
Submitted:
30 April 2023
Posted:
01 May 2023
You are already at the latest version
Abstract
The mycotoxin alternariol (AOH) can be found in the food products infected by Alternaria spp. and is considered an endocrine-disruptive mycotoxin. The main mechanism of AOH toxicity is associated with DNA damage and modulation of the inflammation process. Nevertheless, AOH is considered as one of the emerging mycotoxins. In this study we evaluated how AOH might affect a local steroidogenesis process in prostate, in both normal as well as cancer cells. We observed that AOH itself modulates the cell cycle, inflammation and apoptosis rather than steroidogenesis process in prostate cancer cells; however, in the presence of the other steroidogenic agent, the effect is significant. Therefore, this is the first study to report the effect of AOH on local steroidogenesis in normal and prostate cancer cells. We postulate that AOH might modulate the release of the steroid hormones and expression of the key components by interfering with the steroidogenic pathway and might be considered a steroidogenesis-altering agent.
Keywords:
alternariol
; steroidogenesis
; mycotoxin
; carcinogenesis
; dehydroepiandrosterone
1. Introduction
Mycotoxins are secondary metabolites of Aspergillus, Fusarium, and Penicillium fungi [1]. Food and Agriculture Organization (FAO) estimates, that up to 25% of global food crop is contaminated with mycotoxins [2]. These chemicals contaminate food commodities world-wide, posing a number of significant food safety concerns.
Alternariol (AOH) is one of mycotoxins produced by Alternaria species and often is found in vegetables as well as fruit and processed fruit products (i.e. juices, wine) [3]. The presence of Alternaria mycotoxins has been widely reported, however they still serve as emerging mycotoxins for which a detailed molecular mechanism has not been determined [4]. This fact raises public concern about the relevance of the cytotoxic and genotoxic potential of Alternaria mycotoxins, as well as the need for determination of their health consequences in detail [5].
AOH has been considered as endocrine disruptor chemical (EDC) due to reported estrogen and androgen receptor binding in cells. The acute toxicity of AOH is considered to be low, however little is known about the biological effects of AOH on reproduction and the detailed cellular molecular mechanism still needs to be established [6]. AOH results in an increased expression of progesterone receptor (PR) and modulation of the production of estradiol (E2) and progesterone in H295R cell line [7]. The estrogenicity of AOH is considered weak and partial, moreover AOH is reported to be an androgen receptor (AR) agonist. It was suggested that AOH might modulate the endocrine system through interference of nuclear receptor signalling or upregulation of the expression of steroidogenic receptors [8]. Most importantly, it was also suggested that AOH might mimic cholesterol in cells and thus alter the hormonal balance [9].
Prostate cancer (PCa) is considered as the second leading cause of male cancer death [10]. Increasing age with the average age at the time of diagnosis being 66 years correlate with the incidence and mortality of prostate cancer worldwide [11]. The androgen deprivation therapy is the keystone treatment for men with advanced metastatic PCa as the prostate gland growth and development is dependent on androgens [12]. Majority of death from prostate cancer are the result of castration resistant prostate cancer (CRPC). The potent androgens were confirmed in subsequent studies in patients after the surgical castration [13]. Comparing to pre-treatment level, the residual concentrations of intratumoral dihydrotestosterone (DHT) can reach from 10% to 40%, while reaching the castration concentration of testosterone with tumor response in 80-90% of patients [12].
Steroidogenesis is the multistep process of steroid hormones biosynthesis from cholesterol. The process begins with the transfer of cholesterol from the intracellular store to the inner mitochondrial membrane which is transformed to pregnenolone and into downstream steroids. As the prostate gland tissue is not capable of the de novo steroid synthesis, the intracellular steroid hormone level depends on the blood supply and local biosynthesis [14]. The research showed that tumor growth after androgen deprivation therapy (ADT) is still androgen sensitive and dependent and the resemble of the tumor is caused by local steroidogenesis in the prostate tumor cells [15]. The crucial role of cholesterol in prostate carcinogenesis was also suggested, and it might be confirmed by number of studies addressing usefulness of statins in prostate cancer therapies [16]. An increased level of steroidogenenic enzymes that synthesize androgens from cholesterol or other circulating steroid precursors, such as progesterone or dehydroepiandrosterone (DHEA) was reported in PCa [17]. DHEA is an active androgen precursor (testosterone and dihydrotestosterone). It was reported that DHEA is weakly active as androgen receptor ligand, it is largely inactive as androgen. DHEA might be associated with prostate cancer carcinogenesis although the role of DHEA in prostate cancer progression is unclear [18].
In our previous research, exposure mycotoxin deoxynivalenol (potentially involved in local steroidogenesis modulation) alone or in combination with DHEA had a stimulatory effect on the release of steroid hormones and expression of genes related to steroidogenesis [19]. Therefore, it is highly possible that EDC which modulates the process of steroidogenesis might also modulate the local steroidogenesis in prostate cells and participate in both initiation as well as progression of the tumor.
In this study, we evaluated the ability of AOH to alter local steroidogenesis in prostate cancer and normal cells. In presented experiments, we decided to use androgen independent human prostate cancer cell line PC3, bone-derived metastasis and non-cancer epithelial PNT1A, that has been proved to be a good model for cellular processes analysis (i.e. epithelium proliferation in response to androgens and growth factors).
Thus, the objective of this study was to determine the effect of AOH alone, as well as AOH in combination with the known steroidogenic agent DHEA, on the production of estradiol, testosterone and progesterone in pro state normal epithelial cells as well as adenocarcinoma cells with different reported sensitivities to androgens.
2. Results
2.1. AOH modulates the viability of prostate normal and cancer cell lines in a dose-and time- dependent manner
First, the cytotoxic effect of AOH was verified in a dose- and time- dependent manner. The dose range tested was similar to our previous study [20], however a different cell culture medium modified the response of normal prostate cells to AOH, thus the experiments were conducted, and data is hereby shown once again. It was observed that AOH significantly affected the viability of both normal as well as cancer cells in doses higher than 30 µM upon 24 and 48 h incubation (Figure 1a,b) (p<0.001). Interestingly, we observed that for normal prostate cells, longer incubation time decreased the cytotoxic effect of AOH (Figure 1a), what was not observed for PC3 cells. Based on these results and our previously published data [20] for the rest of experiments two doses of AOH were chosen: 10 µM and 0.1 µM and one exposition time: 48 h.
2.2. AOH modulates the local steroidogenesis in prostate normal and cancer cells in the presence of DHEA
To assess the effect of AOH on local steroidogenesis, cells were treated with both AOH and DHEA and the production of testosterone, estradiol and progesterone was assessed (Figure 2). As expected, the highest changes were found for testosterone production. Furthermore, we observed that in the case of cancer cells, AOH has a higher impact on the chosen hormone production. In both normal as well as cancer cell line, AOH itself did not change the production of testosterone (Figure 2a,b), although in the presence of DHEA which itself significantly increased the production of testosterone (***p<0.001) as compared to control cells, AOH modulated the production of testosterone. In PC3 cells, the statistically significant increase in the production of testosterone was observed for 10 µM AOH + DHEA as compared to control (***p<0.001), 10 µM AOH (***p<0.001), DHEA (***p<0.001) and 0.1 µM AOH + DHEA (***p<0.001). 0.1 µM AOH + DHEA also significantly increased the production of testosterone as compared to control (***p<0.001), 0.1 µM AOH (***p<0.001) but the effect was lower than the one caused by DHEA itself. In the normal prostate cell line, both doses of AOH and DHEA caused a significant increase in testosterone production compared to control (***p<0.001) and treatments with AOH treatments alone (***p<0.001). No significant changes were observed between the treatments with AOH + DHEA and DHEA, indicating that in case of PNT1A cells, the observed effect of AOH + DHEA is primarily dependent on DHEA. Next, we evaluated the production of estradiol and progesterone. We did not observe significant changes in estradiol production in both cancer and normal cells (Figure 2c,d). Only the slight decrease in the production of estradiol was observed in PNT1A cells for 0.1 µM AOH + DHEA treatment, but it was not statistically significant. The last evaluated hormone was progesterone. In PC3 cell line we observed that treatment with AOH insignificantly decreased the production of progesterone as compared to control cells, whereas DHEA itself insignificantly increased (Figure 2e). 10 µM AOH + DHEA significantly decreased the production of progesterone as compared to control (*p<0.05), DHEA alone (***p<0.001) as well as 0.1 µM + DHEA (*p<0.05). Similarly, in normal PNT1A cells, AOH also did not modulate progesterone production significantly as compared to control cells (Figure 2f). A statistically significant decrease was observed for 10 µM AOH compared to 10 µM AOH + DHEA (**p<0.01) and DHEA treatment (*p<0.05).
Next, we evaluated the expression of the mediators of the initial and rate-limiting step in steroidogenesis - cytochrome P450 family 11 subfamily A member 1 (CYP11A1), steroidogenic acute regulatory protein (StAR), cytochrome P450 family 17 subfamily A member 1 (CYP17A1), hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2 (HSD3B2) and androgen receptor (AR) and estrogen receptor beta (ESR2) (Figure 3).
