
Submitted:
28 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
Neuroinflammation is one disease hallmark on the road to neurodegeneration in primary tauopathies. Thus, immunomodulation might be a suitable treatment strategy to delay or even prevent the occurrence of symptoms and thus relieve the burden for patients and caregivers. In the last years, the peroxisome proliferator-activated receptor γ (PPARγ) received increasing attention as it is immediately involved in the regulation of the immune system and can be targeted by the anti-diabetic drug pioglitazone. Previous studies have shown significant immunomodulation in amyloid-β (Aβ) mouse models by pioglitazone. In this study, we performed long-term treatment over six months in P301S mice as a tauopathy model with either pioglitazone or placebo. We performed serial 18 kDa translocator protein positron-emission-tomography (TSPO-PET) imaging and terminal immunohistochemistry to assess microglial activation during treatment. Tau pathology was quantified via immunohistochemistry at the end of the study. Long-term pioglitazone treatment had no significant effect on TSPO-PET, immunohistochemistry read-outs of microglial activation, or tau pathology levels in P301S mice. Thus, we conclude that pioglitazone modifies the time course of Aβ-dependent microglial activation, but does not significantly modulate microglial activation in response to tau pathology.
Keywords:
microglia
; pioglitazone
; TSPO-PET
1. Introduction
Neuroinflammation is a common feature of many neurological diseases [1,2]. In the brain, inflammation is caused by the inherent immune system of the brain, which consists of astrocytes and microglia, both glial cells that support the neuronal network [3]. Microglia form a distinct population among myeloid immune cells of the organism [4] and they are critical to maintain the physiological state of the brain by releasing neurotrophic factors and removing possibly neurotoxic debris like pathological protein aggregates [2,3,5,6]. However, dysregulation leading to chronic inflammation can contribute to neurodegeneration [1,3,7]. Thus, along the disease progression, proinflammatory cytokines might promote amyloid-β (Aβ) accumulation in APPPS1 mice [6]. In a tauopathy mouse model, neurodegeneration was found to be driven by activated microglia rather than by the spread of aggregated tau itself [8]. Hence, modulation of the immune system to temper detrimental phenotypes is an important consideration in the development of therapeutic strategies concerning neurodegenerative diseases.
One well-established option to achieve immunomodulation is targeting the peroxisome proliferator-activated receptor γ (PPARγ) through pioglitazone [9,10]. PPARγ is a transcription factor [11] involved in many different physiological functions amongst others cell differentiation, cell death, glucose metabolism and insulin sensitivity [11,12,13]. Moreover, it has been shown that PPARγ modulates multiple genes associated with the immune response, some also being dysregulated in late-onset Alzheimer’s disease [14,15]. Pioglitazone is a well-known PPARγ agonist and an approved drug for treatment of type 2 diabetes [11,16,17] that enhances PPARγ expression [17,18]. In earlier studies, pioglitazone has been shown to be a promising treatment for Alzheimer’s disease, ameliorating both the pathology as well as the cognition in animal models.
In this study, we investigated the efficacy of long-term pioglitazone treatment to modulate chronic inflammation in a mouse model of tauopathy. Based on previous findings in Aβ mouse models, we tested the hypothesis that decreased microglial activation is detectable by serial 18 kDa translocator protein positron-emission-tomography (TSPO-PET) in pioglitazone-treated P301S mice. Furthermore, we aimed to validate the TSPO-PET results by ionized calcium-binding adapter molecule 1 (Iba1) and Cluster of Differentiation 68 (CD68) immunohistochemistry and we performed AT8 immunohistochemistry to test for alterations of tau accumulation after long-term pioglitazone treatment.
2. Results
2.1. Pioglitazone Treatment Has No Significant Effect on Serial TSPO-PET Signals in P301S Mice
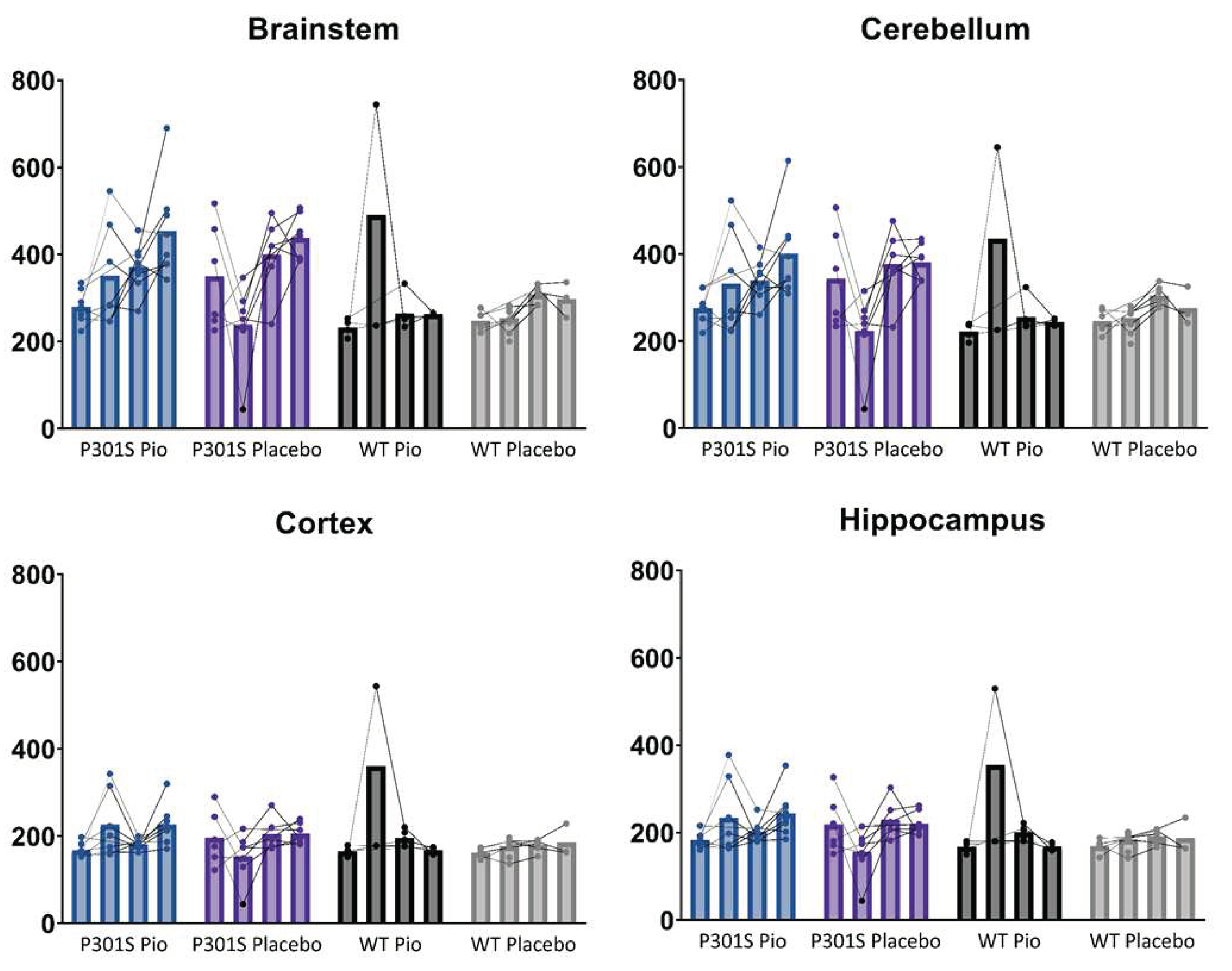
TSPO-PET results of nucleus accumbens normalization are visualized in Figure 1. Mixed-effect models of the different groups revealed significant differences in TSPO-signal between P301S and wild-type mice in all target areas, including brainstem and cerebellum (p<0.0001), hippocampus (p=0.0014), and cortex (p=0.0273). However, no significant differences could be observed for the treatment status (pioglitazone vs. placebo) within P301S and wild-type cohorts, neither as general treatment effect nor with Tukey’s multiple comparisons test, except for wild-type mice at eight months of age (p = 0.0451). Similar results were found for SUV normalized data, myocardium adjusted SUV or %ID (Figure A1,Figure A2,Figure A3).
