
Submitted:
28 April 2023
Posted:
28 April 2023
You are already at the latest version
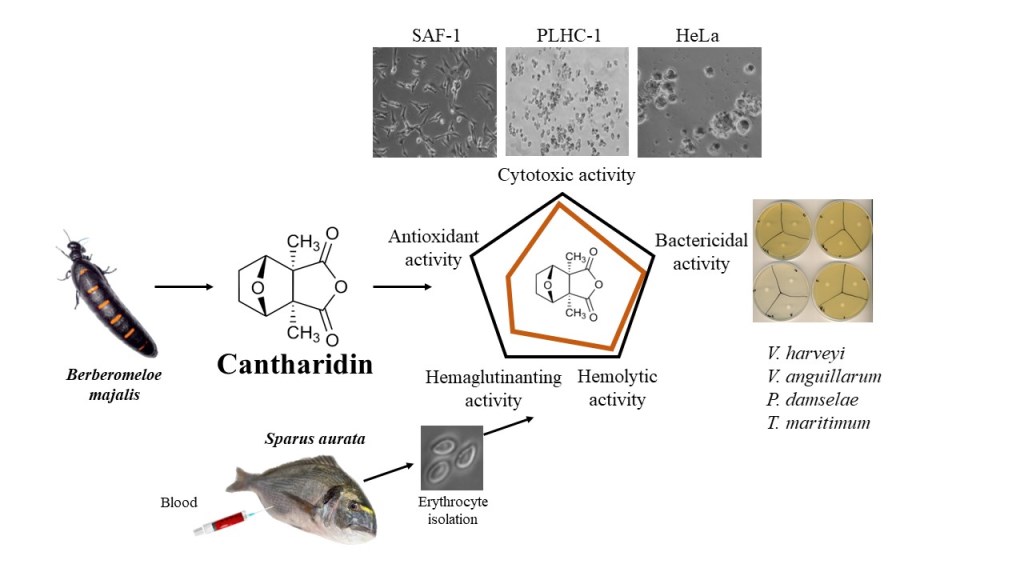
Abstract
Cantharidin, a toxic monoterpene secreted by blister beetles, has long been used in traditional Chinese and modern medicine for its unique properties. However, despite its widespread use, its effects on fish have not been studied in depth. The aim of this study was to evaluate the potential therapeutic applications of cantharidin in fish by examining its antioxidant, hemoagglutinating, hemolytic and cytotoxic activities at different concentrations (0, 0.625, 1.25, 2.5, 5 and 10 μg mL-1) in three different cell lines. In addition, the study explored the bactericidal and bacteriostatic properties of cantharidin against various fish pathogenic bacteria. The results revealed that there were no significant differences in antioxidant, hemagglutinating or hemolytic activities between the different concentrations of cantharidin tested. However, the study found that cantharidin exhibited dose- and time-dependent cytotoxicity in sea bream (Sparus aurata) erythrocytes and in SAF-1, PLHC-1 and Hela cell lines, resulting in morphological changes indicative of apoptosis. Interestingly, the highest dose of cantharidin tested demonstrated potent bactericidal activity against four marine fish opportunistic bacteria, including Vibrio harveyi, V. anguillarum, Photobacterium damselae and Tenacibaculum maritimum, but no statistically significant changes in bacteriostatic activity were observed against any of the bacteria tested. Overall, these results provide valuable information on the potential therapeutic applications of cantharidin in fish aquaculture. Further research is needed to fully understand the mechanisms of action and to explore possible preventive uses of cantharidin in fish.
Keywords:
Cantharidin
; Erythrocytes
; Fish cell lines
; Marine pathogen bacteria
; Gilthead seabream (Sparus aurata)
; Aquaculture.
1. Introduction
Aquaculture is currently one of the fastest growing sectors in the world due to the high demand for seafood products globally, which according to FAO exceeded 82.1 million tons of fish in 2018 alone [1]. The intensive production conditions that farmed animals are subjected to in this industry are sometimes correlated with the weakening of their immune system, as they compromise the welfare of the fish, making them more susceptible to outbreaks of infections and/or related diseases that could be lethal to them [2,3,4,5]. To maintain the health of farmed fish, for many years antibiotics were used in feed (as a preventive measure) to avoid fatal economic repercussions for the industry [6]. However, this practice was banned in the European Union in 2006 due to increasing antibiotic resistance [6]. The search for natural substances that contribute to overcome these problems and can replace antibiotics is one of the hot topics for aquaculture [7,8,9,10].
In this context, cantharidin (from the Greek kantharis), a toxin of terpenoid nature produced by the vesicant beetle of the families Meloidae and Oedemeridae, has been traditionally used in Chinese medicine for its mythical aphrodisiac properties and its topical use as a vesicant [11,12]. In addition, current studies have demonstrated in humans the antitumor, antibiotic, and immunomodulatory properties of cantharidin [13]. In vivo and in vitro assays have been performed in mammals to test the effect of cantharidin [14,15,16]. It appears to exert its function by triggering the release of serine proteases that cleave peptide bonds in proteins and ultimately disrupting cell-cell adhesion and inhibiting serine/threonine phosphatases, such as protein phosphatase 1 (PP1) and 2A (PP2A) [14,15,16]. However, due to the high inhibitory affinity of cantharidin for these proteins and the large number of cellular events controlled by phosphatases, cantharidin overdose by ingestion is associated with the production of skin rashes and redness, irritation of the bladder and dermis, vomiting and diarrhea, nephritis, hematuria, proteinuria, edema, organ failure, and even death, in humans [13]. Therefore, it is essential to use a precise dose of cantharidin to avoid its toxic effects [17,18]. In vivo human trials with cantharidin have not only been mainly based on testing its chemotherapeutic effects, but its role as a potent antitumor agent has also been evidenced, as it is able to inhibit cell proliferation in numerous cancer cell lines, such as leukemic and melanoma cells or in bladder, colorectal, pancreatic, hepatic, lung, and breast cancers, and induce cell apoptosis [19,20,21,22,23,24,25,26,27,28]. Interestingly, because of these toxic effects and its bitter taste, most animals avoid ingesting cantharidin-containing beetles. However, bustards (especially males) are believed to consume them as self-medication to reduce parasites, which deteriorates the appearance of the birds' cloaca and, surprisingly, increases their chances of reproduction [29].
Thus, based on these data, the aim of the present study is to evaluate the potential antioxidant, hemagglutinating, hemolytic and cytotoxic properties of cantharidin in three fish cell lines, as well as the bactericidal and bacteriostatic activities against various fish pathogens. To our knowledge, this is the first study that attempts to elucidate the in vitro antitumor and microbicidal effects of cantharidin on fish cells and pathogenic bacteria. These results help to understand the mechanism of action of cantharidin, which could be applied to fish of commercial interest.
2. Materials and Methods
2.1. Cantharidin
Cantharidin (Sigma) was diluted into dimethyl sulfoxide (DMSO, Sigma) at concentration ranging from 0 to 10 µg mL-1. The cantharidin was resuspended in the appropriate medium depending on the technique used. Thus, cantharidin was resuspended and adjusted to each concentration in phosphate-buffered saline (PBS; 11.9 mM Phosphates, 137 mM NaCl, and 2.7 mM KCl, pH 7.4, Fisher Bioreagents) for antioxidant activity, PBS with sodium chloride and glucose for incubation with erythrocytes, Eagle's Minimum Essential Medium (EMEM, Thermo Fisher) for HeLa and PLHC-1 cell lines, L-15 Leibowitz medium (Gibco) for SAF-1 cell line, Triptic Soy Broth (TSB, Difco Laboratories) for V. harveyi, V. anguillarum and P. damselae subsp. piscicida strain PP3, or Flexibacter maritimus medium (FMM, Conda) for T. maritimum. Erythrocytes, fish cell lines and bacteria were exposed to a final solution of 0 (DMSO diluted in the same volume of culture medium; as control), 0.625, 1.25, 2.5, 5 and 10 μg mL-1 cantharidin. Before performing the assays, the osmolarity of all solutions was measured in an osmometer (Wescor) to avoid effects due to this parameter.
2.2. Total antioxidant activity
The antioxidant capacity of cantharidin was evaluated by the 2,2′-azino-bis-3-ethylbenzothiazoline-6-sulphonic acid (ABTS) method [38]. This method measures the ability of antioxidants in the sample to reduce the radical cation of ABTS, which is determined by the decrease in absorbance of ABTS+, with quenching of absorbance at 730 nm. Activity was calculated by comparing the sample values to an ascorbic acid standard curve and expressed as ascorbic acid equivalents (mM). To perform the assay, 50 μL of each cantharidin concentration, previously diluted and adjusted with PBS, was mixed with 950 μL of ABTS+ solution, and the decrease in absorbance was measured using a spectrophotometer (BOECO S-22 UV/Vis, Germany) with PBS as a blank. Samples were analysed in triplicate.
2.3. Animals
Forty specimens of the seawater teleost gilthead seabream (Sparus aurata L.; mean weight: 403.6 ± 16.5 g; mean length 27.3 ± 0.3 cm, obtained from a local farm (Murcia, Spain), were kept in seawater recirculating aquaria (450 L) at the Marine Fish Facilities at the University of Murcia (Spain) during a quarantine period of one month. The conditions were as follows: water temperature of 20 ± 2º C, flow rate of 900 L h-1, salinity of 28 ‰, photoperiod of 12 h light to 12 h dark and continuous aeration. Fish were fed a commercial diet (Skretting, Spain) at a rate of 2% body weight day−1 and were maintained 24 h without feeding prior to the trial. All experimental protocols were approved by the Ethical Committee of the University of Murcia.
