
Submitted:
29 April 2023
Posted:
30 April 2023
You are already at the latest version
Abstract
Peyronie's disease (PD) is a benign condition caused by plaque formation on the tunica albuginea of the penis. It is associated with penile pain, curvature, and shortening, and contributes to erectile dysfunction, which worsens patient quality of life. In recent years, understanding of the detailed mechanisms and risk factors involved in development of PD has been increasing. In this review, the pathological mechanisms and several closely related signaling pathways, including TGF-β, WNT/β-catenin, Hedgehog, YAP/TAZ, MAPK, ROCK, and PI3K/AKT, are described. Findings regarding cross-talk among these pathways are then discussed to elucidate the complicated cascade behind tunica albuginea fibrosis. Finally, various risk factors including genetic involved in the development of PD are presented and their association with the disease summarized. The purpose of this review is to provide better understanding regarding involvement of risk factors in the molecular mechanisms associated with PD pathogenesis, as well as provide insight into disease prevention and novel therapeutic interventions.
Keywords:
Peyronie's disease
; pathogenesis
; risk factors
; molecular mechanisms
; transforming growth factor-β1
1. Introduction
Peyronie's disease (PD) is a connective tissue disorder characterized by formation of fibrous lesions or plaques in the tunica albuginea (TA) of the penis. The early stage is an active phase, with affected individuals experiencing erectile pain and penile curvature onset. At 12-18 months following PD onset, the disease transitions to a painless chronic phase, with gradual worsening of penile curvature in 20-50% of the cases [1,2,3,4], during which changes in the penis rarely show spontaneous reversal. Therefore, a severely deformed penis may cause sexual intercourse disorders, such as difficulty with insertion, pain during intercourse, body posture restrictions, and erectile dysfunction (ED), which can adversely affect psychological well-being and quality of life (QoL). Notably, it has been reported that PD patients have mental distress and a distorted image of themselves, and depression is suspected in a high proportion [5,6,7]. Furthermore, a large-scale survey of more than 8000 PD patients conducted in Sweden showed that they had increased risk of self-harm, as well as anxiety and depression [8].
Although the exact etiology of PD remains unknown, evidence accumulated over the past two decades shows several molecular alterations and aberrant signaling pathways involved in the pathogenesis. In addition, epidemiological studies have indicated several risk factors possibly associated with its development, such as smoking, hypertension (HT), diabetes mellitus (DM), Dupuytren’s disease (DD), and elderly age [9,10]. Possible connections to certain genetic predispositions have also been proposed as involved in PD development, including single nucleotide polymorphisms (SNPs), though exact susceptibility factors have yet to be established [11,12,13]. The prevalence of PD has been reported to range from 0.6% to 20%, with wide variations among regions and ethnic groups, though it is particularly low in Asians [14,15,16,17,18,19]. One reason for differences may be the influence of certain genetic differences among regions and races. Such intrinsic and extrinsic risk factors, as well as comorbidities may independently increase susceptibility for PD or function synergistically to increase risk for disease development. A better understanding of both related molecular mechanisms and multifactorial factors associated with PD pathogenesis will have major influence on treatment options as well as prevention strategies. The present review summarizes current findings related to the molecular mechanisms of PD pathogenesis and the putative roles of several risk factors, including genetic, associated with its development.
2. Pathogenesis of Plaque in PD Cases
The hypothesis by Devine et al. that PD is initiated by repetitive micro-trauma to the penis during intercourse has become widely accepted [20]. Rats that received repeated local injections of chlorhexidine ethanol to produce repetitive microtrauma to the TA developed fibrous plaque in the penis [21]. For affected patients, the primary site of injury is thought to be the TA, with disruption of the inner and outer layers creating a milieu for inflammation, elastic fiber disruption, and extravascular blood accumulation [13]. This serves as a scaffold for fibrin and fibronectin accumulation, then infiltration by inflammation-associated cells that release various types of cytokines and subsequently show cell-type conversion, including tissue-resident fibroblasts and smooth muscle cells, into myofibroblasts characterized by α-smooth muscle actin (α-SMA) expression and collagen secretion. Transforming growth factor-β1 (TGF-β1) is a key factor for myofibroblast activation and major player in fibrosis in all organs [11,12,13,22]. Myofibroblasts are a crucial element for the fibrotic process by causing excessive accumulation of collagen fibers, fibronectin, and other components of the extracellular matrix (ECM). An imbalance between matrix metalloproteinase (MMP), which removes collagen fibers, and its inhibitor tissue inhibitor of metalloproteinase (TIMP) in myofibroblasts, along with evasion of apoptosis are thought to be key factors in development of fibrotic diseases [13,23,24]. Interestingly, recent studies have revealed involvement of persistent immunological features involving mainly macrophages with fibrosis including PD [12,24,25,26,27,28,29]. Based on the latest findings, following are details of the mechanisms of fibrotic tissue remodeling and postulated functions of each cell type in different stages of PD.
2.1. Role of Immune System in Inflammatory Response
In the tissue fibrosis process, innate and adaptive immune systems are both intricately involved in inflammatory events. Macrophages and monocytes are key immune-related cells related to the innate immune system, while T cells and B cells are involved in the adaptive immune system.
At the site of injury, platelets become activated, then release granules and stop blood loss first by forming an initial provisional matrix composed of fibrin and fibronectin, which has also been identified in PD [30]. Fibrin and fibronectin also function to alter leukocyte recruitment to the wound site and promote fibrosis [31]. The chemokine monocyte chemoattractant protein 1 (MCP-1), another factor that mediates recruitment and activation of macrophages and monocytes, is produced and secreted from monocytes/macrophages, fibroblasts, and vascular endothelial cells by stimulation of lipopolysaccharide and inflammatory cytokines upon tissue injury [32,33]. A gene expression profile study using PD-derived lesions demonstrated significantly elevated expression of MCP-1 in PD plaque over a normal TA condition [34], while Lin et al. confirmed higher levels of MCP-1 mRNA in PD than non-PD cells [35]. Other factors involved in recruitment and stimulation of immune-related cells include pathogen-associated molecular patterns and damage-associated molecular patterns (DAMPs), which stimulate through various receptors on immune cells such as toll-like receptors (TLR) (mostly-2 and -4) [12,29,36]. DAMPs are released following stress or injury to various cells, including dead cells, proliferating neutrophils, macrophages, lymphocytes, natural killer cells, mesenchymal stem cells (MSCs), and resident cells [37,38,39]. In 2000, Mills et al. proposed a new classification for macrophages as either M1, pro-inflammatory, or M2, anti-inflammatory/profibrotic [40]. Thereafter, M2 macrophages were further classified into M2a, M2b, M2c, and M2d subtypes based on secretion of distinct cytokines, presence of certain cell surface proteins, gene expression profiles, and other biological activities, while they commonly express interleukin (IL)-10 [41,42].
There are different subtypes of T cells, each with unique regulatory effects on inflammation and fibrosis, including through cytokine release. Among those, helper T cells are a subpopulation of lymphocytes that express the CD4 antigen on the cell surface and now mainly divided into Th1, Th2, and Th17 types [43]. Th1 cells differentiate generally in the presence of IL-12 and mainly produce interferon gamma (IFN-γ) after differentiation, whereas Th2 cells differentiate by IL-4 and continue to produce mainly IL-4 after differentiation, with each having influence on macrophage activation [44]. In addition to granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor-alpha (TNF-α), the Th1 cell cytokine IFN-γ, activates polarization of M1 macrophages via various pathways, including the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and nuclear factor (NF)-κB pathways [28,29,45]. The Th2 cytokine IL-4, like IL-13 and TGF-β1, activates M2 macrophages through STAT6 or interferon regulatory factor 4-mediated signaling [46,47,48], with this macrophage plasticity known as M1/M2 polarization. M1 macrophages activated in this manner exhibit high levels of antigen-presenting activity and production of pro-inflammatory cytokines such as IL-6 and TNF-α, as well as production of nitric oxide (NO) and reactive oxygen species (ROS) [28,29,47]. In contrast, M2 macrophages produce less of the inflammatory cytokines IL-6 and TNF-α. In particular, the M2c type is characterized by high expression levels of the anti-inflammatory cytokine IL-10 and profibrotic factor TGF-β1, thus contributing to tissue remodeling and fibrosis promotion [28,29,47,48,49].
B cells contribute to several different fibrotic diseases via a number of mechanisms, including direct cell-cell contact, and production of profibrotic and pro-inflammatory cytokines (e.g., IL-6 and TGF-β1) [50,51,52]. Furthermore, decreased IL-10 production by B cells is possibly a factor associated with fibrosis, based on previous examinations of systemic sclerosis patients [52,53,54]. Those studies also showed that increased IL-6 and reduced IL-10 levels in B cells are induced through activation of the JAK/STAT and TLRs pathways.
M1/M2 macrophage phenotypes are interchangeable depending on the microenvironment, while an M1/M2 imbalance expressed by excessive M2 activity is associated with different types of fibrosis through excessive fibroblast activation [28,55,56]. Using a cisplatin-induced rat renal fibrosis model, Nakagawa et al. showed that M1 macrophages begin to increase with upregulation of M1-related cytokines (IFN-γ, TNF-α, IL-6) in the middle stage and the decrease in the late stage [57]. In contrast, M2 macrophages exhibit progressively increased numbers in the middle and late stages, accompanied by increased profibrotic factor TGF-β1 expression. That study also found increased CD4+ and CD8+ T cells in the late stage of the fibrotic process. Thus, M1 macrophages are involved in relatively early stages of wound response and thought to be replaced at later stages by specific repair populations of M2 macrophages via T cell responses. Regarding PD, serum IL-6 levels were found to be significantly elevated in patients with PD in the acute phase as compared to healthy controls [58]. In contrast, a study that compared 91 PD patients with healthy controls showed that patients in the chronic stage had elevated serum levels of TGF-β1 and IFN-γ, and decreased levels of TNF-α, while IL-6 was undetectable [59]. In addition, a recent study by Cui et al. that used integrated bioinformatic analysis provided interesting findings regarding the association of PD with immune-related cells [60]. Those based on immune cell infiltration analysis demonstrated that various immune-related cells, such as T cells, B cells, macrophages, neutrophils, and plasma cells, are widely involved in fibrosis in PD patients. Additionally a number of genes were also identified, including inflammatory cytokines such as IL-6, as important immune-related factors in PD and their correlations were shown. Notably, functional enrichment analysis showed that these genes have potential regulatory roles in several key biological processes, including the JAK/STAT pathway. Thus, complicated mechanisms that arise through M1/M2 macrophage interactions and T cell responses may contribute to PD development.
2.2. Differentiation and Proliferation of Myofibroblasts in Profibrotic Environment
Myofibroblasts, which function in tissue contraction and ECM secretion, have an important role during impaired healing. The profibrotic milieu, formed by infiltration and activation of leukocytic tissue, as well as Th2 cell-M2 macrophage polarization, induces mesenchymal transition (conversion to myofibroblasts) in various cell types, such as tissue-resident fibroblasts, fibrocytes of different origin, smooth muscle and endothelial cells, and mesenchymal stem cells [13]. Various factors are involved in conversion to myofibroblasts, among which TGF-β1 plays a central role. Others include pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β), growth factors [e.g. platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF)], myostatin, and ROS.
PDGF is primarily secreted by endothelial cells and macrophages, and also released from platelets upon degranulation, with its production as well as expression of related receptors maintained by TGF-β1 signaling [12]. Hamid et al. showed that pro-inflammatory macrophages promote myofibroblast transformation of cardiac MSCs through activation of PDGF and its receptor PDGFR-β [61], while another study using a rat PD model revealed significantly elevated expression levels of PDGF-α and PDGF-β, and their receptors PDGFR-α and PDGFR-β [62]. Interestingly, a recent study that used single-cell RNA sequencing (scRNA-Seq) with samples from patients with DD, a widely known risk factor for PD, showed that the endothelium acts to promote immune regulatory fibroblast phenotype through PDGF signaling in development of fibrosis [63]. bFGF, one of the factors expressed in the early stage of tissue injury that plays an important role in wound healing [64,65], functions not only acts as a fibroblast mitogen, but also promotes myofibroblast proliferation and migration after TGF-β1-induced mesenchymal transition [4]. It was also shown that production of bFGF by PD plaque-derived myofibroblasts is significantly greater as compared to that by normal TA myofibroblasts [66]. Furthermore, myostatin, a member of the TGF-β family, may promote fibrous tissue formation in muscle tissue by transforming fibroblasts into myofibroblasts to replace damaged muscle fibers [67]. A study of the effect of myostatin deficiency on alterations in left ventricular function after myocardial infarction showed that α-SMA activity was less intense in myostatin knockout than wild-type mice, suggesting the possibility of fewer myofibroblasts [68]. Also, myostatin was found to be overexpressed in PD plaque, and stimulated myofibroblast generation and collagen expression in rat TA cells [69].
Accumulation of ROS is induced by hypoxia and produces oxidative stress, which has detrimental effects on various cellular components. ROS-induced myofibroblast transformation involves a pathway through ROS-mediated HIF-1α stabilization [70,71]. For example, HIF-1α was shown to induce abnormal fibroblast activity via activation of the extracellular signal-related kinase signaling pathway in cutaneous wound healing [72]. Another study found that rats with PD-like lesions expressed high levels of HIF-1α and inducible NO synthase (iNOS) via activation of NF-κB [62]. More recently, results of bioinformatic approaches identified a number of potential hypoxia-associated genes associated with PD [73].
2.3. Extracellular Matrix Deposition
Following the initial inflammatory event, fibrosis progression is facilitated by accumulation of ECM components secreted by myofibroblasts, leading to fibrotic tissue remodeling. The predominant components include collagen, especially types I, III, and IV, in penile plaque, proteoglycans, fibronectin, and fibromodulin [11,74,75], with excessive accumulation caused by disruption of the complex interactions of various factors.
MMPs, neutral proteases, play a central role in ECM degradation and when activated lead to ECM degradation during normal wound healing. Their activation is regulated by various factors, including TGF-β1, PDGFs, IL-1, TNF-α, and bFGF. For example, TGF-β1 activates collagenase MMPs (MMP-1, -8, and -13, which degrade types I, II, and III collagen) and gelatinase MMPs (MMP-2 and -9, which degrade types IV, V, and VII collagen, as well as gelatin and elastin) [76]. However, a previous study found that administration of TGF-β1 to a fibroblast cell line established from PD plaque did not alter MMP-1, -8 and -13 expressions, and only mildly increased MMP-9, though IL-1β induced production of MMP-1, -3, -10, and -13 [77]. Another study compared samples from PD plaque and normal TA using immunostaining, and also found no differences in MMP-2 and -9 expression between them [78]. Instead, that previous study confirmed that administration of TGF-β1 induced excessive accumulation of TIMP1-4, which suppresses the activity of MMPs, in fibroblast cell lines established from PD plaque [77]. TIMPs are mainly produced in macrophages, fibroblasts, and myofibroblasts, and their expression is also positively regulated by PDGF and bFGF. Plasminogen activator inhibitor type 1 (PAI-1), synthesized in various cell types including endothelial cells, fibroblasts, and macrophages, is also known to inhibit plasmin-mediated MMP activation and promote fibrosis [79]. Davila et al. found that both mRNA and protein levels of PAI-1 were significantly increased in human PD plaque samples and respective fibroblast cultures as compared to normal TA [80]. Therefore, excessive ECM accumulation during PD plaque development may be driven by altered MMP levels caused by inhibitory effects of TIMP and PAI-1 on MMP activation.
2.4. Progression of Fibrosis in Environment with Continuous Inflammation
In normal wound healing, myofibroblasts contract via increased expression of matrix protein receptor integrins to regulate excessively deposited ECM, then continue to support mechanical loads until collagen cross-linking for formation of striated scar tissue [31]. After the wound is repaired, myofibroblasts undergo apoptosis and the repair response is terminated [74]. However, in fibrotic diseases including PD, tissue remodeling and fibroblast activation persist as chronic, uncontrolled processes, with structural changes in fibrotic tissue thought to play an important role in formation of this chronic extracellular profibrotic milieu [81]. Progressive deposition of ECM proteins creates a hypoxic environment by increasing stiffness around the affected TA and inhibiting oxygen diffusion. Lack of oxygen, the terminal electron acceptor of the electron transport chain, increases ROS generation, further promoting cellular and tissue damage and myofibroblast activation [73,82]. ROS-induced damage then induces release of DAMPs from damaged cells, resulting in further continued recruitment and stimulation of macrophages and lymphocytes. Continuous recruitment and stimulation of immune-related cells is also maintained by fibronectin and MCP-1 released from TGF-β1-activated myofibroblasts [83]. Thus, structural changes in fibrous tissue due to excessive ECM deposition may lead to sustained myofibroblast activation, contributing to continuation of the profibrotic milieu.
