe cases. All this is probably due to the confounding factors and biases that inevitably occur, such as age, sex and the presence of comorbidities like hypertension, diabetes, cardiovascular disease and chronic kidney disease of the examined populations. As reported in another study by Wu et al. mortality from severe COVID was increased 34-fold compared to a normal infection102 and very high levels of coagulation markers were correlated with an 11-fold increase in death. These observations underline the importance of an early stratification of disease severity.

Table 3.
Incidence of Thrombotic Events in Patients with SARS-CoV-2 Infections.
| Study | Sample size | Thrombotic event reported | Confirmatory diagnostic test | Incidence |
|---|---|---|---|---|
| Klok et al.8 | N = 184 ICU patients | Venous arterial thrombosis | CTPA or Ultrasound | 31% |
| Leonard-Lorant et al.103 | N = 106 (48 ICU and 58 non-ICU) | Acute PE | CTPA | 30% of all COVID-19 patients developed PE irrespective of ICU status |
| Helms et al.9 | N = 150 ICU patients | Clinically significant thrombosis | CTPA | 43% |
| Wichmann et al.104 | N = 12 (5 ICU and 7 non-ICU) | DVT | Autopsy | 58% of all COVID-19 patients autopsied had evidence of PE, irrespective of ICU status |
| Demelo-Rodríguez et al.105 | N = 156 non-ICU patients | DVT | Ultrasound | 15% |
| Nahum et al.106 | N = 34 ICU patients | DVT | Ultrasound | 79% |
| Middeldorp et al.107 | N = 198 (123 non-ICU and 75 ICU) | VTE in non-ICU vs ICU | Ultrasound | 9.2% in non-ICU vs 59% in ICU |
| Shah et al.108 | N = 187 (182no n-ICU and 5 ICU) | Acute PE | CTPA | 23% |
| Cui et al.109 | N = 81 non ICU | DVT | Ultrasound | 25% |
ICU, intensive Care Unit; CTPA, computed tomography pulmonary angiogram; DVT, deep vein thrombosis; PE, pulmonary embolism; VTE, venous thromboembolism.
3.3. New clinical evidences of anticoagulant therapy in COVID-19
Data on anticoagulant therapy appear to be associated with a better outcome in moderate to severe COVID-19 patients with altered coagulative parameters (elevated D-dimer, elevated FIB, and low levels of anti-thrombin)13, 14,110. A retrospective study by Shi et al. have shown that these treatments can mitigate cytokine storm exerting an anti-inflammatory effect (reduction of IL-6 and increasing of lymphocytes) and improving coagulation dysfunction111. A number of substances are used for COVID-19 VTE such as heparins, direct oral anticoagulants (DOAK), aggregation inhibitors, factor XII inhibitors, thrombolytic agents, anti-complement, anti-NET drugs and IL-1 receptor antagonists.
Heparins, including unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) have several anti-coagulant and anti-inflammatory effects112. Among the various properties of heparin, it has been observed a beneficial effect on endothelium. A dysfunctional endothelium leads to an inflammatory status through the production of vasoconstrictor factors and the recruitment of immune cells113. Histones released from damaged cells may be responsible for endothelial injury114. Heparin exerts its action through an effect on histone methylation and MAPK and NF-κB signaling pathways115. In this way heparin can antagonize histones and therefore "protect" the endothelium29,30. It was proved to have a beneficial effect, related to its anticoagulant function on COVID-19116 and anti-inflammatory properties117. Proposed mechanisms include the binding to inflammatory cytokines, inhibition of neutrophil chemotaxis and leukocyte migration, neutralization of complement factor C5a, and sequestration of acute phase proteins such as P-selectin and L-selectin and the induction of cell apoptosis through the TNF-α and NF-κB pathways118,119. Another potential direct antiviral role of heparin is related to its polyanionic allowing the binding to various proteins thus acting as an effective inhibitor of viral adhesion120. This condition mechanism was also described in other viral diseases120,121 as well as in SARS-CoV. As Mycroft-West et al.122 demonstrated, surface plasmon resonance and circular dichroism were used and it was demonstrated that the receptor binding domain of the Spike S1 SARS-CoV-2 protein interacts with heparin. Finally, in a report by Tang14, a favorable outcome was highlighted with the use of LMWHs in severe patients with COVID-19 who meet the criteria of SCI (sepsis-induced coagulopathy) or with markedly elevated D-dimer. However, several studies could not identify this relationship. As demonstrated by C. Coligher et al. in a randomized control trial, in COVID-19 critically ill patients, an initial strategy of therapeutic-dose anticoagulation with heparin failed to show a greater probability of survival to hospital discharge or a major number of days free of cardiovascular or respiratory organ support than did usual-care pharmacologic thromboprophylaxis123. Interim results from multiplatform RCTs on VTE prophylaxis show that in moderate COVID-19 disease (hospitalized, not intensive), therapeutic doses of LMWH appear to be better than prophylactic doses-with positive effects on morbidity and mortality and less than 2% severe bleeding124. In patients at low or intermediate risk of thrombotic phenomena, treated with prophylactic doses of LMWH has been noticed concomitant reduction of developing severe ARDS and venous thromboembolism, that may reduce the need for mechanical ventilation and consequentially the lower cardiovascular death125. Treatment with heparin did not improve the course in severe COVID-19) and it seems to be inferior to prophylactic doses. The first observational cohort study has examined previous prophylactic anticoagulation versus no anticoagulation in hospitalized COVID-19 patients (not intensive). Early treatment with prophylactic heparin was associated with a 34% reduction in relative 30-day mortality risk and an absolute risk reduction of 4.4%. There was no increased risk of bleeding under prophylactic anticoagulation126. Guidelines of the medical societies currently recommend VTE prophylaxis, preferably with LMWH, for every inpatient COVID-19 patient127. The guidelines do not recommend VTE prophylaxis for COVID-19 outpatients. A prophylactic anticoagulation for 1-2 weeks is recommended by some guidelines in patients discharged by hospital if there are additional risk factors128. Globally the use of heparin is recommended but needs to be titrated against the risk of bleeding and individualized especially in patients affecting by pre-existing endothelial dysfunction (diabetes, hypertension, obesity) at higher risk of adverse outcome during COVID-19 infection129. Also, antiplatelets have been considered as antithrombotic treatment for COVID-19 even though, a rationale for aspirin use in COVID-19 is still uncertain. A recent review130recommend a low-dose aspirin regimen for primary prevention of arterial thromboembolism in patients aged 40-70 years with intermediate or high atherosclerotic cardiovascular risk and a low risk of bleeding. This opens a perspective on aspirin’s protective role in COVID-19 with associated lung injury and vascular thrombosis even in absence of previous known cardiovascular disease. As soon as the data from the RCTs are available, the therapy and prophylaxis recommendations will certainly be adapted and reissued.