In particular, the brainstem TSPO-PET signal showed a time-dependent increase in P301S mice treated with pioglitazone relative to baseline (7.3 months: +21%, p=0.01; 8.2 months: +23%, p=0.002). However, placebo-treated P301S mice indicated a similar TSPO-PET signal increase in the brainstem relative to baseline (7.3 months: +22%, p=0.006; 8.2 months: +23%, p=0.003). Similar TSPO-PET signal increases in P301S mice with and without pioglitazone treatment were observed for the cerebellum. Again, the comparison of TSPO-PET results with different normalization approaches did not indicate long-term pioglitazone related treatment effects on the rate of TSPO-PET change over time in P301S or wild-type mice (Figure A1,Figure A2,Figure A3).
2.2. Pioglitazone Treatment Has No Significant Effect on Abundance of Tau-Positive Cells in P301S Mice
Immunohistochemical stainings with AT8 allowed for the quantification of tau-positive cells in the cortex and the brainstem of P301S mice treated with pioglitazone or placebo. On average, P301S mice receiving placebo had 143.0 ± 42.3 tau-positive cells in 0.014 mm2 of the cortex and 99.0 ± 33.2 tau-positive cells in 0.011 mm2 of the brainstem, whereas P301S mice treated with pioglitazone had 120.9 ± 45.9 (-15%) tau-positive cells in the cortex and 112.8 ± 39.1 (+14%) tau-positive cells in the brainstem (Figure 2). None of these comparisons reached statistical significance (cortex: p=0.35, brainstem: p=0.44).
2.3. Pioglitazone Has No Significant Effect on Iba1 and CD68 Expression in P301S Mice
The immunohistochemical assessment of Iba1 and CD68 expression in 0.002 mm2 of the cortex and the brainstem of P301S mice did not show significant differences between pioglitazone treated mice compared to placebo (Figure 3). While a trend towards lower Iba1 and CD68 reactivity was observed for the cortex of pioglitazone treated P301S mice compared to placebo (p=0.26 and p=0.08, respectively), the brainstem indicated similar abundance of both markers between treatment and placebo groups (p=0.51 and p=0.50, respectively).
3. Discussion
Pioglitazone is considered as a potential treatment for neurodegenerative diseases as several preclinical trials showed beneficial effects on different disease-related hallmarks. In particular, pioglitazone enhanced memory and learning as well as ameliorated behavior [19,20,21], although in the study of Seok et al. (2019) [22] only at lower doses. On a mechanistic level, the activation of PPARγ by pioglitazone hindered the emergence and promoted the clearing of pathological protein aggregation like Aβ [9,17,18,23,24] and tau [24], while some effects were dose- and area-dependent [22]. Moreover, the drug contributed to immunosuppression through acting on PPARγ [17,18,23]. Hence, it reduced the number of reactive astrocytes and microglia in APP/PS1 mice, thereby acting anti-inflammatory while still promoting phagocytosis of Aβ-plaques [9]. Also, more recent studies showed a positive effect of pioglitazone treatment on behavior [25] as well as a reduction of inflammation as measured by attenuation of TSPO-PET signal in PS2APP and APPNL-G-F mice [26].
Of note, there are also some preclinical studies showing no effect or even detrimental effects of pioglitazone on the brain. Thus, despite improved cerebral blood flow, Aβ pathology kept progressing in different amyloid mouse models [27,28], and these studies did not find any change in memory and cognition [27,28]. Late treatment initiation in these studies needs to be considered, with treatment starting at 10 months of age [27] and 10-12 months of age [28].
Given the body of preclinical evidence, several clinical trials have investigated the effectiveness of pioglitazone in humans. Generally, diabetes patients indicated to have a lower risk and later onset of dementia when treated with pioglitazone [29,30]. However, pioglitazone treatment was unable to improve cognition or alter the age of onset of mild cognitive impairment in non-diabetic volunteers [31]. Moreover, one clinical trial showed no adverse, but also no beneficial treatment effects of pioglitazone in patients with probable Alzheimer’s Disease [32], and another recent phase III clinical trial with individuals with high-risk for Alzheimer’s Disease was terminated early due to inefficacy of pioglitazone treatment [33].
Since most preclinical studies focused on Aβ mouse models, we intended to investigate pioglitazone in presence of tau pathology related neuroinflammation and used the previously characterized P301S mouse model [34]. As expected, we were able to reproduce a significant time-dependent increase of TSPO-PET signals in P301S mice. However, we could not observe a significant impact of pioglitazone treatment on the rate of change in serial TSPO-PET results nor in the immunohistological assessment of tau-positive neurons and the microglia markers Iba1 and CD68. Thus, our results in a tau mouse model are in contrast to previous studies that found a decrease in microglial activation after pioglitazone treatment in APPV717I [23], A/T [28], PS2APP, and APPNL-G-F Aβ mouse models [26]. We conclude that the difference in underlying neuropathology might be the reason for effectiveness of PPARγ related modulation of microglial activation. We speculate that this could be one important reason for failing of pioglitazone in clinical trials of Alzheimer’s disease because Alzheimer’s disease comprises an Aβ-plaque mediated secondary tauopathy [35,36,37]. In case of both disease hallmarks present, pioglitazone might has modulate inflammation caused by amyloidosis, but based on our results seems to be an ineffective modulator of tau-induced inflammation. This hypothesis is further supported by our immunohistochemistry analysis of AT8-positive cells and Iba1 and CD68 expression levels, which did not show a difference between long-term treated and untreated P301S mice. We note that future studies could use multiplex panels or single cell RNA of isolated microglia for comparison of long-term pioglitazone treatment between Aβ and tau mouse models to elucidate the underlying mechanisms of ineffective treatment in P301S mice.
Concluding we find that long-term pioglitazone had no significant effect on microglial activation in P301S mice as measured by TSPO-PET and immunohistochemistry. Our results lead to the hypothesis that pioglitazone may has no beneficial impact on tau-mediated neuroinflammation, possibly explaining the failure of this treatment strategy in clinical trials of Alzheimer’s disease. Further studies regarding the mechanistic differences between PPARγ stimulation of Aβ- and tau-related microglial activation will provide novel insights into pathomechanisms of neurodegenerative diseases and possible new treatment strategies.
4. Materials and Methods
4.1. Animals and Study Design
All experiments were carried out in compliance with the National Guidelines for Animal Protection, Germany, and with the approval of the regional animal care committee (Regierung von Oberbayern) and were overseen by a veterinarian. The experiments complied with the ARRIVE guidelines and were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments. Animals were housed in a temperature- and humidity-controlled environment with a 12 h light-dark cycle, with free access to food and water.
