
Submitted:
20 April 2023
Posted:
02 May 2023
You are already at the latest version
Abstract
Beyond plasma glucose-lowering in patients with type 2 diabetes, sodium- glucose cotransporter 2 inhibitors (SGLT2i) significantly reduced the hospitalization for heart failure (HF) and retarded the progression of chronic kidney disease (CKD). Endothelial dysfunction is not only involved in the development and progression of cardiovascular disease (CVD), and is also associated with the progression of CKD. In patients with type 2 diabetes, hyperglycemia, insulin resistance, hyperinsulinemia and dyslipidemia induce the development of endothelial dysfunction. SGLT2i have shown an improvement in endothelial dysfunction, as assessed by flow-mediated vasodila-tion, in individuals at high risk for CVD. Along with an improvement of endothelial dysfunction, SGLT2i showed improvements in oxidative stress, inflammation, mitochondrial dysfunction, glucotoxicity such as advanced glycation end products signaling and nitric oxide bioavailability. The improvements in endothelial dysfunc-tion and such endothelium-derived factors may play an important role in preventing the development of coronary artery disease, coronary microvascular dysfunction and diabetic cardiomyopathy which cause HF and in retarding CKD. The suppression of the development of HF and the progression of CKD achieved by SGLT2i might have been largely induced by the improvement of vascular endothelial function by SGLT2i.
Keywords:
endothelial dysfunction
; chronic kidney disease
; heart failure
; sodium-glucose cotransporter 2 inhibitors
1. Introduction
Beyond plasma glucose-lowering, sodium-glucose cotransporter 2 inhibitors (SGLT2i) significantly reduced the major adverse cardiovascular events (MACE) with the history of cardiovascular disease (CVD) or multiple risk factors for CVD in patients with type 2 diabetes [1,2]. The EMPA-REG OUTCOME using empagliflozin, one of SGLT2i, showed that empagliflozin significantly reduced 3 point-MACE including death from CV causes, nonfatal myocardial infarction, or nonfatal stroke, by 14% compared to placebo [1]. What the EMPA-REG OUTCOME surprised physicians was that empagliflozin reduced the hospitalization for heart failure (HF) by 35%, as compared with placebo. Reduction of the hospitalization for HF was also observed in the CANVAS Program which used canagliflozin [2]. Interestingly, the difference in the hospitalization for HF between placebo and SGTL2i appeared from the early phase after SGLT2i administration in both trials. What did these results mean? Most of type 2 diabetic patients included in the EMPA-REG OUTCOME had the history of CVD, and approximately 65% of patients in the CANVAS Program had the history of CVD [1,2,3]. The difference in the hospitalization for HF between placebo and SGTL2i from the early phase after administration was induced by high CVD risk background of studied patients? The DECLARE–TIMI 58 included about 40% of type 2 diabetic patients with CVD showed that one of SGLT2i, dapagliflozin, reduced the hospitalization for HF by 27% [4]. The DECLARE–TIMI 58 also showed the difference in the hospitalization for HF between placebo and SGTL2i from the early phase after SGLT2i administration. The result obtained from the DECLARE–TIMI 58 challenged that the high-risk background of studied patients induced an early separation of the hospitalization for HF curves between SGLT2i- and placebo-treated patients. Another reasonable answer is that patients with type 2 diabetes are likely to develop HF, and SGLT2i improves the factors that exacerbate HF in patients with type 2 diabetes.
The advantage brought by SGLT2i is that they are effective not only in suppressing the onset of HF, but also in suppressing the development and progression of chronic kidney disease (CKD). Empagliflozin reduced incident or worsening nephropathy by 39% as compared with placebo, and decreased doubling of serum creatinine level and renal-replacement therapy by 44% and 55%, respectively [6]. Canagliflozin also lowered the renal-specific composite of end-stage renal disease (ESRD), a doubling of the creatinine level, or death from renal causes by 34% [2]. Dapagliflozin reduced the composite of a sustained decline in the estimated glomerular filtration rate (eGFR) of at least 50%, ESRD, or death from renal causes by 44% [6]. SGLT2i such as empagliflozin, canagliflozin and dapagliflozin retarded a decline of eGFR in patients with type 2 diabetes. A short-term (12 weeks) empagliflozin treatment reduced urinary albumin-to-creatinine ratio (UACR) by 7% in patients with normo-albuminuria, by 25% in patients with microalbuminuria, and by 32% in patients with macroalbuminuria, as compared with those who used placebo. The reductions in UACR were maintained with empagliflozin in all three groups compared with placebo during a long-term treatment (164 weeks) [7].
SGLT2i reduced the development of atherosclerotic CVD (ASCVD) and HF and retarded CKD. Endothelial dysfunction is a very early event in atherosclerosis. Cardiomyocyte is the main player in cardiac function and development of HF, however, its function is underpinned by non-cardiomyocytes such as vascular endothelial cells. In particular, vascular endothelial cells are important cells for maintaining blood perfusion to myocardial cells. CKD has been found to be an important risk factor not only for ESRD but also for CVD, and the concept of cardiorenal syndrome (CRS) has attracted attention. In CKD patients, systemic vascular endothelial damage is observed from an early stage, which can explain the frequent development of CVD in CKD patients [8].
Endothelial dysfunction is a crucial determinant for the development and progression of ASCVD, HF and CKD, all of which were improved by SGLT2i use in patients with type 2 diabetes. Here, we discuss the effects of SGLT2i on endothelial dysfunction, and the influence of an improvement of endothelial function by SGLT2i on the pathogenesis of ASCVD, HF and CKD.
2. Endothelial Dysfunction Due to Diabetes and/or Insulin Resistance
The endothelium-derived molecules and their effects, induced by endothelial dysfunction due to diabetes or insulin resistance on atherosclerosis were shown in Figure 1.
Vascular endothelial dysfunction is an important early stage of atherosclerosis development. Endothelial nitric oxide synthase (eNOS) produces the nitric oxide (NO) in endothelial cells, and eNOS is closely associated with regulation of anti-atherogenetic processes such as vasorelaxation, an inhibition of the adhesion between leukocyte and endothelial cells, the suppression of migration and proliferation of vascular smooth muscle cells, and the inhibition of platelet aggregation [9,10,11]. NO promotes vasodilation, and suppresses proliferation and migration of vascular smooth muscle cells, and suppresses the expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). Further, NO contributes to the inhibition of cytokine activity such as tumor necrosis factor-α (TNF-α) and platelet aggregation, and reduction in procoagulant factors. NO also suppresses the adhesion of monocytes and macrophages to the vascular wall. Elevated TNF-α levels and hyperglycemia are implicated in endothelial dysfunction in patients with diabetes [12,13,14]. TNF-α and hyperglycemia have been reported to elevate plasminogen activator inhibitor-1 (PAI-1) and ICAM-1 and VCAM-1 expression in endothelial cells. PAI-1 and vascular adhesion molecules are elevated in patients with diabetes, which may largely contribute to the pathogenesis of atherosclerosis in diabetic patients [15,16]. Therefore, reduced NO production by endothelial cells induces inflammatory proliferative changes in the vascular wall and allow monocytes to enter the vascular wall, leading to atherosclerotic lesions. Actually, endothelium-dependent vasorelaxation response is attenuated and vascular endothelial function is impaired due to decreased activity of eNOS, in vascular walls of patients with insulin resistance [17]. The experiments with endothelial cells have shown that eNOS is activated to produce NO by insulin-mediated activation of phosphatidylinositol3 (PI3) kinase and the phosphorylation of its downstream Akt [18,19]. Insulin induces NO production by eNOS.
There is growing evidence that elevated expression of the eNOS inhibitor, asymmetric dimethylarginine (ADMA), is associated with the development of endothelial dysfunction [20,21,22]. Further, elevation of ADMA is associated with an increased risk of CVD. Plasma ADMA levels are positively correlated with insulin resistance in nondiabetic, normotensive people, suggesting a significant association between ADMA and insulin resistance [23].
Endothelial dysfunction is characterized by the enhancement of endothelin-1 (ET-1) expression and reduced expression of eNOS in endothelial cells. ET-1 is a potent vasoconstrictor whereas eNOS induces strong vasodilatation by production of NO [24,25]. Diabetic status induces the formation and accumulation of advanced glycation end products (AGEs). The receptor for AGEs (RAGE) plays a crucial role in the promotion of inflammation and the activation of endothelial cells, which is closely associated with the development and progression of atherosclerosis in patients with diabetes [26,27].
Hyperglycemia may cause the overproduction of mitochondrial reactive oxygen species (ROS), leading to the feed-forward redox stimulation of NADPH oxidases. This vicious cycle may contribute to the development of pathological conditions and facilitate organ damage in diabetes [28]. Such an oxidative stress increases production of oxidized low-density lipoprotein (LDL) which is easily up-taken by macrophages via scavenger receptor (SR), resulting in foam cell formation.
Endothelial progenitor cells (EPCs) are derived from bone marrow, and can enter blood and differentiate into mature endothelial cells [29], and plays an important role in repairing vascular endothelial damage [30]. Lower level of EPCs was significantly associated with a higher CVD incidence in diabetic patients [31].
