
You are currently viewing a beta version of our website. If you spot anything unusual, kindly let us know.
Preprint
Review
Eradication of Drug-Tolerant Mycobacterium tuberculosis 2022: Where We Stand
Altmetrics
Downloads
133
Views
73
Comments
0
A peer-reviewed article of this preprint also exists.



This version is not peer-reviewed
Abstract
The lungs of tuberculosis (TB) patients contain a spectrum of granulomatous lesions ranging from solid and well vascularized cellular granulomas, to avascular caseous granulomas. In solid granulomas, current therapy kills actively replicating (AR) intracellular bacilli, while in low vascularized caseous granulomas the low oxygen tension stimulates aerobic and microaerophilic AR bacilli to transit into non-replicating (NR), drug-tolerant, extracellular stages. These stages, which do not have genetic mutations and are often referred to as persisters, are difficult to eradicate due to low drug penetration inside caseum and mycobacterial cell walls. The sputum of TB patients contains also viable bacilli called differentially detectable (DD) cells that, unlike persisters, grow in liquid, but not in solid media. This review provides a comprehensive update on drug combinations killing in vitro AR and drug-tolerant bacilli (persisters and DD cells), and sterilizing Mycobacterium tuberculosis-infected BALB/c and caseum-forming C3HeB/FeJ mice. These observations have been important for testing new drug combinations in noninferiority clinical trials, in order to shorten duration of current regimens against TB. In 2022, the World Health Organization, based on one of this trial, supported the use of a 4-month regimen for treatment of drug-susceptible TB as a possible alternative to the current 6-month regimen.
Keywords:
Subject: Biology and Life Sciences - Immunology and Microbiology
1. Introduction
The World Health Organization (WHO) estimated that in 2021 approximately 10.6 million people developed tuberculosis (TB), and about 1.6 million died from the active disease, making TB one of the leading causes of death worldwide [1]. Furthermore, an estimated 1.7 billion people have latent TB infection (LTBI) [2]. The current antibiotic treatment of active, drug-susceptible (DS)-TB, requires administration of a combination therapy for 6 months, including the first-line drugs rifampin (R), isoniazid (H), ethambutol (E) and pyrazinamide (Z) (R-H-E-Z) for 2 months, followed by R and H (R-H) for 4 months. To prevent LTBI reactivation, a long treatment is also used, consisting of at least 6 months of H, or 3 to 4 months of R-H.
The lungs of patients with active TB and LTBI contain a spectrum of granulomatous lesions ranging from solid and well vascularized cellular granulomas, to avascular caseous granulomas [3,4,5]. In these lesions, heterogeneous subpopulations of Mycobacterium tuberculosis (Mtb) cells ranging from actively replicating (AR) to nonreplicating (NR) dormant stages, coexist. In solid granulomas, current therapy kills AR intracellular bacilli inside the macrophages, while in low vascularized caseous granulomas the low oxygen tension stimulates aerobic and microaerophilic AR bacilli to transit into NR, hypoxic, drug-tolerant stages. Hypoxic bacilli use host triacylglycerol and cell wall mycolates to accumulate lipid droplets in lipid-loaded (foamy) macrophages [6,7,8]. The foamy macrophages die via inflammatory and necrotic processes, and release lipid droplets into the hypoxic necrotic core of closed caseous granulomas, which contain extracellular, slowly replicating or NR, phenotypically drug-resistant bacilli. Eradication of these bacilli by current TB therapy is difficult due to the low penetration of drugs inside caseous granulomas and NR Mtb cell walls [4,9]. Tubercle bacilli surviving drug treatment in the absence of genetic mutations inside necrotic foci of caseous granulomas are often referred to as persisters, and are thought to be responsible for the long duration of TB therapy [4]. For unclear reasons, in about 10% of LTBI-patients the closed caseous granulomas expand, fuse with the structures of the bronchial tree, and form cavities in which the caseum liquefies after coming into contact with the air. In the liquefied material, the NR cells rapidly multiply and are released into the airways as a mixture of AR and NR bacilli, which are detected in the sputum of contagious pulmonary TB patients as colony forming units (CFU) on solid media such us Middlebrook 7H10 agar plates and Löwenstein-Jensen slants [10,11].
However, the sputum of drug-untreated patients was found to contain also viable Mtb bacilli that were not detectable as CFU [12,13,14,15,16,17,18]. The number of these viable organisms in the sputum was estimated by a limiting dilution (LD) technique in liquid medium, using the most probable number (LD-MPN) method. These subpopulations were called with various terms, including viable but nonculturable cells (VBNC), differentially culturable TB (DCTB) cells and, perhaps the most recently used term, differentially detectable (DD) cells [16]. Several other bacterial species were found to exist in a VBNC state [19]. When the number of Mtb CFU is subtracted from the number of Mtb cells obtained by the LD-MPN method, the difference is the DD Mtb number [16]. Tubercle bacilli growing in liquid but not on solid media were also found in chronic, murine TB [20]. Some studies showed that DD cells decreased less rapidly than CFU after initiation of TB treatment, suggesting that they are also drug-tolerant cells [15,18].
Overall, these observations on dormant, drug-tolerant bacilli (persister cells and DD cells), are important for understanding the heterogeneous nature of the Mtb infection, and may aid in finding new drugs and in designing shorter drug combination-containing regimens. The goal of this review is to give an overview on the reported in vitro and in vivo combinations eradicating all dormant Mtb stages (persister cells and DD cells), and on their impact on the therapeutic strategies endorsed by the WHO to shorten the microbiological and therapeutical management of DS- and drug-resistant (DR)-TB.
2. Persister cells
2.1. Overview
Persister cells (persisters) were reported in several Gram-positive and Gram-negative bacteria [21] and in many eukaryotes including yeasts (Candida spp) [22] and protozoa (Plasmodium, Leishmania, Trypanosoma and Toxoplasma spp) [23], where they contribute to establish persistent infections and treatment failures, without genetic drug resistance. Persisters were also described in human cancer, where they survive chemotherapy and promote development of drug resistance [24,25]. All investigators studying persisters uniformly highlight the issue that their eradication could lead to a strong reduction of treatment failures and relapses in several clinical settings. The underlying reasons for persisters development seem to be multifactorial, involving non-genetic transient mechanisms switched on in microorganisms and cancer cells, as well as sub-optimal pharmacodynamics and immune responses in the host.
2.2. Bacterial persisters
The term “persisters” was used for the first time in 1944 by Joseph Bigger, who realized that penicillin failed to sterilize Staphylococcus pyogenes cultures due to generation of small number of cocci “individually insensitive to penicillin, but developing normally when penicillin is destroyed” [21,26,27]. Well in advance, he specified that “persisters are not resisters”, i.e., cells that we now call antibiotic resistant mutants. Bigger also observed that “persisters are believed to be insensitive to penicillin because they are in a dormant, non-dividing phase”.
In the search for persisters genes, starting from the 1980s, Harris Moyed and coworkers isolated mutants of Escherichia coli with vastly increased frequency of persistence (Hip mutants) [28]. The hip locus affects the frequency of persistence to the lethal consequences of selective inhibition of either DNA or peptidoglycan synthesis [29]. The hipA7 strain produced about 1% persisters and, after exposure to ampicillin, it generated about 1,000 times more persisters than the wild-type strain [27].
In 2019, Nathalie Balaban et al., pushed by increased concerns on antibiotic resistance and several papers published on the persistence phenomenon, released an international consensus statement on definitions and guidelines for research on antibiotic persistence [30]. Antibiotic resistance was defined as the ability of bacteria to replicate, and not just survive, in the presence of a drug, with the minimum inhibitory concentration (MIC) being the most common measure of the resistance level. Resistance is inherited and may be acquired by genetic mutations, or horizontal gene transfer of resistance-encoding genes. Instead, antibiotic persistence is the ability of a subpopulation to survive exposure to a bactericidal concentration [30]. The hallmark of antibiotic persistence is the biphasic killing curve. When persisters regrow in the absence of antibiotics, they give rise to a population of drug-sensitive cells. The size of the persister subpopulation is related to the class of drug and not to the drug concentration. Persisters cannot replicate in the presence of a drug, and are killed at a lower rate than the susceptible population from which they arise. The terms persistence and tolerance refer both to increased survival in the presence of an antibiotic, with no MIC increase. However, persistence refers to a cell subpopulation, while tolerance deals with a general ability of a population to survive longer drug treatments. Thus, persisters are a subpopulation of drug-tolerant bacteria, and tolerance and persistence may arise without dormancy [30,31]. The time required to kill these populations is quantified as the minimum duration for killing, and is useful to compare their survival fractions. The international consensus statement also showed that there are two kinds of persistence, namely type I, triggered by a stress signal (nutrient starvation, high cell number, acid stress, immune factors), and type II (spontaneous), which develops stochastically without any trigger, at a rate that is constant during growth [30,32,33,34]. The spontaneous and drug-induced persistence are difficult to distinguish and can be studied by direct observations of single bacterial cells by microfluidic devices [30,32,33]. Persistent bacterial infections cause death of millions of people every year. In 2022, a bibliometric analysis on the top 100 cited studies on bacterial persisters was reported [35].
2.3. Mycobacterium tuberculosis persisters
M. tuberculosis persisters are NR multidrug-tolerant bacilli genetically identical to drug-susceptible cells, which survive in the hostile environments of granulomas and in the sputum and adipose tissues of TB patients [36,37,38]. These cells are primarily involved in LTBI and TB relapses. Microenvironments with low oxygen tension and pH, nutrient starvation, reactive oxygen and nitrogen stresses, lead to formation of reservoirs of phenotypically drug-resistant cells with reduced antibiotic uptake and increased drug efflux, which renders antibiotics ineffective without genetic resistance [9,36,39]. Responses against drug-treatment of Mtb under hypoxic conditions and other stresses were modeled and/or reviewed in several papers [11,40,41,42,43,44,45].
The primary reason for the 6-month duration of the first-line TB therapy (R-H-E-Z) is due to the ability of Mtb to generate heterogeneous subpopulations adapting to different microenvironments within granulomas. Microfluidic devices for single-cell analysis revealed that the wide diversity of tubercle bacilli can be due to the fact that the mycobacterial cell division is asymmetrical, with the old pole elongating more than the new pole [46,47]. Asymmetric distribution of irreversibly oxidated proteins generates Mtb sister cells with different growth properties and drug susceptibility. In particular, sister cells with a low amount of these proteins are less sensitive to drugs, likely contributing to persisters formation. Asymmetric growth in mycobacteria is regulated mainly by the division protein Wag31 and the growth inhibitor LamA [46].
Other single-cells studies reported that Mtb persisters are rare low-energy cells formed stochastically during normal growth [48]. Stochastic variations may occur in the expression of an energy-generating component. Indeed, Mtb cells cultivated in a minimal medium with acetate showed cell-to-cell variations in the level of mRNA coding for the acetate kinase (AckA), a low energy-generating mechanism. This led to the formation of persister cells with decreased AckA, which showed low ATP levels and inactive drug targets. The knowledge that Staphylococcus aureus and E. coli persisters also have low ATP levels is in keeping with these observations [49,50]. In this view, it was hypothesized that all culture conditions leading to persistence converge in the inability to re-initiate a new round of DNA replication caused by insufficient level of the initiator complex ATP-DnaA, resulting in the lack of formation of a functional orisome [51]. Mycobacteria heterogeneity generated in part by stochastic processes may be amplified by innate asymmetric growth and division patterns [46].
Of importance, persister cells may generate genetically resistant mutants. Indeed, prolonged exposure to lethal concentrations of R or the fluoroquinolone moxifloxacin (M) induced the formation of Mtb and Mycobacterium smegmatis persisters, which carried elevated levels of the reactive oxygen species (ROS) hydroxyl radical, superoxide radical and hydrogen peroxide [52,53,54]. These radicals inflicted extensive genome-wide DNA mutations, as shown by generation of R- and M-resistant mutants carrying clinically relevant mutations in the rpoB and gyrA genes, respectively [52,53]. It is known that Mtb and other pathogens showing high frequency of mutation-based antibiotic resistance, produce ROS-generating enzymes (e.g., NADH dehydrogenase, succinate dehydrogenase, aspartate oxidase), while bacteria with low mutation frequency lack these enzymes [51].
Many bacteria react to ROS-generated DNA lesions caused by the bactericidal drugs quinolones, aminoglycosides and beta-lactams by inducing the SOS gene regulon, which is regulated by the transcriptional repressor LexA and the protease RecA. In Mtb, we found that treatment with M induced overexpression of SOS genes (e.g., recA, lexA, dnaE2) and DNA repair genes [55]. Induction of the error-prone DNA polymerase dnaE2, which potentially drives increased mutation rates, is considered to be a link between phenotypic persistence and genotypic resistance. Other investigators reported that M-mediated killing of Mtb involved accumulation of NADH-dependent ROS, and that repair of M-mediated lesions and ROS detoxification mechanisms contributed to survival [56,57]. Looking for a therapeutic significance of these observations, they found that Mtb treatment with the respiration stimulator N-acetyl cysteine accelerated respiration, and that ROS production increased M-induced lethality, and lowered the mutant prevention concentration. A study proposed that certain metabolic enzymes of Mtb (e.g., isocitrate lyase) serve antioxidant functions and facilitate drug tolerance [58]. Overall, these observations documented the links among bactericidal antibiotics, ROS production and formation of drug-tolerant persisters, i.e of specialized survivors characterized by anoxic metabolism and reduced ATP and ROS synthesis [51].
Low numbers of Mtb persisters are present in the lag and early exponential phases, but reach about 1% in the stationary phase [36,59]. Transcriptome analysis of D-cycloserine-formed Mtb persisters showed a distinct pattern of dormancy characterized by general metabolic downshift and upregulation of a small number of genes overexpressed also in other in vitro dormancy models, including the alpha crystallin heat shock protein Acr2 and the sigma factors SigG and SigF. Transcriptome analysis of high persisters (hip) Mtb mutants, selected with a lethal dose of R and streptomycin, showed upregulation of some toxin-antitoxins, i.e., the modules in which the toxins lead to growth arrest when labile antitoxins are degraded by proteases [36,60]. hip-mutants showed up to 1,000 times more persisters than the wild-type strain, a decrease in the phospholipid biosynthesis, and mutations in the cell wall lipid phthiocerol dimycocerosate, an important cell wall lipid and virulence factor. These and other genetic mechanisms suggested that Mtb persisters formation involves multiple pathways, and that hip-mutants, obtained using both the reference strain H37Rv and human clinical isolates, may contribute to the tolerance of Mtb to drug exposure. Research on Mtb persisters genes will facilitate development of more effective therapies to eradicate them from TB patients.
3. Differentially detectable (DD) Mtb cells
Several studies reported the presence of DD Mtb cells in the sputum produced by patients with active TB. These cells are not able to form colonies on standard glycerol-based solid media (7H10 agar plates and Löwenstein-Jensen slants) but can grow in liquid media supplemented with mycobacterial culture filtrates (CF) [12,13,14,15,16,17,18,61,62] or other substances, such as cyclic-AMP [17] and fatty acids [63,64]. The growth stimulatory effect of CF was ascribed to the five resuscitation promoting factors (Rpfs) of Mtb, which have the ability to resuscitate non-culturable cells [12,65]. However, the sputum contains also Rpfs-independent DD cells, which do not require Rpfs for growth, and CF-independent DD cells, which do not need CF to resuscitate in liquid media [65]. Thus, the growth stimulatory effect observed with CF is most likely the result of a combination of factors [17].
Dormant DD Mtb cells, which retain stable low-abundant mRNAs [66], may play a role in disease persistence due to their phenotypic resistance to anti-TB drugs [18,67]. An in vitro DD Mtb cells model was recently developed by Saito et al., allowing development of ≥90% DD cells after nutrient starvation in phosphate-buffered saline (PBS) followed by exposure to high R concentrations (PBS-R model) [14]. This model generated DD cells similar to those found in TB patients, and their transcriptional profiles may be useful for monitoring DD populations in the sputum [18].
It is known that DD-Mtb cells represented the majority of bacilli in the sputum of 21% of patients with DS-TB, and that this level increased to 65% after 2 weeks of treatment with first-line drugs [16]. In a study which enrolled subjects with DR-TB (94% MDR patients, i.e., resistant at least to H and R), DD-Mtb cells were found in the sputum of 29% of patients prior of treatment, and this amount stabilized at 31% after 2 weeks of treatment with a multidrug regimen that included Z, but not R, H and E. However, after 2 months of therapy, DD cells decreased to 13% and 0% in the sputa of DS- and DR-TB, respectively 16]. Another study reported that CF devoid of rpfs yielded a greater amount of DD cells in sputum from patients with MDR-TB, compared with those with R-monoresistant TB, suggesting that CF is dispensable for detection of DD cells from DR-strains [65].
In order to explore the biological mechanisms generating DD cells, Saito et al. found that Mtb can enter into a DD state after a sub-lethal oxidative stress damaging DNA, proteins and lipid components [68]. This conclusion was in line with the observations of Hong et al., who reported that ROS formation in response to drugs could prevent E. coli cells from growing as CFU, without killing them, and that ROS accumulation mediated by self-amplifying mechanisms continued after antibiotic removal [69]. Transcriptomic analysis showed that, after RIF treatment, DD Mtb cells underwent a partial loss of the oxidative stress response that promoted the formation of the DD phenotype [68]. Thus, Mtb must be able to mitigate oxidative stress to survive in the DD state. However, the capacity of Mtb to do so is limited, so that only intermediate ROS levels lead to DD Mtb. For instance, Mycobacteriun bovis and its vaccine derivative, the Bacillus Calmette-Guérin (BCG), are not able to do it, and do not generate DD cells because they succumb to ROS [68]. The observation that treatment of R-containing cultures in the presence of the antioxidant catalase increased the ability of DD Mtb to form CFU on agar plates, reinforced the relationship between oxidative stress and return to replicative capacity. Hong et al. showed that, above a certain host threshold, E. coli cells died, but below that threshold, provisions of anti-oxidants revealed a CFU population [68,69].
In summary, after treatment of Mtb with bactericidal antibiotics, increasing, self-amplifying, ROS levels may be generated. Surviving bacilli may then be detected as CFU (persister cells) if ROS levels are low, LD-MPN (DD cells) if ROS levels are intermediate, or not detected (dead cells) if ROS levels are high. M. tuberculosis cells are likely to exist in the host at various levels of oxidative stress and ability to replicate [68]. Overall, these observations indicate that, to eliminate Mtb persisters and DD cells from the host, it would be important to find new drugs/drug combinations inducing oxidative damage under aerobic conditions, and/or other stresses under hypoxic conditions such as the reactive nitrogen species (RNS) nitric oxide (NO). For instance, NO is induced by pretomanid (Pa, a new anti-TB drug formerly known as PA-824) inside NR Mtb cells [70]. Combinations of Pa with cytochrome bc1 inhibitors showed strong bactericidal activity against hypoxic NR Mtb [71].
Finally, besides LD-MPN and CFU counting, in the sputum of TB patients, the presence of a greater mycobacterial load in liquid than in solid media may be detected by the mycobacterial growth indicator tube (MGIT) time to positivity (TTP) assay, a semiquantitative measure of viable Mtb that showed an inverse relationship with the CFU [72,73,74]. The MGIT 960 instrument is an automated system monitoring the fluorescence of an oxygen sensor to detect growth of mycobacteria in culture tubes containing a modified Middlebrook 7H9 liquid medium, in which the TTP values are reported as days. A study found a strong correlation between CF+ LD-MPN and TTP values for smear-positive and smear-negative sputa [13]. These data indicate that both LD-MPN and MGIT TTP assays are useful to detect tubercle bacilli growing on liquid but not on solid media.
4. In Vitro Killing of AR+NR Mtb Cells by Drug Combinations
Tools to identify in vitro promising drug regimens for rapid clinical trial evaluation are needed. One such tool is the hollow fibre system model of TB (HFS-TB), a bioreactor containing tubular hollow fibres made of semi-permeable membranes, which showed a 94% predictive accuracy for clinical response rates and optimal exposures [75,76]. In 2015, the European Medicines Agency supported the use of HFS-TB for several drugs, to perform pharmacokinetics-pharmacodynamics monotherapy studies on microbial killing, and to design new combination regimens. After drug exposure, residual viability was evaluated by various tests, including the MGIT TTP assay. Using the HFS-TB model, combinations containing faropenem, linezolid (L) and Z sterilized Mtb cultures in 28 days, as shown by lack of re-growth of drug-treated cells in MGIT 960 tubes after 56 days of incubation (TTP >56 days) [77].