2.4. Cantharidin hemagglutinating and haemolytic activities
For erythrocyte isolation, six fish were randomly selected and anesthetized with clove oil (20 mg L-1, Guinama®). Immediately, 1 mL blood samples were drawn in a heparinized syringe from the caudal vein from each fish. All fish survived, recovered from anaesthesia and were returned to the aquaria. Blood samples were placed in 7 mL of PBS, containing 0.35 % sodium chloride (to adjust the osmolarity of the medium to the osmolarity of gilthead seabream serum), and 10 mM glucose (hereafter referred to as PBS-glu) [39]. Blood samples were layered on a 51 % Percoll density gradient (Pharmacia) and centrifuged (400 x g, 30 min, 4 °C) to separate leucocytes from erythrocytes. After centrifugation, erythrocytes, which were in the pellets, were collected, washed twice with PBS-glu, counted and adjusted to 5 x 108 cells mL-1.
The hemagglutination properties of cantharidin were studied by incubating the isolated erythrocytes in a 96-well U-shaped plate (Nunc). Briefly, 50 µL aliquots of varying concentrations of cantharidin diluted in PBS-glu were placed in sextuplicate and 25 µL of the erythrocyte suspension was added to each well. Erythrocyte hemagglutination was visualized on the plate after 1.5 h of incubation at room temperature. After that time, the erythrocytes in the wells considered blank (without cantharidin) had completely sedimented [40]. Positive or negative controls consisted of erythrocyte suspension (25 µL) and 50 µL Concanavalin A (a lectin that strongly binds erythrocytes) or PBS-glu, respectively. Macroscopic images of the microplates were taken from above and below with a Canon 7D camera with a 22 mm 4.5 wide-angle lens (Canon EF) attached to a ring flash with tripod and with a scanner (CanoScan-4400F), respectively.
Erythrocyte cell death causes the release of their contents, which is hemoglobin, either in the form of oxyhemoglobin (HbO2) or deoxyhemoglobin, the most abundant protein. Thus, erythrocyte viability was determined by hemolysis and subsequent release of oxyhemoglobin to the medium [39]. After 3, 6, 12 and 24 h of exposure of erythrocytes to varying concentrations of cantharidin in PBS-glu, samples were centrifuged (10,000 x g, 1 min) and 100 µL of the supernatant was transferred to 96-well flat-bottom plates (Nunc) and the absorbance at 414 nm (the maximum absorbance for oxyhemoglobin) was measured on a plate reader (BMG, Fluoro Star Galaxy). Positive (maximum hemolysis and absorbance) and negative (minimum hemolysis and absorbance) controls consisted of 100 µL of erythrocytes in 1 mL of sterile distilled water or PBS-glu, respectively.
To study cell morphology, 500 µL aliquots containing 200,000 cells well-1 were seeded in 24-well plates with 500 µL well-1 of 0, 5, and 10 μg mL-1 cantharidin in duplicate, and the cells were incubated for 12 and 24 h at 25°C. Cells were observed and photographed using a phase contrast microscope (Zeiss).
2.5. Cytotoxic activity of cantharidin
The established SAF-1 cell line (ECACC no. 00122301), obtained from gilthead seabream fin, was seeded in 25 cm2 plastic tissue culture flasks (Nunc) in Leibowitz L-15 medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Life Technologies), 2 mM L-glutamine, 100 i.u. mL-1 penicillin and 100 mg mL-1 streptomycin (Life Technologies). Cells were grown at 25°C in a humidified atmosphere (85% humidity). Exponentially growing cells were detached from the culture flasks by brief exposure to trypsin (0.25% in PBS, pH 7.2-7.4) according to standard trypsinization methods. The detached cells were collected by centrifugation (200 x g, 5 min, 25°C) and cell viability was determined by trypan blue exclusion assay.
The established cell line PLHC-1 (ATCC® CRL2406™), a Poeciliopsis lucida hepatocellular carcinoma, was seeded in 25 cm2 plastic tissue culture flasks in EMEM supplemented with 5% FBS, 2 mM mL-1 glutamine, 100 i.u. mL-1, penicillin/streptomycin, 0.1 mM non-essential amino acids and 1.0 mM sodium pyruvate, 95% (Life Technologies). Cells were grown at 30°C in a humidified atmosphere (85% humidity) and 5% CO2. Exponentially growing cells were detached from the culture flasks by brief exposure to trypsin (0.05% in PBS, pH 7.2-7.4) according to standard trypsinization methods. Detached cells were collected by centrifugation (200 x g, 5 min, 30°C) and cell viability was determined as previously described.
The established HeLa cell line (ECACC no. 93021013), the oldest human cell line derived from cervical cancer cells, was seeded in 25 cm2 plastic tissue culture flasks (Nunc) in EMEM (Thermo Fisher) supplemented with 10 % FBS (Life Technologies), 2 mM mL-1 glutamine, 100 i.u. mL-1 penicillin/streptomycin and 0.1 mM non-essential amino acids (Life Technologies). Cells were grown at 37°C in a humidified atmosphere (85% humidity) and 5% CO2. Exponentially growing cells were detached from the culture flasks by brief exposure to trypsin (0.05 % in PBS, pH 7.2-7.4) according to standard trypsinization methods. The detached cells were collected by centrifugation (200 x g, 5 min, 37°C) and cell viability was determined as previously described.
Cells from the three cell lines were detached from the culture flasks with trypsin (as described above) at approximately 80% confluence, and 100 µL aliquots were dispensed into 96-well tissue culture plates (containing 30,000 cells well-1) and incubated (24 h, at the temperature corresponding to each cell line). The cell concentration was determined beforehand to obtain satisfactory absorbance values in the cytotoxic assay and to avoid cell overgrowth. The culture medium was then replaced with 200 µL well-1 of the cantharidin concentrations to be tested. Cells were then incubated for 3, 6, 12 and 24 h and their viability was determined by the MTT assay, which is based on the reduction of the soluble yellow tetrazolium salt (3-(4,5-dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium bromide) (Sigma) to an insoluble blue formazan product by mitochondrial succinate dehydrogenase [27,28]. For this purpose, the medium was removed and 200 µL well-1 of MTT (1 mg mL-1 in culture medium for each cell line) was added. After 4 h of incubation at the corresponding temperature for each cell line tested, the formazan crystals were solubilized with 100 µL well-1 of DMSO. Plates were shaken (5 min, 100 rpm) under dark conditions and absorbance was determined at 570 nm and 690 nm in a microplate reader (BMG Fluostar Omega, USA). A cytotoxicity assay of each cell line was performed in three replicates for each cantharidin concentration.
To study cell morphology, the cell lines were observed and photographed directly from the culture plates using a phase contrast microscope (Zeiss). For this purpose, SAF-1, PLHC-1 and HeLa cells were cultured as described above to 80% confluence and detached from the culture flasks with trypsin. Aliquots of 100 µL were seeded in 24-well plates (containing 200,000 cells well-1) for 24 h at the appropriate temperature for each cell line. The culture medium was then replaced with 500 µL well-1 of cantharidin (0, 5, and 10 μg mL-1) in duplicate, and the cells were incubated again for 12 and 24 h. The doses and experimental time were selected taking into account the previous cytotoxic assay performed in the present study. The cells were observed and photographed using a phase contrast microscope.
2.6. Cantharidin bactericidal and bacteriostatic activity
Four opportunist marine pathogenic bacteria (V. harveyi, V. anguillarum, P. damselae and T. maritimum) were used in the bactericidal and bacteriostatic assays. All bacterial strains were grown from 1 mL of stock culture that had been previously frozen at −80 °C. Before use, aliquots of stock cultures from V. harveyi, V. anguillarum and P. damselae were incubated with TSB (Difco Laboratories) medium supplemented with 1.5% NaCl, whilst an aliquot of stock culture from T. maritimum was incubated with FMM (Conda) in flasks which 90 % empty. The samples were incubated overnight at 25ºC with continuous shaking (100 rpm). Exponentially growing bacteria were resuspended in the sterile corresponding culture medium and adjusted to 108 colony-forming units (c.f.u.) mL-1, according to the McFarland standard curve.
To determine bactericidal activity, 20 µL samples of cantharidin dilutions, previously adjusted in PBS, were added (in triplicate) to the wells of a 96-well U-shaped plate (Nunc). Aliquots of 20 µL of the previously cultured bacteria were added and the plates were incubated for 5 h at 25°C. In some wells PBS solution was added instead of cantharidin and served as a positive control, while in other wells only culture medium was added to ensure sterility of the tests [41]. The control samples were incubated under the same experimental conditions described above. Next, 25 µL of MTT (1 mg mL-1) was added to each well and the plates were incubated again (10 min, 25°C) to allow formazan formation. The plates were centrifuged (2,000 x g, 10 min) and the precipitates were dissolved in 200 µL of DMSO, of which 100 µL were transferred to a 96-well flat-bottom plate (Nunc). The absorbance of dissolved formazan was measured in a spectrophotometer (BMG, SpectroStarnano) at 570 nm and 690 nm. Bactericidal activity was expressed as the percentage of non-viable bacteria, calculated as the difference between the absorbance of surviving bacteria and the absorbance of bacteria from positive controls (100%).
To determine the bacteriostatic activity of cantharidin, 100 µL samples of cantharidin dilutions, previously adjusted in PBS, were added (in triplicate) to the wells of a 96-well flat-bottom plate (Nunc). Aliquots of 100 µL of the previously cultured bacteria were added and the plates were incubated at 25 °C. The turbidity of the samples was measured in a spectrophotometer (BMG, SpectroStarnano) at 620 nm every 120 min for 24 h [42]. In some wells, PBS solution was added instead of cantharidin and served as a positive control to evaluate the growth of untreated bacteria, while in other wells only culture medium was added to ensure sterility of the tests. Control samples were incubated under the same experimental conditions as described above.
2.7. Statistical analysis
The results were expressed as mean ± standard error of the mean (SEM). Data were analysed by One-way ANOVA (followed by Tukey tests). Normality of the data was previously assessed using a Shapiro-Wilk test and homogeneity of variance was also verified using the Levene test. All statistical analyses were conducted using the computer package SPSS (25.0 version; SPSS Inc., Chicago, IL, USA) for Windows. The level of significance used was p < 0.05 for all statistical tests.
