Submitted:
27 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
Despite the strong anticancer activity of SN38 (7-ethyl-10-hydroxy-camptothecin), some defects prevent its clinical application, such as severe side effects and loss of anticancer activity due to lack of selectivity to cancer cells and hydrolysis of ring E, respectively. To address the issue, herein a multifunctional SN38 derivative (compound 9) containing biotin (tumor-targeting group) and valproic acid (histone deacetylase inhibitor, HDACi) was synthesized via click chemistry and evaluated using MTT assay. The in vitro cytotoxicity study showed that compound 9 exhibited superior cytotoxicity as irinotecan against human cervical cancer HeLa cells, albeit inferior to SN38. More significantly, compound 9 significantly reduced toxicity in mouse embryonic fibroblast NIH3T3 cells, indicating that compound 9 had the capacity to enhance tumor targeting due to its cell selectivity. Further studies demonstrated that compared with irinotecan, compound 9 induced similar apoptosis of cancer cells. Consequently, compound 9 can not only improve its tumor-targeting ability mediated by biotin but also exert the potent anticancer activity through the synergism of SN38 and valproic acid, indicating that the design concept is an effective strategy for structural modification of SN38.
Keywords:
7-ethyl-10-hydroxy-camptothecin
; valproic acid
; biotin
; conjugate
; synthesis
; anticancer
1. Introduction
Since camptothecin was isolated from the bark and stem of Camptotheca acuminata in 1966, the alkaloid has received extensive attention from chemists and pharmacologists. Camptothecin-based compounds usually exert their anticancer effect by inhibiting DNA topoisomerase I (topo 1)[1,2,3]. Topo 1 plays an important role in transcription and replication of cells through relieving the stress of DNA supercoiling[4]. Some camptothecin derivatives such as topotecan and irinotecan have been approved by the US Food and Drug Administration for the treatment of several cancers[5]. However, severe side effects due to lack of selectivity to cancer cells and loss of anticancer activity caused by hydrolysis of E-lactone is still a problem to be solved for camptothecin-based compounds, not excepting 7-ethyl-10-hydroxy-camptothecin (SN38)[6,7]. SN38, an active metabolite of irinotecan, is one of the most potent camptothecin derivatives. Though anticancer activity of SN38 is 100-1000 times stronger than that of irinotecan, only2-8% of dosed irinotecan can be converted into SN38 to perform its therapeutic action[8]. Hence, further study on SN38 is in demand. Some research achievements on SN38 have been reported in recent years[9,10,11,12]. For example, HSP90 inhibitor-SN38 conjugate showed excellent in vitro and in vivo antitumor activity[13]. In addition, conjugation of SN38 with artesunate also demonstrated the potent anticancer activity through the formation of reactive radical species by cleavage of endoperoxide bridge in artesunate and inhibition of topo 1 by SN38[14]. Thus, the investigation on SN38 is still a good source of developing new drugs.
Histone deacetylase (HDAC) belongs to proteinase family and plays an important role in structural modification of chromosome and regulation of gene expression[15]. Inhibition of HDAC can suppress the growth of cancer cells. Therefore, histone deacetylase inhibitors (HDACis) had been widely explored for the treatment of cancer[16,17]. For example, Belinostat and panobinostat have been approved to cure several tumors by FDA. Chidamide was approved to cure peripheral T-cell lymphoma by National Medical Products Administration[18,19]. Some literatures reported that HDACis can sensitize chemotherapeutic drugs including camptothecin-based drugs[20,21]. For example, histone deacetylase class IIa inhibitor with lenvatinib synergistically inhibited the growth of hepatocellular carcinoma cells[22]. Panobinostat synergistically enhanced the cytotoxicity of microtubule destabilizing drugs in ovarian cancer cells[23]. In addition, hybrids of HDACis and camptothecin also exhibited potent antitumor activity in A549 and HCT-116 cells[24]. Valproic acid, a classic antiepileptic drug, also exerts its inhibitory activity against HDAC and sensitizes chemotherapeutic drugs[25,26,27]. However, higher concentration of valproic acid is required for suppressing the growth of cancer cells through inhibition of HDAC[27,28,29]. Therefore, combination of valproic acid with other chemotherapeutic agents is a good strategy for the development of anticancer drugs.
Decoration of SN38 with tumor-targeting group is an excellent strategy to reduce side effects caused by SN38. These tumor-targeting groups are usually antibodies or small molecular compounds[30,31,32,33]. For instance, Trastuzumab deruxtecan, an antibody-drug conjugate, was approved by FDA to cure refractory HER2-positive metastatic breast cancer[34]. Biotin is also known as vitamin H and vitamin B7 and is responsible for various normal cellular functions. Biotin is used as a tumor-targeting agent due to overexpression of biotin receptors on several kinds of cancer cells. Incorporation of biotin to SN38 will be benefit for cell selectivity of SN38 to reduce side effects[35,36,37]. It is well established that ester bond is one of the most commonly used linker for design of prodrug and can be hydrolyzed to release parent drug by esterase in cells. The design of prodrug based on ester bond was applied to the study of SN38[24,38,39]. Based on these findings, a novel SN38 derivative containing three valproic acids (HDACis) and biotin (tumor-targeting group) (compound 9) was synthesized as prodrugs via click chemistry under mild conditions. Cytotoxicity test showed that compound 9 had higher anti-proliferative activity toward HeLa cells and lower inhibition toward NIH3T3 cells compared with irinotecan.
2. Results and Discussion
2.1. Chemistry
It is well established that the opening of lactone E-ring for camptothecin analogues will result in the decreased anticancer activity and unexpected side effects, and the lactone E-ring of compound 9 can be stabilized by the introduction of groups with high steric hindrance C-20 of SN38[40]. Hence, as many valproic acid moieties as possible are in need for the design of SN38 derivatives. Here, we initially selected three valproic acid moieties to conjugate with SN38 to prepare the target compound 9 for facilitating its stability. Owing to lack of cell selectivity for SN38, biotin was used as a tumor-targeting unit of compound 9. However, on the premise of ensuring the structural integrity of SN38, there are not enough hydroxyl groups for one molecule of SN38 to link three valproic acid molecules and one biotin molecule via ester bond. Hence, SN38-based dendritic compound was designed to meet the requirement of link between five molecules (biotin: SN38: valproic acid=1:1:3). The synthetic routes of compound 9 was described in scheme 1. Firstly, two key intermediates, compound 3 and 4 need to be completed before synthesis of compound 9. After then, SN38 was protected with (Boc)2O/ pyridine at C10-OH to prepare compound 5, followed by esterification of compound 5 at C20-OH with compound 3. The obtained compound 6 underwent removal of protecting group under the condition of trifluoroacetic acid and dichloromethane to synthesize compound 7. Then the key intermediate compound 8 was synthesized by click reaction of Cu-catalytic Azide-Alkyne Click (CuAAC) between compound 7 and compound 4 in presence of sodium ascorbate and CuSO4. Finally, esterification of compound 8 and biotin was conducted under the condition of EDCI and DMAP to obtain target compound 9. These compounds were verified by HPLC, NMR and MS (Figure S1- Figure S14).
Scheme 1.