Evasion of myofibroblast apoptosis is also critically involved in maintaining a chronic extracellular fibrous milieu. Various types of programmed cell death, including apoptosis, are known to be closely associated with organ fibrosis [84]. Apoptotic pathways include intrinsic pathways activated by cellular stress and DNA damage, as well as others, and also extrinsic pathways activated via detection of extracellular death signals from other cells. The intrinsic apoptotic pathway is tightly regulated by a balance of the activities of Bcl-2 family proteins, consisting of pro-apoptotic (Bax, Bak, Bad, Bid, Puma, Bim, Noxa) and anti-apoptotic (Bcl-2, Bcl-xL, Bcl-w, Mcl-1) proteins. Furthermore, both intrinsic and extrinsic apoptotic pathways ultimately depend on the protease activity of the initiator (e.g., caspase-2, -8, -9, -10, -12) and executioner (e.g., caspase-3, -6, -7). Interestingly, Zorba et al. found high levels of Bcl-2 expression, and decreased levels of caspase-3 and -8 expression in PD plaque, indicating apoptosis suppression [85]. In contrast, Loreto et al. showed that PD tissues had lower expression of Bcl-2, and overexpression of Bax, as well as caspase-3 and -9 as compared to normal TA [86]. They also reported that a tumor necrosis factor-related apoptosis-inducing ligand and its receptor DR5 were overexpressed in PD tissues, suggesting that apoptosis may be induced by intrinsic and extrinsic pathways [87]. These different results may be partially explained by a new concept of apoptosis evasion in myofibroblasts termed “mitochondrial priming” [88], which refers to the proximity of mitochondria to the apoptotic threshold, and is determined by relative expression of pro-apoptotic and pro-survival members of the Bcl-2 family of proteins. Myofibroblasts are “primed” and thus ready to die at the time of conversion from fibroblasts, and become dependent on pro-survival proteins to sequester pro-apoptotic proteins and ensure survival. Therefore, various types of apoptosis-associated proteins are expressed at different ratios in the mitochondria of myofibroblasts that continue to survive while avoiding apoptosis, which might have been a factor contributing to differences in those results. Additionally, epigenetic modifications may lead to sustained myofibroblast activation and consequent fibrosis, details of which will be discussed later.
PD plaques formed in this way evolve towards calcification in 20-25% of affected cases [89]. PDGF functions as an osteoblast recruiter, and contributes to calcification and ossification of PD plaques [62]. Following the initial inflammatory phase, the disease moves to the second phase (chronic), during which the disease stabilizes. Absence of pain and inflammation have long been thought to be hallmarks of the chronic phase. However, a recent study by Milenkovic et al. using immunohistochemistry and scRNA-Seq indicated the possibility of a sustained inflammatory reaction even in the chronic PD stage [90]. Such findings showing inflammation in PD may pave the way for development of new treatments for patients in the chronic phase. Findings related to the suggested pathophysiology of PD based on data presently available are shown in Figure 1.
3. Core signaling Pathways and Their Crosstalk Involved in Development of Peyronie's Disease
TGF-β, a central regulator of tissue inflammation, repair, remodeling, and fibrosis, has three isoforms; TGF-β1, TGF-β2, and TGF-β3 [91]. TGF-β is stored in a latent form in most normal tissues, while upon tissue injury de novo synthesis of TGF-β isoforms and latent stores of TGF-β are activated. In particular, deregulation of TGF-β1 activity and thereby activation of TGF-β signaling have been strongly linked to the pathogenesis of PD, as described above. Indeed, activation of TGF-β signaling by subtunical injection of TGF-β1 in rat and rabbit models has been demonstrated to induce PD-like plaque formation in the penis [92,93]. In addition, there are other critical target molecules and signaling cell pathways related to fibrosis, which develops when these interact around the TGF-β signaling pathway. Those potentially associated with PD include the Wingless/int1 (WNT)/β-catenin, Hedgehog, Yes-associated protein 1(YAP)/transcriptional coactivator with PDZ-binding motif (TAZ), mitogen-activated protein kinase (MAPK), RAS homologue gene family (RHO)-associated kinase (ROCK), and phosphoinositide 3-kinase (PI3K)/RAC-α serine/threonine protein kinase (AKT) signal pathways. In this section, detailed mechanisms of TGF-β1 synthesis and activation in the pro-fibrotic environment are reviewed, along with interactions of the TGF-β signaling pathway involved with other core signaling pathways in PD development.
3.1. TGF-β1 Synthesis and Aberrant Activation of Latent TGF-β1
TGF-β1 is produced by platelets, fibroblasts, and all leukocyte lineage cells [94]. Among those, M2 macrophages play an important role in production and activation of TGF-β1 in fibrotic diseases including PD. PAI-1, which promotes fibrosis through inhibition of MMP activation, is also a factor that promotes TGF-β1 synthesis, while its expression is enhanced by TGF-β1 [95]. Enhanced serotonin signaling mediated by 5-hydroxytryptamine receptors, resulting in increased transcription of TGF-β1, has been identified in some fibrotic diseases [96,97]. TGF-β1 is synthesized in a linked state with latency-associated polypeptide (LAP), then forms a complex with latent TGF-β-binding protein (LTBP) or a leucine-rich repeat containing (LRRC) protein, and subsequently released outside the cell [98]. Thus, synthesized TGF-β1 (as well as the remaining variants) is secreted extracellularly as biologically inactive latent TGF-β1. Released latent TGF-β1 is anchored using LTBP to ECM and LRRC protein molecules on the cell surface to maintain its inactivated state. Several factors, including integrins, thrombospondin 1, ROS, and several proteases (plasmin, cathepsin D, MMP-2, MMP-9), activate latent TGF-β1 by inducing LAP degradation, denaturation, or conformational changes [76,99,100]. Among them, expression of thrombospondin 1 is known to be promoted by the renin-angiotensin system (RAS) [101], which is involved in regulation of fibrosis, inflammation, and oxidative stress, and plays an important role in aging and age-related chronic diseases [102]. Notably, inhibition of this pathway with an angiotensin II receptor blocker has been shown to attenuate penile fibrosis in a rat cavernous nerve injury model [103,104]. Another study found that protein expression of angiotensin-converting enzyme, AT1 receptor, and Mas receptor were upregulated in aged rat penile tissue characterized by increased collagen deposition [105]. Putative mechanisms of TGF-β1 activation during development of PD based on these findings are shown in Figure 2.
3.2. Aberrant TGF-β Signaling Pathway in Pathogenesis of Peyronie's Disease
TGF-β signaling includes SMAD-mediated canonical and non-canonical TGF-β signaling pathways. Canonical TGF-β signaling is initiated when a TGF-β ligand (TGF-β1, 2, 3) binds to the TGF-β type II receptor (TGF-βRII). TGF-βRII subsequently promotes phosphorylation of the TGF-β type I receptor (TGF-βRI), which signals through phosphorylation of SMAD2/SMAD3, triggering a cascade response. Phosphorylated SMAD2/SMAD3 proteins in a complex with SMAD4 then translocate to the nucleus, where the complex binds to specific DNA regions, SMAD-binding elements, to induce transcription of profibrotic genes (αSMA, Collagen, PAI-1, TGF, TGF-βR1, MMPs, fibronectin, etc.). Following TGF-β1 stimulation, PD plaque-derived fibroblasts show faster nuclear translocation of SMAD2/SMAD3, and higher expression levels of SMAD3 and SMAD4 as compared to normal cells [106]. In addition to excessive TGF-β1 activation, several factors may cause such aberrant TGF-β/SMAD signaling activation and promote PD fibrosis.
The TGF-β/SMAD signaling pathway is regulated by feedback inhibition to control cellular homeostasis. SMAD7, an inhibitory SMAD, provides competition for TGF-βRI and inhibits the TGF-β/SMAD signaling pathway by blocking SMAD2 phosphorylation and activation [107]. Magee et al. assessed gene expression differences between PD plaque tissue and normal TA using DNA microarray findings, and reported that SMAD7 was the most significantly downregulated gene in PD plaque tissue [34], suggesting that downregulation of SMAD7 expression activates the TGF-β/SMAD signaling pathway in PD. These observations were supported by other later studies. Specifically, Choi et al. found that human PD plaque-derived fibroblasts with forced expression of the SMAD7 gene inhibited phosphorylation and nuclear translocation of SMAD2 and SMAD3, and suppressed ECM protein production by TGF-β1 [108]. They also confirmed that SMAD7 induces apoptosis and suppresses cell cycle entry in PD plaque-derived fibroblasts. Wang et al. reported that the anti-fibrotic effect of rat bone marrow MSC treatment in PD rat models was associated with increased SMAD7 expression, while that was offset by knockdown of Smad7 expression [109]. Another study suggested that connective tissue growth factor (CTGF) promotes TGF-β1 activity by reducing SMAD7 expression and activating SMAD2 phosphorylation [110]. CTGF itself is also a factor with expression promoted by TGF-β1 that contributes to maintenance of the fibrotic process [111]. Although fibroblasts derived from PD patients did not show upregulation of CTGF [35], a pilot study of prostacyclin 12 analog, a drug for PD patients that suppresses CTGF expression, noted that penile curvature was improved in approximately 30% of examined patients [112]. Insulin-growth factor 1 (IGF-1), a major mediator of growth hormone signaling, is also associated with various fibrotic diseases. Sarenac et al. showed that its administration to human keratinocytes inhibited transition of SMAD3 to nucleation, increased Smad7 expression, and inhibited the TGF-β/SMAD pathway leading to fibrosis [113]. Interestingly, PD plaque shows a significantly lower expression of all IGF-1 isoforms than normal TA, suggesting that suppression of the TGF-β/SMAD pathway by IGF-1 may be reduced in PD [114].
Apart from SMAD signaling, TGF-β ligands can activate multiple alternative pathways, commonly referred to as “non-canonical” TGF-β signaling pathways, relevant to fibrosis pathogenesis. Activation of the MAPK signaling pathways mediated by extracellular signal-regulated kinase (ERK), JUN N-terminal kinase (JNK), and p38 has been implicated in SMAD-independent production of a mediator that promotes development of fibrotic disease. An in vivo experiment performed by Chatzifrangkeskou et al. showed that ERK1/2 acts directly on induction of CTGF expression to mediate myocardial fibrosis and left ventricular dysfunction [115]. Activated by profibrotic factors such as TGF-β1, JNK is subsequently translocated into the nucleus where it regulates transcription of genes involved in inflammatory and fibrotic responses through phosphorylation of c-Jun and SMAD3, leading to kidney inflammation and fibrosis [116]. In the liver, activation of the TGF-β1-mediated p38 MAPK signaling pathway may be associated with liver fibrosis by accumulating ECM through transcriptional activity of α-SMA and collagen [117]. As for penile tissues, studies of animal models showed JNK involvement in corpus cavernosum apoptosis in the acute phase after corpus cavernosum nerve crush injury, and that activation of the p38 MAPK pathway induces penile vascular dysfunction in association with diabetes [118,119].
PI3K/AKT signaling is also induced by TGF-β1 and their dysregulation is associated with various degenerative conditions, including fibrotic diseases. Inhibition of this signaling pathway suppresses TGF-β-induced differentiation of human lung or cardiac fibroblasts into desmoplastic myofibroblasts for escape from the fibrotic process [120,121]. A profibrotic mechanism of AKT activated by TGF-β1 stimulation is inactivation of the inhibitory effect of nuclear receptor subfamily 4 group A member 1 (NR4A1), which inhibits profibrotic gene expression in the nucleus [29]. Another study found that NR4A1 agonists prevented NR4A1 inactivation and exerted antifibrotic effects in many organs of experimental fibrosis models [122]. Furthermore, Jung et al. showed that inhibition of PI3K/AKT signaling by PI3K inhibitors suppress fibrotic responses, such as cell proliferation and collagen synthesis, in PD-derived primary fibroblasts [123].
3.3. Another Core Signaling Pathway and Crosstalk in Peyronie's Disease
The WNT/β-catenin signaling pathway is essential for organ development and generation, though its pathological activation is associated with several fibrotic diseases. Thus, targeted inhibition of this pathway results in antifibrotic effects in a variety of organs [124,125,126]. Under inactivation of WNT signaling, β-catenin is normally phosphorylated in NH2-terminal residues, in which a glycogen synthase kinase 3β (GSK-3β) consensus motif is present, with the aid of a scaffolding complex composed of axin and adenomatous polyposis proteins. Phosphorylated β-catenin is then degraded by the ubiquitin-proteasome system. On the other hand, with activation of WNT signaling, loss of GSK-3β function prevents phosphorylation of β-catenin. Subsequently, the unphosphorylated form of β-catenin accumulates in the cytoplasm, enters the nucleus, and finally activates WNT target genes as a transcriptional activator with help from transcription factor (TCF)/lymphoid enhancer-binding factor (LEF) family proteins. Previous studies have shown that β-catenin is significantly upregulated in cells derived from PD plaque as compared to normal TA tissue [127]. Ten Dam et al. performed immunostaining of PD plaque and normal TA tissues, and found significant upregulation of several WNT ligands in the PD samples, including WNT2, 4, 5a, and 7b, as well as β-catenin [128]. These results suggest that activation of the WNT/β-catenin signaling pathway is associated with PD development.
The WNT/β-catenin signaling pathway activated in fibrotic diseases involves extensive crosstalk between TGF-β and Hedgehog signaling pathways. The dickkopf-related protein 1 (DKK1) inhibits WNT/β-catenin signaling by forming a complex with low-density lipoprotein receptor-related proteins, which inhibits interactions between WNT and the Frizzled receptor. Secreted frizzled-related protein1 (SFRP1) is also a WNT/β-catenin signaling antagonist, which unlike DKK1 directly binds to WNT and inhibits its binding to the Frizzled receptor. TGF-β1 also inhibits transcription of DKK1 and SFRP1 in a p38-dependent manner, and induces epigenetic silencing through DNA methylation, leading to further activation of the WNT/β-catenin signaling pathway [129,130,131]. Hedgehog signaling stimulates fibroblast-to-myofibroblast transition and promotes fibrosis. Indeed, pharmacological or genetic inactivation of hedgehog signaling has been shown to ameliorate experimental fibrosis in several mouse models of organ fibrosis [132,133,134]. Activation of hedgehog signaling is caused, at least in part, by TGF-β1 signaling, which induces transcription of the profibrotic Hedgehog transcription factor GLI family zinc finger 2 (GLI2) [135]. In addition, activation of Hedgehog signaling may upregulate Wnt-2b and Wnt-5a, shown to be overexpressed in PD, and also activate WNT/β-catenin signaling [128,136].
The YAP/TAZ signaling pathway is regulated by both the HIPPO signaling pathway and a mechanically regulated HIPPO-independent mechanism [137]. In addition to HIPPO control, deregulation of YAP and TAZ driven by convergence of different activating signals, such as RHOA/ROCK, WNT/β-catenin, and TGF-β, is associated with fibrosis in various organs including PD [138]. Increased ECM stiffness due to fibrotic tissue remodeling is sensed by αVβ integrins, which are upregulated upon injury and activate intracellular signaling pathways involving RHOA/ROCK signaling. Subsequently, formation of F-actin stress fibers and translocation of YAP/TAZ to the nucleus are activated, where these proteins promote profibrotic gene expression through activation of the TEAD family member transcription enhancer factor (TEF). Furthermore, RHOA/ROCK signaling activates AKT, which may activate p300 in the nucleus via AKT-induced phosphorylation, resulting in further enhanced transcription of profibrotic genes [139]. Gene expression comparisons between normal TA and PD plaque performed by Qian et al. showed increased expression of β integrin and RhOA12 in PD [140]. Additionally, YAP1 expression was found upregulated in PD plaque samples as compared to normal TA tissues in a previous immunohistochemical study [128]. Milenkovic et al. reported that simvastatin and an RHO kinase inhibitor prevented myofibroblast transformation in PD-derived fibroblasts through inhibition of YAP/TAZ nuclear translocation, and reduced CTGF and collagen expression [141]. Also, integrin-linked kinase (ILK) is involved in integrin-mediated signaling, promotes inhibition of phosphorylation of myosin phosphatase target subunit 1, and inactivates Merlin, a key upstream regulator of the Hippo pathway, which activates YAP/TAZ in a large tumor suppressor kinase (LATS)-1/2-dependent manner [142], while ILK-mediated deregulation of the Hippo-YAP/TAZ pathway plays an important role in formation of keloids and hypertrophic scars [143]. In human corpus cavernosum smooth muscle cells, ILK may be a key regulator of biological functions, such as cell attachment, spreading, migration, and microfilament dynamics [144].