3. Results
3.1. Total antioxidant activity
Cantharidin dilutions had no antioxidant activity (data no shown).
3.2. Hemagglutinating and haemolytic activity of cantharidin
Cantharidin did not significantly affect either hemagglutination or hemolytic activities of erythrocytes at any of the concentrations tested compared with negative controls (Figure S1 and S2). In sharp contrast, however, obvious changes in erythrocyte cell morphology were found in a time- and dose-dependent manner after incubation with 5 and 10 μg mL-1 cantharidin at 12 and 24 h. More specifically, gilthead seabream erythrocytes decreased in size and shrank after incubation with cantharidin compared with unexposed erythrocytes (Figure 1).
3.3. Cytotoxic activity of cantharidin
The cytotoxic activity of cantharidin on the SAF-1 cell line was observed after 12 or 24 h of exposure. After 12 h of incubation, a dose response on cytotoxicity was observed in this cell line, although the increase was only statistically significant (p< 0.05) when the highest concentration of cantharidin (10 μg mL-1) was used, compared to the results observed in cells incubated with the control solution (Figure 2). However, all doses of cantharidin elicited similar cytotoxic activity in SAF-1 (p< 0.05) after 24 h of incubation. The morphology of SAF-1 cells showed obvious changes after incubation with cantharidin. The cells detached from the culture flask and acquired a rounded shape, as compared to the characteristic elongated spindle shape of these cells with very large fine processes when attached to the culture cell surface (Figure 3).
Furthermore, although no cytotoxic activity was detected in the PLHC-1 cell line by cantharidin at 3 h of incubation (Figure 4A), dose- and time-dependent increases in cytotoxic activity were observed in these cells after incubation with cantharidin. Thus, cytotoxic activity increased significantly (p< 0.05) at 6 h only after incubation with the highest dose of cantharidin (10 μg mL-1) (Figure 4B). However, after 12 h of incubation, cytotoxic activity increased significantly (p< 0.05) with the two highest doses tested (5 and 10 μg mL-1) (Figure 4C). Finally, after 24 h of incubation with cantharidin, the cytotoxic effects on PLHC-1 cells were increased with the three highest doses tested (2.5, 5 and 10 μg mL-1) (Figure 4D), compared to cells incubated with the other tested concentrations of cantharidin at each corresponding time. Concomitantly, the morphology of these cells showed severe changes after incubation with cantharidin in a time- and dose-dependent manner. The control cells studied by phase contrast microscopy appeared as bright adherent and heterogeneous cells with a characteristic epithelial-like morphology but, after incubation, the cells detached and appeared as dark (dull) cells with a small, rounded shape and a large number of cell debris was evident in the culture medium (Figure 5).
In the case of HeLa cells, no cytotoxic activity was observed after incubation with cantharidin for 3 or 6 h. However, significantly increased cytotoxic activity (p< 0.05) was observed in the cells incubated 12 h with 10 μg mL-1 cantharidin and 24 h with the two highest concentrations (5 and 10 μg mL-1), compared to unexposed cells (Figure 6). As for cell morphology, HeLa cells changed their heterogeneous large oval morphology to small round detached cells. Furthermore, many cell debris were again observed in the culture medium of cell cultures incubated with cantharidin (Figure 7).
3.4. Bactericidal and bacteriostatic activity
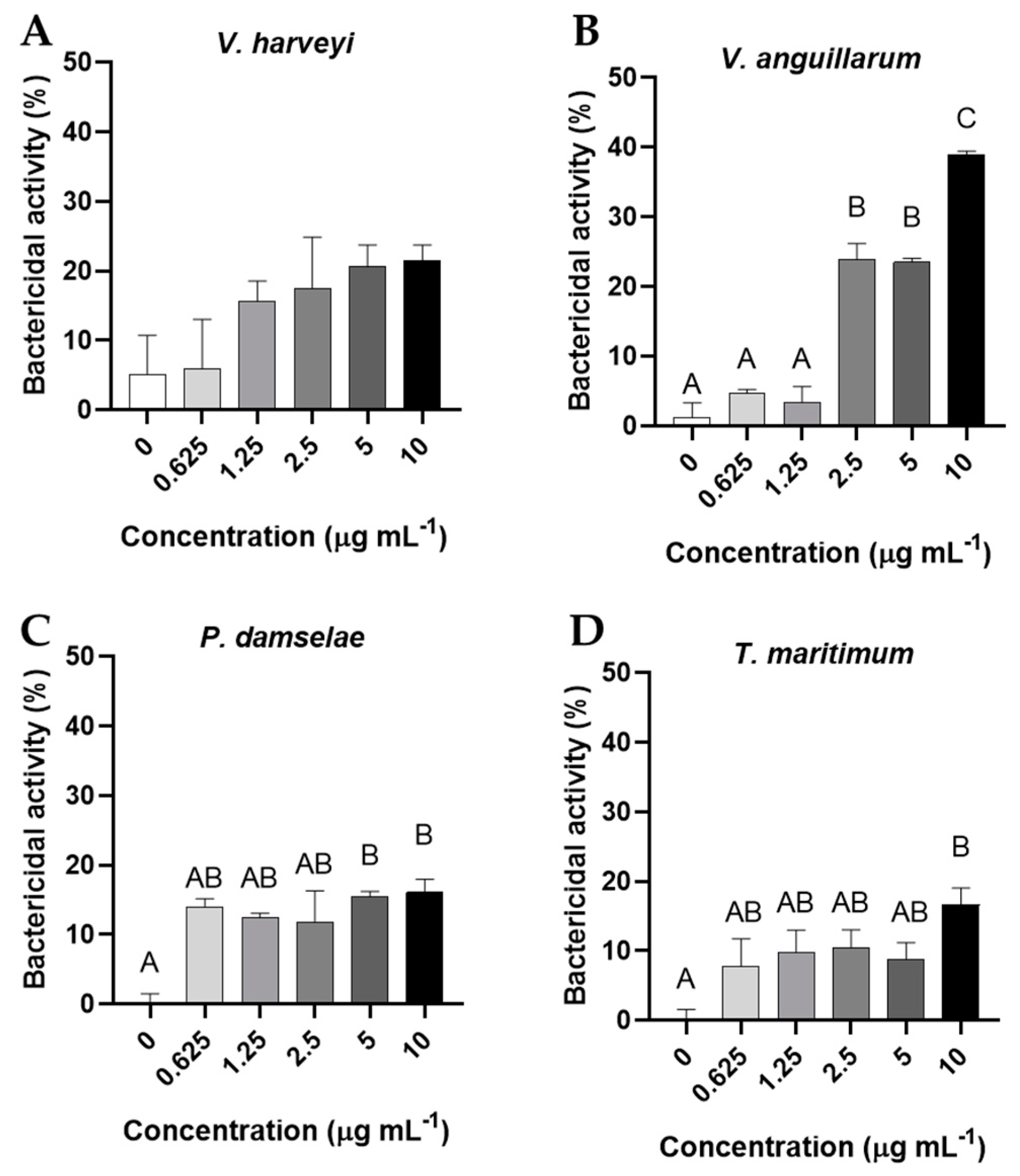
Regarding bactericidal activities, any dose of cantharidin significantly affected the viability of V. harveyi (Figure 8A). However, the three highest concentrations tested (2.5, 5 and 10 μg mL-1) showed bactericidal activity against V. anguillarum compared to all other concentrations (p< 0.05) (Figure 8B). In the case of P. damselae, the two highest concentrations of cantharidin (5 and 10 μg mL-1) showed bactericidal activity with respect to the control dose (Figure 8C). Similarly, only the highest concentration of cantharidin (10 μg mL-1) had bactericidal activity against T. maritimum (p < 0.05), compared to the values obtained for bacteria incubated without cantharidin (Figure 8D). Finally, no significant variations were observed in the bacteriostatic activity of the bacteria assayed after incubation with any dose of cantharidin (data not shown).
4. Discussion
Cantharidin (C10H12O4) is a defensive metabolite stored in the hemolymph and genitalia of male beetles, whose biosynthesis derives from the degradation of the sesquiterpene farnesol [45,46]. It is a colorless, odorless monoterpene, soluble in organic solvents and oils, whose symmetrical structure is composed of a tricyclic skeleton that includes in one ring a carboxylic acid dihydride group (-CO-O-CO-) and a bridging ether in the bicyclic ring [47]. Thus, the relationship between structure and biological activity could explain the high specificity of cantharidin for serine/threonine protein phosphatases, although further studies are needed to understand this molecular interaction [14,46]. Protein phosphatases can control many cellular events and pathways and are widely conserved among eukaryotes, their inhibition by cantharidin being an event of great importance that can be applied for medicinal reasons, as already discussed [48]. Therefore, the doses and experimental times used in this study were selected considering previous studies with human cell lines [19,49,50] and the previous assay developed by our research group in gilthead seabream head kidney leucocytes [37]. Moreover, taking into account the native structure of other terpenes and the various properties and activities they usually show, such as antioxidant, cytotoxic or antimicrobial activities [45,51,52], these biological activities of cantharidin were evaluated in the present assay.
Antioxidant activity refers to the ability of certain compounds to react with free radicals and reduce their harmful effects caused by oxidative stress [53,54,55]. However, the structure of cantharidin and the chemical groups present on it do not seem to have direct scavenging ability for free radicals, as evidenced by our study's results. Therefore, the antitumor effects of cantharidin are likely attributed solely to its function as an inhibitor of serine/threonine protein phosphatases [14]. Similar to other terpenes like farnesol, cantharidin exhibits cytotoxic activity by inhibiting cell proliferation and inducing apoptosis (programmed cell death) [14,56]. Studies with the cervical cancer HeLa cell line demonstrate that cantharidin alters the sorting of glycosylphosphatidylinositol anchored proteins, induces apoptosis, and modifies cell wall integrity [57,58]. Our results support these findings, as cantharidin significantly reduced the viability of HeLa, SAF1, and PLHC1 cell lines in a time dose-dependent manner. Interestingly, both SAF1 and PLHC1 cells were more affected by cantharidin than the HeLa cell line, with PLHC1 cells being the most affected. These differences suggest that fish cells may be more susceptible to cantharidin than human cells and that tumoral cells may be more susceptible to this type of terpene than non-tumoral cells. Such variations could be due to differences in the composition of the plasma membrane of the cells, which could facilitate cantharidin's diffusion through the lipids of the cell membrane to a greater or lesser extent, thereby affecting its action [57].