4. Closing Remarks
COVID-19 can be considered as a systemic disease characterized by dysregulation of the immune system and a hypercoagulable status, consequence of the direct virus-induced endothelial damage, amplified by leukocyte and cytokine-mediated activation of the platelets, release of TF and NETosis and intensified by activation of the complement system. The strong activation of the immune system by the SARS-CoV-2 infection leads to a non-regulatable thrombosis, which can present with many microthrombi in the micro-vascularization, VTE and arterial events. Coagulopathy is a crucial aspect of the disease and its early identification, prevention and treatment may limit the evolution towards potentially irreversible pulmonary and systemic conditions. Scientific evidences suggest that coagulopathy is not to be considered only as a disease complication but may be a real primitive pathogenetic element of the SARS-CoV-2 infection. Recent data from the literature also seem to suggest a favorable prognostic effect of anticoagulant treatment with low molecular weight heparin in patients with COVID-19 manifestations. The latter aspect is particularly pertinent in patients with cardiovascular and/or neurological diseases, obesity, diabetes because they have higher risk to develop vascular thrombosis. In conclusion, however, we underline that available data concerning the anticoagulant treatment in COVID-19 disease are not completely supported by several randomized trials and, therefore, there is an objective difficulty to choose the most indicated therapy, which justify a real advantage of a full-dose anticoagulant treatment in patients with severe disease, considering the potential risk of bleeding increase.
References
- Malerba M, Ragnoli B, Puca E, Pipero P. Supporting healthcare workers on front lines of the Covid-19 fight. Acta Biomed. 2020, 91, e2020157. [CrossRef]
- Coronavirus disease (COVID-19) – World Health Organization. Accessed April 20, 2023. https://www.who.int/emergencies/diseases/novel-coronavirus-2019.
- Harapan H, Itoh N, Yufika A, Winardi W, Keam S, Te H, Megawati D, Hayati Z, Wagner AL, Mudatsir M. Coronavirus disease 2019 (COVID-19): A literature review. J Infect Public Health. 2020, 13, 667-673. [CrossRef]
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet. 2020, 395, 1054-1062. [CrossRef]
- Ragnoli B, Cena T, Radaeli A, Pochetti P, Conti L, Calareso A, Morjaria J, Malerba M. Pneumothorax in hospitalized COVID-19 patients with severe respiratory failure: Risk factors and outcome. Respir Med. 2023, 211:107194. [CrossRef]
- Obi AT, Tignanelli CJ, Jacobs BN, Arya S, Park PK, Wakefield TW, Henke PK, Napolitano LM. Empirical systemic anticoagulation is associated with decreased venous thromboembolism in critically ill influenza A H1N1 acute respiratory distress syndrome patients. J Vasc Surg Venous Lymphat Disord. 2019, 7, 317-324. [CrossRef]
- Fraissé M, Logre E, Pajot O, Mentec H, Plantefève G, Contou D. Thrombotic and hemorrhagic events in critically ill COVID-19 patients: A French monocenter retrospective study. Crit Care. 2020, 24, 275. [CrossRef]
- Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D a. MPJ, Kant KM, Kaptein FHJ, van Paassen J, Stals M a. M, Huisman MV, Endeman H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020, 191:145-147. [CrossRef]
- Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Fagot Gandet F, Fafi-Kremer S, Castelain V, Schneider F, Grunebaum L, Anglés-Cano E, Sattler L, Mertes PM, Meziani F, CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis). High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089-1098. [CrossRef]
- Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD, Sacco C, Bertuzzi A, Sandri MT, Barco S, Humanitas COVID-19 Task Force. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020, 191:9-14. [CrossRef]
- Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020, 395, 1417-1418. [CrossRef]
- Danzi GB, Loffi M, Galeazzi G, Gherbesi E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur Heart J. 2020, 41, 1858. [CrossRef]
- Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020, 18, 844-847. [CrossRef]
- Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020, 18, 1094-1099. [CrossRef]
- Han H, Yang L, Liu R, Liu F, Wu KL, Li J, Liu XH, Zhu CL. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. 2020, 58, 1116-1120. [CrossRef]
- Ren B, Yan F, Deng Z, Zhang S, Xiao L, Wu M, Cai L. Extremely High Incidence of Lower Extremity Deep Venous Thrombosis in 48 Patients With Severe COVID-19 in Wuhan. Circulation. 2020, 142, 181-183. [CrossRef]
- Gralinski LE, Bankhead A, Jeng S, Menachery VD, Proll S, Belisle SE, Matzke M, Webb-Robertson BJM, Luna ML, Shukla AK, Ferris MT, Bolles M, Chang J, Aicher L, Waters KM, Smith RD, Metz TO, Law GL, Katze MG, McWeeney S, Baric RS. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio. 2013, 4, e00271-13. [CrossRef]
- Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, Mou HM, Wang LH, Zhang HR, Fu WJ, Luo T, Liu F, Guo QN, Chen C, Xiao HL, Guo HT, Lin S, Xiang DF, Shi Y, Pan GQ, Li QR, Huang X, Cui Y, Liu XZ, Tang W, Pan PF, Huang XQ, Ding YQ, Bian XW. [A pathological report of three COVID-19 cases by minimal invasive autopsies]. Zhonghua Bing Li Xue Za Zhi. 2020, 49, 411-417. [CrossRef]
- Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S, Nydam TL, Moore PK, McIntyre RC. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J Am Coll Surg. 2020, 231, 193-203.e1. [CrossRef]
- Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: Immunity, inflammation and intervention. Nat Rev Immunol. 2020, 20, 363-374. [CrossRef]
- Baldanzi G, Purghè B, Ragnoli B, Sainaghi PP, Rolla R, Chiocchetti A, Manfredi M, Malerba M. Circulating Peptidome Is Strongly Altered in COVID-19 Patients. International Journal of Environmental Research and Public Health. 2023, 20, 1564. [CrossRef]
- Purghè B, Manfredi M, Ragnoli B, Baldanzi G, Malerba M. Exosomes in chronic respiratory diseases. Biomed Pharmacother. 2021, 144:112270. [CrossRef]
- Wool GD, Miller JL. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology. 2021, 88, 15-27. [CrossRef]
- Malerba M, Clini E, Malagola M, Avanzi GC. Platelet activation as a novel mechanism of atherothrombotic risk in chronic obstructive pulmonary disease. Expert Review of Hematology. 2013, 6, 475-483. [CrossRef]
- Malerba M, Nardin M, Radaeli A, Montuschi P, Carpagnano GE, Clini E. The potential role of endothelial dysfunction and platelet activation in the development of thrombotic risk in COPD patients. Expert Rev Hematol. 2017, 10, 821-832. [CrossRef]
- Violi F, Cammisotto V, Pignatelli P. Thrombosis in Covid-19 and non-Covid-19 pneumonia: Role of platelets. Platelets. 2021, 32, 1009-1017. [CrossRef]
- Polosa R, Malerba M, Cacciola RR, Morjaria JB, Maugeri C, Prosperini G, Gullo R, Spicuzza L, Radaeli A, Di Maria GU. Effect of acute exacerbations on circulating endothelial, clotting and fibrinolytic markers in COPD patients. Intern Emerg Med. 2013, 8, 567-574. [CrossRef]
- Wiedmer T, Esmon CT, Sims PJ. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood. 1986, 68, 875-880.
- JCI Insight - The complement system in COVID-19: Friend and foe? Accessed April 20, 2023. https://insight.jci.org/articles/view/140711.
- Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res. 2019, 181:77-83. [CrossRef]
- Leyfman Y, Erick TK, Reddy SS, Galwankar S, Nanayakkara PWB, Di Somma S, Sharma P, Stawicki SP, Chaudry IH. Potential Immunotherapeutic Targets for Hypoxia Due to COVI-Flu. Shock. 2020, 54, 438-450. [CrossRef]
- Kichloo A, Dettloff K, Aljadah M, Albosta M, Jamal S, Singh J, Wani F, Kumar A, Vallabhaneni S, Khan MZ. COVID-19 and Hypercoagulability: A Review. Clin Appl Thromb Hemost. 2020, 26:1076029620962853. [CrossRef]
- Harzallah I, Debliquis A, Drénou B. Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost. 2020, 18, 2064-2065. [CrossRef]
- Iba T, Connors JM, Levy JH. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm Res. 2020, 69, 1181-1189. [CrossRef]
- Levi M, van der Poll T, Schultz M. Infection and inflammation as risk factors for thrombosis and atherosclerosis. Semin Thromb Hemost. 2012, 38, 506-514. [CrossRef]
- Ekholm M, Kahan T. The Impact of the Renin-Angiotensin-Aldosterone System on Inflammation, Coagulation, and Atherothrombotic Complications, and to Aggravated COVID-19. Front Pharmacol. 2021, 12:640185. [CrossRef]
- Goeijenbier M, van Wissen M, van de Weg C, Jong E, Gerdes VEA, Meijers JCM, Brandjes DPM, van Gorp ECM. Review: Viral infections and mechanisms of thrombosis and bleeding. J Med Virol. 2012, 84, 1680-1696. [CrossRef]
- Branchford BR, Carpenter SL. The Role of Inflammation in Venous Thromboembolism. Front Pediatr. 2018, 6:142. [CrossRef]
- Iba T, Levy JH, Levi M, Thachil J. Coagulopathy in COVID-19. J Thromb Haemost. 2020, 18, 2103-2109. [CrossRef]
- Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012, 32, 1777-1783. [CrossRef]
- Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020, 31, 107602. [CrossRef]
- Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, Woods RJ, Kanthi Y, Knight JS. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020, 5, 138999. [CrossRef]
- Papageorgiou C, Jourdi G, Adjambri E, Walborn A, Patel P, Fareed J, Elalamy I, Hoppensteadt D, Gerotziafas GT. Disseminated Intravascular Coagulation: An Update on Pathogenesis, Diagnosis, and Therapeutic Strategies. Clin Appl Thromb Hemost. 2018, 24(9_suppl):8S-28S. [CrossRef]
- van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW, Brandjes DP. Review: Infectious diseases and coagulation disorders. J Infect Dis. 1999, 180, 176-186. [CrossRef]
- Caci G, Albini A, Malerba M, Noonan DM, Pochetti P, Polosa R. COVID-19 and Obesity: Dangerous Liaisons. J Clin Med. 2020, 9, 2511. [CrossRef]
- Sanchis-Gomar F, Lavie CJ, Mehra MR, Henry BM, Lippi G. Obesity and Outcomes in COVID-19: When an Epidemic and Pandemic Collide. Mayo Clin Proc. 2020, 95, 1445-1453. [CrossRef]
- Lafontan M. Fat cells: Afferent and efferent messages define new approaches to treat obesity. Annu Rev Pharmacol Toxicol. 2005, 45:119-146. [CrossRef]
- Ragnoli B, Pochetti P, Raie A, Malerba M. Comorbid Insomnia and Obstructive Sleep Apnea (COMISA): Current Concepts of Patient Management. Int J Environ Res Public Health. 2021, 18, 9248. [CrossRef]
- Ragnoli B, Pochetti P, Pignatti P, Barbieri M, Mondini L, Ruggero L, Trotta L, Montuschi P, Malerba M. Sleep Deprivation, Immune Suppression and SARS-CoV-2 Infection. International Journal of Environmental Research and Public Health. 2022, 19, 904. [CrossRef]
- Spyropoulos AC, Levy JH, Ageno W, Connors JM, Hunt BJ, Iba T, Levi M, Samama CM, Thachil J, Giannis D, Douketis JD, Subcommittee on Perioperative, Critical Care Thrombosis, Haemostasis of the Scientific, Standardization Committee of the International Society on Thrombosis and Haemostasis. Scientific and Standardization Committee communication: Clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. 2020, 18, 1859-1865. [CrossRef]
- Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J Clin Virol. 2020, 127:104362. [CrossRef]
- Avnon LS, Munteanu D, Smoliakov A, Jotkowitz A, Barski L. Thromboembolic events in patients with severe pandemic influenza A/H1N1. Eur J Intern Med. 2015, 26, 596-598. [CrossRef]
- Fridlender ZG, Khamaisi M, Leitersdorf E. Association between cytomegalovirus infection and venous thromboembolism. Am J Med Sci. 2007, 334, 111-114. [CrossRef]
- Kahn SR, Lim W, Dunn AS, Cushman M, Dentali F, Akl EA, Cook DJ, Balekian AA, Klein RC, Le H, Schulman S, Murad MH. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012, 141(2 Suppl):e195S-e226S. [CrossRef]
- Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020, 135, 2033-2040. [CrossRef]
- Lazzaroni MG, Piantoni S, Masneri S, Garrafa E, Martini G, Tincani A, Andreoli L, Franceschini F. Coagulation dysfunction in COVID-19: The interplay between inflammation, viral infection and the coagulation system. Blood Rev. 2021, 46:100745. [CrossRef]
- Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet. 2020, 395, 1033-1034. [CrossRef]
- Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. 2020, 111:102452. [CrossRef]
- Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020, 368, 473-474. [CrossRef]
- Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, Xie C, Ma K, Shang K, Wang W, Tian DS. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020, 71, 762-768. [CrossRef]
- Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect. 2020, 80, 607-613. [CrossRef]
- Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID-19: Interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents. 2020, 55, 105954. [CrossRef]
- Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. 2017, 149:38-44. [CrossRef]
- Folman CC, Linthorst GE, van Mourik J, van Willigen G, de Jonge E, Levi M, de Haas M, von dem Borne AE. Platelets release thrombopoietin (Tpo) upon activation: Another regulatory loop in thrombocytopoiesis? Thromb Haemost. 2000, 83, 923-930.
- Levi M, van der Poll T, Schultz M. Systemic versus localized coagulation activation contributing to organ failure in critically ill patients. Semin Immunopathol 2012, 34:167–79 - Cerca con Google. Accessed April 5, 2023. https://www.google.com/search?q=Levi+M%2C+van+der+Poll+T%2C+Schultz+M.+Systemic+versus+localized+coagulation+activation+contributing+to+organ+failure+in+critically+ill+patients.+Semin+Immunopathol+2012%3B+34%3A167%E2%80%9379&oq=Levi+M%2C+van+der+Poll+T%2C+Schultz+M.+Systemic+versus+localized+coagulation+activation+contributing+to+organ+failure+in+critically+ill+patients.+Semin+Immunopathol+2012%3B+34%3A167%E2%80%9379&aqs=chrome..69i57.248j0j4&sourceid=chrome&ie=UTF-8.
- Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir Med. 2020, 8, 681-686. [CrossRef]
- Yang M, Ng MH, Li CK. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematology. 2005, 10, 101-105. [CrossRef]
- Stouthard JM, Levi M, Hack CE, Veenhof CH, Romijn HA, Sauerwein HP, van der Poll T. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost. 1996, 76, 738-742.