3. A Significance of Endothelial Dysfunction for Development of HF in Patients with Type 2 Diabetes
3.1. Patients with Type 2 Diabetes Are Likely to Develop HF?
Diabetes as well as obesity is one of crucial risk factors for HF [32]. The association of glucose metabolism with CV outcome, left ventricular mass (LVM) and LV hypertrophy (LVH) were investigated by using 15,010 subjects with euglycemia, prediabetes and type 2 diabetes in the population-based Gutenberg Health Study [33]. The prevalence of LVH was higher in the order of type 2 diabetes (23.8%), prediabetes (17.8%), and euglycemia (10.2%). Prediabetes and type 2 diabetes were associated with increased LVM, independent of age, sex, and CV risk factors. The co-prevalence of type 2 diabetes with LVH reduced life expectancy. The patients with type 2 diabetes without hypertension, albuminuria, and ischemic heart disease showed significantly higher LVM than healthy controls [34]. LVM was significantly and positively associated with the duration of diabetes and HbA1c levels in patients with type 2 diabetes. The development of symptomatic HF, HF hospitalization, and CV death in asymptomatic left ventricular systolic dysfunction patients with and without diabetes were examined [35]. Patients with diabetes had a higher risk of development of HF (hazard ratio [HR], 1.53; 95% confidence interval [95%CI], 1.32 to 1.78; P < 0.001), HF hospitalization (HR, 2.04; 95%CI, 1.65 to 2.52; P < 0.0001), and the composite outcome of development of HF or cardiovascular death (HR, 1.48; 95%CI, 1.30-1.69; P < 0.001). It was determined whether the risk of adverse CV outcomes associated with diabetes differs in patients with reduced and preserved ejection fraction HF (HFrEF and HFpEF) [36]. The prevalence of diabetes was 28.3% in patients with HFpEF and 28.5% in those with HFrEF. Diabetes was associated with a greater relative risk of CV death or HF hospitalization in patients with HFpEF (HR, 2.0; 95%CI, 1.70 to 2.36) than in patients with HFrEF (HR, 1.60; 95%CI, 1.44 to 1.77). In short, diabetes was an independent predictor of CV morbidity and mortality in patients with HF, regardless of EF. Surprisingly, 28% of patients with type 2 diabetes who were not diagnosed with HF had HF such as HFrEF (5%) and HFpEF (23%) [37]. Such HF-prone property of diabetic patients might have brought an early separation of the curve for HF hospitalization between SGLT2i- and placebo-treated patients in various trials.
3.2. The Pathological Conditions Leading to the Development of HF in Patients with Type 2 Diabetes
3.2.1. The Mechanisms Leading to Coronary Artery Disease (CAD) in Patients with Type 2 Diabetes
The pathological conditions leading to the development of HF in patients with type 2 diabetes were shown in Figure 2.
In patients with diabetes, hyperglycemia, insulin resistance, hyperinsulinemia and dyslipidemia induce the development of endothelial dysfunction and atherosclerosis. Coronary arterial stenosis by atherosclerosis causes obstructive CAD such as angina pectoris. Coronary endothelial dysfunction is thought to be a precursor of obstructive CAD, and is also adversely associated with CV outcomes [38]. In the setting of coronary artery spasm, several clinical studies have demonstrated reduced NO activity which is observed in endothelial dysfunction [39]. The observation that animal models with mutations of the eNOS gene are predisposed to developing coronary artery spasm further supports the contribution of coronary endothelial dysfunction in the pathogenesis of coronary artery spasm [40].
3.2.2. The Mechanisms Leading Coronary Microvascular Dysfunction (CMD) in Patients with Type 2 Diabetes
Diabetics are often affected by coronary microvascular dysfunction (CMD). This is a condition that consists of a combination of vasomotor changes and long-term structural changes in the coronary arterioles, leading to dysregulation of blood flow in response to changes in the oxygen demand of myocardial cells [41]. Hyperglycemia, insulin resistance may play a central role in leading to oxidative stress, inflammatory activation, and altered endothelial barrier function. CMD contributes significantly to CV events without obstructive CAD, and the development of HF, especially HFpEF, in patients with diabetes.
3.2.3. The Mechanisms Leading to Diabetic Cardiomyopathy (DCM) in Patients with Type 2 Diabetes
Multiple mechanisms including hyperglycemia contribute to the development of DCM [42,43,44,45]. In patients with diabetes, the presence of myocardial dysfunction in the absence of overt CAD, valvular disease and other conventional CV risk factors has led to the descriptive terminology, “DCM” [43]. Impaired cardiac insulin resistance, mitochondrial dysfunction, increases in oxidative stress, reduced NO bioavailability, accumulation of AGEs, impaired mitochondrial and cardiomyocyte calcium handling, inflammation, renin angiotensin-aldosterone system (RAAS) activation, cardiac autonomic dysfunction, endoplasmic reticulum (ER) stress have been implicated in the development and progression of DCM. Exposure to increased serum lipid levels including fatty acids (FA) and triglycerides (TG) causes cardiac lipotoxicity which is also associated with the development of DCM [44].
Endothelial dysfunction plays a critical role in the onset, development and progression of DCM [46]. Hyperglycemia, hyperinsulinemia, and insulin resistance induce the endothelial dysfunction, including the reduced function of barrier, impairment of NO bioavailability, excessive production of ROS, oxidative stress, and inflammation. Endothelial dysfunction induces an impairment of myocardial metabolism, a mishandling of intracellular Ca2+, ER stress, mitochondrial dysfunction, excess production of AGEs, and extracellular matrix deposit. Such various hazardous factors induced by endothelial dysfunction lead to cardiac stiffness, fibrosis, and remodeling, resulting in cardiac diastolic and systolic dysfunction, and the development of HF.
3.2.4. A Significance of Endothelial Dysfunction for Development of Pathogenic Conditions for HF in Patients with Type 2 Diabetes
Endothelial dysfunction plays an important role in the development of CAD, CMD and DCM which cause HF.
4. A Significance of Endothelial Dysfunction for Development of CKD in Patients with Type 2 Diabetes
It is known that endothelial dysfunction is not only involved in the onset and progression of CVD, but also an aggravating factor for albuminuria and progression of renal damage, and the severity of endothelial damage increases with the progression of CKD. Endothelial dysfunction plays a central role in pathology of CRS. Endothelial dysfunction is deeply involved in renal microvascular hemodynamics, such as regulation of glomerular filtration and interstitial blood flow, and maintenance of the vascular network, and also plays an important role in the tubulo-glomerular feedback (TGF) and natriuresis. In CKD patients, systemic endothelial dysfunction is observed from an early stage, which may explain why CVD occurs frequently in patients with CKD [7]. The sub-analysis of the Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria (IRMA 2) study showed that endothelial dysfunction was the predicting factor for the progression to diabetic nephropathy in microalbuminuria patients with type 2 diabetes, independently of the traditional risk factors [47]. The flow-mediated vasodilation (FMD) as the marker for endothelial dysfunction was significantly impaired in the patients with elevated urinary albumin excretion compared to normoalbuminuric subjects [48], suggesting a significant association between endothelial dysfunction and albuminuria.
The possible mechanisms leading to CKD in patients with type 2 diabetes were show in Figure 3.
Elevated levels of oxidative stress and ADMA represent the novel risk factors for endothelial dysfunction [49]. There are substantial data demonstrating that elevation in ADMA and oxidative stress markers in CKD patients [50,51]. Brachial artery endothelium-dependent vasodilatation which reflects endothelial function, oxidative stress, and ADMA levels are associated with stage of CKD [7]. Elevation of plasma and tissue ADMA levels in CKD are induced by both reduced renal excretion and reduced catabolism by dimethylarginine dimethylaminohydrolase (DDAH), which is inhibited by oxidative stress in CKD [52].
ADMA is closely associated with the loss of glomerular capillary and glomerular sclerosis, leading to the progression of CKD [53]. DDAH regulates L-arginine: methylarginine levels in specific renal cells [54], thereby regulating cell-specific L-arginine uptake and NO generation in renal tubular epithelium. The TGF sensitivity is coupled to NO in the macula densa. The TGF was enhanced by ADMA. ADMA has been found to accumulate in the erythrocytes of patients with renal failure [55]. Serum ADMA levels were significantly decreased in CKD patients with anemia whom were treated with recombinant human erythropoietin (Epo) [56], which may indicate that the activated erythrocyte turnover reduced accumulation of ADMA in erythrocytes. In such patients, urinary protein levels, carotid intima-media thickness (IMT), pulse wave velocity (PWV), plasma brain natriuretic peptide (BNP) were also significantly decreased. Furthermore, recent studies have shown that erythropoietin protects endothelial function and integrity [57]. Erythropoietin could therefore prevent renal tissue injury and CKD progression.
The ADMA/DDAH may play an important role in the epithelial-mesenchymal transition (EMT) of tubular epithelial cells, which was investigated by using diabetic mice [58]. In the kidneys of diabetic mice, the loss of DDAH induced higher degree of renal interstitial fibrosis and collagen deposition, and larger induction of EMT-related changes and oxidative stress than in the kidney of wild-type mice. Excess oxidative stress induces the injury of epithelial cells of renal tubules, and injured epithelial cells produce endothelial dysfunction-associated molecules and inflammatory cytokines [59]. The injury of renal tubules induces inflammation by myeloid cells and also induces transformation of interstitial fibroblasts into myofibroblasts, which leads to renal fibrosis. Such a myofibroblastic transformation induces the impaired Epo production by renal interstitial fibroblasts, which causes renal anemia. Such anemia and inflammation induced by epithelial dysfunction of renal tubules further increase oxidative stress in the kidney, which thus contributes to unfavorable cycle for the progression of CKD.
Endothelial dysfunction is deeply associated with the development and progression of CKD and diabetic kidney disease (DKD).