Several studies have shown that human erythrocytes, which lack mitochondria and nuclei, can undergo a specific type of suicidal cell death called eryptosis after exposure to cantharidin, without producing appreciable hemolysis [59]. Although the molecular signalling pathways are not identical to those in other nucleated cells, the activation of protein phosphatases by cantharidin is able to promote the translocation of phosphatidylserine to the cell surface, resulting in membrane scrambling and cell shrinkage, and ultimately inducing eryptosis [59]. Our present results are consistent with these findings, as cantharidin did not induce hemolysis in gilthead seabream erythrocytes, but did cause evident changes in their cellular morphology, suggesting suicidal erythrocyte death and the conservation of cantharidin's action mechanism from fish to humans [59]. Once again, the nature of the cell membrane, in this case of erythrocytes, appears to play a critical role in the action mechanisms of cantharidin. It should be considered that the biological significance of eryptosis is still not fully understood, but it is thought to play a role in the regulation of erythrocyte number and lifespan in the body. By undergoing eryptosis, damaged or old erythrocytes can be selectively eliminated from the circulation, preventing their accumulation and potential damage to tissues. In addition, eryptosis may contribute to the pathogenesis of various diseases, including anemia, by promoting the premature removal of erythrocytes from the circulation [60]. Further research is needed to fully understand its biological significance in health and disease.
Cantharidin has been documented to have bactericidal activity against various bacteria that may be pathogen for humans, such as Escherichia coli, Staphylococcus aureus, Kocuria sp., Bacillus sp. and Clostridium sp., as well as anti-parasitic activity against protozoans and nematodes [13,29], but there is no data available on its activity against marine pathogens. In our study, the highest doses of cantharidin tested showed bactericidal activity against P. damselae, T. maritimum and V. anguillarum (with the latter being the most affected). However, we did nor observe bactericidal activity against V. harveyi. Effects of cantharidin on bacteria depending on the specie and the cantharidin concentration used in the assay. In this sense, our understanding of the molecular level effects of cantharidin, which is used in anticancer therapy, is still limited. Since numerous serine proteases are widespread throughout evolution and some bacteria use them in their own metabolism, as well as a pathogenic source [61,62,63,64], we hypothesize that cantharidin could increase the release of these bacterial proteases causing self-harming. On the other hand, some studies have shown that a small molecule AdoMet-dependent methyltransferase (CaCrg1) in the pathogenic fungus Candida albicans can interact with cantharidin and affect virulence-related processes such as adhesion, hyphal elongation, and membrane trafficking in response to the terpene. These findings suggest that further studies are necessary to understand the action mechanism of cantharidin on pathogens [65].
5. Conclusions
The results obtained in the present study offer a new approach to understanding the effects of cantharidin on fish cells and bacterial marine pathogens, which could extend its use not only as an antitumoral substance in humans but also for various purposes in the aquaculture sector due to its demonstrated properties.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Hemagglutination activity of gilthead seabream erythrocytes exposed to different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for 1.5 h. Macroscope image of the incubation plate: (A) Top view and (B) bottom view. PBS (0.35 % sodium chloride, 10 mM glucose) and Concanavalin A were used as a negative and negative controls (C- and C+), respectively. The results are representative of at least three independent experiments. Figure S2: Hemolytic activity (expressed as percentage of oxyhaemoglobin release) of gilthead seabream erythrocytes exposed to different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). No significant differences between experimental groups were observed (ANOVA; p > 0.05).
Author Contributions
Jose Carlos Campos-Sánchez: Methodology, Investigation, Writing – original draft, Writing – review & editing, Visualization. Francisco A. Guardiola: Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing, Visualization. María Ángeles Esteban: Term, Conceptualization, Validation, Investigation, Resources, Writing – review & editing, Visualization, Supervision, Project administration, Funding acquisition.
Funding
This research was funded by the proyecto de investigación PID2020-113637RB-C21 financiado por MCIN/AEI/10.13039/501100011033 and is part of the ThinkInAzul program supported by the MCIN with funding from European Union Next Generation EU (PRTR-C17.I1) and by the Comunidad Autónoma de la Región de Murcia-Fundación Séneca (Spain).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available on the DIGITUM institutional repository from the University of Murcia: http://hdl.handle.net/10201/130428 (accessed on 27 April 2023).
Conflicts of Interest
The authors declare no conflict of interest.
References
- FAO, FAO Aquaculture Newsletter, 61 (2020).
- Salinas, I.; Díaz-Rosales, P.; Cuesta, A.; Meseguer, J.; Chabrillón, M.; Moriñigo, M. .; Esteban, M.. Effect of heat-inactivated fish and non-fish derived probiotics on the innate immune parameters of a teleost fish (Sparus aurata L.). Veter- Immunol. Immunopathol. 2006, 111, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Imsland, A.K.; Foss, A.; Dinis, M.T.; Delbare, D.; Schram, E.; Kamstra, A.; Rema, P.; White, P.; Conceição, L.E.C. A review of the culture potential of Solea solea and S. senegalensis. Rev. Fish Biol. Fish. 2003, 13, 379–408. [Google Scholar] [CrossRef]
- Mohanty, B.; Sahoo, P. Immune responses and expression profiles of some immune-related genes in Indian major carp, Labeo rohita to Edwardsiella tarda infection. Fish Shellfish. Immunol. 2010, 28, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Girón-Pérez, M.I.; Santerre, A.; Gonzalez-Jaime, F.; Casas-Solis, J.; Hernández-Coronado, M.; Peregrina-Sandoval, J.; Takemura, A.; Zaitseva, G. Immunotoxicity and hepatic function evaluation in Nile tilapia (Oreochromis niloticus) exposed to diazinon. Fish Shellfish. Immunol. 2007, 23, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Pineiro, M.; Stanton, C. Probiotic Bacteria: Legislative Framework—Requirements to Evidence Basis1, J. Nutr. 2007, 137, 850S–853S. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, F.; Bahi, A.; Jiménez-Monreal, A.; Martínez-Tomé, M.; Murcia, M.; Esteban, M. Dietary administration effects of fenugreek seeds on skin mucosal antioxidant and immunity status of gilthead seabream (Sparus aurata L.). Fish Shellfish. Immunol. 2018, 75, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, J.M.G.; Espinosa, C.; Guardiola, F.A.; Esteban, M. . In vitro effects of Origanum vulgare leaf extracts on gilthead seabream (Sparus aurata L.) leucocytes, cytotoxic, bactericidal and antioxidant activities. Fish Shellfish. Immunol. 2018, 79, 1–10. [Google Scholar] [CrossRef]
- Messina, C.M.; Panettieri, V.; Arena, R.; Renda, G.; Ruiz, C.E.; Morghese, M.; Piccolo, G.; Santulli, A.; Bovera, F. The Inclusion of a Supercritical Fluid Extract, Obtained From Honey Bee Pollen, in the Diet of Gilthead Sea Bream (Sparus aurata), Improves Fish Immune Response by Enhancing Anti-oxidant, and Anti-bacterial Activities. Front. Veter- Sci. 2020, 7, 95. [Google Scholar] [CrossRef]
- Cámara-Ruiz, M.; Balebona, M.C.; Moriñigo, M. .; Esteban, M.. Probiotic Shewanella putrefaciens (SpPdp11) as a Fish Health Modulator: A Review. Microorganisms 2020, 8, 1990. [Google Scholar] [CrossRef]
