
Submitted:
27 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
The kidney and the heart work together in balancing the body circulation and although their physiology is based on strict inter dependence, their performance fulfills different aims. While the heart can rapidly increase its own oxygen consumption to comply with the wide changes in metabolic demand linked to body function, the kidney physiology is primarily designed to maintain a stable metabolic rate and has limited capacity to cope with any steep increase in renal metabolism. In the kidneys the glomerular population filters a large amount of blood and the tubular system has been programmed to reabsorb 99% of filtrate by reabsorbing sodium together with other filtered substances including all glucose molecules. Glucose reabsorption involves the sodium-glucose cotransporters SGLT2 and SGLT1 on the apical membrane in the proximal tubular section; it also enhances bicarbonate formation so as to preserve the acid-base balance. The complex work of reabsorption in the kidney is the main factor in renal oxygen consumption; analysis of renal glucose transport in disease states is providing better understanding of the renal physiology changes that occur when clinical conditions alter the neurohormonal response leading to an increase in glomerular filtration pressure. In this circumstance glomerular hyperfiltration occurs, imposing a higher metabolic demand on kidney physiology and causing progressive renal impairment. Albumin urination is the warning signal of renal engagement over-exertion and most frequently heralds heart failure development, regardless of disease etiology. The review analyzes the mechanisms linked to renal oxygen consumption, focusing on sodium-glucose management.
Keywords:
glucose transport
; SGLT2 inhibitor
; diabetic nephropathy
; heart failure
; chronic kidney disease
1. Introduction
Sodium glucose cotransporter 2 inhibitors (SGLT2i) are a relatively novel class of molecules named gliflozines that were originally designed to control glycemia, but over and above their effect on the glycemic metabolism, they unexpectedly improved the management of clinical conditions involving heart and kidney function. Importantly, the SGLT2i benefits went beyond guidelines-directed medical therapy, in that the drug’s metabolic effects on the kidney and heart ameliorated the whole cardio-circulatory outcome.
Since research began, SGLT2 has proved to decrease hemoglobin A1c by ≈0.7% to 1.0% and to induce weight loss through glycosuria; through concomitant natriuresis and osmotic diuresis it succeeds in contracting plasma volume by ≈7%, contributing ≈5/2–mm Hg antihypertensive effects; lastly it has also proved to reduce albuminuria by 30% - 40% through the intrarenal hemodynamic effect [1]. Notwithstanding the apparently limited range of such benefits, a wealth of investigations performed with SGLT2i have revealed that this class of drug can affect the cardiovascular outcome in diabetics with [2] and without cardiovascular damage [3], diabetics and non-diabetics with advanced CKD with and without renal albuminuric insult [4,5,6] and diabetic and non-diabetic heart failure (HF) patients especially via the ventricular ejection fraction spectrum [7,8,9,10].
Although the mechanisms responsible for SGLT2 inhibition benefits have only partly been elucidated and are still a matter for research, the results of many pivotal studies have inevitably affected cardiology practice and the point has been recognized in the most recent international guideline releases, with a special focus on HF management [11]. Note that gliflozines have displayed high efficacy in decreasing HF onset and exacerbation following optimal therapy, results found within weeks of drug administration. The benefits of these molecules have also proved to affect the overall cardiovascular outcome in subjects with atherosclerosis as their most frequent background pathology, displaying a time-related association with the restraining of CKD progression. Since the principal pharmacologic action of these drugs occurs by blocking SGLT 2 function in the glomerular tubule proximal segment, preventing reabsorption of filtered glucose coupled with Na+ and thus decreasing overall renal glucose reabsorption by approximately 60% and overall kidney Na+ reabsorption by less than 5% [1], the question that arises is, do those pharmacologic effects provide a reasonable explanation of the observed extension of clinical benefits. The question is an important one: SGLT2i is such a potent class of drug, that prescribing it entails responsibility in deciding therapy and demands we understand the manifold action this class of drug can provide in clinical conditions where kidney function is involved and corrupted to a varying extent.
2. Renal physiology, a unique example based on efficient control of oxygen consumption
It is commonly recognized that heart and kidney share pathophysiological mechanisms that generate clinical conditions in both, but little attention is paid to the evidence that their performance is designed to satisfy opposing demands. The heart has to provide each organ and apparatus with oxygen and nutrition on the basis of intercurrent demand without any predefined schedule, while the kidney has to adapt body fluid and electrolyte content to physiological needs in order to cope with changes in the internal and external environment. The heart and kidney therefore cope with two different physiological needs, organ energy requirements vs organ function balance.
When one compares the renal and cardiac physiology, the similarities and differences become apparent as the cardiac and the renal mass are approximately the same (0.4-0.5% of body mass), and likewise the metabolic rate (Ki) which is the highest in the body Ki 440 (in Kcal/Kg/day), while O2 consumption is higher in the heart, 11% vs 7% [12,13,14], though the blood flow is much in favor of the kidney: 25% vs 7% [14,15,16].
The peculiar heart structure, through the engagement of organ-specific mechanisms such as heart rate and the Frank-Starling law - prominent contributors [17] besides contractility and Laplace’s law -, can swiftly increase the cardiac output upon demand via a large range of performances exploiting the strict relation to the phasic coronary flow which may rise as much as 400% in trained subjects. The unmatched capacity of the coronary microvasculature to increase tissue delivery to the energy-craving cardiomyocyte metabolism is provided by physical training through increased arteriolar densities and/or diameters together with the formation of new capillaries that maintain the vasculature at a level commensurate with the degree of exercise-induced physiological myocardial hypertrophy. On top of this, physical training alters the distribution of coronary vascular resistance so that more capillaries are recruited, resulting in an increase in the permeability-surface area product, regardless of numerical capillary density [16].
Kidney physiology works differently and despite the much higher arterial blood flow, the unique renal structure and physiology combine to maintain the filtrate around 180 L daily with an approximate glomerular filtration rate (GFR) of 125 mL/min, while the daily urine output may vary from 0.5 to 2.5 l/day in relation to the dryness of the ambience. It has been shown that renal O2 extraction remains stable over a wide range of renal blood flow [18], indicating that the high renal arterial flow undergoes strict regulation at different levels. The extensive renal sympathetic innervation largely governs the neurohumoral system response which ensures kidney perfusion, affecting the circulation pressure [19], while peculiarities of the vascular architecture trigger preglomerular diffusional shunting of oxygen from arteries to veins, regulating the arterial flow directed toward the nephrons. Besides the above-mentioned regulatory systems in the kidneys, the glomerular hemodynamics and/or transport processes are balanced by local release of various autacoids (nitric oxide, bradykinin, endothelin, angiotensin II, and prostanoids, to name a few) [20] .
In the kidney the wealth of regulatory mechanisms can provide continuous adjustment of vasoconstriction and vasodilation in the afferent and efferent arterioles, counteracting wide-ranging changes in the blood pressure and keeping intraglomerular pressure stable. These regulatory mechanisms are the main factors behind GFR generation, but the complexity of renal physiology regulation makes it largely impossible to predict or assess O2 consumption [21]. One is struck by how the kidney labors to provide continuous checks and balances on body fluid and circulatory pressure. Indeed, the physiology of the kidney is specifically designed to provide efficient blood clearing while maintaining the whole circulatory balance without compromising O2 distribution to its own highly specialized tissues as they perform their endocrine, metabolic and reabsorbing functions.
These highly specific renal activities are sustained by adenosine triphosphate (ATP) production using largely aerobic mechanisms (roughly 95%), whereas some nephron segments, particularly in the less perfused medulla, can use anaerobic metabolism more efficiently [19,20,21].