- Conti P, Caraffa A, Gallenga CE, Ross R, Kritas SK, Frydas I, Younes A, Ronconi G. Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: A promising inhibitory strategy. J Biol Regul Homeost Agents. 2020, 34, 1971-1975. [CrossRef]
- Magro G. SARS-CoV-2 and COVID-19: Is interleukin-6 (IL-6) the “culprit lesion” of ARDS onset? What is there besides Tocilizumab? SGP130Fc. Cytokine X. 2020, 2, 100029. [CrossRef]
- van de Veerdonk FL, Netea MG. Blocking IL-1 to prevent respiratory failure in COVID-19. Crit Care. 2020, 24:445. [CrossRef]
- Zhao Y, Qin L, Zhang P, Li K, Liang L, Sun J, Xu B, Dai Y, Li X, Zhang C, Peng Y, Feng Y, Li A, Hu Z, Xiang H, Ogg G, Ho LP, McMichael A, Jin R, Knight JC, Dong T, Zhang Y. Longitudinal COVID-19 profiling associates IL-1RA and IL-10 with disease severity and RANTES with mild disease. JCI Insight. 2020, 5, e139834, 139834. [CrossRef]
- Satış H, Özger HS, Aysert Yıldız P, Hızel K, Gulbahar Ö, Erbaş G, Aygencel G, Guzel Tunccan O, Öztürk MA, Dizbay M, Tufan A. Prognostic value of interleukin-18 and its association with other inflammatory markers and disease severity in COVID-19. Cytokine. 2021, 137:155302. [CrossRef]
- Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: The vasculature unleashed. Nat Rev Immunol. 2020, 20, 389-391. [CrossRef]
- Martini R. The compelling arguments for the need of microvascular investigation in COVID-19 critical patients. Clin Hemorheol Microcirc. 2020, 75, 27-34. [CrossRef]
- Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, Romero JP, Wirnsberger G, Zhang H, Slutsky AS, Conder R, Montserrat N, Mirazimi A, Penninger JM. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020, 181, 905-913.e7. [CrossRef]
- Stern D, Nawroth P, Handley D, Kisiel W. An endothelial cell-dependent pathway of coagulation. Proc Natl Acad Sci U S A. 1985, 82, 2523-2527. [CrossRef]
- Guang C, Phillips RD, Jiang B, Milani F. Three key proteases – angiotensin-I-converting enzyme (ACE), ACE2 and renin – within and beyond the renin-angiotensin system. Arch Cardiovasc Dis. 2012, 105, 373-385. [CrossRef]
- Scialo F, Daniele A, Amato F, Pastore L, Matera MG, Cazzola M, Castaldo G, Bianco A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung. 2020, 198, 867-877. [CrossRef]
- Marshall RP. The Pulmonary Renin-Angiotensin System. Current Pharmaceutical Design. 9, 715-722. [CrossRef]
- Wu YP, Wei R, Liu ZH, Chen B, Lisman T, Ren DL, Han JJ, Xia ZL, Zhang FS, Xu WB, Preissner KT, de Groot PG. Analysis of thrombotic factors in severe acute respiratory syndrome (SARS) patients. Thromb Haemost. 2006, 96, 100-101. [CrossRef]
- Reheman A, Gross P, Yang H, Chen P, Allen D, Leytin V, Freedman J, Ni H. Vitronectin stabilizes thrombi and vessel occlusion but plays a dual role in platelet aggregation. J Thromb Haemost. 2005, 3, 875-883. [CrossRef]
- Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, Wang C, Zhao J, Sun X, Tian R, Wu W, Wu D, Ma J, Chen Y, Zhang D, Xie J, Yan X, Zhou X, Liu Z, Wang J, Du B, Qin Y, Gao P, Qin X, Xu Y, Zhang W, Li T, Zhang F, Zhao Y, Li Y, Zhang S. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N Engl J Med. 2020, 382, e38. [CrossRef]
- Borghi MO, Beltagy A, Garrafa E, Curreli D, Cecchini G, Bodio C, Grossi C, Blengino S, Tincani A, Franceschini F, Andreoli L, Lazzaroni MG, Piantoni S, Masneri S, Crisafulli F, Brugnoni D, Muiesan ML, Salvetti M, Parati G, Torresani E, Mahler M, Heilbron F, Pregnolato F, Pengo M, Tedesco F, Pozzi N, Meroni PL. Anti-Phospholipid Antibodies in COVID-19 Are Different From Those Detectable in the Anti-Phospholipid Syndrome. Front Immunol. 2020, 11:584241. [CrossRef]
- Gómez-Mesa JE, Galindo-Coral S, Montes MC, Muñoz Martin AJ. Thrombosis and Coagulopathy in COVID-19. Curr Probl Cardiol. 2021, 46, 100742. [CrossRef]
- Li Q, Cao Y, Chen L, Wu D, Yu J, Wang H, He W, Chen L, Dong F, Chen W, Chen W, Li L, Ran Q, Liu Q, Ren W, Gao F, Chen Z, Gale RP, Hu Y. Hematological features of persons with COVID-19. Leukemia. 2020, 34, 2163-2172. [CrossRef]
- Liu Y, Sun W, Guo Y, Chen L, Zhang L, Zhao S, Long D, Yu L. Association between platelet parameters and mortality in coronavirus disease 2019: Retrospective cohort study. Platelets. 2020, 31, 490-496. [CrossRef]
- Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, Garraud O. Human platelets and their capacity of binding viruses: Meaning and challenges? BMC Immunol. 2015, 16:26. [CrossRef]
- Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front Immunol. 2014, 5:649. [CrossRef]
- Seyoum M, Enawgaw B, Melku M. Human blood platelets and viruses: Defense mechanism and role in the removal of viral pathogens. Thromb J. 2018, 16:16. [CrossRef]
- Aleem A, Nadeem AJ. Coronavirus (COVID-19) Vaccine-Induced Immune Thrombotic Thrombocytopenia (VITT). In: StatPearls. StatPearls Publishing; 2023. Accessed April 20, 2023. http://www.ncbi.nlm.nih.gov/books/NBK570605/.