Firstly, it was observed that AOH modulates the expression of CYP11A1 in PNT1a cells: 10 µM of AOH decreased its expression, whereas 0.1 µM AOH increased it and the effect was significantly different as compared between these groups (*p<0.05). The observed contradictory effect was potentiated by addition of DHEA: treatment of cells with 10 µM AOH + DHEA significantly increased the expression of CYP11A1 as compared to 10 µM AOH alone (**p<0.01) as well as 0.1 µM AOH + DHEA (**p<0.01). In case of PC3 cells, a similar, although not statistically significant, effect was observed. The higher tested dose of AOH decreased the expression of CYP11A1 and addition of DHEA increased that effect. For a lower dose of AOH no such effect was observed. In PNT1A cells AOH insignificantly increased the expression of StAR, whereas a significant increase was observed for 10 µM AOH + DHEA as well as 0.1 µM AOH + DHEA as compared to DHEA treatment alone (p<0.05 and p<0.01, respectively). The highest increase was observed for 0.1 µM AOH + DHEA, which was significantly different from control (*p<0.05). In PC3 cells DHEA significantly reduced the expression of StAR as compared to control (***p< 0.001). The higher tested dose of AOH slightly decreased, whereas lower dose increased, the expression of StAR as compared to non-treated cells. A different effect was observed for treatment with 10 µM AOH + DHEA which significantly increased the expression of StAR as compared to AOH treatment alone, as well as DHEA (***p< 0.001 and *p<0.05, respectively). A contradictory and significant effect was observed for 0.1 µM AOH + DHEA (***p< 0.001 and **p<0.01, respectively). In case of CYP17A1 expression evaluated in PNT1A cells, the expression in all treatments was decreased as compared to control cells, although significant only for separated treatments of AOH (*p<0.05). A contradictory, yet not significant effect was observed in PC3 cells, where treatment with DHEA as well as AOH + DHEA resulted in the increased expression of CYP17A1. The expression of HSD3B2 was modulated by AOH. In PNT1A cells increased expression for higher tested dose of AOH was observed as compared to control and DHEA alone, however not statistically significant. In PC3 cells a contradictory effect was observed- all tested dosed decreased the expression of HSD3B2, whereas the statistical significance was observed for 0.1 µM AOH, DHEA and 0.1 µM AOH + DHEA as compared to control (*p<0.05). In the last analysis, we evaluated the expression of AR and ESR2 genes. In PNT1A a statistically significant increase in the expression of AR was observed after treatment with 10 µM AOH as compared to control as well as DHEA (***p<0.001). Addition of DHEA to 10 µM AOH resulted in significant decrease in the expression of AR as compared to 10 µM AOH and control (*p<0.05). A contradictory effect was observed for lower tested dose of AOH, which itself only slightly increased the expression of AR whereas simultaneous treatment with DHEA resulted in a significant increase in the expression of AR as compared to 0.1 µM AOH (p<0.001), DHEA (***p<0.001) as well as control (***p<0.001). In cancer cells, AOH at a dose of 10 µM of AOH caused highest increase in the expression of AR as compared to control and DHEA treatment (***p<0.001). Addition of DHEA to 10 µM of AOH resulted in a decrease in the expression of tested gene, but still presented a significantly higher expression as compared to control (***p<0.001) and DHEA (***p<0.001). A similar effect was observed for lower tested dose of AOH, but in lower extent as the one for 10 µM as compared to 0.1 µM AOH (**p<0.001) and DHEA (***p<0.001). The evaluation of ESR2 gene also showed that AOH in a dose of 10 µM affected the tested gene in both normal as well as cancer cell lines. In PNT1A cells we observed a non-significant increase after treatment with 10 µM of AOH compared to control, whereas treatment with 0.1 µM AOH + DHEA resulted in a significant increase in ESR2 expression of ESR2 as compared to 0.1 µM AOH alone (***p<0.001), DHEA (***p<0.001) as well as control (***p<0.001). In PC3 cells the effect of AOH was also observed both in separated treatments as well as co-treatments with AOH. Both doses of AOH resulted in an increased expression of ESR2 as compared to control (***p<0.001) and DHEA (***p<0.001 as compared to DHEA). The treatment with AOH + DHEA also increased the expression of ESR2, but in a significantly lower extent than AOH alone. Moreover, a similar effect of DHEA in PC3 cell was observed in case of the expression of both tested receptors- DHEA significantly decreased the expression of AR and ESR2 as compared to not treated cells.
2.3. AOH affects the expression of caveolin-1 (CAV-1) in prostate cells
Caveolins (CAVs) are components of caveolae, a part of lipid rafts which bind cholesterol and participate both in the process of steroidogenesis and signal transduction. CAV-1 is involved in many cellular processes such as cell cycle regulation, endocytosis, signal transduction and cholesterol trafficking and efflux [21]. It also has been suggested that CAV-1 may act as a scafffolding protein responsible for organization and concentration of signaling molecules within caveloases [22]. In prostate cancer progression an increased level of CAV-1 was reported in tumor epithelial cells [23]. The reverse effect was observed in stromal cells in advanced metastatic prostate cancer, where the expression of CAV-1 was decreased [24].
CAV-1 allows to bind to cholesterol and regulates the intracellular transport of cholesterol to and from the plasma membrane [25]. The structure of AOH resembles with endogenous molecules like cholesterol [9]. This may provide the key for understanding its complex biological functions including the modulation of the process of steroidogenesis. Thus, during evaluation of the effect of AOH on prostate cells steroidogenesis we also decided to evaluate the possible changes in CAV-1 expression and localization, due to the known role of CAV-1 in prostate cancer [26]. Firstly, we evaluated the changes in the gene expression and observed that in PNT1A cells 10 µM of AOH significantly reduced the expression of CAV-1 as compared to control (**p<0.01) as well as 0.1 µM AOH (*p<0.05) and 10 µM AOH + DHEA (***p<0.001) (Figure 4a). DHEA itself decreased the expression of CAV-1 and similar effect was observed for 0.1 µM AOH + DHEA as compared to 0.1 µM AOH, however the differences were not statistically significant. In case of PC3 cells almost no changes in the expression of CAV-1 were observed, beside the insignificant decrease upon 0.1 µM AOH + DHEA treatment. A similar tendency was observed for protein expression of CAV-1 obtained in Western blot analysis (Figure 4b). For both cell lines the expression of CAV-1 was slightly decreased after AOH treatment and increased after AOH + DHEA treatment. The altered expression might be associated with the observed different localization of AOH in cells (Figure 4c). In PNT1A cells, fluorescent staining of CAV-1 after AOH treatment showed a cell membrane and nuclear localization instead of predominantly cytosolic localization in control cells. In case of PC3 cells, a similar effect was observed for lower AOH treatment rather than the higher dose of AOH.
2.4. AOH modulates cell cycle and apoptosis in prostate cells
Next, we evaluated the process of apoptosis and cell cycle in prostate cells, due to the fact that all of them are associated with steroidogenesis process both in normal as well as cancer cells [27]. Firstly, the induction of apoptosis was evaluated with flow cytometry (Figure 5a,b) and it was revealed that in both normal as well as cancer cell lines, AOH in a dose of 10 µM induced a significant increase in the number of apoptotic cells (***p<0.001) as compared to control. In case of lower tested dose (0.1 µM AOH) no such effect was observed, similarly to DHEA treatment. Simultaneous treatment of AOH + DHEA resulted in a significant increase in the number of apoptotic PC3 cells as compared to AOH treatment alone (**p<0.01) and DHEA (***p<0.001). In case of PNT1A cells a slight, yet not significant decrease in the number of apoptotic cells caused by AOH + DHEA was observed as compared to 10 µM of AOH treatment alone. We also verified the expression of caspase 3 (Casp3) with Western blot (Figure 5c) and observed no significant changes in its expression, indicating that observed changes on flow cytometry are too low to be observed on protein level.
We also observed that AOH differently modulated the cell cycle progression in normal and cancer prostate cells. In normal PNT1A cells, a significant increase in the number of G0/G1 cells was observed for both tested doses of AOH (***p<0.001 and *p<0.05, respectively) compared to not treated cells, DHEA as well as DHEA + AOH increased the number of cells in S cell cycle phase with simultaneous decrease in the number of cells in G2/M cell cycle phase (Figure 6a). The increase in the number of cells in the S phase of cell cycle, as the well as decrease in the G2/M cell cycle phase, was statistically significant for 0.1 µM AOH + DHEA compared to 0.1 µM AOH alone (**p<0.01) and DHEA alone (**p<0.01). In PC3 cells, a different effect was observed: AOH in a dose of 10 µM induced a significant increase in the number of cells in the G2/M cell cycle phase compared to control (**p<0.01). Simultaneous treatment with DHEA resulted in a similar effect. For lower tested dose of AOH, a significant decrease in the number of cells in S cell cycle phase was observed as compared to not treated cells (**p<0.01) with no significant increase in the number of G0/G1 cells (Figure 6b).
Modulation of apoptosis and cell cycle in cells caused by AOH was also verified with RTq-PCR (Figure 7). First of all, we observed that both tested doses of AOH caused a different modulation of the expression of ANXA5, TP53 and CDKN1A and addition of DHEA to AOH treatment resulted in contradictory effects. In PNT1A cells 10 µM AOH caused a significant decrease in the expression of ANXA5 as compared to control cells (***p<0.001), whereas 10 µM AOH + DHEA resulted in no change in the expression as compared to control. A contradictory effect was observed for a lower dose of AOH (Figure 7a). In the case of PC3 cells, modulation of ANXA5 expression was not significant, although a similar trend was observed at the lower extent (Figure 7a). In case of the expression of TP53 in both cell lines a similar effect was observed (Figure 7b). Treatment with 10 µM AOH resulted in decreased expression, whereas the higher dose did not cause any change or increase in expression as compared to control in PC3 cells (**p<0.01). In the case of TP53 we observed that AOH lead to insignificant expression decrease in PNT1A cells, while the addition of DHEA resulted in a significant increase in expression in case of higher dose of AOH (***p<0.001) and significant decrease in the expression in case of lower dose (*p<0.05). DHEA itself caused a significant decrease in the expression of TP53 as compared to control cells (**p<0.01). A similar effect was observed to PC3 cells, but in a higher extent (Figure 7b). Additionally, a similar effect was observed in modulating the expression of CDKN1A, and similar to TP53 expression, a higher modulatory effect was observed in the prostate cancer cell line (Figure 7c). The observed changes in cell cycle progression might be also associated with modulation of the expression of CDK2, especially in case of PNT1A cell in which G0/G1 cell cycle arrest was observed. As suspected, 10 µM of AOH decreased, whereas 0.1 µM increased, CDK2 expression, and the observed effect was significantly different (**p<0.01). Similarly to the other genes expression evaluated, the addition of DHEA to AOH treatment resulted in a significant contradictory effect as observed for AOH alone (***p<0.001). In PC3 cells a similar effect was observed, but not statistically significant (Figure 7d).
3. Discussion
The role of androgens in both physiological as well as pathological processes is already known, mainly due to the fact that testosterone suppression therapy is still the most effective strategy in PCa. Nevertheless, castration-resistant PCa still constitutes a major health problem and suggests that part of the patients re-express the AR [28]. One of the possible explanations might be the fact that prostate might itself produce or support the steroidogenesis process. As observed previously, the presence of the transcript of all important enzymes for testosterone synthesis seems to confirm this hypothesis [29]. Moreover, we hypothesized that environmental pollutants or toxins which might influence the process of steroidogenesis, might also affect the local steroidogenesis process in both normal as well as prostate cancer cells.
AOH is mainly considered as genotoxic due to its inhibitory effect on topoisomerase [30]. It was also reported as a weak estrogenic agent, mostly affecting ERβ [31], as well AR agonist in high concentrations [32]. Although the observed effect of AOH on the steroidogenesis process is low, more interesting is the fact that AOH in the presence of other estrogenic agents might trigger a myriad of different effect [33]. Thus, in this study we firstly evaluated the effect of AOH on local prostate steroidogenesis itself, but also in combination with known androgenic agent (DHEA). We observed that AOH alone did not affect the production of testosterone, estradiol, or progesterone, but combination with DHEA exacerbated its effect in normal as well as prostate cancer cells.