5. The Effects of SGLT2i on Endothelial Dysfunction
5.1. The Effects of SGLT2i on Vascular Function Tests
The noninvasive vascular function tests such as FMD and PWV have been performed to evaluate vascular dysfunction and to identify the individuals at high risk for CVD [60,61,62]. FMD has been used as a method to assess endothelial function, and PWV has been used as the marker for arterial stiffness.
The addition of dapagliflozin to metformin (16 weeks) improved endothelial function assessed by FMD, in patients with poorly controlled early-stage type 2 diabetes [63]. In this study, a reduction in oxidative stress contributed to an improvement in FMD. The 2-day treatment with dapagliflozin decreased systolic blood pressure (BP) and oxidative stress [64]. FMD was significantly increased, and PWV was reduced, even after correction for mean BP. Canagliflozin reduced BP and improved arterial stiffness, as assessed by PWV after 6 months, independently of the BP-lowring effect [65]. The effect of SGLT2i on diastolic function and FMD were evaluated in 184 patients with type 2 diabetes and HFpEF [66]. Short-term (12 weeks) SGLT2i-treatment improved diastolic function, and on multiple regression statistically significant associations were seen between the marker for diastolic function and the change in FMD [67]. The 12-month canagliflozin-treatment improved diastolic function and FMD in patients with type 2 diabetes and chronic HF (CHF) [67]. The effect of treatment with tofogliflozin for 6 months on cardiac and vascular endothelial function in patients with type 2 diabetes and heart diseases was evaluated. Tofogliflozin treatment (6 months) significantly decreased left ventricular end-diastolic dimensions and significantly increased FMD [68]. An improvement of diastolic function was significantly correlated with the increase in acetoacetic acid and 3-hydroxybutyrate levels, suggesting that the elevation of ketone bodies by SGLT2i might improve left ventricular dilatation. FMD was significantly improved after the six-month treatment of SGLT2i [69], and multiple regression analysis demonstrated that the change in serum TG was the strongest predictive factor for an improvement of FMD. The switching to SGLT2i was associated with a statistically significant improvement in endothelial function in diabetic patients with CHF after 3 months, and SGLT2i treatment was significantly associated with an improvement of FMD even by multivariable stepwise regression analysis [70]. The meta-analysis including 26 clinical studies assessing the effects of dipeptidyl peptidase-4 (DPP-4) inhibitors, GLP-1 RAs, and SGLT2i on FMD showed that only SGLT2i significantly improved FMD (mean difference [MD], 1.14%; 95% CI, 0.18 to 1.73, p = 0.016), but neither DPP-4 inhibitors (MD, 0.86%; 95% CI: -0.15 to 1.86, p = 0.095) nor GLP-1 RA (MD, 2.37%; 95% CI, -0.51 to 5.25, p = 0.107) improved FMD [71]. Another meta-analysis including 4 trials demonstrated that SGLT2i significantly increased FMD by 1.66% (95%CI, 0.56 to 2.76; p = 0.003) as compared with placebo or active comparator [72].
5.2. The Effects of SGLT2i on Factors-Associated with Endothelial Dysfunction
The effects of SGLT2i on factors-associated with endothelial dysfunction were shown in Figure 4.
In addition to reducing plasma glucose, empagliflozin normalized endothelial function and ROS in aorta and blood of diabetic rats [73]. In addition, SGLT2i ameliorated the pro-inflammatory phenotype and glucotoxicity such as AGE/RAGE signaling in diabetic animals. Ipragliflozin ameliorated impaired eNOS in the abdominal aorta and reduced ROS generation in diabetic mice [74]. Furthermore, ipragliflozin decreased the expression of VCAM-1 and ICAM-1 in the abdominal aorta. In vitro studies showed dapagliflozin-mediated attenuation of TNF-α- and hyperglycemia-induced increases in ICAM-1, VCAM-1, PAI-1 and nuclear factor-kappa B (NFκB) expression [75]. Phlorizin ameliorated the endothelial dysfunction link with the activation of the PI3K/AKT/eNOS signaling pathway and augmentation of the release of NO, in palmitic acid-induced human umbilical vein endothelial cells [76]. L-arginine is a physiological precursor to the formation of NO. SGLT2i treatment increased L-arginine/ADMA ratio [77]. Reduced cardiac production of NO and elevated oxidative stress were observed in the ob/ob−/− mice. An increase in L-arginine/ADMA ratio increased NO bioavailability, improving cardiac contractile function and coronary microvascular function in the ob/ob-/- mice [77]. Empagliflozin and dapagliflozin restored NO bioavailability by inhibiting ROS production rather than affecting eNOS expression/signaling, barrier function and ICAM-1 and VCAM-1 expression in TNFα-induced endothelial cells [78].
SGLT2i reduced BP, and significantly reduced norepinephrine, and improved endothelial function [79], suggesting a beneficial effect of SGLT2i-mediated improvement in activation of the sympathetic nervous system (SNS) on endothelial function. Empagliflozin reduced frailty in diabetic and hypertensive elderly patients, most likely by decreasing the mitochondrial generation of ROS in endothelial cells [80]. The disruption of the endothelial cell glycocalyx leads to cellular dysfunction promoting inflammation and CVD progression. Empagliflozin mitigated endothelial inflammation and attenuated ER stress signaling caused by sustained glycocalyx disruption [81]. Luseogliflozin ameliorated FA-induced endothelial dysfunction by increasing super oxide dismutase 2 (SOD2) expression and decreasing ROS production in the thoracic aorta of high-fat diet-induced obese mice [82]. Along with an improvement in kidney function, oxidized LDL, and diastolic function, FMD significantly increased by canagliflozin in type 2 diabetic patients with CHF [67].
6. The Effects of SGLT2i on Causative Pathological Conditions Leading to HF in Patients with Type 2 Diabetes
6.1. The Effects of SGLT2i on CAD
In the meta-analysis including 22 clinical trials, SGLT2i did not result in any significant differences in the incidence rate of angina pectoris (relative risk [RR], 0.98; 95%CI, 0.83 to 1.14; p = 0.92), angina unstable (RR, 0.95; 95% CI, 0.84 to 1.07; p = 0.84), or myocardial infarction (RR, 0.94; 95%CI, 0.79 to 1.11; p = 0.98) between SGLT2i and control groups [83]. Another meta-analysis showed that SGLT2i significantly reduced MACE, including hospitalization and all-cause mortality in patients with or without ASCVD, and showed a beneficial trend in patients with HFpEF, and no benefits in patients with stroke or myocardial infarction [84]. The meta-analysis including 15,301 patients with CAD showed that SGLT2i were associated with a risk reduction of MACE (HR, 0.84; 95% CI, 0.74 to 0.95), hospitalization for HF (HR, 0.69; 95%CI, 0.58 to 0.83) and a composite of CV death or hospitalization for HF (HR, 0.78; 95%CI, 0.71 to 0.86) in CAD patients [85]. Although no data has been shown so far that SGLT2i suppresses the onset of CAD, it has been demonstrated that SGLT2i suppresses the hospitalization for HF in CAD patients.
6.2. The Effects of SGLT2i on CMD
SGLT2i treatment ameliorated both cardiac contractile function and coronary microvascular function as assessed by fractional area change (FAC) and coronary flow velocity reserve (CFVR), respectively, in prediabetic ob/ob-/- mice [77]. Coronary flow reserve (CFR) is regulated not only by focal stenosis but also by diffuse atherosclerosis and CMD in patients with CAD. The CFR was reduced in the db/db group, however, empagliflozin significantly increased CFR [86]. The number and microvascular coverage of cardiac pericytes were reduced in the db/db mice, however, which were improved by empagliflozin. In short, empagliflozin improved CMD and reduced cardiac pericyte loss in diabetic mice.
6.2. The Beneficial Effects of SGLT2i on DCM
The beneficial effects of SGLT2i on DCM were shown in Table 1. SGLT2i have multiple beneficial factors to improve DCM [87,88,89,90,91,92,93,94,95,96,97,98]. SGLT2i decreased fibrosis, reduced inflammation and improved systolic function. SGLT2i improved diastolic function and reduced mortality in a model of DCM [90]. SGLT2i may be a promising therapeutic option for DCM.
7. The Effects of SGLT2i on CKD
There are several renal protective mechanisms due to SGLT2i [99,100,101]. An improvement in metabolic factors including reduction of body weight and BP, an increase in insulin sensitivity, and a decrease in serum uric acid, may be associated with SGLT2i-mediated renal protection [102]. An increased ketone bodies utilization by diabetic failing renal cells may be beneficially associated with amelioration of renal function. Ketone bodies are also used by the diabetic failing myocardium as a super fuel to improve heart function, and an improvement in CRS leads to further improvement of renal function. SGLT2i decreases the overload of the proximal tubules and improves the tubulointerstitial hypoxic milieu, leading to the recovery of Epo production by fibroblasts [103]. Therefore, increased hematocrit by SGLT2i suggests the recovery of tubulointerstitial function in DKD [103]. Elevated Epo may also contribute to the renal protective effect of SGLT2i [104]. The treatment with human erythropoietin protected the kidney of streptozotocin-induced diabetic rats [105]. Epo was reported to protect podocytes from the injury by AGEs in mice [106]. Epo ameliorated the injury of podocyte in advanced DKD in db/db mice [107]. An elevated expression of SGLT2 increases the renal NaCl reabsorption in the proximal tubule, inducing a significant reduction in distal NaCl delivery to the macula densa [108]. The decreased NaCl delivery to macula densa is sensed as a decrease in plasma volume, which leads to maladaptive glomerular afferent arterial vasodilatation, such abnormally enhanced TGF increases intraglomerular pressure [109], worsening renal function. SGLT2i restores normal TGF [110], which may reduce albuminuria and maintain eGFR. The above mentioned renal protective mechanisms by SGLT2i are significantly associated with an improvement of endothelial dysfunction.