We must remember that the renal blood flow is the main contributor to determining the glomerular filtration rate (GFR), and as the plasma Na+ concentration is relatively constant in normal subjects, the GFR directly determines the Na+ load in the glomerular filtrate. Given that the kidney reabsorbs 99.5% of filtered Na+ via activation of the ATP-dependent Na+/K+ pump, the renal O2 consumption driven by Na+ reabsorption is proportionate to the GFR [21,22]. The point deserves consideration: if we assume a renal filtrate of 180 L/day, in the presence of 140 mmol of Na+ plasma concentration, the kidneys have to reabsorb ≈ 25 moles of Na+. As one mole of O2 can generate 3 moles of ATP and 1 mole of ATP is needed to reabsorb 9 moles of Na+, the kidneys need ≈3 moles of O2 (corresponding to 96 gr of O2) to reabsorb 99.5% of the Na+ content in 180 L of filtrate [23]. It has been estimated that the energy required to reabsorb 1 mole of Na+ against an electric potential of −70 mV in the cytoplasm and chemical gradient is about the same as required to lift 1 mole of Na+ (≈23 g) to a height of approximately 70 km [21]. As Na+ reabsorption mainly occurs in the renal cortex and any increase in filtrate leads to an increase in Na+ load reabsorption, if one increases filtrate production the O2 consumption increases mainly in the cortex, but may also affect O2 delivery to the medulla.
3. The source of glomerular filtration pressure and generation of the filtration fraction
Among the factors involved in generating the glomerular filtrate, glomerular ultrafiltration pressure plays the leading role. The pressure of glomerular filtration results from the relative combination of the hydrostatic and oncotic pressures across the capillary wall; this provides the energy for water exchange between the plasma and the interstitial fluid. Despite its precapillary origin, it is roughly twice the rate (60 mm Hg) [24] of precapillary pressure elsewhere, due to the energy delivery needed to generate and maintain glomerular filtration over a wide range of renal hemodynamic changes.
Since the filtration fraction (FF) is the renal functional index measured by the ratio between the GFR ml/min (renal O2 consumption index; nominator) and the renal blood flow ml/min (RBF, renal O2 delivery index; denominator) x100, in expressing the percentage of filtrate formed in the Bowman space, it represents the main index of kidney function balance which normally hovers around 20%. If the FF represents space relative to renal blood flow and does not vary to any large extent in humans under physiological conditions [21], this implies a near-constant tissue O2 tension (PO2) as normally occurs in the tissue. It also explains why a relatively mild GFR increase beyond 135 ml/min/1.73m2 is commonly considered as the hyperfiltration condition affecting glomerulus integrity [25,26]. Taking all these factors together, if GFR generation is intimately tied to the RBF and 99.5% of glomerular filtrate together with the Na+ solute has to be reabsorbed through a high energy-consuming process, the way to restore the kidney from a negative energy balance cannot be based on increasing the blood flow to the organ. On the contrary, one needs to lower the filtration production, since this will lessen the number of sodium ions that must be transported according to the oxygen delivered. From the physical forces that generate glomerular filtration, it is apparent that the simplest way to lower filtration production is to reduce the glomerular filtration pressure and thus decrease the FF.
4. The role of adenosine
4.1. Adenosine receptors
Adenosine (ADO) is commonly recognized as the precursor to adenosine triphosphate (ATP), but it also serves as an autacoid that binds to cell surface receptors in the kidney, heart, brain, retina, and skeletal muscle, mediating differentiated organ functions. The ADO autacoid action on the membrane cell receptor consists in matching cell energy consumption with the O2 available [19,20]. According to this relation, the interstitial concentration of ADO rises when the energy balance in the cell tissue turns negative, leading to adenosine A2 receptor (A2R) activation. A2R stimulation activates local vasodilation, adjusting the blood flow to meet demand. The ADO action is based on a short half-life and diffusion, so it is a fast and efficient way of matching supply and demand over a short range within an organ [19,20,27]. Due to the intricacy of renal physiology, it is not surprising that ADO can play a more complex role including differential effects on the renal cortical and medullary vascular structures; it has a specific role in tubuloglomerular feedback (TGF), being involved in governing renin secretion and transport processes in the tubular and collecting duct system (Figure 1).
As the glomerular filtration pressure is the main force involved in GFR generation, the main contributor to the kidney’s energy outlay, ADO can efficiently decrease the filtration pressure by combining its action on vasoconstrictor adenosine 1 receptor (A1R) in the afferent arteriole and on vasodilatory A2R in the efferent arteriole. In the individual nephron the response to ADO stimulation is based on domain nuances driving activity around each of the two receptors. The wide range of differentiated responses provided by the two adenosine receptors has significant conceptual implications for kidney physiology. The vasoconstrictive action exerted by ADO with its A1R activation ends by decreasing the glomerular flow and hence decreasing the renal O2 burden for Na+ reabsorption, but it also implies an overall decline in blood flow to the glomerular structure, including the portion located in the medulla which is supplied by a limited flow, meaning that O2 delivery to the medulla may only withstand a more limited degree of variation. The point is consistent with the observation that A1R-mediated afferent arteriolar constriction prevails in surface nephrons whereas deep cortical nephrons, which supply the blood flow to the renal medulla, can respond to ADO by A2R-mediated vasodilation [19,28]. The differentiated nuance of glomerular adenosine receptor response provides an insight into the sensitive balance that presides over renal nutrition and O2 delivery, particularly in the medullary tubule segments that are more vulnerable to hypoxia and play a crucial role in urine concentration. This aspect seems consistent with data stemming from genetic studies performed in a mouse model with AT II-induced CKD. In those studies ADO stimulation of the red blood cell A2B receptor succeeded in enhancing AMPK activation, leading to a release of 2.3-biphosphoglycerate (2.3-BPG), a specific erythrocyte-metabolite which, by promoting delivery of tissue oxygen, counteracts kidney hypoxia and CKD progression [29] (Figure 1).
4.2. Adenosine action, Macula-Densa and Renin-Angiotensin-Aldosterone System activation
While rapid minor shifts in blood volume and arteriolar tone are managed via the baroreceptor reflex, in the long term regulation is based on the renin-angiotensin-aldosterone system (RAAS) which has a central action in the kidney and can alter blood volume and circulation balance chronically. Although the renin-angiotensin system is present in several organs including the peripheral vessels, the primary source of renin is the juxtaglomerular apparatus (JGA), packed specialized cells, commonly named macula-densa, lining the wall of the distal tubule at the point of glomerulus contact (Figure 2). This cell pack contains the largest store of renin involving the kidney as the center regulating RAAS activation [30], since these specialized cells complement JGA intervention on single nephron function and structure. The macula-densa senses the increase in luminal Na-Cl-K delivery via an NKCC2-based process, leading to an increase in basolateral release of ATP, which in turn is converted by endonucleotidases CD73/39 to adenosine (ADO) [22]. The ADO generated activates A1R in the adjacent afferent arteriole, decreasing the vessel section and the glomerular filtration pressure. The ADO effect on glomerular filtration increases the distal delivery of sodium chloride which also increases the hydrostatic pressure in the Bowman space, further attenuating the filtration pressure [19,21].
In the proximal tubule ADO also stimulates the transport of Na+, decreasing the Na+ load to the tubule segments that extend into the less oxygenated medulla. In these tubule sections ADO locally activates A1R inhibiting Na+ transport up to the thick ascending limb of the medulla, and enhances the medullary blood flow via A2R activation which locally increases O2 delivery and limits O2-consuming Na+ transport. The ADO action in decreasing the glomerular filtration pressure exerts its effects not only by decreasing O2 consumption in the renal cortex through lowering the GFR, but also by partially inhibiting the function of other transporters such as the Na+- H+- exchanger NHE3 [19]. To accomplish the ordinary check and balance system of renal physiology, ADO activation of A1R also causes prolonged inhibition of renal renin secretion into the bloodstream [31], preventing angiotensin (AT) II with its vasoconstrictor action [32,33] (Figure 2).
ADO’s inhibitory action on renin release has considerable weight in the overall circulatory balance, as transpires when dysregulation occurs with the onset of HF. When HF sets in, leading to decreased renal perfusion, the JGA cells react by swiftly spilling renin into the bloodstream which activates the metabolic chain leading to AT II production. The principal effect of AT II in the kidney is to restrict the section of renal vasculature, its main action being on the lesser-sized efferent arteriole, leading to a hydraulic filtration pressure increase. In this setting ATII outstrips the ADO action on the afferent arteriole and becomes the principal contributor to a filtration fraction increase, the leading mechanism involved in GFR maintenance as the HF syndrome evolves [34,35]. In the course of HF progression the increasingly low renal blood flow increases GFR generation dependence by the action of AT II which raises the glomerular filtration pressure gradient, curbing the plasma volume that is exposed to the pressure gradient at any given time per area unit of the capillary wall. The net effect is an increased plasma protein concentration which raises the oncotic pressure at the level of the efferent arteriole and hence causes a rise in FF. The rise in FF will attenuate the absolute drop in the single nephron GFR, even without neurohumoral interference or ultrafiltration pressure change. However, since an ultrafiltration equilibrium is reached when the maximum FF is achieved at ∼60%, a further decrease in renal blood flow causes a linear GFR dip since the ultrafiltration pressure gradient cannot be maintained over the entire length of the glomerular capillary, which results in part of this capillary no longer being used for ultrafiltration and restricting the glomerular filtration area [36]. With HF in progress, AT II counteracts the inhibitory ADO effect on NHE3, leading to retention of Na+ which increases the osmolarity of the blood, and to a shift of fluid into the blood volume and extracellular space [37]. Other unfavorable actions linked to high ATII renal concentration are not only increased production of superoxide anion due to angiotensin I receptor activation [38], which in turn triggers metabolic oxidative stress leading to loss of passive Na+ reabsorption, but also impairment of the paracellular permeability in the tubule [23] (Figure 2).
Taking one thing with another, AT II actions result in increased kidney O2 consumption leading to relative intrarenal hypoxia with counteracting activation of the hypoxia-inducible (HIF) system, this being based on two inducible key mediators in cellular oxygen homeostasis, HIF-1 and HIF-2. The two factors have functions that only partially overlap as the glycolytic genes are predominantly regulated by HIF-1, whereas HIF-2 is the main regulator of hypoxic vascular endothelial growth factor and EPO induction in tissues that express both HIF-1 and HIF-2. The HIF system is designed to provide a cytoprotective effect in acute ischemia-reperfusion injury while in chronic renal hypoxia its response to inflammation takes the form of a profibrotic role [39].
Consistently, in an experimental animal model based on knocking out SGLT2, the prolonged exposure of the medullary tract to increased Na+ concentration proved critical for the induction of renal growth and caused markers of renal injury, inflammation, and fibrosis to appear [40]. This condition seems expressed in type 2 diabetic patients who are hospitalized with acute kidney injury while they are receiving SGLT2 inhibitors. In these subjects there have been reports of an increased finding of blood and urine levels of neutrophil gelatinase–associated lipocalin, originating from distal tubular segments, whereas in the blood and urine, kidney injury molecule 1, a biomarker of proximal tubular damage, is unaltered [41]. The transport shift may therefore “simulate systemic hypoxia” and the lower oxygenation in the corticomedullary junction of the kidneys may trigger the hypoxia-driven hypoxia-inducible factor 2α in interstitial cells, activating an observable increase in erythropoietin expression [42], which may improve oxygen delivery to the outer medulla and facilitate oxygenation of the heart and other organs.
As the emunctory function of the kidney has to preside over the vital regulation of body fluid and electrolyte content, the renal effect of AT II reflects a nephrocentric reaction designed to preserve the GFR, providing an explanation for why it is immediately preserved in the low output state. The kidney reacts to a decrease in its own blood supply in part like other organs by releasing renin as a local factor, but it also unleashes an AT II renal and systemic circulatory response that bolsters preservation of the GFR [43]; unfortunately this occurs at a soaring cost of O2 consumption which impacts on the strict kidney balance in terms of energy expenditure. In the end it is not surprising that kidney consumption of O2 may independently steer the HF outcome.
5. SGLT2 activity and Sympathetic Drive
5.1. Effects on sympathetic activity
In the clinical progression of HF the renal sympathetic tone is greatly enhanced and the kidney is the main contributor to the norepinephrine spillover, directly affecting the clinical outcome of HF [44]. The progression of CKD is also intensively marked by renal sympathetic traffic enhancement, partly related to activation of α-2 adrenoreceptors and partly to release of intrarenal AT II. Renally synthesized AT II regulates organ function in a paracrine fashion by modulating Na+ and water reabsorption within the proximal tubule in concert with systemic AT II [45]; consistently with this, renal sympathetic denervation abrogates the reabsorption of Na+ in the proximal tubule [46] where active natriuretic peptides are fast degraded by highly concentrated neprilysin, which is also responsible for degradation of other peptide hormones such as angiotensin I and II, endothelin, etc. [47]. As well as direct kidney management of Na+ reabsorption, the increased sympathetic traffic in the proximal tubule augments the expression of both NHE3 and SGLT2, playing a further role in the avid sodium reabsorption involved in HF progression [48]. SGLT2 inhibitors interfere with both SGLT2 and NHE3 in the proximal tubule [28] yielding short-term increases in the fractional excretion of sodium [49]. On the plus side, SGLT2 inhibition attenuates renal sympathetic activity and reduces the renal norepinephrine content in states of experimental nutrient excess [28,50], while in animals with HF renal denervation attenuates the magnitude of response to SGLT2 inhibition [51]. These observations place SGLT2 inhibitors as functional antagonists to renal sympathetic nerve hyperactivity in HF, which is the main contributor to norepinephrine spillover [52] and closely linked to outcome in HF patients [53].
5.2. SGLT2 and SGLT1 synergy and “off target” implications
In renal physiology glucose reabsorption is based on a highly efficient architecture placing SGLT2 in the early proximal tubule to perform the bulk of glucose reabsorption (~80-90%) and positioning sodium glucose cotransporter 1 (SGLT1) in the late proximal tubule, where it reabsorbs the amount of glucose that escapes SGLT2. In the final section of the tubule the fluid in the glucose concentration falls below the line of stoichiometric reabsorption of 1 glucose molecule to 1 Na+ performed by SGLT2. Since Na+-glucose cotransport is electrogenic, in the later section of the tubule glucose reabsorption requires SGLT1 to increase its sugar concentration strength, exploiting the electrical power provided by 2 Na+ ion, so as to transfer 1 glucose molecule from the filtrate into the bloodstream, thereby doubling the energy expense of reabsorption.
As the normal daily glomerular filtrate contains ~1 mol of glucose (~180 g) and the combined action of cotransporters has the capacity to reabsorb ~2.5 mol glucose per day (~450 gr per day), it suggests that in nature the SGLT2 function has been dimensioned to cater for a broad variation in glucose concentration that can greatly affect Na+ reabsorption, having implications for renal O2 consumption [19]. Persistent augmenting of Na+ glucose reabsorption leads to proximal tubule hypertrophy, a primary cause and effect of glomerular hypertrophy driven by hyperfiltration [1,21,26].
The peculiar extensibility of SGLT2 and SGLT1 action has important “off-target” effects that stem from their effect on proximal tubular Na+ transport where bicarbonate accounts for approximately 80% of Na+ reabsorption in this portion of the nephron. The rise in sodium-glucose reabsorption may affect body fluid retention such as sodium-bicarbonate, driving additional passive sodium chloride reabsorption. Note that in diabetics the effect of SGLT2 pharmacologic inhibition is partially offset by enhanced SGLT1 activation, while investigation in animals has provided conclusive evidence that SGLT1 can reabsorb ~30% of filtered glucose, explaining why SGLT2 inhibitors never produce the amount of glucosuria to be expected if SGLT2 were completely inhibited (preventing 80–90% reabsorption of the filtered glucose load) [54]. This point focuses attention on the peculiar role SGLT1 plays in the kidney since SGLT1 engagement can prevent or just limit glucose loss in the urine, at the cost of high energy expenditure to the kidney.
Intriguingly, two studies performed in Akita mice support the role of SGLT1, expressed in the membrane of the tubuloglomerular apparatus, in increasing nitric oxide (NO) S1-dependent NO formation by sensing the glucose concentration reaching the macula-densa, thereupon reducing the vasoconstrictor tone set by TGF and contributing to glomerular hyperfiltration [55]. In Akita mice the absence of SGLT1 in the tubuloglomerular apparatus not only lowers glomerular hyperfiltration, but also reduces kidney weight, glomerular size, and albuminuria [56]. These findings suggest that SGLT1 may have implications for renal structure and performance besides the reabsorption of glucose.
The inhibition of SGLT2 can reduce glycemia and insulin resistance, and can lower the availability of cellular glucose, while regardless of basal hyperglycemia it can stimulate a starvation-like response. The response includes SIRT1/AMPK (Sirtuin1/adenosine monophosphate–activated protein kinase) activation and inhibition of the protein kinase b /mTOR1 (mammalian Target Of Rapamycin) pathway [57]. This specific activation, in inducing autophagy, promotes cellular defense and pro-survival mechanisms that counteract the primary pathophysiological mechanism of proximal tubule hypertrophy in diabetes and in conditions involving insulin resistance [20,26,28]. In experimental models and in patients with T2DM, urine metabolomics have indicated that inhibition of SGLT2 induces a metabolic shift from glycolysis to more mitochondrial oxidation [20,22].
Studies in non-diabetic mice suggest that the kidney’s metabolic response to SGLT2 inhibition compensates (a) for the partial inhibition of tubular NHE3 and the glucose uptake/urinary glucose loss, including renal gluconeogenesis upregulation, and (b) for the urinary Na+ loss, by inducing tubular secretion of the tricarboxylic acid cycle intermediate, alpha-ketoglutarate, which communicates to the distal nephron the need for compensatory Na+ reabsorption [58].
SGLT2 inhibition shifts some of the glucose, Na+ and fluid reabsorption downstream, providing a more equal distribution of transport work and mimicking systemic hypoxia to the renal oxygen sensor, triggering upregulation of renal NHE3. This effect in HF and on the remaining nephrons in CKD could enhance the natriuretic efficacy and renal hemodynamic effect of SGLT2 inhibition and thereby contribute to kidney and cardio-protection in nondiabetic patients.
The inhibition of SGLT2 lowers body weight by coupling the initial natriuretic effect with the renal glucose loss, which shifts substrate utilization from carbohydrates to lipids and reduces body fat, lessening visceral and subcutaneous adiposity [57]. This effect also augments the release of free fatty acids leading to ketone body formation, which can be used as an additional more efficient energy substrate both in the kidney and in the failing heart [58]. At the same time, the transport shift to the straight proximal tubule and thick ascending limb in the renal outer medulla could reduce the O2 availability, endangering medullary structures as mentioned above [40,41,42].
As gliflozins are the only class of hypoglycemic drugs combining glycosuric and natriuretic actions, they play a joint role in vascular fluid restriction and hemoconcentration. By inhibiting glucose renal reuptake this class of drug induces significant osmotic diuresis which selectively decreases the volume of the interstitial space between cells (known as the third space) and affects body weight beyond what nutrient loss can provide [1,20,28]. Osmotic diuresis obtained with SGLT2i has been proved in humans by a double-blind randomized study conducted on 59 type 2 diabetics [61]. It has been postulated that SGLT2i may regulate both the volume between the interstitial space and the vascular bed (interstitial> intravascular) in HF, thus reducing the neurohumoral stimulation generated by the signal denoting decreased vascular filling activated by the baroreceptors [62]. Note that, unlike loop diuretics that promote natriuresis by inhibiting carbonic anhydrase in the thick ascending limb, SGLT2 inhibition halts Na+ and glucose reabsorption driven by the activation of the Na+/K+ pump in the brush border, curbing the O2 consumption rate in the critical cortex area without any impact on the electrolyte balance. The peculiarity and sequence of pharmacologic mechanisms activated by SGLT2 inhibitors and by loop diuretics suggests a reason why their combined action generates reciprocal potentiation of natriuresis [63] and supports their combined use in acute decompensated HF.
5.3. Other Implications of SGLT2 inhibition and RAAS interaction
One should note that a significant increase in kidney sensitivity to adenosine-induced vasoconstrictive action is caused by inhibiting the synthesis of local vasodilatory molecules like NO or prostaglandins [19,20,27], so that non-steroidal anti-inflammatory drugs (NSAIDs) are among the substances that can lead to acute kidney injury, among other things, through potentiation of ADO action on A1R.
Inhibition of SGLT2 not only decreases the renal cortex O2 consumption, as a consequence of lowering GFR, but through partial functional inhibition of other transporters such as the Na- H- exchanger NHE3 [21,28] in the brush border, the density of this transporter is augmented by Na+ retention via the increase in GFR prompted by the JGA signal. The co-inhibition of NHE3 contributes to augmenting natriuresis and blood pressure, lowering the effect of SGLT2 inhibitors in the non-diabetic setting [20,63]. It has to be noted that SGLT2 inhibitors enhance renin levels [64] and vasopressin (or copeptin) levels [65,66,67] and reduce renal free-water clearance in animal models and humans [65,66]. This effect is associated with increased renal protein expression of vasopressin V2 receptors and phosphorylated aquaporin-2 in rats [64], indicating active compensation to counter the diuretic and natriuretic effects and highlighting the intricate check and balance system of the renal emunctory function.
At the same time, inhibition of SGLT2 also significantly increases urate excretion, bringing about an indirect effect on the urate transporter URAT1 in the proximal tubule brush border [28,68].
5. Conclusions
We have recently achieved evidence that the renal oxygen demand is critically linked to the circulation neurohormonal balance, and conditions that increase glomerular filtration pressure affect the renal oxygen balance, leading to progressive glomerular damage and loss of filtration power which primarily affect the cardiovascular outcome. In kidney physiology the reabsorption of filtered glucose is primarily taken up into peritubular capillaries and returned to the systemic circulation or supplied as an energy source to further distal tubular segments. Recent studies have provided insights as to the coordination of renal glucose reabsorption, formation, and usage. Moreover, a better understanding of renal glucose transport in disease states is shedding light on the complex interaction between mechanisms of sodium reabsorption and the renal oxygen demand [1,19,20,21]. The kidney’s capacity to provide complete glucose reabsorption is a remnant of the ancient evolutionary tendency to avoid glucose loss, a scanty energy resource until the industrial revolution.
A high daily intake of glucose leads to maladaptive kidney response in the effort to reuptake the excess of filtered glucose. This effort reveals the pathophysiological potential of SGLT2, which links the retention of glucose to the reabsorption of sodium, affecting the volume status and impairing the TGF by increasing the glomerular flow through the vasodilation of the afferent arteriole and the unbalancing the kidney oxygen demand.
As the body’s evolutionary ability adapted the body to long survival in environments with scanty energy resources, the extra-calory loss following the inhibition of renal glucose reabsorption inducing sugar spilling into the urine can activate a mechanism resembling fasting. This action not only results in hypoglycemia but also reduces the exceedingly high O2 that is the prime vulnus of kidney biology.
While this concept contributes to the unexpected rationale of SGLT2 inhibition in the diabetic kidney, the action of SGLT2 can efficiently affect renal O2 consumption whenever cardiac or intrinsic kidney disorders result in a need to increase the glomerular filtration pressure so as to maintain the filtrate.
The benefits observed after administration of SGLT2 inhibitors in diabetics and non-diabetics with and without HF and or CKD [2,3,4,5,6,7,8,9,10] are probably a culmination of the primary metabolic/renal and secondary myocardial benefits as described above. Among them the rebalancing of the renin angiotensin system and the stimulation of erythropoietin may contribute to our understanding of the kidney’s need to maintain its function and preserve the organism’s physiological balance.
Although many questions remain unanswered, such as the systemic metabolic consequences of losing glucose into the urine and the metabolic responses of the early proximal tubule and the down-stream segments, a new avenue in the treatment of cardiovascular and metabolic disorders has now opened and deserves close attention.
Author Contributions
Conceptualization, E.G.; writing—original draft preparation, E.G., M.I., A.P.; writing—review and editing, M.B., D.G., A.A.; supervision, A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest for this manuscript.
References
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Mosenzon, O.; Wiviott, S.D.; Heerspink, H.J.; Dwyer, J.P.; Cahn, A.; Goodrich, E.L.; Rozenberg, A.; Schechter, M.; Yanuv, I.; Murphy, S.A.; et al. The Effect of Dapagliflozin on Albuminuria in DECLARE-TIMI 58. Diabetes Care 2021, 44, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; et al. ; DAPA-CKD Trial Committees and Investigators. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 2020, 383, 1436–1446. [Google Scholar] [PubMed]
- The EMPA-KIDNEY Collaborative Group Empagliflozin in Patients with Chronic Kidney Disease. New Engl. J. Med. 2023, 388, 117–127. [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; et al. Canaglifozin and renal out-comes in type 2 diabetes and nephropathy. N Engl J Med 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; et al.; EMPEROR-Reduced Trial Investigators Cardio-vascular and renal outcomes with empagliflozin in heart failure. N Eng J Med 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B. ; DELIVER Trial Committees and Investigators. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Bayés-Genís, A.; Aimo, A.; Metra, M.; Anker, S.; Seferovic, P.; Rapezzi, C.; Castiglione, V.; Núñez, J.; Emdin, M.; Rosano, G.; et al. Head-to-head comparison between recommendations by the ESC and ACC/AHA/HFSA heart failure guidelines. Eur. J. Hear. Fail. 2022, 24, 916–926. [Google Scholar] [CrossRef]
- Rolfe, D.F.S.; Brown, G.C.; Merdzo, I.; Rutkai, I.; Sure, V.N.L.R.; Katakam, P.V.G.; Busija, D.W.; Karbowski, J.; Gordon, K.; Blondin, D.P.; et al. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef]
- Carlström, M.; Wilcox, C.S.; Arendshorst, W.J. Renal Autoregulation in Health and Disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [CrossRef] [PubMed]
- Duncker, D.J.; Bache, R.J. Regulation of Coronary Blood Flow During Exercise. Physiol. Rev. 2008, 88, 1009–1086. [Google Scholar] [CrossRef] [PubMed]
- Goodwill AG, Dick GM, Kiel AM, Tune JD. Regulation of Coronary Blood Flow. Compr Physiol 2017, 7, 321–382.
- Saks, V.; Dzeja, P.; Schlattner, U.; Vendelin, M.; Terzic, A.; Wallimann, T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J. Physiol. 2006, 571, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.N.; Evans, R.G.; Harrop, G.K.; Ngo, J.P.; Ow, C.P.C.; Gardiner, B.S.; Smith, D.W.; O'Connor, P.M.; Eppel, G.A.; Michaels, S.; et al. Effect of variations of blood flow on renal oxygen extraction. Am. J. Physiol. Content 1960, 199, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Mühlbauer, B.; Osswald, H. Adenosine and Kidney Function. Physiol. Rev. 2006, 86, 901–940. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pfl?gers Arch. Eur. J. Physiol. 2020, 472, 1345–1370. [Google Scholar] [CrossRef] [PubMed]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2012, 40, 123–137. [Google Scholar] [CrossRef]
- Liu, Z.Z.; Bullen, A.; Li, Y.; Singh, P. Renal Oxygenation in the Pathophysiology of Chronic Kidney Disease. Front. Physiol. 2017, 8, 385. [Google Scholar] [CrossRef]
- Layton, A.T.; Vallon, V. SGLT2 inhibition in a kidney with reduced nephron number: modeling and analysis of solute transport and metabolism. Am. J. Physiol. Physiol. 2018, 314, F969–F984. [Google Scholar] [CrossRef]
- Jessup, M.; Costanzo, M.R. The cardiorenal syndrome: do we need a change of strategy or a change of tactics? J Am Coll Cardiol 2009 53, 597–599.
- Magee, G.M.; Bilous, R.W.; Cardwell, C.R.; Hunter, S.J.; Kee, F.; Fogarty, D.G. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 2009, 52, 691–697. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Porrini, E.L.; Gaspari, F.; Motterlini, N.; Cannata, A.; Carrara, F.; Cella, C.; Ferrari, S.; Stucchi, N.; Parvanova, A.; et al. Glomerular Hyperfiltration and Renal Disease Progression in Type 2 Diabetes. Diabetes Care 2012, 35, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Miracle, C.; Thomson, S. Adenosine and kidney function: Potential implications in patients with heart failure. Eur. J. Hear. Fail. 2008, 10, 176–187. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2020, 83, 503–528. [Google Scholar] [CrossRef]
- Peng, Z.; Luo, R.; Xie, T.; Zhang, W.; Liu, H.; Wang, W.; Tao, L.; Kellems, R.E.; Xia, Y. Erythrocyte Adenosine A2B Receptor-Mediated AMPK Activation: A Missing Component Counteracting CKD by Promoting Oxygen Delivery. J. Am. Soc. Nephrol. 2019, 30, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Peti-Peterdi, J.; Harris, R.C. Macula Densa Sensing and Signaling Mechanisms of Renin Release. J. Am. Soc. Nephrol. 2010, 21, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.S.; Steinhausen, M. Differential reactivity of cortical and juxtaglomerullary glomeruli to adenosine-1 and adenosine-2 receptor stimulation and angiotensin converting-enzyme inhibition. Microvasc Res 1993, 45, 122–33. [Google Scholar] [CrossRef]
- Spielman, W.S.; Osswald, H.; Patinha, D.; Fasching, A.; Pinho, D.; Albino-Teixeira, A.; Morato, M.; Palm, F.; Lee, D.L.; Bell, T.D.; et al. Blockade of postocclusive renal vasoconstriction by an angiotensin II antagonists: evidence for an angiotensin-adenosine interaction. Am. J. Physiol. Physiol. 1979, 237, F463–F467. [Google Scholar] [CrossRef]
- Holz, F.G.; Steinhausen, M. Renovascular Effects of Adenosine Receptor Agonists. Kidney Blood Press. Res. 1987, 10, 272–282. [Google Scholar] [CrossRef]
- Vander, A.J.; Malvin, R.L.; Wilde, W.S.; Sullivan, L.P. Re-examination of salt and water retention in congestive heart failure: Significance of renal filtration fraction. Am. J. Med. 1958, 25, 497–502. [Google Scholar] [CrossRef]
- Fliser, D.; Zeier, M.; Nowack, R.; Ritz, E. Renal functional reserve in healthy elderly subjects. J. Am. Soc. Nephrol. 1993, 3, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Dupont, M.; Steels, P. The kidney in congestive heart failure: 'are natriuresis, sodium, and diuretics really the good, the bad and the ugly? Eur J Heart Fail 2014 16, 133–42. [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.S.; Wilcox, C.S.; M. D.; Yue, L.; Wang, W.; Wang, Y.; Du, T.; Shen, W.; Tang, H.; Wang, Y.; et al. NADPH Oxidases in the Kidney. Antioxidants Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H.; Hodrea, J.; Balogh, D.B.; Hosszu, A.; Lenart, L.; Besztercei, B.; Koszegi, S.; Sparding, N.; Genovese, F.; Wagner, L.J.; et al. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef]
- Nespoux, J.; Patel, R.; Zhang, H.; Huang, W.; Freeman, B.; Sanders, P.W.; Kim, Y.C.; Vallon, V. Gene knockout of the Na+-glucose cotransporter SGLT2 in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol. Physiol. 2020, 318, F1100–F1112. [Google Scholar] [CrossRef]
- Darawshi, S.; Yaseen, H.; Gorelik, Y.; Faor, C.; Szalat, A.; Abassi, Z.; Heyman, S.N.; Khamaisi, M. Biomarker evidence for distal tubular damage but cortical sparing in hospitalized diabetic patients with acute kidney injury (AKI) while on SGLT2 inhibitors. Ren. Fail. 2020, 42, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef]
- Packer, M. Why do the kidneys release renin in patients with congestive heart failure? Eur Heart J 1990, 11 Suppl D, 44–52. [Google Scholar] [CrossRef]
- Florea, V.G.; Cohn, J.N. The Autonomic Nervous System and Heart Failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Quan, A.; Baum, M. Regulation of Proximal Tubule Transport by Endogenously Produced Angiotensin II. Nephron 2000, 84, 103–110. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, X.; Katsurada, K.; Patel, K.P. Renal denervation improves sodium excretion in rats with chronic heart failure: effects on expression of renal ENaC and AQP2. Am. J. Physiol. Circ. Physiol. 2019, 317, H958–H968. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Nair, A.P.; Misra, A.; Scott, C.Z.; Mahar, J.H.; Fedson, S. Neprilysin Inhibitors in Heart Failure: The Science, Mechanism of Action, Clinical Studies, and Unanswered Questions. JACC Basic Transl Sci 2022, 8, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Katsurada, K.; Nandi, S.S.; Sharma, N.M.; Patel, K.P. Enhanced Expression and Function of Renal SGLT2 (Sodium-Glucose Cotransporter 2) in Heart Failure: Role of Renal Nerves. Circ. Hear. Fail. 2021, 14, e008365–e008365. [Google Scholar] [CrossRef]
- Scholtes, R.A.; Muskiet, M.H.; van Baar, M.J.; Hesp, A.C.; Greasley, P.J.; Karlsson, C.; Hammarstedt, A.; Arya, N.; van Raalte, D.H.; Heerspink, H.J. Natriuretic Effect of Two Weeks of Dapagliflozin Treatment in Patients With Type 2 Diabetes and Preserved Kidney Function During Standardized Sodium Intake: Results of the DAPASALT Trial. Diabetes Care 2020, 44, 440–447. [Google Scholar] [CrossRef]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; et al. SGLT2 inhibitor-induced sympatho-inhibition: a novel mechanism for cardiorenal protection. JACC Basic Transl Sci 2020, 5, 169–179. [Google Scholar] [CrossRef]
- Katsurada, K.; Nandi, S.S.; Sharma, N.M.; Patel, K.P. Enhanced Expression and Function of Renal SGLT2 (Sodium-Glucose Cotransporter 2) in Heart Failure: Role of Renal Nerves. Circ. Hear. Fail. 2021, 14, e008365–e008365. [Google Scholar] [CrossRef] [PubMed]
- Hasking, G.J.; Esler, M.D.; Jennings, G.L.; Burton, D.; A Johns, J.; I Korner, P. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circ. 1986, 73, 615–621. [Google Scholar] [CrossRef]
- Petersson, M.; Friberg, P.; Eisenhofer, G.; Lambert, G.; Rundqvist, B. Long-term outcome in relation to renal sympathetic activity in patients with chronic heart failure. Eur. Hear. J. 2005, 26, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Novel Hypothesis to Explain Why SGLT2 Inhibitors Inhibit Only 30–50% of Filtered Glucose Load in Humans. Diabetes 2013, 62, 3324–3328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wei, J.; Jiang, S.; Xu, L.; Wang, L.; Cheng, F.; Buggs, J.; Koepsell, H.; Vallon, V.; Liu, R. Macula Densa SGLT1-NOS1-Tubuloglomerular Feedback Pathway, a New Mechanism for Glomerular Hyperfiltration during Hyperglycemia. J. Am. Soc. Nephrol. 2019, 30, 578–593. [Google Scholar] [CrossRef]
- Song, P.; Huang, W.; Onishi, A.; Patel, R.; Kim, Y.C.; van Ginkel, C.; Fu, Y.; Freeman, B.; Koepsell, H.; Thomson, S.; et al. Knockout of Na+-glucose cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase NOS1 in the macula densa and glomerular hyperfiltration. Am. J. Physiol. Physiol. 2019, 317, F207–F217. [Google Scholar] [CrossRef]
- Packer, M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic CKD Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. 2020, 31, 907–919. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; et al. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 2016, 60, 215–225. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Karg, M.V.; Bosch, A.; Kannenkeril, D.; Striepe, K.; Ott, C.; Schneider, M.P.; Boemke-Zelch, F.; Linz, P.; Nagel, A.M.; Titze, J.; et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: a randomised controlled trial. Cardiovasc. Diabetol. 2018, 17, 1–8. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes, Obes. Metab. 2017, 20, 479–487. [Google Scholar] [CrossRef]
- Griffin, M.; Rao, V.S.; Ivey-Miranda, J.; et al. Empagliflozin in Heart Failure: Diuretic and Cardiorenal Effects. Circulation 2020, 142, 1028–1039. [Google Scholar] [CrossRef]
- Eickhoff, M.K.; Dekkers, C.C.J.; Kramers, B.J.; Laverman, G.D.; Frimodt-Møller, M.; Jørgensen, N.R.; Faber, J.; Danser, A.H.J.; Gansevoort, R.T.; Rossing, P.; et al. Effects of Dapagliflozin on Volume Status When Added to Renin–Angiotensin System Inhibitors. J. Clin. Med. 2019, 8, 779. [Google Scholar] [CrossRef]
- Masuda, T.; Watanabe, Y.; Fukuda, K.; Watanabe, M.; Onishi, A.; Ohara, K.; Imai, T.; Koepsell, H.; Muto, S.; Vallon, V.; et al. Unmasking a sustained negative effect of SGLT2 inhibition on body fluid volume in the rat. Am. J. Physiol. Physiol. 2018, 315, F653–F664. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Muto, S.; Fukuda, K.; Watanabe, M.; Ohara, K.; Koepsell, H.; Vallon, V.; Nagata, D. Osmotic diuresis by SGLT2 inhibition stimulates vasopressin-induced water reabsorption to maintain body fluid volume. Physiol. Rep. 2020, 8, e14360. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Bjornstad, P.; Katz, A.; Singh, S.; Godoy, L.; Chung, L.; Vinovskis, C.; Pyle, L.; Roussel, R.; Perkins, B.; et al. SGLT2 inhibition increases serum copeptin in young adults with type 1 diabetes. Diabetes Metab. 2019, 46, 203–209. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The kidney’s largest O2 consumption (approximately 85%) [19,20,21] occurs in the cortex, where the reabsorption of Na+ and water mainly occurs in the proximal tubule. a. The figure summarizes the cascade of effects occurring with SGLT2 and SGLT1 engagement in sodium and glucose reabsorption in the early proximal tubule and in the renal medulla. In a normal glycemic environment the Na+ reabsorption linked to glucose reuptake corresponds approximately to 5% of overall Na+ reabsorbed in the kidney1. It can undergo fluctuating variation linked to timely changes in plasma glucose concentration, and affect Na+ concentration in the filtrate which is sensed by the juxtaglomerular apparatus through Na+ reabsorption via activation of the ATP dependent Na+/K+ pump, leading to adenosine (ADO) release [19,20,21]. b. The amount of liberated ADO engages specialized adenosine 1 receptor (A1R), located in the afferent arteriole, and adenosine 2 receptor (A2R), placed in the efferent arteriole and in the vasa recta situated in the medulla. The A1R in the afferent arteriole causes local vasoconstriction decreasing blood flow in the glomerulus and thus affecting the glomerular filtration pressure and the rate of filtrate production [19,20,21]. c. The A2R located in the efferent arteriole and in the vasa-recta causes a vasodilating action in the vasculature with a bidirectional effect: by vasodilating the efferent arteriole it contributes to lowering the glomerular filtration pressure, and by coupling vasodilation of the vasa recta it preserves the flow to the renal medulla where oxygen delivery is less copious. The simultaneous ADO action on both receptors constantly regulates the local O2 consumption through the juxtaglomerular feed-back, avoiding imbalance in the kidney’s metabolic needs [19,20,21]. d. Recently a further action of ADO has been observed on the AR2 receptor located on the red blood cell membrane where ADO stimulation enhances AMPK activation, leading to a release of 2.3-biphosphoglycerate (2.3-BPG), a specific erythrocyte-metabolite that promotes delivery of oxygen to tissue, counteracting local kidney hypoxia and CKD progression [29].
Figure 1.
The kidney’s largest O2 consumption (approximately 85%) [19,20,21] occurs in the cortex, where the reabsorption of Na+ and water mainly occurs in the proximal tubule. a. The figure summarizes the cascade of effects occurring with SGLT2 and SGLT1 engagement in sodium and glucose reabsorption in the early proximal tubule and in the renal medulla. In a normal glycemic environment the Na+ reabsorption linked to glucose reuptake corresponds approximately to 5% of overall Na+ reabsorbed in the kidney1. It can undergo fluctuating variation linked to timely changes in plasma glucose concentration, and affect Na+ concentration in the filtrate which is sensed by the juxtaglomerular apparatus through Na+ reabsorption via activation of the ATP dependent Na+/K+ pump, leading to adenosine (ADO) release [19,20,21]. b. The amount of liberated ADO engages specialized adenosine 1 receptor (A1R), located in the afferent arteriole, and adenosine 2 receptor (A2R), placed in the efferent arteriole and in the vasa recta situated in the medulla. The A1R in the afferent arteriole causes local vasoconstriction decreasing blood flow in the glomerulus and thus affecting the glomerular filtration pressure and the rate of filtrate production [19,20,21]. c. The A2R located in the efferent arteriole and in the vasa-recta causes a vasodilating action in the vasculature with a bidirectional effect: by vasodilating the efferent arteriole it contributes to lowering the glomerular filtration pressure, and by coupling vasodilation of the vasa recta it preserves the flow to the renal medulla where oxygen delivery is less copious. The simultaneous ADO action on both receptors constantly regulates the local O2 consumption through the juxtaglomerular feed-back, avoiding imbalance in the kidney’s metabolic needs [19,20,21]. d. Recently a further action of ADO has been observed on the AR2 receptor located on the red blood cell membrane where ADO stimulation enhances AMPK activation, leading to a release of 2.3-biphosphoglycerate (2.3-BPG), a specific erythrocyte-metabolite that promotes delivery of oxygen to tissue, counteracting local kidney hypoxia and CKD progression [29].
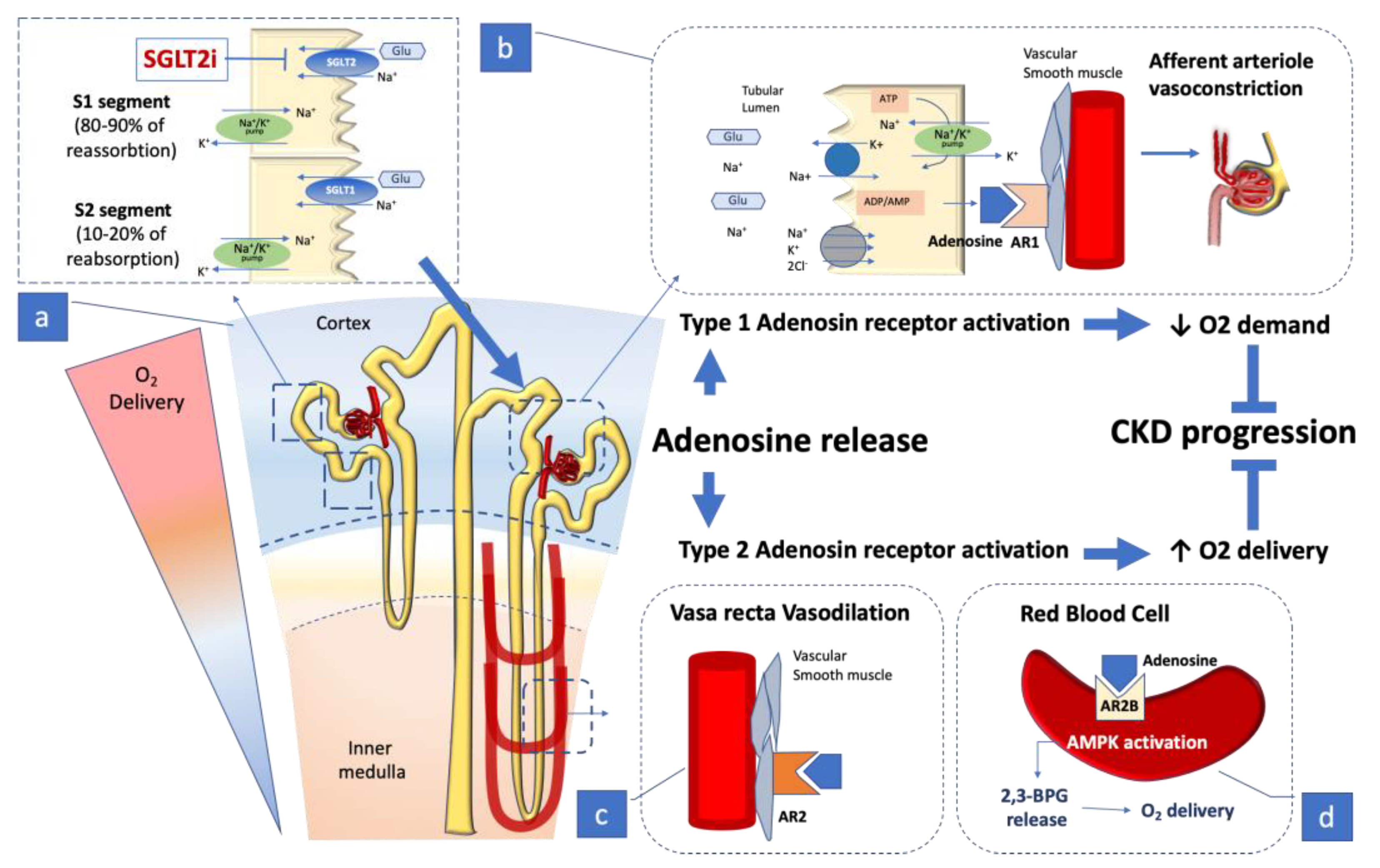
Figure 2.
The figure shows how ADO constantly regulates glomerular filtration in each nephron. In the presence of euglycemia the Na+ amount (a) in the filtrate reaches the juxtaglomerular apparatus and transits across the macula-densa cells. (b) These cells sense the Na+ concentration by re-uptaking the electrolyte via activation of the Na+/K+ energy based by breaking down adenosine triphosphate (ATP) to adenosine (ADO). The ADO generated binds the adjacent adenosine type 1 receptor (AR1) in the afferent arteriole leading to vessel section restriction. This action balances the intraglomerular pressure and, therefore, the filtration fraction which normally does not vary to any large extent from 20% under physiological conditions (see section 3 in the text for details). (c) In the juxta-glomerular apparatus ADO AR1 stimulation through inhibitory G (Gi) protein action mediates the inhibition of renin secretion via cAMP dependent adenylate cyclase (AC) inhibition. This effect not only prevents systemic activation of the renin angiotensin (AT) aldosterone axis, but also affects efferent arteriole vasoconstriction maintaining filtration pressure stable and (d) limits the AT I and AT II detrimental action on cell metabolism and on Na+ retention (see text for details). On note (e) AT II stimulates strong catecholamines release that in turn via stimulatory G (Gs) activates cAMP leading to AC activation and renin release by the juxtaglomerular apparatus, and in turn further enhancing the neurhormonal response. ACE: Angiotensin converter enzyme; ADO: adenosine; AR1: Type 1 adenosine receptor; AT: Angiotensin; Gi: inhibitory protein G; Gs: stimulatory protein G; ATP: adenosine triphosphate.
Figure 2.
The figure shows how ADO constantly regulates glomerular filtration in each nephron. In the presence of euglycemia the Na+ amount (a) in the filtrate reaches the juxtaglomerular apparatus and transits across the macula-densa cells. (b) These cells sense the Na+ concentration by re-uptaking the electrolyte via activation of the Na+/K+ energy based by breaking down adenosine triphosphate (ATP) to adenosine (ADO). The ADO generated binds the adjacent adenosine type 1 receptor (AR1) in the afferent arteriole leading to vessel section restriction. This action balances the intraglomerular pressure and, therefore, the filtration fraction which normally does not vary to any large extent from 20% under physiological conditions (see section 3 in the text for details). (c) In the juxta-glomerular apparatus ADO AR1 stimulation through inhibitory G (Gi) protein action mediates the inhibition of renin secretion via cAMP dependent adenylate cyclase (AC) inhibition. This effect not only prevents systemic activation of the renin angiotensin (AT) aldosterone axis, but also affects efferent arteriole vasoconstriction maintaining filtration pressure stable and (d) limits the AT I and AT II detrimental action on cell metabolism and on Na+ retention (see text for details). On note (e) AT II stimulates strong catecholamines release that in turn via stimulatory G (Gs) activates cAMP leading to AC activation and renin release by the juxtaglomerular apparatus, and in turn further enhancing the neurhormonal response. ACE: Angiotensin converter enzyme; ADO: adenosine; AR1: Type 1 adenosine receptor; AT: Angiotensin; Gi: inhibitory protein G; Gs: stimulatory protein G; ATP: adenosine triphosphate.
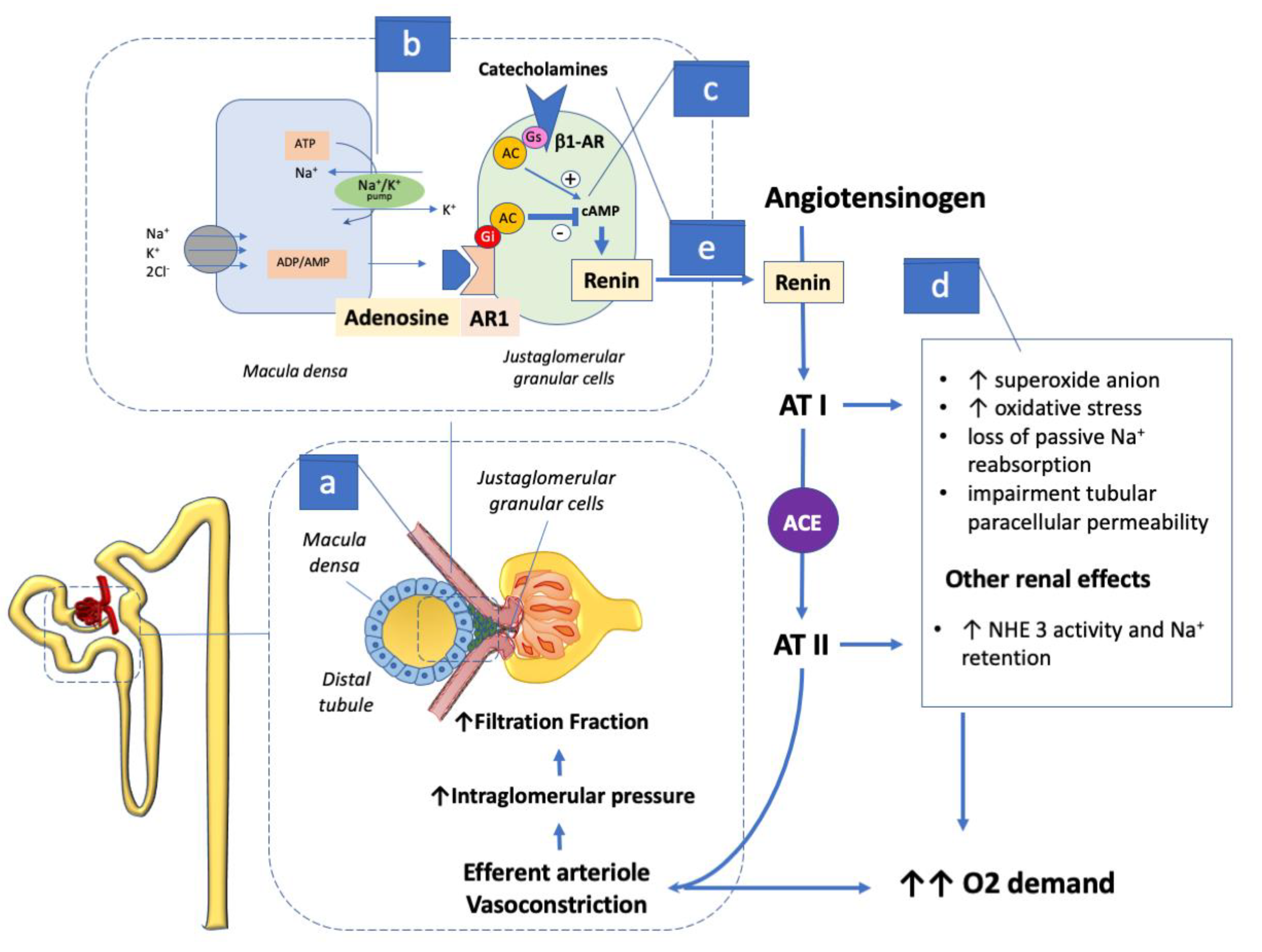

Table 1.
See the text for the details. * The coronary microvasculature can increase tissue delivery by physical training through increased arteriolar densities and/or diameters together with the formation of new capillaries; it can alter the distribution of coronary flow, increasing the permeability-surface area product. **Based on: Starling Law, Contractility, Laplace Law and Heart Rate CO: cardiac output; GFR: glomerular filtration rate; OMR: organ metabolic rate; RBF: renal blood flow.
Table 1.
See the text for the details. * The coronary microvasculature can increase tissue delivery by physical training through increased arteriolar densities and/or diameters together with the formation of new capillaries; it can alter the distribution of coronary flow, increasing the permeability-surface area product. **Based on: Starling Law, Contractility, Laplace Law and Heart Rate CO: cardiac output; GFR: glomerular filtration rate; OMR: organ metabolic rate; RBF: renal blood flow.
| Organ | Total Blood flow (L/min) | Parenchimal flow at rest (ml/min) | Parenchimal flowReserve (ml/min) | Proportion of total body O2 consumption at rest (%) | OMR Ki(kcal/Kg/day) | O2 extraction (%) | Organ O2 consumption(%)Na/ ATPase Ca ATPase Other | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heart | 4/6 | Coronaric220/260 | Coronaric450/600 | 1112 | 440 13 | 75 5 | 1–512 | 15-3012 | - Actinomyosin ATPase– 40-5012- Proton leak- 1512 | |||||||
| Kidney | 1,2 | Glomerular90/120 | Glomerular100/140 | 6-712 | 440 13 | 10-1512 | 70 12 | _ | Gluco-neogenesis12 | |||||||
| Organ | Flow Regulationin Delivering O2to the Organ 14,21 | Maximumincrease of O2Consumption (%) | Prominent Regulator of Performance Maintenance | |||||||||||||
| Heart | Phasic coronary flow16:- Strictly dependent on the magnitude of the difference between arterial and tissue pressure.- This reflects the combined effects of cavity-induced extracellular pressure and shortening-induced intramyocyte pressure (tissue pressure) leading to vascular compression | ~ 400% 16-*- The increased O2 demand** is followed by increased perfusion and delivery of O2 strictly correlated to the increased coronary flow. | Filling Volume Change(Frank O.-Starling E. Law)17- Normal blood circulation under a wide range of workloads, depending on the preload reserve. | |||||||||||||
| Kidney | Inhomogeneous blood flow 14,21:- Renal cortex highly perfused due to as the highest rate NA-K ATPase dependent Na reabsorption.- Only 10 to 15% of blood perfusion is directed toward the medulla to preserve osmotic gradients and enhance urinary concentration by exploiting the countercurrent mechanism. | Difficult to assess21:- O2 delivery is related to renal blood flow responsible for GFR.- In normal subjects the plasma Na+ concentration is relatively constant and determines the Na load in the glomerular filtrate (around 125 ml/min/1,73m2).- The reabsorption of 99,5% of filtered Na+ is dependent on cortical Na/K ATPase. - As the Na+ reabsorption is proportionate to the GFR, the filtrate amount is mainly responsible for O2 consumption. When the GFR exceeds 135 ml/min/1,73m2 a condition of hyperfiltration occurs, entailing a steep rise in kidney O2 consumption. | Filtration Pressure Change(Starling E Law)21 - The renal high precapillary pressure provides the hydrostatic pressure reserve to preserve the GFR over a wide range of renal hemodynamic changes.- The ratio between GFR (expressing the O2 consumption related to active transport) and RBF (expressing the O2 delivery) is the filtration fraction, which does not vary to any large extent in humans under physiological conditions, hovering around 20%. | |||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