A more stringent TTP threshold value was used by our group, who defined Mtb killing of both AR and NR cells (the latter obtained in the Wayne model) as lack of regrowth of drug-treated bacilli after 100 days of incubation in MGIT tubes (TTP >100 days) [74,78,79]. Using this approach, we found that the 4-drug combinations R-M-metronidazole (Mz)-amikacin (Ak) and R-M-Mz-capreomycin (Cp) killed AR+NR Mtb in 21 days [74]. Furthermore, we found that under aerobic and hypoxic acidic conditions (pH of 5.8) likely mimicking those present inside the phagolysosomes of activated macrophages, the 4-drug combinations R-M-Ak-Pa killed AR+NR Mtb in 14 days, while R-M-Ak-nitazoxanide (Nz) killed them in 21 days [78]. Finally, we found that R-Nz-containing combinations, but not R-H-E-Z (currently used for TB therapy), killed NR Mtb in ≥28 days in hypoxia at pH of 7.3, i.e., under conditions likely mimicking those present inside caseous granulomas [79]. Using the MGIT TTP assay, we also reported that the combinations bedaquiline (B)-Ak-rifabutin (Rb)-clarithromycin (Cl)-nimorazole (Nm) and B-Ak-Rb-Cl-Mz-colistin (Cs) killed AR+NR cells of Mycobacterium abscessus in 42 and 56 days, respectively [80]. Overall, our data indicated that the nitro-compounds Mz, Pa, Nz and Nm were important components of combinations killing AR+NR mycobacterial cells. Nitro-compounds are known to induce RNS in anaerobic organisms like Giardia spp [81,82], and in NR Mtb [70]. It is possible that combinations containing ROS-inducing drugs like M [52,53,56,57] and R [52,53,54], and RNS-inducing agents like nitro-compounds, are essential to kill AR and NR stages, which contain drug-tolerant persisters and DD cells.
5. Drug Combinations Eradicating Mtb from BALB/c and C3HeB/FeJ Mice
The ability of a drug combination to sterilize TB lesions and preventing relapses due to drug-tolerant persisters and DD cells is determined by the synergistic activities of drug components. In the current regimen used for treatment of DS TB (R-H-E-Z), R and Z have higher sterilizing activity than H and E [83]. In the last decades, several efforts were done in order to find new combinations eradicating drug-tolerant bacilli residing in low vascularized, hypoxic lesions. Such hypoxic conditions are much more present in caseous granulomas of TB patients, and in Mtb-infected caseum-forming animals (e.g., guinea pigs, rabbits, C3HeB/FeJ mice), than in cellular granulomas of commonly used mice (e.g., Mtb-infected BALB/c mice), which do not form caseous granulomas, the hallmark of human TB. The guinea pig and rabbit models were not used for the study of combination chemotherapy regimens because of their prohibitive costs [83]. Instead, the C3HeB/FeJ mice, a strain recognized to develop necrotic granulomas mimicking those of human TB, were used since 2012 for testing activity of drugs and drug combinations [84,85,86].
However, the low-cost non caseum-forming BALB/c mice model was the most extensively used for testing drug combinations. In several studies, treatment efficacy was assessed on the basis of lung CFU counts at selected time points during treatment (a measure of bactericidal activity), and on the proportion of mice with culture positive relapse at least 3 months after completion of treatment (a measure of sterilizing activity) [83,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. Table 1 (divided into parts A and B) shows a list of 65 combinations sterilizing Mtb H37Rv-infected BALB/c mice. A more synthetic view of Table 1 is given in Figure 1 (also divided into parts A and B), which shows the percentages of sterilizing combinations containing the drugs indicated, stratified by monthly periods. Overall, these data have been useful for identifying pivotal drugs/drug combinations eradicating Mtb from BALB/c mice. Some of these drugs were also tested in combination experiments for ability to sterilize caseum-forming C3HeB/FeJ mice (Table 2), and for efficacy to shorten human TB therapy in well controlled clinical trials (Table 3).
5.1. Eradication of Mtb from Balb/c mice
As anticipated above, Table 1 (parts A and B) shows a list of 65 combinations, stratified by the minimum length of time required for sterilization (1.5-2.0, 2.5-3.0, 3.5-4.0 and 4.5-6.0 months) [83,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. Table 1 also shows information including the months of treatment with each combination, the drug dose in mg/kg of body weight, and some treatment details (5 days/week, etc) [83,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. All mice were aerosol infected with Mtb H37Rv, with the exception of one study in which animals were infected intravenously [98]. Figure 1 shows the percentages of sterilizing combinations reported in Table 1, containing the drug indicated.
The first 6 combinations (regimens 1 to 6) sterilized BALB/c mice after 4.5 to 6 months of treatment [83,91,94,97,99]. Pyrazinamide was contained in 100% of these combinations, followed by R (83%), H (67%), M (33%) and Pa and E (17%); no combinations contained B, L, rifapentine (P), clofazimine (C) and sutezolid (S) (Figure 1). Overall, administration of R10H25Z150 (regimens 1-3) or R10H10E100Z150 (regimen 6) for 2 months, followed by 4 months of R10H25 (regimens 1-3) or R10H10 (regimen 6) sterilized mice in 4.5 to 6 months. These regimens mimicked those currently used for treatment of human TB (R-H-E-Z), but only regimen 6 contained E, a bacteriostatic drug. A 5-month sterilization occurred when H was replaced with M in the intensive and continuation therapy (regimen 4), or when the combination contained the new anti-TB drug Pa (Pa50M100Z150, regimen 5).
Sterilization occurred after 3.5 to 4 months of treatment with the 16 regimens (regimens 7 to 22) listed in Table 1, part A. Overall, none contained R10H25Z150 and R10H10E100Z150 during the entire period of treatment. In some cases, R, H and Z were administered at high doses. For instance, in regimen 7, after initial 0.5 months of administration of 25 mg/kg of H (H25), the drug was later given at 75 mg/kg (H75). Furthermore, R was administered at 50 mg/kg (R50) in regimen 22, and Z was given at 300 mg/kg (Z300) in the intensive phase of regimen 9. A common characteristic of regimens 7-22 was the use of drugs other than R, H and Z, including i) 15 mg/kg of the long-acting rifamycin P (P15) (regimens 7-9), that was substituted for 10 mg/kg of rifampin (R10), ii) 25 mg/kg of B (B25, regimens 13-19), iii) 50 or 100 mg/kg of the nitroimidazole Pa (Pa50 or Pa100, regimens 10, 12-13, 15-19), iv) 100 mg/kg of M (M100, regimens 8-10, 14 and 16), v) 12.5 or 25 mg/kg of C (C12.5, C25, regimens 20-21), vi) 50 mg/kg of S (S50, regimen 15). Overall, Z was contained in 81% of these combinations, followed by Pa (50%), B and R (44%), M (31%), P (25%), H, C and E (13%), S (6%) (Figure 1, parts A and B).
Sterilization was observed also after 2.5 to 3 months of treatment with the 32 regimens (regimens 23 to 54) listed in Table 1 (parts A and B). The top-6 drugs used were Z (72%), followed by P (44%), B (41%), Pa and H (38%), and M (28%) (Figure 1, parts A and B). Rifapentine was used at 7.5, 10, 15 and 20 mg/kg; Pa was used at 50 or 100 mg/kg.
Finally, the most potent activity was shown by 11 combinations (regimens 55 to 65) that sterilized mice in 1.5-2 months. Ten of them contained both B25 and Z150, followed by Pa (6/11), P and C (3/11), M and L (2/11) (Figure 1, parts A and B). In this group, the oxazolidinone L, a protein synthesis inhibitor, was used in regimens 64 and 65, that sterilized mice in 1.5 months when combined with B25, Z150 and Pa100.
All these data, reported from 2006 to 2022 mostly by the Center for TB research of the Johns Hopkins University, Baltimore, Maryland, USA, coordinated by Dr Eric Nuermberger [83,86,87,88,89,90,91,92,93,94,95,96,97,99,100,101,102,103], indicated that all Mtb H37Rv bacilli (AR cells and NR cells containing drug tolerant persisters and DD cells) were eradicated from BALB/c mice, as shown by lack of relapses (culture-negative lungs) 3 or 6 months after treatment completion. Overall, it appeared that the ranking of the top-6 drugs contained in combinations sterilizing BALB/c mice in 1.5 to 3 months by most regimens (regimens 23 to 65) was Z > B > Pa > P > M > L. It is possible that reactive species such as ROS (induced by M) and RNS (induced by Pa) contributed to the sterilizing effect. Furthermore, it was evident that the use of R and H sharply decreased as components of the most powerful combinations (Figure 1A and B, respectively), and that they were not present in the 1.5 to 2 months sterilizing combinations. Rifampin was replaced with the long-acting rifamycin P [104], which was present in several rapidly sterilizing combinations (Figure 1, part A). This was likely due to the knowledge that P has a longer half-life than R in human serum (10–15 h versus 2–3 h, respectively) [105].
5.2. Eradication of Mtb from C3HeB/FeJ mice
The results shown in BALB/c mice were important in designing drug combinations eradicating Mtb also in the caseous lesions of C3HeB/FeJ mice, a strain which develops lung necrotic granulomas similar to those found in human TB [84,85]. In 2012, a paper on the dose-ranging comparison of R and P in BALB/c and C3HeB/FeJ mice [86], reported that the long-acting P was roughly four times more potent than R in both mouse strains.
Table 2 shows a list of 7 combinations published in 2015-2022 that sterilized C3HeB/FeJ mice after 2.5, 3.0, 4.0, 4.5 and 6.0 months (Table 2) [97,102,103]. Administration of R10H10E100Z150 for 2 months, followed by R10H10 for 4 months (regimens 1, corresponding to the first-line therapy of human TB) sterilized mice in 6 months. Addition of Z at 150 mg/kg (Z150) in the continuation phase sterilized mice in 4.5 months (regimen 2). Thus, continuing Z beyond the first two months was beneficial to prevent relapses in caseum-forming mice. However, this is not applicable to humans, because the use of R-Z beyond 2 months is hepatotoxic, as shown during the prolonged use of this combination for treatment of LTBI [106].
In regimen 3, replacement of R10 with P10, and E100 with M100, sterilized C3HeB/FeJ mice in 4 months [103]. Sterilization was observed after 2.5 to 3 months of treatment with regimens 4-7. In particular, mice were sterilized after 3 months when R10 was replaced with high dose of P (P20), and was added 12.5 mg/kg of the anti-leprosy drug C (C12.5) (regimen 4). Clofazimine is a drug which seems in large part to function by inducing ROS in Mtb [102]. Administration of high dose of the long-acting drug P was important because, in comparison to regimen 1, mice were sterilized by P20H10E100Z150 in 2.5 months (regimen 7).
Finally, due to the knowledge that strong induction of the cytochrome P450 3A (CYP3A) caused by R and P, but not rifabutin (Rb), reduced the concentration of B [107], it was found that C3HeB/FeJ mice were sterilized after 3 months of B25 M100Z150Rb10 (regimen 5), or 2 months of B25 M100Z150Rb10 followed by 1 month of B25M100Rb10, (regimen 6) [103]. Overall, in these 7 regimens, Z was contained in 7 out of 7 combinations tested, followed by H (5/7), E (4/7), P and M (3/7), B, R and Rb (2/7) and C (1/7). So far, no papers with Pa- and/or L-containing combinations sterilizing C3HeB/FeJ mice were apparently reported.
6. New drug combinations for treatment of human TB
The frequent presence of Z in combinations sterilizing BALB/c and C3HeB/FeJ mice is likely related to the knowledge that this drug penetrates all TB lung lesions and kills persisters residing in caseum [108]. This information makes Z an irreplaceable component of regimens for treating DS-TB [36,109]. In support of this view is that inclusion of Z into TB treatment shortened the length of therapy from 9-12 months to 6 months [36,109]. Pyrazinamide and R, the two “sterilizing” drugs that contributed most to treatment shortening, distribute favorably into all lesion compartments, including avascular caseum [110]. However, in the combinations sterilizing BALB/c and C3HeB/FeJ mice, the use of R progressively decreased in the most rapidly sterilizing combinations because it was replaced by the long-acting rifamycin P (Table 1 and Figure 1, part A), or the low cytochrome P450 3A-inducer rifamycin Rb (Table 2, regimens 5-6).
Overall, the murine TB data were useful for the choice of drugs to be combined in human TB clinical trials, in order to shorten the current 6-month regimen (R-H-E-Z). This regimen, finalized during the 1980s, and based on seminal studies conducted by the British Medical Research Council in the second half of 20th century, was widely adopted worldwide for more than four decades. It cured more than 95% of persons, but in some cases long-term adherence to therapy was difficult to sustain for patients and national resource constraints, contributing to development of genetic drug-resistance [108,111,112].
Table 3 shows three papers published on the high impact factor magazine The New England Journal of Medicine (NEJM), which reported the results of all-oral regimen trials against DS-TB. All trials included Z-containing combinations with 6-months [113], 4-months [114] and 2-months [115] efficacy as effective as, or noninferior to, the 6-month control regimen (R-H-E-Z).
The first paper [113] was published in 2014 as a report of the noninferiority trial ISRCTN44153044 (RIFAQUIN trial). It included the control regimen (2 months of intensive therapy with R600H300Z1500E1200 and 4 months of continuation therapy with R600H300) and a 6-month regimen as effective as the control regimen in which H was replaced by daily dose of 400 mg of M (M400) for 2 months, followed by one weekly dose of both M400 and high-dose (1200 mg) of P (P1200) for 4 months.
The second paper [114] was published in 2021 as a report of phase 3 noninferiority trial NCT02410772 (Study 31), and included the control regimen (R600H300Z1500E1200), and a 4-months regimen with 2 months (indicated as 8 weeks in the paper) of once-daily P1200H300Z1500M400 (R replaced by P, and E replaced by M), followed by 2 months (indicated as 9 week in the paper) of once-daily of P1200H300M400. This 4-month regimen was supported by the WHO as a possible alternative to current 6-month regimen for DS-TB, because it showed similar performance in terms of efficacy and safety [112,116]. In 2022, the guideline development group of the WHO stated that Study 31 was the first and only phase 3 trial to demonstrate the noninferiority of this 4-month regimen for treatment of DS-TB when compared to the standard of care [112,116].
The third paper [115] was published in 2023 as a report of the noninferiority trial NCT03474198 (TRUNCATE-TB trial). The authors chose a treatment strategy, rather than focusing on a regimen alone, to be compared with control regimen [117]. This approach was selected because, in clinical trials, it is known that at least 85% of participants are cured with 3- and 4-month regimens. Thus, the 6-month standard regimen may lead to overtreatment in the majority of persons, in order to prevent relapse in a minority of persons [115]. Thus, exploration of new therapeutic approaches is important. It was adopted a 5-arms strategy involving initial treatment for 2 months (indicated as 8-week in the paper) of R-susceptible TB patients with 5 different regimens, followed up for 96 weeks for extended treatment for persistent clinical disease and prompt re-treatment for the minority of persons with a relapse. Briefly, the final results showed that only the 2-month regimen that contained B, L, H, Z, E (B400/200L600H300Z1500E1100) (Table 3) was noninferior to the control 6-month regimen, with respect to the risk of a composite clinical outcome at week 96.
7. Discussion
In the framework of the WHO vision to eliminate TB as a public health problem by 2035 [1], the three clinical trials published in the NEJM (Table 3) provided robust data towards the realistic possibility of shortening DS-TB treatment using combinations containing Z (found in 3 trials), M, H and E (found in 2 trials), B, R, P, L (found in 1 trial). The observation that all these drugs were contained in BALB/c and C3HeB/FeJ mice sterilizing combinations (Table 1 and Table 2 and Figure 1), indicated the usefulness of these small animal models for testing new TB regimens.
It is difficult to correlate the physicochemical properties of the drugs considered (Table 4) with in vivo activities shown in the Table 1, Table 2 and Table 3 and Figure 1. However, some observations may be drawn, concerning, for instance, the interplay among the half-life in serum or tissues [118], the unbound fraction (fu) (free drug) in caseum, and the bactericidal activity in caseum, a lipidic niche for Mtb drug-tolerant persisters and DD cells [42,119,120,121]. In rabbit caseum, the most potent bactericidal drugs were the rifamicins (R, P, Rb: 3.1-4.0 log CFUmax decrease in 7 days, Table 4), followed by B, M and Pa (1.6-1.9 log CFUmax decrease) [121].
The drugs with half-lives of ≥10 hours (h) were, in decreasing order, B (5.5 months) > C (about 25 days) >Rb (45 h) >Pa (18 h) > M (11.5-15.6 h) > P (10-15 h) > Z (9-10 h) [118]. In spite of the small number of studies still published, Z, B, P, M, Rb and C were major components of combinations sterilizing caseum-forming C3HeB/FeJ mice in ≤ 3 months (Table 3). Of these, Z, B, P, and M were also components of the noninferiority combinations curing DS-TB in ≤ 6 months (Table 3).
As to the unbound fraction in caseum (fu, Table 4), this parameter is inversely related to the lipophilic character of a drug, that is mostly expressed as hydrophobicity (logP), i.e., the octanol/water partition coefficient, commonly reported as experimental or calculated (clogP) value [118]. Drugs with clogP <0 are considered to be hydrophilic [11]. In Mtb-infected rabbits, only the unbound fraction can penetrate necrotic material or caseum via passive diffusion [119]. The hydrophobicity and aromatic ring count of a drug were shown to be proportional to caseum binding, and compounds with clogP <1 had a high chance of achieving fu >10% [119]. Indeed, the fu values of the hydrophilic H and Z (clogP -0.71) (Table 4) were >99.9%, i.e., they can penetrate caseum, while the fu of highly lipophilic drugs like C and B (clogP of 7.39 and 6.37, respectively) were <0.01%, i.e., they do not penetrate caseum [11,119]. Using a caseum binding assay and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MSI) imaging of TB drugs in Mtb-infected rabbits, it was shown that binding to caseum inversely correlated with passive diffusion into the necrotic core [119]. Among the major positive drivers of binding were high lipophilicity and poor solubility. Thus, highly lipophilic drugs like B and C nonspecifically bind to caseum macromolecules at the outer edge of the caseum core, preventing further passive diffusion toward the center of the necrotic core [119]. Drugs with intermediate clogP values such as P, R, Pa, and S (clogP of 4.83, 3.85, 2.8, and 1.31, respectively) showed rising fu values (fu of 0.5, 5.13, 7.31, and 30.1%, respectively). In this view, a rational design of combinations should involve drugs that complement each other in their ability to penetrate caseous granulomas and to kill drug tolerant persisters and DD cells living inside them.
The active combination of the Study 31 trial (P1200H300Z1500M400) (Table 3), which cured DS-TB in 4 months, and the combination P20H10E100Z150, that sterilized caseum-forming C3HeB/FeJ mice in 2.5 months (Table 2) are quite in keeping with this rationale, because they contain Z, M and P, which are active against persisters, and show an unbound fraction (fu) in rabbit caseum of >99.9%, 13.5% and 0.5%, respectively (Table 4). However, it is also important to consider the half-life and the extent of Mtb killing in caseum (Table 4) [110,120,121]. For instance, P, which showed a longer half-life than R in human serum (10–15 h versus 2–3 h, respectively) [105 and Table 4], killed Mtb at a similar extent than R and Rb in rabbit caseum after 7-day of incubation [121]. We also found that Mtb was selectively killed by R and P in hypoxia at pH of 7.3 (the pH of caseum) [44], and others reported that only the rifamycins (P, R, Rb and rifalazil) showed high bactericidal activity in rabbit caseum [121] and fully sterilized it [110]. All these studies indicated that rifamycins may play a pivotal role to eradicate drug tolerants persisters and DD cells from caseum [110,120,121]. Also half-lives of M and Z (≥ 9-10 h) were in the range of the P half-life value (Table 4). However, it is difficult to explain the sterilizing activity of Z, that shortened the length of therapy from 9 months to 6 months, but that is active only at acidic pH [36,108,109]. Indeed, in rabbit caseum (pH 7.0-7.5), the bactericidal activity of Z was low (0.5 log CFU decrease in 7 days) [110, 121 and Table 4]. A possible solution of this enigma was proposed by Sarathy et al. [110], who pointed out that, in closed human granulomas, hypoxia induces secretion of large amounts of succinate in Mtb. This may generate a “halo” of low pH around Mtb cells, which favors Z cidality. The observations of Kempker et al. [122] that cavity caseum samples obtained from resected lung tissue of 8 out of 10 TB patients had a pH of ≤ 5.5 is in favor of this hypothesis.
The active combination of the TRUNCATE-TB trial (B400/200L600H300Z1500E1100) (Table 3), that cured DS-TB in 2 months [115], and the two B-containing combinations (3 months of B25M100Z150Rb10, and 2 months of B25M100Z150Rb10 followed by 1 month of B25M100Rb10) that sterilized caseum-forming C3HeB/FeJ mice in 3 months (Table 2, regimens 5 and 6), are more difficult to explain in terms of caseum penetration, because B showed a fu value of <0.01 in rabbit caseum (Table 4). However, B has a very long terminal elimination half-life (about 5.5 months) [123], due to the great ability of this drug to bind to the intracellular phospholipids and, consequently, to accumulate in tissues. Half-lives of B in TB lung lesions of BALB/c and C3HeB/FeJ mice were also high (85.5 and 104.4 h, respectively) [124]. In C3HeB/FeJ mice, B accumulated within the highly cellular regions in the lungs, but it was also present at reduced but still biologically relevant levels within the central caseum, where it was most likely not detected by the MALDI-MSI system used [124]. Thus, it is possible that the very long half-life of B and/or the synergism with other drugs may contribute to the potent sterilizing activity of B-containing combinations. Some support to this hypothesis comes from the knowledge that, in recent trials, several B-containing combinations were very active also against DR-TB, including patients treated with B-Pa-L for 6 months (ZeNix, trial NCT03086486) [125], with B-Pa-L-M for 6 months (TB-PRACTECAL, trial NCT02589782) [126], and with two other B-containing regimens for 6 or 9 months (STREAM stage 2, trial ISRCTN18148631) [127]. In 2022, by taking into account the evidences reported in these and other trials, the WHO published consolidated guidelines for treatment of DR-TB [128].