That effect was associated with modulated expression of most important steroidogenic enzymes: CYP11A1, STAR, CYP17A1, HSD3B2, as well AR and ESR2. Our observation is in line with the previous ones suggesting the endocrine modulatory effect of AOH in cells. Kalayou et al. observed that AOH increased the expression of HSD3B and CYP21A2 in the human adenocarcinoma cell line H295R [8]. The same cell line was used by Frizzell et al. to assess the endocrine disrupting effect of AOH and they found a modulation of the expression of CYP11A1, CYP17 [34]. In our study, AOH itself increased the expression of the StAR transcript responsible for cholesterol transport to the inner mitochondrial membrane, HSD3B2, as well AR and ESR2, whereas a contradictory effect was observed for the CYP11A1 transcript responsible for conversion of cholesterol to pregnolone by mitochondrial enzymes and CYP17A1 that participates in testosterone production. One of the possible explanations of modulation of steroidogenesis is the fact that AOH has been postulated to possess a similarity to cholesterol and possibly intercalate the cholesterol-rich membrane domains e.g. caveolae [9]. To elaborate on that, we evaluated the expression of CAV-1 and its cellular localization after treatment with AOH. Our results were different from those observed by Del Favero et al. in THP-1 cells, but a modulation of the expression and localization was observed after AOH treatment [9]. On the other hand, a higher tested dose of AOH in another study (10 µM) resulted in a decrease in the expression of CAV-1 in intestinal cells, which is in line with our results [35].
In previous studies AOH was reported to modulate the process of apoptosis and cell cycle process in cells, i.e. colon carcinoma, murine hepatoma, colorectal adenocarcinoma, Abelson murine leukemia [36,37]. Kalayou et al. observed that AOH modulates the proliferation and cell cycle progression of cells depending on the dose, with arrest in G0/G1 or G2/M cell cycle phase [8]. In this study we observed that AOH modulated the cell cycle progression differently in the different prostate cell lines. Findings of Kalayou might explain a different cellular effects of PNT1A and PC3 cells, also observed in this study. The induction of cell cycle arrest was associated with modulation of the genes responsible for cell cycle progression CDKN1A, CDK2 and TP53. Solhaug at al. suggested previously that modulation of cell cycle by AOH is associated with phosphorylation of histone H2AX, activation of p53 and increased expression of p21, cyclin B1and other cell cycle regulators, although he tested higher doses of AOH than in this study (60 µM) AOH [3].
Nevertheless, in both cell lines the higher tested dose induced apoptosis in cells and DHEA did not abolish that effect. Modulation of cell cycle progression by AOH and apoptosis was also observed in Caco-2 cells in which AOH induced cell cycle arrest in the G2/M cell cycle phase with simultaneous induction of apoptosis and necrosis [37]. The concentration range in that study was higher than in our indicating the switch between apoptosis and necrosis in cells. In HCT116 cell line AOH also induced necrosis/apoptosis with modulation of the expression of caspase 3 expression [38], similar to our observations, but the suggested mechanism was associated with DNA damage and therefore with AOH genotoxicity. Hereby, we report that the induction of apoptosis and cell cycle progression could be different in normal and cancer cell lines, even for the same doses of tested mycotoxins and might be associated with modulation of steroidogenesis, not only genotoxicity.
To our knowledge this is the first study to evaluate the effect of AOH as the well as combinatory effect of AOH and DHEA on a local steroidogenesis process in both normal and cancer prostate cells. We observed that the effect of AOH on the process of steroidogenesis is low, but in combination with other steroidogenic agents it triggers significant changes in both normal and cancer cells. Moreover, this study showed that AOH might also affect apoptosis, cell cycle in cells what in consequence might exacerbate its effect on steroidogenesis in cells.
4. Materials and Methods
4.1. Cell culture and experimental treatments
The normal human prostate epithelial cell line PNT1A and the prostate adenocarcinoma cell lines PC3, were obtained from the European Collection of Authenticated Cell Cultures (ECACC) (Sigma- Aldrich, Saint Louis, MO, USA) and maintained in a humidified incubator (37°C, 5% CO2). The cells were cultured in RMPI medium with 10% of heat inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES and 1% of PenStrep (5000 units/mL of penicillin and 5000 µg/mL of streptomycin). All media and supplements were purchased from Thermo Fisher Scientific Inc., Walthan, MA, USA.
Alternariol (AOH) was obtained from Sigma-Aldrich, Saint Louis and dissolved in dimethyl sulfoxide (DMSO) and considered a stock solution. The dehydroepiandrosterone (DHEA) stock solution (Avanti Polar Lipids, Inc., Alabaster, USA) was prepared in ethanol. The stocks were dissolved in experimental medium each time before use. In the experimental model, cells were treated with 0.1 or 10 µM AOH and/or 100 nM DHEA for 48 h. Non-treated cells were used as a control. The experimental doses of AOH and DHEA were based on our previous observations and research [19,20]. All experiments were performed in triplicates.
4.2. Cell viability assay
The cell viability was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Merck Millipore, Burlington, USA) assay according to the manufacturer’s instructions. The cells were seeded in 96-well plate at the concentration of 1x105 in 100 µl of culture media. The following day, culture medium was changed to experimental medium in the following combinations: 0.1 µM AOH, 10µM AOH, 0.1 µM AOH + 100 nM DHEA, 10 µM AOH + 100 nM DHEA or 48 h. Four hours before the end of the incubation time, 5 mg/ml MTT solution was added to the each well (10 µl per well). The formed formazan crystals were dissolved in DMSO (100 µl of solvent added per well). The absorbance at 570 nm was measured using and ELX 80IU microplate reader (BioTek, Winooski, VT, USA).
4.3. Annexin V Staining Assay
Flow cytometry assays Muse® Annexin V and Dead Cell Kit (Merck Millipore, Burlington, MA, USA) were conducted to assess the number of apoptotic cells. The cells were seeded on 6-well plate at density of 3x105/well. After reaching ca. 90% of confluence, the experimental treatment was performed. Cells were exposed to 0.1 µM AOH, 10 µM AOH, 0.1 µM AOH + 100 nM DHEA, 10 µM AOH + 100 nM DHEA. After 48h, the cells were detached and suspended in 100 µL of culture medium. The assays were performed according to the manufacturer’s instructions. The cells were analyzed on Muse™ Cell Analyzer (Merck Millipore, Burlington, MA, USA). The experiment was carried out in triplicate.
4.4. Cell Cycle
Muse® Cell Cycle Assay Kit (Merck Millipore, Burlington, MA, USA) based on propidium iodide (PI) staining was used to evaluate the percentage of cells in the G0/G1, S and G2 phase of cell cycle. The cells were seeded on 6-well plate at density of 3x105/well. After reaching ca. 90% of confluence, the experimental treatment was performed. Cells were exposed to 0.1 µM AOH, 10 µM AOH, 0.1 µM AOH + 100 nM DHEA, 10 µM AOH + 100 nM DHEA. After 48h, the cells were detached and suspended in 100 µL of culture medium. The assays were performed according to the manufacturer’s instructions. The cells were analyzed on Muse™ Cell Analyzer (Merck Millipore, Burlington, MA, USA). The experiment was carried out in triplicate.
4.4. Steroid assays
Enzyme-linked immunosorbent assay (ELISA) was used to assess the concentration of steroid hormone: progesterone (sensitivity 8,57 pg/ml), testosterone (sensitivity 5,67 pg/ml), 17-β-estradiol (sensitivity 28,5 pg/ml). Cells were seeded on 6-well plate (3x105/well) to reach satisfactory density ca. 90%. After 24 h the culture medium was changed to experimental media containing 1 µM AOH, 10 µM AOH, 1 µM AOH + 100 nM DHEA, 10 µM AOH + 100 nM DHEA for 48h. 100 nM DHEA was used as a positive control. The media collected from the experiments were used to perform ELISA assays (Enzo Chemicals Inc., Farmingdale, NY, USA) according to the manufacturer’s instructions. The absorbance was measured at 405 nm. All the assays were run in a duplicate.
4.5. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted with the use of TRIzol reagent (Thermo Fisher Scientific Inc., Walthan, MA, USA). The cells were cultured in 60 nm Petri dishes at the density of 2x105 cells per dish. After reaching the confluence of 80 %, the experimental treatment was performed. The culture medium was replaced with the experimental ones in the following combinations: 1 µM AOH, 10 µM AOH, 1 µM AOH + 100 nM DHEA, 10 µM AOH + 100 nM DHEA; 100 nM DHEA was used as a positive control. After 48 h, the medium was removed, and the RNA isolation procedure was performed according to the manufacturer’s instructions. BioDrop DUO spectrophotometer (BioDrop, Cambridge, UK) was used to measure the RNA concentration. cDNA synthesis was performed using Prom RT-IITM reverse transcriptase (Promega, Madison, WI, USA). Analyzed targeted genes included: cytochrome P450 Family 11 Subfamily A Member 1 (CYP11A1), cytochrome P450 Family 17 Subfamily A Member 1 (CYP17A1), 3 Beta- And Steroid Delta- Isomerase 2 (HSD3B2), hydroxysteroid 17-Beta Dehydrogenase 2 (HSD17B2), steroidogenic acute regulatory protein (StAR), annexin 5 (ANXA5), estrogen receptor 1 (ESR1), estrogen receptor 2 (ESR2), androgen receptor (AR) caveolin 1 (CAV-1), Cyclin Dependent Kinase Inhibitor 1 (CDK1N1A), Cyclin-dependent kinase 2 (CDK2), tumor protein p53 (TP53). As the calibrator, we used the human reference RNA (Stratagene, San Diego, CA, USA). Primers’ design and validation was performed with Primer BLAST software. Sequences and products sizes are collected in Table 1. Housekeeping genes were used for relative expression normalization as calibrators: ribosomal protein S17 (RPS17), ribosomal protein P0 (RPLP0), and histone H3.3A (H3F3A). The obtained values were calculated using ΔΔCt method. Melting curve analyses were performed to verify the identity of the product for each reaction.
4.6. Western Blot
Firstly, the cells were cultured on Petri dishes (100 nm) at the density 3x105 to reach the satisfactory density 90% and then the experimental treatment was performed. For the isolation of total protein extracts, RIPA protein extraction buffer was used, supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich) and 1 mM PMSF (Sigma Aldrich, Saint Louis, MO, USA). The protein concentration was measured using Direct Detect® (Merck Millipore, Burlington, MA, USA).