- Oldenburg J, Klamroth R, Langer F, Albisetti M, von Auer C, Ay C, Korte W, Scharf RE, Pötzsch B, Greinacher A. Diagnosis and Management of Vaccine-Related Thrombosis following AstraZeneca COVID-19 Vaccination: Guidance Statement from the GTH. Hamostaseologie. 2021, 41, 184-189. [CrossRef]
- Miller E. Rapid evaluation of the safety of COVID-19 vaccines: How well have we done? Clin Microbiol Infect. 2022, 28, 477-478. [CrossRef]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016, 315, 801-810. [CrossRef]
- McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437-e445. [CrossRef]
- Asakura H, Ogawa H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int J Hematol. 2021, 113, 45-57. [CrossRef]
- Rahman S, Montero MTV, Rowe K, Kirton R, Kunik F. Epidemiology, pathogenesis, clinical presentations, diagnosis and treatment of COVID-19: A review of current evidence. Expert Rev Clin Pharmacol. 2021, 14, 601-621. [CrossRef]
- Zhang X, Yang X, Jiao H, Liu X. Coagulopathy in patients with COVID-19: A systematic review and meta-analysis. Aging (Albany NY). 2020, 12, 24535-24551. [CrossRef]
- Fiorentino G, Benincasa G, Coppola A, Franzese M, Annunziata A, Affinito O, Viglietti M, Napoli C. Targeted genetic analysis unveils novel associations between ACE I/D and APO T158C polymorphisms with D-dimer levels in severe COVID-19 patients with pulmonary embolism. J Thromb Thrombolysis. 2023, 55, 51-59. [CrossRef]
- Xiang G, Hao S, Fu C, Hu W, Xie L, Wu Q, Li S, Liu X. The effect of coagulation factors in 2019 novel coronavirus patients. Medicine (Baltimore). 2021, 100, e24537. [CrossRef]
- Xiong M, Liang X, Wei YD. Changes in blood coagulation in patients with severe coronavirus disease 2019 (COVID-19): A meta-analysis. Br J Haematol. 2020, 189, 1050-1052. [CrossRef]
- Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020, 323, 1239-1242. [CrossRef]
- Léonard-Lorant I, Delabranche X, Séverac F, Helms J, Pauzet C, Collange O, Schneider F, Labani A, Bilbault P, Molière S, Leyendecker P, Roy C, Ohana M. Acute Pulmonary Embolism in Patients with COVID-19 at CT Angiography and Relationship to d-Dimer Levels. Radiology. 2020, 296, E189-E191. [CrossRef]
- Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Schröder AS, Burdelski C, de Heer G, Nierhaus A, Frings D, Pfefferle S, Becker H, Bredereke-Wiedling H, de Weerth A, Paschen HR, Sheikhzadeh-Eggers S, Stang A, Schmiedel S, Bokemeyer C, Addo MM, Aepfelbacher M, Püschel K, Kluge S. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann Intern Med. 2020, 173, 268-277. [CrossRef]
- Demelo-Rodríguez P, Cervilla-Muñoz E, Ordieres-Ortega L, Parra-Virto A, Toledano-Macías M, Toledo-Samaniego N, García-García A, García-Fernández-Bravo I, Ji Z, de-Miguel-Diez J, Álvarez-Sala-Walther LA, Del-Toro-Cervera J, Galeano-Valle F. Incidence of asymptomatic deep vein thrombosis in patients with COVID-19 pneumonia and elevated D-dimer levels. Thromb Res. 2020, 192:23-26. [CrossRef]
- Nahum J, Morichau-Beauchant T, Daviaud F, Echegut P, Fichet J, Maillet JM, Thierry S. Venous Thrombosis Among Critically Ill Patients With Coronavirus Disease 2019 (COVID-19). JAMA Netw Open. 2020, 3, e2010478. [CrossRef]
- Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MCA, Bouman CCS, Beenen LFM, Kootte RS, Heijmans J, Smits LP, Bonta PI, van Es N. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. 2020, 18, 1995-2002. [CrossRef]
- Shah A, Donovan K, McHugh A, Pandey M, Aaron L, Bradbury CA, Stanworth SJ, Alikhan R, Von Kier S, Maher K, Curry N, Shapiro S, Rowland MJ, Thomas M, Mason R, Holland M, Holmes T, Ware M, Gurney S, McKechnie SR. Thrombotic and haemorrhagic complications in critically ill patients with COVID-19: A multicentre observational study. Crit Care. 2020, 24, 561. [CrossRef]
- Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020, 18, 1421-1424. [CrossRef]
- Carfora V, Spiniello G, Ricciolino R, Di Mauro M, Migliaccio MG, Mottola FF, Verde N, Coppola N, Coppola N, Sagnelli C, De Pascalis S, Stanzione M, Stornaiuolo G, Cascone A, Martini S, Macera M, Monari C, Calò F, Bianco A, Russo A, Gentile V, Camaioni C, De Angelis G, Marino G, Astorri R, De Sio I, Niosi M, Borrelli S, Celia B, Ceparano M, Cirillo S, De Luca M, Mazzeo G, Paoli G, Russo MG, Carfora V, Di Mauro M, Migliaccio MG, Mottola FF, Ricciolino R, Spiniello G, Verde N, Vanvitelli COVID-19 group. Anticoagulant treatment in COVID-19: A narrative review. J Thromb Thrombolysis. 2021, 51, 642-648. [CrossRef]
- Shi C, Wang C, Wang H, Yang C, Cai F, Zeng F, Cheng F, Liu Y, Zhou T, Deng B, Vlodavsky I, Li JP, Zhang Y. The potential of low molecular weight heparin to mitigate cytokine storm in severe COVID-19 patients: A retrospective clinical study. Published online April 15, 2020:2020.03.28.20046144. [CrossRef]
- Godino C, Scotti A, Maugeri N, Mancini N, Fominskiy E, Margonato A, Landoni G. Antithrombotic therapy in patients with COVID-19? -Rationale and Evidence-. International Journal of Cardiology. 2021, 324:261-266. [CrossRef]
- Mangana C, Lorigo M, Cairrao E. Implications of Endothelial Cell-Mediated Dysfunctions in Vasomotor Tone Regulation. Biologics. 2021, 1, 231-251. [CrossRef]
- Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009, 15, 1318-1321. [CrossRef]
- Zhang X, Li X. The Role of Histones and Heparin in Sepsis: A Review. J Intensive Care Med. 2022, 37, 319-326. [CrossRef]
- Mangiafico M, Caff A, Costanzo L. The Role of Heparin in COVID-19: An Update after Two Years of Pandemics. J Clin Med. 2022, 11, 3099. [CrossRef]
- Mousavi S, Moradi M, Khorshidahmad T, Motamedi M. Anti-Inflammatory Effects of Heparin and Its Derivatives: A Systematic Review. Adv Pharmacol Sci. 2015, 2015:507151. [CrossRef]
- Thachil J. The versatile heparin in COVID-19. J Thromb Haemost. 2020, 18, 1020-1022. [CrossRef]
- Oduah EI, Linhardt RJ, Sharfstein ST. Heparin: Past, Present, and Future. Pharmaceuticals (Basel). 2016, 9, 38. [CrossRef]
- Shukla D, Spear PG. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J Clin Invest. 2001, 108, 503-510. [CrossRef]
- Ghezzi S, Cooper L, Rubio A, Pagani I, Capobianchi MR, Ippolito G, Pelletier J, Meneghetti MCZ, Lima MA, Skidmore MA, Broccoli V, Yates EA, Vicenzi E. Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells. Antiviral Res. 2017, 140:13-17. [CrossRef]
- Mycroft-West C, Su D, Elli S, Guimond S, Miller G, Turnbull J, Yates E, Guerrini M, Fernig D, Lima M, Skidmore M. The 2019 coronavirus (SARS-CoV-2) surface protein (Spike) S1 Receptor Binding Domain undergoes conformational change upon heparin binding. Published online March 2, 2020:2020.02.29.971093. [CrossRef]
- REMAP-CAP Investigators, ACTIV-4a Investigators, ATTACC Investigators, Goligher EC, Bradbury CA, McVerry BJ, Lawler PR, Berger JS, Gong MN, Carrier M, Reynolds HR, Kumar A, Turgeon AF, Kornblith LZ, Kahn SR, Marshall JC, Kim KS, Houston BL, Derde LPG, Cushman M, Tritschler T, Angus DC, Godoy LC, McQuilten Z, Kirwan BA, Farkouh ME, Brooks MM, Lewis RJ, Berry LR, Lorenzi E, Gordon AC, Ahuja T, Al-Beidh F, Annane D, Arabi YM, Aryal D, Baumann Kreuziger L, Beane A, Bhimani Z, Bihari S, Billett HH, Bond L, Bonten M, Brunkhorst F, Buxton M, Buzgau A, Castellucci LA, Chekuri S, Chen JT, Cheng AC, Chkhikvadze T, Coiffard B, Contreras A, Costantini TW, de Brouwer S, Detry MA, Duggal A, Džavík V, Effron MB, Eng HF, Escobedo J, Estcourt LJ, Everett BM, Fergusson DA, Fitzgerald M, Fowler RA, Froess JD, Fu Z, Galanaud JP, Galen BT, Gandotra S, Girard TD, Goodman AL, Goossens H, Green C, Greenstein YY, Gross PL, Haniffa R, Hegde SM, Hendrickson CM, Higgins AM, Hindenburg AA, Hope AA, Horowitz JM, Horvat CM, Huang DT, Hudock K, Hunt BJ, Husain M, Hyzy RC, Jacobson JR, Jayakumar D, Keller NM, Khan A, Kim Y, Kindzelski A, King AJ, Knudson MM; et al. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with Covid-19. N Engl J Med. 2021, 385, 777-789. [CrossRef]
- Stasi C, Fallani S, Voller F, Silvestri C. Treatment for COVID-19: An overview. Eur J Pharmacol. 2020, 889:173644. [CrossRef]
- Mennuni MG, Renda G, Grisafi L, Rognoni A, Colombo C, Lio V, Foglietta M, Petrilli I, Pirisi M, Spinoni E, Azzolina D, Hayden E, Aimaretti G, Avanzi GC, Bellan M, Cantaluppi V, Capponi A, Castello LM, D’Ardes D, Corte FD, Gallina S, Krengli M, Malerba M, Pierdomenico SD, Savoia P, Zeppegno P, Sainaghi PP, Cipollone F, Patti G, COVID-UPO Clinical Team. Clinical outcome with different doses of low-molecular-weight heparin in patients hospitalized for COVID-19. J Thromb Thrombolysis. 2021, 52, 782-790. [CrossRef]
- Rentsch CT, Beckman JA, Tomlinson L, Gellad WF, Alcorn C, Kidwai-Khan F, Skanderson M, Brittain E, King JT, Ho YL, Eden S, Kundu S, Lann MF, Greevy RA, Ho PM, Heidenreich PA, Jacobson DA, Douglas IJ, Tate JP, Evans SJW, Atkins D, Justice AC, Freiberg MS. Early initiation of prophylactic anticoagulation for prevention of coronavirus disease 2019 mortality in patients admitted to hospital in the United States: Cohort study. BMJ. 2021, 372:n311. [CrossRef]
- ISTH interim guidance on recognition and management of coagulopathy in COVID-19 - Journal of Thrombosis and Haemostasis. Accessed April 24, 2023. https://www.jthjournal.org/article/S1538-7836(22)00324-5/fulltext.