8. Conclusions
All data on drug activity against drug-tolerant Mtb, ranging from the most studied in vitro Wayne dormancy model [40] up to the papers on sterilization of non caseum- and caseum-forming mice, shed a new light on TB biology [129,130]. These and other studies showed that the greatest difficulties in curing TB do not depend only on intracellular AR bacilli and of their mutants but, above all, on extracellular NR drug-tolerant bacilli living in the caseous tuberculomas, the hallmark of TB.
In the last two decades, this new way of seeing the TB boosted the search for novel drugs/drug combinations killing in vitro and in vivo both AR cells and NR drug tolerant cells, containing persisters and DD cells. More recently, some of these combinations were tested in several DS- and DR-TB clinical trials, so far achieving in various cases the goal of shortening duration of current drug regimens. In 2022, the WHO, based on one of these trials, supported the use of a 4-month regimen for treatment of DS-TB as a possible alternative to the current 6-month regimen [112,116].
Author Contributions
F.G. and L.F. wrote the manuscript; A.L. and A.I. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. WHO Global tuberculosis report; ; WHO: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1. [Google Scholar]
- WHO. WHO Global tuberculosis report; ; WHO: Geneva, Switzerland, 2019; ISBN 978-92-4-156571-4. [Google Scholar]
- Barry, C.E. 3rd; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, RJ.; Young, D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–55. [Google Scholar] [CrossRef] [PubMed]
- Dartois, V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 2014, 12, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lenaerts, A.; Barry, C.E. 3rd.; Dartois, V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol. Rev. [CrossRef]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–32. [Google Scholar] [CrossRef]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–74. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.; Dartois, V.; Dick, T.; Gengenbacher, M. Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 1648–53. [Google Scholar] [CrossRef] [PubMed]
- Garton, N.J.; Waddell, S.J.; Sherratt, A.L.; Lee, S.M.; Smith, R.J.; Senner, C.; Hinds, J.; Rajakumar, K.; Adegbola, R.A.; Besra, G.S.; et al. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 2008, 5, e75. [Google Scholar] [CrossRef]
- Iacobino, A.; Piccaro, G.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Fighting tuberculosis by drugs targeting nonreplicating Mycobacterium tuberculosis bacilli. Int. J. Mycobacteriol. 2017, 6, 213–221. [Google Scholar] [CrossRef]
- Mukamolova, G.V.; Turapov, O.; Malkin, J.; Woltmann, G.; Barer, M.R. Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am. J. Respir. Crit. Care Med. 2010, 181, 174–80. [Google Scholar] [CrossRef]
- Chengalroyen, M.D.; Beukes, G.M.; Gordhan, B.G.; Streicher, E.M.; Churchyard, G.; Hafner, R.; Warren, R.; Otwombe, K.; Martinson, N.; Kana, B.D. Detection and quantification of differentially culturable tubercle bacteria in sputum from patients with tuberculosis. Am. J. Respir. Crit. Care Med. 2016, 194, 1532–1540. [Google Scholar] [CrossRef]
- Saito, K.; Warrier, T.; Somersan-Karakaya, S.; Kaminski, L.; Mi, J.; Jiang, X.; Park, S.; Shigyo, K.; Gold, B.; Roberts, J.; et al. Rifamycin action on RNA polymerase in antibiotic-tolerant Mycobacterium tuberculosis results in differentially detectable populations. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E4832–E4840. [Google Scholar] [CrossRef]
- McAulay, K.; Saito, K.; Warrier, T.; Walsh, K.F.; Mathurin, L.D.; Royal-Mardi, G.; Lee, M.H.; Ocheretina, O.; Pape, J.W.; Fitzgerald, D.W.; et al. Differentially detectable Mycobacterium tuberculosis cells in sputum from treatment-naive subjects in Haiti and their proportionate increase after initiation of treatment. mBio. 2018, 9, e02192–e18. [Google Scholar] [CrossRef]
- Zainabadi, K.; Walsh, K.F.; Vilbrun, S.C.; Mathurin, L.D.; Lee, M.H.; Saito, K.; Mishra, S.; Ocheretina, O.; Pape, JW.; Nathan, C.; et al. Characterization of differentially detectable Mycobacterium tuberculosis in the sputum of subjects with drug-sensitive or drug-resistant tuberculosis before and after two months of therapy. Antimicrob. Agents Chemother. 2021, 65, e0060821. [Google Scholar] [CrossRef]
- Gordhan, B.G.; Peters, J.S.; McIvor, A.; Machowski, E.E.; Ealand, C.; Waja, Z.; Martinson, N.; Kana, B.D. Detection of differentially culturable tubercle bacteria in sputum using mycobacterial culture filtrates. Sci. Rep. 2021, 11, 6493. [Google Scholar] [CrossRef] [PubMed]
- Zainabadi, K.; Saito, K.; Mishra, S.; Walsh, K.F.; Mathurin, L.D.; Vilbrun, S.C.; Ocheretina, O.; Pape, J.W.; Fitzgerald, D.W.; Nathan, C.F.; et al. Transcriptional biomarkers of differentially detectable Mycobacterium tuberculosis in patient sputum. mBio. 2022, 13, e0270122. [Google Scholar] [CrossRef]
- Li, L.; Mendis, N.; Trigui, H.; Oliver, J.D.; Faucher, S.P. The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol. 2014, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, J.; Fourie, P.B.; Mitchison, D.A. Persister populations of Mycobacterium tuberculosis in sputum that grow in liquid but not on solid culture media. J. Antimicrob. Chemother. 2014, 69, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, J.; Van Dijck, P.; Holtappels, M. Fungal persister cells: The basis for recalcitrant infections? PLoS Pathog. 2018, 14, e1007301. [Google Scholar] [CrossRef]
- Barrett, M.P.; Kyle, D.E.; Sibley, L.D.; Radke, J.B.; Tarleton, R.L. Protozoan persister-like cells and drug treatment failure. Nat. Rev. Microbiol. 2019, 17, 607–620. [Google Scholar] [CrossRef]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-tolerant persister cells in cancer therapy resistance. Cancer Res. 2022, 82, 2503–2514. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Bigger, J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilization. The Lancet. 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–75. [Google Scholar] [CrossRef] [PubMed]
- Black, D.S.; Irwin, B.; Moyed, H.S. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 1994, 176, 4081–91. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, JJ.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Orman, M.A.; Brynildsen, M.P. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 2013, 57, 3230–9. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science. 2004, 305, 1622–5. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Balaban, N.Q. Quantitative measurement of Type I and Type II persisters using ScanLag. In: Bacterial Persistence, Methods and Protocols. Methods in Molecular Biology. Volume 1333. Edited by Michiels J and Fauvart M. Humana Press. 2016, Pages 75-81. [CrossRef]
- Carvalho, G.; Guilhen, C.; Balestrino, D.; Forestier, C.; Mathias, J.D. Relating switching rates between normal and persister cells to substrate and antibiotic concentrations: a mathematical modelling approach supported by experiments. Microb. Biotechnol. 2017, 10, 1616–1627. [Google Scholar] [CrossRef]
- Ju, Y.; Long, H.; Zhao, P.; Xu, P.; Sun, L.; Bao, Y.; Yu, P.; Zhang, Y. The top 100 cited studies on bacterial persisters: A bibliometric analysis. Front. Pharmacol. 2022, 13, 1001861. [Google Scholar] [CrossRef]
- Mandal, S.; Njikan, S.; Kumar, A.; Early, J.V.; Parish, T. The relevance of persisters in tuberculosis drug discovery. Microbiology (Reading). 2019, 165, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, J.; Fourie, P.B.; Mitchison, D.A. Persister populations of Mycobacterium tuberculosis in sputum that grow in liquid but not on solid culture media. J. Antimicrob. Chemother. 2014, 69, 437–40. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, J.P.; Vinnard, C.; Subbian, S.; Nagajyothi, J.F. Effect of Mycobacterium tuberculosis infection on adipocyte physiology. Microbes Infect. 2018, 20, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Iacobino, A.; Fattorini, L.; Giannoni, F. Drug-resistant tuberculosis 2020. Where we stand. Appl. Sci. 2020, 10, 2153. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996, 64, 2062–9. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002, 43, 717–31. [Google Scholar] [CrossRef]
- Piccaro, G.; Poce, G.; Biava, M.; Giannoni, F.; Fattorini, L. Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J Antibiot (Tokyo). 2015, 68, 711–4. [Google Scholar] [CrossRef]
- Iona, E.; Pardini, M.; Mustazzolu, A.; Piccaro, G.; Nisini, R.; Fattorini, L.; Giannoni, F. Mycobacterium tuberculosis gene expression at different stages of hypoxia-induced dormancy and upon resuscitation. J Microbiol. 2016, 54, 565–72. [Google Scholar] [CrossRef]
- Iacobino, A.; Piccaro, G.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Mycobacterium tuberculosis is selectively killed by rifampin and rifapentine in hypoxia at neutral pH. Antimicrob. Agents Chemother. [CrossRef]
- Gold, B.; Nathan, C. Targeting Phenotypically Tolerant Mycobacterium tuberculosis. Microbiol. Spectr. 2017. [Google Scholar] [CrossRef]
- Chung, E.S.; Johnson, W.C.; Aldridge, B.B. Types and functions of heterogeneity in mycobacteria. Nat. Rev. Microbiol. 2022, 20, 529–541. [Google Scholar] [CrossRef]
- Vaubourgeix, J.; Lin, G, Dhar, N. ; Chenouard, N.; Jiang, X.; Botella, H.; Lupoli, T.; Mariani, O.; Yang, G.; Ouerfelli, O.; et al. Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe. 2015, 17, 178–90. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.; Lewis, K. Noise in a metabolic pathway leads to persister formation in Mycobacterium tuberculosis. Microbiol. Spectr. 2022, 10, e0294822. [Google Scholar] [CrossRef]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016; 18, 16051. [Google Scholar] [CrossRef]
- Manuse, S.; Shan, Y.; Canas-Duarte, S.J.; Bakshi, S.; Sun, W.S.; Mori, H.; Paulsson, J.; Lewis, K. Bacterial persisters are a stochastically formed subpopulation of low-energy cells. PLoS Biol. 2021, 19, e3001194. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. Link between antibiotic persistence and antibiotic resistance in bacterial pathogens. Front. Cell. Infect. Microbiol. 2022; 12, 900848. [Google Scholar] [CrossRef]
- Sebastian, J.; Swaminath, S.; Nair, R.R.; Jakkala, K.; Pradhan, A.; Ajitkumar, P. De novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro. Antimicrob. Agents Chemother. 2017, 61, e01343–16. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Nair, R.R.; Jakkala, K.; Pradhan, A.; Ajitkumar, P. Elevated levels of three reactive oxygen species and Fe(II) in the antibiotic-surviving population of mycobacteria facilitate de novo emergence of genetic resisters to antibiotics. Antimicrob. Agents Chemother. 2022, 66, e0228521. [Google Scholar] [CrossRef]
- Piccaro, G.; Pietraforte, D.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 7527–33. [Google Scholar] [CrossRef]
- Iacobino, A.; Piccaro, G.; Pardini, M.; Fattorini, L.; Giannoni, F. Moxifloxacin activates the SOS response in Mycobacterium tuberculosis in a dose- and time-dependent manner. Microorganisms. 2021, 9, 255. [Google Scholar] [CrossRef]
- Shee, S.; Singh, S.; Tripathi, A.; Thakur, C.; Kumar, T.A.; Das, M.; Yadav, V.; Kohli, S.; Rajmani, R.S.; Chandra, N.; et al. Moxifloxacin-mediated killing of Mycobacterium tuberculosis involves respiratory downshift, reductive stress, and accumulation of reactive oxygen species. Antimicrob. Agents Chemother. 2022, 66, e0059222. [Google Scholar] [CrossRef]
- Singh, A.; Zhao, X.; Drlica, K. Fluoroquinolone heteroresistance, antimicrobial tolerance, and lethality enhancement. Front. Cell. Infect. Microbiol. 2022, 12, 938032. [Google Scholar] [CrossRef]
- Ehrt, S.; Schnappinger, D.; Rhee, K.Y. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 496–507. [Google Scholar] [CrossRef]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio. 2011, 2, e00100–11. [Google Scholar] [CrossRef]
- Torrey, H.L.; Keren, I.; Via, L.E.; Lee, J.S.; Lewis, K. High persister mutants in Mycobacterium tuberculosis. PLoS One. 2016, 11, e0155127. [Google Scholar] [CrossRef]
- Hu, Y.; Pertinez, H.; Ortega-Muro, F.; Alameda-Martin, L.; Liu, Y.; Schipani, A.; Davies, G.; Coates, A. Investigation of elimination rate, persistent subpopulation removal, and relapse rates of Mycobacterium tuberculosis by using combinations of first-line drugs in a modified Cornell mouse model. Antimicrob. Agents Chemother. 2016, 60, 4778–85. [Google Scholar] [CrossRef]
- Salina, E.G.; Makarov, V. Mycobacterium tuberculosis dormancy: how to fight a hidden danger. Microorganisms. 2022, 10, 2334. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.R.; Venugopal, U.; Chandra, G.; Bharti, S.; Maurya, R.K.; Krishnan, M.Y. Effect of various drugs on differentially detectable persisters of Mycobacterium tuberculosis generated by long-term lipid diet. Tuberculosis (Edinb). 2019, 115, 89–95. [Google Scholar] [CrossRef]
- Mesman, A.W.; Baek, S.H.; Huang, C.C.; Kim, Y.M.; Cho, S.N.; Ioerger, T.R.; Barreda, N.N.; Calderon, R.; Sassetti, C.M.; Murray, M.B. Characterization of drug-resistant lipid-dependent differentially detectable Mycobacterium tuberculosis. J. Clin. Med. 2021, 10, 3249. [Google Scholar] [CrossRef] [PubMed]
- Gordhan, B.G.; Sewcharran, A.; Letsoalo, M.; Chinappa, T.; Yende-Zuma, N.; Padayatchi, N.; Naidoo, K.; Kana, B.D. Detection of differentially culturable tubercle bacteria in sputum from drug-resistant tuberculosis patients. Antimicrob. Agents Chemother. 2022, 12, 949370. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, D.V.; Salina, E.G.; Fursov, M.V.; Skvortsov, T.A.; Azhikina, T.L.; Kaprelyants, A.S. Dormant non-culturable Mycobacterium tuberculosis retains stable low-abundant mRNA. BMC Genomics. 2015, 16, 954. [Google Scholar] [CrossRef]
- Turapov, O.; O’Connor, B.D.; Sarybaeva, A.A.; Williams, C.; Patel, H.; Kadyrov, A.S.; Sarybaev, A.S.; Woltmann, G.; Barer, M.R.; Mukamolova, G.V. Phenotypically adapted Mycobacterium tuberculosis populations from sputum are tolerant to first-line drugs. Antimicrob Agents Chemother. 2016, 60, 2476–83. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Mishra, S.; Warrier, T.; Cicchetti, N.; Mi, J.; Weber, E.; Jiang, X.; Roberts, J.; Gouzy, A.; Kaplan, E.; et al. Oxidative damage and delayed replication allow viable Mycobacterium tuberculosis to go undetected. Sci Transl Med. 2021, 13, eabg2612. [Google Scholar] [CrossRef]
- Hong, Y.; Zeng, J.; Wang, X.; Drlica, K.; Zhao, X. Post-stress bacterial cell death mediated by reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 10064–10071. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.Y.; Kim, P.; Zhang, L.; Kang, S.; Keller, T.H.; Jiricek, J.; Barry, C. E 3rd. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008, 322, 1392–5. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, J.; Sun, M.; Zhang, X.; Cook, G.M.; Zhang, T. Nitric oxide-dependent electron transport chain inhibition by the cytochrome bc1 inhibitor and pretomanid combination kills Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2021, 65, e0095621. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Maritz, J.S.; Venter, A.; van Helden, P.D.; Andries, K.; McNeeley, D.F.; Donald, P.R. Time to detection of the growth of Mycobacterium tuberculosis in MGIT 960 for determining the early bactericidal activity of antituberculosis agents. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1561–5. [Google Scholar] [CrossRef]
- Bowness, R.; Boeree, M.J.; Aarnoutse, R.; Dawson, R.; Diacon, A.; Mangu, C.; Heinrich, N.; Ntinginya, N.E.; Kohlenberg, A.; Mtafya, B.; et al. The relationship between Mycobacterium tuberculosis MGIT time to positivity and cfu in sputum samples demonstrates changing bacterial phenotypes potentially reflecting the impact of chemotherapy on critical sub-populations. J Antimicrob Chemother. 2015, 70, 448–55. [Google Scholar] [CrossRef]
- Filippini, P.; Iona, E.; Piccaro, G.; Peyron, P.; Neyrolles, O.; Fattorini, L. Activity of drug combinations against dormant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 2712–5. [Google Scholar] [CrossRef]
- Maitra, A.; Solanki, P.; Sadouki, Z.; McHugh, T.D.; Kloprogge, F. Improving the drug development pipeline for mycobacteria: modelling antibiotic exposure in the hollow fibre infection model. Antibiotics 2021, 10, 1515. [Google Scholar] [CrossRef]
- Gumbo, T.; Srivastava, S.; Deshpande, D.; Pasipanodya, J.G.; Berg, A.; Romero, K.; Hermann, D.; Hanna, D. Hollow-fibre system model of tuberculosis reproducibility and performance specifications for best practice in drug and combination therapy development. J. Antimicrob. Chemother. 2023, dkad029. [Google Scholar] [CrossRef]
- Gumbo, T.; Sherman, CM.; Deshpande, D.; Alffenaar, J.W.; Srivastava, S. Mycobacterium tuberculosis sterilizing activity of faropenem, pyrazinamide and linezolid combination and failure to shorten the therapy duration. Int. J. Infect. Dis. 2021, 104, 680–684. [Google Scholar] [CrossRef]
- Piccaro, G.; Giannoni, F.; Filippini, P.; Mustazzolu, A.; Fattorini, L. Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob. Agents Chemother. 2013, 57, 1428–33. [Google Scholar] [CrossRef]
- Iacobino, A.; Giannoni, F.; Pardini, M.; Piccaro, G.; Fattorini, L. The combination rifampin-nitazoxanide, but not rifampin-isoniazid-pyrazinamide-ethambutol, kills dormant Mycobacterium tuberculosis in hypoxia at neutral pH. Antimicrob. Agents Chemother. 2019, 63, e00273–19. [Google Scholar] [CrossRef]
- Lanni, A.; Borroni, E.; Iacobino, A.; Russo, C.; Gentile, L.; Fattorini, L.; Giannoni, F. Activity of drug combinations against Mycobacterium abscessus grown in aerobic and hypoxic conditions. Microorganisms. 2022, 10, 1421. [Google Scholar] [CrossRef]
- Olender, D.; Żwawiak, J.; Zaprutko, L. Multidirectional efficacy of biologically active nitro compounds included in medicines. Pharmaceuticals (Basel). 2018, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Nuermberger, E.; Rosenthal, I.; Tyagi, S.; Williams, K.N.; Almeida, D.; Peloquin, C.A.; Bishai, W.R.; Grosset, J.H. Combination chemotherapy with the nitroimidazopyran PA-824 and first-line drugs in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2006, 50, 2621–5. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Yan, B.S.; Rojas, M.; Shebzukhov, Y.V.; Zhou, H.; Kobzik, L.; Higgins, D.E.; Daly, M.J.; Bloom, B.R.; Kramnik, I. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005, 434, 767–72. [Google Scholar] [CrossRef] [PubMed]
- Driver, E.R.; Ryan, G.J.; Hoff, D.R.; Irwin, S.M.; Basaraba, R.J.; Kramnik, I.; Lenaerts, A.J. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3181–95. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, I.M.; Tasneen, R.; Peloquin, C.A.; Zhang, M.; Almeida, D.; Mdluli, K.E.; Karakousis, P.C.; Grosset, J.H.; Nuermberger, E.L. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 4331–40. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Li, S.Y.; Peloquin, C.A.; Taylor, D.; Williams, K.N.; Andries, K.; Mdluli, K.E.; Nuermberger, EL. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 5485–92. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, I.M.; Williams, K.; Tyagi, S.; Peloquin, C.A.; Vernon, A.A.; Bishai, W.R.; Grosset, J.H.; Nuermberger, E.L. Potent twice-weekly rifapentine-containing regimens in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2006, 174, 94–101. [Google Scholar] [CrossRef]
- Rosenthal, I.M.; Zhang, M.; Williams, K.N.; Peloquin, C.A.; Tyagi, S.; Vernon, A.A.; Bishai, W.R.; Chaisson, R.E.; Grosset, J.H.; Nuermberger, E.L. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 2007, 4, e344. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, I.M.; Zhang, M.; Almeida, D.; Grosset, J.H.; Nuermberger, E.L. Isoniazid or moxifloxacin in rifapentine-based regimens for experimental tuberculosis? Am. J. Respir. Crit. Care Med. 2008, 178, 989–93. [Google Scholar] [CrossRef] [PubMed]
- Nuermberger, E.; Tyagi, S.; Tasneen, R.; Williams, K.N.; Almeida, D.; Rosenthal, I.; Grosset, J.H. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 1522–4. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Tyagi, S.; Williams, K.; Grosset, J.; Nuermberger, E. Enhanced bactericidal activity of rifampin and/or pyrazinamide when combined with PA-824 in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 3664–8. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Minkowski, A.; Amoabeng, O.; Peloquin, CA.; Taylor, D.; Andries, K.; Wallis, R.S.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3114–20. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Tyagi, S.; Minkowski, A.; Peloquin, C.A.; Grosset, J.H.; Nuermberger, E.L. Contribution of moxifloxacin or levofloxacin in second-line regimens with or without continuation of pyrazinamide in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2013, 188, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Williams, K.; Amoabeng, O.; Minkowski, A.; Mdluli, K.E.; Upton, A.M.; Nuermberger, E.L. Contribution of the nitroimidazoles PA-824 and TBA-354 to the activity of novel regimens in murine models of tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 129–35. [Google Scholar] [CrossRef]
- Tasneen, R.; Betoudji, F.; Tyagi, S.; Li, S.Y.; Williams, K.; Converse, P.J.; Dartois, V.; Yang, T.; Mendel, C.M.; Mdluli, K.E.; et al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 60, 270–7. [Google Scholar] [CrossRef]
- Lanoix, J.P.; Betoudji, F.; Nuermberger, E. Sterilizing activity of pyrazinamide in combination with first-line drugs in a C3HeB/FeJ mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 60, 1091–6. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, A.; Ortega-Muro, F.; Alameda-Martin, L.; Mitchison, D.; Coates, A. High-dose rifampicin kills persisters, shortens treatment duration, and reduces relapse rate in vitro and in vivo. Front. Microbiol. 2015, 6, 641. [Google Scholar] [CrossRef]
- Li, S.Y.; Tasneen, R.; Tyagi, S.; Soni, H.; Converse, P.J.; Mdluli, K.; Nuermberger, E.L. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00913–17. [Google Scholar] [CrossRef] [PubMed]
- Ammerman, N.C.; Swanson, R.V.; Bautista, E.M.; Almeida, D.V.; Saini, V.; Omansen, T.F.; Guo, H.; Chang, Y.S.; Li, S.Y.; Tapley, A.; et al. Impact of clofazimine dosing on treatment shortening of the first-line regimen in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00636–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, S.Y.; Almeida, D.V.; Tasneen, R.; Barnes-Boyle, K.; Converse, P.J.; Upton, A.M.; Mdluli, K.; Fotouhi, N.; Nuermberger, E.L. Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00021–19. [Google Scholar] [CrossRef]
- Saini, V.; Ammerman, N.C.; Chang, Y.S.; Tasneen, R.; Chaisson, R.E.; Jain, S.; Nuermberger, E.; Grosset, J.H. Treatment-shortening effect of a novel regimen combining clofazimine and high-dose rifapentine in pathologically distinct mouse models of tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00388–19. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Garcia, A.; Converse, P.J.; Zimmerman, MD.; Dartois, V.; Kurbatova, E.; Vernon, A.A.; Carr, W.; Stout, J.E.; Dooley, K.E.; et al. Novel regimens of bedaquiline-pyrazinamide combined with moxifloxacin, rifabutin, delamanid and/or OPC-167832 in murine tuberculosis models. Antimicrob. Agents Chemother. 2022, 66, e0239821. [Google Scholar] [CrossRef]
- Hibma, J.E.; Radtke, K.K.; Dorman, S.E.; Jindani, A.; Dooley, K.E.; Weiner, M.; McIlleron, HM.; Savic, RM. Rifapentine population pharmacokinetics and dosing recommendations for latent tuberculosis infection. Am. J. Respir. Crit. Care Med. 2020, 202, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Benator, D.; Bhattacharya, M.; Bozeman, L.; Burman, W.; Catanzaro, A.; Chaisson, R.; Gordin, F.; Horsburgh, C.R.; Horton, J.; et al. Rifapentine and isoniazid once a week versus rifampicin and isoniazid twice a week for treatment of drug-susceptible pulmonary tuberculosis in HIV-negative patients: a randomised clinical trial. Lancet. 2002, 360, 528–534. [Google Scholar] [CrossRef]
- 106. Centers for Disease Control and Prevention (CDC); American Thoracic Society. Update: adverse event data and revised American Thoracic Society/CDC recommendations against the use of rifampin and pyrazinamide for treatment of latent tuberculosis infection--United States. MMWR Morb. Mortal. Wkly Rep. 2003; 52, 735-9.