- Sánchez-Barbudo, I.S.; Camarero, P.R.; García-Montijano, M.; Mateo, R. Possible cantharidin poisoning of a great bustard (Otis tarda). Toxicon 2012, 59, 100–103. [Google Scholar] [CrossRef]
- Ghaffarifar, F. Leishmania major: In vitro and in vivo anti-leishmanial effect of cantharidin. Exp. Parasitol. 2010, 126, 126–129. [Google Scholar] [CrossRef]
- Whitman, D.W.; Andrés, M.F.; Martínez-Díaz, R.A.; Ibáñez-Escribano, A.; Olmeda, A.S.; González-Coloma, A. Antiparasitic Properties of Cantharidin and the Blister Beetle Berberomeloe majalis (Coleoptera: Meloidae). Toxins 2019, 11, 234. [Google Scholar] [CrossRef]
- Honkanen, R.E. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett. 1993, 330, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Wisniak, J. Pierre-Jean Robiquet. Educ. Quim 2013, 24, 139–149. [Google Scholar] [CrossRef]
- Moed, L.; Shwayder, T.A.; Chang, M.W. Cantharidin Revisited. Arch. Dermatol. 2001, 137, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Karras, D.J.; E Farrell, S.; A Harrigan, R.; Henretig, F.M.; Gealt, L. Poisoning from “Spanish fly” (cantharidin). Am. J. Emerg. Med. 1996, 14, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Methods Mol. Biol. 2007, 365, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-P.; Tsai, C.-H.; Wu, P.-P.; Hsu, S.-C.; Liu, H.-C.; Huang, Y.-P.; Yang, J.-H.; Chung, J.-G. Cantharidin induces G2/M phase arrest by inhibition of Cdc25c and Cyclin A and triggers apoptosis through reactive oxygen species and the mitochondria-dependent pathways of A375.S2 human melanoma cells. Int. J. Oncol. 2014, 45, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-W.; Ko, S.-W.; Tsai, H.-Y.; Chung, J.-G.; Chiang, J.-H.; Chen, K.-T.; Chen, Y.-C.; Chen, H.-Y.; Chen, Y.-F.; et al. Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. Int. J. Oncol. 2011, 38, 1067–1073. [Google Scholar] [CrossRef]
- Hsia, T.-C.; Yu, C.-C.; Hsu, S.-C.; Tang, N.-Y.; Lu, H.-F.; Huang, Y.-P.; Wu, S.-H.; Lin, J.-G.; Chung, J.-G. Cantharidin induces apoptosis of H460 human lung cancer cells through mitochondria-dependent pathways. Int. J. Oncol. 2014, 45, 245–254. [Google Scholar] [CrossRef]
- Nazim, U.; Yin, H.; Park, S. Downregulation of c-FLIP and upregulation of DR-5 by cantharidin sensitizes TRAIL-mediated apoptosis in prostate cancer cells via autophagy flux. Int. J. Mol. Med. 2020, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Chung; Kuo, J. -H.; Chu, Y.-L.; Yang, J.-S.; Lin, J.-P.; Lai, K.-C.; Kuo, H.-M.; Hsia, T.-C.; Chung, J.-G. Cantharidin induces apoptosis in human bladder cancer TSGH 8301 cells through mitochondria-dependent signal pathways. Int. J. Oncol. 2010, 37, 1243–1250. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, D.; Mao, M.; Lu, Y.; Jiao, N. Roles of p38 and JNK protein kinase pathways activated by compound cantharidin capsules containing serum on proliferation inhibition and apoptosis of human gastric cancer cell line. Exp. Ther. Med. 2017, 14, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Wass, J.; Vincent, P.; Iland, H. Inhibitory effect of norcantharidin on K562 human myeloid leukemia cells in vitro. Leuk. Res. 1991, 15, 883–886. [Google Scholar] [CrossRef]
- Huan, S.K.-H.; Lee, H.-H.; Liu, D.-Z.; Wu, C.-C.; Wang, C.-C. Cantharidin-induced cytotoxicity and cyclooxygenase 2 expression in human bladder carcinoma cell line. Toxicology 2006, 223, 136–143. [Google Scholar] [CrossRef]
- Li, W.; Xie, L.; Chen, Z.; Zhu, Y.; Sun, Y.; Miao, Y.; Xu, Z.; Han, X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010, 101, 1226–1233. [Google Scholar] [CrossRef]
- Yeh, C.-B.; Su, C.-J.; Hwang, J.-M.; Chou, M.-C. Therapeutic effects of cantharidin analogues without bridging ether oxygen on human hepatocellular carcinoma cells. Eur. J. Med. Chem. 2010, 45, 3981–3985. [Google Scholar] [CrossRef]
- Bravo, C.; Bautista, L.M.; García-París, M.; Blanco, G.; Alonso, J.C. Males of a Strongly Polygynous Species Consume More Poisonous Food than Females. PLOS ONE 2014, 9, e111057. [Google Scholar] [CrossRef]
- Shao, H.; Zhang, Y. Non-target effects on soil microbial parameters of the synthetic pesticide carbendazim with the biopesticides cantharidin and norcantharidin. Sci. Rep. 2017, 7, 5521–5521. [Google Scholar] [CrossRef]
- Day, R.M.; Harbord, M.; Forbes, A.; Segal, A.W. Cantharidin blisters: a technique for investigating leukocyte trafficking and cytokine production at sites of inflammation in humans. J. Immunol. Methods 2001, 257, 213–220. [Google Scholar] [CrossRef]
- Dinh, P.H.D.; Corraza, F.; Mestdagh, K.; Kassengera, Z.; Doyen, V.; Michel, O. Validation of the cantharidin-induced skin blister as anin vivomodel of inflammation. Br. J. Clin. Pharmacol. 2011, 72, 912–920. [Google Scholar] [CrossRef]
- Maini, A.A.; George, M.J.; Motwani, M.P.; Day, R.M.; Gilroy, D.W.; O’brien, A.J. A Comparison of Human Neutrophils Acquired from Four Experimental Models of Inflammation. PLOS ONE 2016, 11, e0165502. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, J.; Zhang, Y. Cantharidin impedes the activity of protein serine/threonine phosphatase in Plutella xylostella. Mol. Biosyst. 2013, 10, 240–250. [Google Scholar] [CrossRef]
- Máthé, C.; Garda, T.; Freytag, C.; M-Hamvas, M. The Role of Serine-Threonine Protein Phosphatase PP2A in Plant Oxidative Stress Signaling—Facts and Hypotheses. Int. J. Mol. Sci. 2019, 20, 3028. [Google Scholar] [CrossRef] [PubMed]
- Vanlerberghe, G.C.; Robson, C.A.; Yip, J.Y. Induction of Mitochondrial Alternative Oxidase in Response to a Cell Signal Pathway Down-Regulating the Cytochrome Pathway Prevents Programmed Cell Death. Plant Physiol. 2002, 129, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Campos-Sánchez, J.C.; Guardiola, F.A.; Esteban, M. . In vitro effects of cantharidin on gilthead seabream (Sparus aurata) head-kidney leucocytes. Fish Shellfish. Immunol. 2022, 123, 20–35. [Google Scholar] [CrossRef]
- Arnao, M.B.; Cano, A.; Acosta, M. Methods to Measure the Antioxidant Activity in Plant Material. A Comparative Discussion. Free. Radic. Res. 1999, 31, 89–96. [Google Scholar] [CrossRef]
- Morcillo, P.; Romero, D.; Meseguer, J.; Esteban, M. .; Cuesta, A. Cytotoxicity and alterations at transcriptional level caused by metals on fish erythrocytes in vitro. Environ. Sci. Pollut. Res. 2016, 23, 12312–12322. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Q.; Wang, H.; Ng, T. A novel lectin with potent antitumor, mitogenic and HIV-1 reverse transcriptase inhibitory activities from the edible mushroom Pleurotus citrinopileatus. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2008, 1780, 51–57. [Google Scholar] [CrossRef]
- Stevens, M.G.; Kehrli, M.E.; Canning, P.C. A colorimetric assay for quantitating bovine neutrophil bactericidal activity. Veter- Immunol. Immunopathol. 1991, 28, 45–56. [Google Scholar] [CrossRef]
- Sunyer, J.O.; Tort, L. Natural hemolytic and bactericidal activities of sea bream Sparus aurata serum are effected by the alternative complement pathway. Veter- Immunol. Immunopathol. 1995, 45, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W. , Kirby, W.M., Sherris, J.C., Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef] [PubMed]
- CLSI, Clinical and laboratory standards institute 2018: performance standards for antimicrobial susceptibility testing. 28th edition, CLSI Supplement M100. Clinical and Laboratory Standard Institute, Wayne, PA, 2018, n.d.
- M. Gronquist, F.C. M. Gronquist, F.C. Schroeder, Insect natural products, Compr. Nat. Prod. II Chem. Biol. 2 (2010) 67–108. [CrossRef]
- McCormick, J.P.; Carrel, J.E.; Doom, J.P. Origin of oxygen atoms in cantharidin biosynthesized by beetles. J. Am. Chem. Soc. 1986, 108, 8071–8074. [Google Scholar] [CrossRef]
- Matsuzawa, M.; Graziano, M.J.; Casida, J.E. Endothal and cantharidin analogs: relation of structure to herbicidal activity and mammalian toxicity. J. Agric. Food Chem. 1987, 35, 823–829. [Google Scholar] [CrossRef]
- P. Cohen, The structure and regulation of protein phosphatases, Annu. Rev. Biochem. 58 (1989) 453–508.
