
Submitted:
02 May 2023
Posted:
02 May 2023
You are already at the latest version
Abstract
Non-alcoholic steatohepatitis (NASH), which is the most severe manifestation of non-alcoholic fatty liver disease (NAFLD), has been recognized as a major hepatocellular carcinoma (HCC) catalyst. However, the molecular mechanism of NAFLD-NASH-HCC sequence remains unclear and a specific and effective treatment for NASH has not yet been established. The progress in this field depends on the availability of reliable preclinical models which show the steady progress to NASH, liver cirrhosis, and HCC. However, most of NASH mouse models that have been described to date develop NASH generally for more than 24 weeks and there is an uncertainty of HCC development. To overcome such shortcomings of experimental NASH studies, we established a novel NASH-HCC mouse model with very high reproducibility, generality, and convenience. We treated male C57BL/6J mice with a newly developed choline-deficient and methionine-restricted high-fat diet (CDMRHFD) for 60 weeks. Treatment of CDMRHFD for 3 weeks revealed marked steatosis, lobular inflammation, and fibrosis, histologically diagnosed as NASH. Liver cirrhosis was observed in all mice with 36-week treatment. Liver nodule emerged at 12 weeks of the treatment, and diameter >2 mm liver tumors developed in all mice at 24 weeks of the treatment. In conclusion, our rapidly progressive and highly reproducible NASH-liver cirrhosis-HCC model is helpful for preclinical development and research on the pathogenesis of human NAFLD-NASH-HCC. Our mouse model would be useful for the development of novel chemistry for NASH-HCC-targeted therapies or HCC prevention strategies.
Keywords:
non-alcoholic fatty liver disease
; non-alcoholic steatohepatitis
; hepatocellular carcinoma
; cirrhosis
; fibrosis
; mouse model
1. Introduction
Hepatocellular carcinoma (HCC) is one of the most common and fatal cancers in the world [1,2,3]. In developed countries, the incidence rate of HCC has increased 3-fold in the past 30 years, becoming the fastest rising cause of cancer-related deaths [3,4,5]. Originally attributed to the emergence and spread of hepatitis C virus (HCV), however, due to excessive calorie intake and sedentary lifestyle, excess lipids are stored in hepatocytes, leading to obesity and metabolic syndrome, which are increasing in clinical importance [6]. Non-alcoholic fatty liver disease (NAFLD) is a disease related to these lifestyle changes. It has been calculated that between 4% and 22% of HCC cases can be attributed to NAFLD [7]. Currently, NAFLD is one of the leading causes of the increase in persistent liver abnormalities worldwide. It is estimated that at least 30% of adults in developed countries suffer from NAFLD, with up to 15% of these patients exhibiting some degree of NASH (non-alcoholic steatohepatitis). In Japan, about 30% of Japanese people are found to have NAFLD during their annual health check-ups [8]. Worryingly, about 25% of NASH patients progress to cirrhosis, a severe precancerous condition that may require liver transplantation or lead to liver failure [1].
According to liver histology, NAFLD is divided into simple steatosis (SS) and non-alcoholic steatohepatitis (NASH) leading to liver fibrosis, hepatocellular carcinoma, and ultimately death [9,10,11,12,13]. Currently, NASH is the second indication for orthotopic liver transplantation and has become the main indication for liver transplantation in developing countries [9,14,15,16]. However, the molecular mechanisms of NAFLD-NASH-HCC progression are not fully understood. Understanding the pathogenesis of NAFLD/NASH is essential for developing new therapeutic intervention strategies. Advances in this field depend on reliable preclinical models. Therefore, it is crucial to develop a reliable mouse model of NAFLD-NASH-HCC.
Currently, NASH mouse models can be divided into 4 categories: (1) diet-induced models, (2) chemical-induced models, (3) gene editing models, (4) the former two, or a combination of the three methods [17].
For evaluating the drug efficiency using NASH-HCC animal models, rapid, easy, and absolute occurrence of NASH, liver fibrosis, and liver tumor is mandatory [18,19]. However, simple HFD typically develop dyslipidemia, fatty liver, and mild NASH without appreciable fibrosis and HCC [4,20,21,22]. DMBA (7,12-Dimethylbenz[a]anthracene) injection to HFD can develop HCC after 1 year, but liver fibrosis is rare [23]. Furthermore, Matsumoto, et al. have reported that a choline-deficient, L-amino acid-defined, high-fat diet (CDAAHFD) mouse model develops steatosis, steatohepatitis, and hepatic fibrosis more rapidly and severely than the conventional models, but the processes is slow (24 weeks resulted in NASH, the occurrence of liver tumors after 36 weeks) [24,25]. In addition, several specific mouse knockout models exist about for studding the development of HCC in NAFLD, including mutations in phosphatase and tens homologue (PTEN), enhancer of liver regeneration (ALR), and melanocortin 4 receptor (MC4R) [26,27,28]. All these models suggest that the formation time of HCC is more than 70 weeks, and it cannot develop into HCC 100%. According to these shortcomings, rapid, easy, and convenient NASH-HCC model is eagerly desired.
In the current study, we created a novel mouse model of NASH-HCC similar with the natural course of human pathology with very high reproducibility, generality, and convenience.
2. Materials and methods:
2.1. Animals and Experimental Design
Specific pathogen-free male C57BL/6J mice were purchased from The Jackson Laboratory (Yokohama, Japan) and maintained with AIN93M regular chow and tap water ad libitum. The AIN93M and OYC-NASH2 diets were purchased from ORIENTAL YEAST CO., LTD. Components of OYC-NASH2 diets showed in Supplementary Table S1. All animal experiments were conducted using the methods outlined in the “Guide to the Care and Use of Experimental Animals” approved by the School of Medicine of Shinshu University. At 5 weeks old, the mice were randomly divided into two groups, and maintained under the same controlled clean environment (25°C, 12-h light/dark cycle and tap water available ad libitum) [13]. One group (n=41) was fed AIN93M diet, which was identical to the control group in the previous study. The OYC-NASH2 group (n=74, two mice died in period of 60w) was fed a choline-deficient, L-amino-acid-defined (CDMRHFD). Samples were taken at 0, 3, 6, 9, 12, 24, 36, 48, and 60 weeks after feeding. All mice were maintained under the above-mentioned conditions for 0-60 weeks. At each sampling point (0, 3, 6, 9, 12, 24, 36, 48, and 60 weeks), mice were weighed and then sacrificed by Isoflurane anesthesia. Blood samples were collected and maintained at -80°C until assayed. The liver was quickly removed and weighed. To assess the extent of macroscopic liver nodules (diameter > 2 mm), we scored each liver on a scale of 0-3 as follows: stage 0 shows no visible nodules on the liver; stage 1, one to three nodules; stage 2, four to six nodules; and stage 3, seven or more nodules. After that, part of the liver tissue was snap-frozen on dry ice for mRNA analysis, and another small piece of liver was immediately fixed in 10% neutral-buffered formalin (Wako Pure Chemical Industries, Osaka, Japan) for further histological analysis.