- Hasan SS, Radford S, Kow CS, Zaidi STR. Venous thromboembolism in critically ill COVID-19 patients receiving prophylactic or therapeutic anticoagulation: A systematic review and meta-analysis. J Thromb Thrombolysis. 2020, 50, 814-821. [CrossRef]
- Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, Clark C, Iba T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. Journal of Thrombosis and Haemostasis. 2020, 18, 1023-1026. [CrossRef]
- Sayed Ahmed HA, Merrell E, Ismail M, Joudeh AI, Riley JB, Shawkat A, Habeb H, Darling E, Goweda RA, Shehata MH, Amin H, Nieman GF, Aiash H. Rationales and uncertainties for aspirin use in COVID-19: A narrative review. Fam Med Community Health. 2021, 9, e000741. [CrossRef]
Figure 1.
Schematic representation of endothelium activation towards a pro-thrombotic status. Pathogens and inflammatory mediators from injured host tissue activate monocytes and induce the expression of tissue factor on monocytes and endothelial cell surfaces. Subsequently, activated monocytes release inflammatory cytokines and chemokines amplifying the inflammatory response and stimulating vascular endothelial cells changing their properties to a procoagulant state. NETs: Neutrophil Extracellular Traps; TNF𝜶: Tumor Necrosis Factor alpha; IFN𝛾: Interferon gamma; MCP-1: monocyte chemotactic protein -1; IL: Interleukin.
Figure 1.
Schematic representation of endothelium activation towards a pro-thrombotic status. Pathogens and inflammatory mediators from injured host tissue activate monocytes and induce the expression of tissue factor on monocytes and endothelial cell surfaces. Subsequently, activated monocytes release inflammatory cytokines and chemokines amplifying the inflammatory response and stimulating vascular endothelial cells changing their properties to a procoagulant state. NETs: Neutrophil Extracellular Traps; TNF𝜶: Tumor Necrosis Factor alpha; IFN𝛾: Interferon gamma; MCP-1: monocyte chemotactic protein -1; IL: Interleukin.
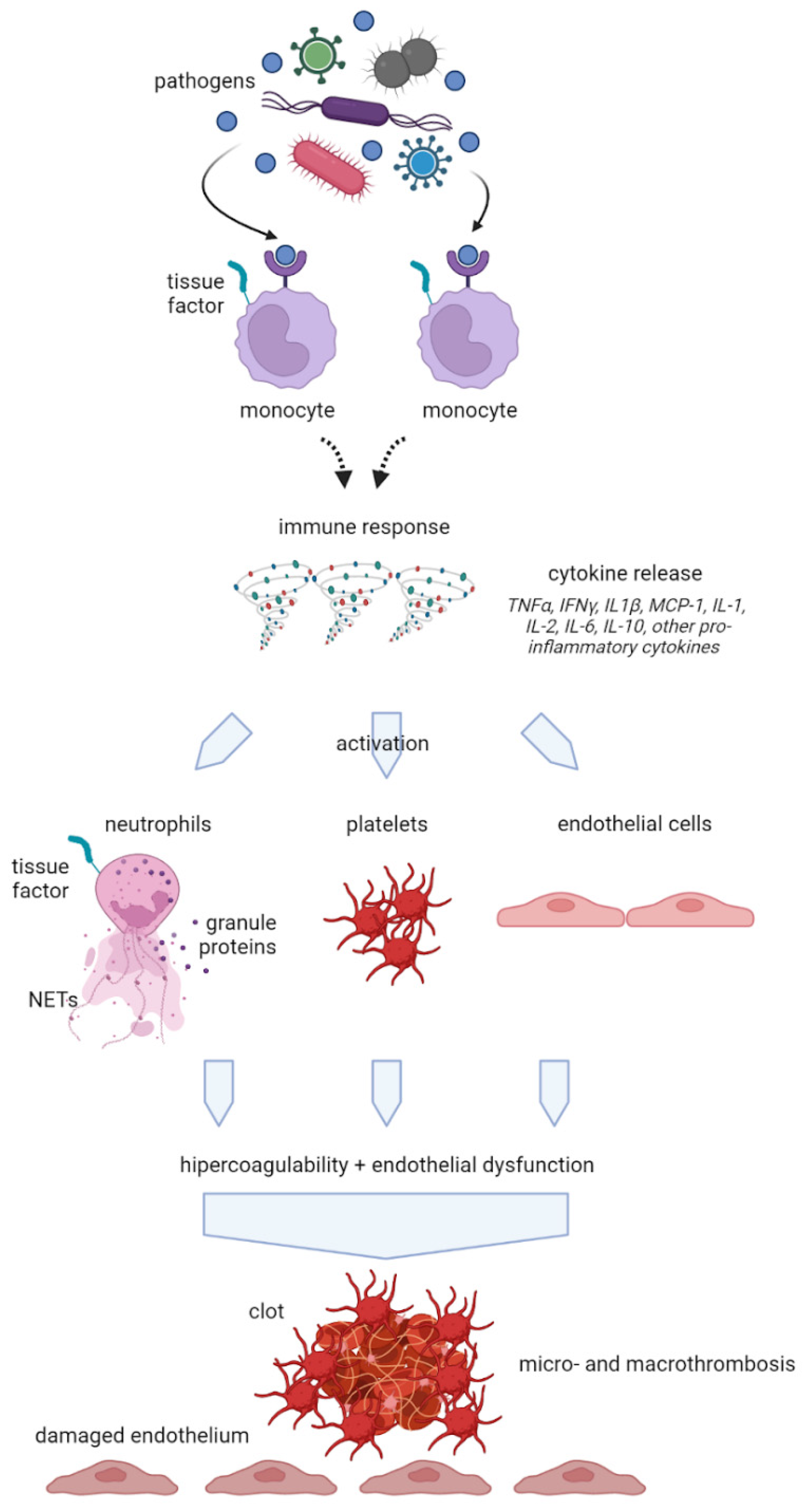
Figure 2.