YAP and TAZ also bind phosphorylated SMAD2/SMAD3 and SMAD4 complexes formed in the nucleus through activation of the TGF-β1 pathway. Subsequently, transcription of Serpin family E member 1 (SERPINE1), also known as PAI-1, is induced, which leads to excessive accumulation of ECM and contributes to development of fibrotic diseases including PD [145]. Activation of YAP/TAZ signaling via RHOA/ROCK signaling is also induced by TGF-β1 as one of the non-canonical TGF-β signaling pathways. Jiang et al. showed that suppression of both TGF-β1-mediated SMAD and the RHOA/ROCK signaling pathway mediated by estrogen may suppress collagen secretion and self-contraction of TA-derived myofibroblasts [146].
Therefore, parallel non-regulated activity of several core pathways, with TGF-β signaling pathway as the main axis, that simultaneously leads to altered signaling crosstalk is conceivable, resulting in development of PD via aberrant tissue repair and fibrosis in TA tissues. A proposed diagram summarizing the core pathways possibly involved in development of PD and their interrelationships is shown in Figure 3.
4. Risk Factors for Pathogenesis of Peyronie's Disease
Multifactorial diseases, characterized by inter-individual variations in etiology, onset age, and penetrance, are relatively common, and arise from the combined activities of genetic and environmental factors [147]. Multifactorial diseases develop when quantitative fluctuations of biological risk factors exceed certain thresholds due to interactions of mutations in causative genes, such as polygenes and major genes, as well as the influence of environmental factors [148]. Accumulating evidence suggests that various genetic and environmental factors are involved in several molecular alterations that contribute to PD pathogenesis (Figure 4). PD is thus considered to be a multifactorial disease and integrated understanding of the complex mechanisms underlying gene-environment interactions associated with PD risk and progression could improve various aspects of patient care. In this section, genetic and precipitating risk factors associated with PD based on their speculated role in its pathogenesis are summarized. Each examined risk factor and its predicted impact on PD development are shown in Table 1.
4.1. Intrinsic Risk Factors
Genetic factors have been reported as representative intrinsic factors possibly associated with PD development. Several studies have revealed involvement of chromosomal aberrations, gene mutations, and epigenetic modifications in this disease. Although no genome-wide association study (GWAS) studies to date have specifically examined genetic susceptibility to PD, presented findings provide several possibly associated candidate genes. Other known intrinsic risk factors include aging and low testosterone level, while associations of the ABO blood group with congenital penile curvature (CPC) and PD have also been reported [149,150].
4.1.1. Genetics of Peyronie's Disease
A pedigree analysis of three families affected by both PD and DD provided the first clue of genetic susceptibility to PD, as an autosomal dominant mode inheritance with incomplete penetrance was identified in all three, with one family exhibiting three generations of father to-son transmission [151]. Chromosomal abnormalities associated with PD have also been shown using fibroblast culture models. Somers found that karyotypic abnormalities in PD plaque-derived fibroblasts included duplication of chromosome 7 and 8, deletion of chromosome Y, and reciprocal translocations of 46XY, t(11;12)(q11,p11) and 46XY, t(1;5)(q25;q11), as well as inversion of 46XY, inv(7)(p22q36) [152]. These chromosomal abnormalities have been proposed to be acquired changes, because they have been found to occur only in PD-derived fibroblasts and not other tissue-derived cells. Mulhall et al. detected chromosomal aberrations involving chromosomes 7, 8, 17, 18, Y, and X in PD plaque-derived fibroblasts, and confirmed that such fibroblasts showed earlier chromosomal instability than those derived from normal TA [153].
A relationship between heritable SNPs and PD that can affect gene expression has been shown by several studies. We previously described that elevated TGF-β1 level and activation of TGF-β signaling are the most important factors for development of PD. It is speculated that elevated TGFβ-1 level is partly due to the presence of SNPs within the TGF-β1 gene. The SNP rs1800471 (p.A35P), which results in substitution of arginine to proline at position 25 in the TGF-β1 protein, might be a PD-associated SNP, as it has been found at a significantly higher frequency in PD patients than healthy subjects [154]. A study by Dolmans et al., which assessed allele frequencies of SNPs associated with the WNT/β-catenin pathway, found that rs4730775 in the WNT2 gene locus on chromosome 7 was associated with PD pathogenesis [155]. As noted previously, WNT2 is one of the WNT ligands significantly upregulated in PD [128]. Furthermore, intriguing results were recently obtained by whole genome sequencing using blood samples from three PD patients with DD performed by Dullea et al. [156], as a protein encoding mutations in the ALMS1 gene including the SNPs rs45501594 (p.T3445S), rs34071195 (p.K3435E), and rs41291187 (p.H624R) was present in all three. Compound heterozygous mutations of the ALMS1 gene are known to cause Alström syndrome, a rare autosomal recessive disorder related to a variety of symptoms, such as cone-rod retinal dystrophy, sensorineural hearing loss, obesity, insulin resistance, DM, hypertriglyceridemia, and dilated cardiomyopathy [157]. Notably, ALMS1-deficient fibroblasts were shown to acquire apoptosis resistance and overexpress ECM components [158]. Based on reports that an ALMS1 mutation is associated with aberrant TGF-β signaling [159,160], Dullea et al. speculated that such a mutation leads to increased TGF-β level, resulting in fibrosis and the PD phenotype [156].
Epigenetic modifications, such as DNA methylation, histone acetylation/deacetylation, and non-coding RNAs [e.g., microRNAs (miRNAs)], have a crucial role in fibrotic disease pathogenesis [29,161]. Such epigenetic changes may contribute to development of fibrotic diseases by leading to sustained myofibroblast activation [162,163]. Specifically, histone deacetylases (HDACs) promote deacetylation of histones, major constituents of the chromatin structure, through TGF-β signaling and promote fibrosis via downregulation of the anti-fibrotic gene PPARGC1A [163]. Interestingly, Kang et al. showed that HDACs were more highly expressed in PD plaque samples as compared to normal TA. They also found that silencing of HDAC7 attenuated TGF-β1-induced profibrinolytic responses in PD plaque-derived fibroblasts [164]. Among the various miRNAs associated with fibrosis, a recent study showed that the miRNA-29 family (miR-29a, b, c) has a key role in the process of multi-organ fibrosis [161]. Members of this family may be closely associated with regulation of expression of multiple cytokines, key fibrotic pathways such as TGF-β1/SMAD, PI3K/AKT, and DNA methylation, and epithelial-mesenchymal transition of fibroblasts [161]. Dos Santos and colleagues also recently found that miR-29b was significantly downregulated in fibrous plaque, TA, and corpus cavernosum samples from PD patients as compared with those from controls [165]. In addition, results from studies that investigated the role of immunological contributors in PD suggest that several miRNAs have regulatory roles in the immune system of affected individuals [60].
4.1.2. Aging
Aging, characterized by progressive loss of physiological integrity, is a major risk factor for human pathologies such as DM, cardiovascular disorders, and neurodegenerative diseases, while fibrosis is also considered as a universal age-related pathological process in various organs. The prevalence of PD increases in the third decade and peaks in the fifth, suggesting aging as risk factor for this disease [10,166]. A decline in alpha-1-antitrypsin, a key protease inhibitor that regulates tissue degradation, was revealed to be an age-related phenomenon in PD-affected patients based on comparisons with age-matched healthy controls [167]. Another possible reason for the increased prevalence of PD with aging is unique microtrauma effects on the penis that differ as compared to young men because erections are less rigid [168]. Furthermore, in older men, the tissue becomes less elastic and deforming force with intercourse can cause bending, leading to minor trauma [169]. Aging has numerous functionally interrelated hallmarks, including stem cell depletion, genomic instability, telomere shortening, epigenetic changes, and loss of protein homeostasis [70]. Thus, genetic alterations and epigenetic modifications associated with PD noted in the previous section may also be induced by aging.
4.1.3. Testosterone Level
Testosterone deficiency (TD) has been found to cause changes in TA and affect collagen metabolism abnormalities associated with PD [171,172]. Subsequent clinical studies also indicated that TD may influence PD etiology and severity [173,174,175,176]. However, any association between testosterone and PD remains controversial, as various presented findings have denied that [177,178,179]. In prostate cancer cells, the androgen receptor (AR), activated by testosterone binding, may attenuate TGF-β signaling by suppressing TGF-βRII expression [180]. AR is also expressed in penile tissues [181] and we speculate that decreased serum testosterone levels may induce TGF-βRII expression through decreased activation of AR, which activates TGF-β signaling associated with PD development.
4.1.4. ABO Blood Type
We recently compared 202 Japanese PD patients with 846 non-PD male patients and found that ABO blood type may be associated with PD risk [149]. Interestingly, as compared with individuals with blood type B, those with type O are at greater risk of PD with an odds ratio of 2.018, and also have higher serum levels of the profibrotic cytokines TNF-α and IL-6 as compared to others [182,183]. TGF-βR1 is located on chromosome 9q22 near the ABO locus (9q34), thus a relatively high rate of recombination between TGF-β1R1 and ABO-type genes is speculated, increasing the likelihood that these will be inherited together with ABO.
4.1.5. Congenital Penile Curvature
CPC is a genetically inherited condition characterized by penis curvature that is present at birth without obvious organic pathological changes in the organ. Thus, CPC is considered to have a completely different pathology from PD, an acquired condition that causes obvious pathological changes in the TA. Recently, Paulis et al. conducted a retrospective analysis of a clinical database from a single andrology clinic and found a history of CPC in 73 of 519 patients with PD (14.1%), whereas only 15 (0.7%) of 2166 in a comparator population without PD had such results [150]. Those authors speculated that the presence of CPC is a risk factor for subsequent development of PD, though the mechanisms underlying the association between the two diseases remain unclear. It is possible that penile curvature causes increased damage to the penis during intercourse, facilitating accumulation of continuous TA microtrauma that triggers PD.
4.2. Extrinsic Risk Factors
Extrinsic risk factors are those have adverse effects on the body from outside sources, such as chemical and physical contaminants found in food, or appear as contaminants in the environment. Smoking, alcohol consumption, and perineal and penile trauma are possible extrinsic risk factors associated with PD.
4.2.1. Smoking
Numerous studies have shown that smoking is strongly associated with PD [10,184,185,186]. Interestingly, the prevalence of PD increases with the number of cigarettes smoked per day as well as habit duration, with a particularly high prevalence reported in individuals who have smoked for more than five years [185,186]. An investigation of the systemic effects of chronic smoking on skin architecture confirmed extensive remodeling of the dermal elastic fiber system [187]. Liu et al. showed that cigarette smoke-induced fibrosis in human kidney cell lines was related to inhibition of the ROS pathway, and induction of expression of the profibrotic factors CTGF and PAI-1 [188]. Furthermore, cigarette smoke was confirmed to induce epithelial-to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin, a prognostic factor for lung cancer in smokers [189]. Smoking-induced ROS and epigenetic changes may also be involved in PD development.
4.2.2. Alcohol
Alcohol consumption is a well-known risk factor for development of several diseases in various organs, including fibrosis. For example, DM, dyslipidemia, and cardiovascular disorders, known PD-related comorbidities, have been shown to be strongly associated with alcohol consumption. A previous case-control study found that alcohol consumption was more frequent in PD cases as compared with a control group, though there was no difference related to duration of alcohol consumption or types of beverages consumed [184]. Rat studies have also shown that long-term heavy alcohol consumption increases expression of fibrotic cytokines such as TIMP-1 and SMAD3, leading to kidney tissue fibrosis [190]. Additionally, a study that investigated early effects of alcohol intoxication on liver fibrosis in adolescents reported significantly elevated serum MMP-9 and TIMP-1 concentrations during intoxication [191].
4.2.3. Perineal and Penile Trauma
As noted previously, PD initiation by penile microtrauma is the most widely accepted theory and a number of studies have shown that history of penile trauma may be associated with PD [10,184,192,193]. Bjekic et al. reported that about a quarter of their examined PD patients had incidental genital perineal injuries, including iatrogenic injury caused by a therapeutic procedure such as catheterization or cystoscopy [184]. In addition, a history of penile trauma during intercourse has been reported in 21-40% of examined PD patients [192,193]. Such damage to the penis may cause detachment of septal fibers with extravasation of blood into the intraluminal space, leading to infiltration of inflammatory cells that trigger PD development.
4.3. Comorbidities
DM, HT, dyslipidemia, obesity, ED, and DD are widely recognized comorbidities associated with PD. Although comorbidities are often included in the category of intrinsic risk factors in a broad sense, the present review discusses them separately.
4.3.1. Hypertension, Diabetes Mellitus, Dyslipidemia, and Obesity
DM, cardiovascular disorders including HT, dyslipidemia, and obesity are considered as so-called lifestyle diseases. Many of these chronic conditions develop in middle or later life and, like PD, are considered to be multifactorial and their association with PD has been shown by previous epidemiological studies [9,10,184]. DM, HT, and dyslipidemia are risk factors for systemic vascular disease, and may induce fibrosis in systemic organs through several mechanisms, such as vascular endothelial cell damage or ROS production. A high-fat, high-cholesterol diet leads to development of steatohepatitis and severe fibrosis, which have been reported to be exacerbated by HT through NF-κB and MAPK pathways in rat models [194,195]. Furthermore, MMPs, TIMPs, and TGF-β1 have been identified as common proteins in signaling pathways involved in the fibroproliferative processes of PD and DM, indicating a common link [9].
4.3.2. Erectile Dysfunction
ED is an important symptom of PD, as it impairs QoL, with previous studies have showing prevalence in greater than 50%of PD patients [196]. This may be a symptom as well as cause of PD because of stronger trauma due to a semi-rigid erection or delayed ejaculation. Furthermore, complex factors are associated with development of ED, as those such as aging, DM, HT, and dyslipidemia associated with PD development are also risk factors for ED. Recently, two GWASs identified loci adjacent to single-minded homolog 1, an obesity-related gene, that may confer higher ED risk [197,198]. These findings have led to renewed interest in understanding the genetic basis of ED and PD [199].
4.3.3. Dupuytren’s Disease
PD is known to frequently occur together with DD, and characterized by progressive fibrosis of the palmar and finger fascia [9,140,184]. Both PD and DD are focal manifestations of a systemic fibroproliferative pathological process, and share common risk factors such as aging, smoking, alcohol use, and diabetes [200]. In addition, several commonalities in their genetics and etiology have been reported [11,12,13,140,156]. A GWAS performed by Dolmans et al. identified nine chromosomal loci associated with susceptibility to DD [201], with 17 additional variants subsequently reported, bringing the current known total to 26 [202,203]. Notably, many of these include genes involved in the WNT/β-catenin signaling pathway that have been implicated in PD.