2.2. Plasma and Liver Biochemical Analysis
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG), total cholesterol (T-Chol), non-esterified fatty acid (NEFA), phospholipid (PL), and glucose were measured with commercial assay kits (Wako Pure Chemical Industries, Ltd.). Total liver lipids and fecal lipids were extracted according to the hexane: isopropanol method with a slight modification and quantified using the above-mentioned kit (Wako Pure Chemical Industries Co., Ltd.). We hydrolyze frozen liver tissue (~10 mg) in 6 M HCl at 95 °C for 20 hours. After the samples were cooled to room temperature and centrifuged at 13,000 × g for 10 min, the supernatant was collected, diluted with deionized water to 4 M HCl, and subsequently diluted five-fold with 4 M HCl to avoid matrix effects [29]. Hydroxyproline measurements were performed using the hydroxyproline concentration using the QuickZyme Hydroxyproline Assay Kit (QuickZyme BioSciences, Leiden, The Netherlands) following the manufacturer’s protocol.
2.3. Histological analysis:
Small pieces of liver tissue were fixed in 10% formalin in phosphate-buffered saline and embedded in paraffin. Sections of 3 μm in thickness were stained with hematoxylin and eosin or Azan-Mallory staining using standard methods. We performed a semiquantitative assessment of NASH in HE-stained or Azan-Mallory staining mouse liver tissue (×200 and ×400 magnifications) using the NAFLD Activity Score (NAS) system proposed by Brunt et al. in 1999. We assessed 5 histological features item: steatosis (score 0-3), lobular inflammation (score 0-3), portal inflammation (score 0-2), foam cell (score 0-2) and fibrosis (score 0-4). The details of scoring system definitions and scores are as follows. Steatosis score was defined as low to moderate power assessment of parenchymal involvement of steatosis. <5% is 0, 5%-33% is 1, >33%-66% is 2, and >66% is 3. Lobular inflammation was defined as overall assessment of all inflammatory foci. No foci are 0, <2 foci per 200× field is 1, 2-4 foci per 200× field is 2, >4 foci per 200× field is 3. Foam cells, also called lipid-laden macrophages, are a type of cell that contain cholesterol, which can form a plaque that can lead to atherosclerosis and trigger heart attacks and stroke. Foam cells were scored since we did not observe ballooning of hepatocytes. 0 is no, 1 is few foam cells, 2 is many cells/prominent foam cells. Portal inflammation was assessed from low magnification. none is 0; minimal is 1; greater than minimal is 2. Azan-Mallory staining was used to observe fibrosis, none is 0; perisinusoidal or periportal is 1; perisinusoidal and portal/periportal is 2; bridging fibrosis is 3; cirrhosis is 4 [30].
2.4. Quantification of mRNA levels
Total RNA was isolated from frozen liver tissue using the RNeasy Mini kit (Qiagen, Tokyo, Japan). RNA samples were then reverse transcribed to cDNA using oligo-dT and random primers using the Prime Script RT Reagent Kit (Perfect Real Time, Takara Bio Inc., Shiga, Japan). All mRNA levels were determined by real-time quantitative polymerase chain reaction (qPCR) on a Thermo Fisher Quant Studio 3 Real-Time PCR Instrument (Thermo Fisher Scientific, Waltham, MA) using SYBR qPCR mix (Toyobo Co., Ltd.). mRNA levels were normalized to 18S ribosomal RNA (18S rRNA) levels and expressed as fold change relative to C57BL/6J mice fed a control diet. Primer sequences are listed in Supplementary Table S2.
2.5. Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM). The two-tailed Student’s t-test and chi-squared test were conducted for quantitative and qualitative data, respectively, using SPSS statistics, version 22 (IBM, Armonk, NY). A P value ≤ 0.05 was considered statistically significant.
3. Results
3.1. OYC-NASH2 feeding for 60 weeks resulted in enlarged liver and symptoms of NASH without involving severe loss of body weight.
Male C57BL/6J mice were treated with OYC-NASH2 diet or purified control diet (AIN93M) for 60 weeks. Six-week-old male mice were divided into two groups: control group (n = 41) and NASH group (n = 74, two mice died during 60w). All mice were reared from 0 to 60 weeks. The body weight of mice fed the OYC-NASH2 diet was slightly less than control mice fed AIN93M, but not severely (Figure S1). The body weights of the two groups of mice tended to increase with the feeding period. At the same time, the ratio of liver to body weight in both groups of mice increased with the feeding period, reaching a maximum at 60 weeks, and the ratio of liver to body weight of mice fed the OYC-NASH2 diet was higher than the control mice fed AIN93M. The ratio of spleen to body weight in mice fed the OYC-NASH2 diet was significantly higher than that in mice fed AIN93M, with an increasing trend (0w-36w) and a slight decreasing after 36w. Long-term feeding of the OYC-NASH2 diet induced sustained liver enlargement without significant weight loss. The eWAT/body weight ratio and cecal body weight ratio did not change significantly.
3.2. OYC-NASH2 diet induces the appearance of nodules in the liver of mice at 12 weeks, and the formation of multiple nodules in all mice at 24 weeks.