Synthetic route of compound 9. (i) SOCl2, pyridine, 100°C, 5 h, yield 63%; (ii) NaN3, DMSO, 90°C, 24 h, yield 93%; (iii) succinic anhydride, triethylamine, CH2Cl2, room temperature, monitored by TLC, yield 88%; (iv) propargyl bromide, K2CO3, DMF, room temperature, monitored by TLC, yield 86%; (v) (Boc)2O, pyridine, CH2Cl2, room temperature, monitored by TLC, yield 86%; (vi) compound 3, EDCI, DMAP, CH2Cl2, room temperature, monitored by TLC, yield 73%; (vii) trifluoroacetic acid, CH2Cl2, 30°C, monitored by TLC, yield 82%; (viii) compound 4, sodium ascorbate, CuSO4, t-BuOH/H2O mixture (2:1), 40°C, monitored by TLC, yield 52%; (ix) biotin, EDCI, DMAP, DMF, 24 h, yield 43%.
Scheme 1.
Synthetic route of compound 9. (i) SOCl2, pyridine, 100°C, 5 h, yield 63%; (ii) NaN3, DMSO, 90°C, 24 h, yield 93%; (iii) succinic anhydride, triethylamine, CH2Cl2, room temperature, monitored by TLC, yield 88%; (iv) propargyl bromide, K2CO3, DMF, room temperature, monitored by TLC, yield 86%; (v) (Boc)2O, pyridine, CH2Cl2, room temperature, monitored by TLC, yield 86%; (vi) compound 3, EDCI, DMAP, CH2Cl2, room temperature, monitored by TLC, yield 73%; (vii) trifluoroacetic acid, CH2Cl2, 30°C, monitored by TLC, yield 82%; (viii) compound 4, sodium ascorbate, CuSO4, t-BuOH/H2O mixture (2:1), 40°C, monitored by TLC, yield 52%; (ix) biotin, EDCI, DMAP, DMF, 24 h, yield 43%.
2.2. In Vitro Biological evaluation
2.2.1. In Vitro Cytotoxicity
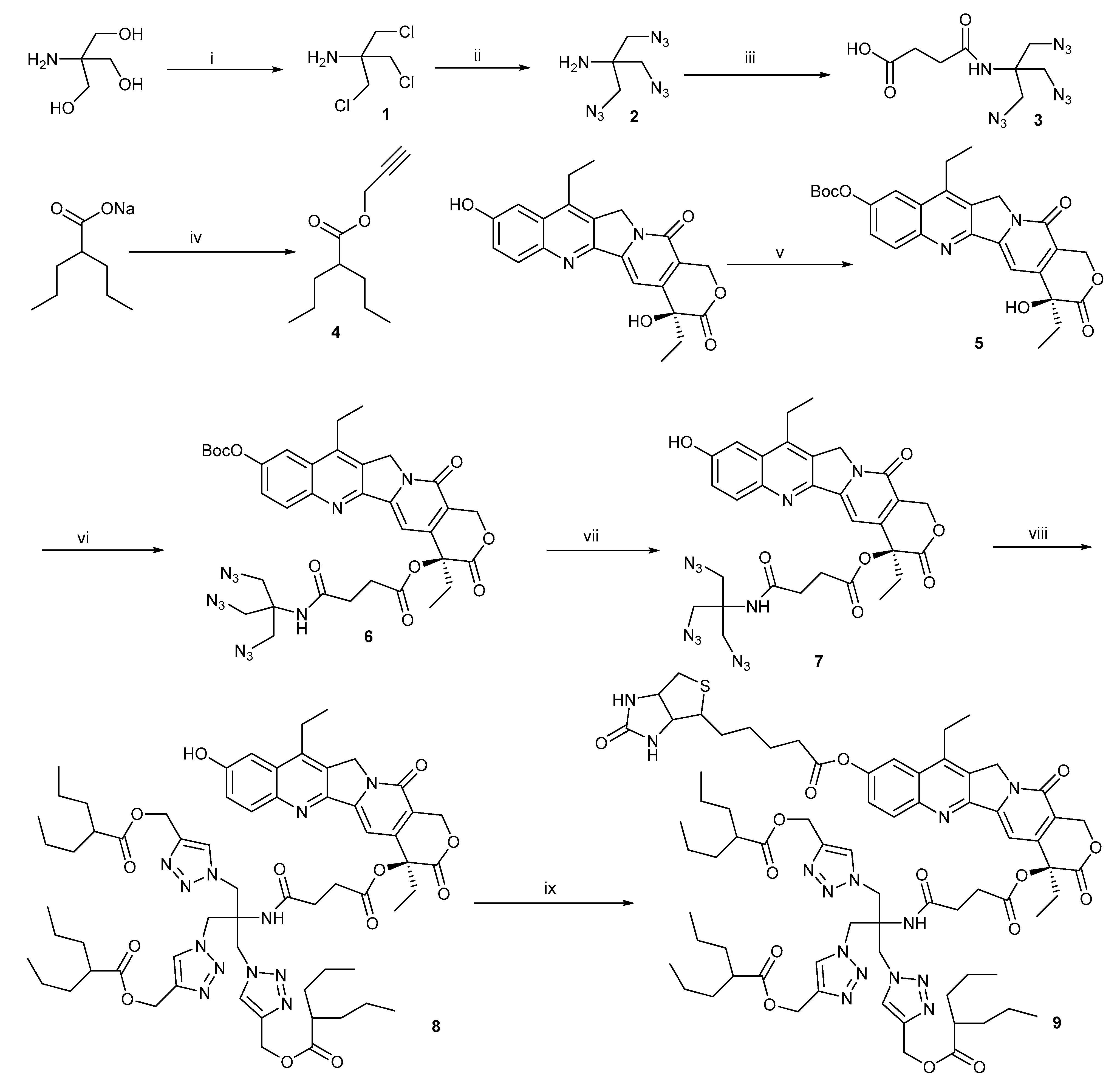
Biotin receptor was overexpressed in various tumor cells[41,42]. Incorporation of biotin in chemotherapeutic agents was good strategy for enhanced selectivity to cells[43,44,45]. The designed compound 9 was expected to possess multi-function, with one biotin molecule on one end to target cancer cells and three releasable valproic acid molecules on the other end to inhibit HDAC. Here, we chose a biotin receptor-positive cancer cell line (HeLa) and a negative normal cell line (NIH3T3) to investigate cytotoxicity and selectivity to cells of compound 9. The half maximal inhibitory concentrations (IC50) indicated that compound 9 was comparable to irinotecan and weaker than SN38 towards HeLa cells (Figure 1). The reason why the cytotoxicity of compound 9 was inferior to SN38 might be that compound 9 only partly released SN38 and valproic acid. Fortunately, compound 9 exhibited lower cytotoxicity than SN38 and irinotecan toward NIH3T3 cells, demonstrating that biotin moiety in compound 9 worked (Figure 1). The survival rate of NIH3T3 cells in compound 9 group was above 60% at the dose of 50 µM, suggesting that compound 9 had the potential of reducing side effects. Moreover, the IC50 values of SN38 and irinotecan to NIH3T3 cells were both under 12.50 µM. There was not different in cytotoxicity for compound 8 toward HeLa cells and NIH3T3 cells at dose of 50 µM, as shown in Figure S15. In addition, valproic acid and biotin both showed only marginal cytotoxicity against HeLa cells and NIN3T3 cells (Figure S16). These findings demonstrated that combination of SN38 with valproic acid and biotin not only retained anticancer activity similar to irinotecan, but also enhanced the selectivity to cells.