- Li, H.-C.; Xia, Z.-H.; Chen, Y.-F.; Yang, F.; Feng, W.; Cai, H.; Mei, Y.; Jiang, Y.-M.; Xu, K.; Feng, D.-X. Cantharidin Inhibits the Growth of Triple-Negative Breast Cancer Cells by Suppressing Autophagy and Inducing Apoptosis in Vitro and in Vivo. Cell. Physiol. Biochem. 2017, 43, 1829–1840. [Google Scholar] [CrossRef]
- Massicot, F.; Dutertre-Catella, H.; Pham-Huy, C.; Liu, X.-H.; Duc, H.T.; Warnet, J.-M. In vitro Assessment of Renal Toxicity and Inflammatory Events of Two Protein Phosphatase Inhibitors Cantharidin and Nor-Cantharidin*. Basic Clin. Pharmacol. Toxicol. 2005, 96, 26–32. [Google Scholar] [CrossRef]
- van Zyl, R.L.; Seatlholo, S.T.; van Vuuren, S.F.; Viljoen, A.M. The Biological Activities of 20 Nature Identical Essential Oil Constituents. J. Essent. Oil Res. 2006, 18, 129–133. [Google Scholar] [CrossRef]
- Paduch, R.; Kandefer-Szerszeń, M.; Trytek, M.; Fiedurek, J. Terpenes: substances useful in human healthcare. Arch. Immunol. et Ther. Exp. 2007, 55, 315–327. [Google Scholar] [CrossRef]
- Kozłowska, M.; Laudy, A.E.; Przybył, J.; Ziarno, M.; Majewska, E. CHEMICAL COMPOSITION AND ANTIBACTERIAL ACTIVITY OF SOME MEDICINAL PLANTS FROM LAMIACEAE FAMILY. Acta Pol. Pharm. -Drug Res. 2015, 72, 757–767. [Google Scholar]
- Yanishlieva, N.V.; Marinova, E.M.; Gordon, M.H.; Raneva, V.G. Antioxidant activity and mechanism of action of thymol and carvacrol in two lipid systems. Food Chem. 1999, 64, 59–66. [Google Scholar] [CrossRef]
- Ruberto, G.; Baratta, M.T. Antioxidant activity of selected essential oil components in two lipid model systems. Food Chem. 2000, 69, 167–174. [Google Scholar] [CrossRef]
- Knapp, J.; Boknı́k, P.; Huke, S.; Gombosová, I.; Linck, B.; Lüss, H.; Müller, F.U.; Müller, T.; Nacke, P.; Schmitz, W.; et al. Contractility and Inhibition of Protein Phosphatases by Cantharidin. Gen. Pharmacol. Vasc. Syst. 1998, 31, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.K.; Tomar, R.S. The natural anticancer agent cantharidin alters GPI-anchored protein sorting by targeting Cdc1-mediated remodeling in endoplasmic reticulum. J. Biol. Chem. 2019, 294, 3837–3852. [Google Scholar] [CrossRef] [PubMed]
- Patzlaff, J.S.; Terrenoire, E.; Turner, B.M.; Earnshaw, W.C.; Paulson, J.R. Acetylation of core histones in response to HDAC inhibitors is diminished in mitotic HeLa cells. Exp. Cell Res. 2010, 316, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.; Egler, J.; Briglia, M.; Fazio, A.; Faggio, C.; Lang, F. Induction of Suicidal Erythrocyte Death by Cantharidin. Toxins 2015, 7, 2822–2834. [Google Scholar] [CrossRef]
- Dreischer, P.; Duszenko, M.; Stein, J.; Wieder, T. Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells 2022, 11, 503. [Google Scholar] [CrossRef]
- Howell, M.; Dumitrescu, D.G.; Blankenship, L.R.; Herkert, D.; Hatzios, S.K. Functional characterization of a subtilisin-like serine protease from Vibrio cholerae. J. Biol. Chem. 2019, 294, 9888–9900. [Google Scholar] [CrossRef]
- Patel, S. A critical review on serine protease: Key immune manipulator and pathology mediator. Allergol. et Immunopathol. 2017, 45, 579–591. [Google Scholar] [CrossRef]
- Kulkarni, N.; Shendye, A.; Rao, M. Molecular and biotechnological aspects of xylanases. FEMS Microbiol. Rev. 1999, 23, 411–456. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Sun, K.; Cheng, S.; Sun, L. Characterization of DegQ Vh, a Serine Protease and a Protective Immunogen from a Pathogenic Vibrio harveyi Strain. Appl. Environ. Microbiol. 2008, 74, 6254–6262. [Google Scholar] [CrossRef]
- Lissina, E.; Weiss, D.; Young, B.; Rella, A.; Cheung-Ong, K.; Del Poeta, M.; Clarke, S.G.; Giaever, G.; Nislow, C. A Novel Small Molecule Methyltransferase Is Important for Virulence in Candida albicans. ACS Chem. Biol. 2013, 8, 2785–2793. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative image of phase contrast microscope of erythrocytes from gilthead seabream exposed to different concentrations of cantharidin (0, 5 and 10 µg mL-1) for 12 and 24 h. Note the morphological changes produced after cantharidin exposure. Scale bar = 50 µm.
Figure 1.
Representative image of phase contrast microscope of erythrocytes from gilthead seabream exposed to different concentrations of cantharidin (0, 5 and 10 µg mL-1) for 12 and 24 h. Note the morphological changes produced after cantharidin exposure. Scale bar = 50 µm.
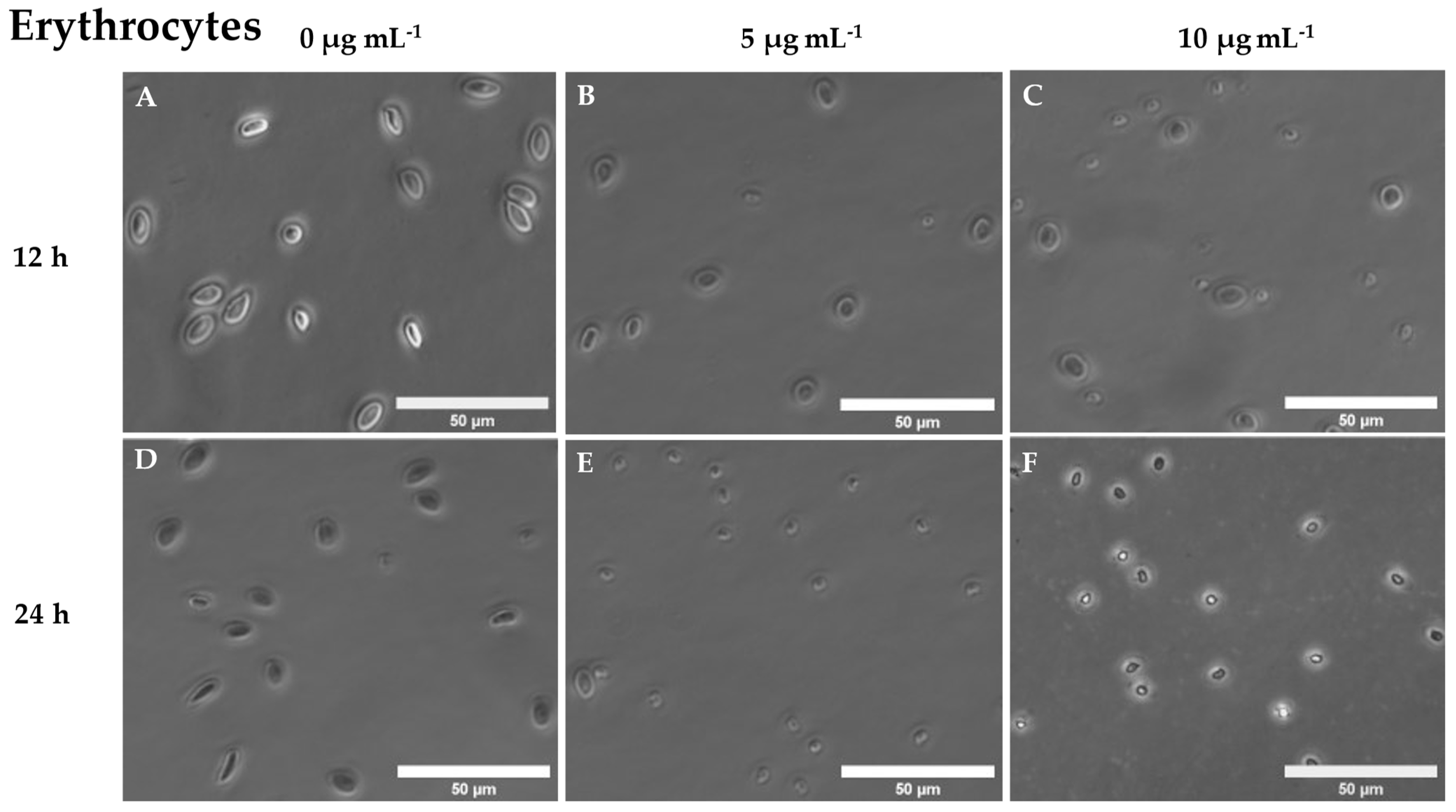
Figure 2.
Cytotoxic activity (expressed as percentage of viable cells) of SAF-1 cells incubated with different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
Figure 2.
Cytotoxic activity (expressed as percentage of viable cells) of SAF-1 cells incubated with different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
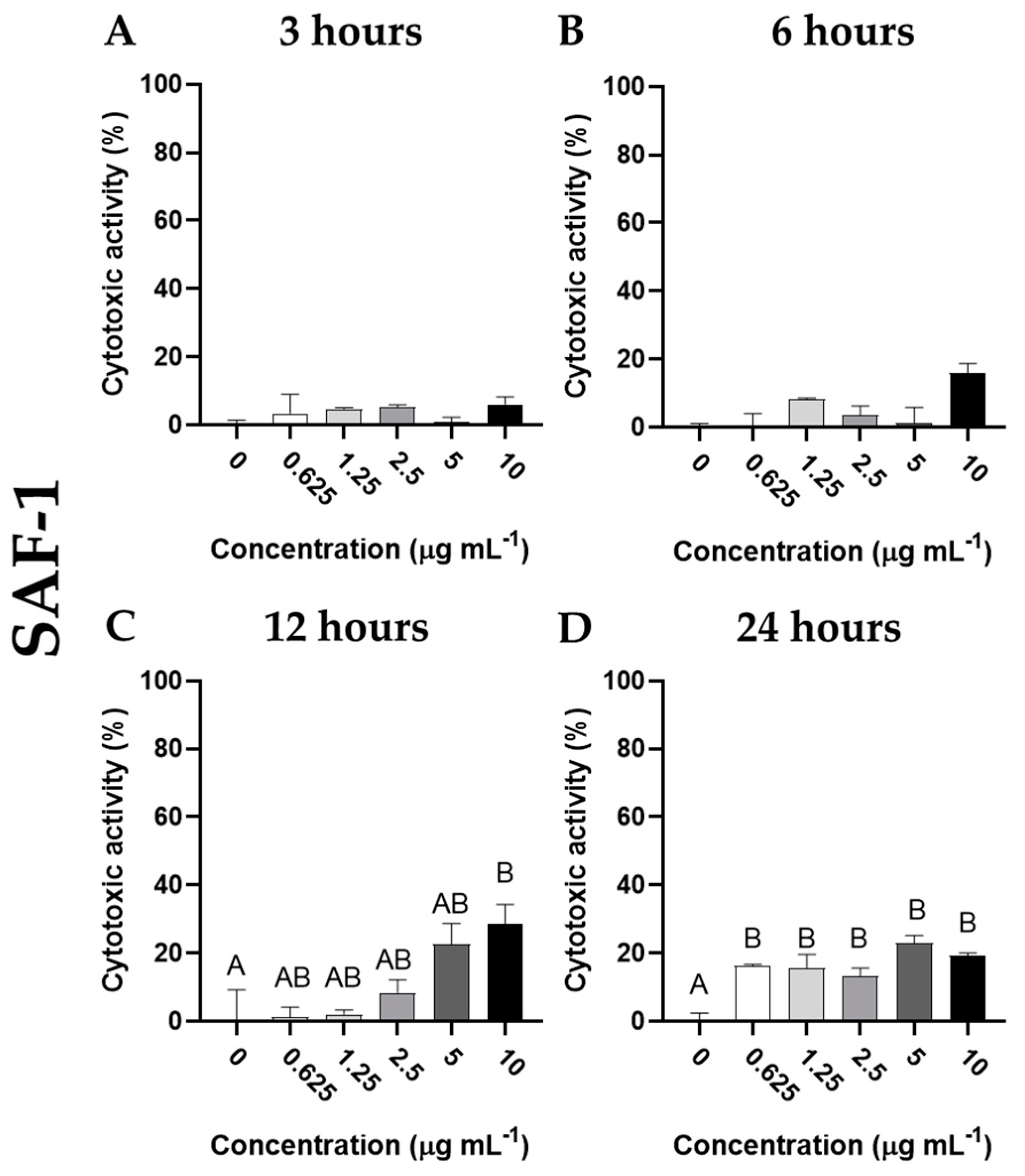
Figure 3.