From the macroscopic appearance of the mouse liver (Figure 1A), it was seen that one nodule appeared in the liver of the mice fed the OYC-NASH2 diet at 12 weeks, and after 24 weeks, multiple nodules filled the liver. We measured the number and size of nodules. The nodules were divided into 3 categories: small nodules (2-5 mm in diameter), middle nodules (5-8 mm in diameter), and large nodules (>8 mm in diameter). Figure 1B is the proportion of mice that developed nodules on the OYC-NASH2 diet. Divided into 0-3 stages according to the number of nodules. Stage 0 shows no visible nodules on the liver. stage 1, one to three nodules; stage 2, four to six nodules; and stage 3, seven or more nodules (we mentioned earlier). In mice fed the OYC-NASH2 diet, no nodules were observed at 3-9 weeks, and 1 nodule (2-5 mm in diameter) was observed at 12 weeks; after that, all mice developed nodules at 24 weeks. Figure 1C is the number of nodules at different nodule sizes in mice fed the OYC-NASH2 diet. The number of small nodules (2-5 mm in diameter) and large nodules (>8 mm in diameter) gradually increased with feeding period. The number of middle nodules (5–8 mm in diameter) decreased slightly at 48 weeks and increased again at 60 weeks. The polymorphism of liver nodules increased from 24 weeks to 60 weeks. No nodules were observed on the livers of control mice fed AIN93M diet during the feeding period. We stained the nodule tissues of mice fed OYC-NASH2 diet at 24w, 36w, 48w, 60w, with hematoxylin and eosin (HE) (Figure 1D).
3.3. C57BL/6J mice fed the OYC-NASH2 diet for 3 weeks induce NASH and fibrosis.
In mice fed the OYC-NASH2 diet, more abundant macrovascular lipid droplets were detected throughout slices. Figure 2A is a photograph of HE-stained liver tissue in mice fed AIN93M diet (control group) and OYC-NASH2 diet (experimental group) for 3 to 60 weeks. We performed histological observation and scoring of HE liver tissue from mice fed the OYC-NASH2 diet using the NAS scoring system (Figure 2B). Severe steatosis was observed in the livers of mice fed the OYC-NASH2 diet for 3 weeks, and the steatosis gradually diminished after 24 weeks. Lobular inflammation began to appear in mice fed the OYC-NASH2 diet for 3 weeks, and gradually increased, reaching a peak at 12 weeks, and the situation was consistent between groups of mice. Lobular inflammation was attenuated at 24 weeks in mice fed the OYC-NASH2 diet, presumably related to fibrosis. However, while hepatocyte ballooning was not observed in the liver tissue of mice fed the OYC-NASH2 diet, foam cells were observed to emerge at 9 weeks and reached score 2 at 24 weeks. Portal inflammation was observed in the liver tissue of mice fed the OYC-NASH2 diet for 24 weeks with score 1 (Figure 2B).
Furthermore, all mice fed the OYC-NASH2 diet developed moderate fibrosis in the liver before multiple nodule formation was observed (Figure 3A). Fibrosis appeared in mice fed the OYC-NASH2 diet for 3 weeks, and gradually increased, with score 4 for fibrosis at 36 weeks. In this model, all mice fed the OYC-NASH2 diet developed fibrosis at 48 weeks (Figure 3B). Expression of Tgfb1 and Col1a1 genes were significantly increased in mice fed the OYC-NASH2 diet for 3 weeks and maintain high expression levels throughout the feeding period compared with mice fed the AIN93M over the time course (Figure 3C). In addition, hepatic hydroxyproline content was significantly higher after 9 weeks of feeding the OYC-NASH2 diet, which markedly enhanced fibrogenesis (Figure 3D).
3.4. Serum/Liver Profile
We detected continuous changes in various biochemical parameters in the serum and liver tissues of mice fed the AIN93M diet or the OYC-NASH2 diet during the feeding period (Figure 4). We observed severe steatosis and inflammation starting at 3 weeks of feeding by HE-stained sections of liver tissue from mice fed the OYC-NASH2 diet. In mice fed the OYC-NASH2 diet, ALT and AST levels were significantly increased at 3 weeks compared with the serum of mice fed the AIN93M diet and remained high levels throughout the feeding period. In addition, the serum TBA level of mice fed the OYC-NASH2 diet continued to increase from 3 weeks, which was significantly higher than that of the control group.
The amount of total cholesterol in the serum of mice fed the OYC-NASH2 diet was slightly lower than that of mice fed the AIN93M diet from 3 weeks to 24 weeks, and higher than that of the control group from 36 weeks to 60 weeks. However, blood glucose levels in mice fed the OYC-NASH2 diet were lower than those fed the AIN93M diet during the feeding period. There was no insulin resistance (Figure 4A). In serum, the levels of T-Chol and NEFA were not altered by OYC-NASH2 diet feeding.
In the liver, the contents of T-Chol, TG, and NEFA started to increase at 3 weeks and were significantly higher than those in the control group throughout the feeding period (Figure 4B). These results suggest that OYC-NASH2 diet may enhances steatosis, inflammation, hepatocellular injury, fibrosis, and hepatic tumorigenesis.