2.2.2. Mitochondrial Membrane Potential Analysis
Apoptosis is a major pathway of camptothecin analogues to inhibit the growth of cancer cells[46,47,48]. Mitochondria plays the key role in apoptosis of cells. The decreased mitochondrial membrane potential (∆Ψm) is a hallmark event in the early stage of cell apoptosis. The cell mitochondrial membrane potential can be detected by using JC-1 staining method. JC-1 can aggregate in normal mitochondria and present red fluorescence. Once the ∆Ψm decrease, JC-1 present green fluorescence due to dispersion of aggregates. The smaller the ratio of red and green fluorescence is, the more the ∆Ψm drops. The results of JC-1 staining method showed that compared with control, valproic acid hardly decreased ∆Ψm at dose of 6 µM while SN38, irinotecan and compound 9 decreased significantly ∆Ψm at dose of 2 µM (Figure 2, Figure S17). The ability of compound 9 to decrease ∆Ψm was comparable to that of irinotecan and weaker than that of SN38, consistent with the results of the cytotoxicity assay. These finding demonstrated that compound 9 inhibited the growth of cancer cells through the same mitochondria-mediated apoptosis as other camptothecin derivatives.
2.2.3. Apoptosis Study
Based on the above findings about decrease of mitochondrial membrane potential caused by compound 9, apoptosis induced by compound 9 was analyzed using flow cytometry. As shown in Figure 3, valproic acid and biotin at dose of 2 µM did not effectively induce apoptosis. Compared with control group, the percentage of apoptotic cells in valproic acid group and biotin group was negligible (< 8%). SN38 still exhibited the strongest ability to induce apoptosis among the three tested camptothecin derivatives and induced 76.6% apoptotic rate toward HeLa cells. Though compound 9 could not induce strong apoptosis like SN38, it also induced 44.7% apoptotic rate toward HeLa cells. Similarly, there were no significantly different in apoptosis induced by compound 9 and irinotecan, consistent with the cell proliferation assay findings. The research on inducing apoptosis showed compound 9 exerted anti-tumor effect mainly by inducing apoptosis of cancer cells.
3. Materials and Methods
SN38 and valproic acid were purchased from Energy Chemical Co., Ltd. Other chemical reagents were obtained from Sinopharm Chemical Reagent Co.,Ltd. Compound 9 was dissolved in 0.05% DMSO. Thin-layer chromatography on 0.25 mm silicon gel plates (GF254) were purchased from Energy Chemical Co., Ltd. DEME culture medium was purchased from Hyclone Co.,Ltd, FBS was purchased from Gibco Co.,Ltd. MTT, JC-1 probe and apoptosis detection kits were purchased from Biyuntian Co.,Ltd. Murine fibroblast NIH3T3 cells and human cervical cancer HeLa cells were purchased from ATCC of US. NMR spectra were recorded on Bruker DRX-300 NMR and Bruker DRX-400 NMR. Mass spectra (MS) were obtained using an Agilent LC-MS (ESI). Fluorescence imaging was observed on Olympus IX71 fluorescence microscope. Apoptosis was analyzed on CytoFLEX flow cytometry.
3.1. Synthesis of Compound 9
3.1.1. The Synthetic Route of Compound 1
Compound 1 was synthesize as described in the literature[49]. In brief, Tris (15 g, 0.124 mol) and SOCl2 (39 mL, 0.519 mol) were placed in 250 mL flask and stirred at 0°C. After pyridine (5 mL,0.062mol) was added dropwise to reaction solution, the reaction temperature was raised slowly to 100℃. Reaction solution was stirred at 100°C for 5 h, followed by another 1 h of reaction at 120°C. Then, the reaction solution was cooled to 20°C and treated with 30 mL H2O. The pH value of aqueous solution was adjusted to 10 with sodium carbonate solution. The obtained aqueous solution was extracted with dichloromethane for three times. The organic layer was dried with anhydrous sodium sulfate. After removal of dichloromethane in vacuo, the obtained mixture was purified using silica gel chromatograph with petroleum ether as elution solvent. The pure compound 1 was a colorless liquid with the yield of 63%. 1H-NMR (300 MHz, Chloroform-d) δ 3.67 (s, 6H), 1.73 (s, 2H).
3.1.2. The Synthetic Route of Compound 2
Compound 2 was synthesized as described in the literature[50]. Compound 1 (3 g, 0.017 mol) and NaN3 (3.65 g, 0.056 mol) were dissolved in 25 mL DMSO and stirred for 24 h at 90°C. Then the reaction solution was cooled to 20°C and poured into 250 mL H2O. The obtained solution was extracted with diethyl ether (3×80 mL). The organic layer was washed with brine and H2O and dried with anhydrous magnesium sulphate. After removal of diethyl ether in vacuo, compound 2 was obtained as a colorless oil with the yield of 93%. 1H-NMR (300 MHz, Chloroform-d) δ 3.34 (s, 6H), 1.53 (s, 2H).
3.1.3. The Synthetic Route of Compound 3
To a solution of compound 2 (1.57 g, 8 mmol) in 10 mL dichloromethane, triethylamine (1 mL) and succinic anhydride (0.8 g, 8 mmol) were added to react at room temperature. The reaction process was monitored using thin layer chromatography (TLC). After the reaction was completed, the reaction solution was washed with saturated KHSO4 and brine. The organic layer was dried with anhydrous magnesium sulphate. After removal of dichloromethane in vacuo, compound 3 was obtained as a white solid with the yield of 88%. 1H-NMR (300 MHz, Chloroform-d) δ 5.91 (s, 1H), 3.70 (s, 6H), 2.71 (t, J=6.6Hz, 2H), 2.52 (t, J=6.6Hz, 2H).
3.1.4. The Synthetic Route of Compound 4
Compound 4 was synthesized in according with the literature[51]. Sodium valproate (6.64 g, 40 mmol), propargyl bromide (7.14 g, 60 mmol) and K2CO3 (1.6 g, 12 mmol) were dissolved in 25 mL DMF and stirred at room temperature. The reaction process was monitored by TLC. After the reaction was completed, the reaction solution was poured into 100 mL H2O and extracted with ethyl acetate. The organic layer was washed with brine and H2O respectively, and dried with anhydrous sodium sulfate. After removal of ethyl acetate in vacuo, compound 4 was obtained as a white solid with the yield of 86%. 1H-NMR (400 MHz, Chloroform-d) δ 4.68 (d, J = 2.5 Hz, 2H), 2.48 (t, J = 2.5 Hz, 1H), 2.45 – 2.35 (m, 1H), 1.72 – 1.53 (m, 2H), 1.50 – 1.38 (m, 2H), 1.37 – 1.19 (m, 4H), 0.90 (t, J = 7.3 Hz, 6H).