5. Conclusions
To date, several basic and clinical studies have revealed precise detailed mechanisms involved in development of PD. Maintenance of the inflammatory environment by macrophages and T cells seems to provide an environment favorable for PD-related plaque formation. In short, a continuous inflammatory environment beginning after penile injury continues to support myofibroblast induction and activation until plaque formation. Furthermore, recent reports suggest that inflammation-associated immune system cells are present within lesions even in chronic-phase PD cases, in which inflammation is thought to have disappeared. Aberrant tissue repair and fibrosis of TA tissue under these circumstances are promoted by the interrelated effects of abnormalities in TGF-β, WNT/β-catenin, Hedgehog, YAP/TAZ, p38 MAPK, ROCK, and PI3K/AKT signaling pathways. PD is manifested in individuals who cross the threshold for disease development, with the influence of various endogenous and extrinsic factors providing pleiotropic stimulation to pathways related with its development. Several reports have elucidated associated genetic abnormalities and identification of PD susceptibility genes by GWAS is expected in the near future. A better understanding of the molecular mechanisms involved in the pathogenesis and how risk translates into disease will aid in earlier recognition of PD and prevention, and also provide insight related to development of adjunct and novel therapeutic options for affected individuals.
Authors’ Contributions
Y.M., F.Y., and K.N. performed conceptualization of the and writing of the manuscript. S.H., M.U., H.K., and another K.N. performed the literature review. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Di Maida, F.; Cito, G.; Lambertini, L.; Valastro, F.; Morelli, G.; Mari, A.; Carini, M.; Minervini, A.; Cocci, A. The natural history of Peyronie's disease. World. J. Mens. Health. 2021, 39, 399–405. [Google Scholar] [CrossRef]
- Kadioglu, A.; Tefekli, A.; Erol, B.; Oktar, T.; Tunc, M.; Tellaloglu, S. A retrospective review of 307 men with Peyronie's disease. J. Urol. 2002, 168, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Mulhall, J.P.; Schiff, J.; Guhring, P. An analysis of the natural history of Peyronie's disease. J. Urol. 2006, 175, 2115–2118. [Google Scholar] [CrossRef] [PubMed]
- Berookhim, B.M.; Choi, J.; Alex, B.; Mulhall. J.P. Deformity stabilization and improvement in men with untreated Peyronie's disease. BJU. Int. 2014, 113, 133–136. [Google Scholar] [CrossRef]
- Nelson, C.J.; Mulhall, J.P. Psychological impact of Peyronie's disease: A review. J. Sex. Med. 2013, 10, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Terrier, J.E.; Nelson, C.J. Psychological aspects of Peyronie's disease. Transl. Androl. Urol. 2016, 5, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.J.; Diblasio. ; Kendirci, M.; Hellstrom, W.; Guhring, P.; Mulhall, J.P. The chronology of depression and distress in men with Peyronie's disease. J. Sex. Med. 2008, 5, 1985–1990. [Google Scholar] [CrossRef]
- Kuja-Halkola, R.; Henningsohn, L.; D'Onofrio, B.M.; Mills, J.; Adolfsson, A.; Larsson, H.; Cederlöf, M. Mental disorders in Peyronie's disease: A Swedish cohort study of 3.5 million men. J. Urol. 2021, 205, 864–870. [Google Scholar] [CrossRef]
- Gelbard, M.K.; Rosenbloom, J. Fibroproliferative disorders and diabetes: Understanding the pathophysiologic relationship between Peyronie's disease, Dupuytren disease and diabetes. Endocrinol. Diabetes. Metab. 2020, 4, e00195. [Google Scholar] [CrossRef]
- Segundo, A.; Glina, S. Prevalence, risk factors, and erectile dysfunction associated with Peyronie's disease among men seeking urological care. Sex. Med. 2020, 8, 230–236. [Google Scholar] [CrossRef]
- Herati, A.S.; Pastuszak, A.W. The genetic basis of Peyronie disease: A review. Sex. Med. Rev. 2016, 4, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, U.; Ilg, M.M.; Cellek, S.; Albersen, M. Pathophysiology and future therapeutic perspectives for resolving fibrosis in Peyronie's disease. Sex. Med. Rev. 2019, 7, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Krakhotkin, D.V.; Chernylovskyi, V.A.; Mottrie, A.; Greco, F.; Bugaev, R.A. New insights into the pathogenesis of Peyronie's disease: A narrative review. Chronic. Dis. Transl. Med. 2020, 6, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Rhoden, E.L.; Teloken, C.; Ting, H.Y.; Lucas, M.L.; Teodosio da Ros, C.; Ary Vargas Souto, C. Prevalence of Peyronie’s disease in men over 50 years old in southern Brazil. Int. J. Impot. Res. 2001, 13, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Tal, R.; Heck, M.; Teloken, P.; Siegrist, T.; Nelson, C.J.; Mulhall, J.P. Peyronie’s disease following radical prostatectomy: Incidence and predictors. J. Sex. Med. 2010, 7, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, K.; Shimabukuro, T.; Matsuyama, H. The prevalence of Peyronie's disease in Japan: A study in men undergoing maintenance hemodialysis and routine health checks. J. Sex. Med. 2012, 9, 2716–2723. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Ralph, D.; Kagioglu, A.; Garaffa, G.; Shamsodini, A.; Bivalacqua, T.; Glina, S.; Hakim, L.; Sadeghi-Nejad, H.; Broderick, G. Evidence-based management guidelines on Peyronie's disease. J. Sex. Med. 2016, 13, 905–923. [Google Scholar] [CrossRef]
- Capoccia, E.; Levine, L.A. Contemporary review of Peyronie’s disease treatment. Curr. Urol. Rep. 2018, 19, 51. [Google Scholar] [CrossRef]
- Cocci, A.; Russo, G.I.; Briganti, A.; Salonia, A.; Cacciamani, G.; Capece, M.; Falcone, M.; Timpano, M.; Cito, G.; Verze, P.; et al. Predictors of treatment success after collagenase Clostridium histolyticum injection for Peyronie's disease: Development of a nomogram from a multicentre single-arm, non-placebo controlled clinical study. BJU. Int. 2018, 122, 680–687. [Google Scholar] [CrossRef]
- Devine, C.J., Jr.; Somers, K.D.; Jordan, S.G.; Schlossberg, S.M. Proposal: Trauma as the cause of the Peyronie's lesion. J. Urol. 1997, 157, 285–290. [Google Scholar] [CrossRef]
- Jiang, H.; Gao, Q.; Che, X.; Zhu, L.; Zhang, Z.; Chen, Y.; Dai, Y. Repeated micro-trauma of the penile tunica albuginea: A new animal model of Peyronie's disease. Urol. Int. 2018, 100, 228–239. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, U.; Ilg, M.; Cellek, S.; Albersen, M.E. What role do pharmaceuticals play in the treatment of Peyronie's disease and is there a need for new emerging drugs? Expert. Opin. Emerg. Drugs. 2019, 24, 1–4. [Google Scholar] [CrossRef] [PubMed]
- O'Dwyer, D.N.; Ashley, S.L.; Moore, B.B. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2016, 311, L590–L601. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell. Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef]
- Wu, L.; Huang, K.; Li, Q.; Wu, H.; Gao, Y. , Xu, X.; Liu, X.; Han, L. Crosstalk between myofibroblasts and macrophages: A regulative factor of valvular fibrosis in calcific aortic valve disease. Cell. Biol. Int. 2023, 47, 754–767. [Google Scholar] [CrossRef]
- Lis-López, L.; Bauset, C.; Seco-Cervera, M.; Cosín-Roger, J. Is the macrophage phenotype determinant for fibrosis development? Biomedicines 2021, 9, 1747. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Van de Water, L. Mechanisms by which fibrin and fibronectin appear in healing wounds: Implications for Peyronie's disease. J. Urol. 1997, 157, 306–310. [Google Scholar] [CrossRef]
- Moretti, L.; Stalfort, J. Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Beller, D.I.; Frendl, G.; Graves, D.T. Monocyte chemoattractant protein 1 regulates adhesion molecule expression and cytokine production in human monocytes. J. Immunol. 1992, 148, 2423–2438. [Google Scholar] [CrossRef]
- Young, L.R.; Gulleman, P.M.; Short, C.W.; Tanjore, H.; Sherrill, T.; Qi, A.; McBride, A.P.; Zaynagetdinov, R.; Benjamin, J.T.; Lawson, W.E.; et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI. Insight. 2016, 1, e88947. [Google Scholar] [CrossRef] [PubMed]
- Magee, T.R.; Qian, A.; Rajfer, J.; Sander, F.C.; Levine, L.A.; Gonzalez-Cadavid, N.F. Gene expression profiles in the Peyronie's disease plaque. Urology. 2002, 59, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Lin, G.; Wang, Z.; Maddah, S.A.; Lue, T.F. Upregulation of monocyte chemoattractant protein 1 and effects of transforming growth factor-beta 1 in Peyronie's disease. Biochem. Biophys. Res. Commun. 2002, 295, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis--A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 373, 96. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. AMJ. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends. Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E. T helper cell subsets: Diversification of the field. Eur. J. Immunol. 2023, 15, e22. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.F.C.; Cardoso, F.G.R.; Ferreira, N.S.; Corazza, B.J.M.; Valera, M.M.C.; Nascimento, G.G.; Martinho, F.C. Effects of calcium hydroxide intracanal medications on T helper (Th1, Th2, Th9, Th17, and Tfh) and regulatory T (Treg) cell cytokines in apical periodontitis: A CONSORT RCT. J. Endod. 2022, 48, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Fu, M.; Xin, H.B. Polarizing macrophages in vitro. Methods. Mol. Biol. 2018, 1784, 119–126. [Google Scholar]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc, Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Todd, N.W.; Scheraga, R.G.; Galvin, J.R.; Iacono, A.T.; Britt, E.J.; Luzina, I.G.; Burke, A.P.; Atamas, SP. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J. Inflamm. Res. 2013, 6, 63–70. [Google Scholar] [CrossRef]
- Bosello, S.; Angelucci, C.; Lama, G.; Alivernini, S.; Proietti, G.; Tolusso, B.; Sica, G.; Gremese, E.; Ferraccioli, G. Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis. Res. Ther. 2018, 20, 75. [Google Scholar] [CrossRef] [PubMed]
- Beesley, C.F.; Goldman, N.R.; Taher, T.E.; Denton, C.P.; Abraham, D.J.; Mageed, R.A.; Ong, V.H. Dysregulated B cell function and disease pathogenesis in systemic sclerosis. Front. Immunol. 2023, 13, 999008. [Google Scholar] [CrossRef] [PubMed]
- Taher, T.E.; Ong, V.H.; Bystrom, J.; Hillion, S.; Simon, Q.; Denton, C.P.; Pers, J.O.; Abraham, D.J.; Mageed, R.A. Association of defective regulation of autoreactive interleukin-6-producing transitional B lymphocytes with disease in patients with systemic sclerosis. Arthritis. Rheumatol. 2018, 70, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Forestier, A.; Guerrier, T.; Jouvray, M.; Giovannelli, J.; Lefèvre, G.; Sobanski, V.; Hauspie, C.; Hachulla, E.; Hatron, P.Y.; Zéphir, H.; et al. Altered B lymphocyte homeostasis and functions in systemic sclerosis. Autoimmun. Rev. 2018, 17, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Ucero, A.C.; Bakiri, L.; Roediger, B.; Suzuki, M.; Jimenez, M.; Mandal, P.; Braghetta, P.; Bonaldo, P.; Paz-Ares, L.; Fustero-Torre, C.; et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J. Clin. Invest. 2019, 129, 3293–3309. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Karim, M.R.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunophenotypical characterization of M1/M2 macrophages and lymphocytes in cisplatin-induced rat progressive renal fibrosis. Cells. 2021, 10, 257. [Google Scholar] [CrossRef]
- Atar, A.; Kural, A.; Yenice, G.; Comez, I.; Tugcu, V.K. Role of interleukin-6 and pentraxin 3 as an early marker in Peyronie's disease. Kaohsiung. J. Med. Sci. 2017, 33, 195–200. [Google Scholar] [CrossRef]
- Zimmermann, R.P.; Feil, G.; Bock, C.; Hoeltl, L.; Stenzl, A. Significant alterations of serum cytokine levels in patient with Peyronie's disease. Int. Braz. J. Urol. 2008, 34, 457–466. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, L.; Wang, X.; Yu, L.; Wu, J. Identifying hub genes, key pathways and key immune-related genes in Peyronie's disease by integrated bioinformatic analysis. Front. Pharmacol. 2022, 13, 1019358. [Google Scholar] [CrossRef]
- Hamid, T.; Xu, Y.; Ismahil, M.A.; Rokosh, G.; Jinno, M.; Zhou, G.; Wang, Q. , Prabhu, S.D. : Cardiac mesenchymal stem cells promote fibrosis and remodeling in heart failure: Role of PDGF signaling. JACC Basic. Transl. Sci. 2022, 7, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Lucattelli, M.; Lunghi, B.; Fineschi, S.; Mirone, V.; d'Emmanuele di Villa Bianca, R.; Longo, N. l Imbimbo, C.; De Palma, R.; Sorrentino, R.; Lungarella, G.; et al A new mouse model of Peyronie's disease: An increased expression of hypoxia-inducible factor-1 target genes during the development of penile changes. Int. J. Biochem. Cell. Biol. 2008, 40, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Layton, T.B.; Williams, L.; Yang, N.; Zhang, M.; Lee, C.; Feldmann, M.; Trujillo, G.; Furniss, D.; Nanchahal, J. A vasculature niche orchestrates stromal cell phenotype through PDGF signaling: Importance in human fibrotic disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2120336119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L. , Dudley, A.C. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Abdelhakim, M.; Lin, X.; Ogawa, R. The Japanese experience with basic fibroblast growth factor in cutaneous wound management and scar prevention: A systematic review of clinical and biological Aspects. Dermatol. Ther (Heidelb). 2020, 10, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Mulhall, J.P.; Thom, J.; Lubrano, T.; Shankey, T.V. Basic fibroblast growth factor expression in Peyronie's disease. Basic fibroblast growth factor expression in Peyronie's disease. J. Urol. 2001, 165, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Burks, T.N.; Cohn, R.D. ; Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle. 2011, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; McMahon, C.D.; Matthews, K.G.; Devlin, G.P.; Elston, M.S.; Conaglen, J.V. Absence of myostatin improves cardiac function following myocardial infarction. Heart. Lung. Circ. 2018, 27, 693–701. [Google Scholar] [CrossRef]
- Cantini, L.P.; Ferrini, M.G.; Vernet, D. , Magee, T.R.; Qian, A.; Gelfand, R.A.; Rajfer, J.; Gonzalez-Cadavid, N.F. Profibrotic role of myostatin in Peyronie's disease. J. Sex. Med. 2008, 5, 1607–1622. [Google Scholar] [CrossRef]
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengellér, Z.K. AP39, a modulator of mitochondrial bioenergetics, reduces antiangiogenic response and oxidative stress in hypoxia-exposed trophoblasts: Relevance for preeclampsia pathogenesis. Am. J. Pathol. 2019, 189, 104–114. [Google Scholar] [CrossRef]
- Li, X.; Xing, J.; Wang, H.; Yu, E. The SLC34A2-ROS-HIF-1-induced up-regulation of EZH2 expression promotes proliferation and chemo-resistance to apoptosis in colorectal cancer. Biosci. Rep. 2019, 39, BSR20180268. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, B.; Kim, S.M.; Yang, C.E.; Song, S.Y.; Lee, W.J.; Lee, J.H. Hypoxia-induced epithelial-to-mesenchymal transition mediates fibroblast abnormalities via ERK activation in cutaneous wound healing. Int. J. Mol. Sci. 2019, 20, 2546. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, Y.; Men, C.; Wu, J.; Liu, L. Bioinformatics-based identification of potential hypoxia-related genes associated with Peyronie's disease. Am. J. Mens. Health. 2022, 16, 15579883221111720. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the "omics" era. Matrix. Biol. 2016, 49, 10–e24. [Google Scholar] [CrossRef] [PubMed]
- Randles, M.; Lennon. R. Applying proteomics to investigate extracellular matrix in health and disease. Curr. Top. Membr. 2015, 76, 171–e196. [Google Scholar] [PubMed]
- Paulis, G.; De Giorgio, G.; Paulis, L. Role of oxidative stress in Peyronie's disease: Biochemical evidence and experiences of treatment with antioxidants. Int. J. Mol. Sci. 2022, 23, 15969. [Google Scholar] [CrossRef] [PubMed]