For gel electrophoresis, 10 µg of protein was mixed with the Laemmli Lysis buffer and heated for 5 min at 100°C. Proteins were separated with 12.5 % SDS-polyacrylamide gels and transferred to PVDF membranes (400 mA, 110 min) (Merck Millipore, Burlington, MA, USA) with wet transfer. The protein visualization after electrophoresis was performed with the use of 0.1% Panceau-S (Sigma Aldrich, Saint Louis, MO, USA) in 1% acetic acid. 5% nonfat milk in TBST for 1 h at RT was used to block membranes which were then incubated overnight at 4°C with selected primary antibodies according to the manufacturer’s instructions: CAV-1 (sc-894, Santa Cruz Biotechnology, Dallas, TX, USA), GAPDH (sc-59540, Santa Cruz Biotechnology, Dallas, TX, USA) and Cleaved Caspase-3 (mAb #9664, CST, Danvers, MA, USA. The following day, the membranes were washed with TBST buffer (3 x 5 min) and incubated with the solution of secondary antibody (1:15000, in 1% nonfat milk in TBST) conjugated with alkaline, phosphatase (A3812, Sigma Aldrich, Saint Louis, MO, USA). After incubation, the membranes were washed again with TBST buffer (3 x 5 min). Protein bands were visualised using Novex® AP Chromogenic Substrate (Life Technologies, Carlsbad).
4.7. Immunohistochemistry staining and confocal microscopy
The cells were seeded on 8-well chamber slide (NuncTM Lab-TekTM II Chamber SlideTM System/ Thermo Fisher Scientific Inc, Waltham, MA, USA). Once reaching the 80% confluence the experimental treatment was performed for 48 h. Cell fixation was performed in 70% ice cold methanol for 15 min in a freezer. The cells were washed three times in DPBS. Subsequently, cells were immersed in the blocking buffer (5% FBS, 0.3% Triton X-100 in DPBS 1X) for 1 h. The incubation primary antibody CAV-1 (sc-894, Santa Cruz Biotechnology, Dallas, TX, USA) was performed overnight (1:100, 1% BSA, 0.3% Triton X-100 in DPBS 1X). The following day, we washed the cells three times with DPBS and incubated for 1.5 h with secondary antibody (1:400, Alexa Fluor Plus® 488, goat anti-rabbit, #A32731, Thermo Fisher Scientific Inc, Waltham, MA, USA). Upon finished incubation, the final washing step was performed (three times in DPBS 1X) and the mounting medium Fluoroshield with DAPI (F6057, Sigma, St. Louis, MO, USA) was used to preserve the fluorescence of cell specimens. The imaging was performed with the use of Olympus iXplore SpinSR ScanR (Olympus, Tokyo, Japan).
4.8. Statistical Analysis
Statistical data analysis was performed with the GraphPad Software (GraphPad Software, San Diego, CA, USA), using one-way ANOVA test. The results are expressed as mean ±SE. p<0.05 was considered statistically significant.
Author Contributions
Conceptualization, K.K and A.W.P.-C.; investigation and data acquisition, K.A.U., K.K., D.E.H.-G., M.J.K. and K.D.; writing—original draft preparation, K.A.U and K.K.; writing—review and editing, K.K. and A.W.P.-C.; supervision, K.K. and A.W.P.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Medical University of Lodz grant no. 503/0-078-03/503-01-001-19-00.
Institutional Review Board Statement
This work was supported by Medical University of Lodz grant no. 503/0-078-03/503-01-001-19-00.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the current article or Supplementary Materials. The raw data or unpublished data that support the findings of this study are available upon request from the corresponding author.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Holanda, D.M.; Kim, S.W. Mycotoxin Occurrence, Toxicity, and Detoxifying Agents in Pig Production with an Emphasis on Deoxynivalenol. Toxins (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Eskola, M.; Kos, G.; Elliott, C.T.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide Contamination of Food-Crops with Mycotoxins: Validity of the Widely Cited ‘FAO Estimate’ of 25%. Crit Rev Food Sci Nutr 2020, 60, 2773–2789. [Google Scholar] [CrossRef] [PubMed]
- Solhaug, A.; Eriksen, G.S.; Holme, J.A. Mechanisms of Action and Toxicity of the Mycotoxin Alternariol: A Review. Basic Clin Pharmacol Toxicol 2016, 119, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Crudo, F.; Aichinger, G.; Dellafiora, L.; Kiss, E.; Mihajlovic, J.; Del Favero, G.; Berry, D.; Dall’Asta, C.; Marko, D. Persistence of the Antagonistic Effects of a Natural Mixture of Alternaria Mycotoxins on the Estrogen-like Activity of Human Feces after Anaerobic Incubation. Toxicol Lett 2022, 358, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Scientific Opinion on the Risks for Animal and Public Health Related to the Presence of Alternaria Toxins in Feed and Food. EFSA Journal 2011, 9, 2407. [CrossRef]
- Fraeyman, S.; Croubels, S.; Devreese, M.; Antonissen, G. Emerging Fusarium and Alternaria Mycotoxins: Occurrence, Toxicity and Toxicokinetics. Toxins (Basel) 2017, 9, 228. [Google Scholar] [CrossRef]
- Frizzell, C.; Ndossi, D.; Verhaegen, S.; Dahl, E.; Eriksen, G.; Sørlie, M.; Ropstad, E.; Muller, M.; Elliott, C.T.; Connolly, L. Endocrine Disrupting Effects of Zearalenone, Alpha- and Beta-Zearalenol at the Level of Nuclear Receptor Binding and Steroidogenesis. Toxicol Lett 2011, 206, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kalayou, S.; Hamre, A.G.; Ndossi, D.; Connolly, L.; Sørlie, M.; Ropstad, E.; Verhaegen, S. Using SILAC Proteomics to Investigate the Effect of the Mycotoxin, Alternariol, in the Human H295R Steroidogenesis Model. Cell Biol Toxicol 2014, 30, 361–376. [Google Scholar] [CrossRef]
- Del Favero, G.; Mayer, R.M.; Dellafiora, L.; Janker, L.; Niederstaetter, L.; Dall’Asta, C.; Gerner, C.; Marko, D. Structural Similarity with Cholesterol Reveals Crucial Insights into Mechanisms Sustaining the Immunomodulatory Activity of the Mycotoxin Alternariol. Cells 2020, 9. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rawla, P. Epidemiology of Prostate Cancer. World J Oncol 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen Receptors in Hormone-Dependent and Castration-Resistant Prostate Cancer. Pharmacol Ther 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Zhu, Z.; Chung, Y.-M.; Sergeeva, O.; Kepe, V.; Berk, M.; Li, J.; Ko, H.-K.; Li, Z.; Petro, M.; DiFilippo, F.P.; et al. Loss of Dihydrotestosterone-Inactivation Activity Promotes Prostate Cancer Castration Resistance Detectable by Functional Imaging. Journal of Biological Chemistry 2018, 293, 17829–17837. [Google Scholar] [CrossRef]
- Frycz, B.A.; Jagodziński, P.P. Expressions of Genes Encoding Steroidogenic Enzymes and Their Role in Prostate Carcinogenesis. J Med Sci 2014, 83, 73–80. [Google Scholar] [CrossRef]
- Fokidis, H.B.; Adomat, H.H.; Kharmate, G.; Hosseini-Beheshti, E.; Guns, E.S.; Soma, K.K. Regulation of Local Steroidogenesis in the Brain and in Prostate Cancer: Lessons Learned from Interdisciplinary Collaboration. Front Neuroendocrinol 2015, 36, 108–129. [Google Scholar] [CrossRef]