TSPO-PET-Scans in P301S and wild-type mice were performed at four different time-points as indicated in Table 1. In the P301S mouse line, which is generated on a C57BL/6 background, the thy1 promotor controls the expression of mutated human tau. Tau deposits are exclusively found in neurons [38]. These mice show the first pathological signs of disease as early as three months of age followed by the formation of neurofibrillary tangles and gliosis of astrocytes and microglia [39,40]. C57BL/6 mice served as wild-type controls. All investigated mice were female.
Cage randomization concerning treatment (pioglitazone) and control (placebo) chow was initiated after the baseline PET-scans and treatment was continued until perfusion of the animals. For transcardial perfusion with PBS, mice were deeply anesthetized. Harvested brains were fixed in 4% paraformaldehyde (12h) and stored in PBS for immunohistochemical analyses.
4.2. PET imaging
Radiochemistry, TSPO-PET image acquisition, and image pre-processing were performed as described previously [41,42]. In brief, mice anesthetized with isoflurane were injected an average dose of 13.6 ± 2.0 MBq of [18F]GE-180. 60 minutes post-injection, TSPO-PET recordings were performed for 30 minutes, leading to an emission window of 60-90 minutes. P301S and wild-type mice were examined simultaneously in a four-mouse chamber imaging setting irrespective of their genotype and treatment in a randomized way. This procedure ensured an equal level of isoflurane anesthesia throughout the whole imaging procedure.
4.3. PET Image Analysis
All image analysis were performed using PMOD (version 3.5, PMOD Technologies, Zurich, Switzerland) as described earlier [42].
Different ways of intensity normalization were used to compare TSPO-PET findings with all commonly applied approaches. Therefore, we assessed the cerebral TSPO-expression after SUV-normalization, myocardial correction [43], intracerebral reference-based SUV ratios (SUVR), and injected dose-adjustment (%ID). As a reference region normalization we used the previously validated nucleus accumbens scaling for generation of standardized uptake value ratios (SUVR) [34]. Furthermore, myocardium adjusted standardized uptake values (SUV), SUV and %injected dose (%ID) were used to account for radiotracer dosing, body weight, and individual physiological differences between mice. Brainstem, cerebellum, frontal cortex and hippocampus served as target regions [34].
4.4. Immunohistochemistry
Immunohistochemistry was performed to assess the number of tau-positive cells in the brains of P301S mice. To this end, paraformaldehyde-fixed 50 µm thick sagittal brain sections were incubated for 48 hours in PBS with 1% BSA, 5% normal goat serum, and 0.3% Triton X-100 containing mouse monoclonal phosphor-tau primary antibody (Ser202, Thr205 (AT8), 1:1000, ThermoFisher MN1020). Afterwards, slices were incubated for 4h at room temperature with a suitable secondary antibody. Imaging was performed on a confocal microscope (LSM 780 Axio invers) with an x20 objective in three sagittal sections. Target areas were selected based on the PET results and consisted of the cortex and the brainstem. Images were processed with the ZEN 3.1 software and image analysis was performed with FiJi/ImageJ [44] by counting the number of tau-positive neurons in the target areas of each section.
To assess the degree of activation of microglia, paraformaldehyde-fixed 50 µm thick sagittal brain sections were incubated overnight at 4°C in PBS with 5% normal goat serum, and 0.5% Triton X-100 containing guinea pig monoclonal anti-Iba1 primary antibody (1:500, Synaptic Systems, 234308) and rat monoclonal anti-CD68 primary antibody (1:500, FA-11, biorad, MCA1957). Afterwards, slices were washed three times with PBS supplemented with 0.5% Triton X-100, and subsequently, slices were incubated for 2 h at room temperature with a suitable secondary antibody. Imaging was performed on a wide-field microscope (Zeiss Axio Vert A1 with ApoTome) with an x20 objective in three sagittal sections. Target areas were selected based on the PET results and consisted of the cortex and the brainstem. Images were processed with the ZEN 3.1 software and image analysis was performed with FiJi/ImageJ [44] by quantifying the area with signal over a certain threshold for Iba1 and CD68.
4.5. Statistics
Relevant group differences (i.e. between genotype or treatment) in longitudinal TSPO-PET data were performed with a mixed-effects model and Tukey’s multiple comparisons test using GraphPad Prism statistical software (version 9.4.1 for Windows, GraphPad Software, San Diego, California, USA). In the same software, an unpaired t-test was used to assess statistically significant differences in the immunohistological data. A threshold of p<0.05 was considered significant to reject the null hypothesis.
Author Contributions
Conceptualization, Peter Bartenstein, Sibylle Ziegler, Lars Paeger, Sabina Tahirovic, Jochen Herms and Matthias Brendel; Formal analysis, Lea Kunze, François Ruch, Gloria Biechele, Florian Eckenweber, Lina Dinkel and Paul Feyen; Funding acquisition, Peter Bartenstein, Sibylle Ziegler, Lars Paeger, Sabina Tahirovic, Jochen Herms and Matthias Brendel; Investigation, Lea Kunze, François Ruch, Gloria Biechele, Florian Eckenweber, Karin Wind-Mark and Lina Dinkel; Methodology, Sabina Tahirovic, Jochen Herms and Matthias Brendel; Project administration, Matthias Brendel; Resources, Sabina Tahirovic, Jochen Herms and Matthias Brendel; Supervision, Matthias Brendel; Visualization, Lea Kunze; Writing – original draft, Lea Kunze; Writing – review & editing, Lina Dinkel, Paul Feyen, Sibylle Ziegler, Sabina Tahirovic and Matthias Brendel. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198). S.T. was supported by the Alzheimer’s Association Grant through the AD Strategic Fund (ADSF-21-831226-C).
Institutional Review Board Statement
The animal study protocol was approved by the regional animal care committee (Regierung von Oberbayern) and all experiments were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments.
Data Availability Statement
All raw data can be obtained by the corresponding author upon reasonable request.
Acknowledgments
We would like to thank Karin Bormann-Giglmaier for her support with the TSPO-PET scans.
Conflicts of Interest
M.B. received speaker honoraria from Roche, GE healthcare and Life Molecular Imaging and is an advisor to Life Molecular Imaging. Other than that, the authors declare no conflict of interest.
Appendix A
Figure A1.
Individual time courses of SUV-scaled TSPO-PET signals of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up) in brainstem, cerebellum, cortex, and hippocampus.
Figure A1.
Individual time courses of SUV-scaled TSPO-PET signals of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up) in brainstem, cerebellum, cortex, and hippocampus.