Several meta-analyses reported beneficial effects of SGLT2i on CKD. SGLT2i was significantly associated with the reduction of albuminuria in patients with type 2 diabetes and CKD [111]. SGLT2i improved the CVD risk and renal outcomes in patients with type 2 diabetes and CKD [112]. In patients with cardiometabolic and kidney disease, SGLT2i improved CV and kidney outcomes, regardless of type 2 diabetes, HF, and/or CKD status [113]. The magnitude of risk reduction was largest for hospitalization for HF and progression of CKD. In the meta-analysis investigated the CVD and renal outcomes with SGLT2i vs. GLP-1RAs in type 2 diabetic patients with CKD, SGLT2i were associated with a decreased risk of CVD and renal events, but GLP-1RAs were not [114]. SGLT2i significantly reduced the risk for renal events as compared with GLP-1RAs. SGLT2i reduced the risk of renal outcomes and MACE for patients with type 2 diabetes and CKD stage 3b-4 [115]. SGLT2i significantly reduced the risk of the primary outcome including worsening kidney function, ESRD, or renal death in CKD patients [116].
7. Conclusions
SGLT2i improve endothelial dysfunction measured by FMD in individuals at high risk for CVD. The improvements in endothelial dysfunction and endothelium-derived factors may play an important role in preventing the development of CAD, CMD and DCM which cause HF and in retarding CKD. The suppression of HF and CKD progression achieved by SGLT2i may be largely due to the improvement of endothelial function by SGLT2i.
Author Contributions
H.Y., M.H., H.A. and H. K. conceived the review; H. Y. wrote the paper; H. K. edited the paper and provided critical guidance. All authors read and approved the final version of this paper.
Funding
This review research received no external funding.
Institutional Review Board Statement
Not applicable because this is a systematic review article and do not include the original data involving humans or animals.
Informed Consent Statement
Not applicable because this is a systematic review article and do not include the original data involving humans.
Data Availability Statement
This is systematic review article and do not include the original data, and we showed all papers we systematically reviewed in the section of “References”.
Conflicts of Interest
The authors declare no conflict of interest in relation to the present review paper.
References
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H. Sodium-glucose cotransporter 2 inhibitors and death and heart failure in type 2 diabetes. Ann. Transl. Med. 2017, 5, 470. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: an exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet. Diabetes. Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; et al. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am. J. Kidney. Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef]
- Yang, Z.; Ming, X.F. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin. Med. Res. 2006, 4, 53–65. [Google Scholar] [CrossRef]
- Bauer, V.; Sotnikova, R. Nitric oxide - the endothelium-derived relaxing factor and its role in endothelial functions. Gen. Physiol. Biophys. 2010, 29, 319–340. [Google Scholar] [CrossRef]
- Stankevicius, E.; Kevelaitis, E.; Vainorius, E.; Simonsen, U. Role of nitric oxide and other endothelium-derived factors. Medicina (Kaunas). 2003, 39, 333–341. [Google Scholar]
- Morigi, M.; Angioletti, S.; Imberti, B.; Donadelli, R.; Micheletti, G.; Figliuzzi, M.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Leukocyte–endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-κB-dependent fashion. J. Clin. Invest. 1998, 101, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Hu, Y.S.; Medcalf, R.L.; Simpson, R.W.; Dear, A.E. Thiazolidinediones inhibit TNFalpha induction of PAI-1 independent of PPARgamma activation. Biochem. Biophys. Res. Commun. 2005, 334, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, A.; Madan, B.; Singhal, V.; Ghosh, B. Isoliquiritigenin inhibits IκB kinase activity and ROS generation to block TNF-alpha induced expression of cell adhesion molecules on human endothelial cells. Biochem. Pharmacol. 2007, 73, 1602–1612. [Google Scholar] [CrossRef]
- Sobel, B.E.; Woodcock-Mitchell, J.; Schneider, D.J.; Holt, R.E.; Marutsuka, K.; Gold, H. Increased plasminogen activator inhibitor type 1 in coronary artery atherectomy specimens from type 2 diabetic compared with nondiabetic patients: a potential factor predisposing to thrombosis and its persistence. Circulation. 1998, 97, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.J.; Mao, S.L.; Taylor, K.L.; Kanjanabuch, T.; Guan, Y.; Zhang, Y.; Brown, N.; et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. 2004, 53, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Balletshofer, B.M.; Rittig, K.; Enderle, M.D.; Volk, A.; Maerker, E.; Jacob, S.; Matthaei, S.; Rett, K.; Häring, H.U. Endothelial dysfunction is detectable in young normotensive first-degree relatives of subjects with type 2 diabetes in association with insulin resistance. Circulation. 2000, 101, 1780–1784. [Google Scholar] [CrossRef]
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Invest. 1996, 98, 894–898. [Google Scholar] [CrossRef]
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000, 101, 1539–1545. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxidesynthesis in chronic renal failure. Lancet. 1992, 339, 572–575. [Google Scholar]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethy-larginine causes hypertension and cardiac dysfunction inhumans and is actively metabolized by dimethylargininedimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Impraim, B.; Simmel, S.; Bode-Böger, S.M.; Tsikas, D.; Frölich, J.C.; Hoeper, M.M.; Haller, H.; Fliser, D. Cardiovascular effectsof systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation. 2004, 109, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Mondon, C.E.; Cooke, J.P. Insulin resistance: potential role of the endogenous nitric oxide synthase inhibitor ADMA. Vasc. Med. 2005, 10 (Suppl 1), S35–S43. [Google Scholar] [CrossRef]
- Madden, J.A. Role of the vascular endothelium and plaque in acute ischemic stroke. Neurology. 2012, 79 (13 Suppl 1), S58–S62. [Google Scholar] [CrossRef]
- Toda, N.; Nakanishi-Toda, M. How mental stress affects endothelial function. Pflugers. Arch. 2011, 462. 779-794.
- Huhtinen, K.; O'Byrne, J.; Lindquist, P.J.; Contreras, J.A.; Alexson, S.E. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 2002, 277, 3424–3432. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox. Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhang, C.; Zhang, G.; Tao, J. Endothelial progenitor cells in cardiovascular diseases. Aging. Med (Milton). 2018, 1, 204–208. [Google Scholar] [CrossRef]
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into endothelial progenitor cells: origin, classification, potentials, and prospects. Stem. Cells. Int. 2018, 2018, 9847015. [Google Scholar] [CrossRef]
- Rigato, M.; Avogaro, A.; Fadini, G.P. Levels of circulating progenitor cells, cardiovascular outcomes and death: a meta-analysis of prospective observational studies. Circ. Res. 2016, 118, 1930–1939. [Google Scholar] [CrossRef]
- Kenchaiah, S.; Narula, J.; Vasan, R.S. Risk factors for heart failure. Med. Clin. North. Am. 2004, 88, 1145–1172. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, V.H.; Billaudelle, A.M.; Schulz, A.; Keller, K.; Hahad, O.; Tröbs, S.O.; Koeck, T.; et al. Disturbed Glucose Metabolism and Left Ventricular Geometry in the General Population. J. Clin. Med. 2021, 10, 3851. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.; Sarkar, P.; Naskar, S.; Biswas, U.; Islam, S.K.S. Asian J Med Sci. 2021;12:96.
- Rørth, R.; Jhund, P.S.; Mogensen, U.M.; Kristensen, S.L.; Petrie, M.C.; Køber, L.; McMurray, J.J.V. Risk of Incident Heart Failure in Patients With Diabetes and Asymptomatic Left Ventricular Systolic Dysfunction. Diabetes. Care. 2018, 41, 1285–1291. [Google Scholar] [CrossRef]
- MacDonald, M.R.; Petrie, M.C.; Varyani, F.; Ostergren, J.; Michelson, E.L.; Young, J.B.; Solomon, S.D.; et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur. Heart. J. 2008, 29, 1377–1385. [Google Scholar] [CrossRef]
- Boonman-de Winter, L.J.; Rutten, F.H.; Cramer, M.J.; Landman, M.J.; Liem, A.H.; Rutten, G.E.; Hoes, A.W. High prevalence of previously unknown heart failure and left ventricular dysfunction in patients with type 2 diabetes. Diabetologia. 2012, 55, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Suwaidi, J.A.; Hamasaki, S.; Higano, S.T.; Nishimura, R.A.; Holmes, D.R. Jr; Lerman, A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000, 101, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Kugiyama, K.; Yasue, H.; Okumura, K.; Ogawa, H.; Fujimoto, K.; Nakao, K.; Yoshimura, M.; Motoyama, T.; Inobe, Y.; Kawano, H. Nitric oxide activity is deficient in spasm arteries of patients with coronary spastic angina. Circulation. 1996, 94, 266–271. [Google Scholar] [CrossRef]
- Nakayama, M.; Yasue, H.; Yoshimura, M.; Shimasaki, Y.; Kugiyama, K.; Ogawa, H.; Motoyama, T.; et al. T-786-->C mutation in the 5'-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation. 1999, 99, 2864–2870. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines. 2022, 10, 2274. [Google Scholar] [CrossRef]
- Marwick TH, Ritchie R, Shaw JE, Kaye D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J Am Coll Cardiol. 2018 Jan 23;71(3):339-351.