Schematic representation of the interlink between inflammatory and thrombotic mechanisms after COVID-19 infection. NETs: Neutrophil Extracellular Traps.
Figure 2.
Schematic representation of the interlink between inflammatory and thrombotic mechanisms after COVID-19 infection. NETs: Neutrophil Extracellular Traps.
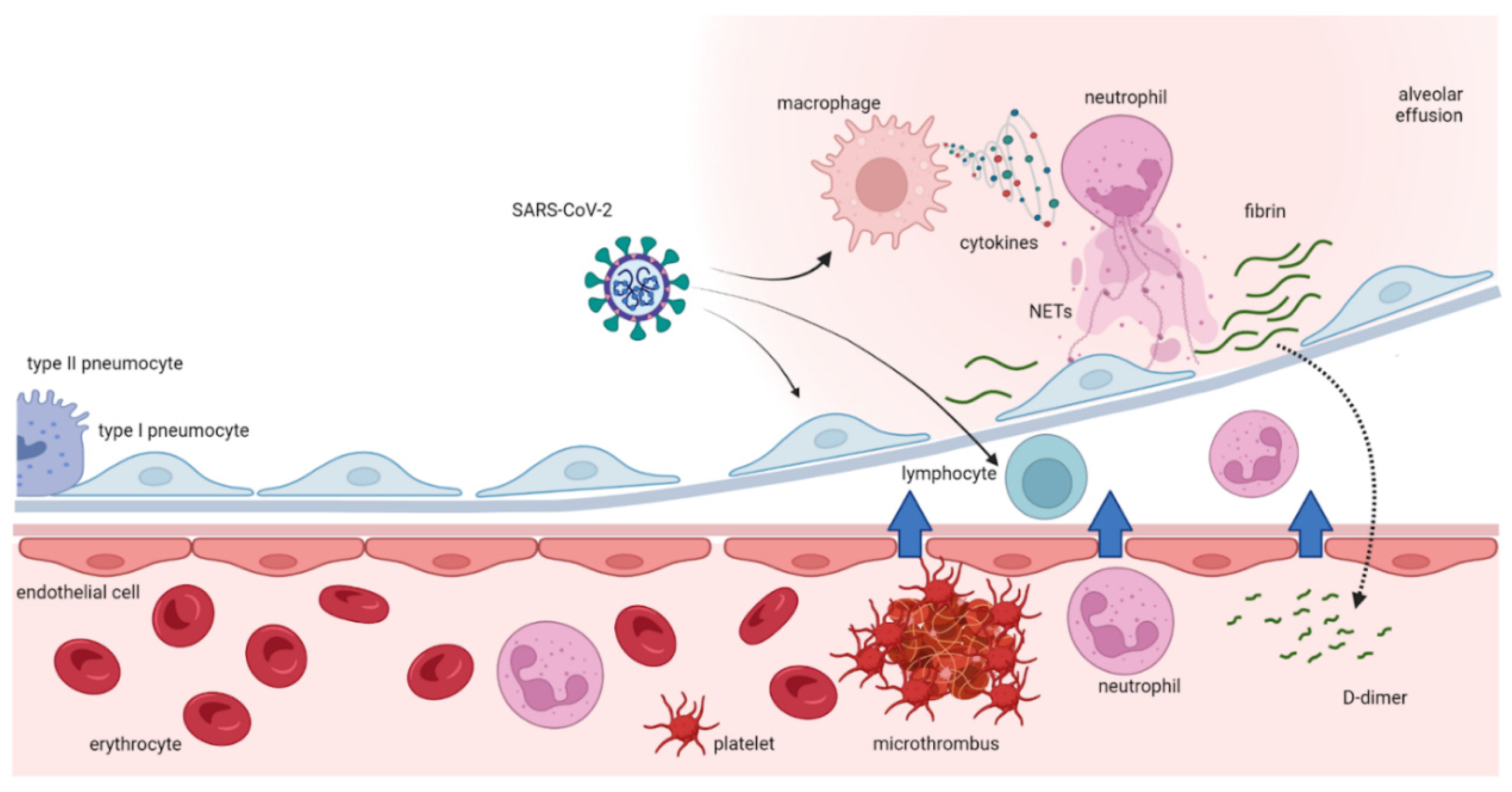
Figure 3.
Effects of the inflammatory response to COVID-19 infection with cytokine release syndrome and dysregulation of immune system with the final effect of a hyper-coagulative state. DAMPs: damage-associated molecular patterns TNF-𝜶: Tumor Necrosis Factor alpha; IFN𝛾: Interferon gamma; MCP-1: monocyte chemotactic protein -1; IL: Interleukin.
Figure 3.
Effects of the inflammatory response to COVID-19 infection with cytokine release syndrome and dysregulation of immune system with the final effect of a hyper-coagulative state. DAMPs: damage-associated molecular patterns TNF-𝜶: Tumor Necrosis Factor alpha; IFN𝛾: Interferon gamma; MCP-1: monocyte chemotactic protein -1; IL: Interleukin.
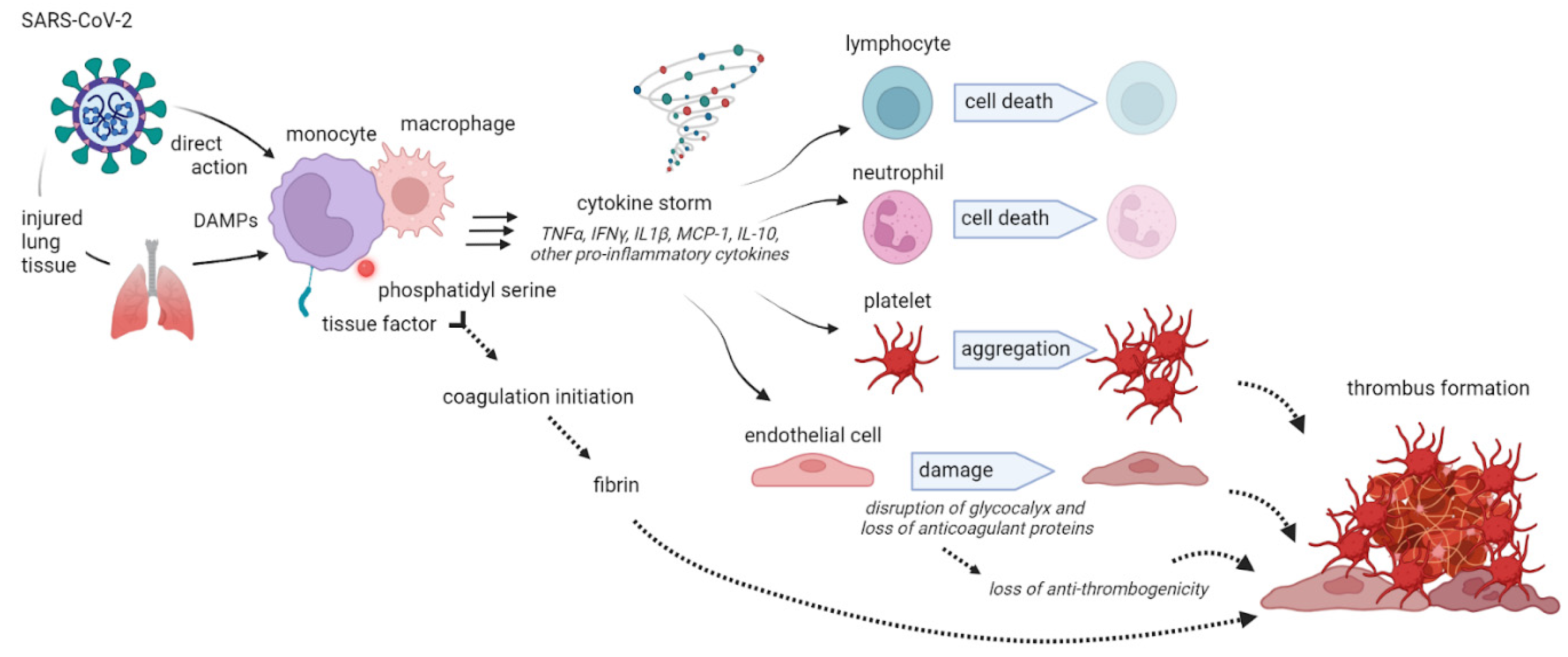
Figure 4.