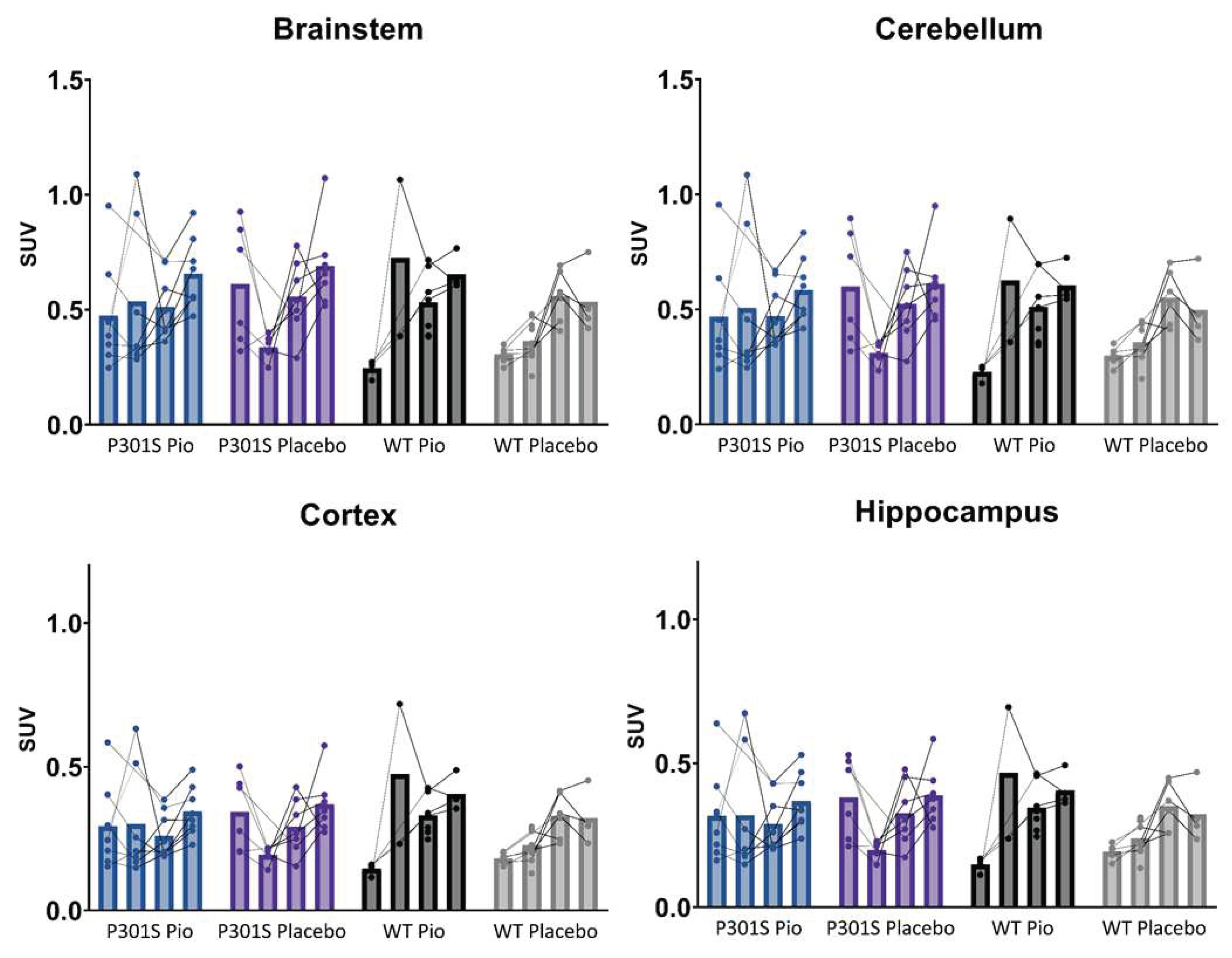
Figure A2.
Individual time courses of %ID-scaled TSPO-PET signals of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up) in brainstem, cerebellum, cortex, and hippocampus.
Figure A2.
Individual time courses of %ID-scaled TSPO-PET signals of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up) in brainstem, cerebellum, cortex, and hippocampus.
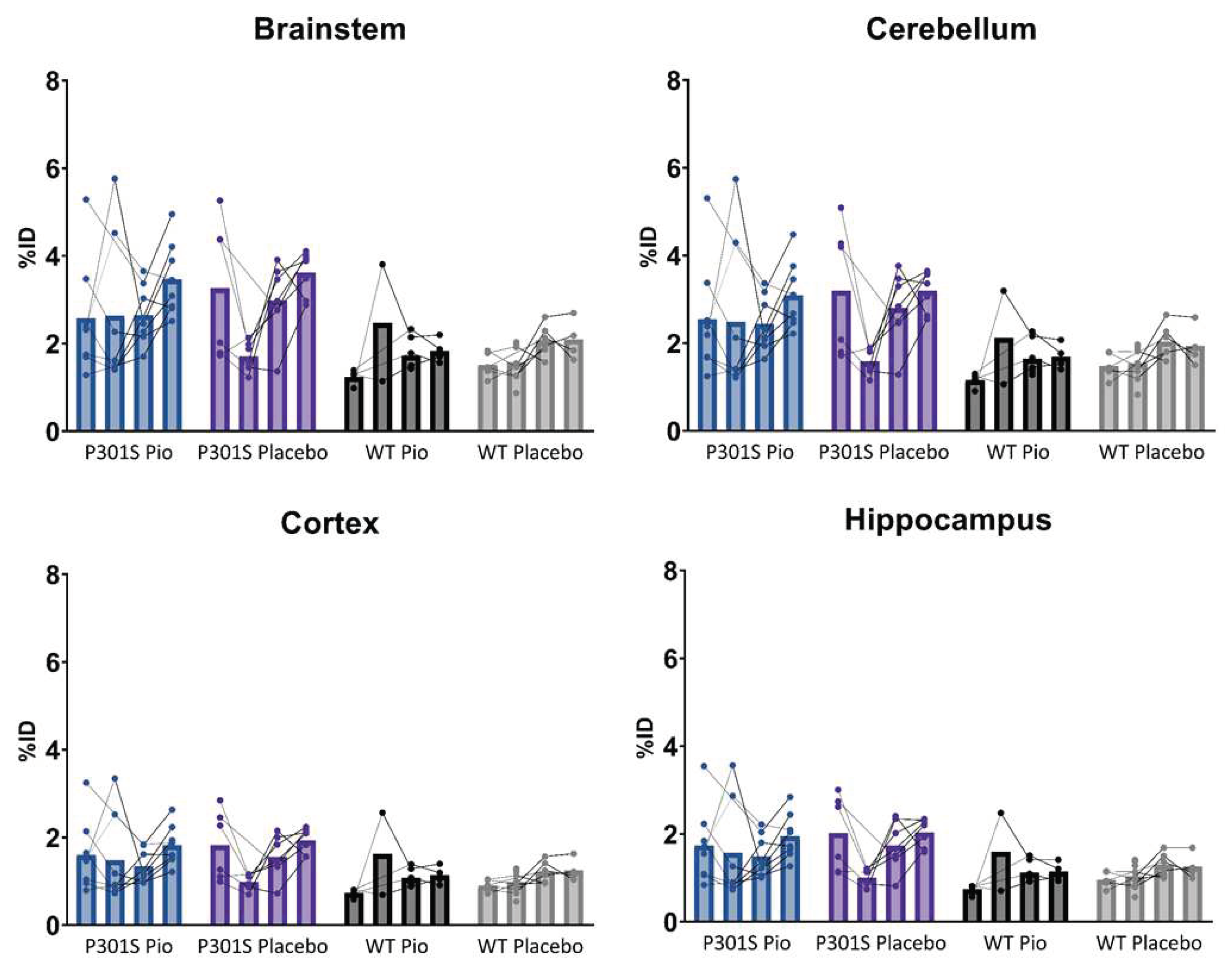
Figure A3.
Individual time courses of myocardium-scaled TSPO-PET signals of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up) in brainstem, cerebellum, cortex, and hippocampus.
Figure A3.
Individual time courses of myocardium-scaled TSPO-PET signals of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up) in brainstem, cerebellum, cortex, and hippocampus.
