
Submitted:
28 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
Chronic stress is a major risk factor for various psychiatric diseases, including depression; it induces a range of cellular and structural adaptations, culminating in altered neurocircuitry and subsequent depression. Accumulating evidence suggests that microglial cells orchestrate stress-induced depression. Preclinical studies of stress-induced depression revealed microglial inflammatory activation in regions of the brain that regulate mood. Although studies have identified several molecules that trigger inflammatory responses in microglia, the pathways that regulate stress-induced microglial activation remain unclear. Understanding the exact triggers that induce microglial inflammatory activation can help find therapeutic targets to treat depression. In the current review, we summarize the recent literature on possible sources of microglial inflammatory activation in animal models of chronic stress-induced depression. In addition, we describe how microglial inflammatory signaling affects neuronal health and causes depressive behavior. Finally, we propose ways to target the microglial inflammatory cascade to treat depressive disorders.
Keywords:
chronic stress
; depression
; microglia
; neuroinflammation
; proinflammatory cytokines
1. Introduction
Major depressive disorder (MDD) is one of the most heterogeneous neuropsychiatric disorders that affects millions worldwide [1]. The disease is characterized by several core behavioral symptoms, including anhedonia, low self-esteem, sleep disturbances, and suicidal ideation [2]. Structural and functional abnormalities have been identified in the depressed brain, including decreased volumes of the prefrontal cortex (PFC) and hippocampus [3]. Various mechanisms have been proposed for decades to regulate depressive behaviors, most of which focused on restoring neuronal health and activity [4,5,6]. The monoamine hypothesis garnered the most attention; therefore, most antidepressant drugs aimed to replenish monoamine levels in the brain. However, poor efficacy of classical anti-depressants in many patients suggests a more complex pathology.
Chronic stress is a major risk factor for MDD and has been associated with increased hypothalamic-pituitary-adrenal (HPA) axis activity and inflammatory activation of immune cells [7]. Increased levels of proinflammatory cytokines in the serum of depressed patients have inspired neuroscientists to investigate the possible role of neuroinflammation in depressive behavior [8]. This ushered in a new era in the neuropsychiatric field, leading to the identification of brain immune cells, particularly microglia, as one of the prominent regulators of inflammation in the depressed brain. Microglia are the second major type of glial cells and primary immune cells that guard the brain parenchyma; these highly receptive cells respond to any changes in the brain microenvironment and adopt various structural and functional phenotypes in a context-dependent manner. In addition to their immune functions, microglia also regulate neuronal functions. For example, microglia aid in formation of neural circuits in the developing brain through synaptic pruning and stripping, secretion of neurotrophic factors, and phagocytosis of dying neurons [9]. Other brain cells such as neurons and astrocytes engage with microglia to maintain brain homeostasis.
Brain samples of patients with MDD have shown that chronic exposure to stressful stimuli affects microglial activation states [10]. Significant inflammatory activation of microglia was found in stress-responsive brain regions, including the PFC, nucleus accumbens, amygdala, and hippocampus [11]. Similar findings were also found in animal models of MDD, where microglial inflammatory activation was positively correlated with depressive-like behaviors [12]. Proinflammatory cytokines released by inflammatory microglia can bind to their cognate neuronal receptors in stress neurocircuitry and regulate behavior [13,14,15]. Moreover, these cytokines can decrease the levels of neurotrophic factors, negatively impacting neuronal health. What exactly triggers microglial activation in stress-related depression remains unclear, though various pathways likely drive microglial immune activation. In the current review, we discuss recent findings on the role of microglia in the pathology of stress-induced depression. We also discuss possible triggers of microglial responses in stress-induced depression. Finally, we identify several potential therapeutic targets that can be exploited to subdue microglial inflammatory activation in stress-induced depression.
2. Possible triggers of microglial inflammatory activation in stress-induced depression
Tissue damage or infection can induce microglial inflammatory activation through the release of proinflammatory mediators [16]. In contrast, the exact triggers of microglial inflammatory activation under sterile inflammatory conditions such as chronic psychosocial stress are unknown. Chronic psychosocial stress causes cellular and structural changes in the brain, resulting in altered neurocircuitry and depressive behavior. In the following sections, we discuss inflammatory factors that trigger activation of microglial cells in response to psychosocial stress (Figure 1).
2.1. Hyperactivity of HPA axis
Neuroendocrine responses to psychosocial stressors are an important compensatory mechanism. The classical “flight and fight response” leads to hyperactivity of the HPA axis, which increases levels of circulating glucocorticoids and catecholamines that return to baseline levels after the threat wanes. In chronic psychosocial stress, however, persistently increased HPA activity exerts deleterious effects on the brain [17]. In vivo models have shown that the upregulation of HPA activity varies according to the type of stressor, which also reflects variable levels of glucocorticoids in a subset of patients with MDD [17].
All major cell types of the brain, including neurons, astrocytes, and microglia, express glucocorticoid receptors [18,19]. These receptors are expressed in distant limbic–midbrain and cortical brain regions including the hippocampus, amygdala, and prefrontal anterior cingulate cortex, suggesting the role of glucocorticoids in stress-related mood disorders [20]. Importantly, distinct regions of the hippocampus show varied sensitivities to glucocorticoid activity [21]. The hippocampus plays diverse roles in memory and behavior due to functional segregation along its longitudinal axis. The dorsal hippocampus primarily contributes to spatial learning and memory, whereas the ventral hippocampus mainly regulates anxiety, which is influenced by stress. Due to its direct connection to the hypothalamus, the ventral hippocampus is more prone to the deleterious effects of glucocorticoids compared to the dorsal hippocampus [22].
Given the impact of the HPA axis on stress neurocircuitry, the increased HPA axis may drive the phenotypic transition of microglia in chronic stress-induced depression. Indeed, recent literature has reported that increased HPA axis activity drives the primed state of microglia and induces the inflammatory phenotype in stress-sensitive brain regions [23,24,25]. Increased serum glucocorticoid levels were found in both preclinical and clinical studies of MDD [26]. Glucocorticoids also increased nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein 3 (NLRP3) inflammasome signaling in the hippocampal region of mice subjected to chronic restraint stress [23]. Increased levels of high-mobility group box 1 (HMGB1) in limbic regions of the rat brain were reported in a model of inescapable tail shock, where subsequent administration of an antagonist blocked glucocorticoid signaling and attenuated the increase of HMGB1 levels [24]. In addition, increased inflammatory signaling in microglia was observed in a mouse model of corticosterone-induced depression [27,28]. Increased levels of proinflammatory cytokines in microglia were accompanied by depressive-like behavior in mice injected with corticosterone [29].