3.1.5. The Synthetic Route of Compound 5
Compound 5 was synthesized as described in the literature[52]. The mixture of SN38 (1.00 g, 2.50 mmol), (Boc)2O (1.00 g, 4.50 mmol), pyridine (5 mL) and dichloromethane (50 mL) was stirred in a flask at room temperature. The reaction process was monitored using TLC. After the reaction was completed, the reaction solution was washed for three times with 1% HCl and water respectively. The resulting dichloromethane fraction was dried with anhydrous magnesium sulfate, filtered and concentrated in vacuum to obtain compound 5, a faint yellow solid, with the yield of 86%. 1H-NMR (300 MHz, Chloroform-d) δ 8.26 (d, J = 9.2 Hz, 1H), 7.91 (d, J = 2.5 Hz, 1H), 7.78 – 7.56 (m, 2H), 5.77 (d, J = 16.4 Hz, 1H), 5.44 – 5.16 (m, 3H), 3.84 (s, 1H), 3.18 (q, J = 7.6 Hz, 2H), 2.07 – 1.80 (m, 2H), 1.63 (s, 9H), 1.42 (t, J = 7.6 Hz, 3H), 1.05 (t, J = 7.4 Hz, 3H). Analytical data were in suitable accordance with the reported data[52]. Judging from the signal of 1.57ppm in NMR, some Boc-OH existed in the product. Since Boc-OH had difficulty in reacting with compound 5 due to its high steric hindrance, compound 5 was directly used in the next step, without further purification.
3.1.6. The Synthetic Route of Compound 6 and 7
Compound 6 and 7 were synthesized by the method reported in the literature[53].Compound 3 (355 mg, 1.2 mmol), compound 5 (518 mg, 1.0 mmol), EDCI (229 mg, 1.2 mmol) and DMAP (15 mg,0.12 mmol) were dissolved in 60 mL dichloromethane and stirred at room temperature. The reaction process was monitored using TLC. After the reaction was over, the reaction solution was washed for three times with 1% HCl and brine respectively. The resulting dichloromethane fraction was dried with anhydrous sodium sulfate, filtered and concentrated in vacuum to obtain compound 6, a faint yellow solid, with the yield of 73%. The resulted compound 6 without any purification and trifluoroacetic acid (9 mL) were dissolved 20 mL dichloromethane and stirred at 30°C to remove Boc protecting group. The reaction process was monitored using TLC. After the reaction was finished, the dichloromethane layer was washed with saturated NaHCO3, brine and H2O respectively. Then organic layer was dried with anhydrous sodium sulfate. After removal of dichloromethane in vacuo, compound 7 was obtained as a faint yellow solid with the yield of 82%. Compound 7 was directly used in the next step, without further purification. 1H-NMR (400 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.44 (d, J = 10.0 Hz, 1H), 7.38 (d, J = 2.4 Hz, 1H), 7.26 (s, 1H), 5.71 (d, J = 17.0 Hz, 2H), 5.39 (d, J = 17.0 Hz, 1H), 5.19 (s, 1H), 3.67 – 3.44 (m, 6H), 3.03 (m, 2H), 2.89 (m, 2H), 2.66 – 2.47 (m, 2H), 2.30 – 2.05 (m, 2H), 1.33 (t, J = 7.7 Hz, 3H), 1.01 (t, J = 7.5 Hz, 3H).
3.1.7. The Synthetic Route of Compound 8
To a solution of compound 4 (300 mg, 1.65 mmol) in t-BuOH/H2O mixture (2:1, 20 mL), sodium ascorbate (594 mg, 0.3 mmol) and CuSO4 (750 mg, 0.3 mmol) were added and stirred at room temperature and under argon atmosphere. When this mixture was yellow, compound 7 (268 mg, 0.4 mmol) was added and stirred at 40 °C. The reaction process was monitored by TLC. After the reaction was over, t-BuOH was removed in vacuum and the residue was lyophilized to prepare the crude compound 8. Then the crude compound 8 was purified using silica gel column chromatography. The final compound 8 was a faint yellow solid, with the yield of 52%. HPLC/Purity: 97.3% (tR = 7.056), 1H-NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 8.39 (s, 1H), 8.01 (s, 2H), 7.93 (d, J= 8.9 Hz, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.34 (t, J = 13.5 Hz, 2H), 7.10 (s, 1H), 5.50 (s, 2H), 5.24 (d, J = 17.0 Hz, 2H), 5.07 (m, 6H), 4.52 (q, J = 8.8 Hz, J = 14.7 Hz, 6H), 3.14 – 2.98 (m, 2H), 2.89 – 2.60 (m, 2H), 2.40 – 2.23 (m, 4H), 2.15 (m, 2H), 1.55 – 1.21 (m,15H), 1.20 – 1.07 (m, 13H), 0.95 (t, J = 7.1 Hz, 3H), 0.75 (t, J = 7.2 Hz, 18H); 13C NMR (101 MHz, DMSO-d6) δ 175.63 , 173.14 , 172.12 , 167.83 , 157.23 , 157.05 , 149.05 , 147.15 , 146.19 , 144.01 , 143.19 , 142.15 , 131.76 , 128.59 , 128.21 , 127.29 , 122.88 , 118.04 , 105.17 , 94.92 , 76.61 , 66.61 , 59.39 , 57.10 , 49.76 , 49.06 , 44.69 , 34.50 , 34.32 , 30.58 , 28.73 , 22.71 , 20.41 , 14.19 , 13.80 , 8.10; ESI-MS: m/z: calcd.1239.62 ([M+Na]+), 1255.59 ([M+K]+); found 1239.70 [M+Na]+, 1255.65 [M+K]+. The peak at 8.39 ppm (s, 1H) showed active hydrogen on amide bond. Due to hydrogen exchange, the integration of active hydrogen was insufficient. In addition, peak splitting of hydrogen seemed to be irregular perhaps due to the different 3D structures caused by these crowded repeating units.