References
- Hickman, S. , Izzy, S., Sen, P., Morsett, L., & El Khoury, J. (2018). Microglia in neurodegeneration. ( 21(10), 1359–1369. [CrossRef] [PubMed]
- Wolf, S. A. , Boddeke, H. W. G. M., & Kettenmann, H. (2017). Microglia in Physiology and Disease. Annu. Rev. Physiol. [CrossRef]
- Fakhoury, M. (2018). Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. [CrossRef]
- Ginhoux, F. , Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., Mehler, M. F., Conway, S. J., Ng, L. G., Stanley, E. R., Samokhvalov, I. M., & Merad, M. (2010). Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science. [CrossRef]
- Bolós, M. , Llorens-Martín, M., Jurado-Arjona, J., Hernández, F., Rábano, A., & Avila, J. (2015). Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimer’s Dis. [CrossRef]
- Hickman, S. E. , Allison, E. K., & El Khoury, J. (2008). Microglial Dysfunction and Defective -Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 8360. [Google Scholar] [CrossRef]
- Sims, R. , van der Lee, S. J., Naj, A. C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., Kunkle, B. W., Boland, A., Raybould, R., Bis, J. C., Martin, E. R., Grenier-Boley, B., Heilmann-Heimbach, S., Chouraki, V., Kuzma, A. B., Sleegers, K., Vronskaya, M., … Schellenberg, G. D. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 1373. [Google Scholar] [CrossRef]
- Shi, Y. , Manis, M., Long, J., Wang, K., Sullivan, P. M., Remolina Serrano, J., Hoyle, R., & Holtzman, D. M. (2019). Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp.Med. 2546. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S. , Karlo, J. C., & Landreth, G. E. (2012). Mechanisms Underlying the Rapid Peroxisome Proliferator-Activated Receptor- -Mediated Amyloid Clearance and Reversal of Cognitive Deficits in a Murine Model of Alzheimer’s Disease. J. Neurosci. 0117. [Google Scholar] [CrossRef]
- Yamanaka, M. , Ishikawa, T., Griep, A., Axt, D., Kummer, M. P., & Heneka, M. T. (2012). PPARγ/RXRα-Induced and CD36-Mediated Microglial Amyloid-β Phagocytosis Results in Cognitive Improvement in Amyloid Precursor Protein/Presenilin 1 Mice. J. Neurosci. 7321. [Google Scholar] [CrossRef]
- Willson, T. M. , Lambert, M. H., & Kliewer, S. A. (2001). Peroxisome Proliferator–Activated Receptor γ and Metabolic Disease. Annu. Rev. Biochem. [CrossRef]
- Chen, Y.-C. , Wu, J.-S., Tsai, H.-D., Huang, C.-Y., Chen, J.-J., Sun, G. Y., & Lin, T.-N. (2012). Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ) and Neurodegenerative Disorders. Mol. Neurobiol. [CrossRef]
- Heneka, M. T. , Reyes-Irisarri, E., Hull, M., & Kummer, M. P. (2011). Impact and Therapeutic Potential of PPARs in Alzheimers Disease. Curr. Neuropharmacol. [CrossRef]
- Bouhlel, M. A. , Derudas, B., Rigamonti, E., Dièvart, R., Brozek, J., Haulon, S., Zawadzki, C., Jude, B., Torpier, G., Marx, N., Staels, B., & Chinetti-Gbaguidi, G. (2007). PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-inflammatory Properties. Cell Metab. [CrossRef]
- Barrera, J. , Subramanian, S., & Chiba-Falek, O. (2018). Probing the role of PPARγ in the regulation of late-onset Alzheimer’s disease-associated genes. PLoS One, 1969. [Google Scholar] [CrossRef]
- Yamasaki, Y. , Ryuzo, K., Wasada, T., Sato, A., Omori, Y., Eguchi, H., Tominaga, M., Sasaki, H., Ikeda, M., Kubota, M., Ishida, Y., Hozumi, T., Baba, S., Uehara, M., Shichiri, M., Kaneko, T., & AD-4833 Glucose Clamp Study Group Japan. (1997). Pioglitazone (AD-4833) Ameliorates Insulin Resistance in Patients with NIDDM. J. Exp. Med.
- Quan, Q. , Qian, Y., Li, X., & Li, M. (2019). Pioglitazone Reduces β Amyloid Levels via Inhibition of PPARγ Phosphorylation in a Neuronal Model of Alzheimer’s Disease. Front. Aging Neurosci. [CrossRef]
- Sastre, M. , Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E., Borghgraef, P., Evert, B. O., Dumitrescu-Ozimek, L., Thal, D. R., Landreth, G., Walter, J., Klockgether, T., van Leuven, F., & Heneka, M. T. (2006). Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. PNAS. [CrossRef]
- Chen, J. , Li, S., Sun, W., & Li, J. (2015). Anti-Diabetes Drug Pioglitazone Ameliorates Synaptic Defects in AD Transgenic Mice by Inhibiting Cyclin-Dependent Kinase5 Activity. PLoS One, 1238. [Google Scholar] [CrossRef]
- Fernandez-Martos, C. M. , Atkinson, R. A. K., Chuah, M. I., King, A. E., & Vickers, J. C. (2017). Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimer’s Dement.: Transl. Res. Clin. Interv. [CrossRef]
- Wong, L. R. , Wong, P., & Ho, P. C.-L. (2020). Metabolic Profiling of Female Tg2576 Mouse Brains Provides Novel Evidence Supporting Intranasal Low-Dose Pioglitazone for Long-Term Treatment at an Early Stage of Alzheimer’s Disease. Biomedicines. [CrossRef]
- Seok, H. , Lee, M., Shin, E., Yun, M. R., Lee, Y., Moon, J. H., Kim, E., Lee, P. H., Lee, B.-W., Kang, E. S., Lee, H. C., & Cha, B. S. (2019). Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci. Rep. [CrossRef]
- Heneka, M. T. , Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O’Banion, K., Klockgether, T., Van Leuven, F., & Landreth, G. E. (2005). Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain, 1453. [Google Scholar] [CrossRef]
- Searcy, J. L. , Phelps, J. T., Pancani, T., Kadish, I., Popovic, J., Anderson, K. L., Beckett, T. L., Murphy, M. P., Chen, K.-C., Blalock, E. M., Landfield, P. W., Porter, N. M., & Thibault, O. (2012). Long-Term Pioglitazone Treatment Improves Learning and Attenuates Pathological Markers in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. [CrossRef]
- Blume, T. , Deussing, M., Biechele, G., Peters, F., Zott, B., Schmidt, C., Franzmeier, N., Wind, K., Eckenweber, F., Sacher, C., Shi, Y., Ochs, K., Kleinberger, G., Xiang, X., Focke, C., Lindner, S., Gildehaus, F.-J., Beyer, L., von Ungern-Sternberg, B., … Brendel, M. (2022). Chronic PPARγ Stimulation Shifts Amyloidosis to Higher Fibrillarity but Improves Cognition. Front. Aging Neurosci. 5403. [Google Scholar] [CrossRef]
- Biechele, G. , Blume, T., Deussing, M., Zott, B., Shi, Y., Xiang, X., Franzmeier, N., Kleinberger, G., Peters, F., Ochs, K., Focke, C., Sacher, C., Wind, K., Schmidt, C., Lindner, S., Gildehaus, F.-J., Eckenweber, F., Beyer, L., von Ungern-Sternberg, B., … Brendel, M. (2021). Pre-therapeutic microglia activation and sex determine therapy effects of chronic immunomodulation. Theranostics, 8964. [Google Scholar] [CrossRef]