2.2. Peripheral signals: Brain-immune axis
The brain is a unique structure segregated from the rest of the body by the blood-brain barrier (BBB), which dictates peripheral access to the brain parenchyma. The loss of BBB integrity has been documented in MDD pathophysiology, but role of peripheral signals in microglial activation in vivo is debatable [30]. Immune dysfunction has been documented in patients with MDD as well as in preclinical models of depression. Peripheral immune cells are the major sources of proinflammatory cytokines in circulation that can induce inflammatory activation of microglia [31]. In addition, peripheral immune cells infiltrate the brain parenchyma in various animal models of depression. Whether these peripheral immune cells trigger microglial activation in depression is unclear.
In a chronic social defeat stress (CSDS) model, susceptible mice exhibited decreased expression of claudin-5, a tight junction protein in the BBB, which allows peripheral immune cells and proinflammatory cytokines to enter the brain [32]. Moreover, transcriptomic analysis of endothelial cells from susceptible mice revealed increased expression of genes associated with the proinflammatory tumor necrosis factor-α (TNF-α) and the nuclear factor kappa-light-chain-enhanced of activated B cells (NF-κB) pathway [32]. The study also found decreased claudin-5 expression in post-mortem samples of patients with MDD [32]. Thus, a compromised BBB can allow proinflammatory signals from the periphery to act on microglia in a depressed brain.
Increased trafficking of monocytes to the perivascular space and parenchyma was also observed in the repeated social defeat model [33]. Using chimeric mice expressing the green fluorescent protein in lysozyme M (LysM)-positive myeloid cells, the study found an increased infiltration of monocytes in various brain regions of defeated mice. Interestingly, significant increases in interleukin-1β (IL-1β), chemokine (C-C motif) ligand 2 (CCL2), and microglial activation were also found in brain regions in which peripheral macrophages infiltrated. Finally, the study found that crosstalk between chemokine receptor-2 (CCR2) and fractalkine receptor (CX3CR1) recruits macrophages to the brain parenchyma under stressful conditions.
Contrary to these findings, another group found that peripheral immune cells do not play a role in microglial inflammatory activation in acute and chronic social defeat stress models [34]. Here, chronic social defeat increased phagocytic microglial cells in the brain without recruiting peripheral immune signals, indicating that microglia are solely responsible for generating inflammation in the brain during chronic stress. Moreover, peripheral signals can attenuate pathways involved in monoamine synthesis. A recent study showed that lipopolysaccharide binding protein (LBP) expression increased both peripherally and centrally in mice following exposure to stressful stimuli [35]. LBP expression also increased in microglial cells and inhibited enzymes involved in the synthesis of monoamine neurotransmitters. These findings suggest a bidirectional communication between neuroendocrine stimuli and the immune system in the pathology of depression.
2.3. Neuronal signals shape microglial responses
Microglia and neurons work together by secreting diverse molecules to regulate brain homeostasis. Particularly, neuronal-derived soluble factors, including colony-stimulating factor 1 (CSF1), CX3CL1, and transforming growth factor-β (TGFβ), play crucial roles in regulating microglial immune functions [36,37]. Dysregulation of neuronal activity and neuronal atrophy following stress alters neuronal-derived factors that maintain microglial activity, leading to increased inflammatory signaling in microglial cells. Mice exposed to the chronic unpredictable stress model displayed increased expression of CSF1 in the PFC as well as CSF1 receptor (CSF1R) in microglial cells in the same region [36]. Augmented CSF1 signaling in microglia increased phagocytosis of neuronal elements, which reduced dendritic spine density. Interestingly, the knockdown of neuronal CSF1 decreased microglial phagocytosis and attenuated behavioral deficits in stressed mice. Impaired CX3CL1-CX3CR1 signaling between neurons and microglia has also been shown in an animal model of chronic stress, causing inflammatory activation of microglial cells [38]. Microglial deletion of CX3CR1 prevented mice from developing depression-like behavior after stress exposure. Ultimately, CX3CR1 deficiency attenuated chronic stress-induced proinflammatory gene expression in microglia and prevented neuronal dysfunction [39].
2.4. Role of damage-associated molecular patterns (DAMPs)
Studies have reported that damage-associated molecular patterns (DAMPs), including heat shock proteins, HMGB1, and S100 proteins, can initiate sterile neuroinflammatory processes in depressed brains [40]. Microglia can recognize these DAMPs and transmit signals to intracellular NLRP3 inflammasomes through toll-like receptors (TLRs) and receptors for advanced glycation end products (RAGE). These immune receptors have been shown to promote microglial inflammatory signaling in an animal model of chronic stress and depression.
In addition to increased mRNA levels of HMGB1 in hippocampal microglia, higher expression of RAGE and activation of NLRP3 inflammasomes were found in the chronic unpredictable mild stress (CUMS) model of depression [41]. The increased HMGB1-RAGE signaling in hippocampal microglia coincided with depressive-like behavior in mice exposed to chronic unpredictable stress. Increased expression of S100a8 and S100a9 was also found in the PFC of susceptible mice subjected to repeated social defeat stress [42]. Microglia-specific reduction of TLR2/4 expression by using a viral infection strategy, however, prevented mice from developing depressive-like behavior after repeated social defeat stress [42]. Increased HMGB1 expression was also observed in the hippocampal region in rat brains following inescapable tail shock. Increased HMGB1 expression positively correlated with heightened NLRP3 inflammasome signaling [43].
Chronic stress can trigger not only previously well-recognized DAMPs but also extracellular nucleosomes and histones. Increased histones and nucleosomes were found in the cerebrospinal fluid of the CUMS mice, which positively correlated with IL-1β levels in PFC [44]. Higher levels of nucleosomes promoted microglial inflammatory signaling in a C-type lectin receptor 2D (Clec2d)-dependent manner, increasing oxidative stress and IL-1β secretion [44]. Knockdown of Clec2d in PFC reduced microglial inflammatory activation and depressive-like behavior in CUMS mice.
The studies discussed in this section show that various DAMPs are released in the brain following chronic exposure to psychosocial stressors, indicating that the sterile neuroinflammatory environment drives depressive-like behavior. However, the cellular sources of these DAMPs are still unclear. Given the multifactorial pathology of depression, neurons or glia may be major sources of DAMPs in a depressed brain. Indeed, increased HMGB1 mRNA has been found in neuronal and microglial cells [41,42] , and microglia release HMGB1 in vivo under chronic stress, which can amplify inflammatory signaling in the stressed brain [43]; however, future research should confirm this.
3. Role of microglial inflammatory signaling in the pathology of stress-induced depression
Hypotrophy of various brain regions, including the PFC and hippocampus, has been observed in depressed patients, indicating altered structure and function of these brain regions [45,46]. Lower brain volumes in patients with MDD are partly due to altered synaptic connectivity and synaptic loss [47]. Microglial inflammatory activation largely contributes to synaptic dysregulation and neuronal atrophy. In a rodent model of stress-induced depression, microglial inflammatory activation positively correlated with neuronal damage [48]. Inflammatory microglia can damage neurons by releasing proinflammatory mediators, increasing phagocytosis of neuronal elements, and reducing the release of neurotrophic factors.