3.1.8. The Synthetic Route of Compound 9
Compound 8 (1.22 g, 1 mmol), biotin (44.8 mg, 2 mmol), EDCI (0.488 g, 2.5 mmol) and DMAP (305 mg, 2.5 mmol) were dissolved in 10 mL DMF, and stirred overnight at 25 °C. After the reaction was completed, 100 mL ethyl acetate was added to obtain light yellow solution. The resulted solution was washed with brine for three times and dried with anhydrous sodium sulfate. After removal of ethyl acetate in vacuo, the crude product was obtained. Then the crude product was purified with silica gel column chromatography to obtain light yellow compound 9 with the yield of 43%. HPLC/Purity: 99.2% (tR = 13.359).1H-NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.23 (s, 1H), 8.21 (s, 1H), 8.13 (d, J = 9.1 Hz, 1H), 8.04 (s, 2H), 7.97 (d, J = 2.0 Hz, 1H), 7.80 (d, J = 4.8Hz, 1H), 7.60 (dt, J = 7.8 Hz, J = 2.3 Hz, 1H), 7.21 (d, J = 17.1 Hz, 1H), 6.51 (s, 1H), 6.41 (s, 1H), 5.52 (s, 2H), 5.37 – 5.16 (m, 4H), 5.10 (m, 5H), 4.98 -4.77 (m, 1H), 4.62 – 4.48 (m, 4H), 4.40 – 4.29 (m, 1H), 4.23 – 4.13 (m, 1H), 3.22 – 3.10 (m, 3H), 2.86 (dd, J = 12.5, 5.0 Hz, 2H), 2.70 (t, J = 7.2 Hz, 3H), 2.61 (d, J = 12.4 Hz, 1H), 2.51 (dt, J = 8.9, 3.8 Hz, 2H), 2.38 – 2.24 (m, 4H), 2.22 – 2.09 (m, 2H), 1.73-1.25 (m, 3H), 1.63 – 1.21(m, 17H), 1.20 – 1.06 (m, 12H), 1.01 – 0.89 (m, 3H), 0.83 – 0.63 (m, 18H). 13C NMR (101 MHz, DMSO-d6) δ 175.64 , 175.33 , 173.16 , 172.26 , 172.14 , 167.75 , 163.21 , 156.99 , 152.17 , 149.68 , 146.86 , 146.61 , 146.15 , 145.77 ,142.17 , 141.75 , 11.50 , 128.76 , 127.48 , 127.29 , 126.14 , 124.73 , 119.05 , 115.67 , 95.77, 76.56 , 66.64 , 62.89 , 61.51 , 59.70 , 59.40 , 57.12 , 55.82 , 49.87 , 49.10 ,44.71 , 34.51 , 34.47, 34.33, 33.85, 30.61, 28.50, 28.45, 24.78, 22.68, 20.43, 20.36, 14.10, 8.11; ESI-MS: m/z: calcd.1465.69 ([M+Na]+), 1481.67 ([M+K]+); found 1465.80 [M+Na]+, 1481.80 [M+K]+. The peaks at 8.40 ppm (s, 1H),8.23 (s, 1H) and 8.21 (s, 1H) showed active hydrogens on amide bonds. Due to hydrogen exchange, the integration of active hydrogens was insufficient. In addition, peak splitting of hydrogen seemed to be irregular perhaps due to the different 3D structures caused by these crowded repeating units.
3.2. In Vitro Biological evaluation
3.2.1. In vitro cytotoxicity assay
Cells were dispersed in the DMEM containing 10% FBS and seeded in 96-well plates at the density of 3000 cells/well. After 12-hour incubation at 37 °C in 5% CO2, cells were treated with 100 µL fresh medium containing varying concentrations of drugs and co-incubated for 48 h. Then the medium containing drugs was replaced with fresh culture medium containing 1 mg/mL MTT, followed by the incubation for 4 h. Finally, the medium containing MTT was removed completely and 150 µL of DMSO was added to very well. After shake for 10 min at 37 °C, the absorbance was measured at 490 nm using a Bio-Rad 680 microplate reader. The IC50 values were calculated using GraphPad Prism software (version 5.01) based on data from three parallel experiments.
3.2.2. Mitochondrial membrane potentials assay
Mitochondrial membrane potentials were assayed in accordance with the instructions of JC-1 Kit. Briefly, HeLa cells were seeded in 6-well plates at the density of 50,000 cells per well and incubated overnight at 37 °C in 5%CO2. Next day, the medium was replaced with fresh medium containing 2 µM of these tested compounds and the cells were co-incubated with these compounds for 24 h. After the medium containing drugs was removed, the cells were rinsed three times with PBS and treated with 1mL JC-1 staining solution for further incubation of 20 min. Then, the cells were washed with buffers provided in JC-1 kit for two times followed by the treatment with 1 mL serum-free medium. Fluorescence imaging of HeLa cells were monitored by determining fluorescent emissions from mitochondrial JC-1 monomers or aggregates using an Olympus fluorescence microscope. Mitochondrial membrane potentials were analyzed through fluorescence intensity.
3.2.3. Cell apoptosis assay
Cell apoptosis were assayed in accordance with the instructions of Annexin V-FITC Apoptosis Detection Kit. Briefly, HeLa cells were seeded in 6-well plates at the density of 100,000 cells per well and incubated overnight at 37 °C in 5%CO2. Next day, cells were treated with 2 µM of these tested compounds. After 48 h of incubation, the medium was harvested and the cells were washed with PBS, followed by trypsinization. Then the cells were resuspended in the resulted medium and centrifugated to harvest these cells. Finally, the cells were processed as described in the AnnexinV-FITC apoptosis detection kit (Beyotime Biotechnology, Nanjing, China). The samples were assayed with CytoFLEX flow cytometer (Beckman Coulter).
3.2.4. Statistical Analysis
All data are presented as means ±SD for three independent experiments. IC 50 value was calculated by GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA).
4. Conclusions
In this study, a novel biotin-SN38-valproic acid conjugate, compound 9 was synthesized via click reaction. Compound 9 was a multifunctional prodrug due to linker of ester between biotin, SN38 and valproic acid. Introduction of valproic acid at C20-OH of SN38 enhanced the stability of E-ring to diminish the inactivation of SN38 while attaching SN38 to biotin could efficiently deliver SN38 into tumor tissues to reduce the harm to normal cells. . The results of MTT assay, mitochondria membrane potentials assay and apoptosis detection all showed that compound 9 exhibited excellent anticancer activity comparable to that of irinotecan. Moreover, compound 9 had lower cytotoxicity toward normal cells, indicating that compound 9 possessed good selectivity for cancer cells. These findings indicated that combination of SN38 with biotin and valproic acid was an effective strategy to design novel camptothecin-based therapeutic agents.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1-S12: NMR and ESI-MS spectrum of compound 1-9; Figure S13: Effects of biotin on cell viability; Figure S14: Effects of the tested compounds on mitochondrial membrane potentials.
Author Contributions
Conceptualization, Y.D. and Y.Z.; methodology, Y.C. and T.Y.; software, Y.D.; validation, Y.Z., Y.D. and Y.C.; formal analysis, Y.Z.; investigation, Y.C and T.Y.; resources, Y.Z.; data curation, Y.Z.; writing—original draft preparation, Y.D.; writing—review and editing, Y.D.; visualization, T.Y.; supervision, Y.D.; project administration, Y.D.; funding acquisition, Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are shown in this paper.