- Svensson, E.M.; Murray, S.; Karlsson, M.O.; Dooley, K.E. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti-TB drug. J. Antimicrob. Chemother. 2015, 70, 1106–14. [Google Scholar] [CrossRef]
- Gopal, P.; Grüber, G.; Dartois, V, Dick, T. Pharmacological and molecular mechanisms behind the sterilizing activity of pyrazinamide. Trends Pharmacol. Sci. 2019, 40, 930–940. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, W. ; Zhang, W, Mitchison, D. Mechanisms of pyrazinamide action and resistance. Microbiol. Spectr. 2014; 18, 10–128. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Via, L.E.; Weiner, D.; Blanc, L.; Boshoff, H.; Eugenin, E.A.; Barry, C.E. 3rd.; Dartois, V.A. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob Agents Chemother. 2018, 62, e02266–17. [Google Scholar] [CrossRef]
- Iseman, M.D. Tuberculosis therapy: past, present and future. Eur. Respir. J. Suppl. 2002, 36, 87s–94s. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO consolidated guidelines on tuberculosis. Module 4: treatment—drug-susceptible tuberculosis treatment. ISBN 978-92-4-004812-6. WHO: Geneva, Switzerland, 2022.
- Jindani, A.; Harrison, T.S.; Nunn, A.J.; Phillips, P.P.; Churchyard, G.J.; Charalambous, S. ; Hatherill, M, Geldenhuys, H.; McIlleron, H.M.; Zvada, S.P.; et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N. Engl. J. Med. 2014; 371, 1599–1608. [Google Scholar] [CrossRef]
- Dorman, S.E.; Nahid, P.; Kurbatova, E.V.; Phillips, P.P.J.; Bryant, K.; Dooley, K.E.; Engle, M.; Goldberg, S.V.; Phan, H.T.T.; Hakim, J.; et al. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med. 2021, 384, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Paton, N.I.; Cousins, C.; Suresh, C.; Burhan, E.; Chew, K.L.; Dalay, V.B. ; Lu, Q, Kusmiati, T.; Balanag, V.M.; Lee, S.L.; et al. Treatment strategy for rifampin-susceptible tuberculosis. N. Engl. J. Med. 2023; 388, 873–887. [Google Scholar] [CrossRef]
- WHO. Treatment of drug-susceptible tuberculosis: rapid communication (June 2021). ISBN 978-92-4-002867-8. WHO: Geneva, Switzerland, 2021.
- Dartois, V.; Rubin, E.J. Shortening tuberculosis treatment—a strategic retreat. N. Engl. J. Med. 2023, 388, 939–941. [Google Scholar] [CrossRef]
- http://www.drugbank.ca.
- Sarathy, J.P.; Zuccotto, F.; Hsinpin, H.; Sandberg, L.; Via, L.E.; Marriner, G.A.; Masquelin, T.; Wyatt, P.; Ray, P.; Dartois, V. Prediction of drug penetration in tuberculosis lesions. ACS Infect Dis. 2016, 2, 552–63. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Dartois, V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev. 2020, 33, e00159-19. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.P.; Xie, M.; Jones, R.M.; Chang, A.; Osiecki, P.; Weiner, D.; Tsao, W.S.; Dougher, M. ; Blanc, L, Fotouhi, N.; Via, L.E, Barry, C.E. 3rd.; et al. A novel tool to identify bactericidal compounds against vulnerable targets in drug-tolerant M. tuberculosis found in caseum. mBio. 2023; 5, e0059823. [Google Scholar] [CrossRef]
- Kempker, R.R.; Heinrichs, M.T.; Nikolaishvili, K.; Sabulua, I.; Bablishvili, N.; Gogishvili, S.; Avaliani, Z.; Tukvadze, N.; Little, B.; Bernheim, A.; et al. Lung tissue concentrations of pyrazinamide among patients with drug-resistant pulmonary tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00226–17. [Google Scholar] [CrossRef]
- McLeay, S.C.; Vis, P.; van Heeswijk, R.P.; Green, B. Population pharmacokinetics of bedaquiline (TMC207), a novel antituberculosis drug. Antimicrob. Agents Chemother. 2014, 58, 5315–5324. [Google Scholar] [CrossRef]
- Irwin, S.M.; Prideaux, B.; Lyon, E.R.; Zimmerman, M.D.; Brooks, E.J.; Schrupp, C.A.; Chen, C.; Reichlen, M.J.; Asay, B.C. ; Voskui,l M.I.; et al. Bedaquiline and pyrazinamide treatment responses are affected by pulmonary lesion eterogeneity in Mycobacterium tuberculosis infected C3HeB/FeJ mice. ACS Infect Dis. 2016; 2, 251–267. [Google Scholar] [CrossRef]
- Conradie, F.; Bagdasaryan, T.R.; Borisov, S.; Howell, P.; Mikiashvili, L.; Ngubane, N.; Samoilova, A.; Skornykova, S.; Tudor, E.; Variava, E.; et al. Bedaquiline-pretomanid-linezolid regimens for drug-resistant tuberculosis. N. Engl. J. Med. 2022, 387, 810–823. [Google Scholar] [CrossRef]
- Nyang’wa, B.T.; Berry, C.; Kazounis, E.; Motta, I.; Parpieva, N.; Tigay, Z.; Solodovnikova, V.; Liverko, I.; Moodliar, R.; Dodd, M.; et al. A 24-week, all-oral regimen for rifampin-resistant ruberculosis. N. Engl. J. Med. 2022, 387, 2331–2343. [Google Scholar] [CrossRef]
- Goodall, R.L.; Meredith, S.K.; Nunn, A.J.; Bayissa, A.; Bhatnagar, A.K.; Bronson, G.; Chiang, C.Y.; Conradie, F.; Gurumurthy, M.; Kirenga, B.; et al. Evaluation of two short standardised regimens for the treatment of rifampicin-resistant tuberculosis (STREAM stage 2): an open-label, multicentre, randomised, non-inferiority trial. Lancet. 2022, 400, 1858–1868. [Google Scholar] [CrossRef]
- WHO. WHO consolidated guidelines on tuberculosis. Module 4: treatment—drug-resistant tuberculosis treatment, 2022 update. ISBN 978-92-4-006312-9. WHO: Geneva, Switzerland, 2022.
- Nuermberger, E.L. Preclinical efficacy testing of new drug candidates. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.; Clary, J.; Hanna, D.; Nuermberger, E.; Lenaerts, A.; Ammerman, N.; Ramey, M.; Hartley, D.; Hermann, D. Model-based meta-analysis of relapsing mouse model studies from the critical path to tuberculosis drug regimens initiative database. Antimicrob. Agents Chemother. 2022, 66, e0179321. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Percentages of sterilizing combinations shown in Table 1 (parts A and B), containing the drugs indicated. Data were stratified by the lenght of time required for sterilization. A) Z, pyrazinamide; B, bedaquiline; Pa, pretomanid (PA-824); P, rifapentine; M, moxifloxacin; R, rifampin; B) H, isoniazid; L, linezolid; C, clofazimine; E, ethambutol; S, sutezolid.
Figure 1.
Percentages of sterilizing combinations shown in Table 1 (parts A and B), containing the drugs indicated. Data were stratified by the lenght of time required for sterilization. A) Z, pyrazinamide; B, bedaquiline; Pa, pretomanid (PA-824); P, rifapentine; M, moxifloxacin; R, rifampin; B) H, isoniazid; L, linezolid; C, clofazimine; E, ethambutol; S, sutezolid.
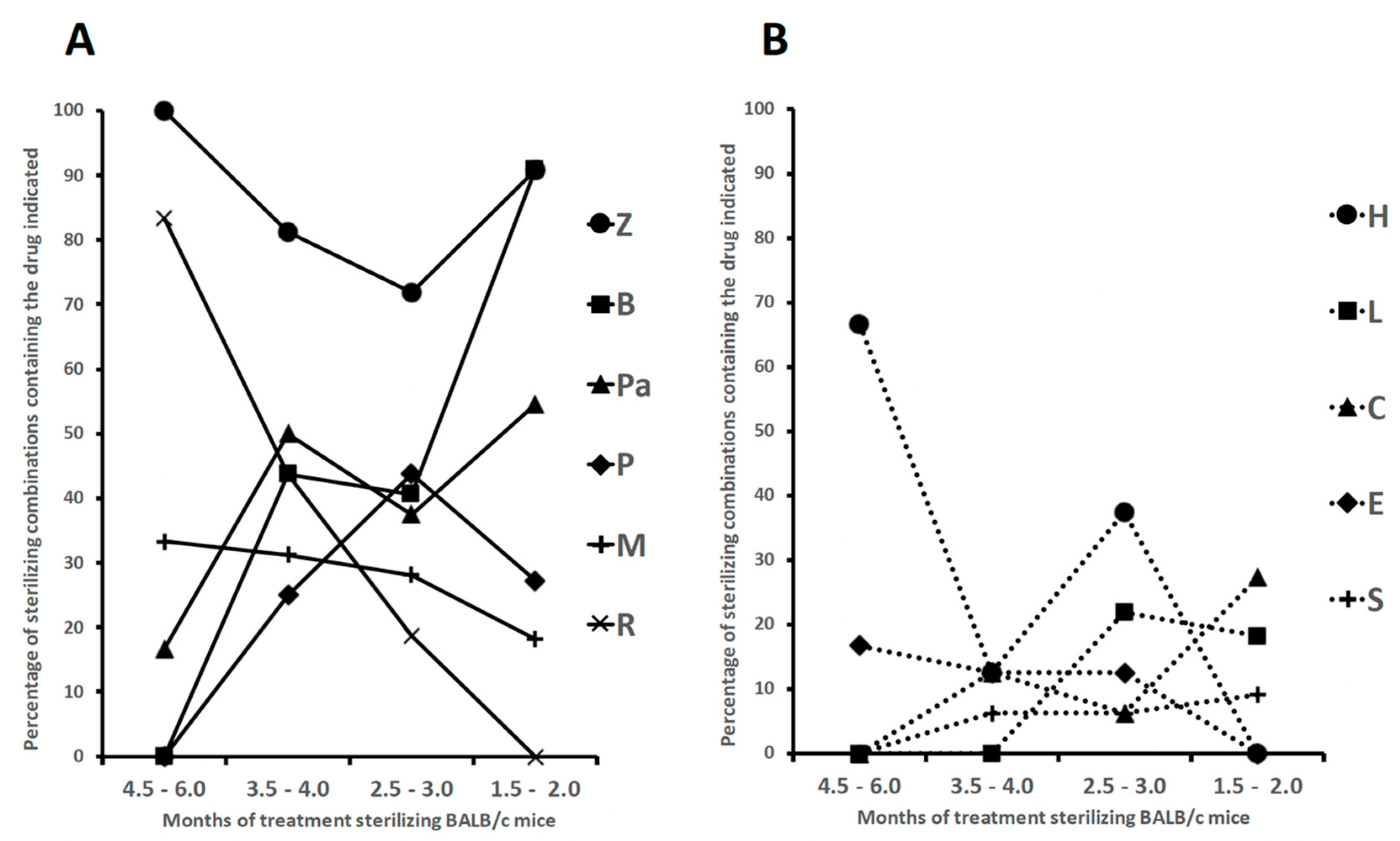

Table 1.
part A. Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 1-35). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment. part B (continued). Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 36-65). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment
Table 1.
part A. Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 1-35). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment. part B (continued). Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 36-65). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment
| part A |
 |
| part B |
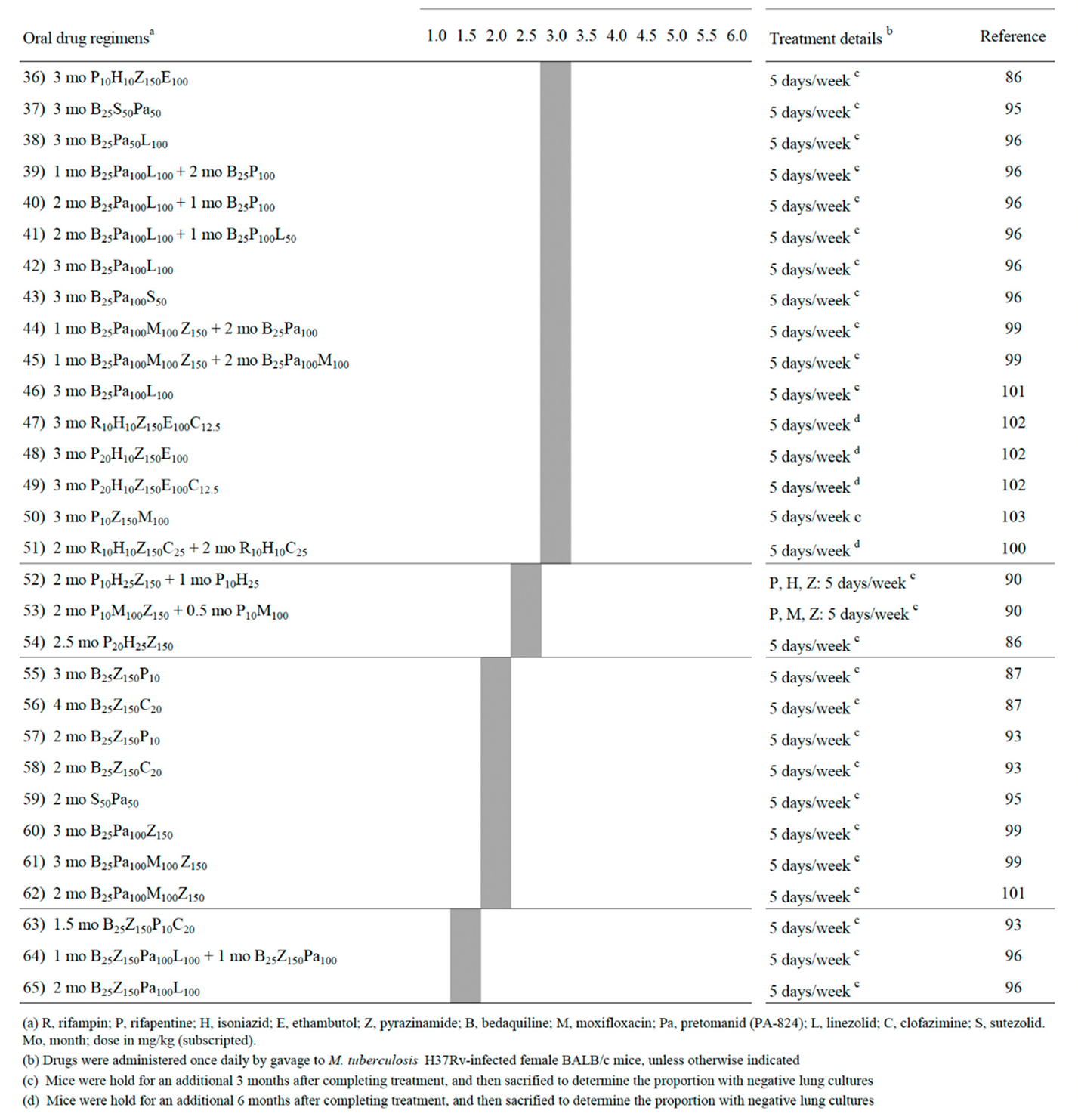 |
Table 2.
Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected C3HeB/FeJ mice Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment.
Table 2.
Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected C3HeB/FeJ mice Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment.