Microglia are active sources of proinflammatory mediators, most of which are implicated in the pathogenesis of MDD. Increased levels of prostaglandin E2 (PGE2) and IL-6 are found in the serum and cerebrospinal fluid of depressed patients [49,50,51]. Microglial inflammatory signaling modulates stress neurocircuitry involved in mood regulation as well as stress-induced behavioral outcomes. A recent study demonstrated the crucial role of microglial inflammatory signaling in depression-like behavior by explicitly controlling microglial activity, where chemogenetic activation of microglia in the dorsal striatum of mice led to aversive behavior [52]. Mechanistically, increased inflammatory signaling in striatal microglia enhanced secretion of IL-6 and PGE2, reducing the excitability of mid-spiny neurons and causing aversive behavior. These findings highlight the critical role of microglial inflammatory signaling and offer mechanistic insights on inflammatory markers and pathways in depression [53].
Interestingly, microglia remained primed for longer periods after the cessation of stressful stimuli in rodents. Following repeated social defeat stress, the primed state of microglia was necessary for prolonged anxiety-like behavior in mice [54]. The elimination of microglia by using a CSF1R antagonist reduced anxiety-like behavior in defeated mice. In addition, the primed microglia were prerequisite to the recruitment of monocytes in the brain, suggesting that microglia initiate brain-immune crosstalk. In the CSDS model, generation of microglial reactive oxygen species (ROS) positively correlated with depressive-like behavior in mice. The depletion of CSF1R inhibitors reduced the overall ROS content in the brain and CSDS-induced behavioral deficits [48]. Furthermore, mice whose microglia exhibit increased inflammatory gene expression are more susceptible to depression, further linking microglia to CSDS-induced behavioral changes [55].
Inflammatory activation of microglia also reduces dendritic spine density in brain regions such as the PFC and hippocampus. Impaired synaptic plasticity was found in preclinical and clinical investigations of stress-induced depression. A recent study observed increased NLRP3 inflammasome and NF-κB signaling in hippocampal microglia of mice exposed to chronic mild stress (CMS) [56]. Activation of NLRP3 inflammasome and NF-κB signaling in microglial cells in the hippocampus of CMS mice increased the release of C1q, IL-1α, and TNF-α, which activate A1 astrocytes. Transcriptomic analysis of astrocyte reactivity revealed increased expression of neurotoxic astrocyte markers, including C3, Amigo2, Fkbp5, and Serping1 in the hippocampus. Consequently, increased C3+ astrocytes in the dentate gyrus (DG) augmented phagocytosis of neuronal elements, resulting in synaptic impairment, including altered synaptic density, and lower levels of pre-and post-synaptic proteins.
Impaired synaptic remodeling, increased synaptic loss, and reduced excitability of pyramidal neurons were observed in the PFC of defeated mice [57]. Interestingly, the increased CD68+ microglia were also found in the PFC of defeated mice. C3-expressing neurons induced the phagocytic microglia, and they were located in close proximity to each other in the PFC, suggesting that stress-induced cortical underconnectivity and behavioral abnormalities are associated with excessive synaptic pruning mediated by microglia.
Microglia are active regulators of adult neurogenesis. The hippocampal DG and subventricular zone are the two brain regions where new neurons are generated throughout adulthood. Neural stem/progenitor cells (NSCs) undergo various processes to develop into new neurons that are integrated into neural networks. However, stressful experiences hinder adult neurogenesis; antidepressant therapies can exert positive effects on neurogenesis. Inflammatory microglia impair neurogenesis in depression through several mechanisms, including the release of microRNA-enriched exosomes and direct engulfment of NSCs. Rats exposed to the CUMS paradigm displayed increased expression of miR-146a-5p in the DG region. Mechanistically, miR-146a-5p decreased the expression of important regulators of neurogenesis, including krüppel-like factor 4 and cyclin-dependent kinase-like 5, reducing neurogenesis and inducing depressive-like behavior thereafter [58]. Another recent study highlighted how microglial phagocytosis alters neurogenesis in the CSDS model [59]. Phosphatidylserine (PS)-expressing NSCs were found in the DG; NSCs express thermosensitive transient receptor potential vanilloid 4 (TRPV4), which was activated by CSDS. Subsequently increased calcium levels exposed PS on NSCs that were engulfed by microglial cells, reducing neurogenesis.
4. Microglia as a potential therapeutic target for treatment of stress-induced depression
Inflammatory activation of microglia in various limbic brain regions are a hallmark of chronic psychosocial stress not only in rodents but also in humans. Patients with MDD exhibit increased proinflammatory cytokines in cerebrospinal fluid, decreased neurogenesis, and impaired synaptic plasticity [61,62]. Inflammatory activation of microglia is strongly linked to neuronal deficits in MDD pathology; therefore, microglial inflammation is a potential therapeutic target for treating depression. Various strategies have been used effectively in in vivo models of depression to mitigate inflammatory activation of microglia (Table 1).
Inhibition of inflammatory signaling in microglia such as NF-κB and NLRP3 has been shown to reduce depressive-like behavior in animal models of chronic stress [63,76]. NLRP3 inflammasome signaling in microglia increased levels of proinflammatory cytokine IL-1β in the PFC and hippocampus in mice exposed to depression-mimicking paradigms. In the CUMS model of depression, mice treated with an NLRP3-specific inhibitor showed decreased inflammatory activation of microglia as shown by lower levels of proinflammatory cytokines and less arborized morphology of microglia in the PFC compared to vehicle-treated animals [63]. Another recent study used astragalin to target the NLRP3 inflammasome pathway in microglia in the CUMS model of depression [64]; astragalin inhibited microglial inflammatory activation mainly by decreasing NLRP3 signaling in CUMS mice, reducing depressive-like behavior in mice. The inhibition of downstream signaling of TLR4 has also shown antidepressant effects in stressed mice [65]. Roflupram, an inhibitor of phosphodiesterase-4, has been shown to decrease TLR4-induced inflammatory signaling, including HMGB1 and NF-κB, in the PFC and hippocampus of CUMS mice, alleviating depressive-like behavior [65].
Microglial-derived BDNF is crucial for neuronal trophic support and neurogenesis. Recombinant adeno-associated virus (rAAV) vectors encoding murine IL-4 were administered in CMS mice to alternatively activate microglia and therefore reduce inflammatory signaling while increasing Arg1-positive microglia in the DG, which increased microglial-derived BDNF. Together, these factors increased neurogenesis and alleviated depressive-like behavior in CMS mice [66]. Sulforaphane, a plant-derived anti-inflammatory compound also reduced depression-like behavior in CSDS mice by increasing BDNF transcription and inhibiting the inflammatory activation of microglial cells [67]. Recently, ketamine has also proved effective in reducing depression in treatment-resistant MDD patients. As shown in an animal model of chronic stress, ketamine ameliorated depression by increasing BDNF transcription in microglial cells [68].