Acknowledgments
This work was supported by Natural Science Research Foundation of the Department of Educa-tion of Anhui Province (No. KJ2021A1166; KJ2019A0873), Scientific Research Team of Anui Xinhua University (No. kytd202211), Pharmaceutical Institute of Anui Xinhua University (No. yjs202107), the National Innovation and Entrepreneurship Training Program for College Students (No. 202012216048) and the Innovation and Entrepreneurship Training Program for College Students in Anhui Province (No. 201912216193).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martino, E.; Della Volpe, S.; Terribile, E.; Benetti, E.; Sakaj, M.; Centamore, A.; Sala, A.; Collina, S. The long story of camptothecin: From traditional medicine to drugs. Bioorganic & medicinal chemistry letters 2017, 27, 701–707. [Google Scholar] [CrossRef]
- Bocian, W.; Naumczuk, B.; Urbanowicz, M.; Sitkowski, J.; Bierczynska-Krzysik, A.; Bednarek, E.; Wiktorska, K.; Milczarek, M.; Kozerski, L. The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Martin-Encinas, E.; Selas, A.; Palacios, F.; Alonso, C. The design and discovery of topoisomerase I inhibitors as anticancer therapies. Expert Opinion on Drug Discovery 2022, 17, 581–601. [Google Scholar] [CrossRef] [PubMed]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Khaiwa, N.; Maarouf, N.R.; Darwish, M.H.; Alhamad, D.W.M.; Sebastian, A.; Hamad, M.; Omar, H.A.; Orive, G.; Al-Tel, T.H. Camptothecin's journey from discovery to WHO Essential Medicine: Fifty years of promise. Eur J Med Chem 2021, 223, 113639. [Google Scholar] [CrossRef] [PubMed]
- Cronin, A.; Barnes, J.; Pedersen, J.; Heathcote, D.; Collins, T. Mitigating exacerbation of Irinotecan-induced gastrointestinal toxicity in combination with an ATM inhibitor in the rat. Toxicology Letters 2016, 258. [Google Scholar] [CrossRef]
- Venditto, V.J.; Simanek, E.E. Cancer Therapies Utilizing the Camptothecins: A Review of the in Vivo Literature. Molecular pharmaceutics 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Bala, V.; Rao, S.; Li, P.; Wang, S.; Prestidge, C.A. Lipophilic Prodrugs of SN38: Synthesis and in Vitro Characterization toward Oral Chemotherapy. Molecular pharmaceutics 2016, 13, 287–294. [Google Scholar] [CrossRef]
- Shi, L.; Wu, X.; Li, T.; Wu, Y.; Song, L.; Zhang, W.; Yin, L.; Wu, Y.; Han, W.; Yang, Y. An esterase-activatable prodrug formulated liposome strategy: potentiating the anticancer therapeutic efficacy and drug safety. Nanoscale Advances 2022, 4, 952–966. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, X.; Wang, H.; Liu, X.; Zhang, Q.; Li, K.; Chen, Y.; Zhu, Q.; Shen, Y.; Sui, M. A nanotherapeutic strategy to overcome chemoresistance to irinotecan/7-ethyl-10-hydroxy-camptothecin in colorectal cancer. Acta Biomater 2022, 137, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Zhao, H.Y.; Lei, H.; Yuan, B.; Mao, S.; Xin, M.; Zhang, S.Q. Synthesis and Biological Evaluation of 10-Substituted Camptothecin Derivatives with Improved Water Solubility and Activity. ChemMedChem 2021, 16, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xie, F.; Xiao, D.; Xu, X.; Su, Z.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Synthesis and evaluation of highly releasable and structurally stable antibody-SN-38-conjugates. Drug Deliv 2021, 28, 2603–2617. [Google Scholar] [CrossRef]
- Zhu, S.; Shen, Q.; Gao, Y.; Wang, L.; Fang, Y.; Chen, Y.; Lu, W. Design, Synthesis, and Biological Evaluation of HSP90 Inhibitor-SN38 Conjugates for Targeted Drug Accumulation. Journal of medicinal chemistry 2020, 63, 5421–5441. [Google Scholar] [CrossRef]
- Botta, L.; Filippi, S.; Zippilli, C.; Cesarini, S.; Bizzarri, B.M.; Cirigliano, A.; Rinaldi, T.; Paiardini, A.; Fiorucci, D.; Saladino, R.; et al. Artemisinin Derivatives with Antimelanoma Activity Show Inhibitory Effect against Human DNA Topoisomerase 1. ACS medicinal chemistry letters 2020, 11, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Autin, P.; Blanquart, C.; Fradin, D. Epigenetic Drugs for Cancer and microRNAs: A Focus on Histone Deacetylase Inhibitors. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef]
- Molife, L.R.; de Bono, J.S. Belinostat: clinical applications in solid tumors and lymphoma. Expert Opinion on Investigational Drugs 2011, 20, 1723–1732. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Panobinostat: First Global Approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, C.H.; Loong, H.H.F. Synergistic activities of the histone deacetylase inhibitors with conventional cytotoxic chemotherapies in angiosarcomas. Invest New Drugs 2022, 40, 868–869. [Google Scholar] [CrossRef]
- Cincinelli, R.; Musso, L.; Artali, R.; Guglielmi, M.B.; La Porta, I.; Melito, C.; Colelli, F.; Cardile, F.; Signorino, G.; Fucci, A.; et al. Hybrid topoisomerase I and HDAC inhibitors as dual action anticancer agents. PLoS One 2018, 13, e0205018. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Miyanishi, K.; Kubo, T.; Hamaguchi, K.; Osuga, T.; Tanaka, S.; Ohnuma, H.; Murase, K.; Takada, K.; Nagayama, M.; et al. Synergistic antitumor effect of histone deacetylase class IIa inhibitor with lenvatinib in hepatocellular carcinoma. Hepatol Int, 2023. [Google Scholar] [CrossRef]
- Ovejero-Sanchez, M.; Asensio-Juarez, G.; Gonzalez, M.; Puebla, P.; Vicente-Manzanares, M.; Pelaez, R.; Gonzalez-Sarmiento, R.; Herrero, A.B. Panobinostat Synergistically Enhances the Cytotoxicity of Microtubule Destabilizing Drugs in Ovarian Cancer Cells. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yu, X.; Shen, Q.; Zhang, Q.; Su, M.; Zhou, Y.; Li, J.; Chen, Y.; Lu, W. A series of camptothecin prodrugs exhibit HDAC inhibition activity. Bioorganic & medicinal chemistry 2018, 26, 4706–4715. [Google Scholar] [CrossRef]
- Chen, J.; Wang, G.Y.; Wang, L.B.; Kang, J.H.; Wang, J.M. Curcumin p38-dependently enhances the anticancer activity of valproic acid in human leukemia cells. European Journal of Pharmaceutical Sciences 2010, 41, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.T.; Lai, H.C.; Lee, H.Y.; Lin, W.H.; Chang, C.C.; Chu, T.Y.; Lin, Y.W.; Lee, K.D.; Yu, M.H. Valproic acid resensitizes cisplatin-resistant ovarian cancer cells. Cancer Science 2008, 99, 1218–1226. [Google Scholar] [CrossRef]