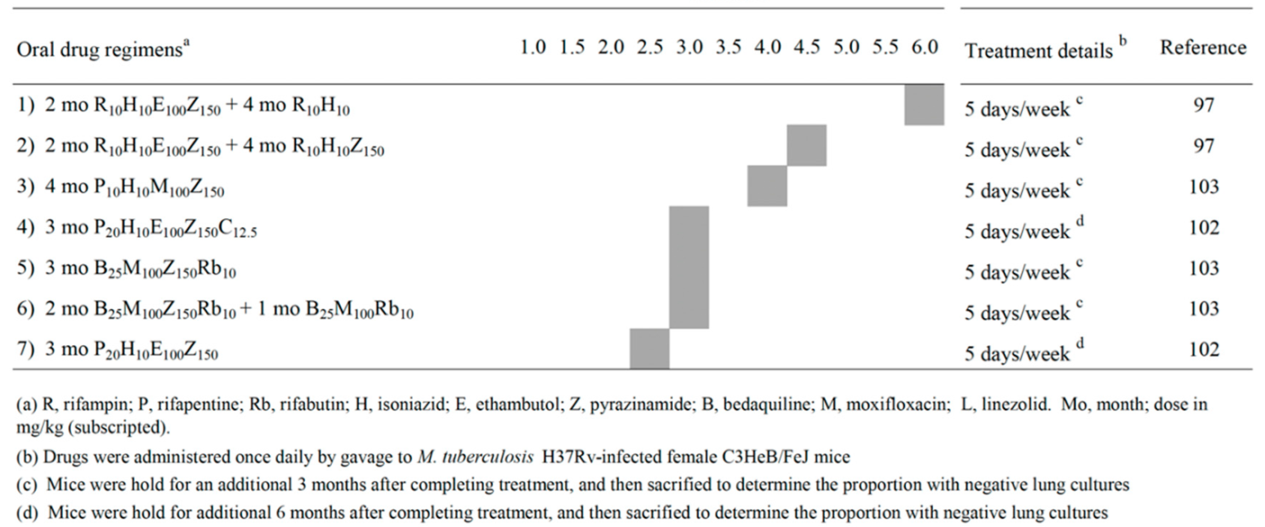 |
Table 3.
Oral regimens for treatment of drug-susceptible TB reported to be as effective as, or noninferior to, the control regimen.
Table 3.
Oral regimens for treatment of drug-susceptible TB reported to be as effective as, or noninferior to, the control regimen.
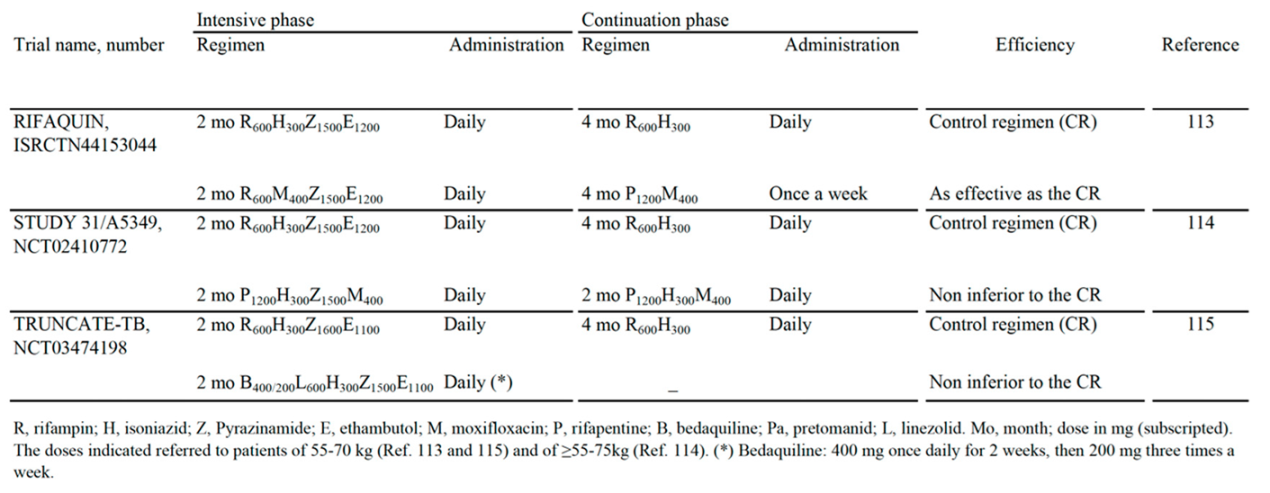 |
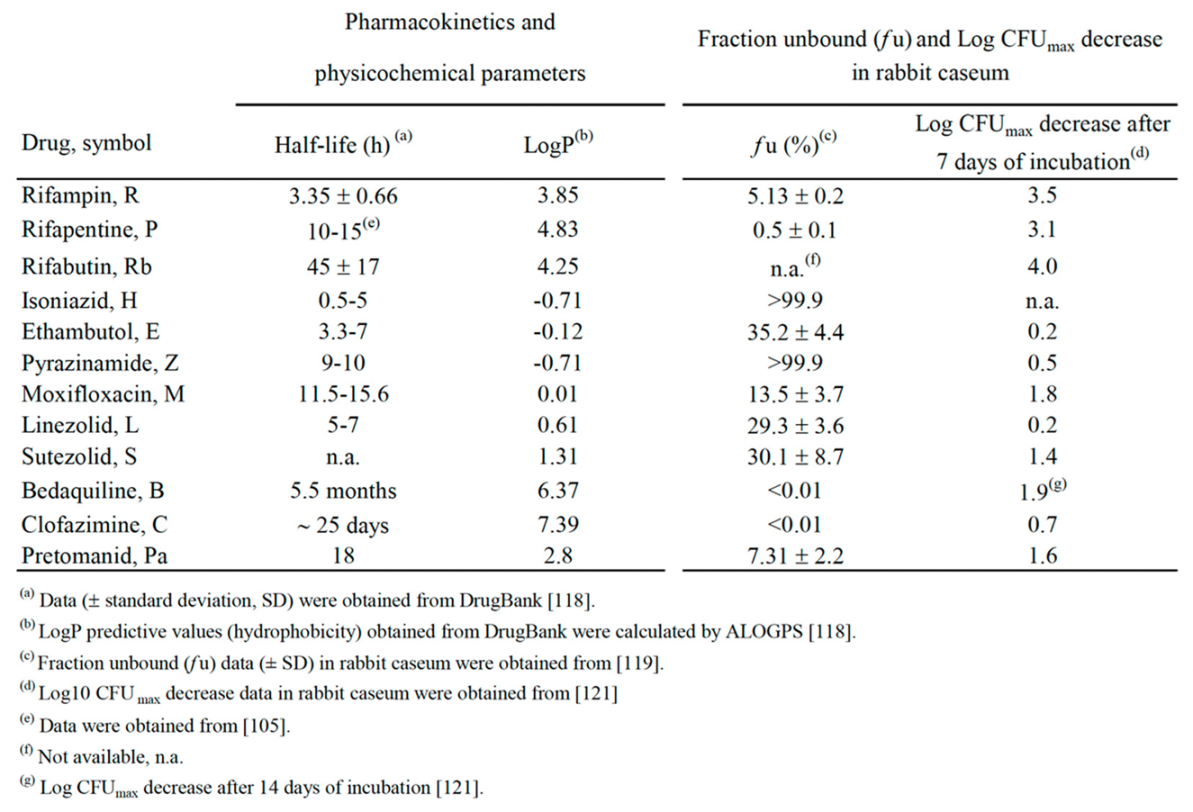
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Submitted:
29 April 2023
Posted:
29 April 2023
You are already at the latest version
Alerts
A peer-reviewed article of this preprint also exists.
This version is not peer-reviewed
Submitted:
29 April 2023
Posted:
29 April 2023
You are already at the latest version
Alerts
Abstract
The lungs of tuberculosis (TB) patients contain a spectrum of granulomatous lesions ranging from solid and well vascularized cellular granulomas, to avascular caseous granulomas. In solid granulomas, current therapy kills actively replicating (AR) intracellular bacilli, while in low vascularized caseous granulomas the low oxygen tension stimulates aerobic and microaerophilic AR bacilli to transit into non-replicating (NR), drug-tolerant, extracellular stages. These stages, which do not have genetic mutations and are often referred to as persisters, are difficult to eradicate due to low drug penetration inside caseum and mycobacterial cell walls. The sputum of TB patients contains also viable bacilli called differentially detectable (DD) cells that, unlike persisters, grow in liquid, but not in solid media. This review provides a comprehensive update on drug combinations killing in vitro AR and drug-tolerant bacilli (persisters and DD cells), and sterilizing Mycobacterium tuberculosis-infected BALB/c and caseum-forming C3HeB/FeJ mice. These observations have been important for testing new drug combinations in noninferiority clinical trials, in order to shorten duration of current regimens against TB. In 2022, the World Health Organization, based on one of this trial, supported the use of a 4-month regimen for treatment of drug-susceptible TB as a possible alternative to the current 6-month regimen.
Keywords:
Subject: Biology and Life Sciences - Immunology and Microbiology
1. Introduction
The World Health Organization (WHO) estimated that in 2021 approximately 10.6 million people developed tuberculosis (TB), and about 1.6 million died from the active disease, making TB one of the leading causes of death worldwide [1]. Furthermore, an estimated 1.7 billion people have latent TB infection (LTBI) [2]. The current antibiotic treatment of active, drug-susceptible (DS)-TB, requires administration of a combination therapy for 6 months, including the first-line drugs rifampin (R), isoniazid (H), ethambutol (E) and pyrazinamide (Z) (R-H-E-Z) for 2 months, followed by R and H (R-H) for 4 months. To prevent LTBI reactivation, a long treatment is also used, consisting of at least 6 months of H, or 3 to 4 months of R-H.
The lungs of patients with active TB and LTBI contain a spectrum of granulomatous lesions ranging from solid and well vascularized cellular granulomas, to avascular caseous granulomas [3,4,5]. In these lesions, heterogeneous subpopulations of Mycobacterium tuberculosis (Mtb) cells ranging from actively replicating (AR) to nonreplicating (NR) dormant stages, coexist. In solid granulomas, current therapy kills AR intracellular bacilli inside the macrophages, while in low vascularized caseous granulomas the low oxygen tension stimulates aerobic and microaerophilic AR bacilli to transit into NR, hypoxic, drug-tolerant stages. Hypoxic bacilli use host triacylglycerol and cell wall mycolates to accumulate lipid droplets in lipid-loaded (foamy) macrophages [6,7,8]. The foamy macrophages die via inflammatory and necrotic processes, and release lipid droplets into the hypoxic necrotic core of closed caseous granulomas, which contain extracellular, slowly replicating or NR, phenotypically drug-resistant bacilli. Eradication of these bacilli by current TB therapy is difficult due to the low penetration of drugs inside caseous granulomas and NR Mtb cell walls [4,9]. Tubercle bacilli surviving drug treatment in the absence of genetic mutations inside necrotic foci of caseous granulomas are often referred to as persisters, and are thought to be responsible for the long duration of TB therapy [4]. For unclear reasons, in about 10% of LTBI-patients the closed caseous granulomas expand, fuse with the structures of the bronchial tree, and form cavities in which the caseum liquefies after coming into contact with the air. In the liquefied material, the NR cells rapidly multiply and are released into the airways as a mixture of AR and NR bacilli, which are detected in the sputum of contagious pulmonary TB patients as colony forming units (CFU) on solid media such us Middlebrook 7H10 agar plates and Löwenstein-Jensen slants [10,11].
However, the sputum of drug-untreated patients was found to contain also viable Mtb bacilli that were not detectable as CFU [12,13,14,15,16,17,18]. The number of these viable organisms in the sputum was estimated by a limiting dilution (LD) technique in liquid medium, using the most probable number (LD-MPN) method. These subpopulations were called with various terms, including viable but nonculturable cells (VBNC), differentially culturable TB (DCTB) cells and, perhaps the most recently used term, differentially detectable (DD) cells [16]. Several other bacterial species were found to exist in a VBNC state [19]. When the number of Mtb CFU is subtracted from the number of Mtb cells obtained by the LD-MPN method, the difference is the DD Mtb number [16]. Tubercle bacilli growing in liquid but not on solid media were also found in chronic, murine TB [20]. Some studies showed that DD cells decreased less rapidly than CFU after initiation of TB treatment, suggesting that they are also drug-tolerant cells [15,18].
Overall, these observations on dormant, drug-tolerant bacilli (persister cells and DD cells), are important for understanding the heterogeneous nature of the Mtb infection, and may aid in finding new drugs and in designing shorter drug combination-containing regimens. The goal of this review is to give an overview on the reported in vitro and in vivo combinations eradicating all dormant Mtb stages (persister cells and DD cells), and on their impact on the therapeutic strategies endorsed by the WHO to shorten the microbiological and therapeutical management of DS- and drug-resistant (DR)-TB.
2. Persister cells
2.1. Overview
Persister cells (persisters) were reported in several Gram-positive and Gram-negative bacteria [21] and in many eukaryotes including yeasts (Candida spp) [22] and protozoa (Plasmodium, Leishmania, Trypanosoma and Toxoplasma spp) [23], where they contribute to establish persistent infections and treatment failures, without genetic drug resistance. Persisters were also described in human cancer, where they survive chemotherapy and promote development of drug resistance [24,25]. All investigators studying persisters uniformly highlight the issue that their eradication could lead to a strong reduction of treatment failures and relapses in several clinical settings. The underlying reasons for persisters development seem to be multifactorial, involving non-genetic transient mechanisms switched on in microorganisms and cancer cells, as well as sub-optimal pharmacodynamics and immune responses in the host.
2.2. Bacterial persisters
The term “persisters” was used for the first time in 1944 by Joseph Bigger, who realized that penicillin failed to sterilize Staphylococcus pyogenes cultures due to generation of small number of cocci “individually insensitive to penicillin, but developing normally when penicillin is destroyed” [21,26,27]. Well in advance, he specified that “persisters are not resisters”, i.e., cells that we now call antibiotic resistant mutants. Bigger also observed that “persisters are believed to be insensitive to penicillin because they are in a dormant, non-dividing phase”.
In the search for persisters genes, starting from the 1980s, Harris Moyed and coworkers isolated mutants of Escherichia coli with vastly increased frequency of persistence (Hip mutants) [28]. The hip locus affects the frequency of persistence to the lethal consequences of selective inhibition of either DNA or peptidoglycan synthesis [29]. The hipA7 strain produced about 1% persisters and, after exposure to ampicillin, it generated about 1,000 times more persisters than the wild-type strain [27].
In 2019, Nathalie Balaban et al., pushed by increased concerns on antibiotic resistance and several papers published on the persistence phenomenon, released an international consensus statement on definitions and guidelines for research on antibiotic persistence [30]. Antibiotic resistance was defined as the ability of bacteria to replicate, and not just survive, in the presence of a drug, with the minimum inhibitory concentration (MIC) being the most common measure of the resistance level. Resistance is inherited and may be acquired by genetic mutations, or horizontal gene transfer of resistance-encoding genes. Instead, antibiotic persistence is the ability of a subpopulation to survive exposure to a bactericidal concentration [30]. The hallmark of antibiotic persistence is the biphasic killing curve. When persisters regrow in the absence of antibiotics, they give rise to a population of drug-sensitive cells. The size of the persister subpopulation is related to the class of drug and not to the drug concentration. Persisters cannot replicate in the presence of a drug, and are killed at a lower rate than the susceptible population from which they arise. The terms persistence and tolerance refer both to increased survival in the presence of an antibiotic, with no MIC increase. However, persistence refers to a cell subpopulation, while tolerance deals with a general ability of a population to survive longer drug treatments. Thus, persisters are a subpopulation of drug-tolerant bacteria, and tolerance and persistence may arise without dormancy [30,31]. The time required to kill these populations is quantified as the minimum duration for killing, and is useful to compare their survival fractions. The international consensus statement also showed that there are two kinds of persistence, namely type I, triggered by a stress signal (nutrient starvation, high cell number, acid stress, immune factors), and type II (spontaneous), which develops stochastically without any trigger, at a rate that is constant during growth [30,32,33,34]. The spontaneous and drug-induced persistence are difficult to distinguish and can be studied by direct observations of single bacterial cells by microfluidic devices [30,32,33]. Persistent bacterial infections cause death of millions of people every year. In 2022, a bibliometric analysis on the top 100 cited studies on bacterial persisters was reported [35].
2.3. Mycobacterium tuberculosis persisters
M. tuberculosis persisters are NR multidrug-tolerant bacilli genetically identical to drug-susceptible cells, which survive in the hostile environments of granulomas and in the sputum and adipose tissues of TB patients [36,37,38]. These cells are primarily involved in LTBI and TB relapses. Microenvironments with low oxygen tension and pH, nutrient starvation, reactive oxygen and nitrogen stresses, lead to formation of reservoirs of phenotypically drug-resistant cells with reduced antibiotic uptake and increased drug efflux, which renders antibiotics ineffective without genetic resistance [9,36,39]. Responses against drug-treatment of Mtb under hypoxic conditions and other stresses were modeled and/or reviewed in several papers [11,40,41,42,43,44,45].
The primary reason for the 6-month duration of the first-line TB therapy (R-H-E-Z) is due to the ability of Mtb to generate heterogeneous subpopulations adapting to different microenvironments within granulomas. Microfluidic devices for single-cell analysis revealed that the wide diversity of tubercle bacilli can be due to the fact that the mycobacterial cell division is asymmetrical, with the old pole elongating more than the new pole [46,47]. Asymmetric distribution of irreversibly oxidated proteins generates Mtb sister cells with different growth properties and drug susceptibility. In particular, sister cells with a low amount of these proteins are less sensitive to drugs, likely contributing to persisters formation. Asymmetric growth in mycobacteria is regulated mainly by the division protein Wag31 and the growth inhibitor LamA [46].
Other single-cells studies reported that Mtb persisters are rare low-energy cells formed stochastically during normal growth [48]. Stochastic variations may occur in the expression of an energy-generating component. Indeed, Mtb cells cultivated in a minimal medium with acetate showed cell-to-cell variations in the level of mRNA coding for the acetate kinase (AckA), a low energy-generating mechanism. This led to the formation of persister cells with decreased AckA, which showed low ATP levels and inactive drug targets. The knowledge that Staphylococcus aureus and E. coli persisters also have low ATP levels is in keeping with these observations [49,50]. In this view, it was hypothesized that all culture conditions leading to persistence converge in the inability to re-initiate a new round of DNA replication caused by insufficient level of the initiator complex ATP-DnaA, resulting in the lack of formation of a functional orisome [51]. Mycobacteria heterogeneity generated in part by stochastic processes may be amplified by innate asymmetric growth and division patterns [46].
Of importance, persister cells may generate genetically resistant mutants. Indeed, prolonged exposure to lethal concentrations of R or the fluoroquinolone moxifloxacin (M) induced the formation of Mtb and Mycobacterium smegmatis persisters, which carried elevated levels of the reactive oxygen species (ROS) hydroxyl radical, superoxide radical and hydrogen peroxide [52,53,54]. These radicals inflicted extensive genome-wide DNA mutations, as shown by generation of R- and M-resistant mutants carrying clinically relevant mutations in the rpoB and gyrA genes, respectively [52,53]. It is known that Mtb and other pathogens showing high frequency of mutation-based antibiotic resistance, produce ROS-generating enzymes (e.g., NADH dehydrogenase, succinate dehydrogenase, aspartate oxidase), while bacteria with low mutation frequency lack these enzymes [51].
Many bacteria react to ROS-generated DNA lesions caused by the bactericidal drugs quinolones, aminoglycosides and beta-lactams by inducing the SOS gene regulon, which is regulated by the transcriptional repressor LexA and the protease RecA. In Mtb, we found that treatment with M induced overexpression of SOS genes (e.g., recA, lexA, dnaE2) and DNA repair genes [55]. Induction of the error-prone DNA polymerase dnaE2, which potentially drives increased mutation rates, is considered to be a link between phenotypic persistence and genotypic resistance. Other investigators reported that M-mediated killing of Mtb involved accumulation of NADH-dependent ROS, and that repair of M-mediated lesions and ROS detoxification mechanisms contributed to survival [56,57]. Looking for a therapeutic significance of these observations, they found that Mtb treatment with the respiration stimulator N-acetyl cysteine accelerated respiration, and that ROS production increased M-induced lethality, and lowered the mutant prevention concentration. A study proposed that certain metabolic enzymes of Mtb (e.g., isocitrate lyase) serve antioxidant functions and facilitate drug tolerance [58]. Overall, these observations documented the links among bactericidal antibiotics, ROS production and formation of drug-tolerant persisters, i.e of specialized survivors characterized by anoxic metabolism and reduced ATP and ROS synthesis [51].