Liver X receptors (LXRs) are nuclear receptors that decrease microglial inflammatory activation by repressing proinflammatory gene expression. Surprisingly, in the CUMS model of depression, LXRβ expression was lower in the hippocampus and basolateral amygdala regions of the mouse brain [75,76]. Intraperitoneal administration of an LXRβ agonist in CUMS mice alleviated CUMS-induced behavioral despair and synaptic deficits [76]. Peroxisome proliferator-activated receptor gamma (PPAR-γ), a ligand-dependent transcription factor, is another pathway that can inhibit microglial inflammatory signaling whose expression decreases in microglial cells in various animal models of depression. Treatment of CMS mice with asperosaponin VI, a natural compound that increased the expression of PPAR-γ in hippocampal microglia, decreased the expression of proinflammatory cytokines; it also increased Arg1 expression, reducing depressive-like behavior in CMS mice [77].
Inhibition of microglial inflammatory signaling has also shown promising results in animal models of depression. Minocycline is another anti-inflammatory antibiotic that inhibits microglial inflammatory activation and exerts antidepressant effects on patients with MDD. A recent study reported that minocycline administration in CUMS mice reduced phagocytic microglia in the DG of the hippocampus, alleviating depressive-like behavior [71]. Minocycline can also inhibit the release of HMGB1 from microglia, attenuating cognitive decline and depressive-like symptoms in CUMS mice [72].
Other molecules have also been used in an animal model of depression to inhibit microglial inflammatory activation and reduce depressive-like behavior. In the learned helplessness (LH) model, microglial inflammatory activation largely contributes to behavioral despair. Administration of IL-10 to LH mice decreased inflammatory activation of microglia and depression-like behavior [78]. Dimethyl fumarate is another anti-inflammatory compound that acts as an antidepressant in the CUMS model of depression. Treatment of mice with dimethyl fumarate reduced proinflammatory cytokine expression as well as microglial immunoreactivity in the hippocampus of the CUMS mice [79].
5. Conclusions and future perspectives
Microglial inflammatory activation plays a key role in MDD pathology. Several preclinical and clinical studies of MDD have reported increased neuroinflammatory signaling in various brain regions that regulate mood and behavior. However, the exact triggers of microglial inflammatory signaling in MDD pathology remain unclear. Recent studies suggest the possible role of elevated glucocorticoids in inducing the inflammatory phenotype of microglia, but the underlying mechanisms are unknown. The brain-immune axis is another factor that can induce microglial inflammatory activation during psychosocial stress.
Despite these ambiguities, inflammatory activation of microglia and neuronal deficits are consistently observed in animal models of stress-induced depression. Increased proinflammatory signaling in microglia hampers neuronal health through phagocytosis of synaptic elements, altered synaptic plasticity, and decreased neurogenesis. Therefore, reducing microglial inflammatory activation is a potential strategy to treat MDD. Further research is needed to elucidate the molecular mechanisms involved in microglia activation and their impact on neurons in stress-induced depression. Future studies should better identify specific therapeutic targets to treat MDD.
Author Contributions
Writing—original draft preparation, R.A.; writing—review and editing K.S. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2017R1A5A2015391, 2020M3E5D9079764).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Konig, H.; Konig, H.H.; Konnopka, A. The excess costs of depression: a systematic review and meta-analysis. Epidemiol Psychiatr Sci 2019, 29, e30. [Google Scholar] [CrossRef]
- Alexopoulos, G.S. Mechanisms and treatment of late-life depression. Transl Psychiatry 2019, 9, 188. [Google Scholar] [CrossRef]
- Koolschijn, P.C.; van Haren, N.E.; Lensvelt-Mulders, G.J.; Hulshoff Pol, H.E.; Kahn, R.S. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp 2009, 30, 3719–3735. [Google Scholar] [CrossRef]
- Wang, B.; Shi, H.; Ren, L.; Miao, Z.; Wan, B.; Yang, H.; Fan, X.; Gustafsson, J.A.; Sun, M.; Xu, X. Ahi1 regulates serotonin production by the GR/ERbeta/TPH2 pathway involving sexual differences in depressive behaviors. Cell Commun Signal 2022, 20, 74. [Google Scholar] [CrossRef]
- Kornhuber, J.; Gulbins, E. New Molecular Targets for Antidepressant Drugs. Pharmaceuticals (Basel) 2021, 14. [Google Scholar] [CrossRef]
- Malki, K.; Keers, R.; Tosto, M.G.; Lourdusamy, A.; Carboni, L.; Domenici, E.; Uher, R.; McGuffin, P.; Schalkwyk, L.C. The endogenous and reactive depression subtypes revisited: integrative animal and human studies implicate multiple distinct molecular mechanisms underlying major depressive disorder. BMC Med 2014, 12, 73. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Gu, J.H.; Wang, C.N.; Guan, W.; Liu, Y.; Tang, W.Q.; Ji, C.H.; Chen, Y.M.; Huang, J., et al. Salt-inducible kinase 1-CREB-regulated transcription coactivator 1 signalling in the paraventricular nucleus of the hypothalamus plays a role in depression by regulating the hypothalamic-pituitary-adrenal axis. Mol Psychiatry 2022. [CrossRef]
- Sukhram, S.D.; Yilmaz, G.; Gu, J. Antidepressant Effect of Ketamine on Inflammation-Mediated Cytokine Dysregulation in Adults with Treatment-Resistant Depression: Rapid Systematic Review. Oxid Med Cell Longev 2022, 2022, 1061274. [Google Scholar] [CrossRef] [PubMed]