- Luna-Palencia, G.R.; Correa-Basurto, J.; Vasquez-Moctezuma, I. Valproic acid as a sensitizing agent for cancer treatment. Gaceta Medica De Mexico 2019, 155, 417–422. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. Profound Inhibitory and Apoptotic Effects of Histone Deacetylase Inhibitor Valproic Acid on Different Cancers. Crescent Journal of Medical and Biological Sciences 2019, 6, 441–448. [Google Scholar]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, L.; Cheng, Z.; Yang, B.; Yu, J.; Chen, Y.; Lu, W. SN38-based albumin-binding prodrug for efficient targeted cancer chemotherapy. J Control Release 2021, 339, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Che, R.; Lu, W. Enhanced cellular uptake efficiency of DCM probes or SN38 conjugating with phenylboronic acids. Bioorganic & medicinal chemistry 2020, 28, 115377. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, J.; Jin, X.; Liu, L.; Tian, X. Folate Receptor Targeting and Cathepsin B-Sensitive Drug Delivery System for Selective Cancer Cell Death and Imaging. ACS medicinal chemistry letters 2020, 11, 1514–1520. [Google Scholar] [CrossRef]
- Kim, H.; Hwang, D.; Choi, M.; Lee, S.; Kang, S.; Lee, Y.; Kim, S.; Chung, J.; Jon, S. Antibody-Assisted Delivery of a Peptide-Drug Conjugate for Targeted Cancer Therapy. Molecular pharmaceutics 2019, 16, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulou, A.; Zografos, E.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. Trastuzumab Deruxtecan (DS-8201a): The Latest Research and Advances in Breast Cancer. Clin Breast Cancer 2021, 21, e212–e219. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.; Bobba, K.N.; Cho, M.Y.; Park, H.S.; Won, M.; Velusamy, N.; Hong, K.S.; Bhuniya, S.; Kim, J.S. Molecular Theranostic Agent with Programmed Activation for Hypoxic Tumors. ACS Applied Bio Materials 2019, 2, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Jangili, P.; Won, M.; Kim, S.J.; Chun, J.; Shim, I.; Kang, C.; Ren, W.X.; Kim, J.S. Binary Drug Reinforced First Small-Molecule-Based Prodrug for Synergistic Anticancer Effects. ACS Applied Bio Materials 2019, 2, 3532–3539. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Xu, X.; Chen, J.; Li, L.; Wang, J.; Fang, T.; Zhou, L.; Wang, H.; Zheng, S. Rational design of multifunctional small-molecule prodrugs for simultaneous suppression of cancer cell growth and metastasis in vitro and in vivo. Chemical communications 2016, 52, 5601–5604. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, J.; Xie, K.; Fang, T.; Chen, C.; Xie, H.; Zhou, L.; Zheng, S. Precise Engineering of Prodrug Cocktails into Single Polymeric Nanoparticles for Combination Cancer Therapy: Extended and Sequentially Controllable Drug Release. ACS Appl Mater Interfaces 2017, 9, 10567–10576. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, W.; He, R.; Ismail, M.; Ling, L.; Yao, C.; Fu, Z.; Li, X. Dual 7-ethyl-10-hydroxycamptothecin conjugated phospholipid prodrug assembled liposomes with in vitro anticancer effects. Bioorganic & medicinal chemistry 2017, 25, 3247–3258. [Google Scholar] [CrossRef]
- de Groot, F.; Busscher, G.; Aben, R.; Scheeren, H. Novel 20-carbonate linked prodrugs of camptothecin and 9-aminocamptothecin designed for activation by tumour-associated plasmin. Bioorganic & medicinal chemistry letters 2002, 12, 2371–2376. [Google Scholar]
- Fam, K.T.; Collot, M.; Klymchenko, A.S. Probing biotin receptors in cancer cells with rationally designed fluorogenic squaraine dimers. Chemical Science 2020, 11, 8240–8248. [Google Scholar] [CrossRef]
- Pei, X.Y.; Huo, F.J.; Yue, Y.K.; Chen, T.G.; Yin, C.X. Cancer cell recognition by a Cys-reactive turn-on significant enhanced fluorescent emission targeting biotin receptors. Sensors and Actuators B-Chemical 2020, 304. [Google Scholar] [CrossRef]
- Maiti, S.; Paira, P. Biotin conjugated organic molecules and proteins for cancer therapy: A review. European Journal of Medicinal Chemistry 2018, 145, 206–223. [Google Scholar] [CrossRef]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Potent Platinum(IV) Prodrugs That Incorporate a Biotin Moiety to Selectively Target Cancer Cells. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Perumal, D.; Golla, M.; Pillai, K.S.; Raj, G.; Krishna, P.K.A.; Varghese, R. Biotin-decorated NIR-absorbing nanosheets for targeted photodynamic cancer therapy. Organic & biomolecular chemistry 2021, 19, 2804–2810. [Google Scholar] [CrossRef]
- Zhang, R.M.; Luo, Y.; Du, C.H.; Wu, L.; Wang, Y.K.; Chen, Y.D.; Li, S.Q.; Jiang, X.; Xie, Y.M. Synthesis and biological evaluation of novel SN38-glucose conjugate for colorectal cancer treatment. Bioorganic & medicinal chemistry letters 2023, 81. [Google Scholar] [CrossRef]
- Zhang, M.; Fu, W.; Zhu, L.-Z.; Liu, X.-F.; Li, L.; Peng, L.-Z.; Kai, G.-Y.; Liu, Y.-Q.; Zhang, Z.-J.; Xu, C.-R. Anti-tumor effects and mechanism of a novel camptothecin derivative YCJ100. Life sciences 2022, 311, 121105. [Google Scholar] [CrossRef]
- Zhang, G.R.; Yin, R.J.; Dai, X.F.; Wu, G.Z.; Qi, X.; Li, J.; Jiang, T. Design, synthesis, and biological evaluation of novel 7-substituted 10,11-methylenedioxy-camptothecin derivatives against drug-resistant small-cell lung cancer in vitro and in vivo. European Journal of Medicinal Chemistry 2022, 241. [Google Scholar] [CrossRef]
- Huang, D.; Liu, Q.; Zhang, M.; Guo, Y.; Cui, Z.; Li, T.; Luo, D.; Xu, B.; Huang, C.; Guo, J.; et al. A Mitochondria-Targeted Phenylbutyric Acid Prodrug Confers Drastically Improved Anticancer Activities. Journal of medicinal chemistry 2022, 65, 9955–9973. [Google Scholar] [CrossRef]
- Joo, Y.H.; Shreeve, J.M. 1,3-Diazido-2-(azidomethyl)-2-propylammonium salts. Inorg Chem 2009, 48, 8431–8438. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Ghosh, A.K.; Hajra, A. Late-stage ortho-C-H alkenylation of 2-arylindazoles in aqueous medium by Manganese(i)-catalysis. RSC Adv 2022, 12, 19412–19416. [Google Scholar] [CrossRef]
- Liu, S.; Hu, Z.; Zhang, Q.; Zhu, Q.; Chen, Y.; Lu, W. Co-Prodrugs of 7-Ethyl-10-hydroxycamptothecin and Vorinostat with in Vitro Hydrolysis and Anticancer Effects. ACS Omega 2019, 5, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, Y.; Kang, S.; Choi, M.; Lee, S.; Kim, S.; Gujrati, V.; Kim, J.; Jon, S. Self-assembled nanoparticles comprising aptide-SN38 conjugates for use in targeted cancer therapy. Nanotechnology 2016, 27, 48LT01. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The in vitro cytotoxicity evaluated by MTT assay on HeLa cells and NIH3T3 cells. Cells were co-incubated with different concentrations of compounds for 48 h.
Figure 1.
The in vitro cytotoxicity evaluated by MTT assay on HeLa cells and NIH3T3 cells. Cells were co-incubated with different concentrations of compounds for 48 h.