- Del Carlo, M.; Cole, A.A.; Levine, L.A. Differential calcium independent regulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases by interleukin-1beta and transforming growth factor-beta in Peyronie's plaque fibroblasts. J. Urol. 2008, 179, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.S.; Theodoro, T.R.; Coelho, N.L.; Mendes, A.; Leonel, M.L.P.; Mader, A.M.; Nader, H.B.; Glina, S.; Pinhal, M.A.S. Extracellular matrix alterations in the Peyronie's disease. J. Adv. Res. 2017, 8, 455–461. [Google Scholar] [CrossRef]
- Flevaris, P.; Vaughan, D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin. Thromb. Hemost. 2017, 43, 169–177. [Google Scholar] [CrossRef]
- Davila, H.H.; Magee, T.R.; Zuniga, F.I.; Rajfer, J.; Gonzalez-Cadavid, N.F. Peyronie's disease associated with increase in plasminogen activator inhibitor in fibrotic plaque. Urology. 2005, 65, 645–648. [Google Scholar] [CrossRef]
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, I.A.; Desmoulière, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis. Tissue. Repair. 2012, 5 (Suppl. 1). [Google Scholar] [CrossRef] [PubMed]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell. Mol. Biol. 2012, 296, 139–185. [Google Scholar] [PubMed]
- Szardening-Kirchner, C.; Konrad, L.; Hauck, E.W.; Haag, S.M.; Eickelberg, O.; Weidner, W. Upregulation of mRNA expression of MCP-1 by TGF-beta1 in fibroblast cells from Peyronie's disease. World. J. Urol. 2009, 27, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Han, X.; Yao, Z.; Zhang, H.; Zhao, M.; Peng, M.; Wang, K.; Shan, Q.; Sang, X.; Wu, X.; et al. The pathogenesis of organ fibrosis: Focus on necroptosis. Br. J. Pharmacol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zorba, O.U.; Sirma, S.; Ozgon, G.; Salabas, E.; Ozbek, U.; Kadioglu, A. Comparison of apoptotic gene expression profiles between Peyronie's disease plaque and tunica albuginea. Clin. Exp. Med. 2012, 21, 607–614. [Google Scholar]
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie's disease. Biomed. Res. Int. 2014, 616149. [Google Scholar] [CrossRef] [PubMed]
- Loreto, C.; Barbagli, G.; Djinovic, R.; Vespasiani, G.; Carnazza, M.L.; Miano, R.; Musumeci, G.; Sansalone, S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and its death receptor (DR5) in Peyronie's disease. a biomolecular study of apoptosis activation. J. Sex. Med. 2011, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. , Lagares, D.N. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Garaffa, G.; Trost, L.W.; Serefoglu, E.C.; Ralph, D.; Hellstrom. W.J. Understanding the course of Peyronie's disease. Int. J. Clin. Pract. 2013, 67, 781–788. [Google Scholar] [CrossRef]
- Milenkovic, U.; Boeckx, B.; Lambrechts, D.; Janky, R.; Hatzichristodoulou, G.; van Renterghem, K.; Gevaert, T.; Cellek, S.; Bivalacqua, T.J.; De Ridder, D.; et al. Single-cell transcriptomics uncover a novel role of myeloid cells and T-lymphocytes in the fibrotic microenvironment in Peyronie's disease. Eur. Urol. Focus. 2022, 8, 814–828. [Google Scholar] [CrossRef]
- Frangogiannis, N. G: Transforming growth factor-β in myocardial disease. Nat. Rev. Cardiol. 2022, 19, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, G.; Nguyen, T.; Namasivayam, A.; Starek, S.; Gelman, J.; Mauney, J.R. Characterization of a novel rabbit model of Peyronie's disease. Int. J. Impot. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Yuan, J.H. [Animal models of Peyronie's disease: An update]. Zhonghua. Nan. Ke. Xue. 2016, 22, 446–449. [Google Scholar] [PubMed]
- Distler, J.H.; Feghali-Bostwick, C.; Soare, A.; Asano, Y.; Distler, O.; Abraham, D.J. Review: Frontiers of antifibrotic therapy in systemic sclerosis. Arthritis, Rheumatol. 2017, 69, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Park, J.; Yu, M.R.; Kim, Y.S.; Ha, H.; Lee, H.B. Positive feedback loop between plasminogen activator inhibitor-1 and transforming growth factor-beta1 during renal fibrosis in diabetes. Am. J. Nephrol. 2009, 30, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S. , Misra, D.P.; Prasad, N.; Rastogi, K.; Singh, H., Rai, M.K.; Agarwal, V. Serotonin 5HT2A receptor antagonism mediated anti-inflammatory and anti-fibrotic effect in adriamycin-induced CKD in rats. Naunyn. Schmiedebergs. Arch. Pharmacol. 2020, 393, 1269–1279. [Google Scholar]
- Chaturvedi, S.; Misra, D.P.; Prasad, N. , Rastogi, K.; Singh, H.; Rai, M.K.; Agarwal, V.I. 5-HT2 and 5-HT2B antagonists attenuate pro-fibrotic phenotype in human adult dermal fibroblasts by blocking TGF-β1 induced non-canonical signaling pathways including STAT3: Implications for fibrotic diseases like scleroderma. Int. J. Rheum. Dis. 2018, 21, 2128–2138. [Google Scholar]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Conroy KP, Kitto LJ, Henderson NC. αv integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Chen, J.; Xu, Y.J. Thrombospondin-1: A key protein that induces fibrosis in diabetic complications. Diabetes Res. 2020, 2020, 8043135. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Chen, F.; Zhang, H.; Tang, P.; Yuan, M.; Wen, J.; Wang, S.; Cai, Z. Role of angiotensin II in aging. Front. Aging. Neurosci. 2022, 14, 1002138. [Google Scholar] [CrossRef] [PubMed]
- Canguven, O.; Lagoda, G.; Sezen, S.F.; Burnett, A.L. Losartan preserves erectile function after bilateral cavernous nerve injury via antifibrotic mechanisms in male rats. J. Urol. 2009, 181, 2816–2822. [Google Scholar] [CrossRef] [PubMed]
- Gur, S.; Kadowitz, P.J.; Hellstrom, W.J. Drugs of the future for Peyronie's disease. Med. Hypotheses. 2012, 78, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Bragina, M.E.; Costa-Fraga, F.; Sturny, M.; Ebadi, B.; Ruoccolo, R.T.; Santos, R.A.S.; Fraga-Silva, R.A.; Stergiopulos, N. Characterization of the renin-angiotensin system in aged cavernosal tissue and its role in penile fibrosis. J. Sex. Med. 2020, 17, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Hauck, E.W.; Szardening-Kirchner, C.; Diemer, T.; Cha, E.S.; Weidner, W.; Eickelberg, O. Alterations in the transforming growth factor (TGF)-beta pathway as a potential factor in the pathogenesis of Peyronie's disease. Eur. Urol. 2007, 51, 255–261. [Google Scholar] [CrossRef]
- de Ceuninck van Capelle, C.; Spit, M.; Ten Dijke, P. Current perspectives on inhibitory SMAD7 in health and disease. Current perspectives on inhibitory SMAD7 in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 691–715. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.J.; Song, K.M.; Park, J.M.; Kwon, M.H.; Kwon, K.D.; Park, S.H.; Ryu, D.S.; Ryu, J.K.; Suh, J.K. Effect of SMAD7 gene overexpression on TGF-β1-induced profibrotic responses in fibroblasts derived from Peyronie's plaque. Asian. J. Androl. 2015, 17, 487–492. [Google Scholar]
- Wang, W.; Ding, W.; Zhang, X.; Wu, S.; Yu, T.; Cui, X.; Xie, Y.; Yang, D.; Lin, C. Intratunical injection of rat-derived bone marrow mesenchymal stem cells prevents fibrosis and is associated with increased Smad7 expression in a rat model of Peyronie's disease. Stem. Cell. Res. Ther. 2022, 13, 390. [Google Scholar] [CrossRef]
- Wahab, N.A.; Weston, B.S.; Mason, R.M. Connective tissue growth factor CCN2 interacts with and activates the tyrosine kinase receptor TrkA. J. Am. Soc. Nephrol. 2005, 16, 340–351. [Google Scholar] [CrossRef]
- Yu, X.Y.; Sun, Q.; Zhang, Y.M.; Zou, L.; Zhao, Y.Y. TGF-β/Smad Signaling Pathway in Tubulointerstitial Fibrosis. Front. Pharmacol. 2022, 13, 860588. [Google Scholar] [CrossRef] [PubMed]
- Pavone, C.; Napoli, G. , Caruana, G.; Alonge, V.; Usala, M.; Abbadessa, D. Safety and tolerability of local treatment with iloprost, a prostacyclin analogue, in patients with Peyronie's disease: A phase I study. BJU. Int. 2012, 110, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Sarenac, T.; Trapecar, M.; Gradisnik, L.; Rupnik, M.S.; Pahor. D. Single-cell analysis reveals IGF-1 potentiation of inhibition of the TGF-β/Smad pathway of fibrosis in human keratocytes in vitro. Sci. Rep. 2016, 6, 34373. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.G.; Psarros, C.; Gekas, A.; Vandoros, G.P.; Philippou, A. , Koutsilieris, M. Alternative splicing of IGF1 gene as a potential factor in the pathogenesis of Peyronie's disease. In Vivo 2016, 30, 251–256. [Google Scholar] [PubMed]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.l; Feng, J.; Lu, X.; Yao, Z.; Liu, Q.; Lv, Y.; Han, Y.; Deng, J.; Zhou, Y. Isorhamnetin inhibits liver fibrosis by reducing autophagy and inhibiting extracellular matrix formation via the TGF-β1/Smad3 and TGF-β1/p38 MAPK pathways. Mediators. Inflamm. 2019, 2019, 6175091. [Google Scholar] [CrossRef]
- Toque, H.A.; Nunes, K.P.; Yao, L.; Liao, J.K.; Webb, R.C.; Caldwell, R.B. Caldwell, R.W. Activated Rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: Possible involvement of p38 MAPK activation. J. Sex. Med. 2013, 10, 1502–1515. [Google Scholar] [CrossRef]
- Song. W.H.; Son, H.; Kim, S.W.; Paick, J.S.; Cho, M.C. Role of Jun amino-terminal kinase (JNK) in apoptosis of cavernosal tissue during acute phase after cavernosal nerve injury. Asian. J. Androl. 2018, 20, 50–55. [Google Scholar] [CrossRef]
- Mercer, P.F.; Woodcock, H.V.; Eley, J.D.; Platé, M.; Sulikowski, M.G.; Durrenberger, P.F.; Franklin, L.; Nanthakumar, C.B.; Man, Y. , Genovese, F.; et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 2016, 71, 701–711. [Google Scholar] [CrossRef]
- Tang, Q.; Markby, G.R.; MacNair, A.J.; Tang, K.; Tkacz, M.; Parys, M.; Phadwal, K.; MacRae, V.E.; Corcoran, B.M. TGF-β-induced PI3K/AKT/mTOR pathway controls myofibroblast differentiation and secretory phenotype of valvular interstitial cells through the modulation of cellular senescence in a naturally occurring in vitro canine model of myxomatous mitral valve disease. Cell. Prolif. 2023, e13435. [Google Scholar] [CrossRef]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Ryu, Y.L.; Lee, H.S.; Lee, H.; Son, M.K.; Yan, H.H.; Hong, S.W.; Ryu, J.K.; Hong, S.; Suh, J.K.; Hong. S.S. A novel PI3K inhibitor alleviates fibrotic responses in fibroblasts derived from Peyronie's plaques. Int. J. Oncol. 2013, 42, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Duspara, K.; Bojanic, K.; Pejic, J.I.; Kuna, L.; Kolaric, T.O.; Nincevic, V.; Smolic, R.; Vcev, A.; Glasnovic, M.; Curcic, I.B.; et al. Targeting the Wnt signaling pathway in liver fibrosis for rug options: An update. J. Clin. Transl. Hepatol. 2021, 9, 960–971. [Google Scholar] [PubMed]
- Tu, M.; Wei, T.; Jia, Y.; Wang, Y.; Wu, J. Molecular mechanisms of alveolar epithelial cell senescence and idiopathic pulmonary fibrosis: A narrative review. J. Thorac. Dis. 2023, 15, 186–203. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Ding, M.; He, W. Emerging therapeutic strategies for attenuating tubular EMT and kidney fibrosis by targeting Wnt/β-catenin signaling. Front. Pharmacol. 2022, 12, 830340. [Google Scholar] [CrossRef]
- De Young, L.X.; Bella, A.J.; O'Gorman, D.B.; Gan, B.S.; Lim, K.B.; Brock, G.B. Protein biomarker analysis of primary Peyronie's disease cells. J. Sex. Med. 2010, 7, 99–106. [Google Scholar] [CrossRef]
- Ten Dam, E.P.M.; van Driel, M.F.; de Jong, I.J.; Werker, P.M.N.; Bank, R.A. Glimpses into the molecular pathogenesis of Peyronie's disease. Aging. Male. 2020, 23, 962–970. [Google Scholar] [CrossRef]
- Sato, M. Upregulation of the Wnt/beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta. Derm. Venereol. 2006, 86, 300–307. [Google Scholar] [CrossRef]
- Chen, J.H.; Chen, W.L.; Sider, K.L.; Yip, C.Y.; Simmons, C.A. β-catenin mediates mechanically regulated, transforming growth factor-β1induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 590–597. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Šumová, B.; Cordazzo, C.; Mallano, T.; Zhang, Y.; Wohlfahrt, T.; Dees, C.; Ramming, A.; Krasowska, D.; Michalska-Jakubus, M.; et al. The transcription factor GLI2 as a downstream mediator of transforming growth factor-β-induced fibroblast activation in SSc. Ann. Rheum. Dis. 2017, 76, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Hu. B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef] [PubMed]
- El-Agroudy, N.N.; El-Naga, R.N.; El-Razeq, R.A.; El-Demerdash, E. Forskolin, a hedgehog signalling inhibitor, attenuates carbon tetrachloride-induced liver fibrosis in rats. Br. J. Pharmacol. 2016, 173, 3248–3260. [Google Scholar] [CrossRef]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis. Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef]
- Hu, L.; Lin, X.; Lu, H.; Chen, B.; Bai, Y. An overview of hedgehog signaling in fibrosis. Mol Pharmacol. 2015, 87, 174–182. [Google Scholar] [CrossRef]
- Petzold, J.; Gentleman, E. Intrinsic mechanical cues and their impact on stem cells and embryogenesis. Front. Cell. Dev. Biol, 2021, 9, 761871. [Google Scholar] [CrossRef]
- Mia, M.M.; Singh, M.K. New insights into Hippo/YAP signaling in fbrotic diseases. Cells. 2022, 11, 2065. [Google Scholar] [CrossRef]
- Dou, C.; Liu, Z.; Tu, K.; Zhang, H.; Chen, C.; Yaqoob, U.; Wang, Y.; Wen, J.; van Deursen, J.; Sicard, D.; et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology 2018, 154, 2209–2221. [Google Scholar] [CrossRef]
- Qian, A.; Meals, R.A.; Rajfer, J.; Gonzalez-Cadavid, N.F. Comparison of gene expression profiles between Peyronie's disease and Dupuytren's contracture. Urology 2004, 64, 399–404. [Google Scholar] [CrossRef]
- Milenkovic, U.; Ilg, M.M.; Zuccato, C.; Ramazani, Y.; De Ridder, D.; Albersen, M. Simvastatin and the Rho-kinase inhibitor Y-27632 prevent myofibroblast transformation in Peyronie's disease-derived fibroblasts via inhibition of YAP/TAZ nuclear translocation. BJU. Int. 2019, 123, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.L.; Choi, S.H.; Mo, J.S. Role of the Hippo pathway in fibrosis and cancer. Cells 2019, 8, 468. [Google Scholar] [CrossRef]
- Petrouj, I.G.; Nikou, S.; Madduri, S.; Nifora, M.; Bravou, V.; Kalbermatten, D.F. The role of Hippo signaling pathway and ILK in the pathophysiology of human hypertrophic scars and keloids: An immunohistochemical investigation. Cells 2022, 11, 3426. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.C.; Yu, L.P.; Li, Q.; Zhang, X.W.; Zhao, Y.P.; He, P.Y.; Xu, T.; Wang, X.F. Effects of integrin-linked kinase on human corpus cavernosum smooth muscle cell cytoskeletal organisation. Andrologia. 2013, 45, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Hiwatashi, N.; Bing, R.; Doyle, C.P.; Branski. R.C. Concurrent YAP/TAZ and SMAD signaling mediate vocal fold fibrosis. Sci. Rep. 2021, 11, 13484. [Google Scholar] [CrossRef]
- Jiang, H.S.; Zhu, L.L.; Zhang, Z.; Chen, H.; Chen, Y.; Dai, Y.T. Estradiol attenuates the TGF-β1-induced conversion of primary TAFs into myofibroblasts and inhibits collagen production and myofibroblast contraction by modulating the Smad and Rho/ROCK signaling pathways. Int. J. Mol. Med. 2015, 36, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.J.; Avramopoulos, D.; Jantzie, L.L.; McCallion, A.S. Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases. Neuroinflammation 2022, 19, 223. [Google Scholar] [CrossRef] [PubMed]
- Sing, C.F.; Haviland, M.B.; Reilly, SL. Genetic architecture of common multifactorial diseases. Ciba. Found. Symp. 1996, 197, 211–229. [Google Scholar]
- Mitsui, Y.; Kobayashi, H.; Yamabe, F.; Nakajima, K.; Nagao, K. ABO blood type and risk of Peyronie's disease in Japanese males. World. J. Mens. Health. 2022, 40, 509–516. [Google Scholar] [CrossRef]
- Paulis, G.; Paulis, A.; Perletti, G.A. Congenital penile curvature as a possible risk factor for the onset of Peyronie's disease, and psychological consequences of penile curvature. Arch. Ital. Urol. Androl. 2023, 95. [Google Scholar] [CrossRef]