3.5. Expression of NASH-related genes
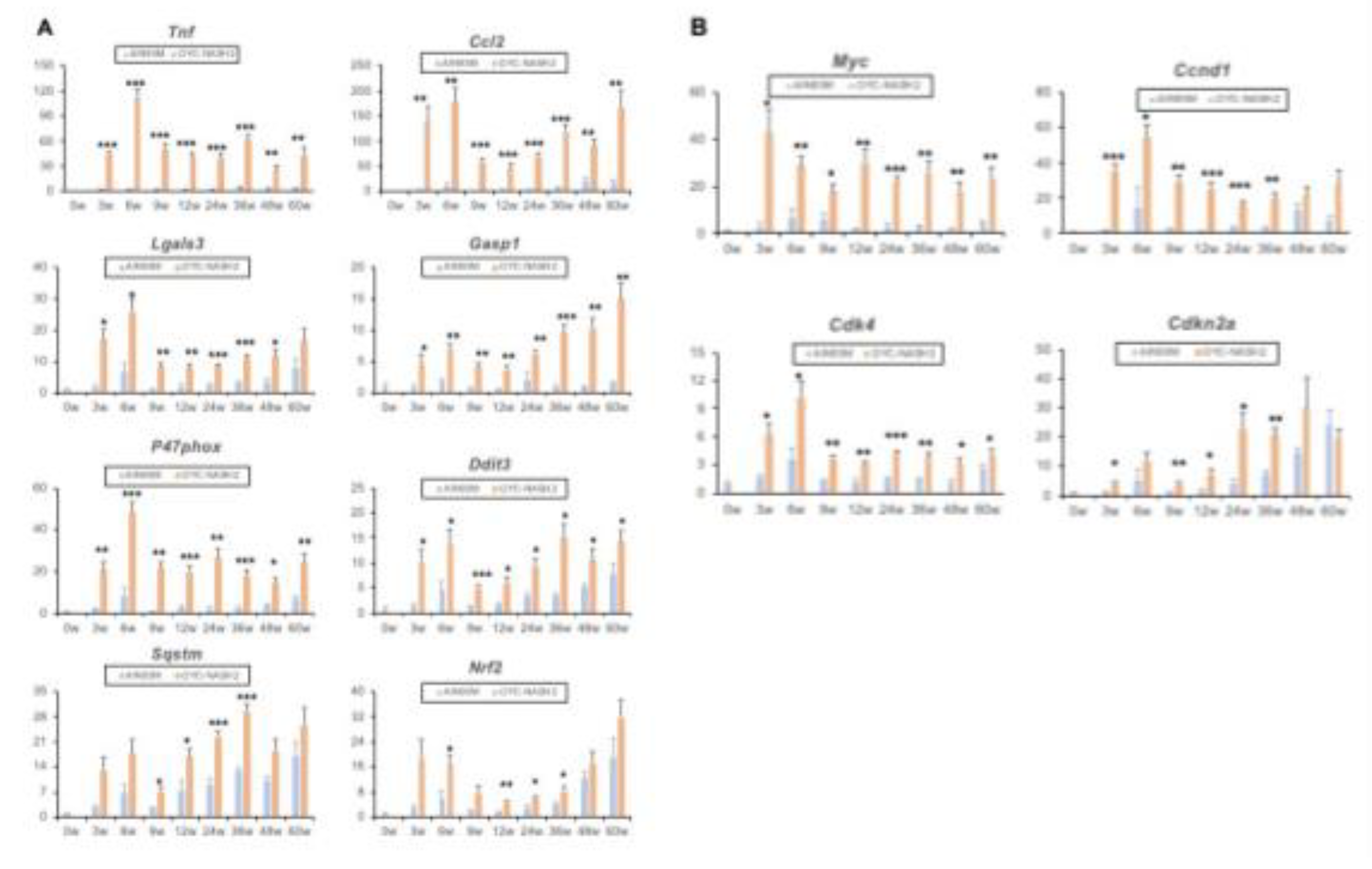
To further clarify the genetic changes in the OYC-NASH2 diet mouse liver from NASH-HCC process. We extracted mouse liver tissue and performed qPCR assay. After 3 weeks of treatment with CDMRHFD, the mice showed severe liver steatosis and inflammatory cell infiltration, and the expression of related genes Tnf, Ccl2, Lgals3, Casp1 increased significantly at 3 weeks. Lgals3 mRNA is induced by reactive oxygen species (ROS) and ER stress. The mRNA level of ROS producing enzymes, such as p47phox (Ncf1), increased. Compared with male C57BL/6J mice fed with AIN93M diet, the mRNA level of ER stress inducible gene was increased in male C57BL/6J mice treated with CDMRHFD, such as DNA damage inducible transcript 3 (Ddit3, also known as CHOP). The expression of genes related to oxidative stress and ER stress, such as Sqstm and Nrf2, also increased significantly at 3 weeks (Figure 5A). In general, the greater oxidative stress and ER stress in the liver of CDMRHFD treated mice may be related to the aggravation of hepatocyte injury and liver inflammation/fibrosis. The expression of Tgfb1 and Col1a1 genes increased significantly (Figure 3C), indicating the change of gene expression during the progression of NASH to HCC. In addition, the expression of cancer fetal markers such as c-Myc, Ccnd1, Cdk4 and Cdkn2a were similarly elevated in mice fed the OYC-NASH2 diet (Figure 5B). These results suggest a possible oncogenic response in the liver of mice fed the OYC-NASH2 diet.
4. Discussion
In this study, we demonstrate a novel NASH-driven mouse model of HCC that closely mimics the natural course of NAFLD/NASH in humans. When we fed male C57BL/6J mice the CDMRHFD, the mice started to develop tumors in the liver at 12 weeks (1/10), and at 24 weeks the incidence of liver tumors in the mice reached 100 % (10/10) (Figure 1A,B). From 24 weeks to 36 weeks, the size of tumors showed diversity, from small tumors to large tumors. At 3 weeks of feeding, mice fed the OYC-NASH2 diet began to develop severe hepatic steatosis (score 3), progressively progressed to steatohepatitis and proliferative fibrosis, and worsened, reaching a fibrosis of 4 at 48 weeks class. Fibrosis developed rapidly, with mild fibrosis occurring at three weeks. HCC formed after 24 weeks (Figure 3A,B). From the observation of liver, HE-stained pathological sections, foam cells were observed in mice fed the OYC-NASH2 diet at 9 weeks (score 1), which gradually increased to score 2 at 24 weeks. Therefore, the mouse model we developed can solve the problems of long-term formation of NASH and incomplete formation of HCC in previous mouse models. This model is likely to be more convenient to use in the study of NAFLD/NASH-HCC disease mechanism.
At present, the literature has been reported many NASH to HCC models, but few of them replicate the whole human phenotype. Most of the published studies use inherited leptin deficiency (ob/ob) or leptin resistance (db/db) mice and dietary methionine/choline deficiency (MCD) models[31]. The MCD model fed a diet deficient in methionine and choline is one of the most used tools in NASH research. One disadvantage of using MCDD to induce the development of NASH is that mice show significant body weight loss[32,33]. Choline deficiency, L-amino acid definition (CDAA) diet model overcame severe weight loss to study the development of NASH induced fibrosis, and this model has been proved to be able to simulate human NASH, liver fibrosis and liver cancer in mice and rats by successively producing steatohepatitis without any weight loss[34,35,36,37]. It is reported that mice fed CDAHFD developed nodules (at least one nodule) and NASH at 24 weeks after feeding, and HCC began to appear at 36 weeks after feeding, with an incidence of 16.7%; After 60 weeks of continuous feeding, the incidence rate of HCC in mice reached 26.7%. Mice fed with CDAHFD did not fully form HCC, and one mouse fed with SD developed HCC[24]. All of mouse model we mentioned above, they have same problem which are the model took too long time for development of NASH and HCC and all mice are uncertain to develop HCC with 100%. However, we solved the above problems by feeding C57BL/6J mice with CDMRHFD making the time shorter to induce NASH (at 3 weeks) and 100% developed into HCC at 24 weeks. In addition, for CDMRHFD, we designed the types and mixing proportion of raw materials to enhance the robustness of feed, to stabilize the palatability of test animals and reduce the variability of pathological data. Diet can also be easily stored at room temperature (no freezer or refrigerator storage space is required), making the establishment of this model more convenient.