- Nicolakakis, N. , Aboulkassim, T., Ongali, B., Lecrux, C., Fernandes, P., Rosa-Neto, P., Tong, X.-K., & Hamel, E. (2008). Complete Rescue of Cerebrovascular Function in Aged Alzheimer’s Disease Transgenic Mice by Antioxidants and Pioglitazone, a Peroxisome Proliferator-Activated Receptor Agonist. J. Neurosci. 9296. [Google Scholar] [CrossRef]
- Papadopoulos, P. , Rosa-Neto, P., Rochford, J., & Hamel, E. (2013). Pioglitazone Improves Reversal Learning and Exerts Mixed Cerebrovascular Effects in a Mouse Model of Alzheimer’s Disease with Combined Amyloid-β and Cerebrovascular Pathology. PLoS One. [CrossRef]
- Heneka, M. T. , Fink, A., & Doblhammer, G. (2015). Effect of pioglitazone medication on the incidence of dementia: Pioglitazone in Dementia. Ann. Neurol. [CrossRef]
- Tseng, C.-H. (2018). Pioglitazone Reduces Dementia Risk in Patients with Type 2 Diabetes Mellitus: A Retrospective Cohort Analysis. J. Clin. Med. [CrossRef]
- Saunders, A. M. , Burns, D. K., & Gottschalk, W. K. (2021). Reassessment of Pioglitazone for Alzheimer’s Disease. Front. Neurosci. [CrossRef]
- Geldmacher, D. S. , Fritsch, T., McClendon, M. J., & Landreth, G. (2011). A Randomized Pilot Clinical Trial of the Safety of Pioglitazone in Treatment of Patients With Alzheimer Disease. Archives of Neurology. [CrossRef]
- Alexander, R. , Burns, D. K., Welsh-Bohmer, K. A., Burke, J. R., Chiang, C., Culp, M., Plassman, B. L., Wu, J., Lutz, M. W., Rubens, R., Evans, R., Saunders, A. M., & Ratti, E. (2019). DT-02-02: TOMMORROW: RESULTS FROM A PHASE 3 TRIAL TO DELAY THE ONSET OF MCI DUE TO AD AND QUALIFY A GENETIC BIOMARKER ALGORITHM. Alzheimer’s & Dementia. [CrossRef]
- Eckenweber, F. , Medina-Luque, J., Blume, T., Sacher, C., Biechele, G., Wind, K., Deussing, M., Briel, N., Lindner, S., Boening, G., von Ungern-Sternberg, B., Unterrainer, M., Albert, N. L., Zwergal, A., Levin, J., Bartenstein, P., Cumming, P., Rominger, A., Höglinger, G. U., … Brendel, M. (2020). Longitudinal TSPO expression in tau transgenic P301S mice predicts increased tau accumulation and deteriorated spatial learning. J. Neuroinflammation. [CrossRef]
- Lichtenthaler, S. F. (2017). Predicting, Preventing, and Treating Alzheimer’s Disease. In M. Gadebusch Bondio, F. Spöring, & J.-S. Gordon (Eds.), Medical ethics, prediction, and prognosis: Interdisciplinary perspectives (1 [edition], pp. 148–155). Routledge/Taylor & Francis Group.
- Förstl, H. , Bickel, H., & Perneczky, R. (2018). Alzheimer-Demenz und andere degenerative Demenzen. In P. Berlit (Ed.), Klinische Neurologie (pp. 1–17). Springer Berlin Heidelberg. [CrossRef]
- Wolfe, M. S. (2021). Probing Mechanisms and Therapeutic Potential of γ-Secretase in Alzheimer’s Disease. Molecules. [CrossRef]
- Allen, B. , Ingram, E., Takao, M., Smith, M. J., Jakes, R., Virdee, K., Yoshida, H., Holzer, M., Craxton, M., Emson, P. C., Atzori, C., Migheli, A., Crowther, R. A., Ghetti, B., Spillantini, M. G., & Goedert, M. (2002). Abundant Tau Filaments and Nonapoptotic Neurodegeneration in Transgenic Mice Expressing Human P301S Tau Protein. The Journal of Neuroscience, 22(21), 9340–9351.
- Hampton, D. W. , Webber, D. J., Bilican, B., Goedert, M., Spillantini, M. G., & Chandran, S. (2010). Cell-Mediated Neuroprotection in a Mouse Model of Human Tauopathy. J. Neurosci. 9983. [Google Scholar] [CrossRef]
- Bellucci, A. , Westwood, A. J., Ingram, E., Casamenti, F., Goedert, M., & Spillantini, M. G. (2004). Induction of Inflammatory Mediators and Microglial Activation in Mice Transgenic for Mutant Human P301S Tau Protein. G. ( 165(5), 1643–1652. [CrossRef]
- Brendel, M. , Probst, F., Jaworska, A., Overhoff, F., Korzhova, V., Albert, N. L., Beck, R., Lindner, S., Gildehaus, F.-J., Baumann, K., Bartenstein, P., Kleinberger, G., Haass, C., Herms, J., & Rominger, A. (2016). Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J. Nucl. Med. [CrossRef]
- Overhoff, F. , Brendel, M., Jaworska, A., Korzhova, V., Delker, A., Probst, F., Focke, C., Gildehaus, F.-J., Carlsen, J., Baumann, K., Haass, C., Bartenstein, P., Herms, J., & Rominger, A. (2016). Automated Spatial Brain Normalization and Hindbrain White Matter Reference Tissue Give Improved [18F]-Florbetaben PET Quantitation in Alzheimer’s Model Mice. Front. Neurosci. [CrossRef]
- Deussing, M. , Blume, T., Vomacka, L., Mahler, C., Focke, C., Todica, A., Unterrainer, M., Albert, N. L., Lindner, S., von Ungern-Sternberg, B., Baumann, K., Zwergal, A., Bartenstein, P., Herms, J., Rominger, A., & Brendel, M. (2018). Coupling between physiological TSPO expression in brain and myocardium allows stabilization of late-phase cerebral [18F]GE180 PET quantification. NeuroImage. [CrossRef]
- Schindelin, J. , Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J.-Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P., & Cardona, A. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods. [CrossRef]
Figure 1.
TSPO-PET signal of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up). (a) Individual time courses of TSPO-PET signals in brainstem, cerebellum, cortex, and hippocampus. P-values derive from a t-test comparing P301S mice with pioglitazone and placebo treatment independently of the time point. (b) Group level TSPO-PET images of pioglitazone or placebo-treated P301S and wild-type mice are shown as sagittal slices upon an MRI template. Data were normalized by average value in nucleus accumbens (SUVR). Extracerebral regions and the pituitary gland were masked.
Figure 1.
TSPO-PET signal of P301S mice treated with pioglitazone (P301S Pio) or placebo chow (P301S Placebo) and the respective wild-type (WT) control groups over time (BL = baseline, FU = follow-up). (a) Individual time courses of TSPO-PET signals in brainstem, cerebellum, cortex, and hippocampus. P-values derive from a t-test comparing P301S mice with pioglitazone and placebo treatment independently of the time point. (b) Group level TSPO-PET images of pioglitazone or placebo-treated P301S and wild-type mice are shown as sagittal slices upon an MRI template. Data were normalized by average value in nucleus accumbens (SUVR). Extracerebral regions and the pituitary gland were masked.