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Dillmann, W.H. Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1160–1162. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Givertz, M.M.; Aguilar, D.; Allen, L.A.; Chan, M.; Desai, A.S.; Deswal, A.; et al. Type 2 Diabetes Mellitus and Heart Failure: A Scientific Statement From the American Heart Association and the Heart Failure Society of America: This statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation. 2019, 140, e294–e324. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol (Lausanne). 2022, 13, 851941. [Google Scholar] [CrossRef] [PubMed]
- Persson, F.; Rossing, P.; Hovind, P.; Stehouwer, C.D.; Schalkwijk, C.G.; Tarnow, L.; Parving, H.H. Endothelial dysfunction and inflammation predict development of diabetic nephropathy in the Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria (IRMA 2) study. Scand. J. Clin. Lab. Invest. 2008, 68, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.; Jensen, J.S.; Jensen, G.; Borch-Johnsen, K.; Feldt-Rasmussen, B. Elevated urinary albumin excretion is associated with impaired arterial dilatory capacity in clinically healthy subjects. Circulation. 2001, 103, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; T. Münzel, T. ADMA and oxidative stress. Atherosclerosis. 2003, 4 (Suppl), 41–51. [Google Scholar] [CrossRef] [PubMed]
- Ghiadoni, L.; Cupisti, A.; Huang, Y.; Mattei, P.; Cardinal, H.; Favilla, S.; Rindi, P.; Barsotti, G.; Taddei, S.; Salvetti, A. Endothelial dysfunction and oxidative stress in chronic renal failure. J. Nephrol. 2004, 17, 512–519. [Google Scholar]
- Kielstein, J.T.; Böger, R.H.; Bode-Böger, S.M.; Frölich, J.C.; Haller, H.; Ritz, E.; Fliser, D. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J. Am. Soc. Nephrol. 2002, 13, 170–176. [Google Scholar] [CrossRef]
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220. [Google Scholar] [CrossRef]
- Ueda, S.; Yamagishi, S.; Matsumoto, Y.; Kaida, Y.; Fujimi-Hayashida, A.; Koike, K.; Tanaka, H.; Fukami, K.; Okuda, S. Involvement of asymmetric dimethylarginine (ADMA) in glomerular capillary loss and sclerosis in a rat model of chronic kidney disease (CKD). Life. Sci. 2009, 84, 853–856. [Google Scholar] [CrossRef]
- Tojo, A.; Welch, W.J.; Bremer, V.; Kimoto, M.; Kimura, K.; Omata, M.; Ogawa, T.; Vallance, P.; Wilcox, C.S. Colocalization of demethylating enzymes and NOS and functional effects of methylarginines in rat kidney. Kidney. Int. 1997, 52, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.; van Hell, A.J.; Visser, M.; Nijveldt, R.J.; van Leeuwen, P.A.; Teerlink, T. Role of the human erythrocyte in generation and storage of asymmetric dimethylarginine. Am. J. Physiol. Heart. Circ. Physiol. 2012, 302, H1762–H1770. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Nakamura, T.; Sato, E.; Kawagoe, Y.; Hikichi, Y.; Ueda, Y.; Node, K. Renovascular protective effects of erythropoietin in patients with chronic kidney disease. Intern. Med. 2011, 50, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D. Perspectives in renal disease progression: the endothelium as a treatment target in chronic kidney disease. J. Nephrol. 2010, 23, 369–376. [Google Scholar]
- Shi, L.; Zhao, C.; Wang, H.; Lei, T.; Liu, S.; Cao, J.; Lu, Z. Dimethylarginine Dimethylaminohydrolase 1 Deficiency Induces the Epithelial to Mesenchymal Transition in Renal Proximal Tubular Epithelial Cells and Exacerbates Kidney Damage in Aged and Diabetic Mice. Antioxid. Redox. Signal. 2017, 27, 1347–1360. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef]
- Greenland, P.; Alpert, J.S.; Beller, G.A.; Benjamin, E.J.; Budoff, M.J.; Fayad, Z.A.; Foster, E.; et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Circulation. 2010, 122, e584–e636. [Google Scholar]
- Vlachopoulos, C.; Xaplanteris, P.; Aboyans, V.; Brodmann, M.; Cifkova, R.; Cosentino, F.; De Carlo, M.; et al. The role of vascular biomarkers for primary and secondary prevention. A position paper from the European Society of Cardiology Working Group on peripheral circulation: endorsed by the Association for Research into Arterial Structure and Physiology (ARTERY) Society. Atherosclerosis. 2015, 241, 507–532. [Google Scholar]
- Maruhashi, T.; Soga, J.; Fujimura, N.; Idei, N.; Mikami, S.; Iwamoto, Y.; Iwamoto, A.; Kajikawa, M.; et al. Endothelial dysfunction, increased arterial stiffness, and cardiovascular risk prediction in patients with coronary artery disease: FMD-J (Flow-Mediated Dilation Japan) Study A. J. Am. Heart. Assoc. 2018, 7, e008588. [Google Scholar] [CrossRef]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: a pilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.J.; Sanchez, M.J.; Sanchez, R.A. Diabetic patients with essential hypertension treated with amlodipine: blood pressure and arterial stiffness effects of canagliflozin or perindopril. J. Hypertens. 2019, 37, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Miura, S. Effects of Sodium-Glucose Cotransporter 2 Inhibitor on Vascular Endothelial and Diastolic Function in Heart Failure With Preserved Ejection Fraction- Novel Prospective Cohort Study. Circ. Rep. 2019, 1, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Sezai, A.; Sekino, H.; Unosawa, S.; Taoka, M.; Osaka, S.; Tanaka, M. Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc. Diabetol. 2019, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Tochiya, M.; Makino, H.; Tamanaha, T.; Matsuo, M.; Hishida, A.; Koezuka, R.; Ohata, Y. Effect of tofogliflozin on cardiac and vascular endothelial function in patients with type 2 diabetes and heart diseases: A pilot study. J. Diabetes. Investig. 2020, 11, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Uzu, K.; Hashimoto, N.; Onishi, T.; Takaya, T.; Shimane, A.; Taniguchi, Y.; Yasaka, Y.; Ohara, T.; Kawai, H. Empagliflozin's Ameliorating Effect on Plasma Triglycerides: Association with Endothelial Function Recovery in Diabetic Patients with Coronary Artery Disease. J. Atheroscler. Thromb. 2020, 27, 644–656. [Google Scholar] [CrossRef]
- Correale, M.; Mazzeo, P.; Mallardi, A.; Leopizzi, A.; Tricarico, L.; Fortunato, M.; Magnesa, M.; et al. Switch to SGLT2 Inhibitors and Improved Endothelial Function in Diabetic Patients with Chronic Heart Failure. Cardiovasc. Drugs. Ther. 2022, 36, 1157–1164. [Google Scholar] [CrossRef]
- Batzias, K.; Antonopoulos, A.S.; Oikonomou, E.; Siasos, G.; Bletsa, E.; Stampouloglou, P.K.; Mistakidi, C.V.; et al. Effects of Newer Antidiabetic Drugs on Endothelial Function and Arterial Stiffness: A Systematic Review and Meta-Analysis. J. Diabetes. Res. 2018, 2018, 1232583. [Google Scholar] [CrossRef]
- Patoulias, D.; Papadopoulos, C.; Kassimis, G.; Vassilikos, V.; Karagiannis, A.; Doumas, M. Meta-Analysis Addressing the Effect of Sodium-Glucose Cotransporter 2 Inhibitors on Flow-Mediated Dilation in Patients With Type 2 Diabetes Mellitus. Am. J. Cardiol. 2022, 165, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS. One. 2014, 9, e112394. [Google Scholar] [CrossRef]
- Salim, H.M.; Fukuda, D.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front. Cardiovasc. Med. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diab. Vasc. Dis. Res. 2018, 15, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Wang, L.X.; Dong, S.S.; Hong, Y.; Zhou, X.H.; Zheng, W.W.; Zheng, C. Phlorizin Exerts Direct Protective Effects on Palmitic Acid (PA)-Induced Endothelial Dysfunction by Activating the PI3K/AKT/eNOS Signaling Pathway and Increasing the Levels of Nitric Oxide (NO). Med. Sci. Monit. Basic. Res. 2018, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.M.; Jönsson-Rylander, A.C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/- mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [PubMed]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.C.; Arcambal, A.; et al. SGLT2 Inhibitor-Induced Sympathoinhibition: A Novel Mechanism for Cardiorenal Protection. JACC. Basic. Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Mone, P.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Lombardi, A.; Frullone, S.; Santulli, G. SGLT2 Inhibition via Empagliflozin Improves Endothelial Function and Reduces Mitochondrial Oxidative Stress: Insights From Frail Hypertensive and Diabetic Patients. Hypertension. 2022, 79, 1633–1643. [Google Scholar] [CrossRef]
- Campeau, M.A.; Leask, R.L. Empagliflozin mitigates endothelial inflammation and attenuates endoplasmic reticulum stress signaling caused by sustained glycocalyx disruption. Sci. Rep. 2022, 12, 12681. [Google Scholar] [CrossRef]
- Kawade, S.; Ogiso, K.; Shayo, S.C.; Obo, T.; Arimura, A.; Hashiguchi, H.; Deguchi, T.; Nishio, Y. Luseogliflozin and caloric intake restriction increase superoxide dismutase 2 expression, promote antioxidative effects, and attenuate aortic endothelial dysfunction in diet-induced obese mice. J. Diabetes. Investig. 2023, 14, 548–559. [Google Scholar] [CrossRef]