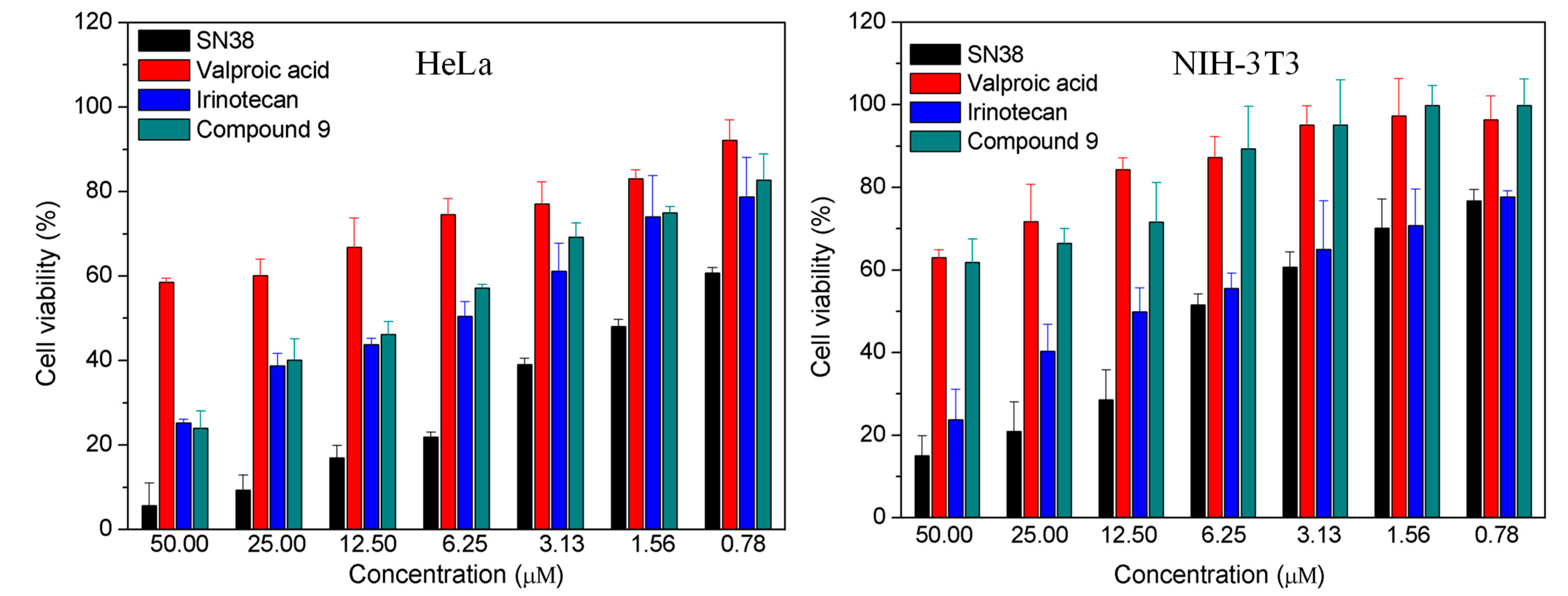
Figure 2.
Mitochondrial membrane potential analyzed by fluorescence microscope. HeLa cells were co-incubated with 6 µM of valproic acid or 2 µM of SN38, irinotecan and compound 9 for 24 h, stained with JC-1 and detected by fluorescence microscope.
Figure 2.
Mitochondrial membrane potential analyzed by fluorescence microscope. HeLa cells were co-incubated with 6 µM of valproic acid or 2 µM of SN38, irinotecan and compound 9 for 24 h, stained with JC-1 and detected by fluorescence microscope.
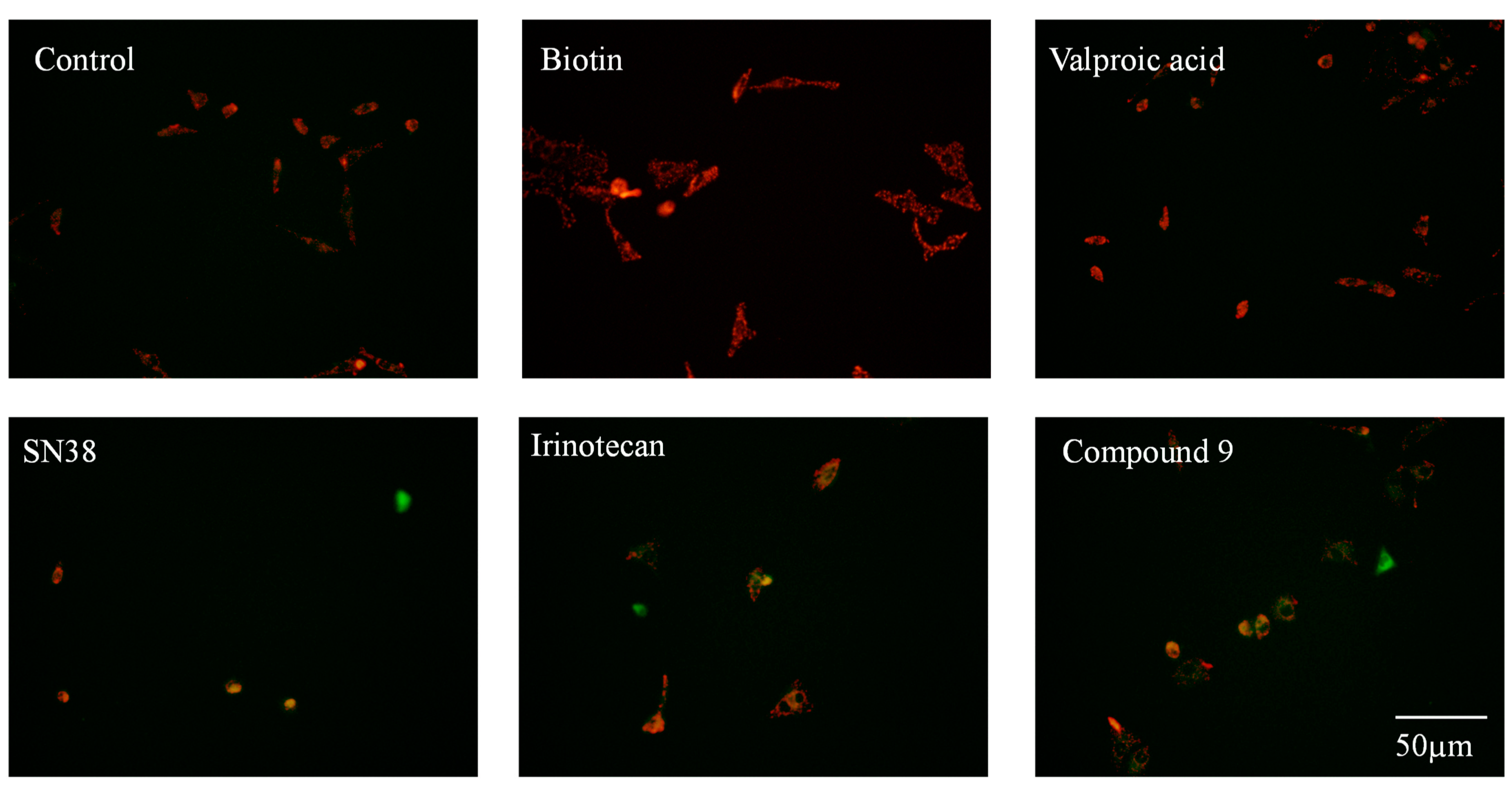
Figure 3.
Cell apoptosis detected by flow cytometry. HeLa cells were co-incubated with 6 µM of valproic acid or 2 µM of SN38, irinotecan and compound 9 for 48 h, stained with Annexin V-FITC/PI and detected by flow cytometry.
Figure 3.
Cell apoptosis detected by flow cytometry. HeLa cells were co-incubated with 6 µM of valproic acid or 2 µM of SN38, irinotecan and compound 9 for 48 h, stained with Annexin V-FITC/PI and detected by flow cytometry.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



