
Submitted:
28 April 2023
Posted:
01 May 2023
You are already at the latest version
Abstract
Plant reproduction is a very important process on Earth from the perspective of biodiversity, biomass gain and crop productivity. It is therefore important to understand sex determination process and many researchers are investigating the molecular basis of this phenomenon. However, information on the influence of transcription factors (TFs) on this process is limited, although cucumber is a model plant in this regard. In the present study, based on RNA-seq data for differential gene expression (DEG) analyses, we aimed to investigate the regulatory TFs that may influence the metabolomic processes in the shoot apex containing the forming flower buds. Therefore, a robust TF database was established for the B10v3 cucumber genome. Sex-specific interactome network maps were generated, indicating the regulatory TFs by their effects on DEGs and further on processes leading to the formation of different sex flowers. The network analysis identified major families of regulatory TFs. The most abundant families were: MYB, AP2/ERF, NAC and bZIP, and those with the greatest impact on developmental processes were identified, namely the AP/ERF family, followed by DOF, MYB, MADS and others. Thus, the central nodes and key regulators in the networks were identified with respect to male, female and hermaphrodite. Here, we proposed the first model of the regulatory network of TFs that influences the metabolism of sex development in cucumber. These findings may help to understand the molecular genetics and functional mechanisms underlying sex determination processes.
Keywords:
transcription factors
; interactome network
; sex development
; sex determination
; Cucumber (Cucumis sativus)
; metabolomic processes
1. Introduction
Cucumber (Cucumis sativus) is a globally significant vegetable crop and is also recognized as a model organism for exploring the intricacies of plant sex determination, encompassing male, female, and hermaphrodite forms. Although the process of sex determination in cucumbers is currently the focus of numerous scientific investigations, the underlying mechanisms of sex determination in this species remain incompletely understood.
The knowledge about the transcription factors that influence the metabolic processes involved in sex determination is also very limited. It will be very interesting to see which transcription factors and which metabolic processes are involved in processes of sex determination in cucumber. With the increasing amount of omics data such as sequenced genomes and transcriptomes, there is a strong basis and the opportunity to construct interactome networks that indicate the influence of regulatory TFs on selected processes.
Among the most well-known and described reference genomes of Cucumis sativus are: 9930 - Chinese line [1] Gy14 North American line [2] and B10v3 European line [3] Recently, genomes were compared using structural data presented in databases as well as previously reported experimental data. It was shown that the B10v3 genome is the longest due to 342.5 Mbz assembled [4]. Among others, in order to have the most complete picture of the relevant elements, in this study we focused on this version of the genome for further analyses. Understanding how genomes are organized is the basis for insight into the functioning of organisms. Knowledge of regulatory mechanisms and their links to metabolic processes is an important part of the interactions that control gene action.
Transcription factors are proteins that control the activity of gene regulatory networks and cell type specification. They represent a class of essential regulatory proteins that are critical for controlling gene expression and modulating various physiological processes in plants, including: development, hormone signaling, and stress responses. TFs play an important role in the regulation of complex metabolic pathways in response to environmental and physiological signals. They are key regulators of plant primary and secondary metabolism, that produce a large number of specialized metabolites with a wide range of functions and applications [5,6]. However, the inferred function of TF can be influenced by the genomic context in which it occurs. Each family of TFs possesses a specific DNA binding domain that recognizes a unique DNA sequence. In addition, knowledge of the genome can be useful for identification of novel TFs [7]. TFs bind to sequences of DNA, usually to motifs in the promoters of their target genes. Together with other proteins, such as transcriptional regulators (TRs) they regulate gene transcription [8]. The regulatory mechanism underlying gene expression mediated by TFs relies upon the fundamental process of binding to cis-regulatory elements located within gene promoters [9] influencing the expression of nearby genes. This regulatory mechanism plays an important role in orchestrating gene expression in plants [5].
In recent years, the identification and characterization of TFs have been made possible by the development of numerous databases, including iTAK [6] and PlantRegMap [10] which encompass 197 and 165 plant organisms, respectively. These databases contain detailed descriptions of broadly classified TF families for each species, thereby facilitating their efficient exploration. Furthermore, the iTAK database offers supplementary information on TRs that function by interacting with the basal transcriptional apparatus, which includes TFs [6]. This additional information is particularly valuable in expanding our understanding of the intricate mechanisms underlying gene expression regulation in plants. By leveraging the comprehensive information provided by these reference databases, researchers can efficiently search for TFs within the results of a transcriptome study experiment and examine their interactions with other components in the system being investigated. This provides a valuable framework for investigating the complex regulatory networks underlying various biological processes in plants among which there is sex determination in cucumber. In the present work, the objectives of the analyses were: (1) to identify and localize of TFs in the B10v3 cucumber genome according to known TFs in the databases, (2) to update functional characteristics of differentially expressed genes (DEGs) pointed in RNA-seq analyses regarded to sex determination, (3) to search for regulatory TFs that may influence genes correlated with sex expression (4) to establish an interactome map of regulatory TFs and their target genes that have been identified as relevant in sex determination analyses (5) to examine which metabolic processes are associated with regulatory TFs that form a sex-specific interactome and have the impact on the DEGs.
In this paper, we present the world's first-ever interactome map of transcription factors influencing metabolic processes linked to sex determination in cucumber, providing their functional characterization. We proposed multi-omics to integrate data and gain a complex view on the interplay between cell signaling and gene regulation in regard to specific sex in plants.
2. Materials and methods
The work presented in the publication consisted of two steps carried out: the search for TFs in the B10v3 cucumber genome [3] and the functional analysis of RNA-seq data from an experiment comparing leaves and shoot apex expression between cucumber lines differing in sex [11]. A schematic representation of the analytical methodology employed for each of these discrete steps is provided in Figure 1.
2.1. Finding the transcription factors in the B10v3 genome.
To identify TFs in the B10v3 genome, data from two databases were used: PlantRegMap and iTAK. The PlantRegMap database contains information on plant TFs and also provides available software to detect TFs in the genome. The iTAK database contains information on TFs, TRs and protein kinases (PKs), as well as software for their detection in a given datasets. To detect the TFs in the B10v3 genome dataset, the longest amino acid sequences per gene were used as an input for TF identification. In order to work with the PlantRegMap database it was necessary to have gene identifiers that were consistent with the identifiers contained in the database. This required the translation of the protein identifiers from the B10v3 genome to those in the database which are the Gy14v1 cucumber genome identifiers. For this purpose, the "ID mapping" tool that is available on the database server of PlantRegMap was used.
2.2. Tanscription factors among the results of RNA-seq experiments
TFs were searched among DEGs (genes with statistically significant differential expression from RNA-seq experiment) from shoot apex between cucumber lines differing in sex [11]. We analyzed DEGs in the leaves vs shoot apex (LvS) and in the shoot apex based on the following three comparisons of flower sex types: female vs male (FvM), female vs hermaphrodite (FvH), and male vs hermaphrodite (MvH). In the previous experiments, the sequencing reads were mapped to one of the first versions of reference cucumber genome. Therefore, it was necessary to update information and connect them using BLAST algorithm with the gene identifiers of the newest version of the genome - B10v3 [3]. Using the results of the RNA-seq experiment together with the information TFs within the B10v3 genome (from the previous step 2.1), DEGs were assigned to the TFs, TRs or PKs family, according to PlantRegMap and iTAK database.
2.3. Ontology analysis among differentially expressed genes
The ontology of the DEGs was examined using the GO Term Enrichment tool from the PlantRegMap database to gain further insight into the metabolic processes which could differ cucumber lines with the varying sex. This tool helps to identify significantly overrepresented GO terms or the parents of these terms in the selected gene set. This analysis was based on the DEGs identified in the LvS and FvM, FvH, MvH comparisons [12].
2.4. KEGG pathways enrichment analysis in DEGs
DEGs were subjected to KEGG pathway analysis to identify enriched KEGG terms. Gene identifiers from the Cucumis sativus 9930 genome were required to use the KEGG database. To obtain these identifiers, the BLASTP program was used to search for the peptides coded by the DEGs among the protein database for 9930 genome datasets. The ShinyGO server [12] was used to perform the KEGG pathways enrichment step. An annotation for the B10v3 genome has been added for each of the genes which are part of the detected enriched KEGG terms.
2.5. Regulatory Transcription Factors enrichment in PlantRegMap for DEGs
The 'TF enrichment' tool in the PlantRegMap database was used to determine the enrichment of regulatory TFs which interact with the DEGs. Retrieved regulatory TFs were identified from literature and ChIP-seq data and also inferred by combining TF binding motifs and regulatory element data, according to PlantRegMap database. Both regulatory TFs and targeted DEGs were used to build the interactome networks. They were prepared using the networkD3 library in the R programming language [13]. The study determined the effect of the interaction relevance of the TF families on the DEGs based on the constructed networks.
3. Results and discussions
3.1. Transcription factor search results in the B10v3 genome.
In order to get a complete view of the TFs across the genome, we carried out a whole genome analysis using B10v3 as a reference. Using the PlantRegMap and iTAK databases, it was possible to find TFs in the B10v3 cucumber reference genome. The results of the matching between the databases differed slightly, but for the most part remained consistent with each other. Based on the consensus rules for TF prediction and classification with the use of data from the PlantTFDB database, iTAK database was created [6]. Annotations from the iTAK database were therefore used in the analyses. Using the "TF prediction" tool available in the PlantRegMap database, 1 045 TFs were assigned to the searched amino acid sequences. The same input file was used to search for TFs in the iTAK database, allowing for annotation of 1 082 TF families, 1 355 TRs and 656 PKs in the B10v3 genome. Of the 16 104 sequences of the B10v3 genome, 14 269 sequence identifiers were assigned from the PlantRegMap database (Supplement S1). TF search results from the PlantRegMap and iTAK databases were added to the B10v3 genome annotation and supplemented with TR and PKs search results. The resulting B10v3 genome annotation is provided in Supplement 2. For the results obtained using the PlantRegMap and iTAK databases, the percentage of detected TFs per family is shown in Figure 2. The prepared annotation of the B10v3 genome, supplemented with information on genes encoding TFs, RFs and PKs, enriches the knowledge of the genome of B10v3 cucumber line. The number of TF families detected in the cucumber genome corresponds to TFs detected in other plants [14]. The distribution of the identified genes itself is consistent with the factors detected in other plants, where the main TF families are: MYB, bHLH, NAC, WRKY [15].
3.2. Transcription factor among DEGs
TFs have been identified among sex specific DEGs sets from RNA-seq analyses [11]. The results of the RNA-seq experiment supplemented with annotation of TFs, TRs, PKs can be found in Supplement S3. Among DEGs from the tested comparisons: LvS, FvM, FvH, MvH, the 8,7%, 10,77%, 5,88% and 10,91% of TFs were detected, respectively (Table 1).