- Ye, G.; Wang, S.; Peng, D. Effects of SGLT2 Inhibitor on Ischemic Events Stemming From Atherosclerotic Coronary Diseases: A Systematic Review and Meta-analysis With Trial Sequential Analysis of Randomized Controlled Trials. J. Cardiovasc. Pharmacol. 2021, 77, 787–795. [Google Scholar] [CrossRef]
- Eraikhuemen, N.; Leung, S.; Warren, S.B.; Lazaridis, D.; Smith, C.H.; Kearson, M.L.; Marcellus, V. Effects of the Sodium-Glucose Cotransporter Inhibitors on Cardiovascular Death and All-Cause Mortality: A Systematic Review and Meta-analysis of Randomized Placebo-Controlled Clinical Trials. Am. J. Cardiovasc. Drugs. 2023, 23, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, J.; Chen, S.; Xu, X.; Guo, D.; He, Y.; Huang, Z.; et al. Sodium Glucose Cotransporter Type 2 Inhibitors Improve Cardiorenal Outcome of Patients With Coronary Artery Disease: A Meta-Analysis. Front Endocrinol (Lausanne). 2022, 13, 850836. [Google Scholar] [CrossRef]
- Tu, Y.; Li, Q.; Zhou, Y.; Ye, Z.; Wu, C.; Xie, E.; Li, Y.; et al. Empagliflozin inhibits coronary microvascular dysfunction and reduces cardiac pericyte loss in db/db mice. Front. Cardiovasc. Med. 2022, 9, 995216. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, W. The Sodium-Glucose Co-Transporter 2 Inhibitor, Empagliflozin, Protects against Diabetic Cardiomyopathy by Inhibition of the Endoplasmic Reticulum Stress Pathway. Cell. Physiol. Biochem. 2017, 41, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Li, T.; Wang, Y.; Chang, Y.; Cheng, Y.; Lu, Y.; Liu, X.; et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci (Lond). 2019, 133, 1705–1720. [Google Scholar] [CrossRef] [PubMed]
- Arow, M.; Waldman, M.; Yadin, D.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Freimark, D.; et al. Sodium-glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy. Cardiovasc. Diabetol. 2020, 19, 7. [Google Scholar] [CrossRef]
- Moellmann, J.; Klinkhammer, B.M.; Droste, P.; Kappel, B.; Haj-Yehia, E.; Maxeiner, S.; Artati, A.; et al. Empagliflozin improves left ventricular diastolic function of db/db mice. Biochim. Biophys. Acta. Mol. Basis. Dis. 2020, 1866, 165807. [Google Scholar] [CrossRef] [PubMed]
- Trang, N.N.; Chung, C.C.; Lee, T.W.; Cheng, W.L.; Kao, Y.H.; Huang, S.Y.; Lee, T.I.; Chen, Y.J. Empagliflozin and Liraglutide Differentially Modulate Cardiac Metabolism in Diabetic Cardiomyopathy in Rats. Int. J. Mol. Sci. 2021, 22, 1177. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef]
- Wang, J.; Huang, X.; Liu, H.; Chen, Y.; Li, P.; Liu, L.; Li, J.; et al. Empagliflozin Ameliorates Diabetic Cardiomyopathy via Attenuating Oxidative Stress and Improving Mitochondrial Function. Oxid. Med. Cell. Longev. 2022, 2022, 1122494. [Google Scholar] [CrossRef]
- Xi, Y.; Chen, D.; Dong, Z.; Zhang, J.; Lam, H.; He, J.; Du, K.; Chen, C.; Guo, J.; Xiao, J. Multi-omics insights into potential mechanism of SGLT2 inhibitors cardiovascular benefit in diabetic cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 999254. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Shi, H.; Xiong, L.; Wang, P.; Shi, Y. Canagliflozin mitigates ferroptosis and improves myocardial oxidative stress in mice with diabetic cardiomyopathy. Front. Endocrinol (Lausanne). 2022, 13, 1011669. [Google Scholar] [CrossRef] [PubMed]
- Kadosaka, T.; Watanabe, M.; Natsui, H.; Koizumi, T.; Nakao, M.; Koya, T.; Hagiwara, H.; et al. Empagliflozin attenuates arrhythmogenesis in diabetic cardiomyopathy by normalizing intracellular Ca2+ handling in ventricular cardiomyocytes. Am. J. Physiol. Heart. Circ. Physiol. 2023, 324, H341–H354. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Moscato, S.; Cufaro, M.C.; Pieragostino, D.; Mattii, L.; Del Boccio, P.; Ghelardoni, S.; Zucchi, R.; De Caterina, R. Empagliflozin inhibits excessive autophagy through the AMPK/GSK3β signaling pathway in diabetic cardiomyopathy. Cardiovasc. Res. 2023, cvad009. [Google Scholar]
- Croteau, D.; Baka, T.; Young, S.; He, H.; Chambers, J.M.; Qin, F.; Panagia, M.; et al. SGLT2 inhibitor ertugliflozin decreases elevated intracellular sodium, and improves energetics and contractile function in diabetic cardiomyopathy. Biomed. Pharmacother. 2023, 160, 114310. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Lang, J.; Xia, P.; Zhao, X.; Wang, L.; Yu, Y.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibition on renal function and albuminuria in patients with type 2 diabetes: a systematic review and meta-analysis. PeerJ. 2017, 5, e3405. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. Multi-Organ Protective Effects of Sodium Glucose Cotransporter 2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 4416. [Google Scholar] [CrossRef] [PubMed]
- Koguchi, A.; Adachi, H.; Yanai, H. The Application of Sodium-Glucose Cotransporter 2 Inhibitors to Chronic Kidney Disease Stage 4. J. Clin. Med. Res. 2017, 9, 1029–1031. [Google Scholar] [CrossRef]
- Yanai, H. Dipeptidyl Peptidase-4 Inhibitor versus Sodium-Glucose Cotransporter-2 Inhibitor in the Management of Type 2 Diabetes. J. Endocrinol. Metab. 2019, 9, 117–119. [Google Scholar] [CrossRef]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased Hematocrit during Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyama, H. A Possible Mechanism for Renoprotective Effect of Sodium-Glucose Cotransporter 2 Inhibitor: Elevation of Erythropoietin Production. J. Clin. Med. Res. 2017, 9, 178–179. [Google Scholar] [CrossRef]
- Toba, H.; Sawai, N.; Morishita, M.; Murata, S.; Yoshida, M.; Nakashima, K.; Morita, Y.; Kobara, M.; Nakata, T. Chronic treatment with recombinant human erythropoietin exerts renoprotective effects beyond hematopoiesis in streptozotocin-induced diabetic rat. Eur. J. Pharmacol. 2009, 612, 106–114. [Google Scholar] [CrossRef]
- Ruester, C.; Franke, S.; Bondeva, T.; Wolf, G. Erythropoietin protects podocytes from damage by advanced glycation end-products. Nephron Exp. Nephrol. 2011, 117, e21–30. [Google Scholar] [CrossRef]
- Loeffler, I.; Ruster, C.; Franke, S.; Liebisch, M.; Wolf, G. Erythropoietin ameliorates podocyte injury in advanced diabetic nephropathy in the db/db mouse. Am. J. Physiol. Renal. Physiol. 2013, 305, F911–918. [Google Scholar] [CrossRef]
- Vallon, V.; Mühlbauer, B.; Osswald, H. Adenosine and kidney function. Physiol. Rev. 2006, 86, 901–940. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Richter, K.; Blantz, R.C.; Thomson, S.; Osswald, H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J. Am. Soc. Nephrol. 1999, 10, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Rieg, T.; Miracle, C.; Mansoury, H.; Whaley, J.; Vallon, V.; Singh, P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R75–R83. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, Y.; Lang, J.; Xia, P.; Zhao, X.; Wang, L.; Yu, Y.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibition on renal function and albuminuria in patients with type 2 diabetes: a systematic review and meta-analysis. PeerJ. 2017, 5, e3405. [Google Scholar] [CrossRef]
- Toyama, T.; Neuen, B.L.; Jun, M.; Ohkuma, T.; Neal, B.; Jardine, M.J.; Heerspink, H.L.; et al. Effect of SGLT2 inhibitors on cardiovascular, renal and safety outcomes in patients with type 2 diabetes mellitus and chronic kidney disease: A systematic review and meta-analysis. Diabetes. Obes. Metab. 2019, 21, 1237–1250. [Google Scholar] [CrossRef]
- Salah, H.M.; Al'Aref, S.J.; Khan, M.S.; Al-Hawwas, M.; Vallurupalli, S.; Mehta, J.L.; Mounsey, J.P.; et al. Effect of sodium-glucose cotransporter 2 inhibitors on cardiovascular and kidney outcomes-Systematic review and meta-analysis of randomized placebo-controlled trials. Am. Heart. J. 2021, 232, 10–22. [Google Scholar] [CrossRef]
- Yamada, T.; Wakabayashi, M.; Bhalla, A.; Chopra, N.; Miyashita, H.; Mikami, T.; Ueyama, H.; et al. Cardiovascular and renal outcomes with SGLT-2 inhibitors versus GLP-1 receptor agonists in patients with type 2 diabetes mellitus and chronic kidney disease: a systematic review and network meta-analysis. Cardiovasc. Diabetol. 2021, 20, 14. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Liu, Y.; Tian, Z.; Lian, Y.; Jia, J.; Liu, M.; Li, D. Sodium-glucose cotransporter 2 inhibitors benefit to kidney and cardiovascular outcomes for patients with type 2 diabetes mellitus and chronic kidney disease 3b-4: A systematic review and meta-analysis of randomized clinical trials. Diabetes. Res. Clin. Pract. 2021, 180, 109033. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lv, D.; Zhu, X.; Wei, P.; Gui, Y.; Liu, S.; Zhou, E.; Zheng, M.; Zhou, D.; Zhang, L. Effects of SGLT2 Inhibitors on Renal Outcomes in Patients With Chronic Kidney Disease: A Meta-Analysis. Front. Med (Lausanne). 2021, 8, 728089. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The endothelium-derived molecules and their effects, induced by endothelial dysfunction due to diabetes or insulin resistance on atherosclerosis. AGE, advanced glycation end products; ADMA, asymmetric dimethylarginine; eNOS, endothelial nitric oxide synthase; EPCs, endothelial progenitor cells; ET-1, endothelin-1; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; SR, scavenger receptor; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1.