NASH is expected to become the main cause of cirrhosis and HCC worldwide. However, there is no drug approved for the treatment of NASH. Unless the pathogenesis is better understood, its treatment can hardly make progress. The CDMRHFD increased cholesterol in the liver of mice (Figure 4B). We have known that, as a major lipotoxic molecule, hepatic free cholesterol (FC) drives the development of progressive liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease (NAFLD), leading to nonalcoholic steatohepatitis (NASH) even progress to liver cirrhosis and hepatocellular carcinoma (HCC)[3]. Cholesterol levels in the livers of mice fed the OYC-NASH2 diet increased at 3 weeks of feeding and remained high throughout the feeding period. Elevated cholesterol can activate Kupffer cells (KCs) to release inflammatory factors. Mice fed with CDMRHFD for 3 weeks showed severe hepatic steatosis and inflammatory cell infiltration, and the expression of its related genes Tnf, Ccl2, Lgals3 was significantly increased at 3 weeks. Cholesterol-induced activation of KCs, can lead to the activation of Hepatic stellate cells (HSCs) through the release of inflammatory cytokines, resulting in the development of fibrosis[38]. The expression of Tgfb1 and Col1a1 fibrosis-related genes were significantly elevated at 3 weeks of feeding (Figure 3C). Recently, it has been proved that the accumulation of free cholesterol in HSCs can directly activate HSCs into fibroblasts and this process may play a role through Toll like receptor 4 (TLR-4) dependent pathway[39,40]. Therefore, we speculate that the formation of NASH is related to the increase of cholesterol. The liver histopathology in mice fed with OYS-NASH2 diet showed many foam cells at 12 weeks. It has been found that KCs can surround and process fatty dead hepatocytes containing cholesterol crystals, and then transform them into activated foam cells containing FC and cholesterol crystals[41,42]. In addition, low density lipoprotein (LDL) molecules are oxidized and taken up by macrophages, which become engorged and form foam cells. These foam cells often become trapped in the walls of blood vessels and contribute to atherosclerotic plaque formation[43,44]. According to the lipid hypothesis, elevated levels of cholesterol in the blood lead to atherosclerosis which may increase the risk of heart attack, stroke, and peripheral artery disease. The expressions of genes related to oxidative stress and ER stress, such as P47phox, Ddit3, Sqstm, Nrf2, were also significantly increased at 3 weeks. Collectively, oxidative stress and ER stress in the livers of CDMRHFD-treated mice may be associated with aggravated hepatocyte damage and liver inflammation and fibrosis.
Insulin resistance and diabetes have been reported to be associated with NAFLD, NAFLD is associated with more severe fibrosis, and impaired glucose metabolism and insulin signaling exacerbate both liver fibrosis and NAFLD[45]. According to serum glucose values, the blood glucose level of mice fed the OYC-NASH2 diet was significantly lower than that of the mice fed the AIN93M diet, which is not conducive to clinical research in NASH patients with diabetes. Studies have shown that db/db mice and foz/foz HF/HC mouse models are helpful for the study of diabetes combined with NASH[31]. Therefore, we also hope to use this new diet in combination with chemical induced mice to improve this model and develop a mouse model that is more suitable for clinical use.
In conclusion, our developed mouse model of NASH-driven HCC will facilitate the development of new NASH-HCC-targeted therapies or HCC prevention strategies. This modified CDMRHFD will allow faster establishment of a NASH-HCC mouse model for studying the mechanism of NASH on HCC. Contributes to the further development of NASH models that are highly imitative to humans.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Naoki Tanaka; Formal analysis, Qianqian Zheng, Pan Diao, Xuguang Zhang, Zhe Zhang, Takero Nakajima, Takanobu Iwadare, Jun Nakayama and Naoki Tanaka; Investigation, Qianqian Zheng, Pan Diao, Xuguang Zhang, Zhe Zhang, Takero Nakajima, Takanobu Iwadare, Takefumi Kimura and Naoki Tanaka; Methodology, Masaya Kawaguchi, Hayato Mikami and Naoki Tanaka; Resources, Masaya Kawaguchi and Hayato Mikami; Supervision, Takefumi Kimura and Jun Nakayama; Writing—original draft, Qianqian Zheng and Naoki Tanaka; Writing—review & editing, Naoki Tanaka.
Funding
This study was partially supported by JST SPRING, Grant Number JPMJSP2144 (Shinshu University).
Acknowledgments
This work was supported by JST SPRING, Grant Number JPMJSP2144 (Shinshu University).
Conflicts of Interest
MK and HM were employed by ORIENTAL YEAST CO., LTD, but the authors declare that the research was conducted in the absence of any financial relationships that could be constructed as a potential conflict of interest.
Abbreviations
ALR, augmenter of liver regeneration; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CD, choline deficiency; CDAAHFD, choline-deficient, L-amino acid-defined, high-fat diet; CDMRHFD, choline-deficient and methionine-restricted high-fat diet; DMBA, 7,12-Dimethylbenz[a]anthracene; eWAT, epididymal white adipose tissue; ELISA, enzyme-linked immunosorbent assay; FA, fatty acid; FC, free cholesterol; HCA, Hepatocellular adenoma; HCC, hepatocellular carcinoma; HCl, hydrochloride; HSCs, Hepatic stellate cells; HCV, hepatitis C virus; HE, hematoxylin and eosin; KCs, Kupffer cells; LDL, low-density lipoprotein; MCDD, methionine/choline deficiency diet; MC4R, melanocortin 4 receptor; NAFLD, non-alcoholic fatty liver disease; NAS, NAFLD activity score; NASH, non-alcoholic steatohepatitis; NEFA, non-esterified fatty acid; PL, phospholipid; PTEN, phosphatase and tens in homolog; qPCR, quantitative polymerase chain reaction; ROS, reactive oxygen species; rRNA, ribosomal RNA; SD, standard deviation; SEM, standard error of the mean; TBA, total bile acid; T-Chol, total cholesterol; TG, triglyceride; TLR-4, Toll like receptor 4; VLDL, very low-density lipoprotein.