The largest number of TFs was detected for the LvS comparison (8,70%). However, this number is due to the largest number of DEGs detected between the leaf and shoot apex and contains genes responsible for the transition from the vegetative to flowering phase. Number of DEGs in the shoot apex among male, female, and hermaphrodite lines was significantly lower, while the percentage of TFs detected for these comparisons remained similar. In comparison MvH, where six TFs were detected, half of them were the MADS-MIKC family. The other assigned families were HB-WOX, NAC, C2C2-YABBY. For comparison FvH, in which only two TFs were detected, the bHLH and C3H families were defined. The following Figure 3 and Figure 4 show graphs with the highest number of the TFs detected in the FvM and LvS comparisons, respectively. For the FvM and MvH comparisons the largest number of differentially expressed factors belonged to the MADS-MIKC family. This represents a significant increase in the proportion of these TFs relative to their contribution to the whole genome. When comparing LvS, the TFs that were found in the highest abundance, i.e., bHLH, MYB, NAC, and C2H2, correspond in abundance to the distribution of TFs across the genome. TFs of the MADS-MIKC family in this comparison also represent an increased proportion in the number of 16 differentially expressed TFs relative to their presence in the reference genome. MADS TFs are a family of DNA-binding proteins that play an essential role in various plant developmental processes, especially floral organ identity and differentiation [16,17] additionally controlling the expression of genes that determine the identity and morphology of sepals, petals, stamens, and carpels. The MADS TFs in cucumber are similar to those in other plants, as they are also involved in the flowering time regulation and the floral organs developement [18]. The detection of the MIKC-MADS family as the most abundant among the TFs detected directing us towards linking MADS to the ABC model of flower development [19].
In addition, the presence of eight differentially expressed TFs of the AP2 family is important notification, due to the link between sex determination processes and ethylene metabolism [20].
3.3. Ontology analysis among differentially expressed genes
For DEGs, an overrepresentation of GO terms was found in all comparisons: LvS, FvH, FvM and MvH, what is presented in Figure 5. For FvH, most of the GO terms were related to processes like pollen sperm differentiation, male gametogenesis, or microgametogensis differentiation, indicating significant relationships to processes involved in sex determination and male organ formation. In this comparison, the female organs are formed in the female as well as in hermaphrodites, while the male organs are present only in hermaphrodite flower.
In the FvM comparison, the most significant enrichment concerned genes that are involved in the flower formation developmental processes, overall organ development and carbohydrate metabolism processes.
Processes related to enzyme activity: monooxygenase, oxidoreductase, hydrolase or pectinesterase were the most enriched in MvH comparison. Genes involved in the development of the carpels and gynoecium were also enriched. In this comparison, the female organs are formed in the hermaphrodite flowers but in male flowers this organ is inhibited in the growth, thus the difference connected to these group is expected. When comparing LvS, the enriched processes differed significantly from the other comparisons. The highest enrichment in this case was for processes related to photosynthesis, light response, plastid or chloroplast organization. Comparing a vegetative organ such as the leaf with a generative organ such as the whole structure of shoot apex with small floral buds, indicates which genes and processes differentiate these two organs mostly. The number of DEGs is the highest in the LvS comparison and significantly exceeds the number of DEGs in the other comparisons. The analysis of the ontology network (Supplements S4-S7) shows that those processes are significantly enriched and are marked in red and yellow. For the FvH and MvH comparisons, the created ontology networks are significantly simpler than the other: FvM and LvS comparisons, which is due to the smaller number of significantly DEGs identified. FvH comparison represents the most enriched final processes, converging to a single final process of pollen germ cell differentiation. Similarly, for the MvH comparison, the structure of the ontology converges on final terms describing gynoecium development and carpel development, although a separate branch indicating metabolic processes and a final term describing oxidation and reduction processes are additionally described. In comparison the FvH ontology network is much more extensive, where three main branches can be observed. The first corresponds to floral organ formation processes, the second indicates metabolic processes taking place while the third describes enriched processes related to transport. In the case of ontological terms describing organ development, processes such as floral organ development and floral organ formation have the greatest enrichment. In addition, we can distinguish terms describing gynoecium development and carpel development. The processes responsible for oxidation-reduction and carbohydrate metabolism have the greatest enrichment within metabolic processes. The different branches of ontology converge to final terms describing processes related to metabolism of pectin, salicylic acid, inositol and fatty acids. A separate branch of the network describes processes related to transport of carbohydrates, saccharides and sucrose.
The next step of the analysis was to check whether there were DEGs in the enriched GO term that were TFs. For this purpose, we checked all differentially expressed TFs in the sex specific comparisons. In Figure 6, the frequency of TFs among DEGs for each ontology term is shown for FvM comparison. It can be seen that TFs are responsible for floral developmental processes, carpel development, and organ formation. This indicates the actual involvement of TFs in processes linked to the plant's sex development. No TFs were detected for the enriched ontology terms in the FvH comparison. For the MvH comparison, two TFs involved in gynoecium and carpel development processes were detected among the enriched ontology terms. They are also directly related to issues of plant sex development which, as can be seen, represents the relevance of TFs in this process. The LvS comparison is the most abundant. Therefore, it consists of the largest number of TFs. The ontological terms to which the TFs were assigned were in relation to metabolic processes and biosynthesis.
3.4. KEGG enrichment results in DEGs
The enrichment analysis of the KEGG pathways, performed for DEGs (LvS, FvM, FvH, and MvH comparisons) allowed for a better understanding of the related functions and networks involved in related to sex determination processes. The KEGG enrichment results are shown in Figure 7. No statistically significant results were obtained for the FvH comparison. For the FvM comparison, the highest enrichment is related to the ABC transporters pathway, which refers to the ATP-binding cassette (ABC) transporters. For both the FvM and MvH comparisons, statistically significant enrichments were found for the pentose and glucuronate interconversion pathways. When comparing LvS, the most significant were pathways included photosynthesis and metabolic processes. The annotated results of the KEGG analysis are presented in Supplement S8. A search for transcription factors among the genes in the KEGG enrichment pathway did not identify any TFs. Only protein-coding genes were present in the KEGG pathways found in the analysis. However, similar to GO analysis, significant differences in overrepresented pathways could be observed between the generative (FvM, MvH comparisons) and vegetative (LvS comparison) organs.
3.6. Regulatory Transcription Factors influencing DEGs.
The study of flowering in cucumber is crucial due to its significant economic importance and vulnerability to both: endogenous and exogenous factors. The impact of these factors collectively determines the expression of genes, which in turn is influenced by the activity of various TFs. The regulatory TFs were assigned to families and functionally curated.
Our study shows the interaction of regulatory TFs and their influence on DEGs thus, taking together, we can answer the question: what TFs influence flower morphogenesis at early stages of growth.
As a result of the TF enrichment program in the PlantRegMap database, a list of enriched regulatory TFs was obtained for each of the FvM, FvH, MvH and LvS comparisons. The retrieved regulatory TFs were annotated according to B10v3 information data sets. (Supplement S9). Analysis of the detected TF families revealed that the NAC family was the most abundant family detected for the FvH and MvH enriched comparisons. Furthermore, genes encoding TF families such as bHLH and MYB showed a higher frequency of detection. Notably, a greater number of enriched TFs were identified in the FvH and MvH comparisons compared to the FvM comparison. For each of the comparisons considered, enriched TFs were detected for a significant number of TF families. The LvS comparison showed the highest number of enriched TFs, with MYB, bZIP, bHLH and DOF being the most frequently detected families.
Created interaction maps between regulatory TFs and their targeted DEGs are highly complex and have therefore been presented in the form of interactive networks available in HTML files (Supplements S10-S13). The advantage of such a network presentation is that it is possible to view the gene of interest together with the genes with which it interacts (DEGs or regulatory TFs), and to read their annotation. A static image of the networks for FvM, FvH and MvH comparisons is presented in Figure 8.
Between FvM, the number of DEGs is significantly higher than that for FvH and MvH comparisons. The FvM interaction network thus contains many more connections, showing that the male and female lines have significant differences in expressed genes. The FvH and MvH lines are thus a simplified model as the number of differentially expressed genes is much smaller but refers to processes associated with sex variation.
The resulting interaction networks between regulatory TFs and DEGs varied in complexity, what was based on the number of DEGs used to construct the network. The largest network was created for the LvS comparison consisted of 3029 nodes. The network for FvM comparison consisted 468 nodes, for FvH comparison consisted of 177 nodes, while for MvH comparison consisted of 191 nodes. The number of individual regulatory TFs and DEGs used to construct the networks is shown in Table 2. The smallest networks were created for the FvH and MvH comparisons, due to the flowers possessing a common element in the flower architecture. The FvM network is more developed due to flower architecture concerning distinction of the generative organ.
In the next step we grouped up regulatory TFs that influence DEGs into 34 families and performed functional characterization (Supplements S14). This allowed as to elucidate which and how the regulatory TFs influence DEGs and thus proteins involved in metabolic processes in lines varying in sex in cucumber. In order to check the force of influence of regulatory TFs families, the links between TFs and their targets were counted (Figure 9).
TFs are proteins that help to 'turn on' or 'turn off' certain genes by binding to the promoter, thereby regulating the functioning of the organism. The present study identified several TF families that majorly influence the expression of DEGs in male, female and hermaphrodite flowers. The most numerous family of regulatory TFs in all three networks were: AP2/ERF (total 101), MYB (73), NAC (44), bZIP (39) and bHLH (35) family. Other families were less numerous in the sum of the three comparisons (Figure 9). However, some TFs are specified only for one comparison, namely FvM: YABBY, LFY, SRS, EIL or only for to comparison of FvH and FvM, such as: WRKY, CPP, FAR1 and for other set, FvM and MvH - HSF family. In terms of edge numbers, that is, family interactivity, which can be translated into power to influence DEGs, the most numerous families totally in three networks were also AP2/ERF (1069), DOF(465), MYB (296), MIKS – MADS (233) and BBR-BBC (219). Other families possess less than 200 connections. Table 3 presents the top 10 TFs that have the most connections in each interaction network.