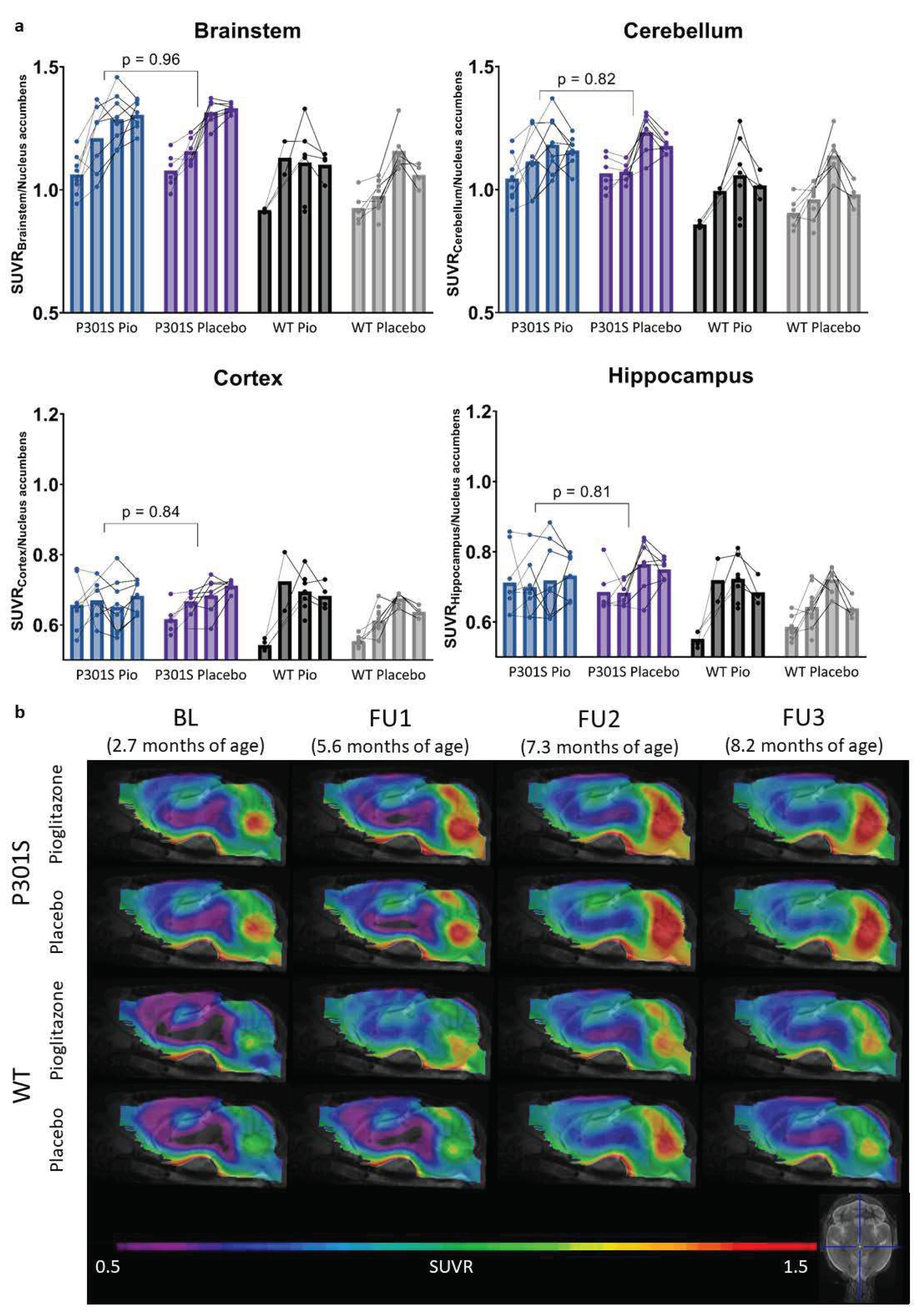
Figure 2.
(a) Abundance of tau-positive cells in 0.014 mm2 of the cortex and 0.011 mm2 of the brainstem of P301S mice treated with pioglitazone (P301S Pio) or placebo (P301S Placebo) as counted from triplicates of immunohistological stainings with AT8. (b) Representative orthogonal projections of AT8 immunohistochemistry for the cortex (left panel) and the brainstem (right panel) of two P301S mice treated with pioglitazone and placebo, respectively.
Figure 2.
(a) Abundance of tau-positive cells in 0.014 mm2 of the cortex and 0.011 mm2 of the brainstem of P301S mice treated with pioglitazone (P301S Pio) or placebo (P301S Placebo) as counted from triplicates of immunohistological stainings with AT8. (b) Representative orthogonal projections of AT8 immunohistochemistry for the cortex (left panel) and the brainstem (right panel) of two P301S mice treated with pioglitazone and placebo, respectively.
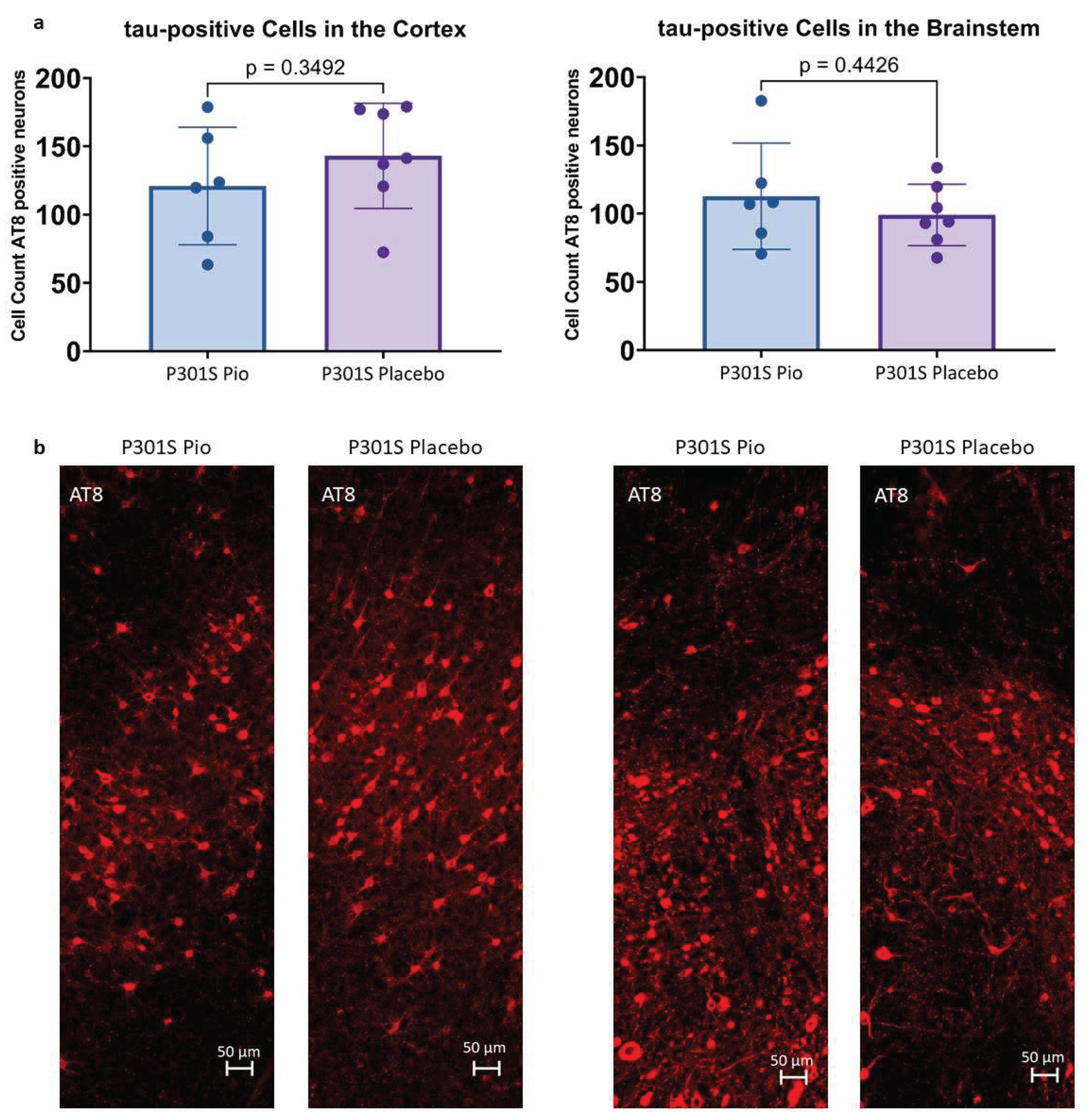
Figure 3.