- Schlegelmilch, T.; Henke, K.; Peri, F. Microglia in the developing brain: from immunity to behaviour. Curr Opin Neurobiol 2011, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Li, Y.; Jiang, Y.; Huang, J.H.; Wang, F. Glymphatic Dysfunction Induced Oxidative Stress and Neuro-Inflammation in Major Depression Disorders. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Tynan, R.J.; Naicker, S.; Hinwood, M.; Nalivaiko, E.; Buller, K.M.; Pow, D.V.; Day, T.A.; Walker, F.R. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 2010, 24, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Nemeth, D.P.; Liu, X.; Witcher, K.G.; O'Neil, S.M.; Oliver, B.; Bray, C.E.; Sheridan, J.F.; Godbout, J.P.; Quan, N. Interleukin-1 receptor on hippocampal neurons drives social withdrawal and cognitive deficits after chronic social stress. Mol Psychiatry 2021, 26, 4770–4782. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Sivakumar, V.; Zou, Z.; Ling, E.A. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct Funct 2014, 219, 151–170. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Varghese, F.P.; Brown, E.S. The Hypothalamic-Pituitary-Adrenal Axis in Major Depressive Disorder: A Brief Primer for Primary Care Physicians. Prim Care Companion J Clin Psychiatry 2001, 3, 151–155. [Google Scholar] [CrossRef]
- Niraula, A.; Wang, Y.; Godbout, J.P.; Sheridan, J.F. Corticosterone Production during Repeated Social Defeat Causes Monocyte Mobilization from the Bone Marrow, Glucocorticoid Resistance, and Neurovascular Adhesion Molecule Expression. J Neurosci 2018, 38, 2328–2340. [Google Scholar] [CrossRef]
- Meijer, O.C.; Buurstede, J.C.; Schaaf, M.J.M. Corticosteroid Receptors in the Brain: Transcriptional Mechanisms for Specificity and Context-Dependent Effects. Cell Mol Neurobiol 2019, 39, 539–549. [Google Scholar] [CrossRef]
- Wang, Q.; Van Heerikhuize, J.; Aronica, E.; Kawata, M.; Seress, L.; Joels, M.; Swaab, D.F.; Lucassen, P.J. Glucocorticoid receptor protein expression in human hippocampus; stability with age. Neurobiol Aging 2013, 34, 1662–1673. [Google Scholar] [CrossRef]
- Sahay, A.; Hen, R. Adult hippocampal neurogenesis in depression. Nat Neurosci 2007, 10, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Levone, B.R.; Codagnone, M.G.; Moloney, G.M.; Nolan, Y.M.; Cryan, J.F.; OF, O.L. Adult-born neurons from the dorsal, intermediate, and ventral regions of the longitudinal axis of the hippocampus exhibit differential sensitivity to glucocorticoids. Mol Psychiatry 2021, 26, 3240–3252. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front Mol Neurosci 2019, 12, 210. [Google Scholar] [CrossRef]
- Frank, M.G.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Glucocorticoids mediate stress induction of the alarmin HMGB1 and reduction of the microglia checkpoint receptor CD200R1 in limbic brain structures. Brain Behav Immun 2019, 80, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Thompson, B.M.; Watkins, L.R.; Maier, S.F. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun 2012, 26, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.A.; Cattaneo, A.; Cattane, N.; Lopizzo, N.; Tojo, L.; Bakunina, N.; Musaelyan, K.; Borsini, A.; Zunszain, P.A.; Pariante, C.M. Glucocorticoids prime the inflammatory response of human hippocampal cells through up-regulation of inflammatory pathways. Brain Behav Immun 2020, 87, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Qiao, Y.; Lo, P.C.; Song, L.; Yang, Y.; Duan, L.; Wei, S.; Li, M.; Huang, S.; Zhang, B. , et al. Anti-depressive effects of Jiao-Tai-Wan on CORT-induced depression in mice by inhibiting inflammation and microglia activation. J Ethnopharmacol 2022, 283, 114717. [Google Scholar] [CrossRef]
- Horchar, M.J.; Wohleb, E.S. Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav Immun 2019, 81, 329–340. [Google Scholar] [CrossRef]
- Mao, Z.F.; Ouyang, S.H.; Zhang, Q.Y.; Wu, Y.P.; Wang, G.E.; Tu, L.F.; Luo, Z.; Li, W.X.; Kurihara, H.; Li, Y.F. , et al. New insights into the effects of caffeine on adult hippocampal neurogenesis in stressed mice: Inhibition of CORT-induced microglia activation. FASEB J 2020, 34, 10998–11014. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: a review of clinical and experimental evidence. J Neuroinflammation 2013, 10, 142. [Google Scholar] [CrossRef]
- Afridi, R.; Seol, S.; Kang, H.J.; Suk, K. Brain-immune interactions in neuropsychiatric disorders: Lessons from transcriptome studies for molecular targeting. Biochem Pharmacol 2021, 188, 114532. [Google Scholar] [CrossRef]
- Dudek, K.A.; Dion-Albert, L.; Lebel, M.; LeClair, K.; Labrecque, S.; Tuck, E.; Ferrer Perez, C.; Golden, S.A.; Tamminga, C.; Turecki, G. , et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc Natl Acad Sci U S A 2020, 117, 3326–3336. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 2013, 33, 13820–13833. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.L.; Cooper, H.A.; Maric, D.; Herkenham, M. Social defeat induces depressive-like states and microglial activation without involvement of peripheral macrophages. J Neuroinflammation 2016, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Li, Y.; Liao, Z.; Wang, G.; Cao, Q.; Li, Y.; Duan, Y.; Han, Y.; Deng, X.; Wu, F. , et al. Lipopolysaccharide-binding protein expression is increased by stress and inhibits monoamine synthesis to promote depressive symptoms. Immunity 2023, 56, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Terwilliger, R.; Duman, C.H.; Duman, R.S. Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol Psychiatry 2018, 83, 38–49. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E. , et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Rimmerman, N.; Schottlender, N.; Reshef, R.; Dan-Goor, N.; Yirmiya, R. The hippocampal transcriptomic signature of stress resilience in mice with microglial fractalkine receptor (CX3CR1) deficiency. Brain Behav Immun 2017, 61, 184–196. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, T.; Meng, D.; Sun, L.; Yang, G.; He, Y.; Zhang, C. Involvement of CX3CL1/CX3CR1 in depression and cognitive impairment induced by chronic unpredictable stress and relevant underlying mechanism. Behav Brain Res 2020, 381, 112371. [Google Scholar] [CrossRef]