References
- Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther 2017, 179, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Ikawa-Yoshida A, Matsuo S, Kato A et al. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int J Exp Pathol 2017, 98, 221–233. [Google Scholar] [CrossRef]
- Anstee QM, Reeves HL, Kotsiliti E et al. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Jia F, Hu X, Kimura T, Tanaka N. Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies. Int J Mol Sci 2021, 22, 10303. [Google Scholar] [CrossRef] [PubMed]
- Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Younossi Z, Anstee QM, Marietti M et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Kojima S, Watanabe N, Numata M et al. Increase in the prevalence of fatty liver in Japan over the past 12 years: analysis of clinical background. J Gastroenterol 2003, 38, 954–961. [Google Scholar] [CrossRef]
- Tanaka N, Ichijo T, Okiyama W et al. Laparoscopic findings in patients with nonalcoholic steatohepatitis. Liver Int 2006, 26, 32–38. [Google Scholar] [CrossRef]
- Tanaka N, Moriya K, Kiyosawa K et al. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: implications for HCV-associated hepatocarcinogenesis. Int J Cancer 2008, 122, 124–131. [Google Scholar] [CrossRef]
- Tanaka N, Yazaki M, Kobayashi K. A lean man with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2007, 5, A32. [Google Scholar] [CrossRef] [PubMed]
- Van Herck MA, Vonghia L, Francque SM. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients 2017, 9, 1072. [CrossRef] [PubMed]
- Tanaka N, Mukaiyama K, Morikawa A et al. Pemafibrate, a novel selective PPARα modulator, attenuates tamoxifen-induced fatty liver disease. Clin J Gastroenterol 2021, 14, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Baran B, Akyüz F. Non-alcoholic fatty liver disease: what has changed in the treatment since the beginning? World J Gastroenterol 2014, 20, 14219–14229. [Google Scholar] [CrossRef] [PubMed]
- Tanaka N, Horiuchi A, Yokoyama T et al. Clinical characteristics of de novo nonalcoholic fatty liver disease following pancreaticoduodenectomy. J Gastroenterol 2011, 46, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Boursier J, Mueller O, Barret M et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Hansen HH, Feigh M, Veidal SS et al. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Teufel A, Itzel T, Erhart W et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 2016, 151, 513–525.e510. [Google Scholar] [CrossRef]
- Tanaka N, Moriya K, Kiyosawa K et al. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Investig 2008, 118, 683–694. [Google Scholar]
- Tanaka N, Takahashi S, Fang ZZ et al. Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim Biophys Acta 2014, 1841, 1596–1607. [Google Scholar] [CrossRef]
- Diao P, Wang X, Jia F et al. A saturated fatty acid-rich diet enhances hepatic lipogenesis and tumorigenesis in HCV core gene transgenic mice. J Nutr Biochem 2020, 85, 108460. [Google Scholar]
- Parlati L, Régnier M, Guillou H, Postic C. New targets for NAFLD. JHEP Rep 2021, 3, 100346. [Google Scholar] [CrossRef]
- Gurses SA, Banskar S, Stewart C et al. Nod2 protects mice from inflammation and obesity-dependent liver cancer. Sci Rep 2020, 10, 20519. [Google Scholar] [CrossRef]
- Matsumoto M, Hada N, Sakamaki Y et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int J Exp Pathol 2013, 94, 93–103. [Google Scholar] [CrossRef]
- Tanaka N, Takahashi S, Matsubara T et al. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J Biol Chem 2015, 290, 3092–3105. [Google Scholar] [CrossRef]
- Wu, J. Utilization of animal models to investigate nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Oncotarget 2016, 7, 42762–42776. [Google Scholar] [CrossRef]
- Tanaka N, Takahashi S, Zhang Y et al. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim Biophys Acta 2015, 1852, 1242–1252. [Google Scholar] [CrossRef]
- Horie Y, Suzuki A, Kataoka E et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Investig 2004, 113, 1774–1783. [Google Scholar] [CrossRef]
- Zhang X, Diao P, Yokoyama H et al. Acidic Activated Charcoal Prevents Obesity and Insulin Resistance in High-Fat Diet-Fed Mice. Front Nutr 2022, 9, 852767. [Google Scholar] [CrossRef]
- Kleiner DE, Brunt EM, Van Natta M et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 2006, 87, 1–16. [Google Scholar] [CrossRef]
- Kashireddy PR, Rao MS. Sex differences in choline-deficient diet-induced steatohepatitis in mice. Exp Biol Med (Maywood) 2004, 229, 158–162. [Google Scholar] [CrossRef]
- Rizki G, Arnaboldi L, Gabrielli B et al. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res 2006, 47, 2280–2290. [Google Scholar] [CrossRef]
- Nakae D, Yoshiji H, Maruyama H et al. Production of both 8-hydroxydeoxyguanosine in liver DNA and gamma-glutamyltransferase-positive hepatocellular lesions in rats given a choline-deficient, L-amino acid-defined diet. Jpn J Cancer Res 1990, 81, 1081–1084. [Google Scholar] [CrossRef]
- Nakae D, Yoshiji H, Mizumoto Y et al. High incidence of hepatocellular carcinomas induced by a choline deficient L-amino acid defined diet in rats. Cancer Res 1992, 52, 5042–5045. [Google Scholar]
- Denda A, Kitayama W, Kishida H et al. Development of hepatocellular adenomas and carcinomas associated with fibrosis in C57BL/6J male mice given a choline-deficient, L-amino acid-defined diet. Jpn J Cancer Res 2002, 93, 125–132. [Google Scholar] [CrossRef]
- Endo H, Niioka M, Kobayashi N et al. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PLoS One 2013, 8, e63388. [Google Scholar]
- Tanaka N, Takahashi S, Hu X et al. Growth arrest and DNA damage-inducible 45α protects against nonalcoholic steatohepatitis induced by methionine- and choline-deficient diet. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 3170–3182. [Google Scholar] [CrossRef]
- Teratani T, Tomita K, Suzuki T et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164.e110. [Google Scholar] [CrossRef]
- Tomita K, Teratani T, Suzuki T et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res 2013, 54, 1326–1334. [Google Scholar] [CrossRef]
- Ioannou GN, Van Rooyen DM, Savard C et al. Cholesterol-lowering drugs cause dissolution of cholesterol crystals and disperse Kupffer cell crown-like structures during resolution of NASH. J Lipid Res 2015, 56, 277–285. [Google Scholar] [CrossRef]
- Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab 2020, 2, 132–141. [Google Scholar] [CrossRef]
- Kazankov K, Jørgensen SMD, Thomsen KL et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Marušić M, Paić M, Knobloch M, Liberati Pršo AM. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can J Gastroenterol Hepatol 2021, 2021, 6613827. [Google Scholar]
Figure 1.