Low numbers of Mtb persisters are present in the lag and early exponential phases, but reach about 1% in the stationary phase [36,59]. Transcriptome analysis of D-cycloserine-formed Mtb persisters showed a distinct pattern of dormancy characterized by general metabolic downshift and upregulation of a small number of genes overexpressed also in other in vitro dormancy models, including the alpha crystallin heat shock protein Acr2 and the sigma factors SigG and SigF. Transcriptome analysis of high persisters (hip) Mtb mutants, selected with a lethal dose of R and streptomycin, showed upregulation of some toxin-antitoxins, i.e., the modules in which the toxins lead to growth arrest when labile antitoxins are degraded by proteases [36,60]. hip-mutants showed up to 1,000 times more persisters than the wild-type strain, a decrease in the phospholipid biosynthesis, and mutations in the cell wall lipid phthiocerol dimycocerosate, an important cell wall lipid and virulence factor. These and other genetic mechanisms suggested that Mtb persisters formation involves multiple pathways, and that hip-mutants, obtained using both the reference strain H37Rv and human clinical isolates, may contribute to the tolerance of Mtb to drug exposure. Research on Mtb persisters genes will facilitate development of more effective therapies to eradicate them from TB patients.
3. Differentially detectable (DD) Mtb cells
Several studies reported the presence of DD Mtb cells in the sputum produced by patients with active TB. These cells are not able to form colonies on standard glycerol-based solid media (7H10 agar plates and Löwenstein-Jensen slants) but can grow in liquid media supplemented with mycobacterial culture filtrates (CF) [12,13,14,15,16,17,18,61,62] or other substances, such as cyclic-AMP [17] and fatty acids [63,64]. The growth stimulatory effect of CF was ascribed to the five resuscitation promoting factors (Rpfs) of Mtb, which have the ability to resuscitate non-culturable cells [12,65]. However, the sputum contains also Rpfs-independent DD cells, which do not require Rpfs for growth, and CF-independent DD cells, which do not need CF to resuscitate in liquid media [65]. Thus, the growth stimulatory effect observed with CF is most likely the result of a combination of factors [17].
Dormant DD Mtb cells, which retain stable low-abundant mRNAs [66], may play a role in disease persistence due to their phenotypic resistance to anti-TB drugs [18,67]. An in vitro DD Mtb cells model was recently developed by Saito et al., allowing development of ≥90% DD cells after nutrient starvation in phosphate-buffered saline (PBS) followed by exposure to high R concentrations (PBS-R model) [14]. This model generated DD cells similar to those found in TB patients, and their transcriptional profiles may be useful for monitoring DD populations in the sputum [18].
It is known that DD-Mtb cells represented the majority of bacilli in the sputum of 21% of patients with DS-TB, and that this level increased to 65% after 2 weeks of treatment with first-line drugs [16]. In a study which enrolled subjects with DR-TB (94% MDR patients, i.e., resistant at least to H and R), DD-Mtb cells were found in the sputum of 29% of patients prior of treatment, and this amount stabilized at 31% after 2 weeks of treatment with a multidrug regimen that included Z, but not R, H and E. However, after 2 months of therapy, DD cells decreased to 13% and 0% in the sputa of DS- and DR-TB, respectively 16]. Another study reported that CF devoid of rpfs yielded a greater amount of DD cells in sputum from patients with MDR-TB, compared with those with R-monoresistant TB, suggesting that CF is dispensable for detection of DD cells from DR-strains [65].
In order to explore the biological mechanisms generating DD cells, Saito et al. found that Mtb can enter into a DD state after a sub-lethal oxidative stress damaging DNA, proteins and lipid components [68]. This conclusion was in line with the observations of Hong et al., who reported that ROS formation in response to drugs could prevent E. coli cells from growing as CFU, without killing them, and that ROS accumulation mediated by self-amplifying mechanisms continued after antibiotic removal [69]. Transcriptomic analysis showed that, after RIF treatment, DD Mtb cells underwent a partial loss of the oxidative stress response that promoted the formation of the DD phenotype [68]. Thus, Mtb must be able to mitigate oxidative stress to survive in the DD state. However, the capacity of Mtb to do so is limited, so that only intermediate ROS levels lead to DD Mtb. For instance, Mycobacteriun bovis and its vaccine derivative, the Bacillus Calmette-Guérin (BCG), are not able to do it, and do not generate DD cells because they succumb to ROS [68]. The observation that treatment of R-containing cultures in the presence of the antioxidant catalase increased the ability of DD Mtb to form CFU on agar plates, reinforced the relationship between oxidative stress and return to replicative capacity. Hong et al. showed that, above a certain host threshold, E. coli cells died, but below that threshold, provisions of anti-oxidants revealed a CFU population [68,69].
In summary, after treatment of Mtb with bactericidal antibiotics, increasing, self-amplifying, ROS levels may be generated. Surviving bacilli may then be detected as CFU (persister cells) if ROS levels are low, LD-MPN (DD cells) if ROS levels are intermediate, or not detected (dead cells) if ROS levels are high. M. tuberculosis cells are likely to exist in the host at various levels of oxidative stress and ability to replicate [68]. Overall, these observations indicate that, to eliminate Mtb persisters and DD cells from the host, it would be important to find new drugs/drug combinations inducing oxidative damage under aerobic conditions, and/or other stresses under hypoxic conditions such as the reactive nitrogen species (RNS) nitric oxide (NO). For instance, NO is induced by pretomanid (Pa, a new anti-TB drug formerly known as PA-824) inside NR Mtb cells [70]. Combinations of Pa with cytochrome bc1 inhibitors showed strong bactericidal activity against hypoxic NR Mtb [71].
Finally, besides LD-MPN and CFU counting, in the sputum of TB patients, the presence of a greater mycobacterial load in liquid than in solid media may be detected by the mycobacterial growth indicator tube (MGIT) time to positivity (TTP) assay, a semiquantitative measure of viable Mtb that showed an inverse relationship with the CFU [72,73,74]. The MGIT 960 instrument is an automated system monitoring the fluorescence of an oxygen sensor to detect growth of mycobacteria in culture tubes containing a modified Middlebrook 7H9 liquid medium, in which the TTP values are reported as days. A study found a strong correlation between CF+ LD-MPN and TTP values for smear-positive and smear-negative sputa [13]. These data indicate that both LD-MPN and MGIT TTP assays are useful to detect tubercle bacilli growing on liquid but not on solid media.
4. In Vitro Killing of AR+NR Mtb Cells by Drug Combinations
Tools to identify in vitro promising drug regimens for rapid clinical trial evaluation are needed. One such tool is the hollow fibre system model of TB (HFS-TB), a bioreactor containing tubular hollow fibres made of semi-permeable membranes, which showed a 94% predictive accuracy for clinical response rates and optimal exposures [75,76]. In 2015, the European Medicines Agency supported the use of HFS-TB for several drugs, to perform pharmacokinetics-pharmacodynamics monotherapy studies on microbial killing, and to design new combination regimens. After drug exposure, residual viability was evaluated by various tests, including the MGIT TTP assay. Using the HFS-TB model, combinations containing faropenem, linezolid (L) and Z sterilized Mtb cultures in 28 days, as shown by lack of re-growth of drug-treated cells in MGIT 960 tubes after 56 days of incubation (TTP >56 days) [77].
A more stringent TTP threshold value was used by our group, who defined Mtb killing of both AR and NR cells (the latter obtained in the Wayne model) as lack of regrowth of drug-treated bacilli after 100 days of incubation in MGIT tubes (TTP >100 days) [74,78,79]. Using this approach, we found that the 4-drug combinations R-M-metronidazole (Mz)-amikacin (Ak) and R-M-Mz-capreomycin (Cp) killed AR+NR Mtb in 21 days [74]. Furthermore, we found that under aerobic and hypoxic acidic conditions (pH of 5.8) likely mimicking those present inside the phagolysosomes of activated macrophages, the 4-drug combinations R-M-Ak-Pa killed AR+NR Mtb in 14 days, while R-M-Ak-nitazoxanide (Nz) killed them in 21 days [78]. Finally, we found that R-Nz-containing combinations, but not R-H-E-Z (currently used for TB therapy), killed NR Mtb in ≥28 days in hypoxia at pH of 7.3, i.e., under conditions likely mimicking those present inside caseous granulomas [79]. Using the MGIT TTP assay, we also reported that the combinations bedaquiline (B)-Ak-rifabutin (Rb)-clarithromycin (Cl)-nimorazole (Nm) and B-Ak-Rb-Cl-Mz-colistin (Cs) killed AR+NR cells of Mycobacterium abscessus in 42 and 56 days, respectively [80]. Overall, our data indicated that the nitro-compounds Mz, Pa, Nz and Nm were important components of combinations killing AR+NR mycobacterial cells. Nitro-compounds are known to induce RNS in anaerobic organisms like Giardia spp [81,82], and in NR Mtb [70]. It is possible that combinations containing ROS-inducing drugs like M [52,53,56,57] and R [52,53,54], and RNS-inducing agents like nitro-compounds, are essential to kill AR and NR stages, which contain drug-tolerant persisters and DD cells.
5. Drug Combinations Eradicating Mtb from BALB/c and C3HeB/FeJ Mice
The ability of a drug combination to sterilize TB lesions and preventing relapses due to drug-tolerant persisters and DD cells is determined by the synergistic activities of drug components. In the current regimen used for treatment of DS TB (R-H-E-Z), R and Z have higher sterilizing activity than H and E [83]. In the last decades, several efforts were done in order to find new combinations eradicating drug-tolerant bacilli residing in low vascularized, hypoxic lesions. Such hypoxic conditions are much more present in caseous granulomas of TB patients, and in Mtb-infected caseum-forming animals (e.g., guinea pigs, rabbits, C3HeB/FeJ mice), than in cellular granulomas of commonly used mice (e.g., Mtb-infected BALB/c mice), which do not form caseous granulomas, the hallmark of human TB. The guinea pig and rabbit models were not used for the study of combination chemotherapy regimens because of their prohibitive costs [83]. Instead, the C3HeB/FeJ mice, a strain recognized to develop necrotic granulomas mimicking those of human TB, were used since 2012 for testing activity of drugs and drug combinations [84,85,86].
However, the low-cost non caseum-forming BALB/c mice model was the most extensively used for testing drug combinations. In several studies, treatment efficacy was assessed on the basis of lung CFU counts at selected time points during treatment (a measure of bactericidal activity), and on the proportion of mice with culture positive relapse at least 3 months after completion of treatment (a measure of sterilizing activity) [83,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. Table 1 (divided into parts A and B) shows a list of 65 combinations sterilizing Mtb H37Rv-infected BALB/c mice. A more synthetic view of Table 1 is given in Figure 1 (also divided into parts A and B), which shows the percentages of sterilizing combinations containing the drugs indicated, stratified by monthly periods. Overall, these data have been useful for identifying pivotal drugs/drug combinations eradicating Mtb from BALB/c mice. Some of these drugs were also tested in combination experiments for ability to sterilize caseum-forming C3HeB/FeJ mice (Table 2), and for efficacy to shorten human TB therapy in well controlled clinical trials (Table 3).
5.1. Eradication of Mtb from Balb/c mice
As anticipated above, Table 1 (parts A and B) shows a list of 65 combinations, stratified by the minimum length of time required for sterilization (1.5-2.0, 2.5-3.0, 3.5-4.0 and 4.5-6.0 months) [83,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. Table 1 also shows information including the months of treatment with each combination, the drug dose in mg/kg of body weight, and some treatment details (5 days/week, etc) [83,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. All mice were aerosol infected with Mtb H37Rv, with the exception of one study in which animals were infected intravenously [98]. Figure 1 shows the percentages of sterilizing combinations reported in Table 1, containing the drug indicated.
The first 6 combinations (regimens 1 to 6) sterilized BALB/c mice after 4.5 to 6 months of treatment [83,91,94,97,99]. Pyrazinamide was contained in 100% of these combinations, followed by R (83%), H (67%), M (33%) and Pa and E (17%); no combinations contained B, L, rifapentine (P), clofazimine (C) and sutezolid (S) (Figure 1). Overall, administration of R10H25Z150 (regimens 1-3) or R10H10E100Z150 (regimen 6) for 2 months, followed by 4 months of R10H25 (regimens 1-3) or R10H10 (regimen 6) sterilized mice in 4.5 to 6 months. These regimens mimicked those currently used for treatment of human TB (R-H-E-Z), but only regimen 6 contained E, a bacteriostatic drug. A 5-month sterilization occurred when H was replaced with M in the intensive and continuation therapy (regimen 4), or when the combination contained the new anti-TB drug Pa (Pa50M100Z150, regimen 5).
Sterilization occurred after 3.5 to 4 months of treatment with the 16 regimens (regimens 7 to 22) listed in Table 1, part A. Overall, none contained R10H25Z150 and R10H10E100Z150 during the entire period of treatment. In some cases, R, H and Z were administered at high doses. For instance, in regimen 7, after initial 0.5 months of administration of 25 mg/kg of H (H25), the drug was later given at 75 mg/kg (H75). Furthermore, R was administered at 50 mg/kg (R50) in regimen 22, and Z was given at 300 mg/kg (Z300) in the intensive phase of regimen 9. A common characteristic of regimens 7-22 was the use of drugs other than R, H and Z, including i) 15 mg/kg of the long-acting rifamycin P (P15) (regimens 7-9), that was substituted for 10 mg/kg of rifampin (R10), ii) 25 mg/kg of B (B25, regimens 13-19), iii) 50 or 100 mg/kg of the nitroimidazole Pa (Pa50 or Pa100, regimens 10, 12-13, 15-19), iv) 100 mg/kg of M (M100, regimens 8-10, 14 and 16), v) 12.5 or 25 mg/kg of C (C12.5, C25, regimens 20-21), vi) 50 mg/kg of S (S50, regimen 15). Overall, Z was contained in 81% of these combinations, followed by Pa (50%), B and R (44%), M (31%), P (25%), H, C and E (13%), S (6%) (Figure 1, parts A and B).
Sterilization was observed also after 2.5 to 3 months of treatment with the 32 regimens (regimens 23 to 54) listed in Table 1 (parts A and B). The top-6 drugs used were Z (72%), followed by P (44%), B (41%), Pa and H (38%), and M (28%) (Figure 1, parts A and B). Rifapentine was used at 7.5, 10, 15 and 20 mg/kg; Pa was used at 50 or 100 mg/kg.
Finally, the most potent activity was shown by 11 combinations (regimens 55 to 65) that sterilized mice in 1.5-2 months. Ten of them contained both B25 and Z150, followed by Pa (6/11), P and C (3/11), M and L (2/11) (Figure 1, parts A and B). In this group, the oxazolidinone L, a protein synthesis inhibitor, was used in regimens 64 and 65, that sterilized mice in 1.5 months when combined with B25, Z150 and Pa100.
All these data, reported from 2006 to 2022 mostly by the Center for TB research of the Johns Hopkins University, Baltimore, Maryland, USA, coordinated by Dr Eric Nuermberger [83,86,87,88,89,90,91,92,93,94,95,96,97,99,100,101,102,103], indicated that all Mtb H37Rv bacilli (AR cells and NR cells containing drug tolerant persisters and DD cells) were eradicated from BALB/c mice, as shown by lack of relapses (culture-negative lungs) 3 or 6 months after treatment completion. Overall, it appeared that the ranking of the top-6 drugs contained in combinations sterilizing BALB/c mice in 1.5 to 3 months by most regimens (regimens 23 to 65) was Z > B > Pa > P > M > L. It is possible that reactive species such as ROS (induced by M) and RNS (induced by Pa) contributed to the sterilizing effect. Furthermore, it was evident that the use of R and H sharply decreased as components of the most powerful combinations (Figure 1A and B, respectively), and that they were not present in the 1.5 to 2 months sterilizing combinations. Rifampin was replaced with the long-acting rifamycin P [104], which was present in several rapidly sterilizing combinations (Figure 1, part A). This was likely due to the knowledge that P has a longer half-life than R in human serum (10–15 h versus 2–3 h, respectively) [105].
5.2. Eradication of Mtb from C3HeB/FeJ mice
The results shown in BALB/c mice were important in designing drug combinations eradicating Mtb also in the caseous lesions of C3HeB/FeJ mice, a strain which develops lung necrotic granulomas similar to those found in human TB [84,85]. In 2012, a paper on the dose-ranging comparison of R and P in BALB/c and C3HeB/FeJ mice [86], reported that the long-acting P was roughly four times more potent than R in both mouse strains.
Table 2 shows a list of 7 combinations published in 2015-2022 that sterilized C3HeB/FeJ mice after 2.5, 3.0, 4.0, 4.5 and 6.0 months (Table 2) [97,102,103]. Administration of R10H10E100Z150 for 2 months, followed by R10H10 for 4 months (regimens 1, corresponding to the first-line therapy of human TB) sterilized mice in 6 months. Addition of Z at 150 mg/kg (Z150) in the continuation phase sterilized mice in 4.5 months (regimen 2). Thus, continuing Z beyond the first two months was beneficial to prevent relapses in caseum-forming mice. However, this is not applicable to humans, because the use of R-Z beyond 2 months is hepatotoxic, as shown during the prolonged use of this combination for treatment of LTBI [106].
In regimen 3, replacement of R10 with P10, and E100 with M100, sterilized C3HeB/FeJ mice in 4 months [103]. Sterilization was observed after 2.5 to 3 months of treatment with regimens 4-7. In particular, mice were sterilized after 3 months when R10 was replaced with high dose of P (P20), and was added 12.5 mg/kg of the anti-leprosy drug C (C12.5) (regimen 4). Clofazimine is a drug which seems in large part to function by inducing ROS in Mtb [102]. Administration of high dose of the long-acting drug P was important because, in comparison to regimen 1, mice were sterilized by P20H10E100Z150 in 2.5 months (regimen 7).
Finally, due to the knowledge that strong induction of the cytochrome P450 3A (CYP3A) caused by R and P, but not rifabutin (Rb), reduced the concentration of B [107], it was found that C3HeB/FeJ mice were sterilized after 3 months of B25 M100Z150Rb10 (regimen 5), or 2 months of B25 M100Z150Rb10 followed by 1 month of B25M100Rb10, (regimen 6) [103]. Overall, in these 7 regimens, Z was contained in 7 out of 7 combinations tested, followed by H (5/7), E (4/7), P and M (3/7), B, R and Rb (2/7) and C (1/7). So far, no papers with Pa- and/or L-containing combinations sterilizing C3HeB/FeJ mice were apparently reported.
6. New drug combinations for treatment of human TB
The frequent presence of Z in combinations sterilizing BALB/c and C3HeB/FeJ mice is likely related to the knowledge that this drug penetrates all TB lung lesions and kills persisters residing in caseum [108]. This information makes Z an irreplaceable component of regimens for treating DS-TB [36,109]. In support of this view is that inclusion of Z into TB treatment shortened the length of therapy from 9-12 months to 6 months [36,109]. Pyrazinamide and R, the two “sterilizing” drugs that contributed most to treatment shortening, distribute favorably into all lesion compartments, including avascular caseum [110]. However, in the combinations sterilizing BALB/c and C3HeB/FeJ mice, the use of R progressively decreased in the most rapidly sterilizing combinations because it was replaced by the long-acting rifamycin P (Table 1 and Figure 1, part A), or the low cytochrome P450 3A-inducer rifamycin Rb (Table 2, regimens 5-6).
Overall, the murine TB data were useful for the choice of drugs to be combined in human TB clinical trials, in order to shorten the current 6-month regimen (R-H-E-Z). This regimen, finalized during the 1980s, and based on seminal studies conducted by the British Medical Research Council in the second half of 20th century, was widely adopted worldwide for more than four decades. It cured more than 95% of persons, but in some cases long-term adherence to therapy was difficult to sustain for patients and national resource constraints, contributing to development of genetic drug-resistance [108,111,112].
Table 3 shows three papers published on the high impact factor magazine The New England Journal of Medicine (NEJM), which reported the results of all-oral regimen trials against DS-TB. All trials included Z-containing combinations with 6-months [113], 4-months [114] and 2-months [115] efficacy as effective as, or noninferior to, the 6-month control regimen (R-H-E-Z).
The first paper [113] was published in 2014 as a report of the noninferiority trial ISRCTN44153044 (RIFAQUIN trial). It included the control regimen (2 months of intensive therapy with R600H300Z1500E1200 and 4 months of continuation therapy with R600H300) and a 6-month regimen as effective as the control regimen in which H was replaced by daily dose of 400 mg of M (M400) for 2 months, followed by one weekly dose of both M400 and high-dose (1200 mg) of P (P1200) for 4 months.
The second paper [114] was published in 2021 as a report of phase 3 noninferiority trial NCT02410772 (Study 31), and included the control regimen (R600H300Z1500E1200), and a 4-months regimen with 2 months (indicated as 8 weeks in the paper) of once-daily P1200H300Z1500M400 (R replaced by P, and E replaced by M), followed by 2 months (indicated as 9 week in the paper) of once-daily of P1200H300M400. This 4-month regimen was supported by the WHO as a possible alternative to current 6-month regimen for DS-TB, because it showed similar performance in terms of efficacy and safety [112,116]. In 2022, the guideline development group of the WHO stated that Study 31 was the first and only phase 3 trial to demonstrate the noninferiority of this 4-month regimen for treatment of DS-TB when compared to the standard of care [112,116].