- Škara, L.; Huđek Turković, A.; Pezelj, I.; Vrtarić, A.; Sinčić, N.; Krušlin, B.; Ulamec, M. Prostate Cancer—Focus on Cholesterol. Cancers (Basel) 2021, 13, 4696. [Google Scholar] [CrossRef]
- Deb, S.; Pham, S.; Ming, D.-S.; Chin, M.; Adomat, H.; Hurtado-Coll, A.; Gleave, M.; Guns, E. Characterization of Precursor-Dependent Steroidogenesis in Human Prostate Cancer Models. Cancers (Basel) 2018, 10, 343. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Uemura, H.; Umemoto, S.; Sakamaki, K.; Taguri, M.; Suzuki, K.; Shibata, Y.; Masumori, N.; Ichikawa, T.; Mizokami, A.; et al. Low Serum Dehydroepiandrosterone Examined by Liquid Chromatography-Tandem Mass Spectrometry Correlates with Poor Prognosis in Hormone-Naïve Prostate Cancer. Prostate 2016, 76, 376–382. [Google Scholar] [CrossRef]
- Urbanek, K.A.; Kowalska, K.; Habrowska-Górczyńska, D.E.; Domińska, K.; Sakowicz, A.; Piastowska-Ciesielska, A.W. In Vitro Analysis of Deoxynivalenol Influence on Steroidogenesis in Prostate. Toxins (Basel) 2021, 13, 685. [Google Scholar] [CrossRef]
- Kowalska, K.; Habrowska-Górczyńska, D.E.; Urbanek, K.A.; Domińska, K.; Sakowicz, A.; Piastowska-Ciesielska, A.W. Estrogen Receptor β Plays a Protective Role in Zearalenone-Induced Oxidative Stress in Normal Prostate Epithelial Cells. Ecotoxicol Environ Saf 2019, 172. [Google Scholar] [CrossRef]
- Yang, G.; Xu, H.; Li, Z.; Li, F. Interactions of Caveolin-1 Scaffolding and Intramembrane Regions Containing a CRAC Motif with Cholesterol in Lipid Bilayers. Biochimica et Biophysica Acta (BBA) - Biomembranes 2014, 1838, 2588–2599. [Google Scholar] [CrossRef]
- Chan, N.N.; Yamazaki, M.; Maruyama, S.; Abé, T.; Haga, K.; Kawaharada, M.; Izumi, K.; Kobayashi, T.; Tanuma, J. Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma. Int J Mol Sci 2023, 24, 6035. [Google Scholar] [CrossRef]
- Thompson, T.C.; Tahir, S.A.; Li, L.; Watanabe, M.; Naruishi, K.; Yang, G.; Kadmon, D.; Logothetis, C.J.; Troncoso, P.; Ren, C.; et al. The Role of Caveolin-1 in Prostate Cancer: Clinical Implications. Prostate Cancer Prostatic Dis 2010, 13, 6–11. [Google Scholar] [CrossRef]
- Ayala, G.; Morello, M.; Frolov, A.; You, S.; Li, R.; Rosati, F.; Bartolucci, G.; Danza, G.; Adam, R.M.; Thompson, T.C.; et al. Loss of Caveolin-1 in Prostate Cancer Stroma Correlates with Reduced Relapse-Free Survival and Is Functionally Relevant to Tumour Progression. J Pathol 2013, 231, 77–87. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Wüstner, D. Intracellular Cholesterol Transport. Journal of Clinical Investigation 2002, 110, 891–898. [Google Scholar] [CrossRef]
- Kowalska, K.; Nowakowska, M.; Dominska, K.; Piastowska-Ciesielska, A.W. Coexpression of CAV-1, AT1-R and FOXM1 in Prostate and Breast Cancer and Normal Cell Lines and Their Influence on Metastatic Properties. Acta Biochim.Pol 2016, 63, 493–499. [Google Scholar] [CrossRef]
- Ali, A.; Kulik, G. Signaling Pathways That Control Apoptosis in Prostate Cancer. Cancers (Basel) 2021, 13, 937. [Google Scholar] [CrossRef]
- Small, E.J.; Ryan, C.J. The Case for Secondary Hormonal Therapies in the Chemotherapy Age. Journal of Urology 2006, 176. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res 2008, 68, 4447–4454. [Google Scholar] [CrossRef]
- Fehr, M.; Pahlke, G.; Fritz, J.; Christensen, M.O.; Boege, F.; Altemöller, M.; Podlech, J.; Marko, D. Alternariol Acts as a Topoisomerase Poison, Preferentially Affecting the IIα Isoform. Mol Nutr Food Res 2009, 53, 441–451. [Google Scholar] [CrossRef]
- Lehmann, L.; Wagner, J.; Metzler, M. Estrogenic and Clastogenic Potential of the Mycotoxin Alternariol in Cultured Mammalian Cells. Food and Chemical Toxicology 2006, 44, 398–408. [Google Scholar] [CrossRef]
- Stypuła-Trębas, S.; Minta, M.; Radko, L.; Jedziniak, P.; Posyniak, A. Nonsteroidal Mycotoxin Alternariol Is a Full Androgen Agonist in the Yeast Reporter Androgen Bioassay. Environ Toxicol Pharmacol 2017, 55, 208–211. [Google Scholar] [CrossRef]
- Dellafiora, L.; Warth, B.; Schmidt, V.; Del Favero, G.; Mikula, H.; Fröhlich, J.; Marko, D. An Integrated in Silico/in Vitro Approach to Assess the Xenoestrogenic Potential of Alternaria Mycotoxins and Metabolites. Food Chem 2018, 248, 253–261. [Google Scholar] [CrossRef]
- Frizzell, C.; Ndossi, D.; Kalayou, S.; Eriksen, G.S.; Verhaegen, S.; Sørlie, M.; Elliott, C.T.; Ropstad, E.; Connolly, L. An in Vitro Investigation of Endocrine Disrupting Effects of the Mycotoxin Alternariol. Toxicol Appl Pharmacol 2013, 271, 64–71. [Google Scholar] [CrossRef]
- Rebhahn, V.I.C.; Kiss, E.; Marko, D.; Del Favero, G. Foodborne Compounds That Alter Plasma Membrane Architecture Can Modify the Response of Intestinal Cells to Shear Stress in Vitro. Toxicol Appl Pharmacol 2022, 446, 116034. [Google Scholar] [CrossRef]
- Huang, C.-H.; Wang, F.-T.; Chan, W.-H. Alternariol Exerts Embryotoxic and Immunotoxic Effects on Mouse Blastocysts through ROS-Mediated Apoptotic Processes. Toxicol Res (Camb) 2021, 10, 719–732. [Google Scholar] [CrossRef]
- Fernández-Blanco, C.; Juan-García, A.; Juan, C.; Font, G.; Ruiz, M.-J. Alternariol Induce Toxicity via Cell Death and Mitochondrial Damage on Caco-2 Cells. Food and Chemical Toxicology 2016, 88, 32–39. [Google Scholar] [CrossRef]
- Bensassi, F.; Gallerne, C.; Sharaf El Dein, O.; Hajlaoui, M.R.; Bacha, H.; Lemaire, C. Cell Death Induced by the Alternaria Mycotoxin Alternariol. Toxicology in Vitro 2012, 26, 915–923. [Google Scholar] [CrossRef]
Figure 1.
AOH decreases the viability of prostate cells in a dose- and time- dependent manner in normal (a) as well as prostate cancer (b) cells obtained with MTT assay. Results are expressed as mean ± SE of % of control cell viability (control= 100%). *** p < 0.001 as compared to the control. AOH – alternariol.
Figure 1.
AOH decreases the viability of prostate cells in a dose- and time- dependent manner in normal (a) as well as prostate cancer (b) cells obtained with MTT assay. Results are expressed as mean ± SE of % of control cell viability (control= 100%). *** p < 0.001 as compared to the control. AOH – alternariol.
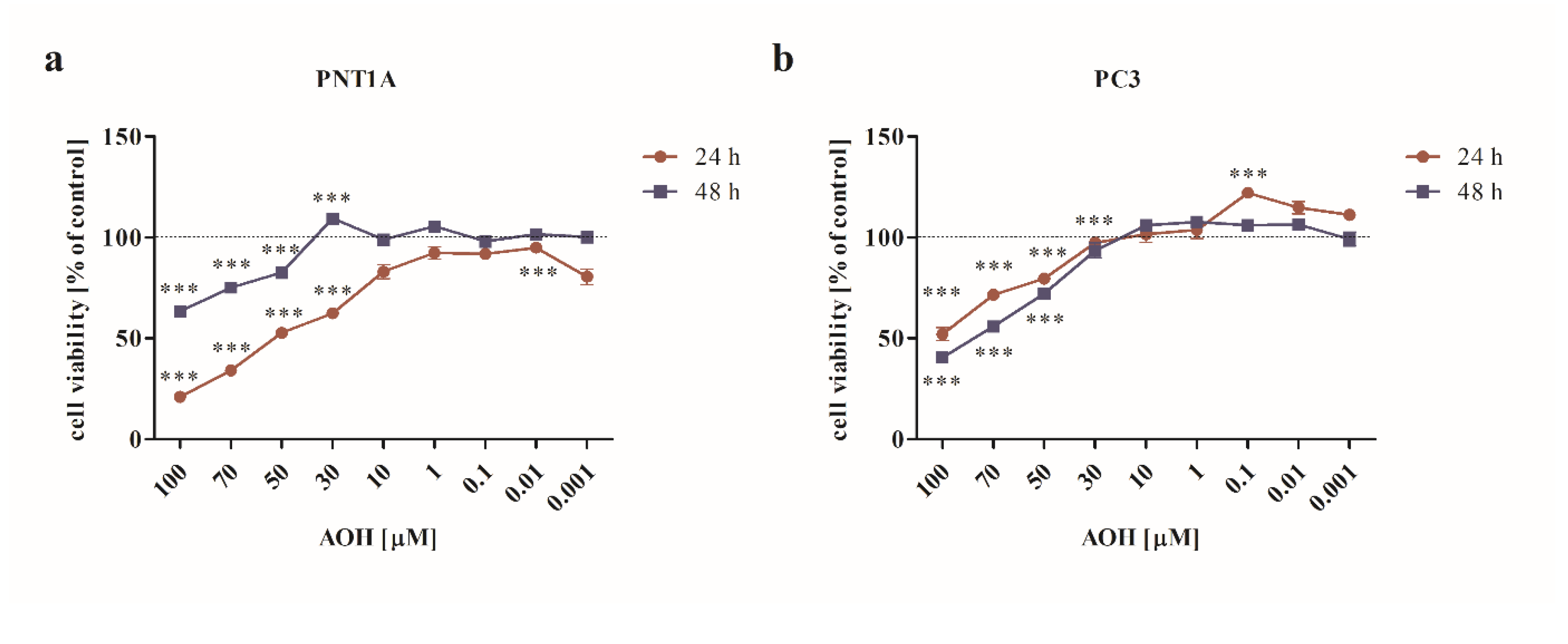
Figure 2.
The secretion of testosterone (a and b), estradiol (c and d) and progesterone (e and f) after 48 h of exposure to AOH and DHEA, evaluated by ELISA tests in PC3 and PNT1A cells. The results are presented and mean ±SE. One way ANOVA was used to perform statistical analysis of the results, p<0.05 was considered as statistically significant. *p<0.05, **p<0.01, ***p<0.001. AOH – alternariol, DHEA - dehydroepiandrosterone, Cnt—control.
Figure 2.
The secretion of testosterone (a and b), estradiol (c and d) and progesterone (e and f) after 48 h of exposure to AOH and DHEA, evaluated by ELISA tests in PC3 and PNT1A cells. The results are presented and mean ±SE. One way ANOVA was used to perform statistical analysis of the results, p<0.05 was considered as statistically significant. *p<0.05, **p<0.01, ***p<0.001. AOH – alternariol, DHEA - dehydroepiandrosterone, Cnt—control.