- Bias, W.B.; Nyberg, L.M. Jr.; Hochberg, M.C.; Walsh, P.C. Peyronie's disease: A newly recognized autosomal-dominant trait. Am. J. Med. Genet. 1982, 12, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Somers, K.D.; Winters, B.A.; Dawson, D.M.; Leffell, M.S.; Wright, G.L. Jr. Devine, C.J. Jr. Gilbert, D.A.; Horton, C.E. Chromosome abnormalities in Peyronie's disease. J. Urol. 1987, 137, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Mulhall, J.P.; Nicholson, B.; Pierpaoli, S.; Lubrano, T.; Shankey, T.V. Chromosomal instability is demonstrated by fibroblasts derived from the tunica of men with Peyronie's disease. Int. J. Impot. Res. 2004, 16, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Hauck, E.W.; Hauptmann, A.; Schmelz, H.U.; Bein, G.; Weidner, W.; Hackstein, H. Prospective analysis of single nucleotide polymorphisms of the transforming growth factor beta-1 gene in Peyronie's disease. J. Urol. 2003, 169, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, G.H.; Werker, P.M.; de Jong, I.J.; Nijman, R.J.; LifeLines Cohort, S.; Wijmenga, C.; Ophoff, R.A. WNT2 locus is involved in genetic susceptibility of Peyronie's disease. J. Sex. Med. 2012, 9, 1430–1434. [Google Scholar] [CrossRef]
- Dullea, A.; Efimenko, I.; Firdaus, F.; Griswold, A.; Arora, H.; Masterson, T.; Ramasamy. R. Whole-genome sequencing identifies novel heterozygous mutation in ALMS1 in three men with both Peyronie's and Dupuytren's disease. Urology 2022, 166, 76–78. [Google Scholar] [CrossRef]
- Dedeoglu, S.; Dede, E.; Oztunc, F.; Gedikbasi, A.; Yesil, G.; Dedeoglu, R. Mutation identification and prediction for severe cardiomyopathy in Alström syndrome, and review of the literature for cardiomyopathy. Orphanet. J. Rare. Dis. 2022, 17, 359. [Google Scholar] [CrossRef]
- Zulato, E.; Favaretto, F.; Veronese, C.; Campanaro, S.; Marshall, J.D.; Romano, S.; Cabrelle, A.; Collin, G.B.; Zavan, B.; Belloni, A.S.; et al. ALMS1-deficient fibroblasts over-express extra-cellular matrix components, display cell cycle delay and are resistant to apoptosis. PLoS ONE 2011, 6, e19081. [Google Scholar] [CrossRef]
- Álvarez-Satta, M.; Lago-Docampo, M.; Bea-Mascato, B.; Solarat, C.; Castro-Sánchez, S.; Christensen, S.T.; Valverde, D. ALMS1 regulates TGF-β signaling and morphology of primary cilia. Front. Cell. Dev. Biol. 2021, 9, 623829. [Google Scholar] [CrossRef]
- Bea-Mascato, B.; Neira-Goyanes, E.; Iglesias-Rodríguez, A.; Valverde, D. Depletion of ALMS1 affects TGF-β signalling pathway and downstream processes such as cell migration and adhesion capacity. Front. Mol. Biosci. 2022, 9, 992313. [Google Scholar] [CrossRef]
- Wang, M.; Huo, Z.; Wu, L.; Liu, F.; Liang, J.; He, X.; Yang, D. The role of miR-29 in the mechanism of fibrosis. Mini. Rev. Med. Chem. 2023. [Google Scholar] [CrossRef] [PubMed]
- Altorok, N.; Tsou, P.S.; Coit, P.; Khanna, D.; Sawalha, A.H. Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann. Rheum. Dis. 2015, 74, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Haak, A.J.; Caporarello, N.; Choi, K.M.; Ye, Z.; Yan, H.; Varelas, X.; Ordog, T.; Ligresti, G.; Tschumperlin, D.J. TGFβ-induced fibroblast activation requires persistent and targeted HDAC-mediated gene repression. J. Cell. Sci. 2019, 132, jcs233486. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Yin, G.N.; Choi, M.J.; Song, K.M.; Ghatak, K.; Minh, N.N.; Kwon, M.H.; Seong, D.H.; Ryu, J.K.; Suh, J.K.W. Silencing histone deacetylase 7 alleviates transforming growth factor-β1-induced profibrotic responses in fibroblasts derived from Peyronie's plaque. World. J. Mens. Health. 2018, 36, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, V.G.; Dos Santos, G.A.; Neto, C.B.; Viana, N.I.; Pimenta, R.; Guimarães, V.R.; Candido, P.; Romão, P.; de Camargo, J.A.; Leite, K.R.M.; et al. Downregulation of miR-29b is associated with Peyronie's disease. Urologia. 2022, 89, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.P.; Christensen, M.B.; Hotaling, J.M.; Pastuszak, A.W. A review of inflammation and fibrosis: Implications for the pathogenesis of Peyronie's disease. World. J. Urol. 2020, 38, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Hauck, E.W.; Hauptmann, A.; Haag, S.M.; Bohnert, A.; Weidner, W.; Bein, G.; Hackstein, H. alpha-1-antitrypsin levels and genetic variation of the alpha-1-antitrypsin gene in Peyronie's disease. Eur. Urol. 2004, 46, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, U.; Sommer, F.; Klotz, T.; Braun, M.; Reifenrath, B.; Engelmann, U. The prevalence of Peyronie's disease: Resultsof a large survey. BJU. Int. 2001, 88, 727–730. [Google Scholar] [CrossRef]
- Perimenis, P.; Athanasopoulos, A.; Gyftopoulos, K.; Katsenis, G.; Barbalias, G. Peyronie's disease: Epidemiology and clinical presentation of 134 cases. Int. Urol. Nephrol. 2001, 32, 691–694. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Shen, Z.J.; Zhou, X.L.; Lu, Y.L.; Chen, Z.D. Effect of androgen deprivation on penile ultrastructure. Asian. J. Androl. 2003, 5, 33–36. [Google Scholar] [PubMed]
- Karavitakis, M.; Komninos, C.; Simaioforidis, V.; Kontos, S.; Lefakis, G.; Politis, V.; Koritsiadis, G.; Konstantellou, K.; Doumanis, G. . The relationship between androgens, regulators of collagen metabolism, and Peyronie's disease: A case control study. J. Sex. Med. 2010, 7, 4011–4007. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.A.; Morgentaler, A. Testosterone deficiency and Peyronie's disease: Pilot data suggesting a significant relationship. J. Sex. Med. 2009, 6, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.J.; Park, H.J.; Park, N.C. Does testosterone deficiency exaggerate the clinical symptoms of Peyronie's disease? Int. J. Urol. 2011, 18, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, G.; Biagiotti, G.; Lo Giudice, C. Association between Peyronie disease and low serum testosterone levels: Detection and therapeutic considerations. J. Androl. 2012, 33, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Yamabe, F.; Hori, S.; Uetani, M.; Aoki, H.; Sakurabayashi, K.; Okawa, M.; Kobayashi, H.; Nakajima, K.; Nagao, K. Significant inverse association of testosterone level with penile deformity severity in Japanese males with Peyronie's disease. Int. J. Urol. 2023, 30, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Kirby, E.W.; Verges, D.; Matthews, J.; Carson, C.C.; Coward, R.M. Low testosterone has a similar prevalence among men with sexual dysfunction due to either Peyronie's disease or erectile dysfunction and does not correlate with Peyronie's disease severity. J. Sex. Med. 2015, 12, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Mulhall, J.P.; Matsushita, K.; Nelson, C.J. Testosterone levels are not associated with magnitude of deformity in men with Peyronie's disease. J. Sex. Med. 2019, 16, 1283–1289. [Google Scholar] [CrossRef]
- Candela, L.; Boeri, L.; Capogrosso, P.; Oreggia, D.; Cazzaniga, W.; Pozzi, E.; Belladelli, F.; Baudo, A.; Abbate, C.; Montorsi, F.; et al. Serum testosterone levels are not associated with the severity of penile curvature in men with Peyronie's disease-findings from a cross-sectional study. Int. J. Impot. Res. 2020, 33, 832–838. [Google Scholar] [CrossRef]
- Mishra, S. , Deng, J.J.; Gowda, P.S.; Rao, M.K.; Lin, C.L.; Chen, C.L.; Huang, T., Sun, L.Z. Androgen receptor and microRNA-21 axis downregulates transforming growth factor beta receptor II (TGFBR2) expression in prostate cancer. Oncogene 2014, 33, 4097–4106. [Google Scholar] [CrossRef]
- Khanna, S.; Shankar Raman, V.; Badwal, S.; Vinu Balraam, K.V. Quantification of the androgen and estrogen receptors in the penile tissues of hypospadias in comparison with normal children. Fetal. Pediatr. Pathol. 2023, 42, 175–186. [Google Scholar] [CrossRef]
- Alkout, A.M.; Blackwell, C.C.; Weir, D.M. Increased inflammatory responses of persons of blood group O to Helicobacter pylori. J. Infect. Dis. 2000, 181, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Bei, J.X.; Li, Y.; Jia, W.H.; Feng, B.J.; Zhou, G.; Chen, L.Z.; Feng, Q.S.; Low, H.Q.; Zhang, H.; He, F.; et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat. Genet. 2010, 42, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Bjekic, M.D.; Vlajinac, H.D.; Sipetic, S.B.; Marinkovic, J.M. Risk factors for Peyronie's disease: A case-control study. BJU. Int. 2006, 97, 570–574. [Google Scholar] [CrossRef] [PubMed]
- La Pera, G.; Pescatori, E.S.; Calabrese, M.; Boffini, A.; Colombo, F.; Andriani, E.; Natali, A.; Vaggi, L.; Catuogno, C.; Giustini, M.; et al. SIMONA Study Group. Peyronie's disease: Prevalence and association with cigarette smoking. a multicenter population-based study in men aged 50-69 years. Eur. Urol. 2001, 40, 525–530. [Google Scholar] [CrossRef] [PubMed]
- El-Sakka, A.I. Prevalence of Peyronie's disease among patients with erectile dysfunction. Eur. Urol. 2006, 49, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Langton, A.K.; Tsoureli-Nikita, E.; Merrick, H.; Zhao, X.; Antoniou, C.; Stratigos, A.; Akhtar, R.; Derby, B.; Sherratt, M.J.; Watson, R.E.; et al. The systemic influence of chronic smoking on skin structure and mechanical function. J. Pathol. 2020, 251, 420–428. [Google Scholar] [CrossRef]
- Liu, W.C.; Chuang, H.C.; Chou, C.L.; Lee, Y.H.; Chiu, Y.J.; Wang, Y.L.; Chiu, H.W. Cigarette smoke exposure increases glucose-6-phosphate dehydrogenase, autophagy, fibrosis, and senescence in kidney cells in vitro and in vivo. Oxid. Med. Cell. Longev. 2022, 2022, 5696686. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Massion, P.P.; Gonzalez, A.L.; Lu, P.; Datta, P.K. Smoking induces epithelial-to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin. Mol. Cancer. Ther. 2012, 11, 2362–2372. [Google Scholar] [CrossRef]
- Wu, Y.; Song, P.; Yuan, X.; Li, D. Exploring the effect of dapagliflozin on alcoholic kidney injury and renal interstitial fibrosis in rats based on TIMP-1/MMP-24 pathway. Evid. Based. Complement. Alternat. Med. 2021, 2021, 6538189. [Google Scholar] [CrossRef]
- Zdanowicz, K.; Kowalczuk-Kryston, M.; Olanski, W.; Werpachowska, I.; Mielech, W.; Lebensztejn, D.M. Increase in serum MMP-9 and TIMP-1 concentrations during alcohol intoxication in adolescents-a preliminary study. Biomolecules. 2022, 12, 710. [Google Scholar] [CrossRef] [PubMed]
- Chilton, C.P.; Castle, W.M.; Westwood, C.A.; Pryor, J.P. Factors associated in the aetiology of peyronie's disease. Br. J. Urol. 1982, 54, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Devine, C.J., Jr.; Somers, K.D.; Jordan, S.G.; Schlossberg, S.M. Proposal: Trauma as the cause of the Peyronie's lesion. J. Urol. 1997, 157, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Kitamori, K.; Naito, H.; Tamada, H.; Kobayashi, M.; Miyazawa, D.; Yasui, Y.; Sonoda, K.; Tsuchikura, S.; Yasui, N.; Ikeda, K.; et al. Development of novel rat model for high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis progression in SHRSP5/Dmcr. Environ. Health. Prev. Med. 2012, 17, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Naito, H.; Jia, X.; Kitamori, K.; Nakajima, T. Combination of hypertension along with a high fat and cholesterol diet induces severe hepatic inflammation in rats via a signaling network comprising NF-κB, MAPK, and Nrf2 pathways. Nutrients 2017, 9, 1018. [Google Scholar] [CrossRef]
- Rhoden, E.L.; Teloken, C. , Ting, H.Y.; Lucas, M.L.; Teodósio da Ros, C.; Ary Vargas Souto, C. Prevalence of Peyronie's disease in men over 50-y-old from Southern Brazil. Int. J. Impot. Res. 2001, 13, 291–293. [Google Scholar] [CrossRef]
- Jorgenson, E.; Matharu, N.; Palmer, M.R.; Yin, J.; Shan, J.; Hoffmann, T.J.; Thai, K.K.; Zhou, X.; Hotaling, J.M.; Jarvik, G.P.; et al. Genetic variation in the SIM1 locus is associated with erectile dysfunction. Proc. Natl. Acad. Sci. USA 2018, 115, 11018–11023. [Google Scholar] [CrossRef]
- Bovijn, J.; Jackson, L.; Censin, J.; Chen, C.Y.; Laisk, T.; Laber, S.; Ferreira, T.; Pulit, S.L.; Glastonbury, C.A.; Smoller, J.W.; et al. GWAS identifies risk locus for erectile dysfunction and implicates hypothalamic neurobiology and diabetes in etiology. Am. J. Hum. Genet. 2019, 104, 157–163. [Google Scholar] [CrossRef]
- Patel, D.P.; Christensen, M.B.; Hotaling, J.M.; Pastuszak, A.W. Erectile dysfunction and Peyronie's disease: Genetic diseases? Eur. Urol. Focus. 2020, 6, 572–574. [Google Scholar] [CrossRef]
- Sasaki, N.; Uesato, R.; Yamauchi, T.; Ishibashi, Y.; Nakaji, S. Epidemiology of Dupuytren's disease in Japanese general population. J. Hand. Surg. Asian. Pac. Vol. 2021, 26, 229–234. [Google Scholar] [CrossRef]
- Dolmans, G.H.; Werker, P.M.; Hennies, H.C.; Furniss, D.; Festen, E.A.; Franke, L.; Becker, K.; van der Vlies, P.; Wolffenbuttel, B.H.; Tinschert, S.; et al. Wnt signaling and Dupuytren’s disease. N. Engl. J. Med. 2011, 365, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ten Dam, E.J.; van Beuge, M.M.; Bank, R.A.; Werker, P.M.J. Further evidence of the involvement of the Wnt signaling pathway in Dupuytren's disease. J. Cell. Commun. Signal. 2016, 10, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Thakkar, D.; Southam, L.; Werker, P.; Ophoff, R.; Becker, K.; Nothnagel, M.; Franke, A.; Nürnberg, P.; Espirito-Santo, A.I.; et al. A genome-wide association study of Dupuytren Disease reveals 17 additional variants implicated in fibrosis. Am. J. Hum. Genet. 2017, 101, 417–427. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed mechanism of molecular regulation of Peyronie’s disease. Repeated microtrauma disrupts the inner and outer layers of the TA. Platelets are activated, then release granules that form an early provisional matrix composed of fibrin and fibronectin. In addition, DAMPs and MCP-1 released from various cells upon tissue damage or mechanical stress promote recruitment of inflammation-related cells to the early provisional matrix. Th1 cells produce IFN-γ, TNF-α, and GM-CSF to activate M1 macrophages, and Th2 cells produce IL-4, IL-13, and TGF-β1 to activate M2 macrophages. Activated M1 macrophages exhibit increased antigen-presenting activity, and high production of pro-inflammatory cytokines, such as IL-6, IL-1b and TNF-α, as well as ROS. M2 macrophages are characterized by increased expressions of the anti-inflammatory cytokine IL-10 and profibrotic factor TGF-β1. An M1/M2 imbalance, expressed by excessive M2 activity, induces fibroblast activation (conversion to myofibroblasts) and promotes fibrosis. Conversion to myofibroblasts involves TGF-β1, TNF-α, IL-6, PDGF, bFGF, myostatin, and ROS. Activated myofibroblasts produce collagen and fibronectin, components of the ECM. On the other hand, expression of MMPs that cause ECM degradation is largely unchanged by overproduction of PAI-1 and TIMP, resulting in excessive accumulation of ECM. Increased ROS due to excessive ECM deposition, which promotes release of DAMPs due to tissue damage, along with continued recruitment and stimulation of immune-related cells by MCP-1 released from myofibroblasts can lead to sustained activation of myofibroblasts. Furthermore, evasion of myofibroblast apoptosis is also critically involved in maintenance of a chronic extracellular fiber environment. As a result, ECM continues to accumulate and is thought to promote fibrosis. TA, tunica albuginea; DAMPs, damage-associated molecular patterns; MCP-1, monocyte chemoattractant protein 1; IFN-γ, interferon gamma; TNF-α, tumor necrosis factor-alpha; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; TGF-β1, transforming growth factor-β1; ROS, reactive oxygen species; PDGF, platelet-derived growth factor; bFGF, basic fibroblast growth factor; ECM, extracellular matrix; MMPs, matrix metalloproteinases; PAI-1, plasminogen activator inhibitor type 1; TIMP, tissue inhibitor of metalloproteinase.