(a,b) Microglial activation (area%) in the cortex and the brainstem of P301S mice treated with pioglitazone (P301S Pio) or placebo (P301S Placebo) as quantified by analyzing triplicates of immunohistochemical stainings with Iba1 (a) and CD68 (b). (c,d) Representative orthogonal projections of immunohistochemical stainings of P301S mice treated with pioglitazone (P301S Pio) or placebo (P301S Placebo) in the cortex (c) and the brainstem (d).
Figure 3.
(a,b) Microglial activation (area%) in the cortex and the brainstem of P301S mice treated with pioglitazone (P301S Pio) or placebo (P301S Placebo) as quantified by analyzing triplicates of immunohistochemical stainings with Iba1 (a) and CD68 (b). (c,d) Representative orthogonal projections of immunohistochemical stainings of P301S mice treated with pioglitazone (P301S Pio) or placebo (P301S Placebo) in the cortex (c) and the brainstem (d).
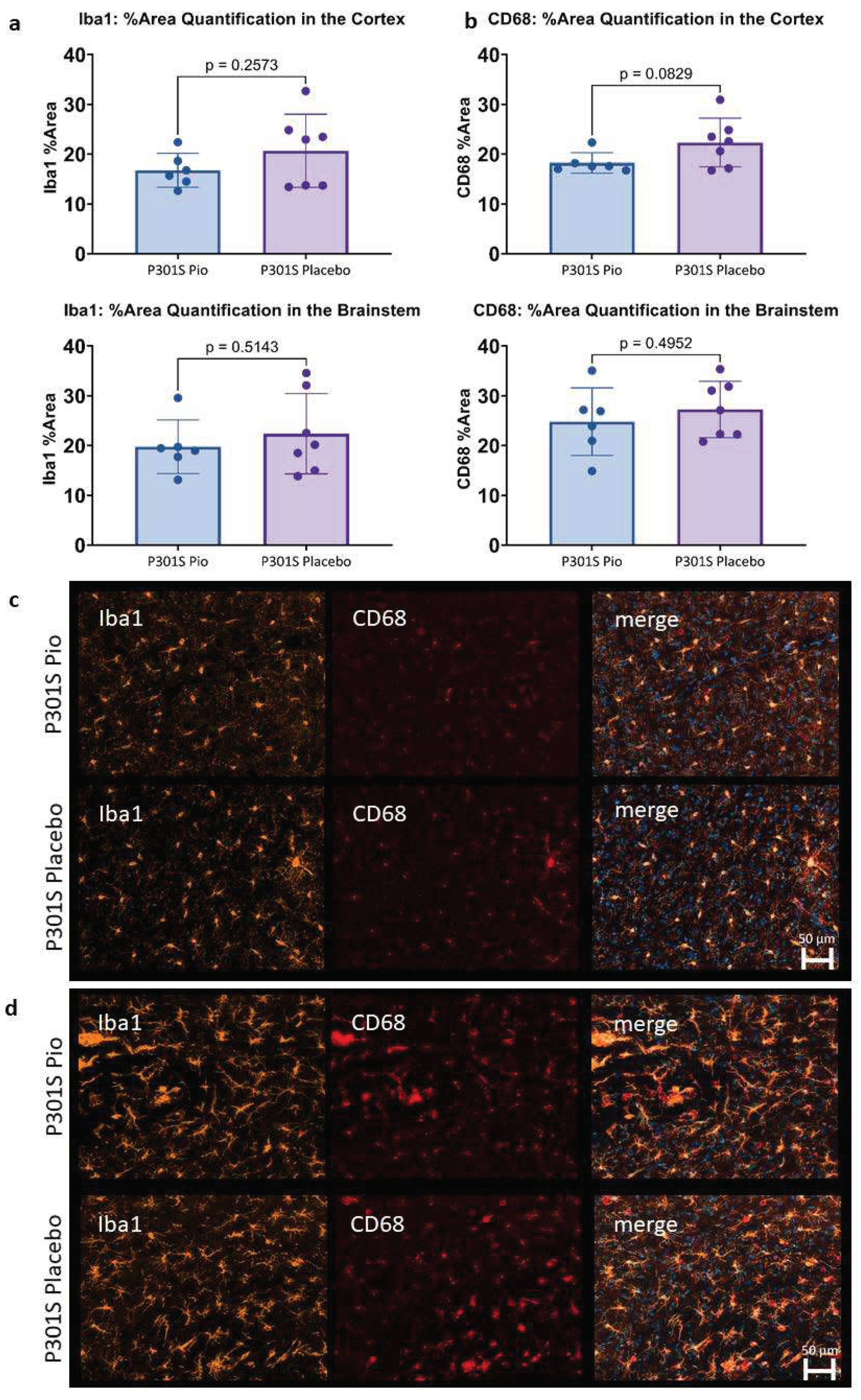

Table 1.
Age in months (Age) and number (N) of the mice with successful TSPO-PET for each group (P301S mice with pioglitazone treatment (P301S + Pio), P301S mice with placebo treatment (P301S + Placebo), wild-type mice with pioglitazone treatment (WT + Pio), and wild-type mice with placebo treatment (WT + Placebo)), as well as number of mice available for immunohistochemistry (IHC). f = female.
Table 1.
Age in months (Age) and number (N) of the mice with successful TSPO-PET for each group (P301S mice with pioglitazone treatment (P301S + Pio), P301S mice with placebo treatment (P301S + Placebo), wild-type mice with pioglitazone treatment (WT + Pio), and wild-type mice with placebo treatment (WT + Placebo)), as well as number of mice available for immunohistochemistry (IHC). f = female.
| BL | FU1 | FU2 | FU3 | IHC | |||||
|---|---|---|---|---|---|---|---|---|---|
| Age | N | Age | N | Age | N | Age | N | N | |
| P301S + Pio | 2.6 | 8 (f) | 5.9 | 7 (f) | 7.3 | 8 (f) | 8.2 | 8 (f) | 6 (f) |
| P301S + Placebo | 2.6 | 6 (f) | 6.0 | 6 (f) | 7.3 | 7 (f) | 8.2 | 7 (f) | 7(f) |
| WT + Pio | 2.8 | 4 (f) | 5.9 | 2 (f) | 7.7 | 6 (f) | 8.2 | 4 (f) | 0 |
| WT + Placebo | 2.9 | 6 (f) | 5.0 | 10 (f) | 7.4 | 6 (f) | 8.4 | 4 (f) | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