Figure 1.
The endothelium-derived molecules and their effects, induced by endothelial dysfunction due to diabetes or insulin resistance on atherosclerosis. AGE, advanced glycation end products; ADMA, asymmetric dimethylarginine; eNOS, endothelial nitric oxide synthase; EPCs, endothelial progenitor cells; ET-1, endothelin-1; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; SR, scavenger receptor; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1.
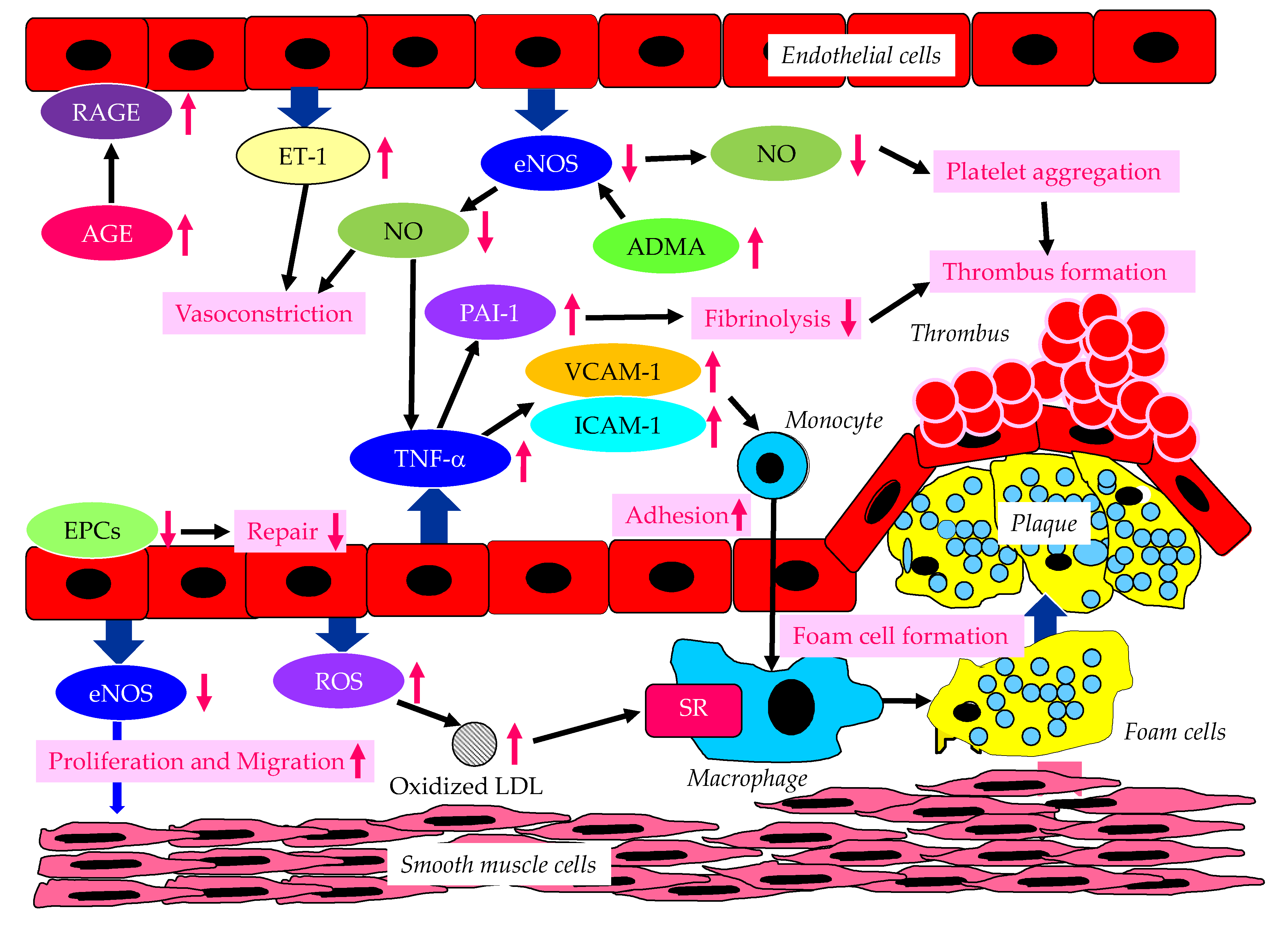
Figure 2.
The pathological conditions leading to the development of heart failure in patients with type 2 diabetes. AGE, advanced glycation end products; ER, endoplasmic reticulum; FA, fatty acids; RAAS, renin-angiotensin-aldosterone system; RAGE, receptor for advanced glycation end products.
Figure 2.
The pathological conditions leading to the development of heart failure in patients with type 2 diabetes. AGE, advanced glycation end products; ER, endoplasmic reticulum; FA, fatty acids; RAAS, renin-angiotensin-aldosterone system; RAGE, receptor for advanced glycation end products.
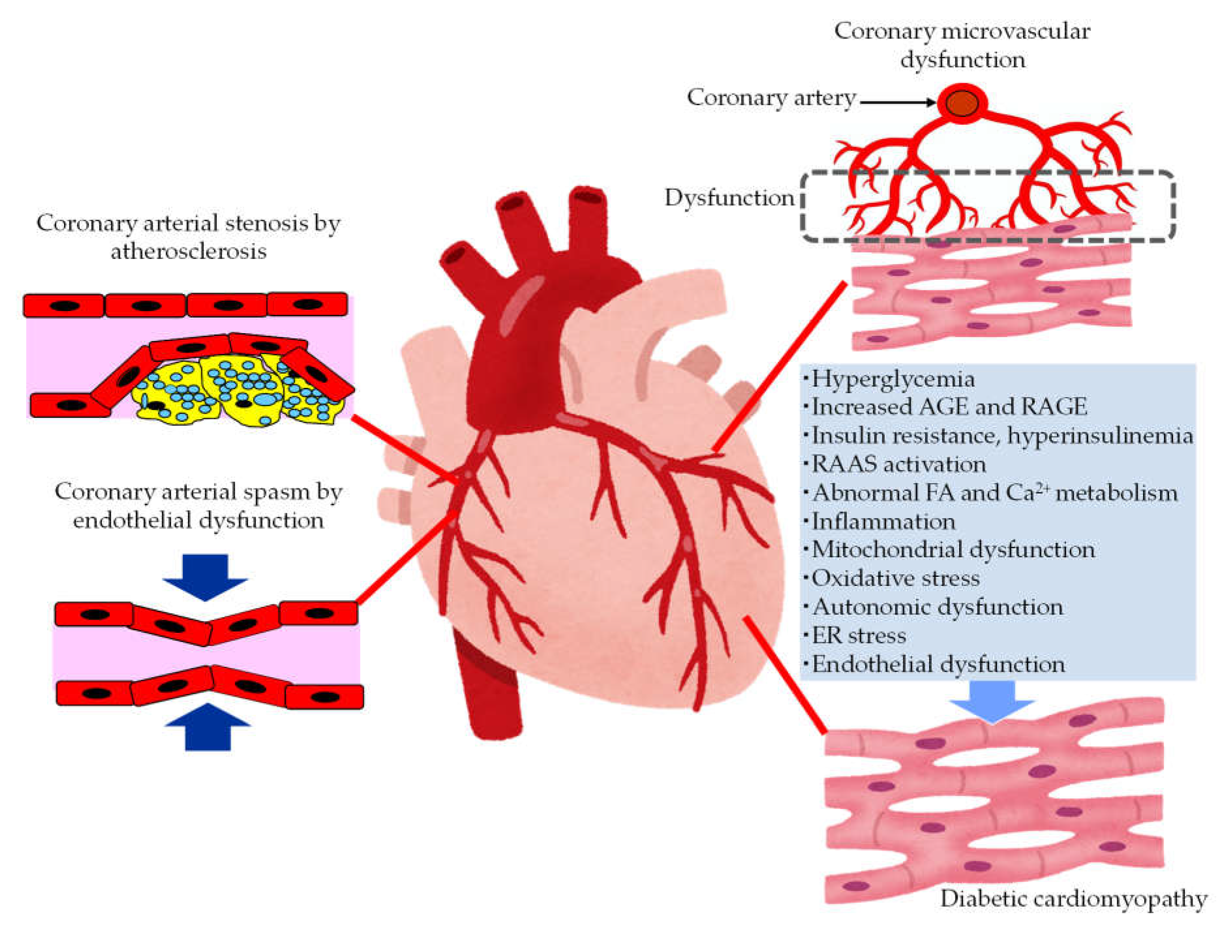
Figure 3.