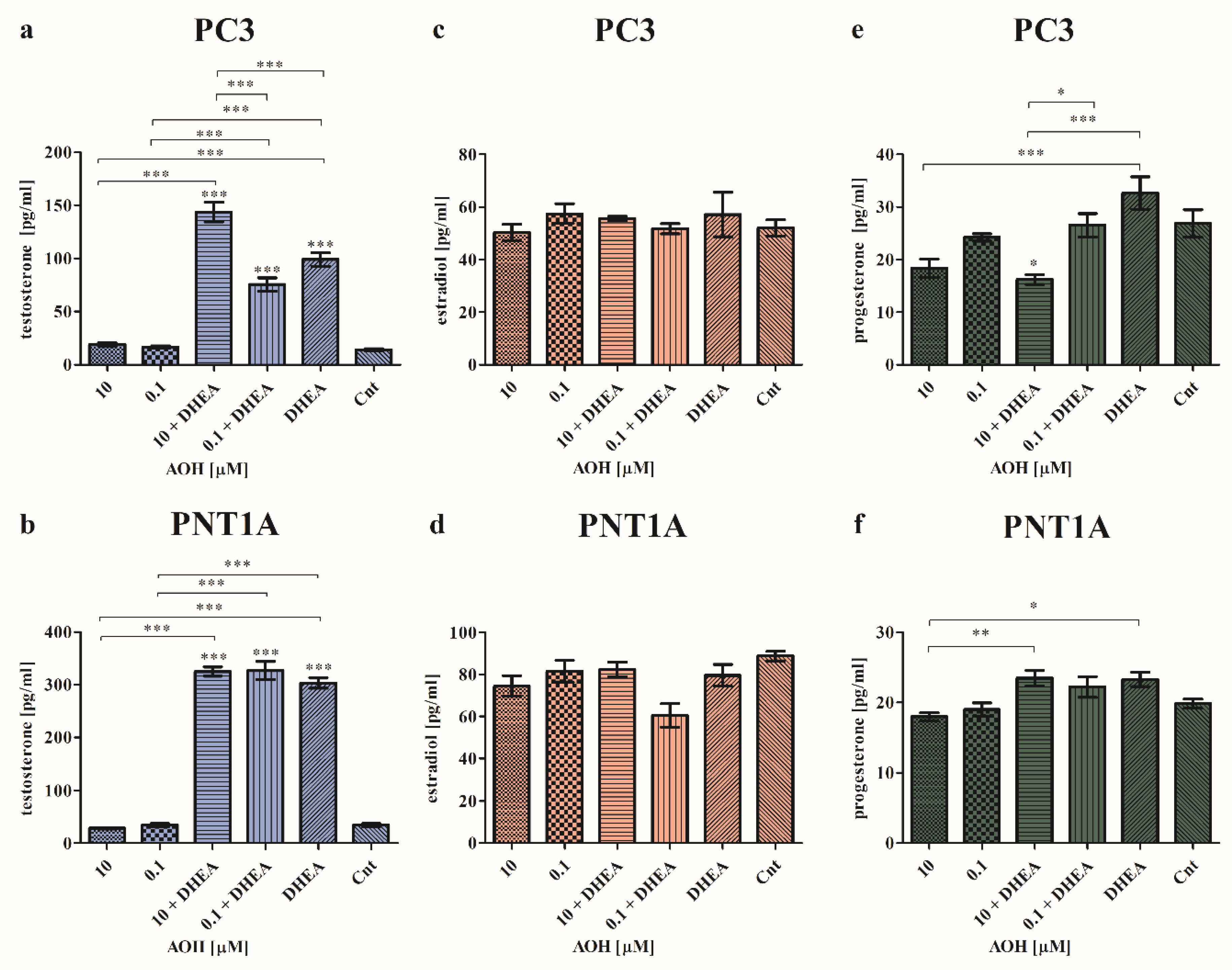
Figure 3.
The relative expression of the mediators of the initial and rate-limiting step in steroidogenesis - CYP11A1, STAR, CYP17A1, HSD3B2 and AR and ESR2 obtained in RT-qPCR. The results are expressed as mean relative expression of three independent experiments. The results are presented and mean ±SE. The results were calculated as ΔΔCt method. AOH - alternariol, DHEA – dehydroepiandrosterone, Cnt – control, CYP11A1- cytochrome P450 family 11 subfamily A member 1, StAR- steroidogenic acute regulatory protein, CYP17A1- cytochrome P450 family 17 subfamily A member 1, HSD3B2- hydroxy-delta-5-steroid dehydrogenase 3 beta- and steroid delta-isomerase 2, AR- androgen receptor, ESR2- estrogen receptor beta. One way ANOVA was used to perform statistical analysis of the results, p<0.05 was considered as statistically significant. *p<0.05, **p<0.01, ***p<0.001.
Figure 3.
The relative expression of the mediators of the initial and rate-limiting step in steroidogenesis - CYP11A1, STAR, CYP17A1, HSD3B2 and AR and ESR2 obtained in RT-qPCR. The results are expressed as mean relative expression of three independent experiments. The results are presented and mean ±SE. The results were calculated as ΔΔCt method. AOH - alternariol, DHEA – dehydroepiandrosterone, Cnt – control, CYP11A1- cytochrome P450 family 11 subfamily A member 1, StAR- steroidogenic acute regulatory protein, CYP17A1- cytochrome P450 family 17 subfamily A member 1, HSD3B2- hydroxy-delta-5-steroid dehydrogenase 3 beta- and steroid delta-isomerase 2, AR- androgen receptor, ESR2- estrogen receptor beta. One way ANOVA was used to perform statistical analysis of the results, p<0.05 was considered as statistically significant. *p<0.05, **p<0.01, ***p<0.001.
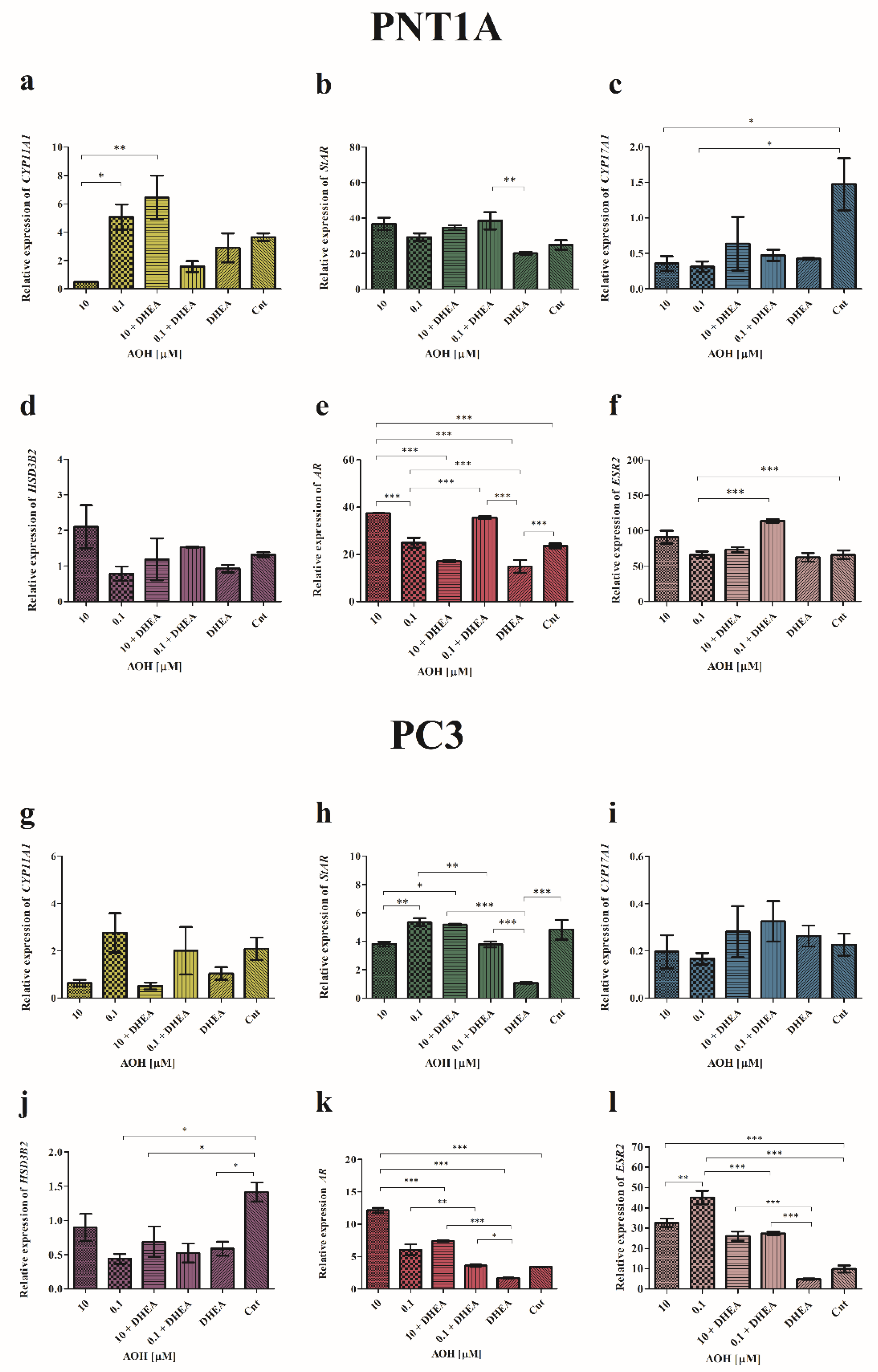
Figure 4.
AOH affects the expression and localization of CAV-1 in prostate cells. Analysis of the relative expression of CAV-1 gene in normal prostate and cancer cell line (a). One-way ANOVA was used for statistical analysis. p<0.05 was considered statistically significant. The results are presented and mean ±SE. The results are expressed as mean relative expression of three independent experiments. Representative results of Western blot analysis of CAV-1 expression (b). Observed different localizations of CAV-1 in cells from confocal microscopy analysis performed in the sequence series of acquisition in the same microscope settings (c). AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control, CAV-1 – caveolin 1.
Figure 4.
AOH affects the expression and localization of CAV-1 in prostate cells. Analysis of the relative expression of CAV-1 gene in normal prostate and cancer cell line (a). One-way ANOVA was used for statistical analysis. p<0.05 was considered statistically significant. The results are presented and mean ±SE. The results are expressed as mean relative expression of three independent experiments. Representative results of Western blot analysis of CAV-1 expression (b). Observed different localizations of CAV-1 in cells from confocal microscopy analysis performed in the sequence series of acquisition in the same microscope settings (c). AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control, CAV-1 – caveolin 1.