- Fleshner, M.; Frank, M.; Maier, S.F. Danger Signals and Inflammasomes: Stress-Evoked Sterile Inflammation in Mood Disorders. Neuropsychopharmacology 2017, 42, 36–45. [Google Scholar] [CrossRef]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaca, M.; Hare, B.; Duman, R.S. Persistent Increase in Microglial RAGE Contributes to Chronic Stress-Induced Priming of Depressive-like Behavior. Biol Psychiatry 2018, 83, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Kitaoka, S.; Tanaka, K.; Segi-Nishida, E.; Imoto, Y.; Ogawa, A.; Nakano, F.; Tomohiro, A.; Nakayama, K.; Taniguchi, M. , et al. The Innate Immune Receptors TLR2/4 Mediate Repeated Social Defeat Stress-Induced Social Avoidance through Prefrontal Microglial Activation. Neuron 2018, 99, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.D.; Frank, M.G.; Tracey, K.J.; Watkins, L.R.; Maier, S.F. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 2015, 35, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Bao, H.; Liu, C.; Zhang, Q.; Huang, A.; Quan, M.; Li, C.; Xiong, Y.; Chen, G.; Hou, L. Extracellular Nucleosomes Accelerate Microglial Inflammation via C-Type Lectin Receptor 2D and Toll-Like Receptor 9 in mPFC of Mice With Chronic Stress. Front Immunol 2022, 13, 854202. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev 2009, 33, 699–771. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Yucel, K.; Taylor, V.H.; Macdonald, K.; Joffe, R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol Psychiatry 2008, 64, 880–883. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K. Synaptic dysfunction in depression: potential therapeutic targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.L.; Weigel, T.K.; Poffenberger, C.N.; Herkenham, M. The Behavioral Sequelae of Social Defeat Require Microglia and Are Driven by Oxidative Stress in Mice. J Neurosci 2019, 39, 5594–5605. [Google Scholar] [CrossRef]
- Wang, A.K.; Miller, B.J. Meta-analysis of Cerebrospinal Fluid Cytokine and Tryptophan Catabolite Alterations in Psychiatric Patients: Comparisons Between Schizophrenia, Bipolar Disorder, and Depression. Schizophr Bull 2018, 44, 75–83. [Google Scholar] [CrossRef]
- Muller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Muller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N. , et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef]
- Linnoila, M.; Whorton, A.R.; Rubinow, D.R.; Cowdry, R.W.; Ninan, P.T.; Waters, R.N. CSF prostaglandin levels in depressed and schizophrenic patients. Arch Gen Psychiatry 1983, 40, 405–406. [Google Scholar] [CrossRef]
- Klawonn, A.M.; Fritz, M.; Castany, S.; Pignatelli, M.; Canal, C.; Simila, F.; Tejeda, H.A.; Levinsson, J.; Jaarola, M.; Jakobsson, J. , et al. Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 2021, 54, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N. , et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand 2017, 135, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.D.; McKim, D.B.; Niraula, A.; Witcher, K.G.; Yin, W.; Sobol, C.G.; Wang, Y.; Sawicki, C.M.; Sheridan, J.F.; Godbout, J.P. The Influence of Microglial Elimination and Repopulation on Stress Sensitization Induced by Repeated Social Defeat. Biol Psychiatry 2019, 85, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.L.; Weigel, T.K.; Cooper, H.A.; Elkahloun, A.G.; Kigar, S.L.; Herkenham, M. Decoding microglia responses to psychosocial stress reveals blood-brain barrier breakdown that may drive stress susceptibility. Sci Rep 2018, 8, 11240. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fang, Y.; Zhang, Y.; Song, M.; Zhang, X.; Ding, X.; Yao, H.; Chen, M.; Sun, Y.; Ding, J. , et al. Microglial NLRP3 inflammasome activates neurotoxic astrocytes in depression-like mice. Cell Rep 2022, 41, 111532. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, H.S.; Li, H.H.; Wang, H.J.; Zou, R.S.; Lu, X.J.; Wang, J.; Nie, B.B.; Wu, J.F.; Li, S. , et al. Microglia-dependent excessive synaptic pruning leads to cortical underconnectivity and behavioral abnormality following chronic social defeat stress in mice. Brain Behav Immun 2023, 109, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Li, Y.; Lan, T.; Wang, W.; Long, Y.; Yu, S.Y. Microglia secrete miR-146a-5p-containing exosomes to regulate neurogenesis in depression. Mol Ther 2022, 30, 1300–1314. [Google Scholar] [CrossRef]
- Hoshi, Y.; Shibasaki, K.; Gailly, P.; Ikegaya, Y.; Koyama, R. Thermosensitive receptors in neural stem cells link stress-induced hyperthermia to impaired neurogenesis via microglial engulfment. Sci Adv 2021, 7, eabj8080. [Google Scholar] [CrossRef]
- Picard, K.; Bisht, K.; Poggini, S.; Garofalo, S.; Golia, M.T.; Basilico, B.; Abdallah, F.; Ciano Albanese, N.; Amrein, I.; Vernoux, N. , et al. Microglial-glucocorticoid receptor depletion alters the response of hippocampal microglia and neurons in a chronic unpredictable mild stress paradigm in female mice. Brain Behav Immun 2021, 97, 423–439. [Google Scholar] [CrossRef]
- Lucassen, P.J.; Stumpel, M.W.; Wang, Q.; Aronica, E. Decreased numbers of progenitor cells but no response to antidepressant drugs in the hippocampus of elderly depressed patients. Neuropharmacology 2010, 58, 940–949. [Google Scholar] [CrossRef]
- Campbell, S.; Marriott, M.; Nahmias, C.; MacQueen, G.M. Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry 2004, 161, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, M.M.; Guo, M.X.; Zhang, Q.P.; Li, N.Z.; Cheng, J.; Wang, S.L.; Xu, G.H.; Li, C.F.; Zhu, J.X., et al. Inhibition of Microglial NLRP3 with MCC950 Attenuates Microglial Morphology and NLRP3/Caspase-1/IL-1beta Signaling In Stress-induced Mice. J Neuroimmune Pharmacol 2022. [CrossRef]
- Tong, Y.; Fu, H.; Xia, C.; Song, W.; Li, Y.; Zhao, J.; Zhang, X.; Gao, X.; Yong, J.; Liu, Q. , et al. Astragalin Exerted Antidepressant-like Action through SIRT1 Signaling Modulated NLRP3 Inflammasome Deactivation. ACS Chem Neurosci 2020, 11, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, B.; Qin, Y.; Dong, W.; Zhong, J.; Li, M.; Cheng, Y.; Xu, J.; Wang, H. Inhibition of phosphodiesterase-4 suppresses HMGB1/RAGE signaling pathway and NLRP3 inflammasome activation in mice exposed to chronic unpredictable mild stress. Brain Behav Immun 2021, 92, 67–77. [Google Scholar] [CrossRef]
- Zhang, J.; Rong, P.; Zhang, L.; He, H.; Zhou, T.; Fan, Y.; Mo, L.; Zhao, Q.; Han, Y.; Li, S. , et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci Adv 2021, 7. [Google Scholar] [CrossRef]
- Tang, R.; Cao, Q.Q.; Hu, S.W.; He, L.J.; Du, P.F.; Chen, G.; Fu, R.; Xiao, F.; Sun, Y.R.; Zhang, J.C. , et al. Sulforaphane activates anti-inflammatory microglia, modulating stress resilience associated with BDNF transcription. Acta Pharmacol Sin 2022, 43, 829–839. [Google Scholar] [CrossRef]