The third paper [115] was published in 2023 as a report of the noninferiority trial NCT03474198 (TRUNCATE-TB trial). The authors chose a treatment strategy, rather than focusing on a regimen alone, to be compared with control regimen [117]. This approach was selected because, in clinical trials, it is known that at least 85% of participants are cured with 3- and 4-month regimens. Thus, the 6-month standard regimen may lead to overtreatment in the majority of persons, in order to prevent relapse in a minority of persons [115]. Thus, exploration of new therapeutic approaches is important. It was adopted a 5-arms strategy involving initial treatment for 2 months (indicated as 8-week in the paper) of R-susceptible TB patients with 5 different regimens, followed up for 96 weeks for extended treatment for persistent clinical disease and prompt re-treatment for the minority of persons with a relapse. Briefly, the final results showed that only the 2-month regimen that contained B, L, H, Z, E (B400/200L600H300Z1500E1100) (Table 3) was noninferior to the control 6-month regimen, with respect to the risk of a composite clinical outcome at week 96.
7. Discussion
In the framework of the WHO vision to eliminate TB as a public health problem by 2035 [1], the three clinical trials published in the NEJM (Table 3) provided robust data towards the realistic possibility of shortening DS-TB treatment using combinations containing Z (found in 3 trials), M, H and E (found in 2 trials), B, R, P, L (found in 1 trial). The observation that all these drugs were contained in BALB/c and C3HeB/FeJ mice sterilizing combinations (Table 1 and Table 2 and Figure 1), indicated the usefulness of these small animal models for testing new TB regimens.
It is difficult to correlate the physicochemical properties of the drugs considered (Table 4) with in vivo activities shown in the Table 1, Table 2 and Table 3 and Figure 1. However, some observations may be drawn, concerning, for instance, the interplay among the half-life in serum or tissues [118], the unbound fraction (fu) (free drug) in caseum, and the bactericidal activity in caseum, a lipidic niche for Mtb drug-tolerant persisters and DD cells [42,119,120,121]. In rabbit caseum, the most potent bactericidal drugs were the rifamicins (R, P, Rb: 3.1-4.0 log CFUmax decrease in 7 days, Table 4), followed by B, M and Pa (1.6-1.9 log CFUmax decrease) [121].
The drugs with half-lives of ≥10 hours (h) were, in decreasing order, B (5.5 months) > C (about 25 days) >Rb (45 h) >Pa (18 h) > M (11.5-15.6 h) > P (10-15 h) > Z (9-10 h) [118]. In spite of the small number of studies still published, Z, B, P, M, Rb and C were major components of combinations sterilizing caseum-forming C3HeB/FeJ mice in ≤ 3 months (Table 3). Of these, Z, B, P, and M were also components of the noninferiority combinations curing DS-TB in ≤ 6 months (Table 3).
As to the unbound fraction in caseum (fu, Table 4), this parameter is inversely related to the lipophilic character of a drug, that is mostly expressed as hydrophobicity (logP), i.e., the octanol/water partition coefficient, commonly reported as experimental or calculated (clogP) value [118]. Drugs with clogP <0 are considered to be hydrophilic [11]. In Mtb-infected rabbits, only the unbound fraction can penetrate necrotic material or caseum via passive diffusion [119]. The hydrophobicity and aromatic ring count of a drug were shown to be proportional to caseum binding, and compounds with clogP <1 had a high chance of achieving fu >10% [119]. Indeed, the fu values of the hydrophilic H and Z (clogP -0.71) (Table 4) were >99.9%, i.e., they can penetrate caseum, while the fu of highly lipophilic drugs like C and B (clogP of 7.39 and 6.37, respectively) were <0.01%, i.e., they do not penetrate caseum [11,119]. Using a caseum binding assay and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MSI) imaging of TB drugs in Mtb-infected rabbits, it was shown that binding to caseum inversely correlated with passive diffusion into the necrotic core [119]. Among the major positive drivers of binding were high lipophilicity and poor solubility. Thus, highly lipophilic drugs like B and C nonspecifically bind to caseum macromolecules at the outer edge of the caseum core, preventing further passive diffusion toward the center of the necrotic core [119]. Drugs with intermediate clogP values such as P, R, Pa, and S (clogP of 4.83, 3.85, 2.8, and 1.31, respectively) showed rising fu values (fu of 0.5, 5.13, 7.31, and 30.1%, respectively). In this view, a rational design of combinations should involve drugs that complement each other in their ability to penetrate caseous granulomas and to kill drug tolerant persisters and DD cells living inside them.
The active combination of the Study 31 trial (P1200H300Z1500M400) (Table 3), which cured DS-TB in 4 months, and the combination P20H10E100Z150, that sterilized caseum-forming C3HeB/FeJ mice in 2.5 months (Table 2) are quite in keeping with this rationale, because they contain Z, M and P, which are active against persisters, and show an unbound fraction (fu) in rabbit caseum of >99.9%, 13.5% and 0.5%, respectively (Table 4). However, it is also important to consider the half-life and the extent of Mtb killing in caseum (Table 4) [110,120,121]. For instance, P, which showed a longer half-life than R in human serum (10–15 h versus 2–3 h, respectively) [105 and Table 4], killed Mtb at a similar extent than R and Rb in rabbit caseum after 7-day of incubation [121]. We also found that Mtb was selectively killed by R and P in hypoxia at pH of 7.3 (the pH of caseum) [44], and others reported that only the rifamycins (P, R, Rb and rifalazil) showed high bactericidal activity in rabbit caseum [121] and fully sterilized it [110]. All these studies indicated that rifamycins may play a pivotal role to eradicate drug tolerants persisters and DD cells from caseum [110,120,121]. Also half-lives of M and Z (≥ 9-10 h) were in the range of the P half-life value (Table 4). However, it is difficult to explain the sterilizing activity of Z, that shortened the length of therapy from 9 months to 6 months, but that is active only at acidic pH [36,108,109]. Indeed, in rabbit caseum (pH 7.0-7.5), the bactericidal activity of Z was low (0.5 log CFU decrease in 7 days) [110, 121 and Table 4]. A possible solution of this enigma was proposed by Sarathy et al. [110], who pointed out that, in closed human granulomas, hypoxia induces secretion of large amounts of succinate in Mtb. This may generate a “halo” of low pH around Mtb cells, which favors Z cidality. The observations of Kempker et al. [122] that cavity caseum samples obtained from resected lung tissue of 8 out of 10 TB patients had a pH of ≤ 5.5 is in favor of this hypothesis.
The active combination of the TRUNCATE-TB trial (B400/200L600H300Z1500E1100) (Table 3), that cured DS-TB in 2 months [115], and the two B-containing combinations (3 months of B25M100Z150Rb10, and 2 months of B25M100Z150Rb10 followed by 1 month of B25M100Rb10) that sterilized caseum-forming C3HeB/FeJ mice in 3 months (Table 2, regimens 5 and 6), are more difficult to explain in terms of caseum penetration, because B showed a fu value of <0.01 in rabbit caseum (Table 4). However, B has a very long terminal elimination half-life (about 5.5 months) [123], due to the great ability of this drug to bind to the intracellular phospholipids and, consequently, to accumulate in tissues. Half-lives of B in TB lung lesions of BALB/c and C3HeB/FeJ mice were also high (85.5 and 104.4 h, respectively) [124]. In C3HeB/FeJ mice, B accumulated within the highly cellular regions in the lungs, but it was also present at reduced but still biologically relevant levels within the central caseum, where it was most likely not detected by the MALDI-MSI system used [124]. Thus, it is possible that the very long half-life of B and/or the synergism with other drugs may contribute to the potent sterilizing activity of B-containing combinations. Some support to this hypothesis comes from the knowledge that, in recent trials, several B-containing combinations were very active also against DR-TB, including patients treated with B-Pa-L for 6 months (ZeNix, trial NCT03086486) [125], with B-Pa-L-M for 6 months (TB-PRACTECAL, trial NCT02589782) [126], and with two other B-containing regimens for 6 or 9 months (STREAM stage 2, trial ISRCTN18148631) [127]. In 2022, by taking into account the evidences reported in these and other trials, the WHO published consolidated guidelines for treatment of DR-TB [128].
8. Conclusions
All data on drug activity against drug-tolerant Mtb, ranging from the most studied in vitro Wayne dormancy model [40] up to the papers on sterilization of non caseum- and caseum-forming mice, shed a new light on TB biology [129,130]. These and other studies showed that the greatest difficulties in curing TB do not depend only on intracellular AR bacilli and of their mutants but, above all, on extracellular NR drug-tolerant bacilli living in the caseous tuberculomas, the hallmark of TB.
In the last two decades, this new way of seeing the TB boosted the search for novel drugs/drug combinations killing in vitro and in vivo both AR cells and NR drug tolerant cells, containing persisters and DD cells. More recently, some of these combinations were tested in several DS- and DR-TB clinical trials, so far achieving in various cases the goal of shortening duration of current drug regimens. In 2022, the WHO, based on one of these trials, supported the use of a 4-month regimen for treatment of DS-TB as a possible alternative to the current 6-month regimen [112,116].
Author Contributions
F.G. and L.F. wrote the manuscript; A.L. and A.I. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. WHO Global tuberculosis report; ; WHO: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1. [Google Scholar]
- WHO. WHO Global tuberculosis report; ; WHO: Geneva, Switzerland, 2019; ISBN 978-92-4-156571-4. [Google Scholar]
- Barry, C.E. 3rd; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, RJ.; Young, D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–55. [Google Scholar] [CrossRef] [PubMed]
- Dartois, V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 2014, 12, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lenaerts, A.; Barry, C.E. 3rd.; Dartois, V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol. Rev. [CrossRef]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–32. [Google Scholar] [CrossRef]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–74. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.; Dartois, V.; Dick, T.; Gengenbacher, M. Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 1648–53. [Google Scholar] [CrossRef] [PubMed]
- Garton, N.J.; Waddell, S.J.; Sherratt, A.L.; Lee, S.M.; Smith, R.J.; Senner, C.; Hinds, J.; Rajakumar, K.; Adegbola, R.A.; Besra, G.S.; et al. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 2008, 5, e75. [Google Scholar] [CrossRef]
- Iacobino, A.; Piccaro, G.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Fighting tuberculosis by drugs targeting nonreplicating Mycobacterium tuberculosis bacilli. Int. J. Mycobacteriol. 2017, 6, 213–221. [Google Scholar] [CrossRef]
- Mukamolova, G.V.; Turapov, O.; Malkin, J.; Woltmann, G.; Barer, M.R. Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am. J. Respir. Crit. Care Med. 2010, 181, 174–80. [Google Scholar] [CrossRef]
- Chengalroyen, M.D.; Beukes, G.M.; Gordhan, B.G.; Streicher, E.M.; Churchyard, G.; Hafner, R.; Warren, R.; Otwombe, K.; Martinson, N.; Kana, B.D. Detection and quantification of differentially culturable tubercle bacteria in sputum from patients with tuberculosis. Am. J. Respir. Crit. Care Med. 2016, 194, 1532–1540. [Google Scholar] [CrossRef]
- Saito, K.; Warrier, T.; Somersan-Karakaya, S.; Kaminski, L.; Mi, J.; Jiang, X.; Park, S.; Shigyo, K.; Gold, B.; Roberts, J.; et al. Rifamycin action on RNA polymerase in antibiotic-tolerant Mycobacterium tuberculosis results in differentially detectable populations. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E4832–E4840. [Google Scholar] [CrossRef]
- McAulay, K.; Saito, K.; Warrier, T.; Walsh, K.F.; Mathurin, L.D.; Royal-Mardi, G.; Lee, M.H.; Ocheretina, O.; Pape, J.W.; Fitzgerald, D.W.; et al. Differentially detectable Mycobacterium tuberculosis cells in sputum from treatment-naive subjects in Haiti and their proportionate increase after initiation of treatment. mBio. 2018, 9, e02192–e18. [Google Scholar] [CrossRef]
- Zainabadi, K.; Walsh, K.F.; Vilbrun, S.C.; Mathurin, L.D.; Lee, M.H.; Saito, K.; Mishra, S.; Ocheretina, O.; Pape, JW.; Nathan, C.; et al. Characterization of differentially detectable Mycobacterium tuberculosis in the sputum of subjects with drug-sensitive or drug-resistant tuberculosis before and after two months of therapy. Antimicrob. Agents Chemother. 2021, 65, e0060821. [Google Scholar] [CrossRef]
- Gordhan, B.G.; Peters, J.S.; McIvor, A.; Machowski, E.E.; Ealand, C.; Waja, Z.; Martinson, N.; Kana, B.D. Detection of differentially culturable tubercle bacteria in sputum using mycobacterial culture filtrates. Sci. Rep. 2021, 11, 6493. [Google Scholar] [CrossRef] [PubMed]
- Zainabadi, K.; Saito, K.; Mishra, S.; Walsh, K.F.; Mathurin, L.D.; Vilbrun, S.C.; Ocheretina, O.; Pape, J.W.; Fitzgerald, D.W.; Nathan, C.F.; et al. Transcriptional biomarkers of differentially detectable Mycobacterium tuberculosis in patient sputum. mBio. 2022, 13, e0270122. [Google Scholar] [CrossRef]
- Li, L.; Mendis, N.; Trigui, H.; Oliver, J.D.; Faucher, S.P. The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol. 2014, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, J.; Fourie, P.B.; Mitchison, D.A. Persister populations of Mycobacterium tuberculosis in sputum that grow in liquid but not on solid culture media. J. Antimicrob. Chemother. 2014, 69, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, J.; Van Dijck, P.; Holtappels, M. Fungal persister cells: The basis for recalcitrant infections? PLoS Pathog. 2018, 14, e1007301. [Google Scholar] [CrossRef]
- Barrett, M.P.; Kyle, D.E.; Sibley, L.D.; Radke, J.B.; Tarleton, R.L. Protozoan persister-like cells and drug treatment failure. Nat. Rev. Microbiol. 2019, 17, 607–620. [Google Scholar] [CrossRef]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-tolerant persister cells in cancer therapy resistance. Cancer Res. 2022, 82, 2503–2514. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Bigger, J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilization. The Lancet. 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–75. [Google Scholar] [CrossRef] [PubMed]
- Black, D.S.; Irwin, B.; Moyed, H.S. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 1994, 176, 4081–91. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, JJ.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Orman, M.A.; Brynildsen, M.P. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 2013, 57, 3230–9. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science. 2004, 305, 1622–5. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Balaban, N.Q. Quantitative measurement of Type I and Type II persisters using ScanLag. In: Bacterial Persistence, Methods and Protocols. Methods in Molecular Biology. Volume 1333. Edited by Michiels J and Fauvart M. Humana Press. 2016, Pages 75-81. [CrossRef]
- Carvalho, G.; Guilhen, C.; Balestrino, D.; Forestier, C.; Mathias, J.D. Relating switching rates between normal and persister cells to substrate and antibiotic concentrations: a mathematical modelling approach supported by experiments. Microb. Biotechnol. 2017, 10, 1616–1627. [Google Scholar] [CrossRef]
- Ju, Y.; Long, H.; Zhao, P.; Xu, P.; Sun, L.; Bao, Y.; Yu, P.; Zhang, Y. The top 100 cited studies on bacterial persisters: A bibliometric analysis. Front. Pharmacol. 2022, 13, 1001861. [Google Scholar] [CrossRef]
- Mandal, S.; Njikan, S.; Kumar, A.; Early, J.V.; Parish, T. The relevance of persisters in tuberculosis drug discovery. Microbiology (Reading). 2019, 165, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, J.; Fourie, P.B.; Mitchison, D.A. Persister populations of Mycobacterium tuberculosis in sputum that grow in liquid but not on solid culture media. J. Antimicrob. Chemother. 2014, 69, 437–40. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, J.P.; Vinnard, C.; Subbian, S.; Nagajyothi, J.F. Effect of Mycobacterium tuberculosis infection on adipocyte physiology. Microbes Infect. 2018, 20, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Iacobino, A.; Fattorini, L.; Giannoni, F. Drug-resistant tuberculosis 2020. Where we stand. Appl. Sci. 2020, 10, 2153. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996, 64, 2062–9. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002, 43, 717–31. [Google Scholar] [CrossRef]
- Piccaro, G.; Poce, G.; Biava, M.; Giannoni, F.; Fattorini, L. Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J Antibiot (Tokyo). 2015, 68, 711–4. [Google Scholar] [CrossRef]
- Iona, E.; Pardini, M.; Mustazzolu, A.; Piccaro, G.; Nisini, R.; Fattorini, L.; Giannoni, F. Mycobacterium tuberculosis gene expression at different stages of hypoxia-induced dormancy and upon resuscitation. J Microbiol. 2016, 54, 565–72. [Google Scholar] [CrossRef]
- Iacobino, A.; Piccaro, G.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Mycobacterium tuberculosis is selectively killed by rifampin and rifapentine in hypoxia at neutral pH. Antimicrob. Agents Chemother. [CrossRef]
- Gold, B.; Nathan, C. Targeting Phenotypically Tolerant Mycobacterium tuberculosis. Microbiol. Spectr. 2017. [Google Scholar] [CrossRef]
- Chung, E.S.; Johnson, W.C.; Aldridge, B.B. Types and functions of heterogeneity in mycobacteria. Nat. Rev. Microbiol. 2022, 20, 529–541. [Google Scholar] [CrossRef]
- Vaubourgeix, J.; Lin, G, Dhar, N. ; Chenouard, N.; Jiang, X.; Botella, H.; Lupoli, T.; Mariani, O.; Yang, G.; Ouerfelli, O.; et al. Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe. 2015, 17, 178–90. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.; Lewis, K. Noise in a metabolic pathway leads to persister formation in Mycobacterium tuberculosis. Microbiol. Spectr. 2022, 10, e0294822. [Google Scholar] [CrossRef]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016; 18, 16051. [Google Scholar] [CrossRef]
- Manuse, S.; Shan, Y.; Canas-Duarte, S.J.; Bakshi, S.; Sun, W.S.; Mori, H.; Paulsson, J.; Lewis, K. Bacterial persisters are a stochastically formed subpopulation of low-energy cells. PLoS Biol. 2021, 19, e3001194. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. Link between antibiotic persistence and antibiotic resistance in bacterial pathogens. Front. Cell. Infect. Microbiol. 2022; 12, 900848. [Google Scholar] [CrossRef]
- Sebastian, J.; Swaminath, S.; Nair, R.R.; Jakkala, K.; Pradhan, A.; Ajitkumar, P. De novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro. Antimicrob. Agents Chemother. 2017, 61, e01343–16. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Nair, R.R.; Jakkala, K.; Pradhan, A.; Ajitkumar, P. Elevated levels of three reactive oxygen species and Fe(II) in the antibiotic-surviving population of mycobacteria facilitate de novo emergence of genetic resisters to antibiotics. Antimicrob. Agents Chemother. 2022, 66, e0228521. [Google Scholar] [CrossRef]
- Piccaro, G.; Pietraforte, D.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 7527–33. [Google Scholar] [CrossRef]
- Iacobino, A.; Piccaro, G.; Pardini, M.; Fattorini, L.; Giannoni, F. Moxifloxacin activates the SOS response in Mycobacterium tuberculosis in a dose- and time-dependent manner. Microorganisms. 2021, 9, 255. [Google Scholar] [CrossRef]
- Shee, S.; Singh, S.; Tripathi, A.; Thakur, C.; Kumar, T.A.; Das, M.; Yadav, V.; Kohli, S.; Rajmani, R.S.; Chandra, N.; et al. Moxifloxacin-mediated killing of Mycobacterium tuberculosis involves respiratory downshift, reductive stress, and accumulation of reactive oxygen species. Antimicrob. Agents Chemother. 2022, 66, e0059222. [Google Scholar] [CrossRef]
- Singh, A.; Zhao, X.; Drlica, K. Fluoroquinolone heteroresistance, antimicrobial tolerance, and lethality enhancement. Front. Cell. Infect. Microbiol. 2022, 12, 938032. [Google Scholar] [CrossRef]
- Ehrt, S.; Schnappinger, D.; Rhee, K.Y. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 496–507. [Google Scholar] [CrossRef]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio. 2011, 2, e00100–11. [Google Scholar] [CrossRef]