Figure 1.
Proposed mechanism of molecular regulation of Peyronie’s disease. Repeated microtrauma disrupts the inner and outer layers of the TA. Platelets are activated, then release granules that form an early provisional matrix composed of fibrin and fibronectin. In addition, DAMPs and MCP-1 released from various cells upon tissue damage or mechanical stress promote recruitment of inflammation-related cells to the early provisional matrix. Th1 cells produce IFN-γ, TNF-α, and GM-CSF to activate M1 macrophages, and Th2 cells produce IL-4, IL-13, and TGF-β1 to activate M2 macrophages. Activated M1 macrophages exhibit increased antigen-presenting activity, and high production of pro-inflammatory cytokines, such as IL-6, IL-1b and TNF-α, as well as ROS. M2 macrophages are characterized by increased expressions of the anti-inflammatory cytokine IL-10 and profibrotic factor TGF-β1. An M1/M2 imbalance, expressed by excessive M2 activity, induces fibroblast activation (conversion to myofibroblasts) and promotes fibrosis. Conversion to myofibroblasts involves TGF-β1, TNF-α, IL-6, PDGF, bFGF, myostatin, and ROS. Activated myofibroblasts produce collagen and fibronectin, components of the ECM. On the other hand, expression of MMPs that cause ECM degradation is largely unchanged by overproduction of PAI-1 and TIMP, resulting in excessive accumulation of ECM. Increased ROS due to excessive ECM deposition, which promotes release of DAMPs due to tissue damage, along with continued recruitment and stimulation of immune-related cells by MCP-1 released from myofibroblasts can lead to sustained activation of myofibroblasts. Furthermore, evasion of myofibroblast apoptosis is also critically involved in maintenance of a chronic extracellular fiber environment. As a result, ECM continues to accumulate and is thought to promote fibrosis. TA, tunica albuginea; DAMPs, damage-associated molecular patterns; MCP-1, monocyte chemoattractant protein 1; IFN-γ, interferon gamma; TNF-α, tumor necrosis factor-alpha; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; TGF-β1, transforming growth factor-β1; ROS, reactive oxygen species; PDGF, platelet-derived growth factor; bFGF, basic fibroblast growth factor; ECM, extracellular matrix; MMPs, matrix metalloproteinases; PAI-1, plasminogen activator inhibitor type 1; TIMP, tissue inhibitor of metalloproteinase.
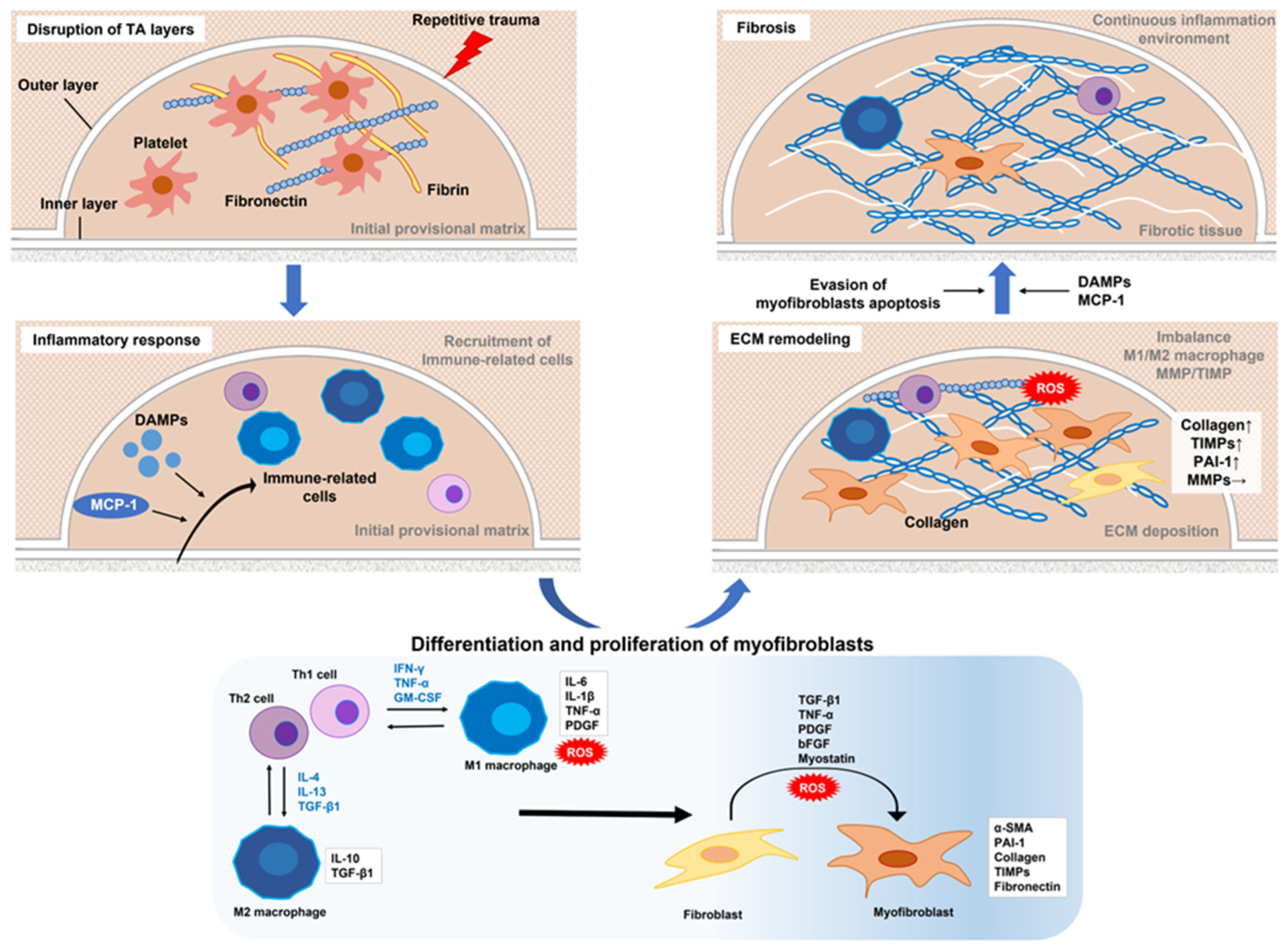
Figure 2.
Putative mechanisms of transforming growth factor-β1 activation in development of Peyronie’s disease. TGF-β1 is synthesized intracellularly in platelets, fibroblasts, and all leukocytes while bound to LAP, then forms a complex with LTBP or LRRC, and released extracellularly. Released latent TGF-β1 uses LTBP to the ECM to anchor LRRC molecules to the cell surface and maintain their inactivated state. Several factors, including integrins, thrombospondin 1, ROS, and several proteases (plasmin, cathepsin D, MMP-2, MMP-9), activate latent TGF-β1 by inducing LAP degradation, denaturation, or conformational changes. Expression of thrombospondin 1 is driven by the renin-angiotensin system. Activated TGF-β1 promotes fibrosis by further activating the TGF-β signaling pathway in macrophages and myofibroblasts. TGF-β1, transforming growth factor-β1; LAP, latency-associated polypeptide; LTBP, latent TGF-β-binding protein; LRRC, leucine-rich repeat containing; ECM, extracellular matrix; ROS, reactive oxygen species; MMP, matrix metalloproteinase.
Figure 2.
Putative mechanisms of transforming growth factor-β1 activation in development of Peyronie’s disease. TGF-β1 is synthesized intracellularly in platelets, fibroblasts, and all leukocytes while bound to LAP, then forms a complex with LTBP or LRRC, and released extracellularly. Released latent TGF-β1 uses LTBP to the ECM to anchor LRRC molecules to the cell surface and maintain their inactivated state. Several factors, including integrins, thrombospondin 1, ROS, and several proteases (plasmin, cathepsin D, MMP-2, MMP-9), activate latent TGF-β1 by inducing LAP degradation, denaturation, or conformational changes. Expression of thrombospondin 1 is driven by the renin-angiotensin system. Activated TGF-β1 promotes fibrosis by further activating the TGF-β signaling pathway in macrophages and myofibroblasts. TGF-β1, transforming growth factor-β1; LAP, latency-associated polypeptide; LTBP, latent TGF-β-binding protein; LRRC, leucine-rich repeat containing; ECM, extracellular matrix; ROS, reactive oxygen species; MMP, matrix metalloproteinase.
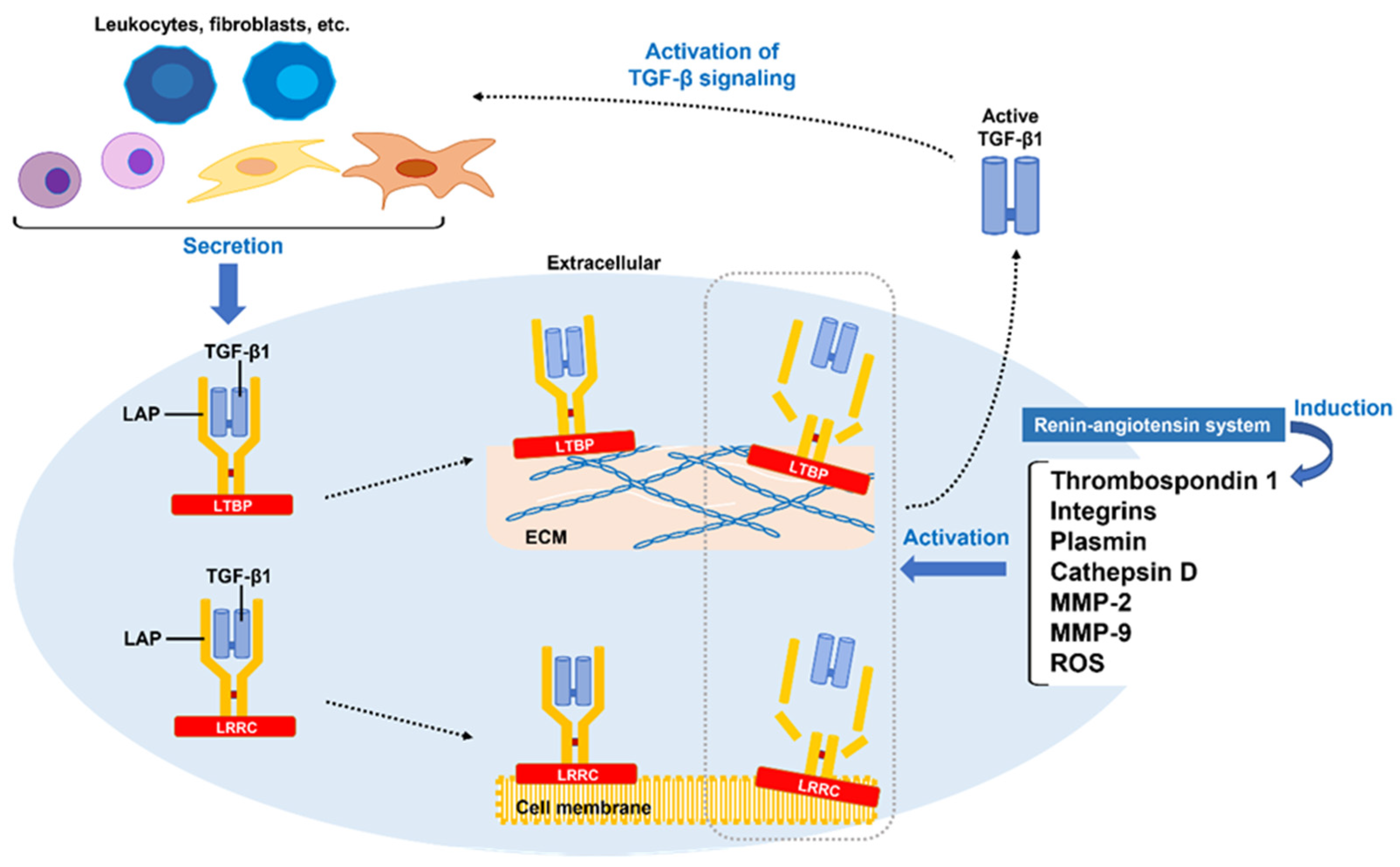
Figure 3.