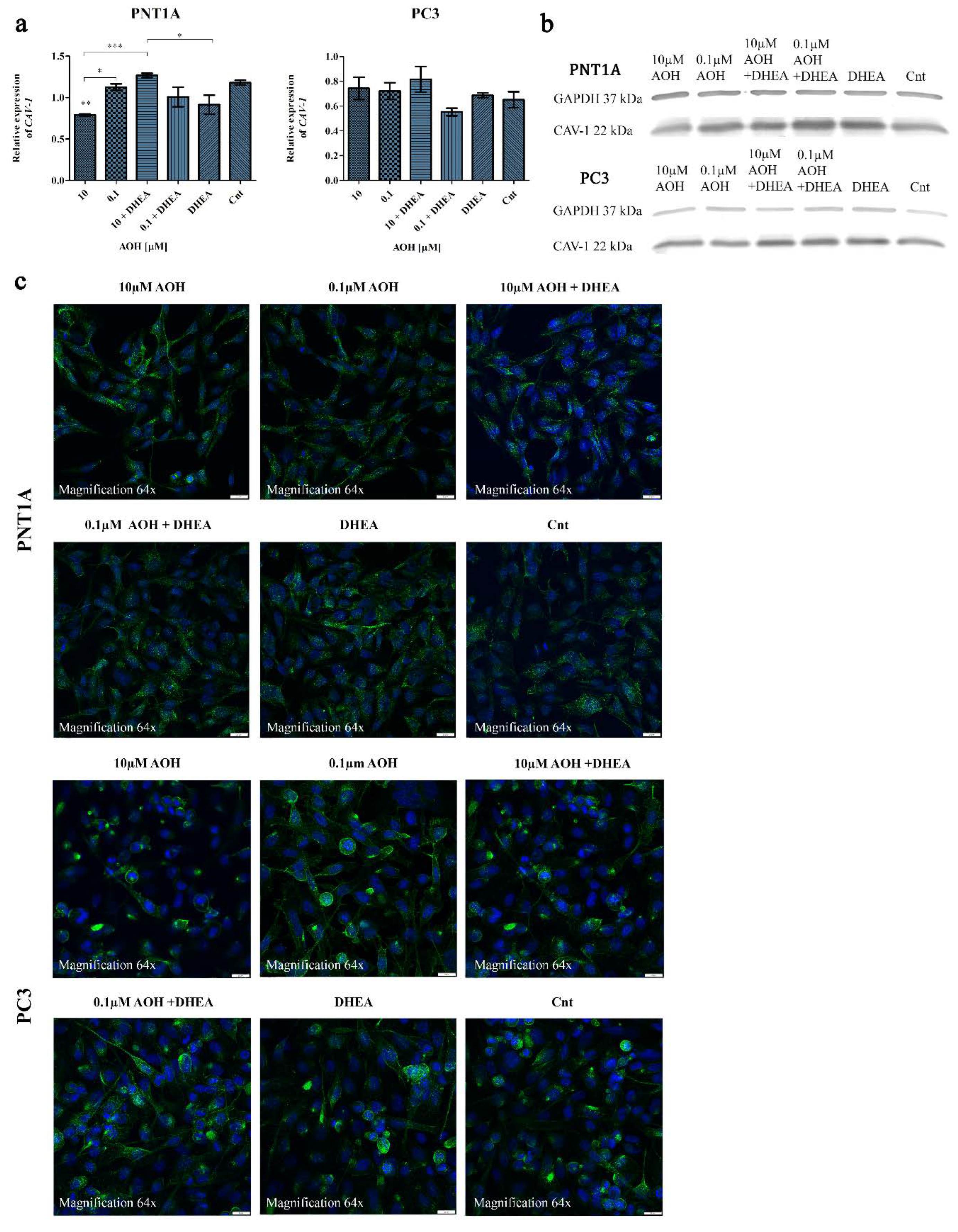
Figure 5.
AOH, DHEA as well as co-treatment induce apoptosis after 48 h exposure in PC3 and PNT1A cells (a). Analysis of apoptotic cells based on flow cytometry with representative results (b). One-way ANOVA was used for statistical analysis. results are presented and mean ±SE. ***p<0.001 as compared to the control. Representative results of Western blot analysis of CASP3 (c). AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control, CASP3 – caspase 3.
Figure 5.
AOH, DHEA as well as co-treatment induce apoptosis after 48 h exposure in PC3 and PNT1A cells (a). Analysis of apoptotic cells based on flow cytometry with representative results (b). One-way ANOVA was used for statistical analysis. results are presented and mean ±SE. ***p<0.001 as compared to the control. Representative results of Western blot analysis of CASP3 (c). AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control, CASP3 – caspase 3.
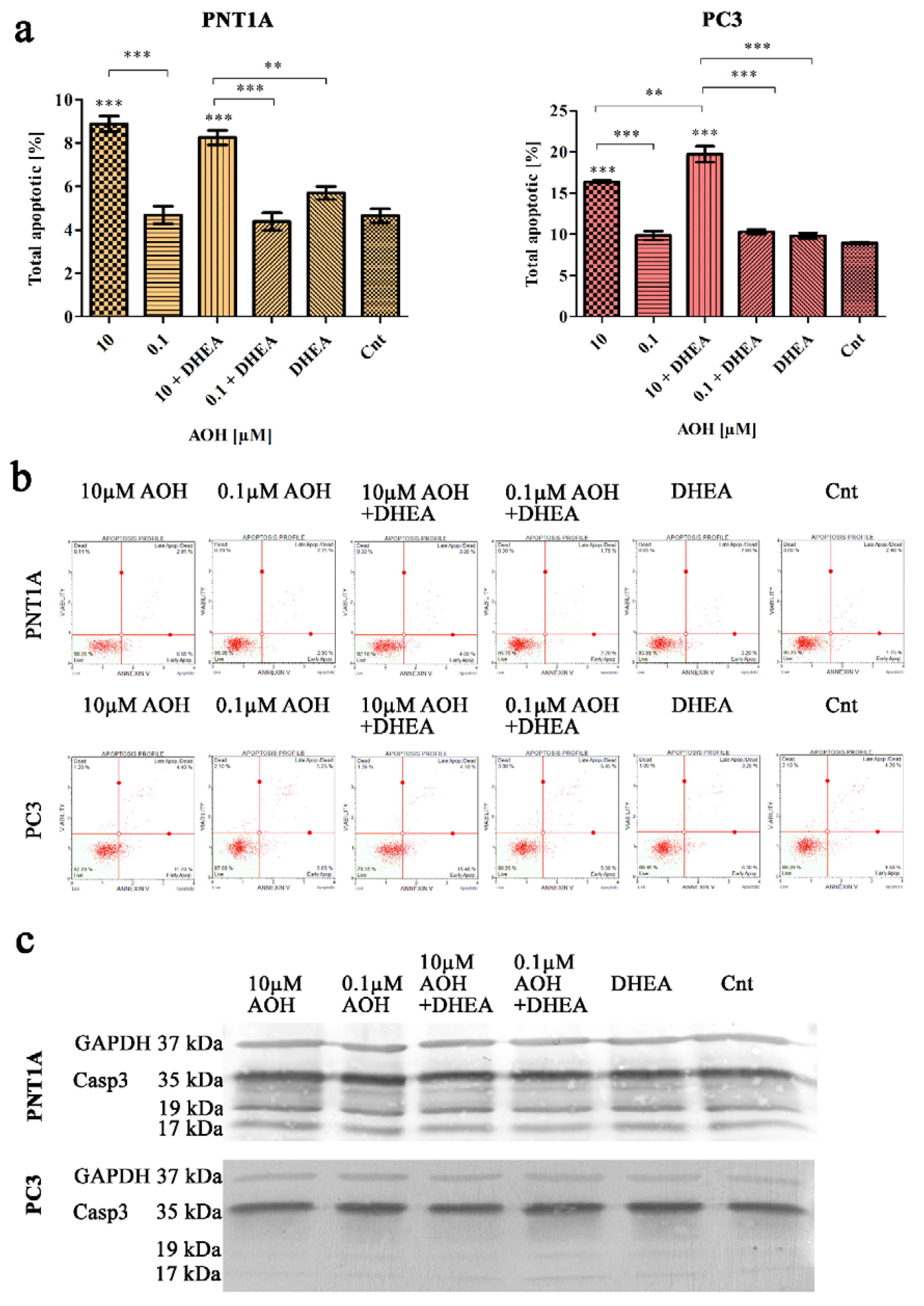
Figure 6.
AOH, DHEA as well as co-treatment induce changes in cell cycle progression after 48 h exposure in PNT1A (a) and PC3 cells (b). Analysis based on the results obtained in flow cytometry. One-way ANOVA was used for statistical analysis. The results are presented and mean ±SE. *p<0.05, **p<0.01, ***p<0.001. AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control. The results acquired in three independent experiments.
Figure 6.
AOH, DHEA as well as co-treatment induce changes in cell cycle progression after 48 h exposure in PNT1A (a) and PC3 cells (b). Analysis based on the results obtained in flow cytometry. One-way ANOVA was used for statistical analysis. The results are presented and mean ±SE. *p<0.05, **p<0.01, ***p<0.001. AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control. The results acquired in three independent experiments.
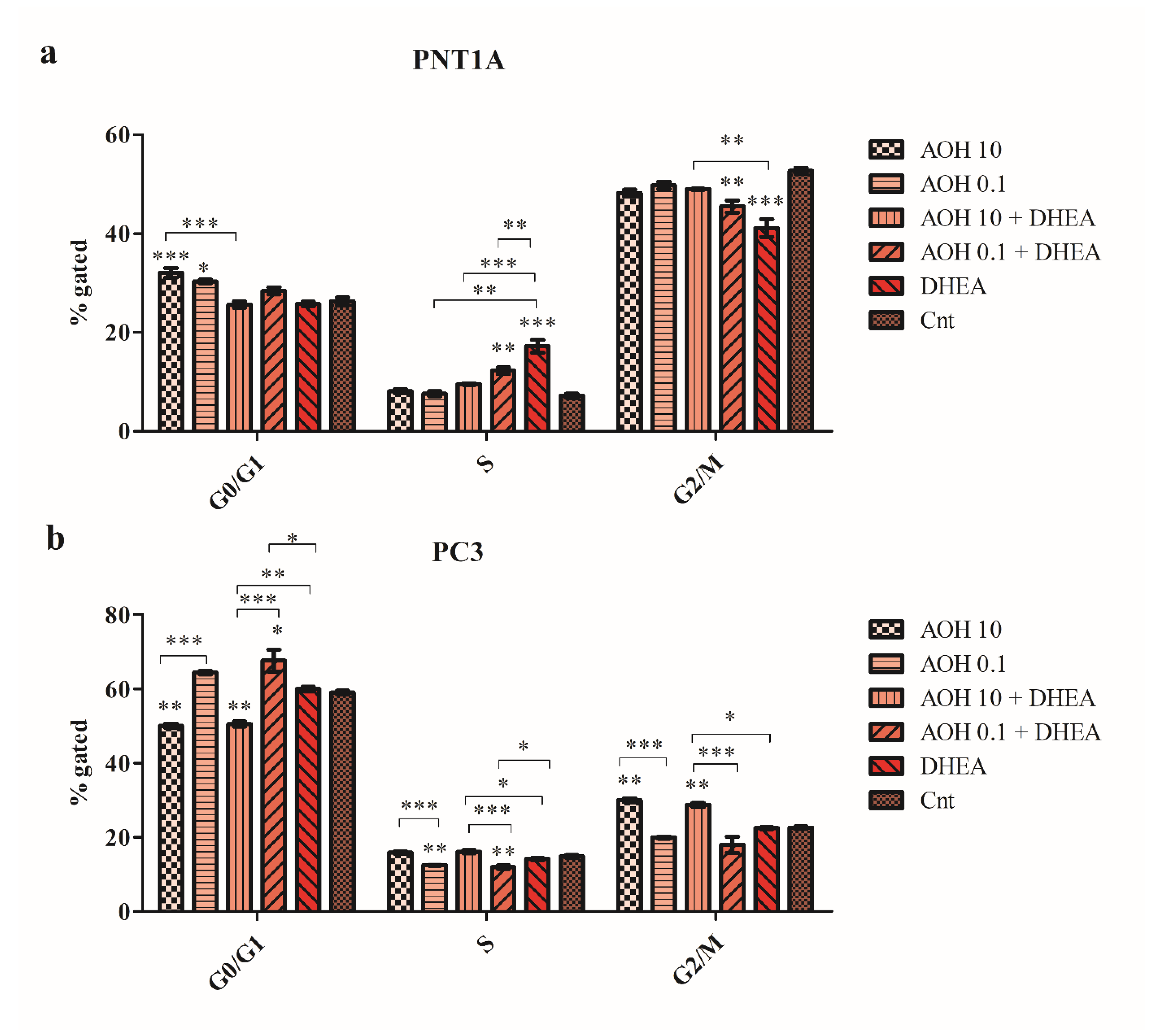
Figure 7.