The question arises as to: how the families of TFs that have been identified affect sex determination; which processes they are involved in and with what interaction. The hormonal regulation plays a crucial role in the process of sex determination, as the genes primarily involved in this process are associated with ethylene synthesis, such as: CsACS1, CsACS2, CsACS11 and they are linked with genetic loci F, M, and A respectively [21,22,23,24]. Expression and interaction among all three genes help in the development of female flower in cucumber. TFs act as a very important factor thatwhich can be either activate or repress the gene expression. Ethylene is the principal hormone that is responsible for the formation of specific organs and genes responsible for ethylene biosynthesis have a direct association with the development of female flowers [25,26]. The ethylene production in shoot apex primordia can readily modify the male to female flower ratio on the plant. It is known that sex in cucumber is linked to hormonal regulation and ethylene plays an important role in the cucumber. Of the identified TF families, ten are associated with the ethylene response in other plants and these are: AP2/ERF [27], MYB [28], NAC [29], bZIP [30], bHLH [31], WRKY [32], TCP [33], C2H2 [34], TALE [35] and MIKC_MADS [36]. Additionally, other hormones such as auxin and cytokinin exert a positive effect on female sex determination through interaction with ethylene biosynthesis and signaling pathways[37,38]. The families connected with other hormones were also identified in this study: auxins – NAC [39], bHLH [40], LFY [41], cytokinin bHLH [40] and BBR-BPC [42,43]. The results of others studies demonstrated that gibberellins (GA) can have dual effects on sex expression in cucumber, inhibiting femaleness and inducing maleness and expression analysis has shown that CsACS1G transcription is promoted by auxins and inhibited by gibberellic acid [44,45]. According to the literature, there were five families: LFY [46], YABBY [47,48], MYB [49], BBR-BPC [42,43] and TALE [50] which were correlated with this hormone. AP2/ERF are identified as one of the largest groups of TFs in this study in all three comparisions. AP2/ERF family members induce ethylene signalling and flowering [51,52,53]. The CsACS11 is one of the ethylene biosynthetic genes [54] and also thought to be a sex gene (a) in cucumber [22]. So far, it is not clear how the hormonal signalling pathways influence sex at the molecular level, so further detailed characteristics of the link between the regulatory TFs is needed. The formation of the complex flower architecture involves the MADS family described above. For the families identified, we found numerous TFs - DEGs links to flower development. Several studies reported role of MICKS-MADS [36,55,56], bHLH [57,58], bZIP [59], and NAC [60,61,62] in promoting or delaying flowering development. TALE family shows interaction with ethylene and cytokinin signalling [35]. Together with floral development, timimg of flowering is also crucial in plants for fruits and seed production. MYB [63], bZIP [64], bHLH [57,58] and WRKY [65] families are involved in regulation of flowering time, as desribed previously. Study in tomato, shows that bHLH acts with SFT or LFY and controls flowering time. It also influences ethylene biosynthesis genes, as the expression of ethylene biosynthesis genes is upregulated in the overexpression line of bHLH [66]. In the forming gynoecium of the Arabidopsis flower, other hormones such as auxin and cytokinin interact with bHLH [40]. BBR-BPC/GAGA has been described in Arabidopsis to regulate the phytohormonal signalling of cytokinins, brassinosteroids and ethylene [42,43]. The DOF TF has been reported to be involved in tissue differentiation, cell expansion, seed development, anther or pollen development and flowering in plants [67,68,69]. DOF has been also implicated in the formation of vascular tissue in reproductive organs [70]. The interaction between the bZIP member and the C2H2 member in melon inhibits the development of the carpel in male flowers [71,72]. It also represses the transcription of ethylene biosynthetic genes [73]. The present study reveals that the transcription factor WRKY is present solely in the FvH and FvM comparisons, while being absent in MvH. WRKY TF interacts with various flowering genes to regulate flowering timing in plants [65,74]. Another transcription factor, YABBY, is exclusively found in FvM comparison. The YABBY TF was desribed to play a crucial role in the development of anthers and pollen sacs in cucumber, Arabidopsis and rice [75,76,77,78]. The TFs from the YABBY family, interacts with MADS-box to control its expression during carpel development [79] In addition, only the LFY TF was identified in the FvM comparison, which is known to respond to auxin and regulate flowering initiation as presented in Arabidopsis [41,80].
A parallel and second approach to network analysis is to explore proteins (encoded by DEGs) to determine their reactivity with TFs regulators. In the compariosns of sex network such nodes were: for the FvH - DNA/RNA binding proteins, oxidoreductase proteins, for the FvM - pectinase, MADS box TFs and lipase proteins, and for the MvH - monooxygenase and triphosphate hydrolases.
Our analyses demonstrated the link between regulatory TFs and various developmental processes, including flower morphogenesis, flowering timing, and interactions with phytohormones. Gene expression governing specific functions involves the action of TFs. By conducting a detailed examination of interaction networks, we have identified regulatory TFs that have the potential to regulate a significant number of DEGs. These regulatory TFs act as central hubs in the network, and can influence a large portion of the nodes, thereby characterizing them as master regulators/hot links. The identification of these master regulators can serve as a valuable hub point for future investigations. By selectively focusing on these factors, their regulation or knockout, may be utilized to observe changes in DEGs within the context of sex comparison in cucumbers. This approach holds considerable potential for expanding our understanding of the complex regulatory networks underlying sex development in cucumber.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
“Conceptualization, M.P..; methodology, S.T. and M.P..; software, S.T..; validation, S.T..,.; formal analysis, S.T, A, M.P.,.; investigation, S.T., A., M.P.,.; resources, M.P..; data curation, M.P..; writing—original draft preparation, S.T. and M.P.,.; writing—review and editing, A.S. and W.P..; visualization, S.T., A., A.S., M.P.,; .; project administration, M.P.,.; funding acquisition, M.P.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a project from the National Science Center UMO-2020/37/B/NZ9/00586.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Publicly available datasets were analyzed in this study, according to cited articles.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author's contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The Genome of the Cucumber, Cucumis Sativus L. Nature Genetics 2009 41:12 2009, 41, 1275–1281. [Google Scholar] [CrossRef]
- Yang, L.; Koo, D.H.; Li, Y.; Zhang, X.; Luan, F.; Havey, M.J.; Jiang, J.; Weng, Y. Chromosome Rearrangements during Domestication of Cucumber as Revealed by High-Density Genetic Mapping and Draft Genome Assembly. The Plant Journal 2012, 71, 895–906. [Google Scholar] [CrossRef]
- Osipowski, P.; Pawełkowicz, M.; Wojcieszek, M.; Skarzyńska, A.; Przybecki, Z.; Pląder, W. A High-Quality Cucumber Genome Assembly Enhances Computational Comparative Genomics. Molecular Genetics and Genomics 2020, 295, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Turek, S.; Pląder, W.; Hoshi, Y.; Skarzyńska, A.; Pawełkowicz, M. Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data. Int J Mol Sci 2023, 24, 4011. [Google Scholar] [CrossRef] [PubMed]
- Biłas, R.; Szafran, K.; Hnatuszko-Konka, K.; Kononowicz, A.K. Cis-Regulatory Elements Used to Control Gene Expression in Plants. Plant Cell, Tissue and Organ Culture (PCTOC) 2016 127:2 2016, 127, 269–287. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. ITAK: A Program for Genome-Wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Fletcher, M. Genome-Scale Characterization of Transcription Factors. Nature Genetics 2023 55:3 2023, 55, 357–357. [Google Scholar] [CrossRef]
- Dai, X.; Sinharoy, S.; Udvardi, M.; Zhao, P.X. PlantTFcat: An Online Plant Transcription Factor and Transcriptional Regulator Categorization and Analysis Tool. BMC Bioinformatics 2013, 14, 1–6. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; López-Vidriero, I.; Carrasco, J.L.; Godoy, M.; Vera, P.; Solano, R. DNA-Binding Specificities of Plant Transcription Factors and Their Potential to Define Target Genes. Proc Natl Acad Sci U S A 2014, 111, 2367–2372. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting Functional Regulatory Maps in Plants. Nucleic Acids Res 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Pawełkowicz, M.; Pryszcz, L.; Skarzyńska, A.; Wóycicki, R.K.; Posyniak, K.; Rymuszka, J.; Przybecki, Z.; Pląder, W. Comparative Transcriptome Analysis Reveals New Molecular Pathways for Cucumber Genes Related to Sex Determination. Plant Reproduction 2019 32:2 2019, 32, 193–216. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- GitHub - Christophergandrud/NetworkD3: D3 JavaScript Network Graphs from R Available online:. Available online: https://github.com/christophergandrud/networkD3 (accessed on 25 April 2023).
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis Transcription Factors: Genome-Wide Comparative Analysis among Eukaryotes. Science (1979) 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Meraj, T.A.; Fu, J.; Raza, M.A.; Zhu, C.; Shen, Q.; Xu, D.; Wang, Q. Transcriptional Factors Regulate Plant Stress Responses Through Mediating Secondary Metabolism. Genes 2020, Vol. 11, Page 346 2020, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Daher, H.; Galien, A.; Hugouvieux, V.; Zubieta, C. Structural Basis for Plant MADS Transcription Factor Oligomerization. Comput Struct Biotechnol J 2019, 17, 946–953. [Google Scholar] [CrossRef]
- Theißen, G.; Gramzow, L. Structure and Evolution of Plant MADS Domain Transcription Factors. Plant Transcription Factors: Evolutionary, Structural and Functional Aspects 2016, 127–138. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, L.; Song, J.; Jiang, L.; Liu, S. Isolation and Characterization of a MADS-Box Gene in Cucumber (Cucumis Sativus L.) That Affects Flowering Time and Leaf Morphology in Transgenic Arabidopsis. http://mc.manuscriptcentral.com/tbeq 2019, 33, 54–63. [Google Scholar] [CrossRef]
- Theißen, G.; Melzer, R.; Ruümpler, F. MADS-Domain Transcription Factors and the Floral Quartet Model of Flower Development: Linking Plant Development and Evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, L.; Chen, P.; Wu, C.; Zhang, H.; Yuan, J.; Zhou, J.; Li, X. Genome-Wide Identification of APETALA2/ETHYLENE RESPONSIVE FACTOR Transcription Factors in Cucurbita Moschata and Their Involvement in Ethylene Response. Front Plant Sci 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Pawełkowicz, M.E.; Skarzyńska, A.; Pląder, W.; Przybecki, Z. Genetic and Molecular Bases of Cucumber (Cucumis Sativus L.) Sex Determination. Molecular Breeding 2019, 39, 1–27. [Google Scholar] [CrossRef]
- Boualem, A.; Troadec, C.; Camps, C.; Lemhemdi, A.; Morin, H.; Sari, M.A.; Fraenkel-Zagouri, R.; Kovalski, I.; Dogimont, C.; Perl-Treves, R.; et al. A Cucurbit Androecy Gene Reveals How Unisexual Flowers Develop and Dioecy Emerges. Science 2015, 350, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Boualem, A.; Troadec, C.; Kovalski, I.; Sari, M.A.; Perl-Treves, R.; Bendahmane, A. A Conserved Ethylene Biosynthesis Enzyme Leads to Andromonoecy in Two Cucumis Species. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Trebitsh, T.; Staub, J.E.; O’Neill, S.D. Identification of a 1-Aminocyclopropane-1-Carboxylic Acid Synthase Gene Linked to the Female (F) Locus That Enhances Female Sex Expression in Cucumber. Plant Physiol 1997, 113, 987. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Fujii, N.; Takahashi, H. Characterization of Ethylene Effects on Sex Determination in Cucumber Plants. Sex Plant Reprod 2003, 16, 103–111. [Google Scholar] [CrossRef]
- BHOWMICK, B.K.; JHA, S.U.M.I.T.A. Dynamics of Sex Expression and Chromosome Diversity in Cucurbitaceae: A Story in the Making. J Genet 2015, 94, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pan, X.; Liu, S.; Lin, W.; Li, Y.; Zhang, X. Genome-Wide Analysis of AP2/ERF Transcription Factors in Pineapple Reveals Functional Divergence during Flowering Induction Mediated by Ethylene and Floral Organ Development. Genomics 2021, 113, 474–489. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Chen, S.; Jiang, J.; Chen, F.; Chen, Y.; Gu, C.; Li, P.; Song, A.; Zhu, X.; Gao, H.; et al. Heterologous Expression of the Chrysanthemum R2R3-MYB Transcription Factor CmMYB2 Enhances Drought and Salinity Tolerance, Increases Hypersensitivity to ABA and Delays Flowering in Arabidopsis Thaliana. Mol Biotechnol 2012, 51, 160–173. [Google Scholar] [CrossRef]
- Pei, H.; Ma, N.; Tian, J.; Luo, J.; Chen, J.; Li, J.; Zheng, Y.; Chen, X.; Fei, Z.; Gao, J. An NAC Transcription Factor Controls Ethylene-Regulated Cell Expansion in Flower Petals. Plant Physiol 2013, 163, 775. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Yu, Y.; Quan, R.; Zhang, Z.; Zhang, H.; Huang, R. The Ethylene Response Factor AtERF11 That Is Transcriptionally Modulated by the BZIP Transcription Factor HY5 Is a Crucial Repressor for Ethylene Biosynthesis in Arabidopsis. Plant J 2011, 68, 88–99. [Google Scholar] [CrossRef]
- Alessio, V.M.; Cavaçana, N.; Dantas, L.L.D.B.; Lee, N.; Hotta, C.T.; Imaizumi, T.; Menossi, M. The FBH Family of BHLH Transcription Factors Controls ACC Synthase Expression in Sugarcane. J Exp Bot 2018, 69, 2511–2525. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Wang, Y.; Sun, M.; Zhao, P.; Wang, Q.; Yang, B.; Li, J.; Jiang, Y.Q. WRKY29 Transcription Factor Regulates Ethylene Biosynthesis and Response in Arabidopsis. Plant Physiol Biochem 2023, 194, 134–145. [Google Scholar] [CrossRef] [PubMed]
- van Es, S.W.; Silveira, S.R.; Rocha, D.I.; Bimbo, A.; Martinelli, A.P.; Dornelas, M.C.; Angenent, G.C.; Immink, R.G.H. Novel Functions of the Arabidopsis Transcription Factor TCP5 in Petal Development and Ethylene Biosynthesis. Plant J 2018, 94, 867–879. [Google Scholar] [CrossRef]
- Ohta, M.; Matsui, K.; Hiratsu, K.; Shinshi, H.; Ohme-Takagi, M. Repression Domains of Class II ERF Transcriptional Repressors Share an Essential Motif for Active Repression. Plant Cell 2001, 13, 1959. [Google Scholar] [CrossRef]
- Hamant, O.; Nogué, F.; Belles-Boix, E.; Jublot, D.; Grandjean, O.; Traas, J.; Pautot, V. The KNAT2 Homeodomain Protein Interacts with Ethylene and Cytokinin Signaling. Plant Physiol 2002, 130, 657. [Google Scholar] [CrossRef]
- Brian, L.; Warren, B.; McAtee, P.; Rodrigues, J.; Nieuwenhuizen, N.; Pasha, A.; David, K.M.; Richardson, A.; Provart, N.J.; Allan, A.C.; et al. A Gene Expression Atlas for Kiwifruit (Actinidia Chinensis) and Network Analysis of Transcription Factors. BMC Plant Biol 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Trebitsh, T.; Rudich, J.; Riov, J. Auxin, Biosynthesis of Ethylene and Sex Expression in Cucumber (Cucumis Sativus). Plant Growth Regul 1987, 5, 105–113. [Google Scholar] [CrossRef]
- Shannon, S.; De La Guardia, M.D. Sex Expression and the Production of Ethylene Induced by Auxin in the Cucumber (Cucumis Sativum L.). Natur 1969, 223, 186. [Google Scholar] [CrossRef]
- He, X.J.; Mu, R.L.; Cao, W.H.; Zhang, Z.G.; Zhang, J.S.; Chen, S.Y. AtNAC2, a Transcription Factor Downstream of Ethylene and Auxin Signaling Pathways, Is Involved in Salt Stress Response and Lateral Root Development. Plant J 2005, 44, 903–916. [Google Scholar] [CrossRef]
- Reyes-Olalde, J.I.; Zúñiga-Mayo, V.M.; Serwatowska, J.; Chavez Montes, R.A.; Lozano-Sotomayor, P.; Herrera-Ubaldo, H.; Gonzalez-Aguilera, K.L.; Ballester, P.; Ripoll, J.J.; Ezquer, I.; et al. The BHLH Transcription Factor SPATULA Enables Cytokinin Signaling, and Both Activate Auxin Biosynthesis and Transport Genes at the Medial Domain of the Gynoecium. PLoS Genet 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Wu, M.F.; Winter, C.M.; Wagner, D. LEAFY and Polar Auxin Transport Coordinately Regulate Arabidopsis Flower Development. Plants 2014, 3, 251. [Google Scholar] [CrossRef] [PubMed]
- Shanks, C.M.; Hecker, A.; Cheng, C.; Brand, L.; Collani, S.; Schmid, M.; Schaller, G.E.; Wanke, D.; Harter, K.; Kieber, J.J. Role of BASIC PENTACYSTEINE Transcription Factors in a Subset of Cytokinin Signaling Responses. Plant J 2018, 95, 458–473. [Google Scholar] [CrossRef]
- Theune, M.L.; Bloss, U.; Brand, L.H.; Ladwig, F.; Wanke, D. Phylogenetic Analyses and GAGA-Motif Binding Studies of BBR/BPC Proteins Lend to Clues in GAGA-MOTIF Recognition and a Regulatory Role in Brassinosteroid Signaling. Front Plant Sci 2019, 10, 466. [Google Scholar] [CrossRef]
- Yin, T.; Quinn, J.A. Tests of a Mechanistic Model of One Hormone Regulating Both Sexes in Cucumis Sativus (Cucurbitaceae). Am J Bot 1995, 82, 1537–1546. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, G.; Li, Y.; Mo, N.; Zhang, J.; Liang, Y. Transcriptomic Analysis Implies That GA Regulates Sex Expression via Ethylene-Dependent and Ethylene-Independent Pathways in Cucumber (Cucumis Sativus L.). Front Plant Sci 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Winter, C.M.; Wu, M.F.; Kanno, Y.; Yamaguchi, A.; Seo, M.; Wagner, D. Gibberellin Acts Positively Then Negatively to Control Onset of Flower Formation in Arabidopsis. Science 2014, 344, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ma, Y.; Li, J. The Rice YABBY4 Gene Regulates Plant Growth and Development through Modulating the Gibberellin Pathway. J Exp Bot 2016, 67, 5545–5556. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Zhao, Y.; Ma, Q.; Hu, Y.; Hedden, P.; Zhang, Q.; Zhou, D.X. The Rice YABBY1 Gene Is Involved in the Feedback Regulation of Gibberellin Metabolism. Plant Physiol 2007, 144, 121. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Liu, B.; Wang, W.; Liu, X.; Chen, C.; Liu, X.; Yang, S.; Ren, H. A GAMYB Homologue CsGAMYB1 Regulates Sex Expression of Cucumber via an Ethylene-Independent Pathway. J Exp Bot 2014, 65, 3201. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Xing, L.; Zhang, C.; Chen, H.; Li, Y.; Zhang, D.; Ma, J.; Zhao, C.; Han, M.; Ren, X.; et al. MdKNOX15, a Class I Knotted-like Transcription Factor of Apple, Controls Flowering and Plant Height by Regulating GA Levels through Promoting the MdGA2ox7 Transcription. Environ Exp Bot 2021, 185. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, X.; Liu, S.; Lin, W.; Li, Y.; Zhang, X. Genome-Wide Analysis of AP2/ERF Transcription Factors in Pineapple Reveals Functional Divergence during Flowering Induction Mediated by Ethylene and Floral Organ Development. Genomics 2021, 113, 474–489. [Google Scholar] [CrossRef]
- Lin, R.C.; Park, H.J.; Wang, H.Y. Role of Arabidopsis RAP2.4 in Regulating Light- and Ethylene-Mediated Developmental Processes and Drought Stress Tolerance. Mol Plant 2008, 1, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhao, L.; Chong, K.; Wang, T. Overexpression of OsERF1, a Novel Rice ERF Gene, up-Regulates Ethylene-Responsive Genes Expression besides Affects Growth and Development in Arabidopsis. J Plant Physiol 2008, 165, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Niu, H.; Wang, Z.; Zhang, W.; Wang, H.; Wang, S.; Zhang, X.; Li, Z. Ethylene Responsive Factor ERF110 Mediates Ethylene-Regulated Transcription of a Sex Determination-Related Orthologous Gene in Two Cucumis Species. J Exp Bot 2018, 69, 2953–2965. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Luo, Y.; Zhang, Z.; Xu, M.; Wang, W.; Zhao, Y.; Zhang, L.; Fan, Y.; Wang, L. ZmSOC1, an MADS-Box Transcription Factor from Zea Mays, Promotes Flowering in Arabidopsis. Int J Mol Sci 2014, 15, 19987. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhuo, S.; Liu, X.; Che, G.; Wang, Z.; Gu, R.; Shen, J.; Song, W.; Zhou, Z.; Han, D.; et al. The MADS-Box Gene CsSHP Participates in Fruit Maturation and Floral Organ Development in Cucumber. Front Plant Sci 2020, 10, 1781. [Google Scholar] [CrossRef]
- Sharma, N.; Xin, R.; Kim, D.H.; Sung, S.; Lange, T.; Huq, E. NO FLOWERING IN SHORT DAY (NFL) Is a BHLH Transcription Factor That Promotes Flowering Specifically under Short-Day Conditions in Arabidopsis. Development (Cambridge) 2016, 143, 682–690. [Google Scholar] [CrossRef]
- Ye, Y.; Xin, H.; Gu, X.; Ma, J.; Li, L. Genome-Wide Identification and Functional Analysis of the Basic Helix-Loop-Helix (Bhlh) Transcription Family Reveals Candidate Ptfbh Genes Involved in the Flowering Process of Populus Trichocarpa. Forests 2021, 12, 1439. [Google Scholar] [CrossRef]
- Li, X.; Tian, X.; He, M.; Liu, X.; Li, Z.; Tang, J.; Mei, E.; Xu, M.; Liu, Y.; Wang, Z.; et al. BZIP71 Delays Flowering by Suppressing Ehd1 Expression in Rice. J Integr Plant Biol 2022, 64, 1352–1363. [Google Scholar] [CrossRef]
- Pei, H.; Ma, N.; Tian, J.; Luo, J.; Chen, J.; Li, J.; Zheng, Y.; Chen, X.; Fei, Z.; Gao, J. An NAC Transcription Factor Controls Ethylene-Regulated Cell Expansion in Flower Petals. Plant Physiol 2013, 163, 775. [Google Scholar] [CrossRef]
- Hendelman, A.; Stav, R.; Zemach, H.; Arazi, T. The Tomato NAC Transcription Factor SlNAM2 Is Involved in Flower-Boundary Morphogenesis. J Exp Bot 2013, 64, 5497–5507. [Google Scholar] [CrossRef]
- Guo, S.; Dai, S.; Singh, P.K.; Wang, H.; Wang, Y.; Tan, J.L.H.; Wee, W.; Ito, T. A Membrane-Bound NAC-like Transcription Factor OsNTL5 Represses the Flowering in Oryza Sativa. Front Plant Sci 2018, 9, 555. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, G.; Jia, J.; Zhao, G.; Xia, C.; Zhang, L.; Li, F.; Zhang, Q.; Dong, C.; Gao, S.; et al. The Wheat MYB-Related Transcription Factor TaMYB72 Promotes Flowering in Rice. J Integr Plant Biol 2016, 58, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Kobayashi, Y.; Yamamoto, S.; Daimon, Y.; Yamaguchi, A.; Ikeda, Y.; Ichinoki, H.; Notaguchi, M.; Goto, K.; Araki, T. FD, a BZIP Protein Mediating Signals from the Floral Pathway Integrator FT at the Shoot Apex. Science 2005, 309, 1052–1056. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Z.; Wang, L.; Kim, S.G.; Seo, P.J.; Qiao, M.; Wang, N.; Li, S.; Cao, X.; Park, C.M.; et al. WRKY71 Accelerates Flowering via the Direct Activation of FLOWERING LOCUS T and LEAFY in Arabidopsis Thaliana. Plant J 2016, 85, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Li, N.; Su, D.; Chen, J.; Li, Z. Overexpression of a Basic Helix-Loop-Helix Transcription Factor Gene, SlbHLH22, Promotes Early Flowering and Accelerates Fruit Ripening in Tomato (Solanum Lycopersicum L.). Planta 2019, 250, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Verhoeff, N.I.; Chen, Z.; Chen, S.; Wang, M.; Zhu, Z.; Ouwerkerk, P.B.F. Functions of OsDof25 in Regulation of OsC4PPDK. Plant Mol Biol 2015, 89, 229. [Google Scholar] [CrossRef] [PubMed]
- Noguero, M.; Atif, R.M.; Ochatt, S.; Thompson, R.D. The Role of the DNA-Binding One Zinc Finger (DOF) Transcription Factor Family in Plants. Plant Sci 2013, 209, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Lorrai, R.; Gandolfi, F.; Boccaccini, A.; Ruta, V.; Possenti, M.; Tramontano, A.; Costantino, P.; Lepore, R.; Vittorioso, P. Genome-Wide RNA-Seq Analysis Indicates That the DAG1 Transcription Factor Promotes Hypocotyl Elongation Acting on ABA, Ethylene and Auxin Signaling. Sci Rep 2018, 8. [Google Scholar] [CrossRef]
- Rojas-Gracia, P.; Roque, E.; Medina, M.; López-Martín, M.J.; Cañas, L.A.; Beltrán, J.P.; Gómez-Mena, C. The DOF Transcription Factor Sldof10 Regulates Vascular Tissue Formation during Ovary Development in Tomato. Front Plant Sci 2019, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, K.; Park, C.; Hwang, H.J.; Lee, I. SUPPRESSOR OF FRIGIDA4, Encoding a C2H2-Type Zinc Finger Protein, Represses Flowering by Transcriptional Activation of Arabidopsis FLOWERING LOCUS C. Plant Cell 2006, 18, 2985–2998. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; You, C.; Li, C.; Long, T.; Chen, G.; Byrne, M.E.; Zhang, Q. RID1, Encoding a Cys2/His2-Type Zinc Finger Transcription Factor, Acts as a Master Switch from Vegetative to Floral Development in Rice. Proc Natl Acad Sci U S A 2008, 105, 12915–12920. [Google Scholar] [CrossRef] [PubMed]
- Han, Y. chao; Fu, C. chun; Kuang, J. fei; Chen, J. ye; Lu, W. jin Two Banana Fruit Ripening-Related C2H2 Zinc Finger Proteins Are Transcriptional Repressors of Ethylene Biosynthetic Genes. Postharvest Biol Technol 2016, 116, 8–15. [Google Scholar] [CrossRef]
- Huang, R.; Liu, D.; Huang, M.; Ma, J.; Li, Z.; Li, M.; Sui, S. CpWRKY71, a WRKY Transcription Factor Gene of Wintersweet (Chimonanthus Praecox), Promotes Flowering and Leaf Senescence in Arabidopsis. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ning, K.; Che, G.; Yan, S.; Han, L.; Gu, R.; Li, Z.; Weng, Y.; Zhang, X. CsSPL Functions as an Adaptor between HD-ZIP III and CsWUS Transcription Factors Regulating Anther and Ovule Development in Cucumis Sativus (Cucumber). Plant J 2018, 94, 535–547. [Google Scholar] [CrossRef]
- Unte, U.S.; Sorensen, A.M.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-Box Gene That Affects Pollen Sac Development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.K.; Skinner, D.J.; Gallagher, T.L.; Gasser, C.S. Integument Development in Arabidopsis Depends on Interaction of YABBY Protein INNER NO OUTER with Coactivators and Corepressors. Genetics 2017, 207, 1489–1500. [Google Scholar] [CrossRef]
- Jang, S.; Hur, J.; Kim, S.J.; Han, M.J.; Kim, S.R.; An, G. Ectopic Expression of OsYAB1 Causes Extra Stamens and Carpels in Rice. Plant Mol Biol 2004, 56, 133–143. [Google Scholar] [CrossRef]
- Gross, T.; Broholm, S.; Becker, A. CRABS CLAW Acts as a Bifunctional Transcription Factor in Flower Development. Front Plant Sci 2018, 9, 835. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Jeong, C.W.; Nole-Wilson, S.; Krizek, B.A.; Wagner, D. AINTEGUMENTA and AINTEGUMENTA-LIKE6/PLETHORA3 Induce LEAFY Expression in Response to Auxin to Promote the Onset of Flower Formation in Arabidopsis. Plant Physiol 2016, 170, 283–293. [Google Scholar] [CrossRef]
Figure 1.
Diagram showing the analysis steps performed.
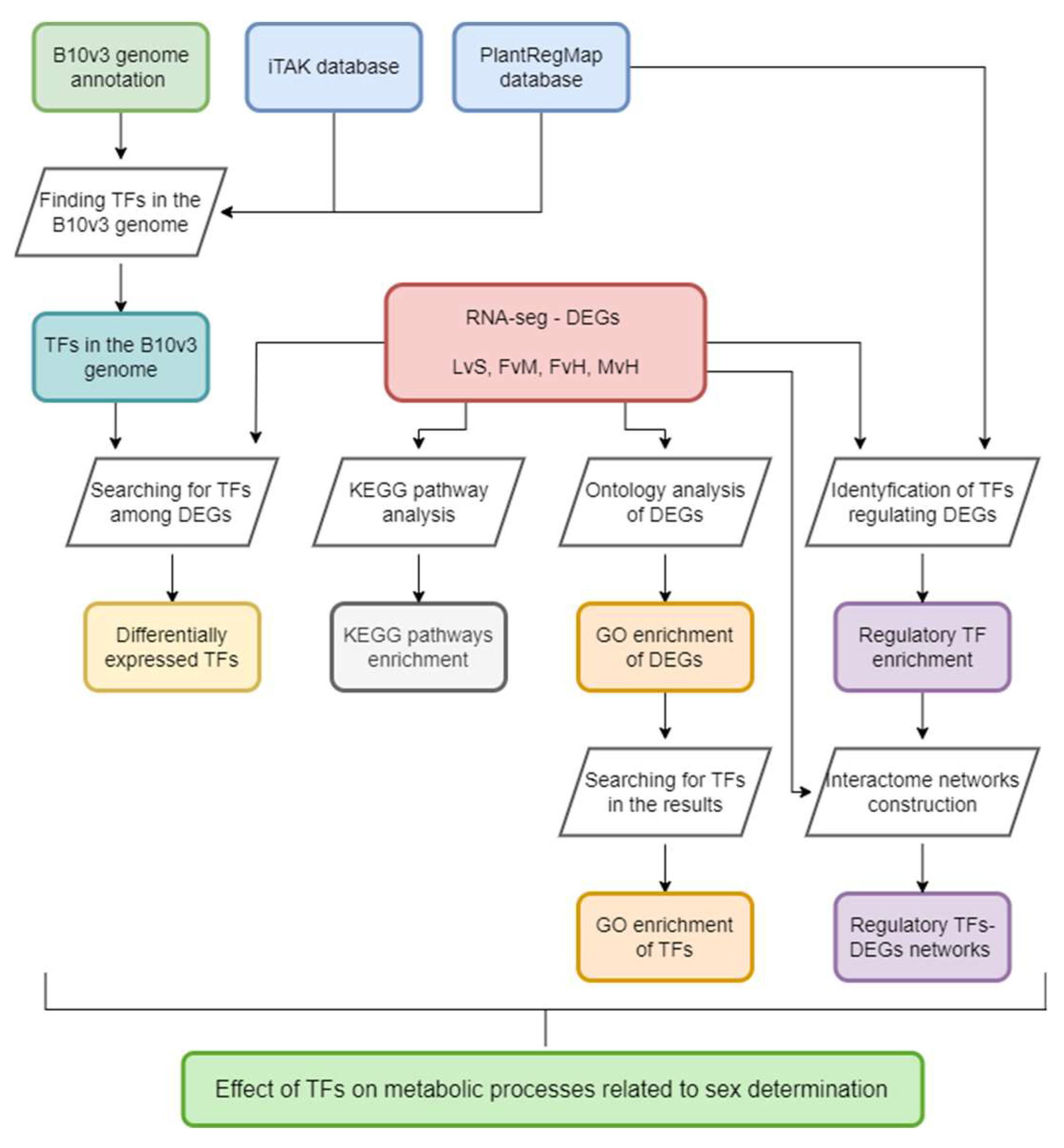
Figure 2.
Transcription factor families detected in the B10v3 genome by the iTAK (a) and PlantRegMap (b) databases, which account for more than 2% of all identified TFs.
Figure 2.
Transcription factor families detected in the B10v3 genome by the iTAK (a) and PlantRegMap (b) databases, which account for more than 2% of all identified TFs.
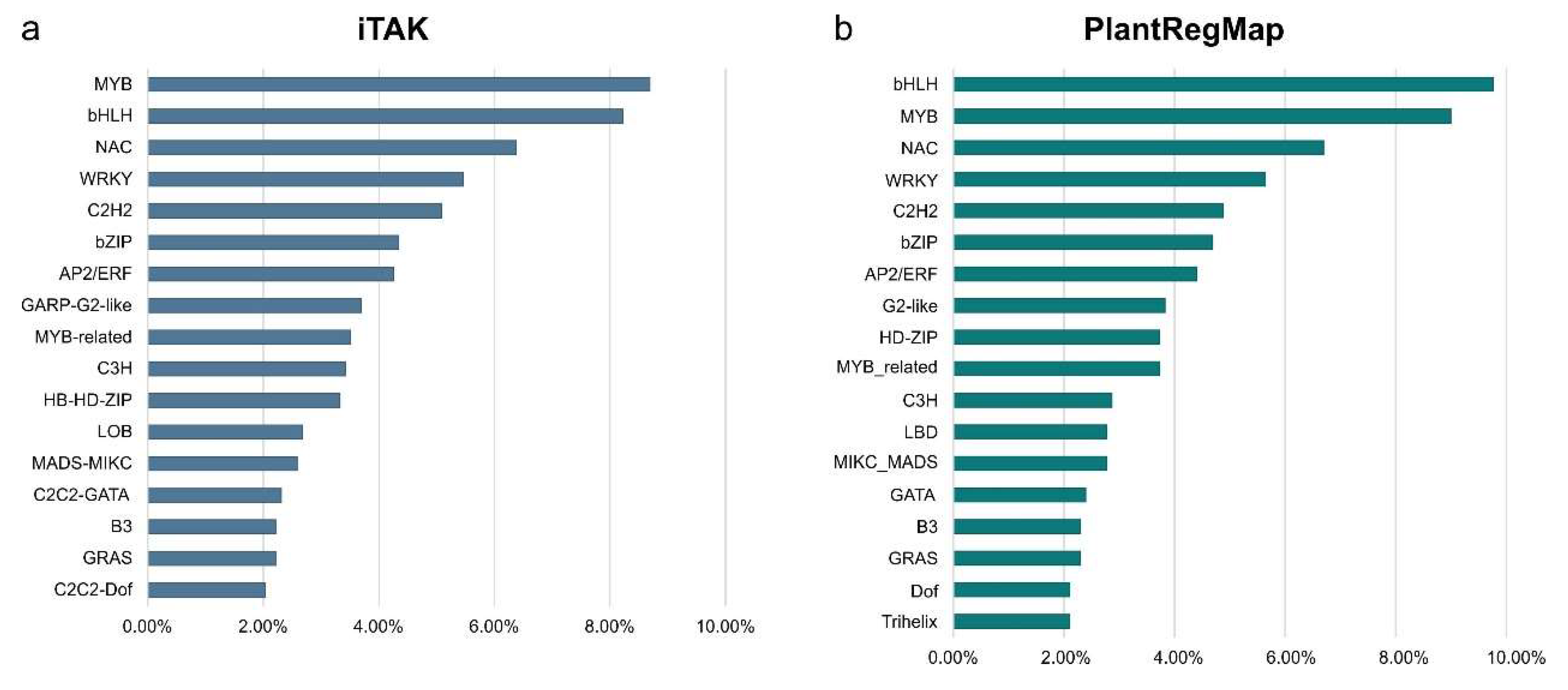
Figure 3.
Percentage of transcription factors families among differentially expressed genes in FvM comparison.
Figure 3.
Percentage of transcription factors families among differentially expressed genes in FvM comparison.
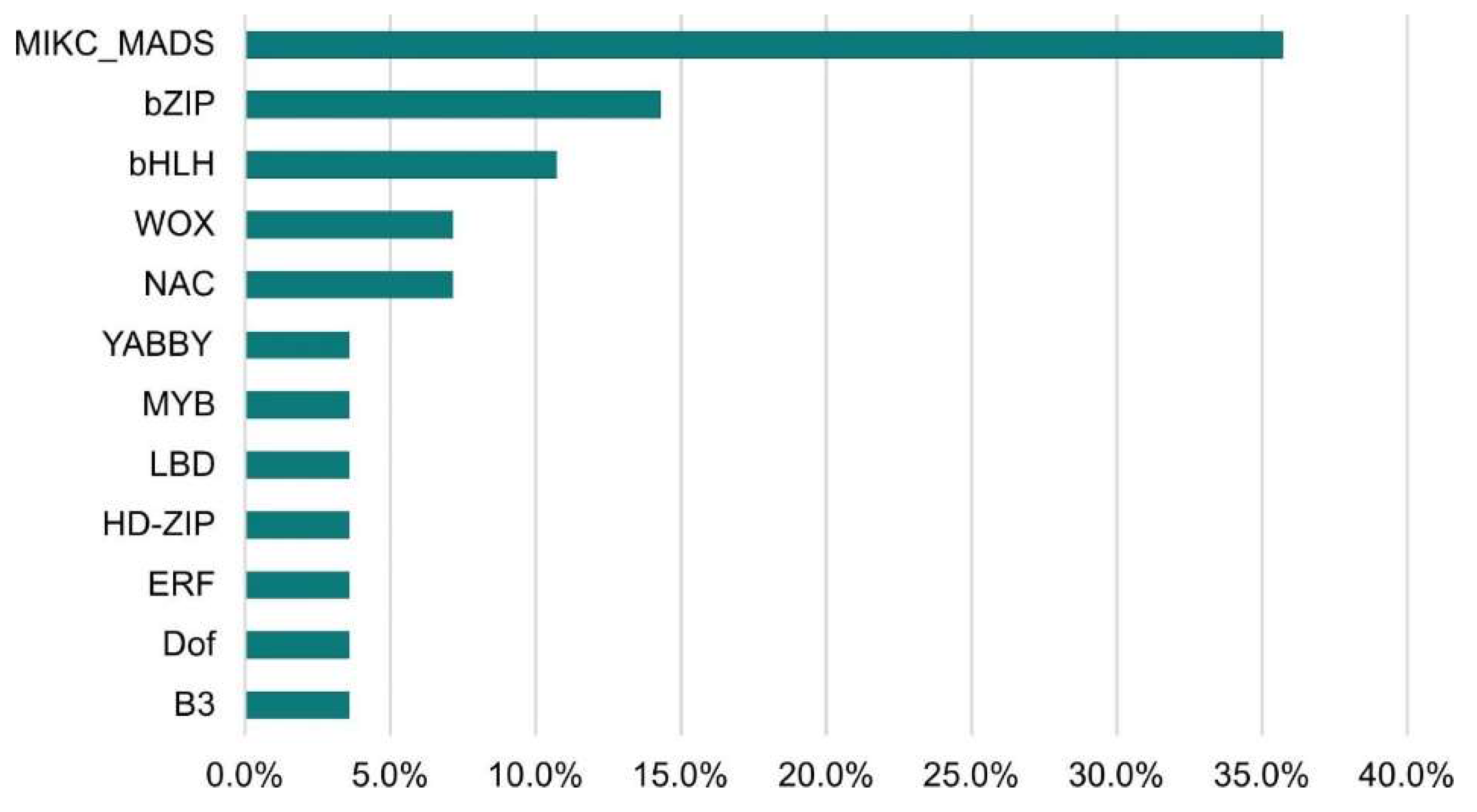
Figure 4.
Percentage of transcription factors among differentially expressed genes in LvS comparison.
Figure 4.
Percentage of transcription factors among differentially expressed genes in LvS comparison.
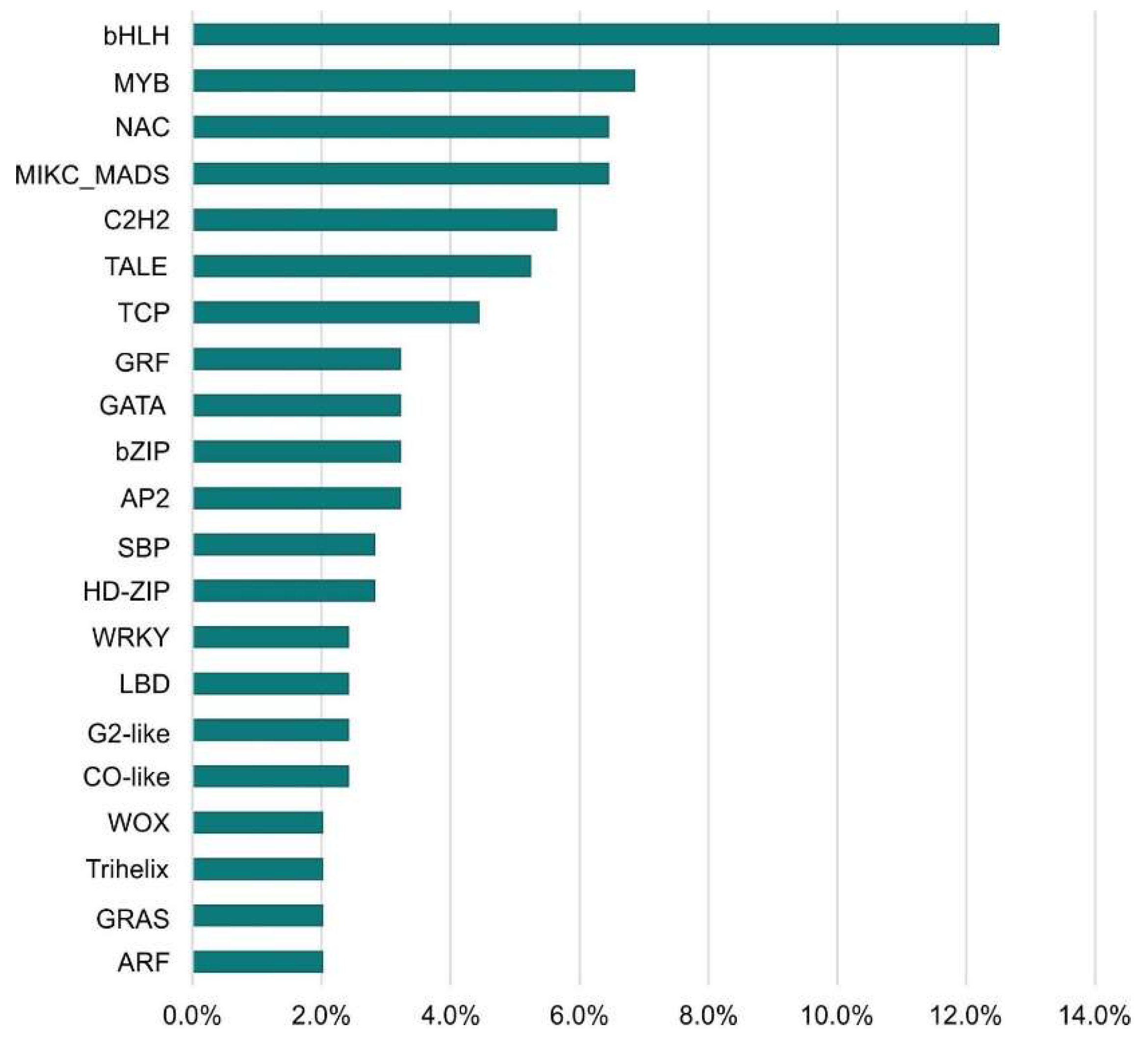
Figure 5.
Box plots of significantly enriched GO terms for genes with significantly differential expressions for LvS, FvM, FvH, MvH comparisons.
Figure 5.
Box plots of significantly enriched GO terms for genes with significantly differential expressions for LvS, FvM, FvH, MvH comparisons.
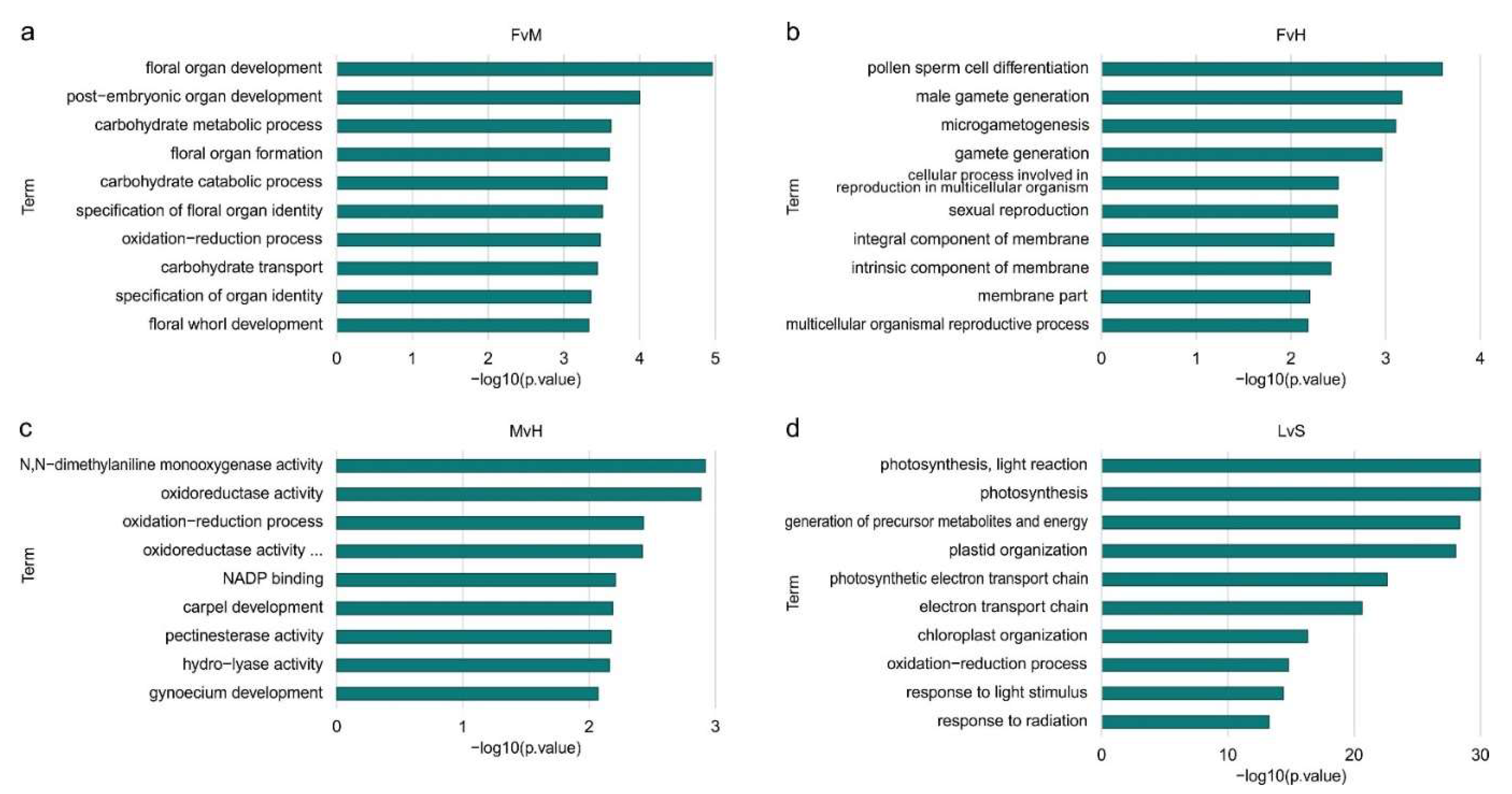
Figure 6.
The number of TFs among DEGs for FvM comparison regard to ontology terms.
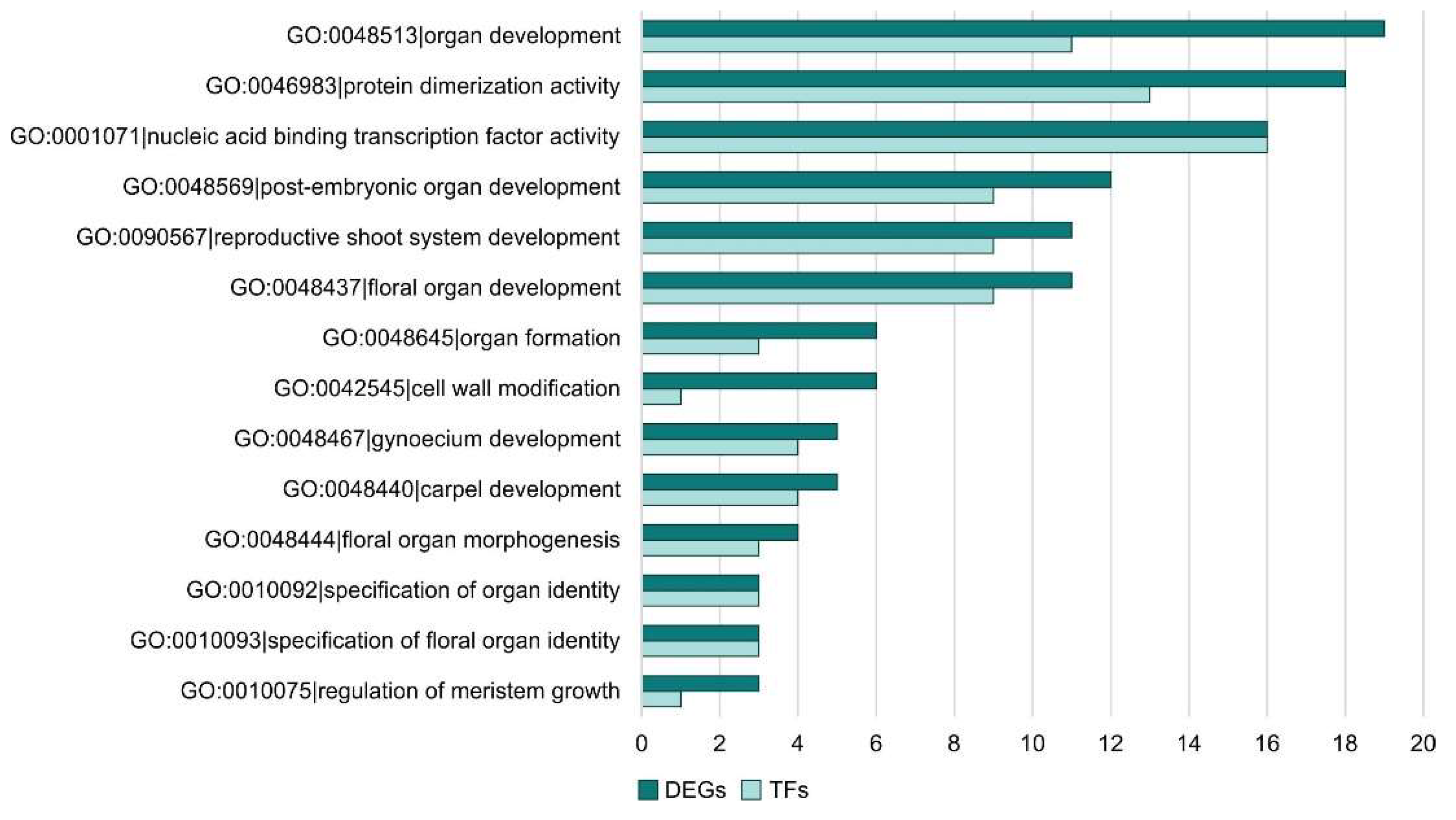
Figure 7.
Graphs showing enriched KEGG pathways for LvS (a), FvM (b) and FvH (c) comparisons.
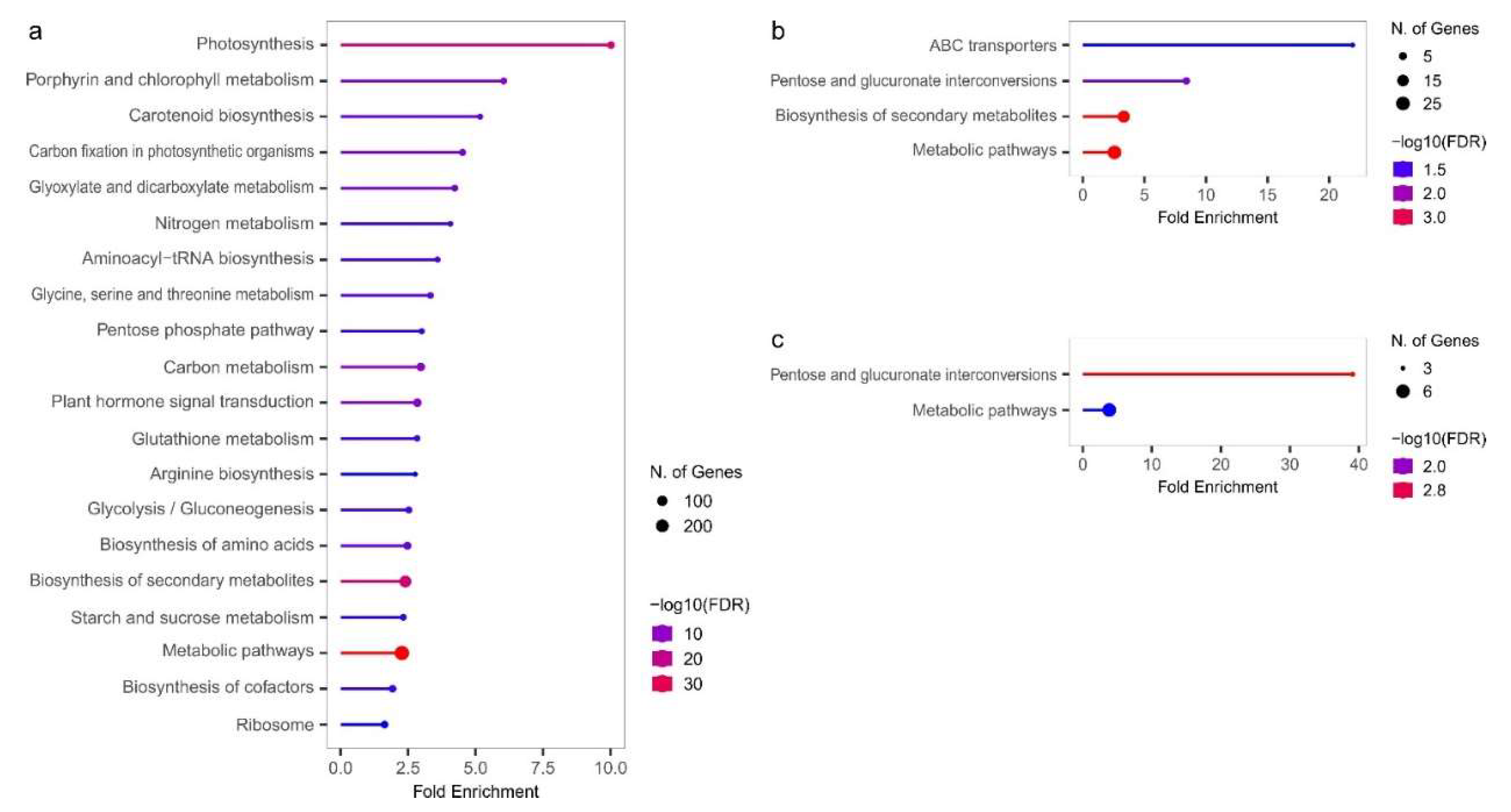
Figure 8.
Static image of interactions networks between transcription factors and differentially expressed gene targets in FvM comparison (a), FvH comparison (b) and MvH comparison (c). Orange color indicates DEGs, while blue color indicates regulatory TFs.
Figure 8.
Static image of interactions networks between transcription factors and differentially expressed gene targets in FvM comparison (a), FvH comparison (b) and MvH comparison (c). Orange color indicates DEGs, while blue color indicates regulatory TFs.
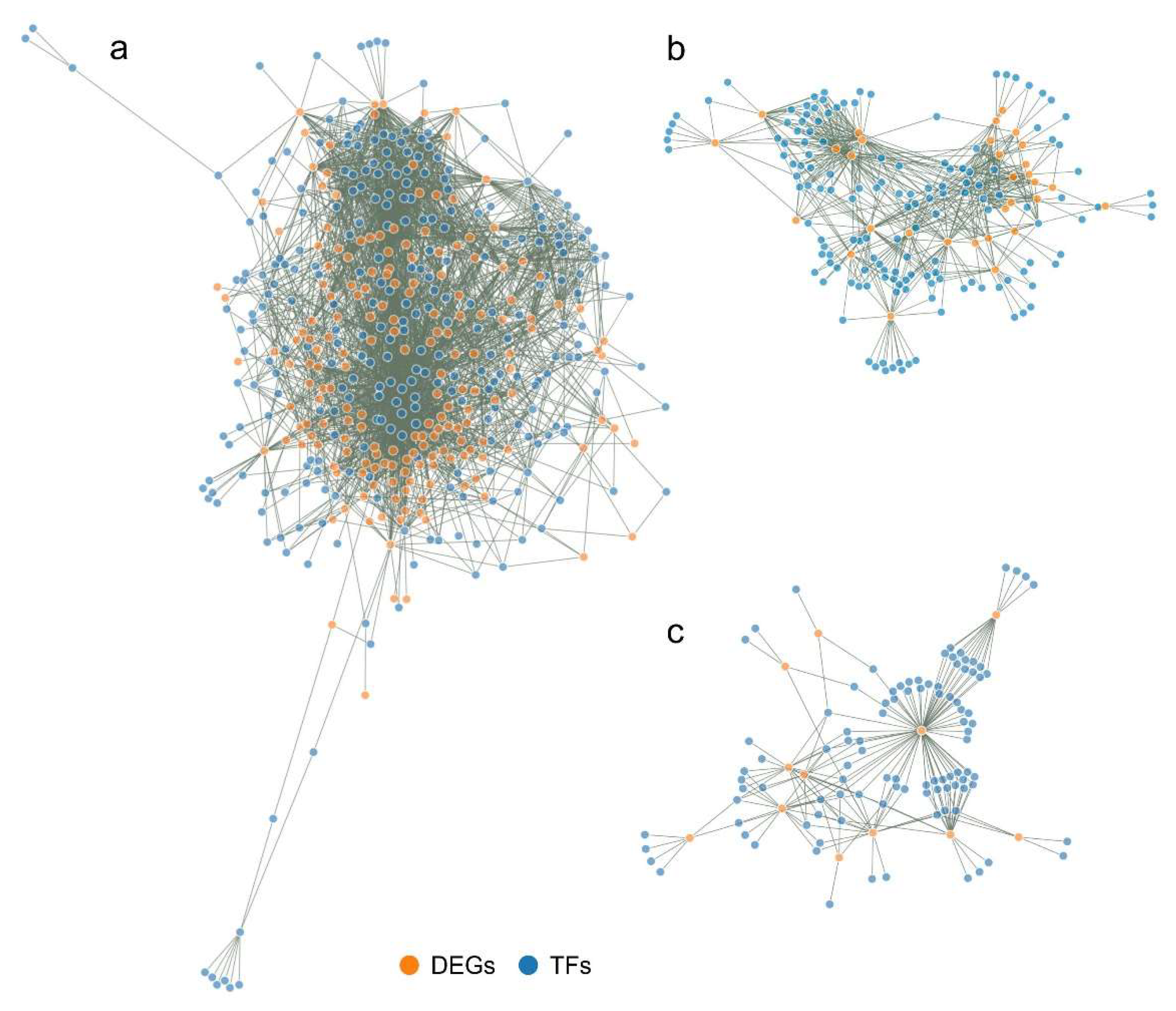
Figure 9.
The number of nodes represents TFs from different family (a) and the number of edges to TFs family (b) among the different comparisons.
Figure 9.
The number of nodes represents TFs from different family (a) and the number of edges to TFs family (b) among the different comparisons.
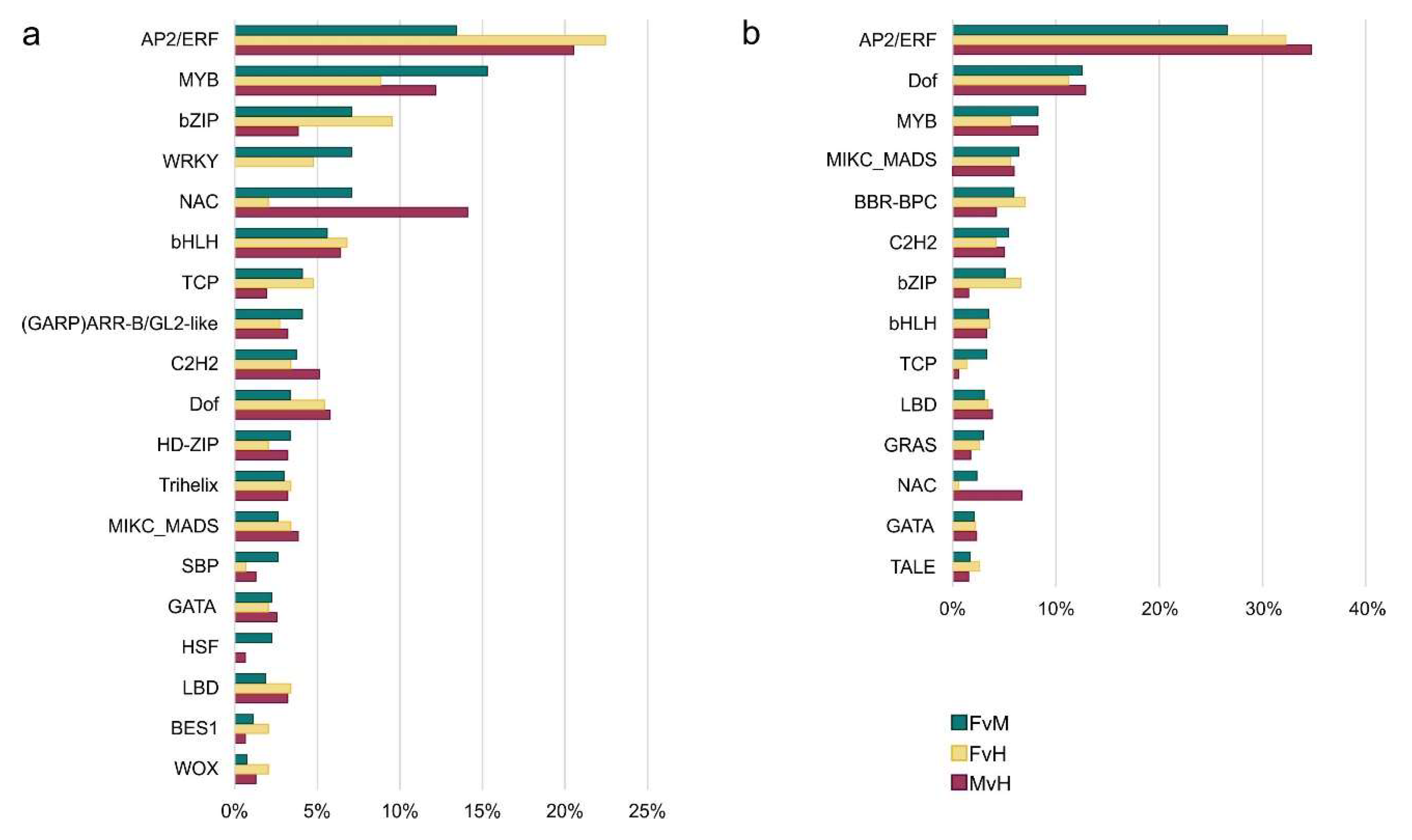

Table 1.
Number and percentage of transcription factors found in the list of genes with significantly differential expression for LvS, FvM, FvH, MvH comparisons.
Table 1.
Number and percentage of transcription factors found in the list of genes with significantly differential expression for LvS, FvM, FvH, MvH comparisons.
| Comparison | Number of DEGs | Number of TFs found for the comparison: | % of all DEGs |
|---|---|---|---|
| LvS | 2852 | 248 | 8,70% |
| FvM | 260 | 28 | 10,77% |
| FvH | 36 | 2 | 5,88% |
| MvH | 55 | 6 | 10,91% |
Table 2.
The number of DEGs and regulatory TFs that were used to create the interactome network for FvM, FvH, MvH, LvS comparisons.
Table 2.
The number of DEGs and regulatory TFs that were used to create the interactome network for FvM, FvH, MvH, LvS comparisons.
| FvM | FvH | MvH | LvS | |
|---|---|---|---|---|
| DEG | 186 | 31 | 35 | 244 |
| TF | 282 | 147 | 156 | 2785 |
Table 3.
Top 10 Number of TFs from established interaction networks that have the most connections to DEGs.
Table 3.
Top 10 Number of TFs from established interaction networks that have the most connections to DEGs.
| Comparison | Transcription factor | No. of edges |
|---|---|---|
| FvM | Cucsa.362960|MIKC_MADS|MADS box transcription factor | 116 |
| Cucsa.277740|AP2|AP2-like ethylene-responsive transcription factor | 109 | |
| Cucsa.026600|BBR-BPC|GAGA-binding transcriptional activator | 93 | |
| Cucsa.307870|C2H2|Transcription factor IIIA | 82 | |
| Cucsa.102120|Dof|Dof zinc finger protein | 81 | |
| Cucsa.280310|GRAS|DELLA protein GAI | 81 | |
| Cucsa.213830|Dof|Dof zinc finger protein | 76 | |
| Cucsa.159750|BBR-BPC|GAGA-binding transcriptional activator | 69 | |
| Cucsa.341290|Dof|Dof zinc finger protein | 60 | |
| Cucsa.098430|Dof|Dof zinc finger protein | 48 | |
| FvH | Cucsa.362960|MIKC_MADS|MADS box transcription factor | 23 |
| Cucsa.026600|BBR-BPC|GAGA-binding transcriptional activator | 21 | |
| Cucsa.277740|AP2|AP2-like ethylene-responsive transcription factor | 19 | |
| Cucsa.307870|C2H2|Transcription factor IIIA | 17 | |
| Cucsa.159750|BBR-BPC|GAGA-binding transcriptional activator | 14 | |
| Cucsa.280310|GRAS|DELLA protein GAI | 13 | |
| Cucsa.353140|TALE|Homeobox protein knotted-1-like 1 | 13 | |
| Cucsa.102120|Dof|Dof zinc finger protein | 12 | |
| Cucsa.213830|Dof|Dof zinc finger protein | 12 | |
| Cucsa.098430|Dof|Dof zinc finger protein | 9 | |
| MvH | Cucsa.362960|MIKC_MADS|MADS box transcription factor | 21 |
| Cucsa.102120|Dof|Dof zinc finger protein | 17 | |
| Cucsa.277740|AP2|AP2-like ethylene-responsive transcription factor | 16 | |
| Cucsa.213830|Dof|Dof zinc finger protein | 12 | |
| Cucsa.307870|C2H2|Transcription factor IIIA | 12 | |
| Cucsa.341290|Dof|Dof zinc finger protein | 12 | |
| Cucsa.026600|BBR-BPC|GAGA-binding transcriptional activator | 11 | |
| Cucsa.159750|BBR-BPC|GAGA-binding transcriptional activator | 11 | |
| Cucsa.136780|ERF|Dehydration responsive element binding transcription factor | 9 | |
| Cucsa.237150|ERF|Ethylene-responsive transcription factor ERF021 | 9 | |
| LvS | Cucsa.362960|MIKC_MADS|MADS box transcription factor | 1579 |
| Cucsa.277740|AP2|AP2-like ethylene-responsive transcription factor | 1485 | |
| Cucsa.026600|BBR-BPC|GAGA-binding transcriptional activator | 1324 | |
| Cucsa.307870|C2H2|Transcription factor IIIA | 1205 | |
| Cucsa.280310|GRAS|DELLA protein GAI | 1090 | |
| Cucsa.102120|Dof|Dof zinc finger protein | 1076 | |
| Cucsa.213830|Dof|Dof zinc finger protein | 1031 | |
| Cucsa.159750|BBR-BPC|GAGA-binding transcriptional activator | 1000 | |
| Cucsa.353140|TALE|Homeobox protein knotted-1-like 1 | 920 | |
| Cucsa.341290|Dof|Dof zinc finger protein | 823 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