Imbalance between coagulation and fibrinolysis: the effects by SARS-CoV2. a. Physiologically, ACE2 reduces the availability of angiotensin II with no effects on coagulation and fibrinolysis. b. SARS-CoV2 reducing ACE2 availability, increases the level of Angiotensin II and the PAI-1 and favors the activation of coagulation system. ACE2: Angiotensin Converting Enzyme 2, PAI-1: plasminogen activator inhibitor 1 .
Figure 4.
Imbalance between coagulation and fibrinolysis: the effects by SARS-CoV2. a. Physiologically, ACE2 reduces the availability of angiotensin II with no effects on coagulation and fibrinolysis. b. SARS-CoV2 reducing ACE2 availability, increases the level of Angiotensin II and the PAI-1 and favors the activation of coagulation system. ACE2: Angiotensin Converting Enzyme 2, PAI-1: plasminogen activator inhibitor 1 .
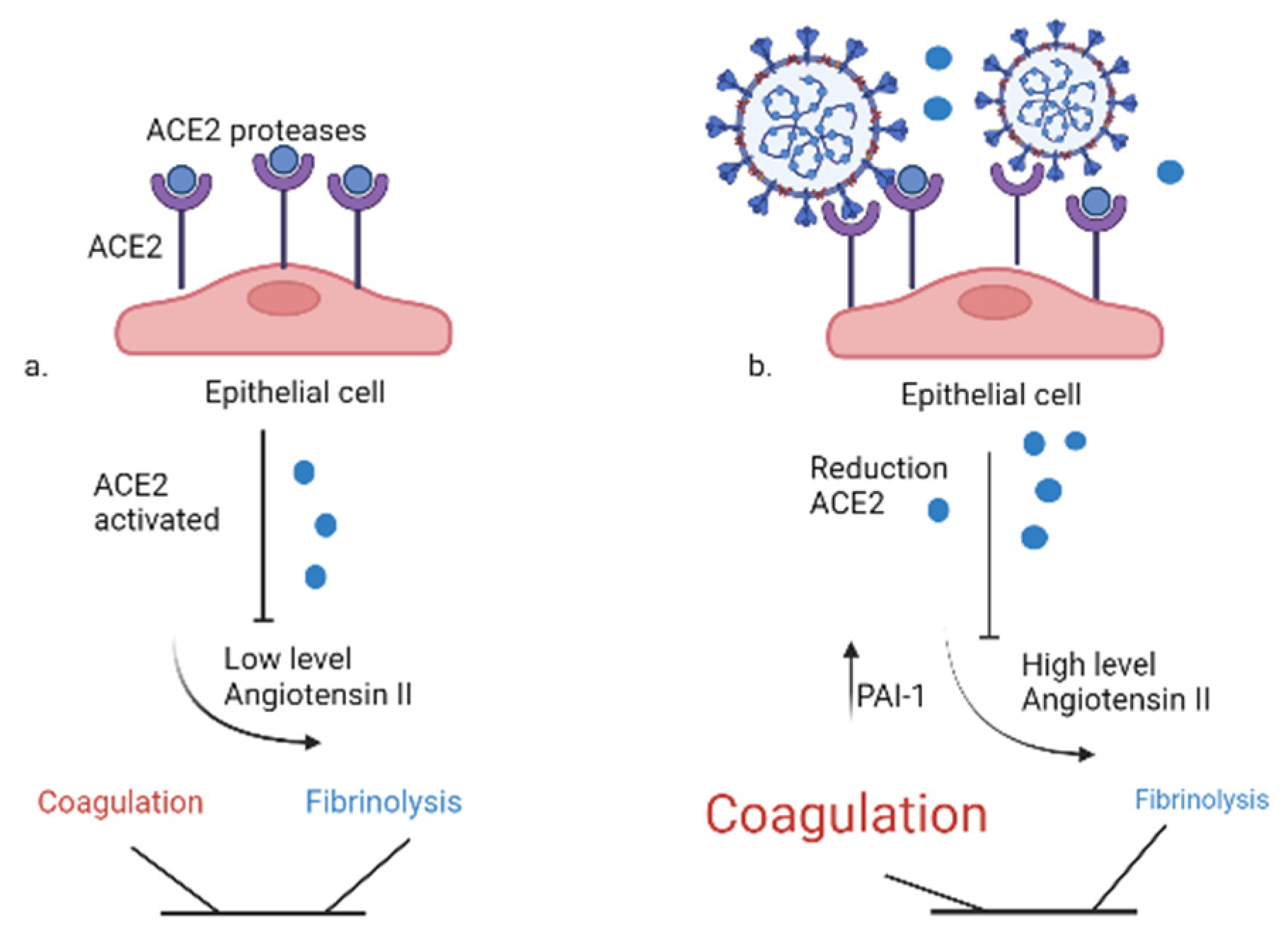
Table 1.
Sepsis induced coagulopathy (SIC) score. ISTH score.
| Item | Value | Score |
|---|---|---|
| SOFA score | 1 ≥2 |
1 2 ≥4 |
| PT-INR | 1.2-1.4 >1.4 |
1 2 |
| Platelet count (x mm3) | 100.000-150.000 < 100.000 |
1 2 |
INR: international normalized ratio; PT: prothrombin time; SOFA: Sequential Organ Failure Assessment.
Table 2.
Increasing of Coagulation and Inflammatory Biomarkers .
| Coagulation biomarkers | D-di D-dimer, PLT, PT, APTT, FIB |
| Inflammatory biomarkers | ES ESR, CRP, Serum ferritin, PCT, IL-2, IL-6, IL8, IL10 |
Platelets (PLT), Prothrombin time (PT), activated partial thromboplastin time (APTT) Fibrinogen (FIB). erythrocyte sedimentation rate (ESR) C-reactive protein (CRP) Procalcitonin (PCT) Interleukin (IL).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