- Torrey, H.L.; Keren, I.; Via, L.E.; Lee, J.S.; Lewis, K. High persister mutants in Mycobacterium tuberculosis. PLoS One. 2016, 11, e0155127. [Google Scholar] [CrossRef]
- Hu, Y.; Pertinez, H.; Ortega-Muro, F.; Alameda-Martin, L.; Liu, Y.; Schipani, A.; Davies, G.; Coates, A. Investigation of elimination rate, persistent subpopulation removal, and relapse rates of Mycobacterium tuberculosis by using combinations of first-line drugs in a modified Cornell mouse model. Antimicrob. Agents Chemother. 2016, 60, 4778–85. [Google Scholar] [CrossRef]
- Salina, E.G.; Makarov, V. Mycobacterium tuberculosis dormancy: how to fight a hidden danger. Microorganisms. 2022, 10, 2334. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.R.; Venugopal, U.; Chandra, G.; Bharti, S.; Maurya, R.K.; Krishnan, M.Y. Effect of various drugs on differentially detectable persisters of Mycobacterium tuberculosis generated by long-term lipid diet. Tuberculosis (Edinb). 2019, 115, 89–95. [Google Scholar] [CrossRef]
- Mesman, A.W.; Baek, S.H.; Huang, C.C.; Kim, Y.M.; Cho, S.N.; Ioerger, T.R.; Barreda, N.N.; Calderon, R.; Sassetti, C.M.; Murray, M.B. Characterization of drug-resistant lipid-dependent differentially detectable Mycobacterium tuberculosis. J. Clin. Med. 2021, 10, 3249. [Google Scholar] [CrossRef] [PubMed]
- Gordhan, B.G.; Sewcharran, A.; Letsoalo, M.; Chinappa, T.; Yende-Zuma, N.; Padayatchi, N.; Naidoo, K.; Kana, B.D. Detection of differentially culturable tubercle bacteria in sputum from drug-resistant tuberculosis patients. Antimicrob. Agents Chemother. 2022, 12, 949370. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, D.V.; Salina, E.G.; Fursov, M.V.; Skvortsov, T.A.; Azhikina, T.L.; Kaprelyants, A.S. Dormant non-culturable Mycobacterium tuberculosis retains stable low-abundant mRNA. BMC Genomics. 2015, 16, 954. [Google Scholar] [CrossRef]
- Turapov, O.; O’Connor, B.D.; Sarybaeva, A.A.; Williams, C.; Patel, H.; Kadyrov, A.S.; Sarybaev, A.S.; Woltmann, G.; Barer, M.R.; Mukamolova, G.V. Phenotypically adapted Mycobacterium tuberculosis populations from sputum are tolerant to first-line drugs. Antimicrob Agents Chemother. 2016, 60, 2476–83. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Mishra, S.; Warrier, T.; Cicchetti, N.; Mi, J.; Weber, E.; Jiang, X.; Roberts, J.; Gouzy, A.; Kaplan, E.; et al. Oxidative damage and delayed replication allow viable Mycobacterium tuberculosis to go undetected. Sci Transl Med. 2021, 13, eabg2612. [Google Scholar] [CrossRef]
- Hong, Y.; Zeng, J.; Wang, X.; Drlica, K.; Zhao, X. Post-stress bacterial cell death mediated by reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 10064–10071. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.Y.; Kim, P.; Zhang, L.; Kang, S.; Keller, T.H.; Jiricek, J.; Barry, C. E 3rd. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008, 322, 1392–5. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, J.; Sun, M.; Zhang, X.; Cook, G.M.; Zhang, T. Nitric oxide-dependent electron transport chain inhibition by the cytochrome bc1 inhibitor and pretomanid combination kills Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2021, 65, e0095621. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Maritz, J.S.; Venter, A.; van Helden, P.D.; Andries, K.; McNeeley, D.F.; Donald, P.R. Time to detection of the growth of Mycobacterium tuberculosis in MGIT 960 for determining the early bactericidal activity of antituberculosis agents. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1561–5. [Google Scholar] [CrossRef]
- Bowness, R.; Boeree, M.J.; Aarnoutse, R.; Dawson, R.; Diacon, A.; Mangu, C.; Heinrich, N.; Ntinginya, N.E.; Kohlenberg, A.; Mtafya, B.; et al. The relationship between Mycobacterium tuberculosis MGIT time to positivity and cfu in sputum samples demonstrates changing bacterial phenotypes potentially reflecting the impact of chemotherapy on critical sub-populations. J Antimicrob Chemother. 2015, 70, 448–55. [Google Scholar] [CrossRef]
- Filippini, P.; Iona, E.; Piccaro, G.; Peyron, P.; Neyrolles, O.; Fattorini, L. Activity of drug combinations against dormant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 2712–5. [Google Scholar] [CrossRef]
- Maitra, A.; Solanki, P.; Sadouki, Z.; McHugh, T.D.; Kloprogge, F. Improving the drug development pipeline for mycobacteria: modelling antibiotic exposure in the hollow fibre infection model. Antibiotics 2021, 10, 1515. [Google Scholar] [CrossRef]
- Gumbo, T.; Srivastava, S.; Deshpande, D.; Pasipanodya, J.G.; Berg, A.; Romero, K.; Hermann, D.; Hanna, D. Hollow-fibre system model of tuberculosis reproducibility and performance specifications for best practice in drug and combination therapy development. J. Antimicrob. Chemother. 2023, dkad029. [Google Scholar] [CrossRef]
- Gumbo, T.; Sherman, CM.; Deshpande, D.; Alffenaar, J.W.; Srivastava, S. Mycobacterium tuberculosis sterilizing activity of faropenem, pyrazinamide and linezolid combination and failure to shorten the therapy duration. Int. J. Infect. Dis. 2021, 104, 680–684. [Google Scholar] [CrossRef]
- Piccaro, G.; Giannoni, F.; Filippini, P.; Mustazzolu, A.; Fattorini, L. Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob. Agents Chemother. 2013, 57, 1428–33. [Google Scholar] [CrossRef]
- Iacobino, A.; Giannoni, F.; Pardini, M.; Piccaro, G.; Fattorini, L. The combination rifampin-nitazoxanide, but not rifampin-isoniazid-pyrazinamide-ethambutol, kills dormant Mycobacterium tuberculosis in hypoxia at neutral pH. Antimicrob. Agents Chemother. 2019, 63, e00273–19. [Google Scholar] [CrossRef]
- Lanni, A.; Borroni, E.; Iacobino, A.; Russo, C.; Gentile, L.; Fattorini, L.; Giannoni, F. Activity of drug combinations against Mycobacterium abscessus grown in aerobic and hypoxic conditions. Microorganisms. 2022, 10, 1421. [Google Scholar] [CrossRef]
- Olender, D.; Żwawiak, J.; Zaprutko, L. Multidirectional efficacy of biologically active nitro compounds included in medicines. Pharmaceuticals (Basel). 2018, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Nuermberger, E.; Rosenthal, I.; Tyagi, S.; Williams, K.N.; Almeida, D.; Peloquin, C.A.; Bishai, W.R.; Grosset, J.H. Combination chemotherapy with the nitroimidazopyran PA-824 and first-line drugs in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2006, 50, 2621–5. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Yan, B.S.; Rojas, M.; Shebzukhov, Y.V.; Zhou, H.; Kobzik, L.; Higgins, D.E.; Daly, M.J.; Bloom, B.R.; Kramnik, I. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005, 434, 767–72. [Google Scholar] [CrossRef] [PubMed]
- Driver, E.R.; Ryan, G.J.; Hoff, D.R.; Irwin, S.M.; Basaraba, R.J.; Kramnik, I.; Lenaerts, A.J. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3181–95. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, I.M.; Tasneen, R.; Peloquin, C.A.; Zhang, M.; Almeida, D.; Mdluli, K.E.; Karakousis, P.C.; Grosset, J.H.; Nuermberger, E.L. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 4331–40. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Li, S.Y.; Peloquin, C.A.; Taylor, D.; Williams, K.N.; Andries, K.; Mdluli, K.E.; Nuermberger, EL. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 5485–92. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, I.M.; Williams, K.; Tyagi, S.; Peloquin, C.A.; Vernon, A.A.; Bishai, W.R.; Grosset, J.H.; Nuermberger, E.L. Potent twice-weekly rifapentine-containing regimens in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2006, 174, 94–101. [Google Scholar] [CrossRef]
- Rosenthal, I.M.; Zhang, M.; Williams, K.N.; Peloquin, C.A.; Tyagi, S.; Vernon, A.A.; Bishai, W.R.; Chaisson, R.E.; Grosset, J.H.; Nuermberger, E.L. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 2007, 4, e344. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, I.M.; Zhang, M.; Almeida, D.; Grosset, J.H.; Nuermberger, E.L. Isoniazid or moxifloxacin in rifapentine-based regimens for experimental tuberculosis? Am. J. Respir. Crit. Care Med. 2008, 178, 989–93. [Google Scholar] [CrossRef] [PubMed]
- Nuermberger, E.; Tyagi, S.; Tasneen, R.; Williams, K.N.; Almeida, D.; Rosenthal, I.; Grosset, J.H. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 1522–4. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Tyagi, S.; Williams, K.; Grosset, J.; Nuermberger, E. Enhanced bactericidal activity of rifampin and/or pyrazinamide when combined with PA-824 in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 3664–8. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Minkowski, A.; Amoabeng, O.; Peloquin, CA.; Taylor, D.; Andries, K.; Wallis, R.S.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3114–20. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Tyagi, S.; Minkowski, A.; Peloquin, C.A.; Grosset, J.H.; Nuermberger, E.L. Contribution of moxifloxacin or levofloxacin in second-line regimens with or without continuation of pyrazinamide in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2013, 188, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Williams, K.; Amoabeng, O.; Minkowski, A.; Mdluli, K.E.; Upton, A.M.; Nuermberger, E.L. Contribution of the nitroimidazoles PA-824 and TBA-354 to the activity of novel regimens in murine models of tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 129–35. [Google Scholar] [CrossRef]
- Tasneen, R.; Betoudji, F.; Tyagi, S.; Li, S.Y.; Williams, K.; Converse, P.J.; Dartois, V.; Yang, T.; Mendel, C.M.; Mdluli, K.E.; et al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 60, 270–7. [Google Scholar] [CrossRef]
- Lanoix, J.P.; Betoudji, F.; Nuermberger, E. Sterilizing activity of pyrazinamide in combination with first-line drugs in a C3HeB/FeJ mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 60, 1091–6. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, A.; Ortega-Muro, F.; Alameda-Martin, L.; Mitchison, D.; Coates, A. High-dose rifampicin kills persisters, shortens treatment duration, and reduces relapse rate in vitro and in vivo. Front. Microbiol. 2015, 6, 641. [Google Scholar] [CrossRef]
- Li, S.Y.; Tasneen, R.; Tyagi, S.; Soni, H.; Converse, P.J.; Mdluli, K.; Nuermberger, E.L. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00913–17. [Google Scholar] [CrossRef] [PubMed]
- Ammerman, N.C.; Swanson, R.V.; Bautista, E.M.; Almeida, D.V.; Saini, V.; Omansen, T.F.; Guo, H.; Chang, Y.S.; Li, S.Y.; Tapley, A.; et al. Impact of clofazimine dosing on treatment shortening of the first-line regimen in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00636–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, S.Y.; Almeida, D.V.; Tasneen, R.; Barnes-Boyle, K.; Converse, P.J.; Upton, A.M.; Mdluli, K.; Fotouhi, N.; Nuermberger, E.L. Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00021–19. [Google Scholar] [CrossRef]
- Saini, V.; Ammerman, N.C.; Chang, Y.S.; Tasneen, R.; Chaisson, R.E.; Jain, S.; Nuermberger, E.; Grosset, J.H. Treatment-shortening effect of a novel regimen combining clofazimine and high-dose rifapentine in pathologically distinct mouse models of tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00388–19. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Garcia, A.; Converse, P.J.; Zimmerman, MD.; Dartois, V.; Kurbatova, E.; Vernon, A.A.; Carr, W.; Stout, J.E.; Dooley, K.E.; et al. Novel regimens of bedaquiline-pyrazinamide combined with moxifloxacin, rifabutin, delamanid and/or OPC-167832 in murine tuberculosis models. Antimicrob. Agents Chemother. 2022, 66, e0239821. [Google Scholar] [CrossRef]
- Hibma, J.E.; Radtke, K.K.; Dorman, S.E.; Jindani, A.; Dooley, K.E.; Weiner, M.; McIlleron, HM.; Savic, RM. Rifapentine population pharmacokinetics and dosing recommendations for latent tuberculosis infection. Am. J. Respir. Crit. Care Med. 2020, 202, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Benator, D.; Bhattacharya, M.; Bozeman, L.; Burman, W.; Catanzaro, A.; Chaisson, R.; Gordin, F.; Horsburgh, C.R.; Horton, J.; et al. Rifapentine and isoniazid once a week versus rifampicin and isoniazid twice a week for treatment of drug-susceptible pulmonary tuberculosis in HIV-negative patients: a randomised clinical trial. Lancet. 2002, 360, 528–534. [Google Scholar] [CrossRef]
- 106. Centers for Disease Control and Prevention (CDC); American Thoracic Society. Update: adverse event data and revised American Thoracic Society/CDC recommendations against the use of rifampin and pyrazinamide for treatment of latent tuberculosis infection--United States. MMWR Morb. Mortal. Wkly Rep. 2003; 52, 735-9.
- Svensson, E.M.; Murray, S.; Karlsson, M.O.; Dooley, K.E. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti-TB drug. J. Antimicrob. Chemother. 2015, 70, 1106–14. [Google Scholar] [CrossRef]
- Gopal, P.; Grüber, G.; Dartois, V, Dick, T. Pharmacological and molecular mechanisms behind the sterilizing activity of pyrazinamide. Trends Pharmacol. Sci. 2019, 40, 930–940. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, W. ; Zhang, W, Mitchison, D. Mechanisms of pyrazinamide action and resistance. Microbiol. Spectr. 2014; 18, 10–128. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Via, L.E.; Weiner, D.; Blanc, L.; Boshoff, H.; Eugenin, E.A.; Barry, C.E. 3rd.; Dartois, V.A. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob Agents Chemother. 2018, 62, e02266–17. [Google Scholar] [CrossRef]
- Iseman, M.D. Tuberculosis therapy: past, present and future. Eur. Respir. J. Suppl. 2002, 36, 87s–94s. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO consolidated guidelines on tuberculosis. Module 4: treatment—drug-susceptible tuberculosis treatment. ISBN 978-92-4-004812-6. WHO: Geneva, Switzerland, 2022.
- Jindani, A.; Harrison, T.S.; Nunn, A.J.; Phillips, P.P.; Churchyard, G.J.; Charalambous, S. ; Hatherill, M, Geldenhuys, H.; McIlleron, H.M.; Zvada, S.P.; et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N. Engl. J. Med. 2014; 371, 1599–1608. [Google Scholar] [CrossRef]
- Dorman, S.E.; Nahid, P.; Kurbatova, E.V.; Phillips, P.P.J.; Bryant, K.; Dooley, K.E.; Engle, M.; Goldberg, S.V.; Phan, H.T.T.; Hakim, J.; et al. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med. 2021, 384, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Paton, N.I.; Cousins, C.; Suresh, C.; Burhan, E.; Chew, K.L.; Dalay, V.B. ; Lu, Q, Kusmiati, T.; Balanag, V.M.; Lee, S.L.; et al. Treatment strategy for rifampin-susceptible tuberculosis. N. Engl. J. Med. 2023; 388, 873–887. [Google Scholar] [CrossRef]
- WHO. Treatment of drug-susceptible tuberculosis: rapid communication (June 2021). ISBN 978-92-4-002867-8. WHO: Geneva, Switzerland, 2021.
- Dartois, V.; Rubin, E.J. Shortening tuberculosis treatment—a strategic retreat. N. Engl. J. Med. 2023, 388, 939–941. [Google Scholar] [CrossRef]
- http://www.drugbank.ca.
- Sarathy, J.P.; Zuccotto, F.; Hsinpin, H.; Sandberg, L.; Via, L.E.; Marriner, G.A.; Masquelin, T.; Wyatt, P.; Ray, P.; Dartois, V. Prediction of drug penetration in tuberculosis lesions. ACS Infect Dis. 2016, 2, 552–63. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Dartois, V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev. 2020, 33, e00159-19. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.P.; Xie, M.; Jones, R.M.; Chang, A.; Osiecki, P.; Weiner, D.; Tsao, W.S.; Dougher, M. ; Blanc, L, Fotouhi, N.; Via, L.E, Barry, C.E. 3rd.; et al. A novel tool to identify bactericidal compounds against vulnerable targets in drug-tolerant M. tuberculosis found in caseum. mBio. 2023; 5, e0059823. [Google Scholar] [CrossRef]
- Kempker, R.R.; Heinrichs, M.T.; Nikolaishvili, K.; Sabulua, I.; Bablishvili, N.; Gogishvili, S.; Avaliani, Z.; Tukvadze, N.; Little, B.; Bernheim, A.; et al. Lung tissue concentrations of pyrazinamide among patients with drug-resistant pulmonary tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00226–17. [Google Scholar] [CrossRef]
- McLeay, S.C.; Vis, P.; van Heeswijk, R.P.; Green, B. Population pharmacokinetics of bedaquiline (TMC207), a novel antituberculosis drug. Antimicrob. Agents Chemother. 2014, 58, 5315–5324. [Google Scholar] [CrossRef]
- Irwin, S.M.; Prideaux, B.; Lyon, E.R.; Zimmerman, M.D.; Brooks, E.J.; Schrupp, C.A.; Chen, C.; Reichlen, M.J.; Asay, B.C. ; Voskui,l M.I.; et al. Bedaquiline and pyrazinamide treatment responses are affected by pulmonary lesion eterogeneity in Mycobacterium tuberculosis infected C3HeB/FeJ mice. ACS Infect Dis. 2016; 2, 251–267. [Google Scholar] [CrossRef]
- Conradie, F.; Bagdasaryan, T.R.; Borisov, S.; Howell, P.; Mikiashvili, L.; Ngubane, N.; Samoilova, A.; Skornykova, S.; Tudor, E.; Variava, E.; et al. Bedaquiline-pretomanid-linezolid regimens for drug-resistant tuberculosis. N. Engl. J. Med. 2022, 387, 810–823. [Google Scholar] [CrossRef]
- Nyang’wa, B.T.; Berry, C.; Kazounis, E.; Motta, I.; Parpieva, N.; Tigay, Z.; Solodovnikova, V.; Liverko, I.; Moodliar, R.; Dodd, M.; et al. A 24-week, all-oral regimen for rifampin-resistant ruberculosis. N. Engl. J. Med. 2022, 387, 2331–2343. [Google Scholar] [CrossRef]
- Goodall, R.L.; Meredith, S.K.; Nunn, A.J.; Bayissa, A.; Bhatnagar, A.K.; Bronson, G.; Chiang, C.Y.; Conradie, F.; Gurumurthy, M.; Kirenga, B.; et al. Evaluation of two short standardised regimens for the treatment of rifampicin-resistant tuberculosis (STREAM stage 2): an open-label, multicentre, randomised, non-inferiority trial. Lancet. 2022, 400, 1858–1868. [Google Scholar] [CrossRef]
- WHO. WHO consolidated guidelines on tuberculosis. Module 4: treatment—drug-resistant tuberculosis treatment, 2022 update. ISBN 978-92-4-006312-9. WHO: Geneva, Switzerland, 2022.
- Nuermberger, E.L. Preclinical efficacy testing of new drug candidates. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.; Clary, J.; Hanna, D.; Nuermberger, E.; Lenaerts, A.; Ammerman, N.; Ramey, M.; Hartley, D.; Hermann, D. Model-based meta-analysis of relapsing mouse model studies from the critical path to tuberculosis drug regimens initiative database. Antimicrob. Agents Chemother. 2022, 66, e0179321. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Percentages of sterilizing combinations shown in Table 1 (parts A and B), containing the drugs indicated. Data were stratified by the lenght of time required for sterilization. A) Z, pyrazinamide; B, bedaquiline; Pa, pretomanid (PA-824); P, rifapentine; M, moxifloxacin; R, rifampin; B) H, isoniazid; L, linezolid; C, clofazimine; E, ethambutol; S, sutezolid.
Figure 1.
Percentages of sterilizing combinations shown in Table 1 (parts A and B), containing the drugs indicated. Data were stratified by the lenght of time required for sterilization. A) Z, pyrazinamide; B, bedaquiline; Pa, pretomanid (PA-824); P, rifapentine; M, moxifloxacin; R, rifampin; B) H, isoniazid; L, linezolid; C, clofazimine; E, ethambutol; S, sutezolid.
Table 1.
part A. Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 1-35). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment. part B (continued). Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 36-65). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment
Table 1.
part A. Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 1-35). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment. part B (continued). Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected BALB/c mice (combinations 36-65). Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment
| part A |
|
| part B |
|
Table 2.
Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected C3HeB/FeJ mice Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment.
Table 2.
Drug combinations sterilizing Mycobacterium tuberculosis H37Rv-infected C3HeB/FeJ mice Minimum months of treatment (indicated as grey boxes) in which mice lungs were culture negative 3 or 6 months after completing treatment.
|
Table 3.
Oral regimens for treatment of drug-susceptible TB reported to be as effective as, or noninferior to, the control regimen.
Table 3.
Oral regimens for treatment of drug-susceptible TB reported to be as effective as, or noninferior to, the control regimen.
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Optimization of In Vitro Mycobacterium avium and Mycobacterium intracellulare Growth Assays for Therapeutic Development
Lauren Auster
et al.
Microorganisms,
2019
Harnessing Innate Immunity to Treat Mycobacterium tuberculosis Infections: Heat-Killed Caulobacter crescentus as a Novel Biotherapeutic
Nancy Gupta
et al.
Cells,
2023
Antimycobacterial Effects of Everolimus in a Human Granuloma Model
David Ashley
et al.
JCM,
2020



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated