
Submitted:
30 April 2023
Posted:
01 May 2023
You are already at the latest version
Abstract
The enteric nervous system (ENS) is a neural network referred to as "the brain in the gut" because of its similarities to the central nervous system (CNS). The ENS consists of numerous types of neurons and glial cells distributed in two intramuscular plexuses that span the length of the intestine and control coordinated smooth muscle contractile activity and other intestinal functions. It is well-established that reciprocal communication exists between the brain and the gastrointestinal tract. Although the ENS can function independently, it is connected to the CNS through the afferent and efferent pathways of the parasympathetic and sympathetic nervous systems. In addition to regulating ENS function by the CNS, these connections are likely to be critically involved in the pathophysiology of Parkinson's disease (PD). PD is a common neurodegenerative disorder that presents with non-motor and motor symptoms. Surprisingly, ENS lesions have been shown to occur very early in the disease, even before CNS involvement. This has led to the postulation that the ENS may be central to the pathophysiology of PD. Autopsy studies have shown that α-synuclein (αS) aggregates in PD patients are found both in the substantia nigra (SN) and the ENS. Therefore, it has been hypothesized that the pathological process leading to PD may initially occur in the ENS years before the appearance of motor features. This process induces misfolding and aggregation of αS in specific subtypes of neurons in the CNS. Finally, it spreads retrogradely in the CNS through preganglionic vagal fibers to the dorsal motor nucleus of this nerve and to other central nervous structures. In addition to the presumed role in the spread of the disease process, it has also been suggested that pathological changes in the ENS might be involved in the gastrointestinal dysfunction frequently seen in Pd patients. Starting from the evidence in animal models and using a translational point of view, in this review, we aim to summarize the role of the ENS in the pathogenesis of PD and how this system could be modulated for a novel therapeutic approach. While acknowledging the presumed role of the microbiome in the gut-brain axis, we will shift the focus from this point of view to focus more on the neurons of the ENS.
Keywords:
Parkinson's Disease
; translational medicine
; gut-brain axis
; enteric nervous system
; microbiota
; rodent models
; clinical evidence
1. Introduction
Over the past decade, numerous preclinical and clinical publications have shed light on the complex relationship between the gut and the brain in neurological disorders, especially in Parkinson's disease (PD), in which gastrointestinal dysfunction is a prominent feature [1].
PD is the second most common neurodegenerative disorder after Alzheimer’s disease, and its pathological hallmarks are the loss of dopaminergic cells of Substantia Nigra pars compacta (SNpc) and the brain accumulation of Lewy bodies (LB), which are abnormal aggregates of α-synuclein (αS) [2]. In most patients, PD is the result of a synergistic interaction between genetic factors and environmental stressors, a condition referred to as “double hit theory”[2,3].
Therefore, exploring the potential interaction of distinct genetic and environmental factors is essential to identify convergent pathways and potential molecular targets for neuroprotection [4].
Patients with PD are a heterogeneous group, varying in age at disease onset, speed of progression, severity of motor and non-motor symptoms, and extent of central and peripheral inflammation [3].
Although still considered a paradigmatic movement disorder, PD is associated with a broad spectrum of non-motor symptoms, including mood and affective disturbances like apathy, anhedonia and depression, cognitive dysfunction, and complex behavioral disorders [5,6]. Sensory dysfunctions like hyposmia or pain and sleep-wake cycle regulation disturbances are common. In addition, many patients show autonomic dysfunction, including orthostatic hypotension, urogenital dysfunction, and constipation [7]. Constipation, in particular, affects up to 80% of PD patients and may precede the onset of motor symptoms in years [7]. The premotor stage of the disease may occur 5 to 10 years before the onset of motor symptoms [5,6].
In the premotor phase, idiopathic constipation is one of the most decisive risk factors for the onset of PD. It is associated with neurodegenerative changes in the enteric nervous system (ENS) [1,7]. According to Braak's classic hypothesis [8], neurodegenerative diseases, particularly PD, may recognize a peripheral origin when putative pathogens enter the mucosa of the gastrointestinal tract, inducing misfolding and aggregation of the hallmark αS in specific subtypes of central nervous system (CNS) neurons, then spreading retrogradely to the CNS through the vagal preganglionic fibers to the dorsal motor nucleus and finally to other central nervous structures[9,10].
Indeed, two categories of PD patients have recently been identified: a brain-first (top-down) type, in which αS pathology initially arises in the CNS and then in the peripheral autonomic nervous system, and a body-first (bottom-up) type, in which pathology originates in the ENS and then spreads to the CNS [11,12,13].
The ENS, the intrinsic nervous system of the gastrointestinal tract, often referred to as the "second brain," is a complex, and to date not fully understood, network comprising different types of neurons and glial cells[14].
The ENS is essential in regulating many gastrointestinal functions, including motility and fluid secretion. Degeneration of enteric neurons (NEs) could therefore be responsible for the gastrointestinal symptoms commonly observed in neurological disorders [15]. Indeed, numerous articles demonstrate that the pathology of PD is not limited to the CNS and that there is also extensive involvement of the ENS [16,17,18].
Data from patients and animal models suggest that PD affects distinct subsets of neurons and glia in the ENS and that the latter may participate in the pathogenesis of this disorder [15,19]. Although there has been much enthusiasm for the possibility of sampling the ENS for diagnosis or therapeutic monitoring of PD, further work needs to determine which NEs are most affected and how ENS function can be modulated to improve gastrointestinal symptoms in patients.
In this context, we aims to provide a deeper insight into the role of the ENS in PD by describing its normal and pathological physiology in human and animal models.
2. Overview of the Enteric Nervous System (ENS): anatomy and function.
The ENS, the intrinsic innervation of the gastrointestinal tract (GI), is the largest and most complex division of the peripheral and autonomic nervous systems in vertebrates. In humans, the ENS contains 400-600 million neurons and an array of neurotransmitters and neuromodulators similar to those found in the CNS [20]. Unlike the CNS, in which efferent pathways are characterized by pre-ganglionic and post-ganglionic neurons [21], the axons of gut neurons in the ENS project to the sympathetic ganglia, brainstem, spinal cord, pancreas, gallbladder, and trachea [14]. The anatomy and physiology of the ENS have been studied since the 19th century, going so far as to demonstrate early in the last century how the peristaltic reflex (i.e., the pressure-induced propulsive activity of the intestines) is a local nervous mechanism that occurs in the absence of external nerve input [22]. Because of this autonomy and its complexity, Michael D. Gershon likened the ENS to a second brain [20]. Two-way communications between the ENS and the CNS are always active: the CNS can regulate or alter the normal functioning of the ENS and vice versa. For example, certain gut disorders impair the production of psychoactive substances such as serotonin (5-HT, 5-hydroxytryptamine), dopamine (DA), and opiates, which can affect mood [23]. Conversely, emotional states, such as intense anxiety, can cause colitis, constipation, irritable colon, or mucosal ulcers through stimulation of peristalsis and hyperproduction of neurotransmitters [23]. The ENS originates around the eighth day of embryonic life from neural crest progenitor cells, endowed with stem-like properties, which migrate through the forming gastrointestinal tract and colonize it within five days [20]. They subsequently differentiate into neurons and glia by integrating predetermined instructions with information from the microenvironment [24]. In humans, the ENS becomes functional in the last trimester of gestation and continues to develop after birth [24]. The ENS is composed of small aggregations of nerve cells, the enteric ganglia, the neural connections between these ganglia, and the nerve fibers that supply effector tissues, including gut wall muscle, epithelial lining, intrinsic blood vessels, and gastroenteropancreatic endocrine cells [14,20,22,25]. NEs are organized into ganglionic plexuses: the myenteric (Auerbach's) plexus and the submucosal (Meissner's) plexus. Ganglionic plexuses are enveloped by glial cells, like CNS astrocytes, which form a true blood-enteric barrier. Glial cells release enterocyte differentiation factors, participate in gastrointestinal functions, and are involved in the pathogenesis of inflammatory disorders of the GI tract. Auerbach's myenteric plexus, located in the muscle tonaca between the layers of longitudinal and circular muscles, consists of linear chains of numerous interconnected neurons that span the length of the gastrointestinal tract and regulate its movements. Meissner's submucosal plexus, located in the submucosa of the small and large intestines but absent in the esophagus and stomach, consists of ganglia stratified at different levels. It integrates sensory signals from the intestinal epithelium and contributes to the local control of secretion, intestinal absorption, blood flow, and submucosal muscle contraction [14,22,25] (Figure 1).
A: Time course of ENS development. The ENS originates around the eighth day of embryonic life from neural crest progenitor cells (ENCDCs) with stem-like properties, which migrate through the GI tract and colonize it within five days.
After invading the anterior intestine, these pre-ENCDCs migrate rostro-caudally, proliferating and differentiating into neurons and glia. During this process, the intestine elongates, changing shape from a straight line to a single curve, with the middle and small intestine closely adjacent. The cecal appendix grows, and the entire intestine elongates further. At embryonic days 11 and 13, ENCDCs invade the colon by crossing the mesentery and transiting into the cecum. The cecal and trans mesenteric populations then fuse to form the ENS enteric nervous system in the rostral colon. In humans, the ENS becomes functional in the last trimester of gestation and continues to develop after birth. B: Schematic diagram of the human GI tract. C: Organization of the enteric nervous system. NEs are organized into ganglionic plexuses: the myenteric plexus and the submucosal plexus. The ganglionic plexuses are enveloped by glial cells, such as CNS astrocytes, which form a proper blood-enteric barrier.
The myenteric plexus is in the muscle tonaca between the layers of longitudinal and circular muscles. It consists of linear chains of numerous interconnected neurons that span the length of the GI and regulate its movements.
Twenty types of NEs characterized by different morphological, neurochemical, and electrophysiological aspects, connections, and functional roles have been identified [24,26,27]. Based on intracellular electrophysiological recordings, two types of NEs were detected: S and AH neurons. S neurons are characterized by high excitability and can exhibit rapid excitatory postsynaptic potentials, followed by a short-lived hyperpolarizing current (20-100 ms), rapidly restoring the membrane potential [26] [28]. On the other hand, AH neurons exhibit large action potentials followed by a slow hyperpolarizing current (2-30 s) that makes them less excitable. NEs use more than 50 neurotransmitters (NTs) in synaptic communications, from small neurotransmitters (e.g., ACh, AcetylCholine, 5-HT) to neuropeptides (e.g., CGRP, Calcitonin Gene-Related Peptide, somatostatin, substance P, VIP, Vasoactive Intestinal Peptide), to gases (e.g., NO, Nitric Oxide) [27,28]. NEs are grouped into three functional classes: intrinsic sensory neurons called IPANs, muscle motor neurons, and interneurons. IPANs are large and equipped with numerous axons: they can sense mechanical, chemical, and thermal stimuli and transmit information about muscle tension state and endoluminal content to motor neurons [29], triggering reflexes that regulate motility, secretion, and blood flow. They make up about 10-30% of the neurons located in the submucosal and myenteric plexus of the small and large intestines; they are not present in the esophagus (whose motility is controlled by fibers originating from the CNS) and stomach (whose motility is under the control of vagal fibers) [29]. Motor neurons are divided into muscular and secretomotor-vasodilatory. The former (Dogiel's type I) innervate the circular and longitudinal musculature and the muscular mucosae, determining their contraction or relaxation; they have an elongated cell body, numerous dendrites, and a single slender axon; electrophysiologically, they correspond to type S. Neurons innervating circular and longitudinal musculature have their cell bodies in the myenteric plexus and are excitatory (using ACh and TK, TachyKinin, and projecting orally) or inhibitory (using NO and VIP and projecting anally) [29]. Muscle motor neurons generate, following regional stimulation, coordinated and polarized muscle responses that allow the progression of intestinal contents, i.e., induce contraction in the oral direction and relaxation in the anal direction [29]. On the other hand, secretomotor-vasodilator neurons are located mainly in the submucosal ganglia, controlling both the secretion of ions and water via ACh and the vasodilation of submucosal arterioles via VIP [26,27]. Some influence glucose transport across the mucosa of the small intestine [30], a process also regulated by vagal-like reflexes; others modulate acid secretion in the stomach [30]. Interneurons integrate sensory afferents and organize effector responses [27,28]. In the myenteric plexus, they form chains that run in ascending and descending directions. They resemble type I neurons and are S-type [28]. The ENS, in the course of life, undergoes plastic changes as a spatiotemporal adaptive response to external stimuli, which arrive through sensory afferents, and to internal stimuli that come from autonomic innervation [22]. In the complex microenvironment of the gut wall lodge, different types of cells (neurons, glia, Cajal cells, muscle cells, and immune cells) capable of communicating with each other in synaptic or paracrine ways. This interactive plurality modulates the functional state of NEs by influencing the digestive and secretory functions of the GI tract [31]. Changes in diet and perturbations in the gut microbiome, with its metabolites and neuroactive compounds, affect the functioning of the NE and its connections with the CNS since they alter mucosal permeability and the secretion of hormones and immune cells. In addition, NEs are vulnerable to aging-related degeneration [31].
3. Evidence of the role of the ENS in animal models of Parkinson's disease
Gastrointestinal dysfunction is a common non-motor symptom of PD. While in PD patient, gastrointestinal dysfunction is present in 80-90% of cases and has been associated with αS aggregation and neuronal loss in the CNS, reports of gastrointestinal symptoms in animal models of PD are known to vary, and the degree to which pathology in the CNS contributes to gastrointestinal symptoms remains unclear [32].
PD benefits from a wide range of animal models whose diverse pharmacological, toxin, and genetic are essential to study its etiology and neurobiology [33]. Animal models of PD rely on pharmacological or genetic approaches to simulate nigrostriatal neurodegeneration and disease pathogenesis [33]. However, much remains to be discovered and requires continuous questioning by the research community.
The most commonly used pharmacological models are based on neurotoxins administered to mice, rats, and non-human primates [34] (Figure 2).
PD is a heterogeneous disorder with varying ages of onset, symptoms, and progression rates. This heterogeneity requires the use of a variety of animal models to study different aspects of the disease.
(Right) Neurotoxin-based approaches include exposure of rodents or nonhuman primates to 6-OHDA, MPTP, and agrochemicals such as the pesticide rotenone. Acute neurotoxin exposure induces motor deficits and rapid nigro-striatal dopaminergic cell death by disrupting mitochondrial function and increasing oxidative stress. In contrast, chronic neurotoxin administration induces progressive patterns that may include aS aggregates. Genetics-based approaches to modeling Parkinson's disease include transgenic and viral vector-mediated models based on genes linked to monogenic Parkinson's disease. Among these, overexpression, and introduction of preformed α-synuclein fibrils induce toxic protein aggregates, nigro-striatal neurodegeneration, and variable motor deficits, depending on the specific model. (Left) GI dysfunction is the most common non-motor symptom of PD. Symptoms of GI dysmotility in Parkinson's disease include premature satiety and weight loss due to delayed gastric emptying and constipation due to altered colonic transit. We can find numerous alterations in the enteric nervous system in preclinical models of PD: neurodegeneration of NEs, which is the leading cause of behavioral and electrophysiological alterations in mouse models.
Both neurotoxins, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA), consistently affect nigrostriatal dopaminergic pathways [34]. However, their impact on gut function and the CNS varies, depending on the agent, mode of administration, and assays used [35,36,37,38]. Systemic administration of MPTP in mice causes loss of dopaminergic neurons in the myenteric plexus, but does not cause severe defects in gastrointestinal motility [36,37]. Peripheral administration of MPTP in rats does not significantly affect the number of dopaminergic neurons and the expression of dopaminergic markers in the SNpc [39]. However, it significantly reduces Tyrosine Hydroxylase-Immunoreactive (TH-IR) neurons in the GI tract, suggesting that the degeneration of dopaminergic neurons might start earlier than in the SNpc [36,39,40]. Parenteral administration of MPTP, using a dosing paradigm that consistently causes dopaminergic neurodegeneration in the SN of mice, simultaneously induces dopaminergic neurodegeneration in the ENS, associated with behavioral and electrophysiological consequences. The accelerated colonic motility and colonic muscle relaxation defect observed after MPTP intoxication are consistent with the inhibitory nature of dopaminergic neurons in the ENS [41,42]. The finding of a reduction in TH-positive neurons in the myenteric ganglia, in the absence of effects on cholinergic or nitric oxide neurons, confirms that MPTP is selectively toxic to dopaminergic neurons in the ENS, just as in the CNS [43,44,45].
TH is a marker of catecholaminergic neurons, but adrenergic and noradrenergic inputs to the GI tract are mainly extrinsic. Therefore, most TH-positive neurons with cell bodies in the myenteric plexus can be considered dopaminergic [46].
The colon muscle of MPTP-treated mice showed increased contraction and decreased relaxation in response to electric field stimulation of NEs. These results are complementary and indicate an altered function of an inhibitory subpopulation of NEs, in this case DA neurons. This agrees with previous functional assessments of the effect of DA on enteric neuron-mediated muscle contraction [42].
Exogenous DA has been shown to antagonize colonic muscle contractility in a receptor-dependent manner [41,42]. Considering the neuropathological and electrophysiological findings, it is likely that dysfunction and death of dopaminergic neurons cause the transient increase in colonic motility observed after MPTP intoxication. Decreased dopaminergic inhibitory tone results in faster colonic transit due to the relative abundance of stimulatory neuronal input [41,42].
Neurotransmitters related to the gastrointestinal dysfunction of PD could be involved in the intestinal dopaminergic, cholinergic, and oxidergic nitric systems [35]. To investigate the relationship between the gastrointestinal dysfunction of PD and the alteration of gastrointestinal neurotransmitters, 6-OHDA was microinjected into one side of the nigrostriatal system of the brain to generate an animal model of PD through the impairment of rat dopaminergic neurons, and the effect of neurotransmitter alterations in the central nervous system on gastrointestinal function was observed [35].
Gastrointestinal dysfunction and changes in dopaminergic, nitric oxide synthase (NOS), and cholinergic neurons in the myenteric plexus were analyzed. Compared with control samples, 6-OHDA rats had delayed gastric emptying and constipation, which could be related to increased gastrointestinal TH and decreased NOS. These symptoms were not associated with alterations in cholinergic transmitters [38].
Unfortunately, some of these studies did not analyze the submucosal plexus, making a direct comparison with more robust findings in complex PD patients [15]. Rats treated with 6-OHDA show elevated protein levels of TH and dopamine transporter (DAT) (dopaminergic markers) in both the epithelium and neurons of the gastrointestinal tract, resulting in increased dopamine (DA) content in the gut and delayed gastric emptying [39]. Neurodegeneration of the central SN by 6-OHDA increases the expression of TH and DAT proteins in both the epithelium and neurons of the GI- tract. These observations suggest that, in some PD patients, the number of enteric dopaminergic neurons and cells may increase, rather than decrease, due to a compensatory mechanism to attenuate the loss of DA in the SN [39]. In contrast, in 6-OHDA-treated rats, increased protein expression of TH and DAT could lead to increased DA concentration in the colon, which is more likely to cause constipation [39]. Alterations in the monoaminergic system and decreased colonic motility were observed in rats microinjected with 6-OHDA in the bilateral SN.
DA, NE, and 5-HT play essential roles in regulating colonic motility: increased DA content, upregulation of β3-ARs, and decreased 5-HT4 receptors could contribute to the decreased spontaneous colonic contraction and constipation observed in rats with 6-OHDA [35].
Rats with lesions of SN dopaminergic neurons manifest gastrointestinal dysmotility [47,48], including gastroparesis and constipation [48,49].
Animal models do not yet allow for an adequate study of how PD prodromal constipation occurs [50]. To date, there is a paucity of relevant experimental models of GI dysfunction associated with α-syn pathology: α-syn deposition in the ENS of PD patients has been reported in the myenteric and submucosal plexuses of gastrointestinal tracts [51,52]. Transgenic mouse lines expressing a mutant form of human αS (A53T or A30P) under its promoter show colonic disorders similar to constipation and pathology characteristic of αS [53]. In a transgenic mouse model in which mutant human αS (A53T) was expressed under the control of the prion promoter [54], aggregates of αS were observed in the ENS prior to changes in the CNS [53]. This finding suggests that αS pathology may be initiated from the ENS and propagate to the CNS via the vagus nerve [8]. In support of this, in a transgenic mouse model, the accumulation of αS aggregates in the ENS precedes changes in the CNS [53].
Expression of human αS in the dorsal motor nucleus of the vagus nerve (DMV), a region of the brain severely affected by PD, causes an age-related slowing in A53T mice of gastrointestinal motility reminiscent of that observed in patients with PD [8,55]. The symptoms coincide with the disruption of efferent vagal processes that project from the DMV to the GI tract. This pattern parallels the pathology of postmortem specimens of PD patients and implicates the DMV as a possible mediator of GI neuropathology and symptomatology in PD [56].
However, αS mutations are only responsible for rare cases of PD [57]. Mice overexpressing wild-type human αS under the Thy-1 promoter (Thy1-αS) show increased transit time and colonic content compared with wild-type (WT) pups when tested at 12-14 months of age [58]. However, striatal dopamine loss occurs only after 14 months in Thy1-αS mice, manifesting motor and non-motor deficits, such as olfactory disturbances, as early as 2-3 months of age [59,60].
The mechanisms underlying colonic motor impairments may be related to αS overexpression in the colonic myenteric nervous system [58]. The reduced response to defecation stimuli in Thy1-αS could be related to the accumulation of α-S in colonic myenteric plexuses [58].
The gastrointestinal system is one of the most susceptible to environmental since stresses it is in direct contact with environmental agents [61,62,63]. In a recent study, intra-gastric administration of rotenone in mice caused progressive αS deposition in both ENS and CNS neurons affected by PD, such as neurons in the myenteric plexus, the vagus dorsal motor nucleus (DMV), the spinal cord, and the Sympathetic Nervous System (SNS) [64]. These studies suggested that environmental stresses to the gastrointestinal system could lead to αS pathology in the CNS.
Numerous preclinical pieces of evidence associate gastrointestinal symptoms in toxic models of PD based on oral administration of rotenone [61]. In previous studies, it has been shown that orally administered rotenone exposure induces PD-like changes in the ENS and triggers PD progression throughout the nervous system to the SN [62,64]. Interestingly, the latter changes appear as early as the first moments after rotenone administration (2 months) before the onset of motor symptoms (which occur after 3 months of exposure in this animal model), thus mimicking the pattern of progression observed in PD patients.
In two recent studies, rotenone exposure reduced sympathetic noradrenergic [65] and vagal cholinergic gut innervation [66].
The mechanism by which environmental agents induce αS aggregation is unknown. However, a recent study showed that αS expression in the ENS could be upregulated by agents that cause depolarization and increase cyclic AMP levels [67].
An emerging concept in gastroenterology is that a wide range of diseases, such as motility disorders, can be partially considered enteric neuropathies. In particular, aging is associated with various motility or gut disorders, including delayed gastric emptying and longer intestinal transit time [68]. Aged rats show neuronal loss and changes in neurochemical phenotype in the ENS, which may result in motility disorders [69]. Surprisingly, along with neuronal loss, these rats exhibit dystrophic NEs that contain αS aggregates reminiscent of Lewy pathology [70].
Braak et al. suggested that the gastrointestinal tract is an entry point for a second pathogenic hit that goes to the CNS [52]. Transgenic double mice expressing mutant αS offer an opportunity to investigate the hypothesis that early ENS dysfunction is not only an early marker of disease but also that, once triggered, it facilitates the entry of deleterious factors that cause progression and spread to the CNS [53].
A summary of animal models exhibiting each of these characteristics is provided in Table 1.
Parkinson's disease (PD); Enteric Neurons (NEs); 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP); Substantia nigra pars compacta (SNpc), Tyrosine Hydroxylase-Immunoreactive (TH-IR); Gastrointestinal tract (GI), Tyrosine Hydroxylase (TH); Enteric Nervous System (ENS); Central Nervous System (CNS); Sympathetic Nervous System (SNS); Alpha-Synuclein (αS); 6-hydroxydopamine (6-OHDA); Dorsal motor nucleus of the vagus nerve (DMV); Dopamine transporter (DAT); Dopamine (DA); Nitric oxide synthase (NOS).
4. The possible role of the ENS in Parkinson's Disease: clinical evidence
In idiopathic PD, most patients showing PD-related inclusions at CNS sites also have LB and Lewy neurites (LN) in the ENS and sympathetic ganglia [52]. Braak et al. proposed a pathological disease staging based on autopsies performed on PD patients and healthy individuals [71]. According to this staging, PD lesions follow a spatiotemporal pattern that begins in the olfactory bulb (OB) and the ENS and progresses in the CNS through synaptically connected structures. This pathological staging of the disease seems to correlate well with the appearance of early non-motor symptoms in PD patients, including hyposmia, gastrointestinal changes, autonomic dysfunction, and pain [72] (Figure 3).
Diagnosis of PD currently depends on motor deficits, including bradykinesia, rigidity, and tremor. The motor characteristics predominantly result from the loss of dopaminergic neurons in the SNpc. However, the non-motor symptoms of PD often begin before the more visible motor symptoms. These are called "pre-motor symptoms”, such as loss of smell, depression, and constipation which can appear years before diagnosis. The GI symptoms include excessive drooling, dysphagia, impaired gastric emptying, constipation, and impaired defecation. Moreover, alterations of the ENS levels have been reported in PD. The presence of αS aggregations along the GI tract has been proposed as a diagnostic tool that allows early diagnosis of the disease process before the onset of motor symptoms. Indeed, αS is abundantly expressed in all nerve plexuses of the ENS in normal individuals, and its levels increase with age. Thus, its pathological relevance must be carefully evaluated before use as a predictive biomarker of PD.
Little is known about the ENS degenerative process in PD patients. Clinical studies have revealed delayed gastric emptying, external anal sphincter dystonia causing difficult rectal evacuation, and general slow-transit constipation, probably caused by local loss of dopaminergic neurons [73,74,75]. Many groups have recently studied the expression and modifications of enteric αS in PD patients, with controversial results. Although some have found increased inclusions of αS and phosphorylated αS in the above areas compared with control subjects [76], there seems to be high variability among patients [61].
All these hypotheses take advantage of clinical observation of both the prodromal symptoms and the non-motor ones [77,78,79], assuming that the involvement of the dopaminergic system in the PD neurodegenerative process starts at the level of the motor nucleus dorsal vagus with a pattern of periphery-center (bottom-top) [78].
This process would initially occur in the enteric system (signs: constipation slow and transit alteration) and then progress to the brainstem with hypo/anosmia and sleep disturbances up to the mesencephalon (SNpc) with the appearance of the cardinal symptoms (rigidity, bradykinesia, tremor, postural instability); finally, it would involve the cerebral cortex with the appearance of cognitive and behavioral disturbances [80]. Braak predicted six stages of the disease, of which only the 3rd one involves the CNS with the appearance of motor symptoms [8,71], in a sort of dominos game [81]. An interesting and innovative hypothesis has shifted the interest of researchers in the last 20 years toward the discovery of early biomarkers [82]. The big challenge was to identify a potential pathogen capable of passing the mucosal barrier of the GI tract and, via postganglionic ENs, entering the CNS along unmyelinated preganglionic fibers generated from the viscero-motor projection cells of the vagus nerve [83]. There is also epidemiological evidence that complete but not partial vagotomy may protect against later PD [84,85].
However, other studies suggested that not all cases of PD start in the ENS. Autopsy studies have shown that a minority of cases with Lewy pathology do not have pathological inclusions in the dorsal motor nucleus of the vagus and that a fraction of cases display a limbic-predominant distribution of αS inclusions with less pathology in the brainstem [88]. Therefore, different subtypes of PD have been proposed: i) a body first (bottom-up) subtype, in which the pathology starts in the enteric or peripheral autonomic nervous system and arises via vagus nerve sympathetic connectome to CNS [89]. This phenotype shows prolonged intestinal transit and constipation as prodromic symptoms; ii) a brain–first (top-down) in which the αS pathology originates in the brain or via the OB and descends to the peripheral autonomic nervous system. This phenotype shows hyposmia and sleep disturbances as prodromic signs[12]. The gut-brain axis is composed of different functional systems as well as neuroendocrine and neuroimmune systems, including the hypothalamic-pituitary-adrenal axis, ENS through the sympathetic and parasympathetic system, and vagus nerve, gut immune cells, and the gut microbiota [90].
In the human gut, around 100 trillion microbes are involved in food fermentation, metabolic and immune maturity, development of the ENS and CNS, and, as recently revealed, also in modulation of the pathogenesis of many metabolic, neurodevelopmental, and neurodegenerative disorders [91].
The bacteria present in the gut can influence brain function via different pathways. These bottom-up pathways include direct absorption through the gut-blood/lymphatic-brain pathways, as well as local signaling in the gut to prime immune cells and the vagal retrograde transport pathways [92]. Small and lipophilic bacterial products or metabolites can enter the brain by crossing the blood-brain barrier through the first gut-blood/lymphatic-brain communication pathway [93]. Microbial-secreted products like neurotransmitters, including catecholamines, 5-HT, GABA, and gut metabolites, have been demonstrated to transit the gut-blood and blood-brain barriers to elicit an immune response altering brain neurochemistry and plasma proteomic profiles. In the second communication pathway, immune cells are activated by bacterial metabolites [94]. The third pathway is the vagal route. There is much evidence that the vagus nerve transports aS from the gut to the brain [95].
Many studies performed by rRNA gene amplicon surveys or shotgun metagenomic sequencing analysis have revealed changes in the PD patient's gut microbiota compared to healthy controls [96]. Changes in the gut microbiota also correlate with disease progression in PD. A decrease in the short-chain fatty acids-producing microbiota and an increase in pro-inflammatory bacteria correlate with motor and cognitive severity in patients with PD [97]. A 3-year longitudinal follow-up study of PD patients revealed that a reduced amount of Roseburia species predicted faster progression of both motor and non-motor symptoms of PD. A lower abundance of short-chain fatty acids-producing bacteria, including Fusicatenibacter and Faecalibacterium, correlates with elevated fecal inflammatory calprotectin levels in PD patients [98].
Systemic and fecal inflammatory markers IFN-γ, TNF-α, and neutrophil gelatinase-associated lipocalin, were also associated with an elevated expression of Bacteroides and Bifidobacterium in PD patients [99]. It seems, therefore, that the composition of gut microbiota influences the pharmaceutical treatment responses in PD patients [97]. The growing literature has shown the role of the gut microbiome in the pharmacokinetics of prescription drugs and the effects that the drugs can have in turn on the composition of the gut microbiome [100]. These observations lend support to the notion that the composition of the gut microbiome may affect the treatment efficacy and potential side effects of levodopa treatment in patients with PD [97]. In conclusion, in the very early stages of PD prior to CNS pathology, accumulation of enteric αS [101] may promote activation of immune/inflammatory signaling, including: canonical caspase-1- dependent inflammasome pathways [102], resulting in a massive release of IL-1β, which, in turn, alters intestinal epithelial barrier, through the activation of IL-1 receptors on intestinal epithelial cells [102]. In this context, intestinal inflammation and altered intestinal epithelial barrier can induce changes in short-chain fatty acid levels, characterized by alterations in butyrate levels, which could contribute to the impairment of the intestinal epithelial barrier [103]. They can also induce an increase in the concentration of circulating lipopolysaccharide, which, by translocating into the intestinal mucosa, could further contribute to the activation of immune/inflammatory pathways, thus generating a vicious circle that could lead to the chronicization of inflammatory processes and contributing to both intestinal symptoms and brain pathology [104] (Table 2).

Table 2.
Pathological features identified in PD patients.
| PD symptomps | Affected Neuron types | GI Symptoms | Alteration Biomarker | References |
|---|---|---|---|---|
|
Nd |
Nd |
Gastric emptying Difficult rectal evacuation Slow transit constipation |
Nd |
[73,74,75] |
| Hypo/anosmia Sleep disturbances Rigidity, bradykinesia, tremor, postural instability Cognitive and behavioral disturbances |
Neurodegenerative process starts at the level of the DMV with a pattern of periphery-center (bottom-top) |
Nd |
Increased inclusions of αS and phosphorylated αS |
[80] [78] [80] |
|
Nd |
Nd |
Prolonged intestinal transit, constipation | Minority of cases with Lewy pathology do not have pathological inclusions in the DMV. Limbic-predominant distribution of αS inclusions with less pathology in the brainstem |
[86,87] [89] |
| Motor and cognitive symptoms |
Nd |
Nd |
Decrease in the short-chain fatty acids, including Fusicatenibacter and Faecalibacterium Increase in pro-inflammatory bacteria |
[97] [98] |
|
Nd |
Nd |
Nd |
Systemic and fecal inflammatory markers IFN-γ, TNF-α, and neutrophil gelatinase-associated lipocalin, associated with an elevated expression of Bacteroides and Bifidobacterium. | [99] |
|
Nd |
Nd |
Alteration of intestinal epithelial barrier | Accumulation of enteric αS Activation of immune/inflammatory signaling, including canonical caspase-1- dependent inflammasome pathways Massive release of IL-1β |
[103] [102] [101] |
Alpha-Synuclein (αS); Dorsal motor nucleus of the vagus nerve (DMV); Tumor necrosis factor-alpha (TNF-α); Interferon-gamma (IFN-γ); Interleukin -1β (IL-1β).
5. New therapeutic approach targeting the ENS
The lessening of dopaminergic striatal and nigral innervation leads to an alteration in local microcircuits [105]. The emerging scenarios concerning enteric involvement in PD pathogenesis offer a new therapeutic approach.
For example, the European Medicines Agency (2011) and the U.S. FDA (2015) approved levodopa–carbidopa intestinal gel for treating advanced-stage idiopathic PD. This approach is utilized in patients with severe motor fluctuation, but unresponsive to classical Levodopa oral treatments. This gel is administered directly into the duodenum through a percutaneous endoscopic gastrostomy tube and portable infusion pump [106]. This kind of therapy via infusion maintains a more stable dopamine plasma level, contributing to reducing the freezing phenomena. The non-pharmacological approach based on the increase of enteric system motility is well defined in the last few years. For example, a high fiber diet, appropriate fluid intake, and psyllium can represent a good approach to counteract the slowing of bowel pain in many PD patients, as well as exercise and physical activity directed to stimulate autonomic symptoms (impaired gastric motility, dysphagia, constipation, and bowel incontinence) [107]. These approaches are based on the pieces of evidence that exercise may change dopamine receptor availability in animal models of PD and in patients, too [108,109].
Adjustment of anticholinergics and dopaminergic agents used for PD therapy can contribute to relieving intestinal and motor symptoms by demonstrating the connection between enteric and CNS [110]. Many other approaches were proposed to treat the comorbidity of PD, considering that gut dysfunction may contribute to the symptomatic fluctuation in PD patients. However, the discussion of all approaches to the treatment of enteric symptomatology falls outside the scope of this review, whose aim is to emphasize how the enteric system can play a role in the genesis of PD.
The aspect of the microbiome is also well discussed in many other papers that focus on the enteric flora to explain different phenomena. Anyway, to date many different types of microbiomes have been found in different PD patients, which does not allow a unique key for reading.
6. Future perspectives
Following the hypothesis that the pathogenesis of PD may begin in the gut, numerous studies have investigated the role of the ENS within this pathology [52].
On the one hand, Lewy pathology can be induced in the ENS and transported to the CNS via the vagal nerve. On the other hand, the altered gut microbiota composition causes an imbalance between beneficial and harmful microbial metabolites that interacts with increased intestinal permeability, intestinal inflammation, and systemic inflammation. The activated inflammatory state then affects the CNS and promotes PD pathology.
Although the mechanisms underlying the link between PD and gastrointestinal diseases remain unclear, such gastrointestinal dysfunctions may share gut-derived pathogenesis with PD.
Because gastrointestinal dysfunction in PD spans the fields of neurology and gastroenterology, management of these symptoms should ideally include a collaborative multidisciplinary team approach to ensure the best patient outcomes. However, such resources may be limited in real-world clinical practice.
In addition, differences in ethnology, cultural background, and lifestyles may cause heterogeneous results among studies, representing a limitation for further research. Therefore, more studies from different nations and regions are needed to explore the relationship between PD and gastrointestinal diseases in the future.
Until April 2023, there have been 66,523 papers about the etiology of PD on the Pub-Med database. Of these articles, 13,060 are reviews. However, only 0.99 % of the articles support the gut-brain axis theory, with only 546 papers discussing the ENS and PD 1119 works focusing on PD and the microbiome.
According to this evidence, the ENS is a good target for investigating some multifactorial aspects of PD, but it still needs to be explored. Moreover, current research focuses mainly on the microbiome and not on the relationships between the autonomic nervous system and the CNS, which are likely to underlie all etiological processes.
Furthermore, it is now well known that in PD patients, there are several alterations in all nonmotor autonomic functions. GI tract involvement is particularly pronounced and precedes the onset of motor signs. This means that changes occurring in the ENS may subsequently affect the brain via the vagus nerve, where misfolded αS moves retrogradely.
Considering these hypotheses, knowledge of the enteric system and its relationships beyond the microbiome could represent a new scenario to better characterize a disease of which only the final stages are known but probably represents the sum of numerous insults occurring over a lifetime of more than 20 years.
Author Contributions
"Conceptualization, M.M; A.P; and G.M; Resources, P.B; Methnalisys, M.M; A.P; P.B; and G.M; Writing – Original Draft Preparation, M.M; A.P; P.I; and G.M; Writing – Review & Editing, P.I; M.M; PB; and G.M; Visualization, P.I; M.M; PB; A.P; and G.M.; Supervision, P.B; and G.M; Project Administration, M.M; A.P; and G.M.
Funding
This work was partially supported by the Italian Ministry of Health “Ricerca Finalizzata” grants: RF-2019-12370182 to P.B and RF-2021-12374979 to A.P. The funding source was not involved in the design or writing of the report and in the decision to submit the article for publication.
Data Availability Statement
All the data shown in this paper are available in PubMed Library. All representative draws were created appositely by the authors and are available on request.
Acknowledgments
All authors thank Massimo Tolu, and Massimiliano Di Virgilio for their excellent technical assistance.
Conflicts of Interest
The authors declare no conflict of interest. No sponsors participate in the choice of the items; the design of the paper; the collection of literature, the interpretation of analyzed papers; the writing of the manuscript; or in the decision to publish in the Biomedicine journal.
Abbreviation LIST
- Acetylcholine (ach)
- α-synuclein (αS)
- Central nervous system (CNS)
- Dopamine (DA)
- Dopamine transporter (DAT)
- Dorsal motor nucleus of the vagus nerve (DMV)
- Enteric nervous system (ENS)
- Enteric neurons (NEs)
- Gastrointestinal tract (GI)
- Gut-brain axis (GBA)
- G-aminobutyric acid (GAB)
- Interferon-gamma (IFN-γ)
- Interleukin -1β (IL-1β)
- Human αs under the thy-1 promoter (Thy1-αS)
- 6-hydroxydopamine (6-OHDA)
- Lewy bodies (LB)
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
- Nitric oxide (NO)
- Nitric oxide synthase (NOS)
- Olfactory bulb (OB)
- Parkinson's disease (PD)
- Substantia nigra (SN)
- Substantia nigra pars compacta (SNpc)
- 5-hydroxytryptamine or serotonine (5-HT)
- Tumor necrosis factor-alpha (TNF-α)
- Tyrosine hydroxylase (TH)
- Tyrosine Hydroxylase-Immunoreactive (TH-IR)
- Vasoactive intestinal peptide (VIP)
- Wild-type (WT)
References
- Tan, A.H.; Lim, S.Y.; Lang, A.E. The microbiome-gut-brain axis in Parkinson disease - from basic research to the clinic. Nat. Rev. Neurol. 2022, 18, 476–495. [CrossRef]
- Schirinzi, T.; Martella, G.; Pisani, A. Double hit mouse model of Parkinson’s disease. Oncotarget 2016, 7, 80109–80110. [CrossRef]
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and environmental factors in parkinson’s disease converge on immune function and inflammation. Mov. Disord. 2021, 36, 25–36. [CrossRef]
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; Mandolesi, G.; Mercuri, N.B.; Bonsi, P.; Pisani, A. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol. Dis. 2016, 91, 21–36. [CrossRef]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 Suppl 1, 14–20. [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [CrossRef]
- Cersosimo, M.G.; Benarroch, E.E. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 559–564. [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [CrossRef]
- Lomax, A.E.; Fernández, E.; Sharkey, K.A. Plasticity of the enteric nervous system during intestinal inflammation. Neurogastroenterol. Motil. 2005, 17, 4–15. [CrossRef]
- Giaroni, C.; De Ponti, F.; Cosentino, M.; Lecchini, S.; Frigo, G. Plasticity in the enteric nervous system. Gastroenterology 1999, 117, 1438–1458. [CrossRef]
- Leclair-Visonneau, L.; Neunlist, M.; Derkinderen, P.; Lebouvier, T. The gut in Parkinson’s disease: Bottom-up, top-down, or neither? Neurogastroenterol. Motil. 2020, 32, e13777. [CrossRef]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; Sommerauer, M.; Danielsen, E.H.; Bech, E.; Kraft, J.; Munk, O.L.; Hansen, S.D.; Pavese, N.; Göder, R.; Brooks, D.J.; Berg, D.; Borghammer, P. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 2020. [CrossRef]
- Arotcarena, M.-L.; Dovero, S.; Prigent, A.; Bourdenx, M.; Camus, S.; Porras, G.; Thiolat, M.-L.; Tasselli, M.; Aubert, P.; Kruse, N.; Mollenhauer, B.; Trigo Damas, I.; Estrada, C.; Garcia-Carrillo, N.; Vaikath, N.N.; El-Agnaf, O.M.A.; Herrero, M.T.; Vila, M.; Obeso, J.A.; Derkinderen, P.; Bezard, E. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 2020, 143, 1462–1475. [CrossRef]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [CrossRef]
- Chalazonitis, A.; Rao, M. Enteric nervous system manifestations of neurodegenerative disease. Brain Res. 2018, 1693, 207–213. [CrossRef]
- O’Donovan, S.M.; Crowley, E.K.; Brown, J.R.-M.; O’Sullivan, O.; O’Leary, O.F.; Timmons, S.; Nolan, Y.M.; Clarke, D.J.; Hyland, N.P.; Joyce, S.A.; Sullivan, A.M.; O’Neill, C. Nigral overexpression of α-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol. Motil. 2020, 32, e13726. [CrossRef]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2. [CrossRef]
- Ma, C.; Zhang, W.; Cao, M. Role of the peripheral nervous system in PD pathology, diagnosis, and treatment. Front. Neurosci. 2021, 15, 598457. [CrossRef]
- Chalazonitis, A.; Rao, M.; Sulzer, D. Similarities and differences between nigral and enteric dopaminergic neurons unravel distinctive involvement in Parkinson’s disease. npj Parkinsons Disease 2022, 8, 50. [CrossRef]
- Gershon, M.D. The enteric nervous system: A second brain. Hosp Pract (Minneap) 1999, 34, 31–2, 35. [CrossRef]
- The Central Nervous System: Structure and Function - Per Brodal - Google Libri; Oxford University Press, U., 2004, Ed.; ISBN 9780195165609.
- The Enteric Nervous System - John Barton Furness - Google Libri; John Wiley & Sons, 2008, Ed.; ISBN 9781405173445.
- Cussotto, S.; Strain, C.R.; Fouhy, F.; Strain, R.G.; Peterson, V.L.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Differential effects of psychotropic drugs on microbiome composition and gastrointestinal function. Psychopharmacology (Berl) 2019, 236, 1671–1685. [CrossRef]
- Sasselli, V.; Pachnis, V.; Burns, A.J. The enteric nervous system. Dev. Biol. 2012, 366, 64–73. [CrossRef]
- The enteric nervous system and regulation of intestinal motility - ProQuest. Available online: https://www.proquest.com/docview/222539969?pq-origsite=gscholar&fromopenview=true (accessed on 10 August 2022).
- Structure of Enteric Neurons - Axel Brehmer - Google Libri; Springer Science & Business Media, 2006, Ed.; ISBN 9783540328742.
- Costa, M.; Furness, J.B.; Gibbins, I.L. Chapter 15 Chemical coding of enteric neurons. In; Progress in brain research; Elsevier, 1986; Vol. 68, pp. 217–239 ISBN 9780444807625.
- Furness, J.B.; Costa, M. Types of nerves in the enteric nervous system. In Commentaries in the neurosciences; Elsevier, 1980; pp. 235–252 ISBN 9780080255019.
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.-J. The enteric nervous system and gastrointestinal innervation: Integrated local and central control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [CrossRef]
- Shirazi-Beechey, S.P.; Moran, A.W.; Batchelor, D.J.; Daly, K.; Al-Rammahi, M. Glucose sensing and signalling; regulation of intestinal glucose transport. Proc. Nutr. Soc. 2011, 70, 185–193. [CrossRef]
- Saffrey, M.J. Cellular changes in the enteric nervous system during ageing. Dev. Biol. 2013, 382, 344–355. [CrossRef]
- McQuade, R.M.; Singleton, L.M.; Wu, H.; Lee, S.; Constable, R.; Di Natale, M.; Ringuet, M.T.; Berger, J.P.; Kauhausen, J.; Parish, C.L.; Finkelstein, D.I.; Furness, J.B.; Diwakarla, S. The association of enteric neuropathy with gut phenotypes in acute and progressive models of Parkinson’s disease. Sci. Rep. 2021, 11, 7934. [CrossRef]
- Lama, J.; Buhidma, Y.; Fletcher, E.J.R.; Duty, S. Animal models of Parkinson’s disease: A guide to selecting the optimal model for your research. Neuronal Signal. 2021, 5, NS20210026. [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [CrossRef]
- Zhang, X.; Li, Y.; Liu, C.; Fan, R.; Wang, P.; Zheng, L.; Hong, F.; Feng, X.; Zhang, Y.; Li, L.; Zhu, J. Alteration of enteric monoamines with monoamine receptors and colonic dysmotility in 6-hydroxydopamine-induced Parkinson’s disease rats. Transl. Res. 2015, 166, 152–162. [CrossRef]
- Anderson, G.; Noorian, A.R.; Taylor, G.; Anitha, M.; Bernhard, D.; Srinivasan, S.; Greene, J.G. Loss of enteric dopaminergic neurons and associated changes in colon motility in an MPTP mouse model of Parkinson’s disease. Exp. Neurol. 2007, 207, 4–12. [CrossRef]
- Chaumette, T.; Lebouvier, T.; Aubert, P.; Lardeux, B.; Qin, C.; Li, Q.; Accary, D.; Bézard, E.; Bruley des Varannes, S.; Derkinderen, P.; Neunlist, M. Neurochemical plasticity in the enteric nervous system of a primate animal model of experimental Parkinsonism. Neurogastroenterol. Motil. 2009, 21, 215–222. [CrossRef]
- Zhu, H.C.; Zhao, J.; Luo, C.Y.; Li, Q.Q. Gastrointestinal dysfunction in a Parkinson’s disease rat model and the changes of dopaminergic, nitric oxidergic, and cholinergic neurotransmitters in myenteric plexus. J. Mol. Neurosci. 2012, 47, 15–25. [CrossRef]
- Tian, Y.M.; Chen, X.; Luo, D.Z.; Zhang, X.H.; Xue, H.; Zheng, L.F.; Yang, N.; Wang, X.M.; Zhu, J.X. Alteration of dopaminergic markers in gastrointestinal tract of different rodent models of Parkinson’s disease. Neuroscience 2008, 153, 634–644. [CrossRef]
- Singaram, C.; Ashraf, W.; Gaumnitz, E.A.; Torbey, C.; Sengupta, A.; Pfeiffer, R.; Quigley, E.M. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet 1995, 346, 861–864. [CrossRef]
- Li, Z.S.; Schmauss, C.; Cuenca, A.; Ratcliffe, E.; Gershon, M.D. Physiological modulation of intestinal motility by enteric dopaminergic neurons and the D2 receptor: Analysis of dopamine receptor expression, location, development, and function in wild-type and knock-out mice. J. Neurosci. 2006, 26, 2798–2807. [CrossRef]
- Walker, J.K.; Gainetdinov, R.R.; Mangel, A.W.; Caron, M.G.; Shetzline, M.A. Mice lacking the dopamine transporter display altered regulation of distal colonic motility. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G311-8. [CrossRef]
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494. [CrossRef]
- Jackson-Lewis, V.; Jakowec, M.; Burke, R.E.; Przedborski, S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 1995, 4, 257–269. [CrossRef]
- Heikkila, R.E.; Hess, A.; Duvoisin, R.C. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science 1984, 224, 1451–1453. [CrossRef]
- Li, Z.S.; Pham, T.D.; Tamir, H.; Chen, J.J.; Gershon, M.D. Enteric dopaminergic neurons: Definition, developmental lineage, and effects of extrinsic denervation. J. Neurosci. 2004, 24, 1330–1339. [CrossRef]
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Ikuta, F. Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990, 79, 581–583. [CrossRef]
- Colucci, M.; Cervio, M.; Faniglione, M.; De Angelis, S.; Pajoro, M.; Levandis, G.; Tassorelli, C.; Blandini, F.; Feletti, F.; De Giorgio, R.; Dellabianca, A.; Tonini, S.; Tonini, M. Intestinal dysmotility and enteric neurochemical changes in a Parkinson’s disease rat model. Auton. Neurosci. 2012, 169, 77–86. [CrossRef]
- Zheng, L.F.; Song, J.; Fan, R.F.; Chen, C.L.; Ren, Q.Z.; Zhang, X.L.; Feng, X.Y.; Zhang, Y.; Li, L.S.; Zhu, J.X. The role of the vagal pathway and gastric dopamine in the gastroparesis of rats after a 6-hydroxydopamine microinjection in the substantia nigra. Acta Physiol (Oxf) 2014, 211, 434–446. [CrossRef]
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl. Neurodegener. 2019, 8, 5. [CrossRef]
- Qualman, S.J.; Haupt, H.M.; Yang, P.; Hamilton, S.R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Gastroenterology 1984, 87, 848–856. [CrossRef]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [CrossRef]
- Kuo, Y.-M.; Li, Z.; Jiao, Y.; Gaborit, N.; Pani, A.K.; Orrison, B.M.; Bruneau, B.G.; Giasson, B.I.; Smeyne, R.J.; Gershon, M.D.; Nussbaum, R.L. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 2010, 19, 1633–1650. [CrossRef]
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.-W.; Deller, T.; Braak, H.; Auburger, G.; Nussbaum, R.L. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell. Neurosci. 2003, 24, 419–429. [CrossRef]
- Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Parkinsonism Relat. Disord. 2011, 17, 10–15. [CrossRef]
- Noorian, A.R.; Rha, J.; Annerino, D.M.; Bernhard, D.; Taylor, G.M.; Greene, J.G. Alpha-synuclein transgenic mice display age-related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol. Dis. 2012, 48, 9–19. [CrossRef]
- Vance, J.M.; Ali, S.; Bradley, W.G.; Singer, C.; Di Monte, D.A. Gene-environment interactions in Parkinson’s disease and other forms of parkinsonism. Neurotoxicology 2010, 31, 598–602. [CrossRef]
- Wang, L.; Fleming, S.M.; Chesselet, M.-F.; Taché, Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. Neuroreport 2008, 19, 873–876. [CrossRef]
- Lam, H.A.; Wu, N.; Cely, I.; Kelly, R.L.; Hean, S.; Richter, F.; Magen, I.; Cepeda, C.; Ackerson, L.C.; Walwyn, W.; Masliah, E.; Chesselet, M.-F.; Levine, M.S.; Maidment, N.T. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein. J. Neurosci. Res. 2011, 89, 1091–1102. [CrossRef]
- Chesselet, M.-F.; Richter, F. Modelling of Parkinson’s disease in mice. Lancet Neurol. 2011, 10, 1108–1118. [CrossRef]
- Schaffernicht, G.; Shang, Q.; Stievenard, A.; Bötzel, K.; Dening, Y.; Kempe, R.; Toussaint, M.; Gündel, D.; Kranz, M.; Reichmann, H.; Vanbesien-Mailliot, C.; Brust, P.; Dieterich, M.; Funk, R.H.W.; Ravens, U.; Pan-Montojo, F. Pathophysiological Changes in the Enteric Nervous System of Rotenone-Exposed Mice as Early Radiological Markers for Parkinson’s Disease. Front. Neurol. 2021, 12, 642604. [CrossRef]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.-M.; Rodrigo-Angulo, M.; Gille, G.; Funk, R.H.W.; Reichmann, H. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [CrossRef]
- Pan-Montojo, F.J.; Funk, R.H.W. Oral administration of rotenone using a gavage and image analysis of alpha-synuclein inclusions in the enteric nervous system. J. Vis. Exp. 2010. [CrossRef]
- Arnhold, M.; Dening, Y.; Chopin, M.; Arévalo, E.; Schwarz, M.; Reichmann, H.; Gille, G.; Funk, R.H.W.; Pan-Montojo, F. Changes in the sympathetic innervation of the gut in rotenone treated mice as possible early biomarker for Parkinson’s disease. Clin. Auton. Res. 2016, 26, 211–222. [CrossRef]
- Sharrad, D.F.; Chen, B.N.; Gai, W.P.; Vaikath, N.; El-Agnaf, O.M.; Brookes, S.J.H. Rotenone and elevated extracellular potassium concentration induce cell-specific fibrillation of α-synuclein in axons of cholinergic enteric neurons in the guinea-pig ileum. Neurogastroenterol. Motil. 2017, 29. [CrossRef]
- Paillusson, S.; Tasselli, M.; Lebouvier, T.; Mahé, M.M.; Chevalier, J.; Biraud, M.; Cario-Toumaniantz, C.; Neunlist, M.; Derkinderen, P. α-Synuclein expression is induced by depolarization and cyclic AMP in enteric neurons. J. Neurochem. 2010, 115, 694–706. [CrossRef]
- Camilleri, M.; Cowen, T.; Koch, T.R. Enteric neurodegeneration in ageing. Neurogastroenterol. Motil. 2008, 20, 185–196. [CrossRef]
- Phillips, R.J.; Powley, T.L. Innervation of the gastrointestinal tract: Patterns of aging. Auton. Neurosci. 2007, 136, 1–19. [CrossRef]
- Phillips, R.J.; Walter, G.C.; Ringer, B.E.; Higgs, K.M.; Powley, T.L. Alpha-synuclein immunopositive aggregates in the myenteric plexus of the aging Fischer 344 rat. Exp. Neurol. 2009, 220, 109–119. [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [CrossRef]
- Wolters, E.C.; Braak, H. Parkinson’s disease: Premotor clinico-pathological correlations. J. Neural Transm. Suppl. 2006, 309–319. [CrossRef]
- Krogh, K.; Christensen, P. Neurogenic colorectal and pelvic floor dysfunction. Best Pract. Res. Clin. Gastroenterol. 2009, 23, 531–543. [CrossRef]
- Marrinan, S.; Emmanuel, A.V.; Burn, D.J. Delayed gastric emptying in Parkinson’s disease. Mov. Disord. 2014, 29, 23–32. [CrossRef]
- Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2003, 2, 107–116. [CrossRef]
- Lebouvier, T.; Neunlist, M.; Bruley des Varannes, S.; Coron, E.; Drouard, A.; N’Guyen, J.-M.; Chaumette, T.; Tasselli, M.; Paillusson, S.; Flamand, M.; Galmiche, J.-P.; Damier, P.; Derkinderen, P. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE 2010, 5, e12728. [CrossRef]
- Taguchi, T.; Ikuno, M.; Yamakado, H.; Takahashi, R. Animal model for prodromal parkinson’s disease. Int. J. Mol. Sci. 2020, 21. [CrossRef]
- Liepelt-Scarfone, I.; Ophey, A.; Kalbe, E. Cognition in prodromal Parkinson’s disease. Prog. Brain Res. 2022, 269, 93–111. [CrossRef]
- Solla, P.; Wang, Q.; Frau, C.; Floris, V.; Loy, F.; Sechi, L.A.; Masala, C. Olfactory impairment is the main predictor of higher scores at REM sleep behavior disorder (RBD) screening questionnaire in parkinson’s disease patients. Brain Sci. 2023, 13, 599. [CrossRef]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [CrossRef]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J Parkinsons Dis 2017, 7, S71–S85. [CrossRef]
- Yilmaz, R.; Hopfner, F.; van Eimeren, T.; Berg, D. Biomarkers of Parkinson’s disease: 20 years later. J. Neural Transm. 2019, 126, 803–813. [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [CrossRef]
- Kelly, M.J.; Breathnach, C.; Tracey, K.J.; Donnelly, S.C. Manipulation of the inflammatory reflex as a therapeutic strategy. Cell Rep. Med. 2022, 3, 100696. [CrossRef]
- Parkkinen, L.; Pirttilä, T.; Alafuzoff, I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008, 115, 399–407. [CrossRef]
- Frigerio, R.; Fujishiro, H.; Ahn, T.-B.; Josephs, K.A.; Maraganore, D.M.; DelleDonne, A.; Parisi, J.E.; Klos, K.J.; Boeve, B.F.; Dickson, D.W.; Ahlskog, J.E. Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol. Aging 2011, 32, 857–863. [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [CrossRef]
- Macefield, V.G.; Henderson, L.A. Identification of the human sympathetic connectome involved in blood pressure regulation. Neuroimage 2019, 202, 116119. [CrossRef]
- Socała, K.; Doboszewska, U.; Szopa, A.; Serefko, A.; Włodarczyk, M.; Zielińska, A.; Poleszak, E.; Fichna, J.; Wlaź, P. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharmacol. Res. 2021, 172, 105840. [CrossRef]
- Dash, S.; Syed, Y.A.; Khan, M.R. Understanding the role of the gut microbiome in brain development and its association with neurodevelopmental psychiatric disorders. Front. Cell Dev. Biol. 2022, 10, 880544. [CrossRef]
- Baj, A.; Moro, E.; Bistoletti, M.; Orlandi, V.; Crema, F.; Giaroni, C. Glutamatergic Signaling Along The Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2019, 20. [CrossRef]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [CrossRef]
- Caspani, G.; Kennedy, S.; Foster, J.A.; Swann, J. Gut microbial metabolites in depression: Understanding the biochemical mechanisms. Microb. Cell 2019, 6, 454–481. [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; Shen, C.; Lee, H.; Kulkarni, S.; Pasricha, P.J.; Lee, G.; Pomper, M.G.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627-641.e7. [CrossRef]
- Wallen, Z.D.; Demirkan, A.; Twa, G.; Cohen, G.; Dean, M.N.; Standaert, D.G.; Sampson, T.R.; Payami, H. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 2022, 13, 6958. [CrossRef]
- Zhu, M.; Liu, X.; Ye, Y.; Yan, X.; Cheng, Y.; Zhao, L.; Chen, F.; Ling, Z. Gut microbiota: A novel therapeutic target for parkinson’s disease. Front. Immunol. 2022, 13, 937555. [CrossRef]
- Chen, S.-J.; Lin, C.-H. Gut microenvironmental changes as a potential trigger in Parkinson’s disease through the gut-brain axis. J. Biomed. Sci. 2022, 29, 54. [CrossRef]
- Zeng, J.; Wang, X.; Pan, F.; Mao, Z. The relationship between Parkinson’s disease and gastrointestinal diseases. Front. Aging Neurosci. 2022, 14, 955919. [CrossRef]
- Misera, A.; Łoniewski, I.; Palma, J.; Kulaszyńska, M.; Czarnecka, W.; Kaczmarczyk, M.; Liśkiewicz, P.; Samochowiec, J.; Skonieczna-Żydecka, K. Clinical significance of microbiota changes under the influence of psychotropic drugs. An updated narrative review. Front. Microbiol. 2023, 14, 1125022. [CrossRef]
- Horsager, J.; Knudsen, K.; Sommerauer, M. Clinical and imaging evidence of brain-first and body-first Parkinson’s disease. Neurobiol. Dis. 2022, 164, 105626. [CrossRef]
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases. J. Inflamm. Res. 2020, 13, 749–764. [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [CrossRef]
- Pellegrini, C.; D’Antongiovanni, V.; Miraglia, F.; Rota, L.; Benvenuti, L.; Di Salvo, C.; Testa, G.; Capsoni, S.; Carta, G.; Antonioli, L.; Cattaneo, A.; Blandizzi, C.; Colla, E.; Fornai, M. Enteric α-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. npj Parkinsons Disease 2022, 8, 9. [CrossRef]
- Muenter, M.D.; Tyce, G.M. L-dopa therapy of Parkinson’s disease: Plasma L-dopa concentration, therapeutic response, and side effects. Mayo Clin. Proc. 1971, 46, 231–239.
- Poewe, W.; Antonini, A. Novel formulations and modes of delivery of levodopa. Mov. Disord. 2015, 30, 114–120. [CrossRef]
- Amara, A.W.; Memon, A.A. Effects of Exercise on Non-motor Symptoms in Parkinson’s Disease. Clin. Ther. 2018, 40, 8–15. [CrossRef]
- Ouchi, Y.; Kanno, T.; Okada, H.; Yoshikawa, E.; Futatsubashi, M.; Nobezawa, S.; Torizuka, T.; Tanaka, K. Changes in dopamine availability in the nigrostriatal and mesocortical dopaminergic systems by gait in Parkinson’s disease. Brain 2001, 124, 784–792. [CrossRef]
- Bhidayasiri, R.; Phuenpathom, W.; Tan, A.H.; Leta, V.; Phumphid, S.; Chaudhuri, K.R.; Pal, P.K. Management of dysphagia and gastroparesis in Parkinson’s disease in real-world clinical practice - Balancing pharmacological and non-pharmacological approaches. Front. Aging Neurosci. 2022, 14, 979826. [CrossRef]
- Mukherjee, A.; Biswas, A.; Das, S.K. Gut dysfunction in Parkinson’s disease. World J. Gastroenterol. 2016, 22, 5742–5752. [CrossRef]
Figure 1.
Overview of the anatomy and organization of the ENS.
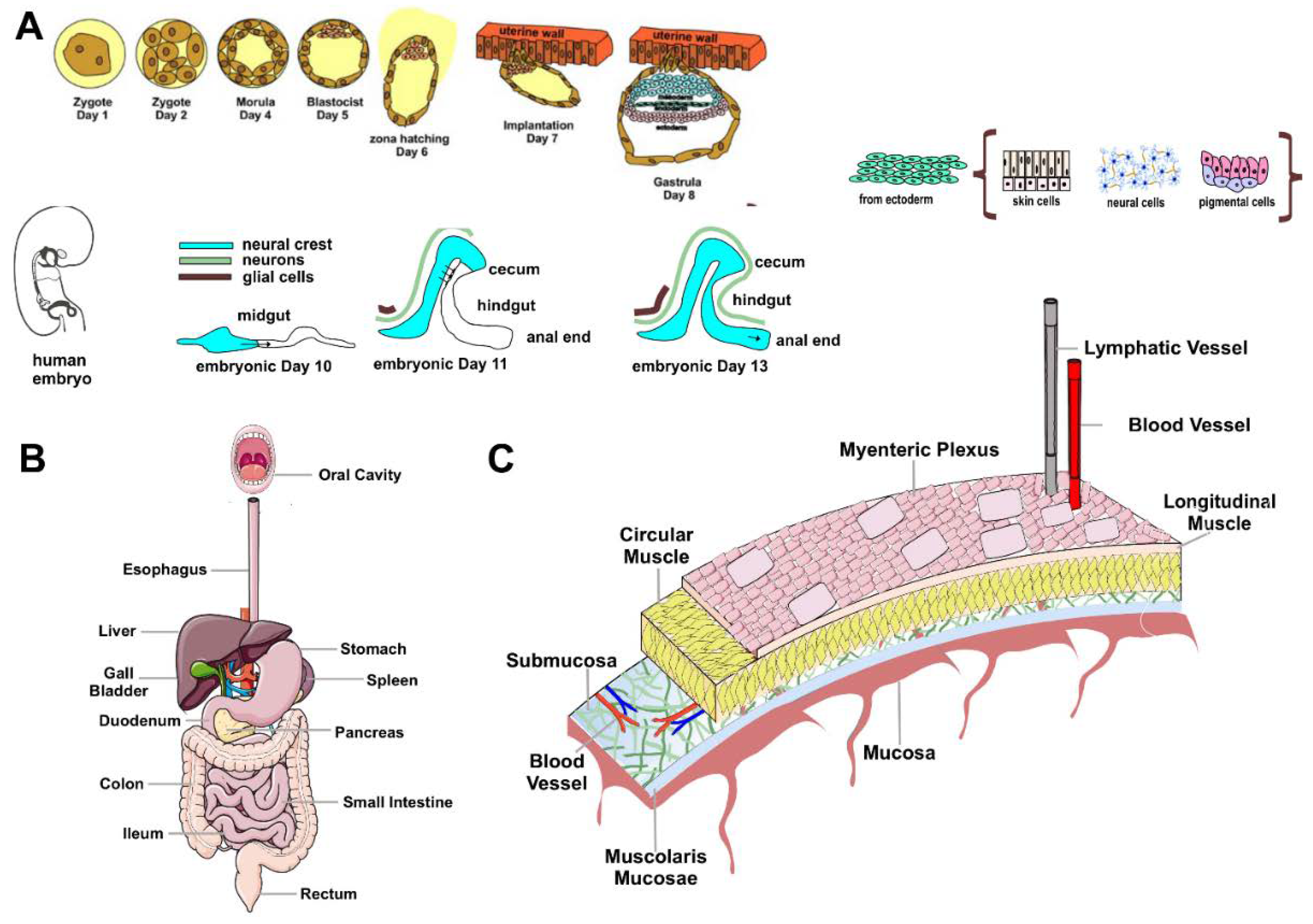
Figure 2.
Schematic representation of the main physiological and behavioral changes in CNS and ENS of preclinical models of PD.
Figure 2.
Schematic representation of the main physiological and behavioral changes in CNS and ENS of preclinical models of PD.
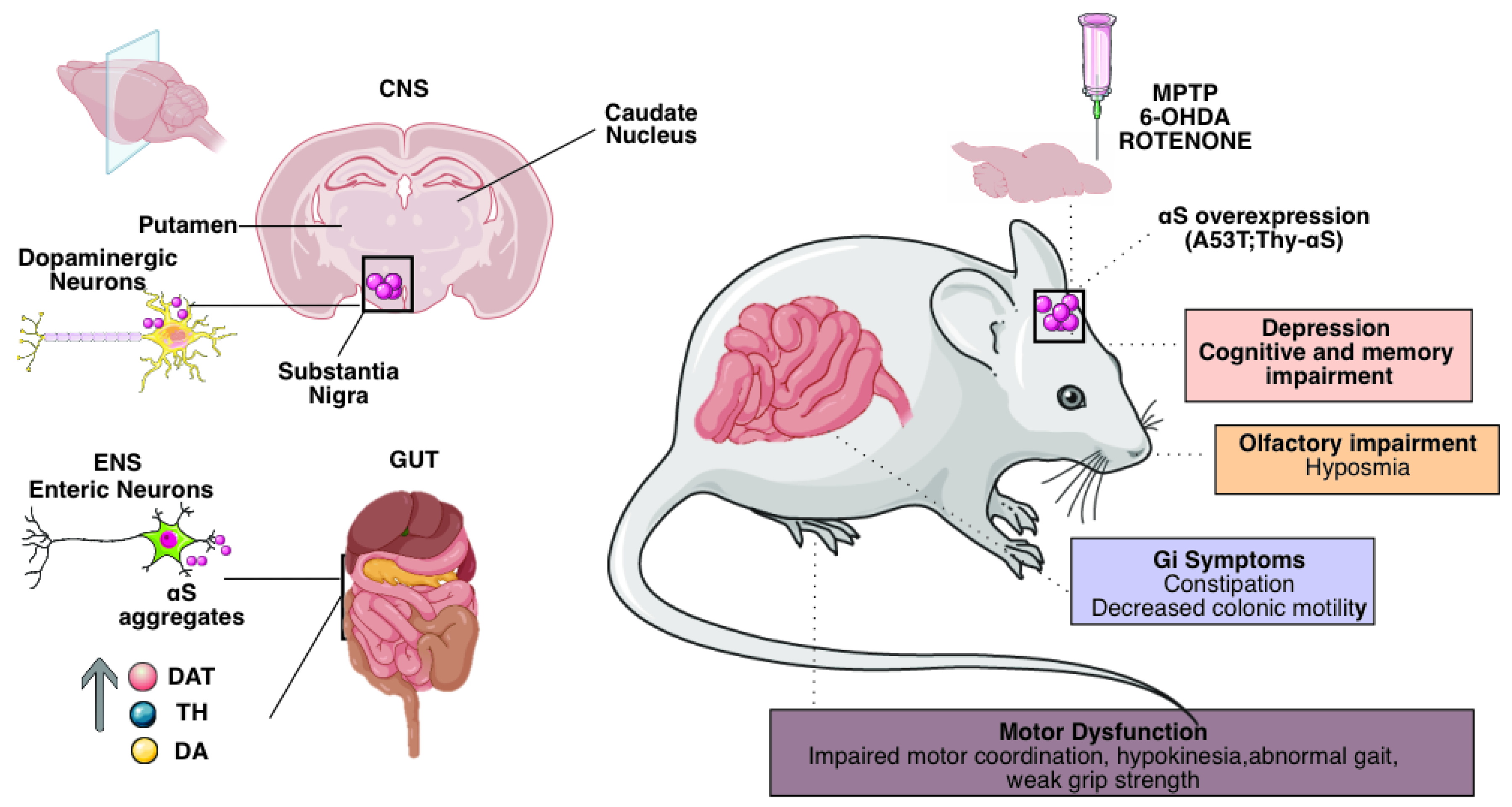
Figure 3.
Non-motor features of Parkinson's disease, focus on gastrointestinal symptoms.
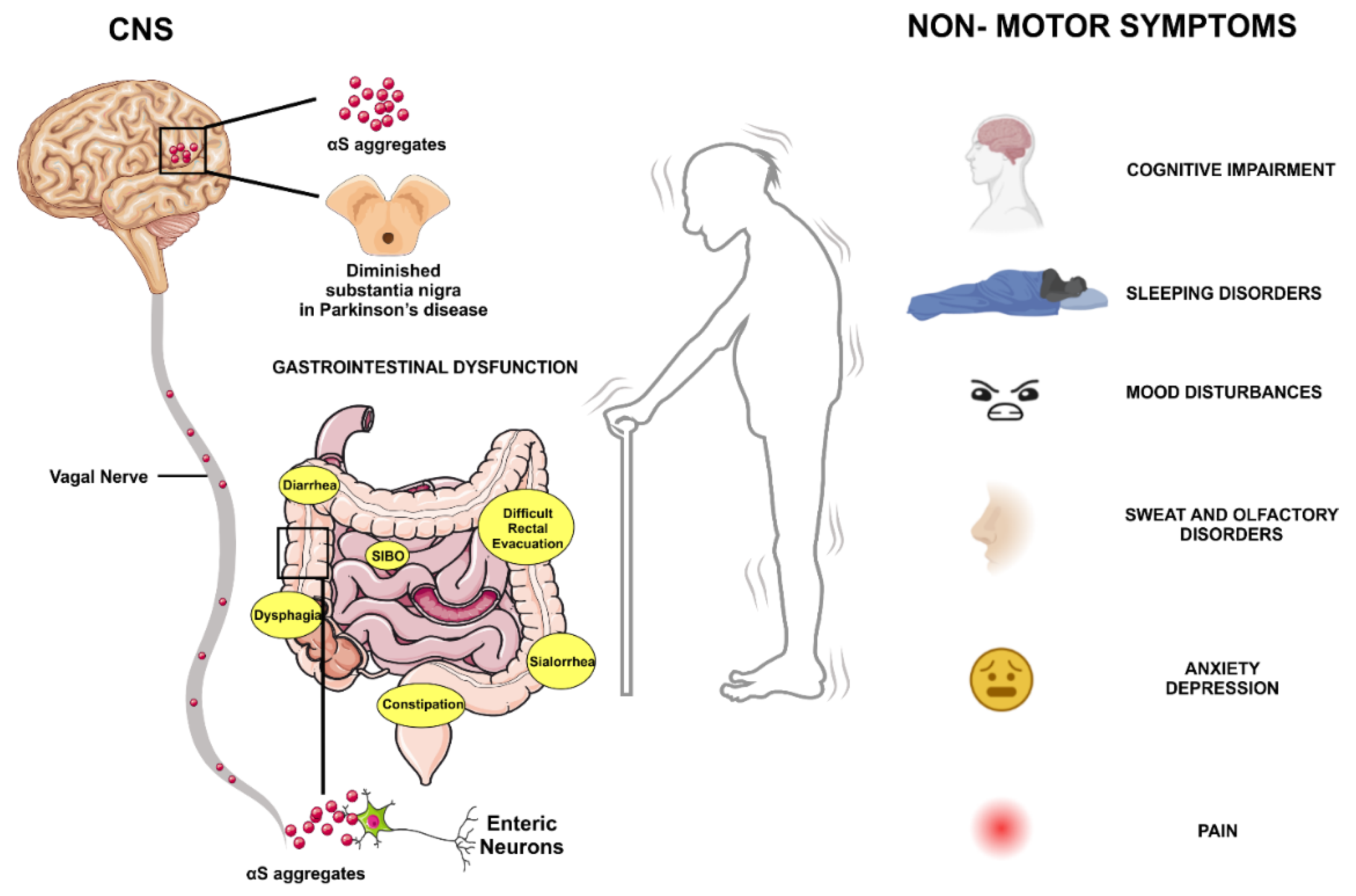
Table 1.
Pathological features identified in animal models of PD. The table summarizes the major alterations found in murine models of PD. The legend of the abbreviations is listed below.
Table 1.
Pathological features identified in animal models of PD. The table summarizes the major alterations found in murine models of PD. The legend of the abbreviations is listed below.
| PD Model | Affected Neuron types | GI Symptoms | Alteration Biomarker | References |
|---|---|---|---|---|
|
MPTP mice |
Loss of dopaminergic neurons in the myenteric plexus | Absence of severe defects in gastrointestinal motility. increased contraction and decreased relaxation of colon muscle in response to electric field stimulation of NEs |
Nd |
[36,37] |
|
MPTP rats (Peripheral administration) |
Unaltered number of dopaminergic neurons in the SNpc Presence of TH-IR neurons in the GI tract |
Nd |
Unaltered expression of dopaminergic markers in the SNpc |
[39] [40] |
|
6-OHDA rats |
Alterations in the monoaminergic and cholinergic system | Delayed gastric emptying and constipation, which could be related to increased gastrointestinal TH and decreased NOS. Increased DA concentration in the colon, which is more likely to cause constipation. Decreased colonic motility. |
Unaltered cholinergic transmitters. Elevated protein levels of TH and DAT both in the epithelium and neurons of the gastrointestinal tract, resulting in increased DA content in the gut and delayed gastric emptying |
[35] [38,39] [47,48,49] |
|
A53T mice (Expressing a mutant form of human αS) |
Disruption of efferent vagal processes that project from the DMV to the gastrointestinal tract | Related slowing of gastrointestinal motility caused by expression of human αS in the DMV | Accumulation of αS aggregates in the ENS before changes in the CNS. | [53] [8,55] [56] |
|
Thy1-αS mice |
Nd |
Striatal dopamine loss only after 14 months: manifesting motor and non-motor deficits, such as olfactory disturbances, as early as 2-3 months of age | Increased transit time and colonic content. Overexpression of αS in the colonic myenteric nervous system. Reduced response to defecation stimuli. |
[57] [58] [59,60] |
|
Rotenone mice model |
Reduced sympathetic noradrenergic and vagal cholinergic gut innervation | Progressive αS deposition both in ENS and CNS neurons affected by PD such as neurons in the myenteric plexus, the DMV, the spinal cord, and the SNS |
Nd |
[61] [64] [65] [66] |
|
Fischer 344 rat |
Neuronal loss and changes in neurochemical phenotype in the ENS | Dystrophic enteric neurons that contain αS aggregates reminiscent of Lewy pathology | Motility disorders | [70] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