Representative image of phase contrast microscope of SAF-1 cell culture after being incubated with different concentrations of cantharidin (0, 5 and 10 µg mL-1) for (A, B, C) 12 and (D, E, F) 24 h. Note the presence of detached and rounded cells after cantharidin exposure. Scale bar = 50 µm.
Figure 3.
Representative image of phase contrast microscope of SAF-1 cell culture after being incubated with different concentrations of cantharidin (0, 5 and 10 µg mL-1) for (A, B, C) 12 and (D, E, F) 24 h. Note the presence of detached and rounded cells after cantharidin exposure. Scale bar = 50 µm.
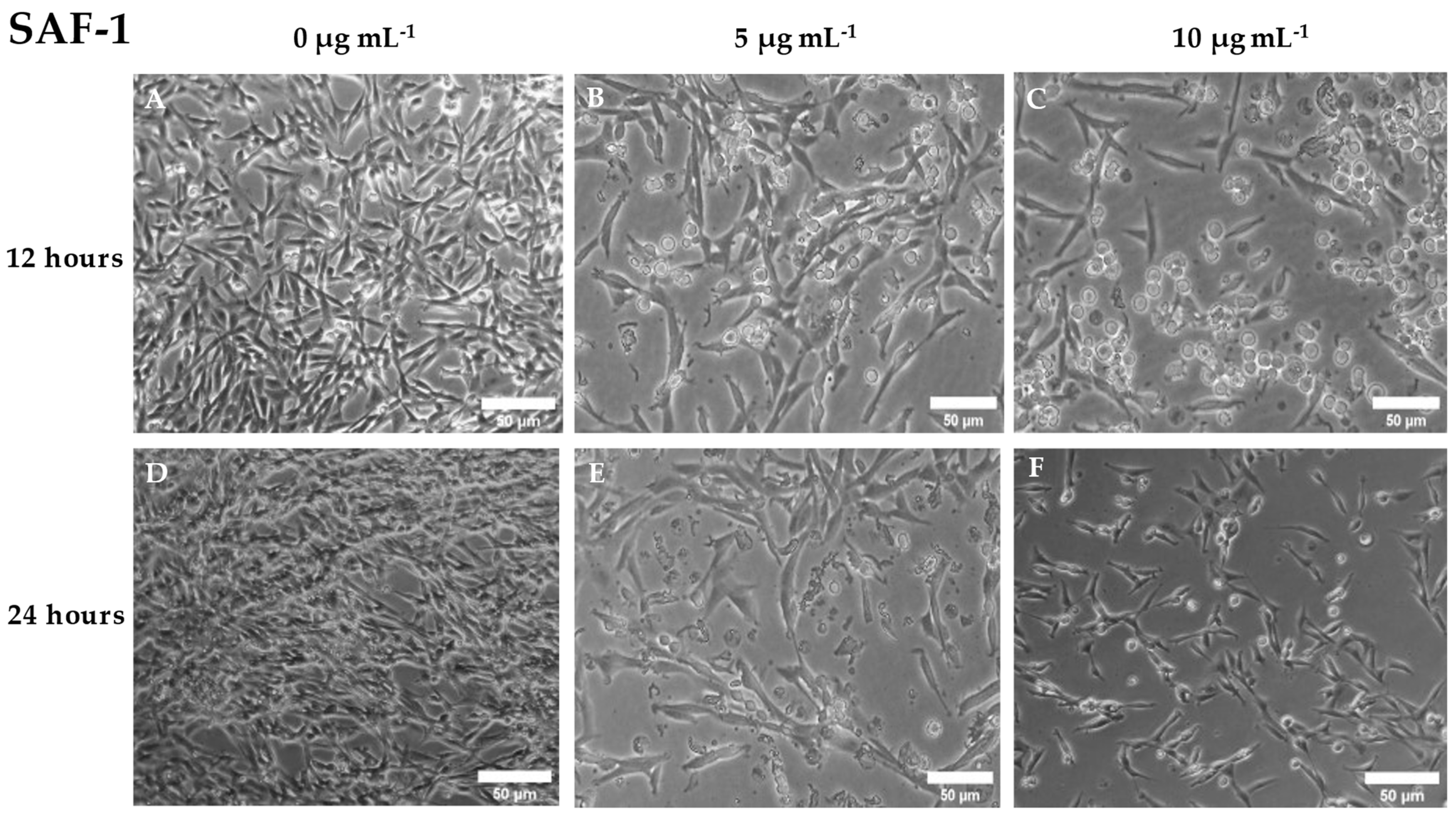
Figure 4.
Cytotoxic activity (expressed as percentage of viable cells) of PLHC-1 cells incubated with different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
Figure 4.
Cytotoxic activity (expressed as percentage of viable cells) of PLHC-1 cells incubated with different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
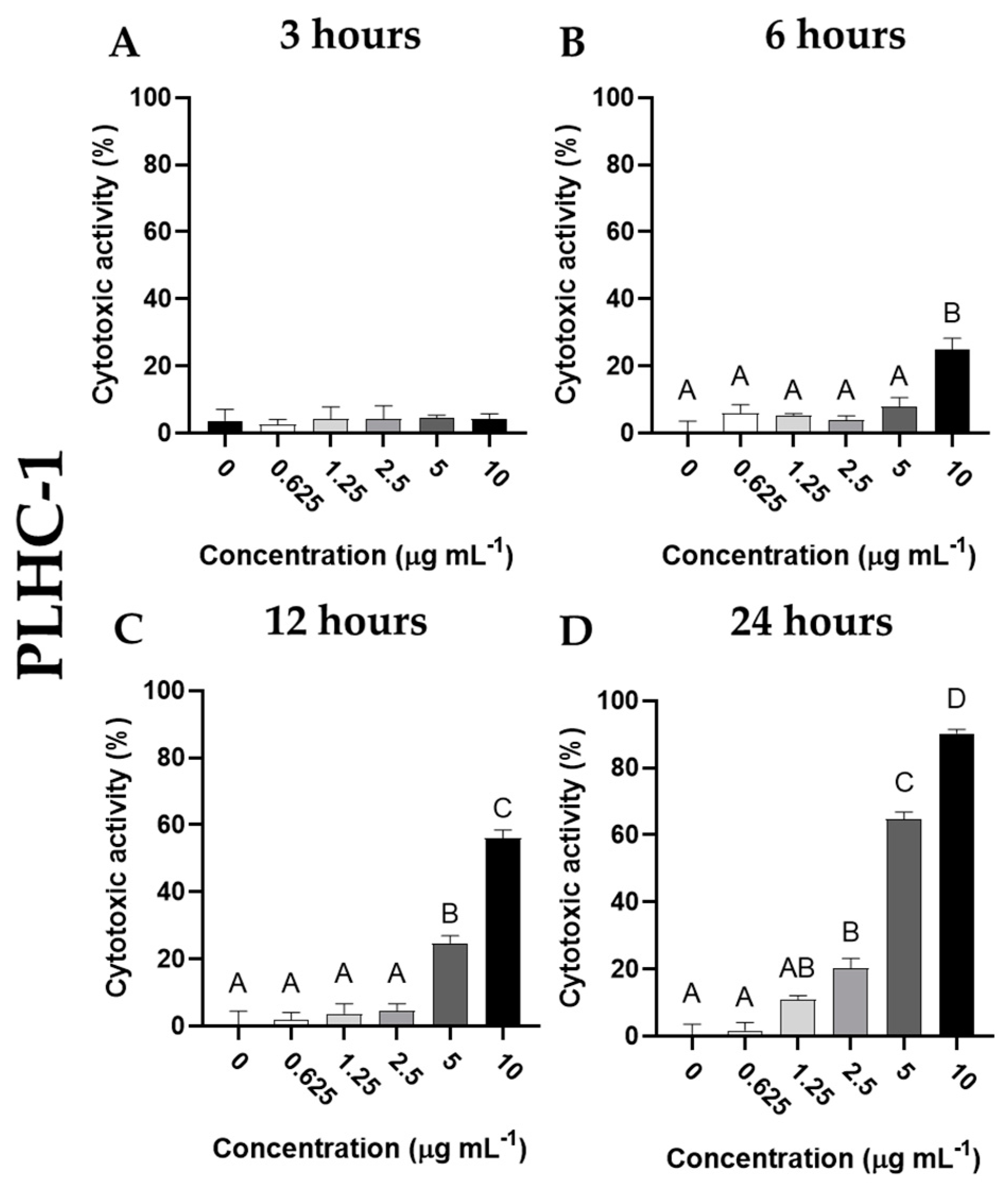
Figure 5.
Representative image of phase contrast microscope of PLHC-1 cells culture after being incubated with different concentrations of cantharidin (0, 5 and 10 µg mL-1) for (A, B, C) 12 and (D, E, F) 24 h. Note as the cells detached and appeared as dark (dull) cells with a small rounded shape after cantharidin exposure. Scale bar = 50 µm.