Time course of gross appearance and nodules formation in the livers of C57BL/6J mice fed OYC-NASH2 diet. (A) Appearance of mouse liver. The control group was fed the AIN93M diet and the OYC-NASH2 group was fed the modified CDMRHFD. (B) Proportion of mice with nodules in different nodules size. Stage 0 shows no visible nodules on the liver. stage 1, one to three nodules; stage 2, four to six nodules; and stage 3, seven or more nodules. (C) Numbers of nodules of different size in mice fed the modified CDMRHFD for 12, 24, 36, 48 or 60 weeks. (D) Representative liver histopathology with HE staining of liver tumor tissues from mice fed the OYC-NASH2 diet at 24 weeks, 36 weeks, 48 weeks, and 60 weeks (×200 magnification). The numbers in parentheses are the number of tested mice at each feeding period. Scale bars = 100 𝛍m.
Figure 1.
Time course of gross appearance and nodules formation in the livers of C57BL/6J mice fed OYC-NASH2 diet. (A) Appearance of mouse liver. The control group was fed the AIN93M diet and the OYC-NASH2 group was fed the modified CDMRHFD. (B) Proportion of mice with nodules in different nodules size. Stage 0 shows no visible nodules on the liver. stage 1, one to three nodules; stage 2, four to six nodules; and stage 3, seven or more nodules. (C) Numbers of nodules of different size in mice fed the modified CDMRHFD for 12, 24, 36, 48 or 60 weeks. (D) Representative liver histopathology with HE staining of liver tumor tissues from mice fed the OYC-NASH2 diet at 24 weeks, 36 weeks, 48 weeks, and 60 weeks (×200 magnification). The numbers in parentheses are the number of tested mice at each feeding period. Scale bars = 100 𝛍m.
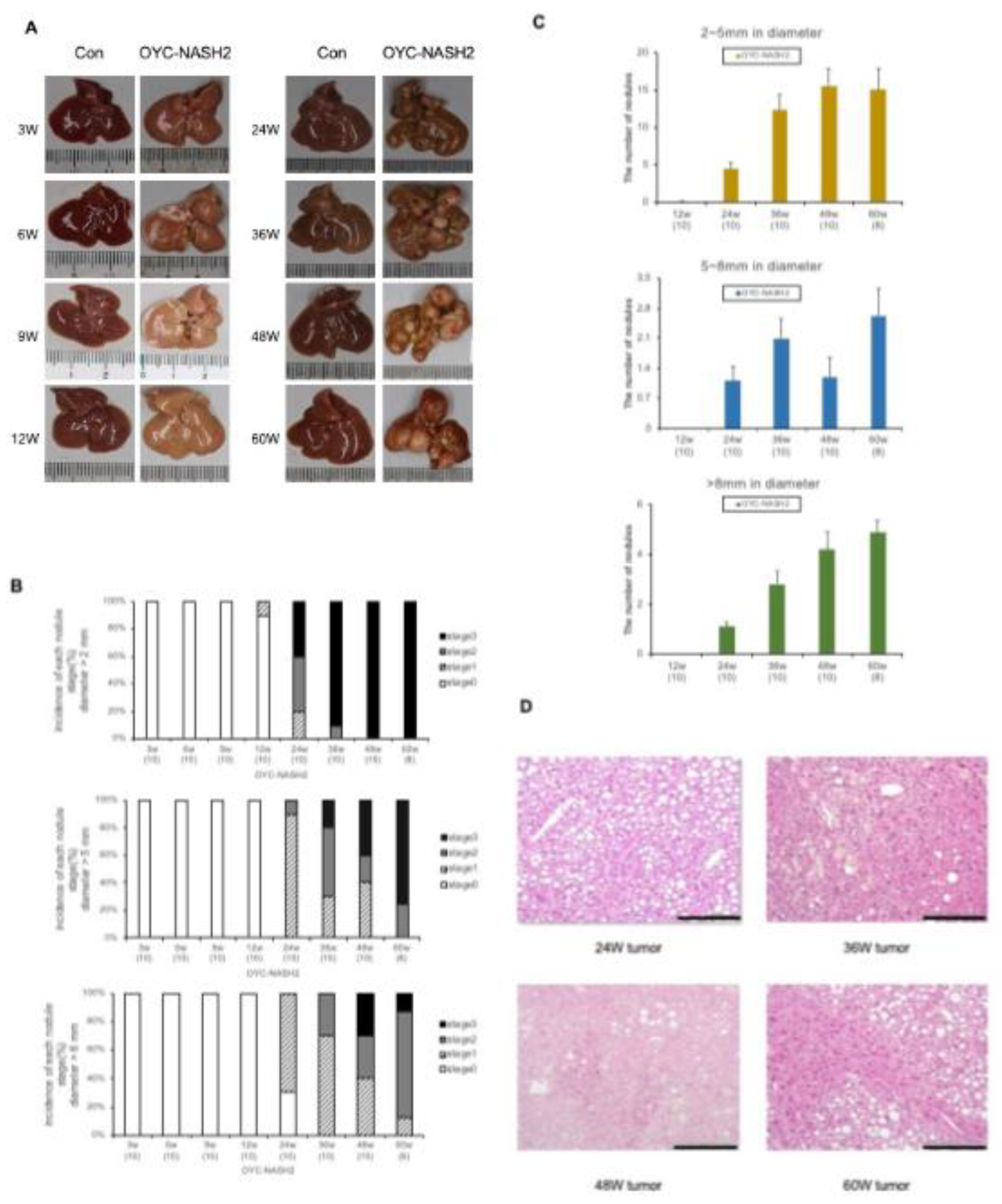
Figure 2.
C57BL/6J mice fed OYC-NASH2 diet for 3 weeks results NASH histopathological changes. (A) HE-stained liver histopathology of OYC-NASH2 diet-fed (×200, ×400 magnification) or AIN93M diet-fed (×200 magnification, as control group) mice for 60 weeks. (B) Histopathological assessment of non-alcoholic fatty liver in C57BL/6J mice fed modified CDMRHFD for 60 weeks. The morphological features of steatosis (score 0–3), lobular inflammation (score 0–3), portal inflammation (score 0–3) and foam cell (score 0–2) were semiquantitative evaluated. Subsequently, the NAFLD activity score was applied (<3, non-NASH; 3–4, possible NASH; and >5, NASH). The numbers in parentheses are the number of tested mice at each feeding period. Scale bars = 100 𝛍m.