Relative expression of genes related to apoptosis and cell cycle. The relative expression of ANXA5 (a), TP53 (b), CDKN1A (c), and CDK2 (d) obtained in RT-qPCR and expressed as mean± SE. H3F3A, RPLP0 and RPS17 were used as housekeeping genes. One- way ANOVA was used for statistical analysis. p < 0.05 was considered as significant. *p<0.05,**p< 0.01, ***p< 0.001 as compared to the control. The experiment was carried out in triplicate. AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control, ANXA5 – annexin 5, TP53 – tumor protein TP-53 - protein p53, CDK1N1A - Cyclin Dependent Kinase Inhibitor 1; CDK2 - Cyclin-dependent kinase 2.
Figure 7.
Relative expression of genes related to apoptosis and cell cycle. The relative expression of ANXA5 (a), TP53 (b), CDKN1A (c), and CDK2 (d) obtained in RT-qPCR and expressed as mean± SE. H3F3A, RPLP0 and RPS17 were used as housekeeping genes. One- way ANOVA was used for statistical analysis. p < 0.05 was considered as significant. *p<0.05,**p< 0.01, ***p< 0.001 as compared to the control. The experiment was carried out in triplicate. AOH – alternariol, DHEA – dehydroepiandrosterone, Cnt—control, ANXA5 – annexin 5, TP53 – tumor protein TP-53 - protein p53, CDK1N1A - Cyclin Dependent Kinase Inhibitor 1; CDK2 - Cyclin-dependent kinase 2.
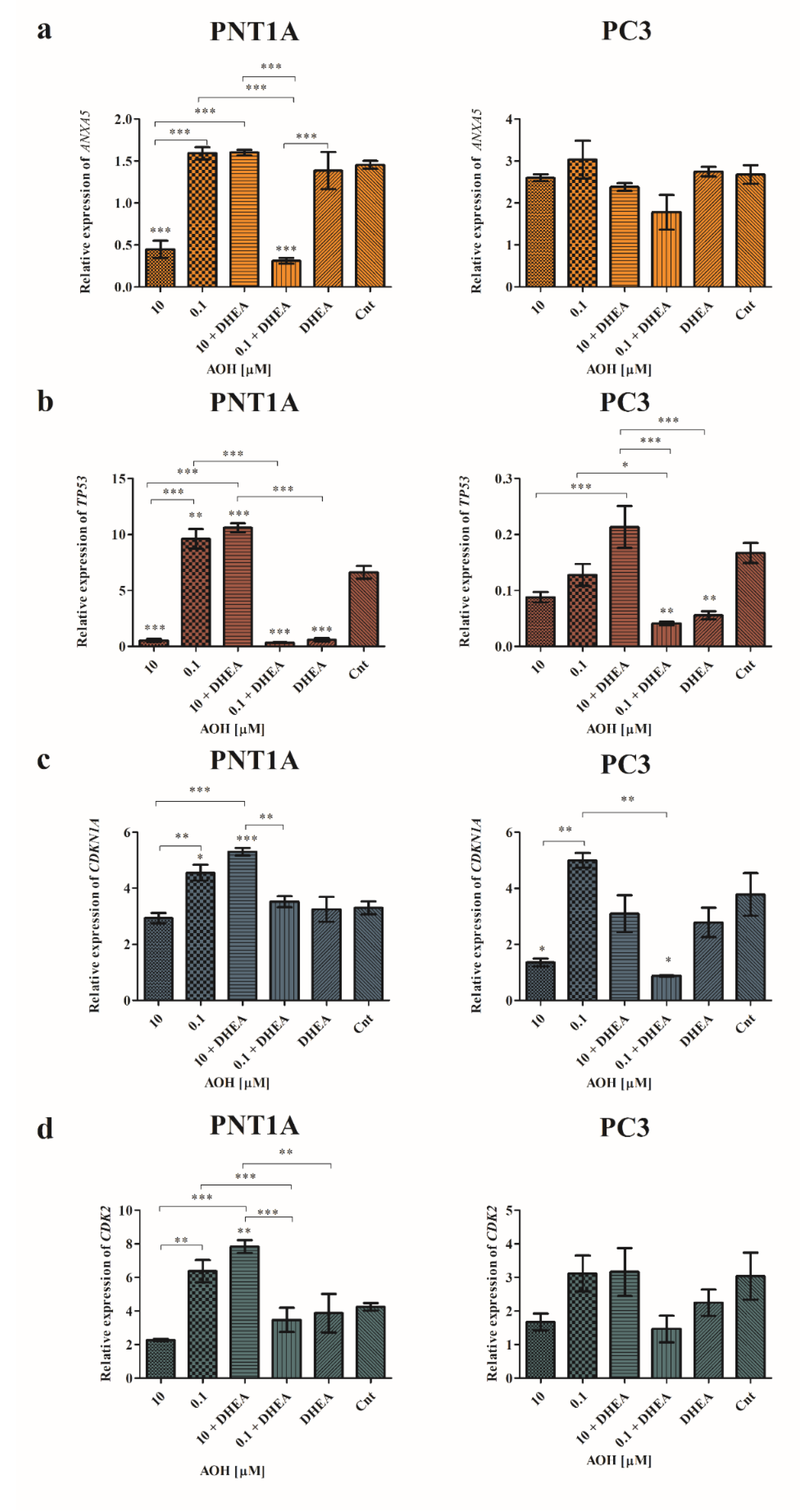

Table 1.
Primers used in RT-qPCR. RPS17 - ribosomal protein S17; RPLP0 - ribosomal protein P0; H3F3A - histone H3.3A; CYP11A1 - cytochrome P450 Family 11 Subfamily A Member 1; CYP17A1 - cytochrome P450 Family 17 Subfamily A Member 1; HSD3B2 - 3 Beta- And Steroid Delta- Isomerase 2, STAR - steroidogenic acute regulatory protein; ANXA5 – annexin 5; ESR2 - estrogen receptor 2, AR - androgen receptor; CAV-1 - caveolin , CDK1N1A - Cyclin Dependent Kinase Inhibitor 1; CDK2 - Cyclin-dependent kinase 2; tumor TP-53 - protein p53.
Table 1.
Primers used in RT-qPCR. RPS17 - ribosomal protein S17; RPLP0 - ribosomal protein P0; H3F3A - histone H3.3A; CYP11A1 - cytochrome P450 Family 11 Subfamily A Member 1; CYP17A1 - cytochrome P450 Family 17 Subfamily A Member 1; HSD3B2 - 3 Beta- And Steroid Delta- Isomerase 2, STAR - steroidogenic acute regulatory protein; ANXA5 – annexin 5; ESR2 - estrogen receptor 2, AR - androgen receptor; CAV-1 - caveolin , CDK1N1A - Cyclin Dependent Kinase Inhibitor 1; CDK2 - Cyclin-dependent kinase 2; tumor TP-53 - protein p53.
| Gene | Sequence (5’- 3’) | Product Size (bp) |
|---|---|---|
| CYP11A1 | For CCAGAACGATTCCTCATCC | 126 |
| Rev CATCACCTCCTGGTTCAG | ||
| CYP17A1 | For GAAGTTATCATCAATCTGTGGG | 119 |
| Rev ACTGACGGTGAGATGAGC | ||
| Rev AAGATGTCTGGTTTGATGAGGAG | ||
| HSD3B2 | For CTTGGTGTCACTCACAGAGAG | 128 |
| Rev GTAGATGAAGACTGGCACACTG | ||
| Rev CACCTCCAATTGTGACATAA | ||
| STAR | For CATGGAGAGGCTCTATGAAGA | 128 |
| Rev CAGCCAGCTCGTGAGTAAT | ||
| ANXA5 | For ACCCTCTCGGCTTTATGATGCT | 116 |
| Rev TGGCTCTCAGTTCTTCAGGTGT | ||
| Rev TGCTGGACAGAAATGTGTACACTCCAGA | ||
| ESR2 | For ACACCTGGGCACCTTTCTCCTTTA | 90 |
| Rev TCTTGCTTCACACCAGGGACTCTT | ||
| AR | For GGGAGGTTACACCAAAGGGC | 102 |
| Rev AGAGACAGGGTAGACGGCAG | ||
| CAV-1 | Rev GAACTTGAAATTGGCACCAGG | 139 |
| For ACCCACTCTTTGAAGCTGTTG | ||
| CDK1N1A | Rev CTGAGACTAAGGCAGAAGATGT | 133 |
| For GACAGATTTCTACCACTCCAA | ||
| CDK2 | Rev GAGCAGAGGCATCCATGAATT | 126 |
| For TGCTTAAGGAGCTTAACCATCC | ||
| TP53 | Rev TTTATGGCGGGAGGTAGA | 102 |
| For TTGGAACTCAAGGATGCC | ||
| For GCAAGACTGTTAGCCCTCAA | ||
| RPLP0 | For ACGGATTACACCTTCCCACTTGCTAAAAGGTC | 69 |
| Rev AGCCACAAAGGCAGATGGATCAGCCAAG | ||
| RPS17 | For AAGCGCGTGTGCGAGGAGATCG | 87 |
| Rev TCGCTTCATCAGAT GCGTGACATAACCTG | ||
| H3F3A | For AGGACTTTAAAAGATCTGCGCTTCCAGAG | 74 |
| Rev ACCAGATAGGCCTCACTTGCCTCCTGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