Figure 5.
Representative image of phase contrast microscope of PLHC-1 cells culture after being incubated with different concentrations of cantharidin (0, 5 and 10 µg mL-1) for (A, B, C) 12 and (D, E, F) 24 h. Note as the cells detached and appeared as dark (dull) cells with a small rounded shape after cantharidin exposure. Scale bar = 50 µm.

Figure 6.
Cytotoxic activity (expressed as percentage of viable cells) of HeLa cells incubated with different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
Figure 6.
Cytotoxic activity (expressed as percentage of viable cells) of HeLa cells incubated with different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1) for (A) 3, (B) 6, (C) 12 and (D) 24 h. Data represent the mean ± standard error of the mean (SEM) (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
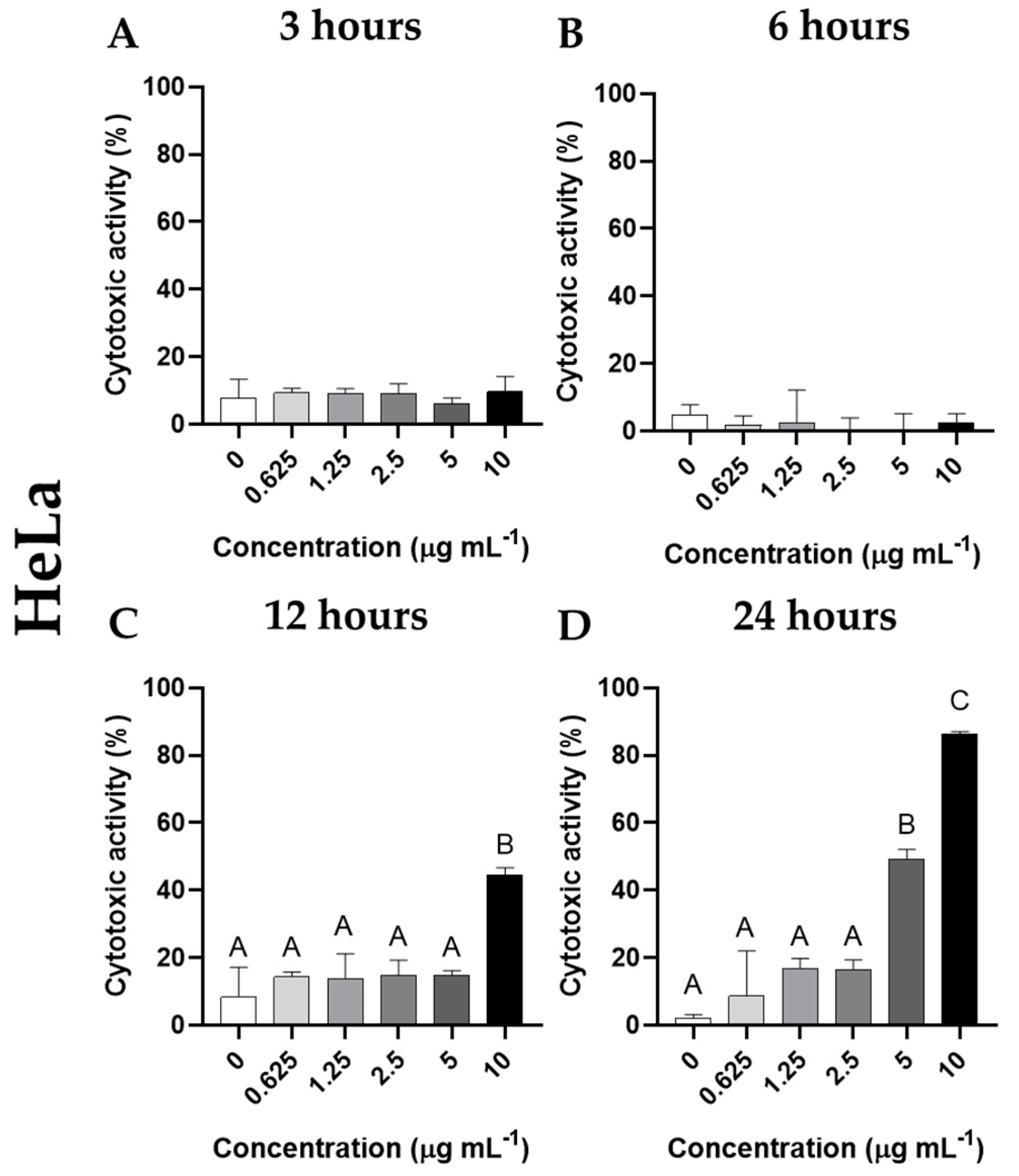
Figure 7.
Representative image of phase contrast microscope of HeLa cell culture after being exposed to different concentrations of cantharidin (0, 5 and 10 µg mL-1) for (A, B, C) 12 and (D, E, F) 24 h. Note the morphological changes produced after cantharidin exposure. *, cell debris. Scale bar = 50 µm.
Figure 7.
Representative image of phase contrast microscope of HeLa cell culture after being exposed to different concentrations of cantharidin (0, 5 and 10 µg mL-1) for (A, B, C) 12 and (D, E, F) 24 h. Note the morphological changes produced after cantharidin exposure. *, cell debris. Scale bar = 50 µm.
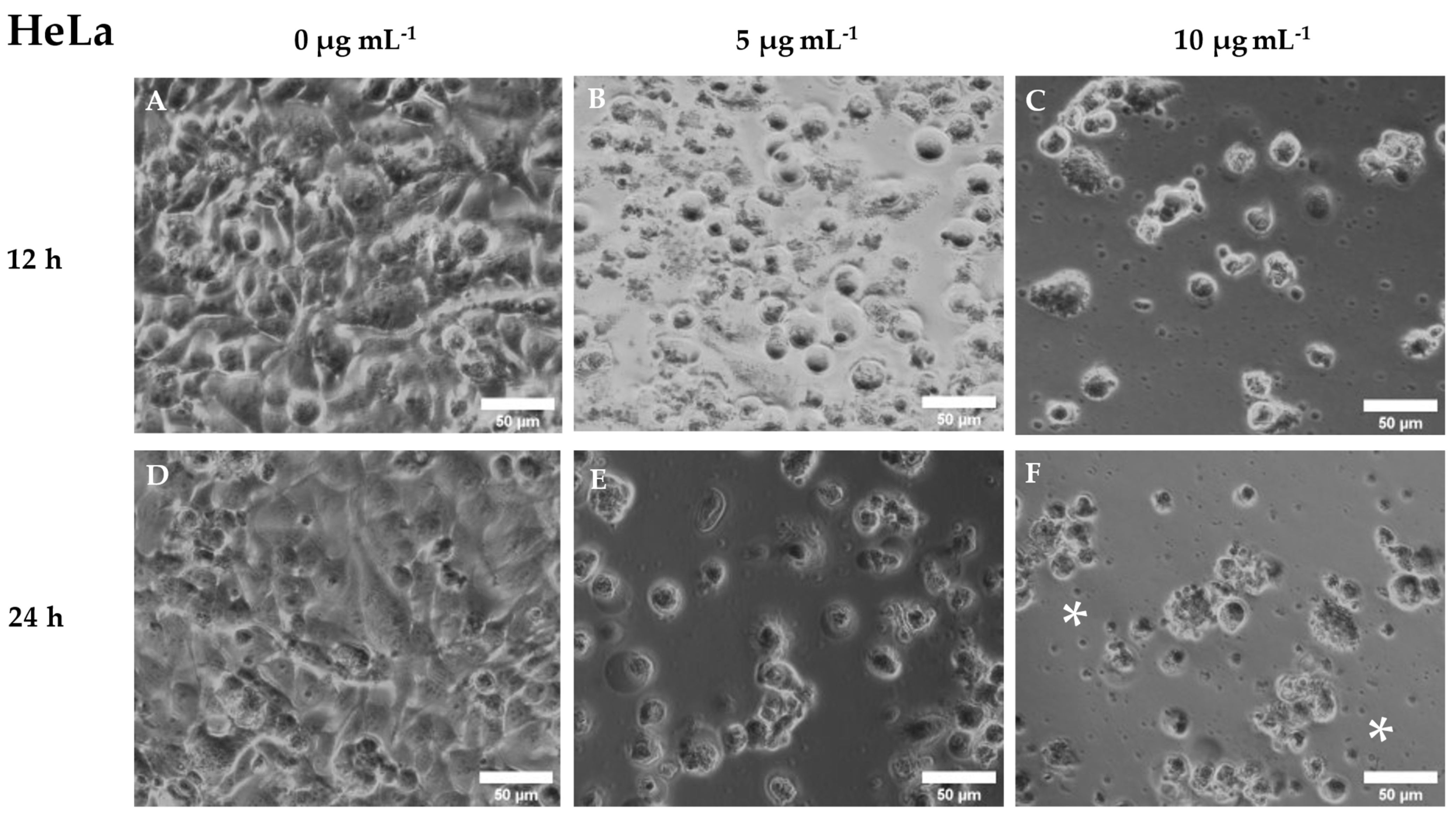
Figure 8.
Bactericidal activity (expressed as percentage of viable bacteria) of (A) Vibrio harveyi, (B) Vibrio anguillarum, (C) Photobacterium damselae and (D) Tenacibaculum maritimum exposed to different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1). The results are representative of at least three independent experiments and are expressed as mean ± SEM (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
Figure 8.
Bactericidal activity (expressed as percentage of viable bacteria) of (A) Vibrio harveyi, (B) Vibrio anguillarum, (C) Photobacterium damselae and (D) Tenacibaculum maritimum exposed to different concentrations of cantharidin (0, 0.625, 1.25, 2.5, 5 and 10 µg mL-1). The results are representative of at least three independent experiments and are expressed as mean ± SEM (n = 6). Different letters denote significant differences between experimental concentrations (ANOVA; p < 0.05).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