The possible mechanisms leading to CKD in patients with type 2 diabetes. ADMA, asymmetric dimethylarginine; DDAH, dimethylarginine dimethylaminohydrolase; Epo, erythropoietin; Glu, glucose; SGLT2, sodium-glucose cotransporter 2; TGF, tubulo-glomerular feedback.
Figure 3.
The possible mechanisms leading to CKD in patients with type 2 diabetes. ADMA, asymmetric dimethylarginine; DDAH, dimethylarginine dimethylaminohydrolase; Epo, erythropoietin; Glu, glucose; SGLT2, sodium-glucose cotransporter 2; TGF, tubulo-glomerular feedback.
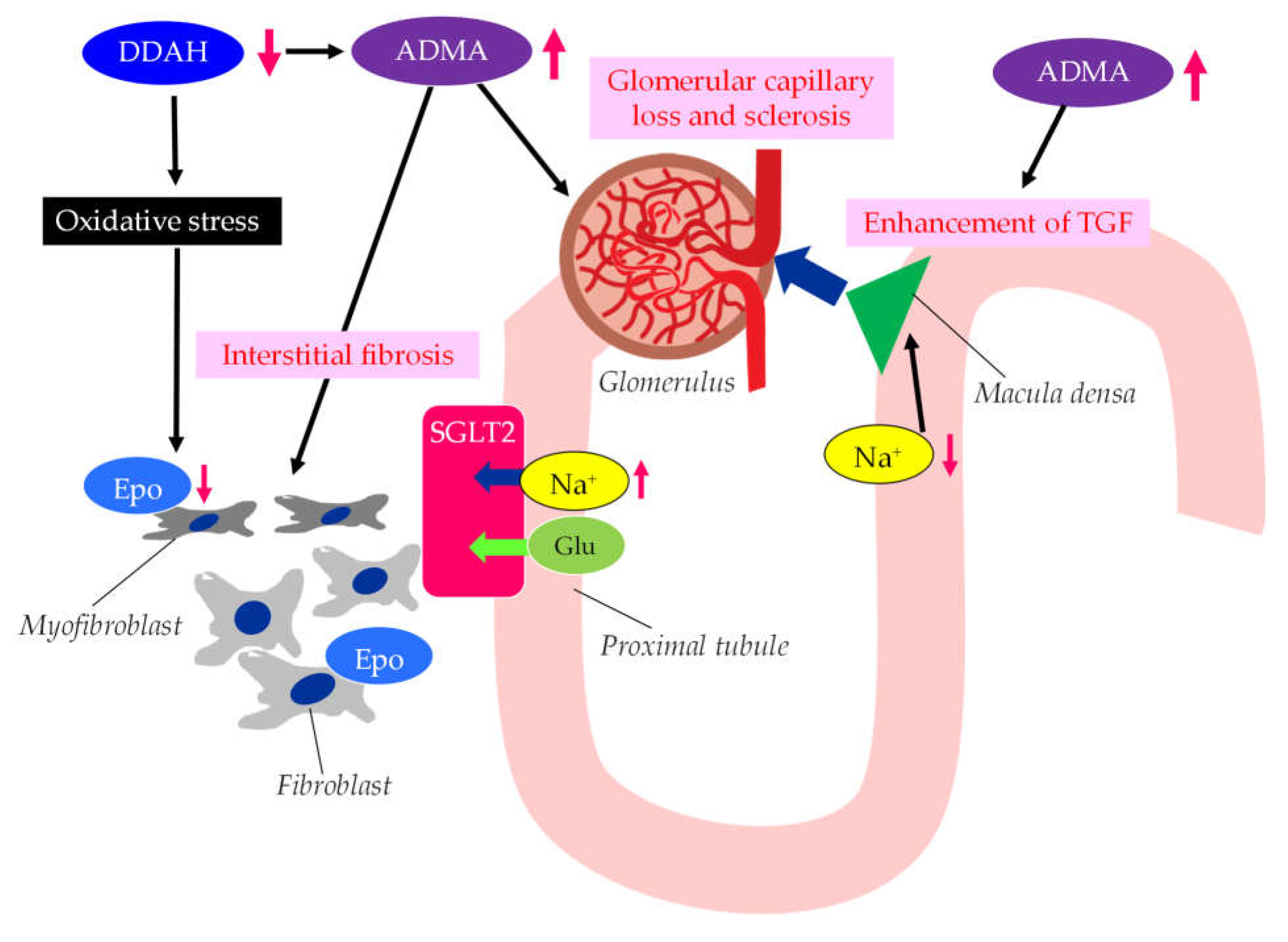
Figure 4.
The effects of SGLT2i on endothelium-derived factors induced by endothelial dysfunction. AGE, advanced glycation end products; ADMA, asymmetric dimethylarginine; eNOS, endothelial nitric oxide synthase; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; NFκB, nuclear factor-kappa B; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; SOD-2, super oxide dismutase-2; SR, scavenger receptor; VCAM-1, vascular cell adhesion molecule-1.
Figure 4.
The effects of SGLT2i on endothelium-derived factors induced by endothelial dysfunction. AGE, advanced glycation end products; ADMA, asymmetric dimethylarginine; eNOS, endothelial nitric oxide synthase; ICAM-1, intercellular adhesion molecule-1; LDL, low-density lipoprotein; NFκB, nuclear factor-kappa B; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; SOD-2, super oxide dismutase-2; SR, scavenger receptor; VCAM-1, vascular cell adhesion molecule-1.
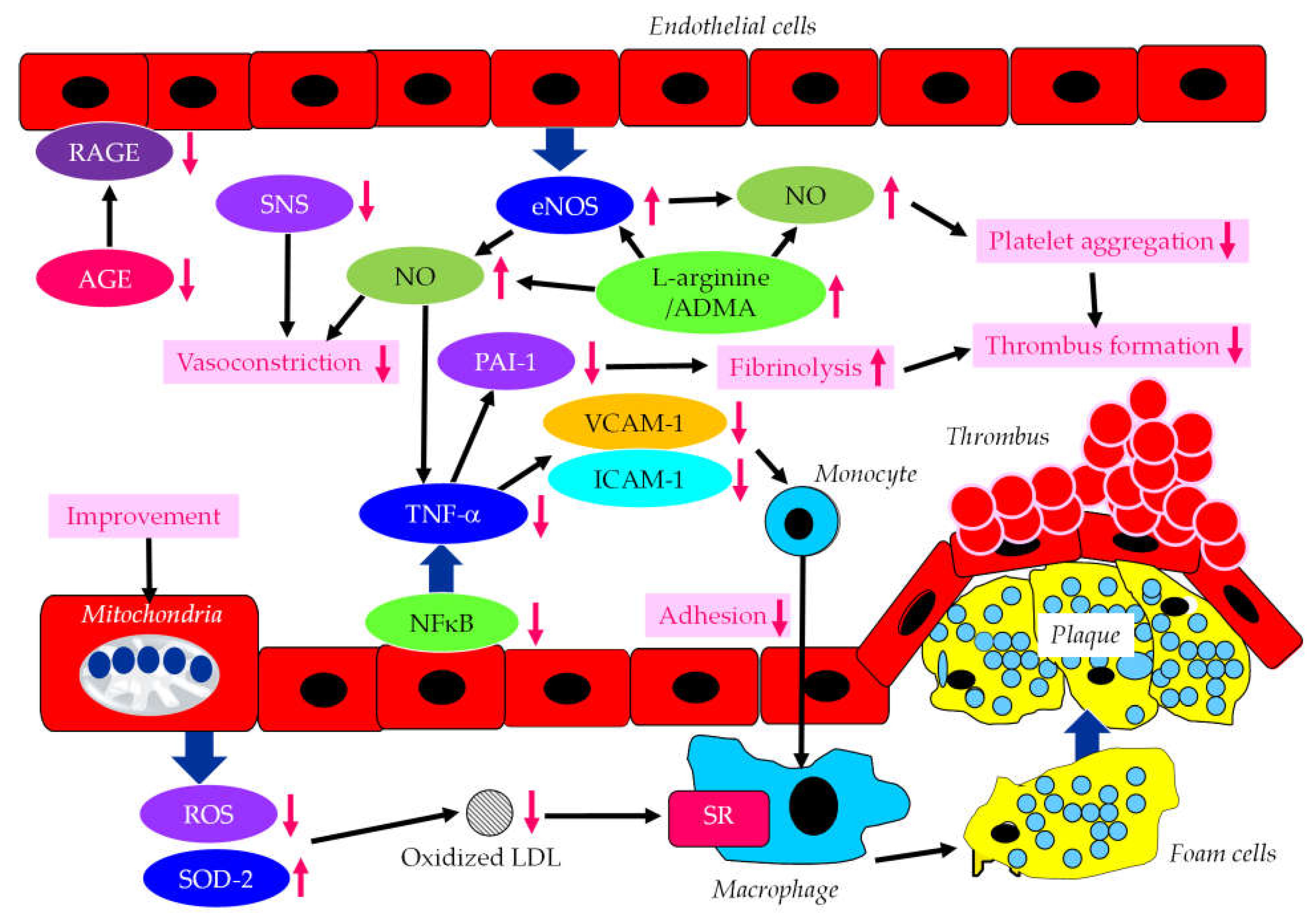

Table 1.
The beneficial effects of SGLT2i on DCM.
| Beneficial factors by SGLT2i to improve DCM |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