Schematic representation of core pathways involved in development of Peyronie's disease and their interrelationships. In canonical TGF-β signaling, binding of TGF-β1 to the receptor leads to phosphorylation of SMAD2/SMAD3, triggering a cascade of responses. Phosphorylated SMAD2/SMAD3 in a complex with SMAD4 is then translocated to the nucleus, leading to increased transcription of profibrotic genes. SMAD7, an inhibitory SMAD, is downregulated in PD and activates the TGF-β/SMAD signaling pathway. CTGF is directly induced by ERK1/2, induced by non-canonical TGF-β signaling, then promotes TGF-β1 activity by decreasing SMAD7 expression and activating SMAD2 phosphorylation. IGF-1, which inhibits the transition of SMAD3 to nucleation, is also decreased in PD and contributes to promotion of the TGF-β/SMAD pathway. TGF-β1 signaling also induces transcription of the profibrotic Hedgehog transcription factor GLI2, upregulates WNT ligands, and activates WNT/β-catenin signaling. Under activation of WNT signaling, GSK-3β loss of function prevents β-catenin phosphorylation. Subsequently, the unphosphorylated form of β-catenin accumulates in cytoplasm and enters the nucleus, then finally activates WNT target genes with the help of TCF/LEF family proteins. TGF-β1 inhibits transcription of the WNT suppressors DKK1 and SFRP1 in a p38-dependent manner, and further activates the WNT/β-catenin signaling pathway involved in fibrosis. The profibrotic mechanism of TGF-β1-induced PI3K/AKT signaling is through inactivation of NR4A1, which inhibits expression of profibrotic genes. αVβ integrins activate intracellular signaling pathways, including RHOA/ROCK signaling. Subsequently, formation of F-actin stress fibers and translocation of YAP/TAZ to the nucleus are activated, promoting profibrotic gene expression via TEAD activation. Additionally, RHOA/ROCK signaling activates p300 in the nucleus through activation of AKT, further promoting the transcription of profibrotic genes. YAP and TAZ also bind to phosphorylated SMAD2/SMAD3 and SMAD4 complexes formed in the nucleus, and induce SERPINE1 transcription, thus leading to excessive accumulation of ECM. TGF-β1, transforming growth factor-β1; CTGF, connective tissue growth factor; ERK1/2, extracellular signal-regulated kinase 1/2; IGF-1, insulin growth factor-1; PD, Peyronie’s disease; GLI2, GLI family zinc finger 2; WNT, wingless/int1; DKK1, dickkopf-related protein 1; SFRP1, secreted frizzled-related protein1; PI3K/AKT, phosphoinositide 3-kinase/RAC-α serine/threonine protein kinase; NR4A1, nuclear receptor subfamily 4 group A member 1; ROCK, RAS homologue gene family-associated kinase; YAP/TAZ, yes-associated protein 1/transcriptional coactivator with PDZ-binding motif; SERPINE1, serpin family E member 1; SHH, sonic hedgehog; SMO, smoothened; JNK, c-Jun N-terminal kinase; GSK3β, glycogen synthase kinase 3; TCF/LEF, T cell factor/lymphoid enhancer factor; LRP5/6, low-density lipoprotein receptor-related protein 5/6.
Figure 3.
Schematic representation of core pathways involved in development of Peyronie's disease and their interrelationships. In canonical TGF-β signaling, binding of TGF-β1 to the receptor leads to phosphorylation of SMAD2/SMAD3, triggering a cascade of responses. Phosphorylated SMAD2/SMAD3 in a complex with SMAD4 is then translocated to the nucleus, leading to increased transcription of profibrotic genes. SMAD7, an inhibitory SMAD, is downregulated in PD and activates the TGF-β/SMAD signaling pathway. CTGF is directly induced by ERK1/2, induced by non-canonical TGF-β signaling, then promotes TGF-β1 activity by decreasing SMAD7 expression and activating SMAD2 phosphorylation. IGF-1, which inhibits the transition of SMAD3 to nucleation, is also decreased in PD and contributes to promotion of the TGF-β/SMAD pathway. TGF-β1 signaling also induces transcription of the profibrotic Hedgehog transcription factor GLI2, upregulates WNT ligands, and activates WNT/β-catenin signaling. Under activation of WNT signaling, GSK-3β loss of function prevents β-catenin phosphorylation. Subsequently, the unphosphorylated form of β-catenin accumulates in cytoplasm and enters the nucleus, then finally activates WNT target genes with the help of TCF/LEF family proteins. TGF-β1 inhibits transcription of the WNT suppressors DKK1 and SFRP1 in a p38-dependent manner, and further activates the WNT/β-catenin signaling pathway involved in fibrosis. The profibrotic mechanism of TGF-β1-induced PI3K/AKT signaling is through inactivation of NR4A1, which inhibits expression of profibrotic genes. αVβ integrins activate intracellular signaling pathways, including RHOA/ROCK signaling. Subsequently, formation of F-actin stress fibers and translocation of YAP/TAZ to the nucleus are activated, promoting profibrotic gene expression via TEAD activation. Additionally, RHOA/ROCK signaling activates p300 in the nucleus through activation of AKT, further promoting the transcription of profibrotic genes. YAP and TAZ also bind to phosphorylated SMAD2/SMAD3 and SMAD4 complexes formed in the nucleus, and induce SERPINE1 transcription, thus leading to excessive accumulation of ECM. TGF-β1, transforming growth factor-β1; CTGF, connective tissue growth factor; ERK1/2, extracellular signal-regulated kinase 1/2; IGF-1, insulin growth factor-1; PD, Peyronie’s disease; GLI2, GLI family zinc finger 2; WNT, wingless/int1; DKK1, dickkopf-related protein 1; SFRP1, secreted frizzled-related protein1; PI3K/AKT, phosphoinositide 3-kinase/RAC-α serine/threonine protein kinase; NR4A1, nuclear receptor subfamily 4 group A member 1; ROCK, RAS homologue gene family-associated kinase; YAP/TAZ, yes-associated protein 1/transcriptional coactivator with PDZ-binding motif; SERPINE1, serpin family E member 1; SHH, sonic hedgehog; SMO, smoothened; JNK, c-Jun N-terminal kinase; GSK3β, glycogen synthase kinase 3; TCF/LEF, T cell factor/lymphoid enhancer factor; LRP5/6, low-density lipoprotein receptor-related protein 5/6.
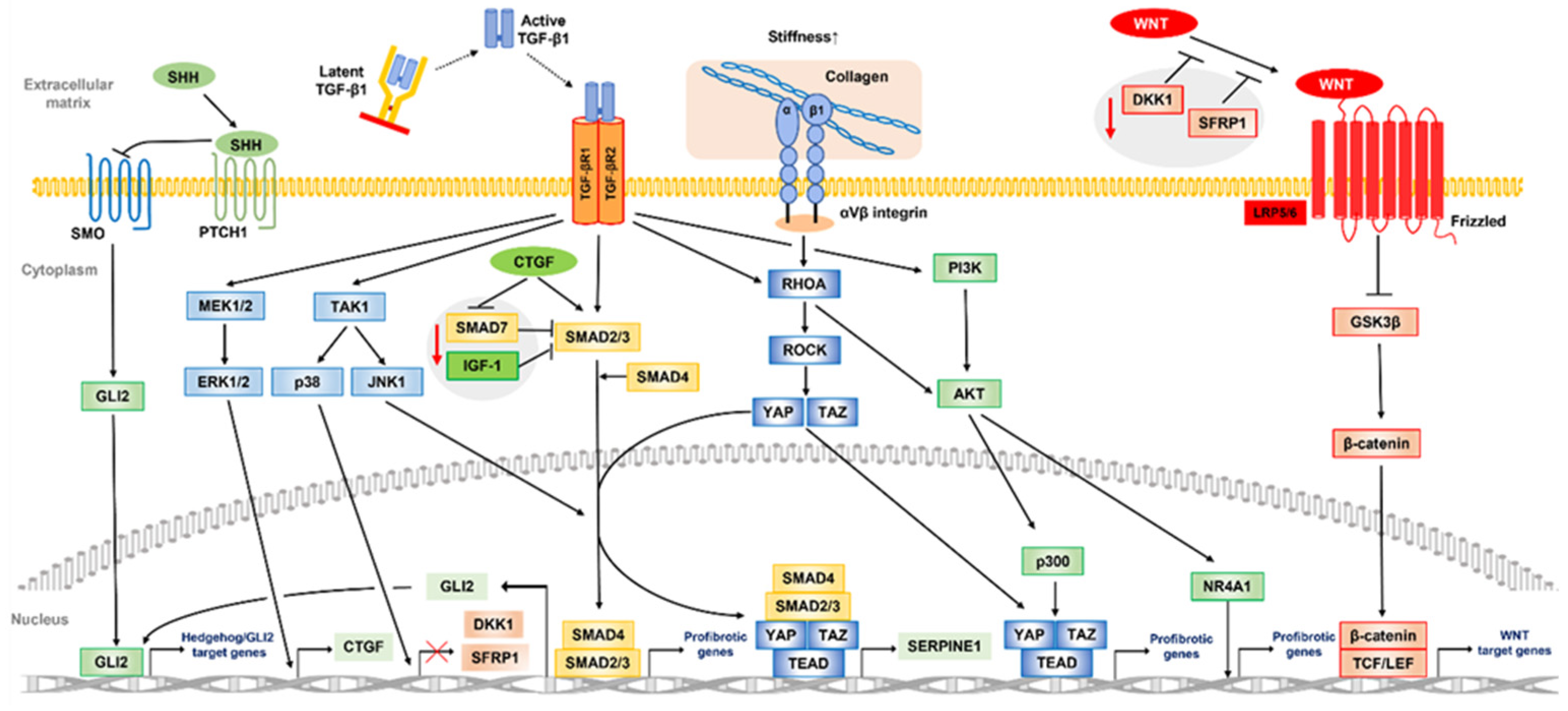
Figure 4.
Risk factors contributing to pathogenesis of PD and their correlation diagram. A variety of comorbidities, and intrinsic and extrinsic factors, both individually and in conjunction with each other, increase the risk for development of Peyronie’s disease. DM, diabetes mellitus; HT, hypertension, ED, erectile dysfunction; DD, Dupuytren’s disease; CPC, congenital penile curvature.
Figure 4.
Risk factors contributing to pathogenesis of PD and their correlation diagram. A variety of comorbidities, and intrinsic and extrinsic factors, both individually and in conjunction with each other, increase the risk for development of Peyronie’s disease. DM, diabetes mellitus; HT, hypertension, ED, erectile dysfunction; DD, Dupuytren’s disease; CPC, congenital penile curvature.
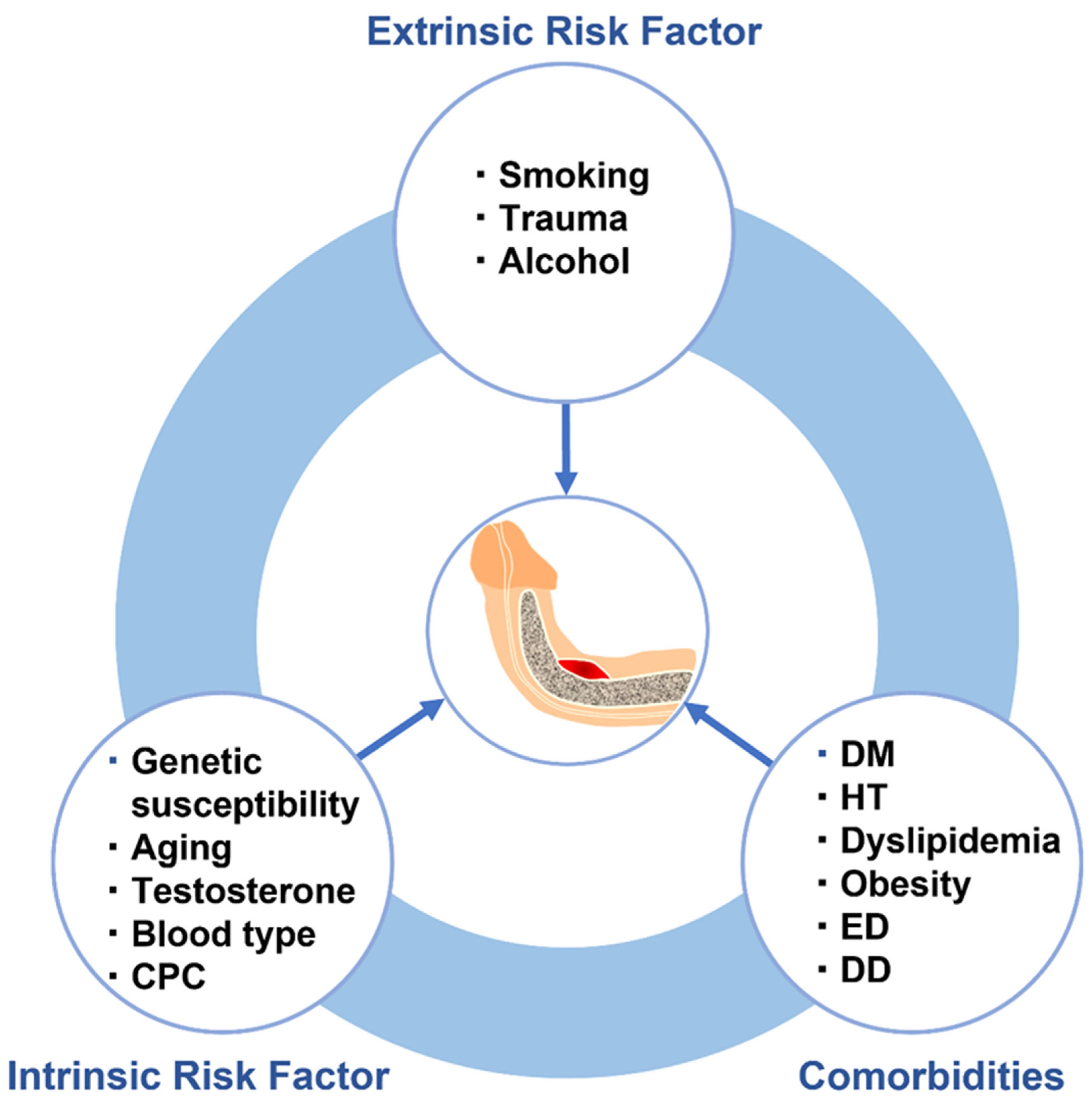

Table 1.
Risk factors associated with development of Peyronie's disease and their possible effects.
| Proposed mechanisms and pathologic effects | Molecular biological changes | |
|---|---|---|
| Intrinsic risk factors | ||
| Genetics | ||
| Chromosomal abnormalities | Familial aggregation | |
| Additions Deletions | ||
| Inversions Reciprocal translocations | ||
| Single nucleotide polymorphisms | ||
| TGF-β1 (rs1800471) | Activation of TGF-β pathway | TGF-β1↑ |
| WNT2 (rs4730775) | Activation of WNT/β-catenin pathway | WNT2↑ |
| ALMS1 ( rs45501594, rs34071195, rs41291187) | Evasion of apoptosis | TGF-β1↑ |
| Aging | Increase in PD-related comorbidities | α1-antitrypsin↓ ROS↑ |
| Semi-rigid erection | ||
| Genetic alterations, epigenetic modifications | ||
| Low testosterone level | Abnormal collagen metabolism in TA | TGF-βRII↑ |
| ABO blood type (type O) | Promotion of inflammatory reaction | TNF-α↑ IL-6↑ |
| Recombination of TGF-β1R1 and ABO gene | ||
| Congenital penile curvature | More penile trauma during intercourse | |
| Extrinsic risk factors | ||
| Smoking | Induction of profibrotic factor | CTGF↑ PAI-1↑ ROS↑ |
| Induction of EMT | ||
| Epigenetic modifications | HDACs↑ | |
| Alcohol | Increase in PD-related comorbidities | |
| Induction of profibrotic factors | TIMP-1↑ SMAD3↑ MMP-9↑ | |
| Perineal and penile trauma | Promotion of detachment of septal fibers | |
| Cormorbidities | ||
| DM, HT, dyslipidemia, obesity | Vascular endothelial cell damage | TGF-β1↑ MMPs↑ TIMPs↑ROS↑ |
| Erectile dysfunction | Semi-rigid erection, delayed ejaculation | |
| (Genetic variation in SIM1 locus) | ||
| Dupuytren’s disease | Genetic connection | WNTs↑ TGF-β1↑ MMPs↑ TIMPs↑ |
| Activation of WNT/β-catenin pathway |
Abbreviations: TGF-β, transforming growth factor-β; WNT, Wingless/int1; ALMS, Alström syndrome; PD, Peyronie's disease; ROS, reactive oxygen species; TGF-βR, TGF-β receptor; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; CTGF, connective tissue growth factor; PAI-1, plasminogen activator inhibitor type 1; HDACs, histone deacetylases; TIMP, tissue inhibitor of metalloproteinase; SIM1, SIM BHLH Transcription Factor 1; MMP, matrix metalloproteinase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