- Yao, W.; Cao, Q.; Luo, S.; He, L.; Yang, C.; Chen, J.; Qi, Q.; Hashimoto, K.; Zhang, J.C. Microglial ERK-NRBP1-CREB-BDNF signaling in sustained antidepressant actions of (R)-ketamine. Mol Psychiatry 2022, 27, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Lyu, D.; Wang, F.; Zhang, M.; Yang, W.; Huang, H.; Huang, Q.; Wu, C.; Qian, N.; Wang, M.; Zhang, H. , et al. Ketamine induces rapid antidepressant effects via the autophagy-NLRP3 inflammasome pathway. Psychopharmacology (Berl) 2022, 239, 3201–3212. [Google Scholar] [CrossRef]
- Bollinger, J.L.; Horchar, M.J.; Wohleb, E.S. Diazepam limits microglia-mediated neuronal remodeling in the prefrontal cortex and associated behavioral consequences following chronic unpredictable stress. Neuropsychopharmacology 2020, 45, 1766–1776. [Google Scholar] [CrossRef]
- Bassett, B.; Subramaniyam, S.; Fan, Y.; Varney, S.; Pan, H.; Carneiro, A.M.D.; Chung, C.Y. Minocycline alleviates depression-like symptoms by rescuing decrease in neurogenesis in dorsal hippocampus via blocking microglia activation/phagocytosis. Brain Behav Immun 2021, 91, 519–530. [Google Scholar] [CrossRef]
- Wang, B.; Huang, X.; Pan, X.; Zhang, T.; Hou, C.; Su, W.J.; Liu, L.L.; Li, J.M.; Wang, Y.X. Minocycline prevents the depressive-like behavior through inhibiting the release of HMGB1 from microglia and neurons. Brain Behav Immun 2020, 88, 132–143. [Google Scholar] [CrossRef]
- Han, Q.Q.; Shen, S.Y.; Chen, X.R.; Pilot, A.; Liang, L.F.; Zhang, J.R.; Li, W.H.; Fu, Y.; Le, J.M.; Chen, P.Q. , et al. Minocycline alleviates abnormal microglial phagocytosis of synapses in a mouse model of depression. Neuropharmacology 2022, 220, 109249. [Google Scholar] [CrossRef]
- Cheng, D.; Qin, Z.S.; Zheng, Y.; Xie, J.Y.; Liang, S.S.; Zhang, J.L.; Feng, Y.B.; Zhang, Z.J. Minocycline, a classic antibiotic, exerts psychotropic effects by normalizing microglial neuroinflammation-evoked tryptophan-kynurenine pathway dysregulation in chronically stressed male mice. Brain Behav Immun 2023, 107, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, H.; Sen Ta Na, H.; Wang, L.; Zhong, C.; Deng, B.; Liu, C.; Bao, H.; Sang, H.; Hou, L. Neuronal-microglial liver X receptor beta activating decrease neuroinflammation and chronic stress-induced depression-related behavior in mice. Brain Res 2022, 1797, 148112. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xiao, X.; Yan, Y.; Zhang, T. Activation of liver X receptors prevents emotional and cognitive dysfunction by suppressing microglial M1-polarization and restoring synaptic plasticity in the hippocampus of mice. Brain Behav Immun 2021, 94, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yi, S.; Liu, Q.; Su, D.; Li, L.; Xiao, C.; Zhang, J. Asperosaponin VI ameliorates the CMS-induced depressive-like behaviors by inducing a neuroprotective microglial phenotype in hippocampus via PPAR-gamma pathway. J Neuroinflammation 2022, 19, 115. [Google Scholar] [CrossRef] [PubMed]
- Worthen, R.J.; Garzon Zighelboim, S.S.; Torres Jaramillo, C.S.; Beurel, E. Anti-inflammatory IL-10 administration rescues depression-associated learning and memory deficits in mice. J Neuroinflammation 2020, 17, 246. [Google Scholar] [CrossRef]
- de Souza, A.G.; Lopes, I.S.; Filho, A.; Cavalcante, T.M.B.; Oliveira, J.V.S.; de Carvalho, M.A.J.; de Lima, K.A.; Juca, P.M.; Mendonca, S.S.; Mottin, M. , et al. Neuroprotective effects of dimethyl fumarate against depression-like behaviors via astrocytes and microglia modulation in mice: possible involvement of the HCAR2/Nrf2 signaling pathway. Naunyn Schmiedebergs Arch Pharmacol 2022, 395, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Triggers and role of microglial inflammatory activation in the pathogenesis of depression. Chronic psychosocial stress can increase hyperactivity of the hypothalamic-pituitary-adrenal axis, activation of peripheral immune cells, and release of damage-associated molecular patterns (DAMPS). Stress can also disturb communication between microglia and neurons that regulate microglial immune responses. Inflammatory signaling in microglia increases the expression of proinflammatory cytokines and generation of reactive oxygen species (ROS). Inflammatory microglia also show decreased neurotrophic signaling, which hampers the release of brain-derived neurotrophic factor (BDNF) from microglia. These changes culminate in neuronal damage including decreased neurogenesis, dendritic spine density, and impaired synaptic plasticity, leading to depression. NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IL, interleukin; C1q, complement component 1q; TNF, tumor necrosis factor; MeCP2, methyl-CpG binding protein 2; CREB, cAMP response element binding protein; ERK, extracellular signal-regulated kinase; PPAR-γ, peroxisome proliferator-activated receptor gamma, NLRP3, nucleotide-binding domain, leucine-rich repeat, pyrin domain-containing protein 3.
Figure 1.
Triggers and role of microglial inflammatory activation in the pathogenesis of depression. Chronic psychosocial stress can increase hyperactivity of the hypothalamic-pituitary-adrenal axis, activation of peripheral immune cells, and release of damage-associated molecular patterns (DAMPS). Stress can also disturb communication between microglia and neurons that regulate microglial immune responses. Inflammatory signaling in microglia increases the expression of proinflammatory cytokines and generation of reactive oxygen species (ROS). Inflammatory microglia also show decreased neurotrophic signaling, which hampers the release of brain-derived neurotrophic factor (BDNF) from microglia. These changes culminate in neuronal damage including decreased neurogenesis, dendritic spine density, and impaired synaptic plasticity, leading to depression. NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IL, interleukin; C1q, complement component 1q; TNF, tumor necrosis factor; MeCP2, methyl-CpG binding protein 2; CREB, cAMP response element binding protein; ERK, extracellular signal-regulated kinase; PPAR-γ, peroxisome proliferator-activated receptor gamma, NLRP3, nucleotide-binding domain, leucine-rich repeat, pyrin domain-containing protein 3.
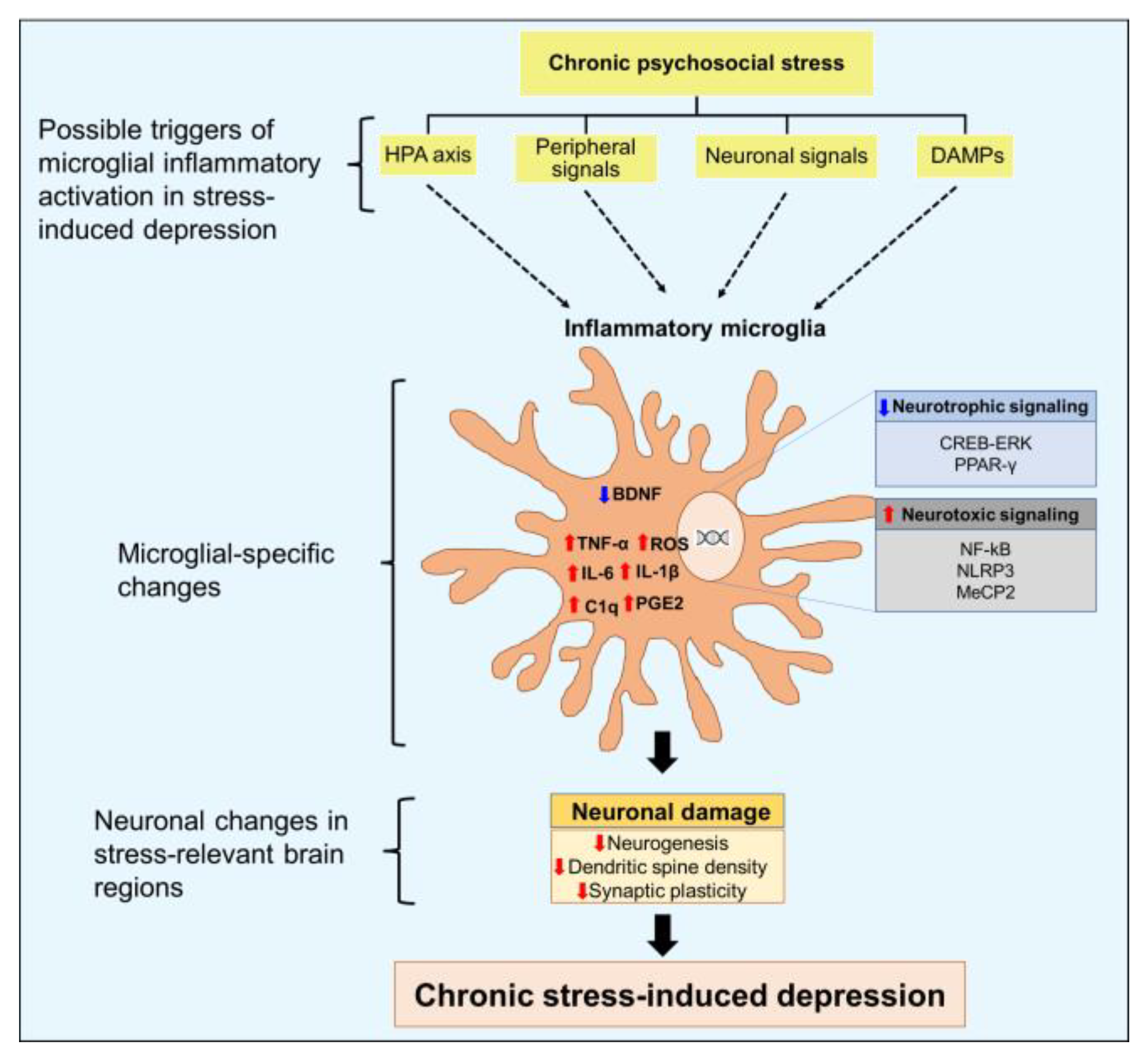

Table 1.
Targeting microglial inflammatory activation in animal models of depression.
| Putative microglial targets | Animal models | Brain regions | Targeting strategies | Outcomes | References |
| ↓ NLRP3 signaling | CUMS (Mice) | PFC | MCC950 | ↓Depressive-like behavior ↓Neuroinflammatory markers ↓IL-1β | [63] |
| ↓ NLRP3 signaling | CUMS (Mice) | Hippocampus | Astragalin | ↓Depressive-like behavior ↓Neuroinflammatory markers ↓IL-1β | [64] |
| ↓p38 MAPK signaling ↓NF-κB signaling ↓HMGB1/RAGE/TLR4 signaling | CUMS (Mice) | Hippocampus PFC | Roflupram | ↓depressive-like behavior ↓proinflammatory cytokines | [65] |
| ↑ BDNF signaling | CMS (Mice) | Hippocampus | Viral-mediated overexpression of IL-4 | ↑ Neurogenesis ↓ Depressive-like behavior ↓ Proinflammatory cytokines ↑ Arg-1 positive microglia |
[66] |
| ↑BDNF by increasing Nrf2 signaling ↓MeCP2 expression | CSDS (Mice) | PFC | Sulforaphane | ↑ Resilience to stress ↑ Synaptic plasticity ↓ Proinflammatory cytokines | [67] |
| ↑ ERK-NRBP1-CREB signaling ↑ microglial BDNF | CSDS (Mice) | PFC | (R)-Ketamine | ↑ Dendritic spine density long-lasting antidepressant action |
[68] |
| ↓ NLRP3 signaling ↑Autophagy | CRS (rats) | PFC Hippocampus | Ketamine | ↑ Synaptic plasticity ↓Depressive-like behavior | [69] |
| ↓CSF1 receptor expression ↓CD11b ↓(CR3)-C3 phagocytic pathway |
CUS (Mice) | PFC | Diazepam | ↑ Dendritic spine density long-lasting antidepressant action |
[70] |
| ↓ERK 1/2 signaling ↓Phagocytic microglia |
CMS (Mice) | Hippocampus |
Minocycline | ↑ Neurogenesis ↓ Depressive-like behavior | [71] |
| ↓ HMGB1 release | CUMS (Mice) | ↑ Cognitive performance ↓ Depressive-like behavior | [72] | ||
| ↓ Phagocytic microglia | CSDS (Mice) | ↓ Proinflammatory cytokines ↓ Synaptic loss ↓ Behavioral despair | [73] | ||
| ↓Phagocytic and inflammatory microglia | CUMS (Mice) | PFC Hippocampus | ↑ Kynurenic acid ↓Behavioral despair | [74] | |
| ↑ LXR- β signaling ↓ NF-κB signaling ↓NLRP3 signaling ↓IL-1β ↓ Phagocytic microglia |
CUMS Corticosterone-induced depression | Basolateral amygdala | TO90137 | ↓ Neuroinflammation ↓Depressive-like behavior | [75] |
| ↑ LXR- β signaling ↓ NF-κB signaling |
CUMS (Mice) | Hippocampus | GW3965 | ↓ Inflammatory markers ↓Synaptic impairment | [76] |
| ↑PPAR-γ signaling ↑ Neuroprotective microglia | CMS (Mice) | Hippocampus | Asperosaponin VI | ↑ Microglial-neuronal interactions ↓ Synaptic deficits | [77] |
| Not discussed | Learned helplessness (mice) | Hippocampus | murine recombinant IL-10 | ↑ Dendritic spine density ↑ Cognitive performance | [78] |
| Not discussed | CUMS (mice) | Hippocampus | Dimethyl fumarate | ↓Neuroinflammatory markers ↓ Cognitive impairment | [79] |
CUMS, chronic unpredictable mild stress; CSDS, chronic social defeat stress; CMS, chronic mild stress; CRS, chronic restraint stress; CUS; chronic unpredictable stress; PFC, prefrontal cortex; NLRP3, nucleotide-binding domain, leucine-rich repeat, pyrin domain-containing protein 3; MAPK, mitogen-activated protein kinases, NF-κB, nuclear factor kappa-light-chain-enhanced of activated B cells, IL-1β, interleukin-1β; HMGB1, high mobility group box 1; RAGE, receptors for advanced glycation end products; TLR, toll-like receptors; BDNF, brain-derived neurotrophic factor; PPAR-γ, peroxisome proliferator-activated receptor gamma, MeCP2, methyl-CpG binding protein 2; CREB, cAMP response element binding protein; ERK, extracellular signal-regulated kinase; LXR, Liver X receptors; CSF1, colony-stimulating factor 1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