Figure 2.
C57BL/6J mice fed OYC-NASH2 diet for 3 weeks results NASH histopathological changes. (A) HE-stained liver histopathology of OYC-NASH2 diet-fed (×200, ×400 magnification) or AIN93M diet-fed (×200 magnification, as control group) mice for 60 weeks. (B) Histopathological assessment of non-alcoholic fatty liver in C57BL/6J mice fed modified CDMRHFD for 60 weeks. The morphological features of steatosis (score 0–3), lobular inflammation (score 0–3), portal inflammation (score 0–3) and foam cell (score 0–2) were semiquantitative evaluated. Subsequently, the NAFLD activity score was applied (<3, non-NASH; 3–4, possible NASH; and >5, NASH). The numbers in parentheses are the number of tested mice at each feeding period. Scale bars = 100 𝛍m.
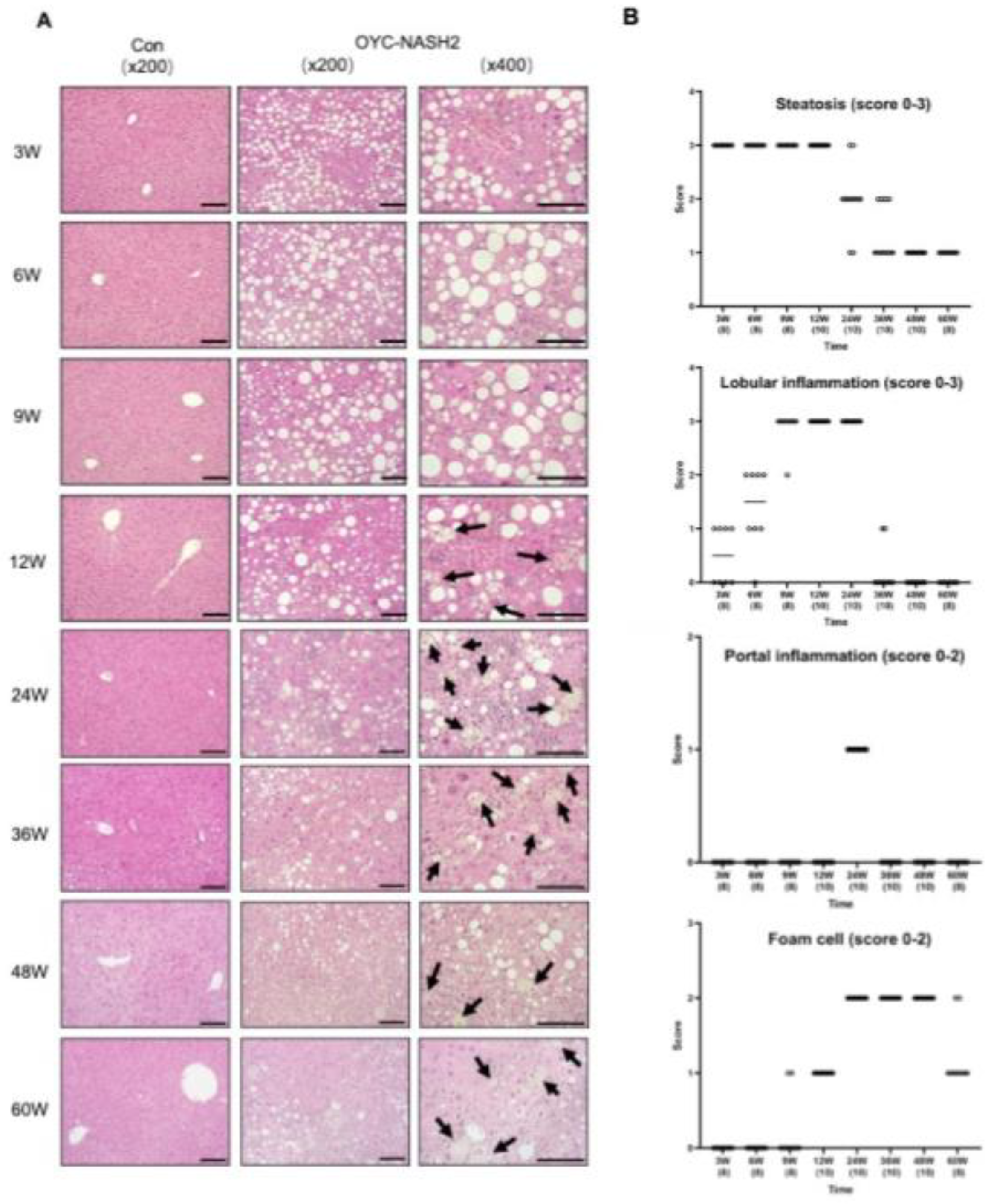
Figure 3.
Development of fibrosis and expression of fibrosis-related genes during the feeding period in C57BL/6J mice fed OYC-NASH2 diet. (A) Azon-stained liver histopathology of OYC-NASH2 diet-fed (×100, ×200 magnification) or AIN93M diet-fed (×100 magnification, as control group) mice for 60 weeks. (B) Time course of fibrosis (stage 0-4) in mice fed OYC-NASH2 diet for 60 weeks. (C) Assessment of mRNA expression levels of the Tgf and Col1a1 genes in the nodule-free areas of liver during feeding period by RT-qPCR. (D) Hepatic hydroxyproline content. The numbers in parentheses are the number of tested mice at each feeding period. Scale bars = 100 𝛍m. Data are means ± SEM (n = 113). *P < 0.05, **P < 0.01 and ***P < 0.001 between AIN93M diet group and OYC-NASH2 diet group.
Figure 3.
Development of fibrosis and expression of fibrosis-related genes during the feeding period in C57BL/6J mice fed OYC-NASH2 diet. (A) Azon-stained liver histopathology of OYC-NASH2 diet-fed (×100, ×200 magnification) or AIN93M diet-fed (×100 magnification, as control group) mice for 60 weeks. (B) Time course of fibrosis (stage 0-4) in mice fed OYC-NASH2 diet for 60 weeks. (C) Assessment of mRNA expression levels of the Tgf and Col1a1 genes in the nodule-free areas of liver during feeding period by RT-qPCR. (D) Hepatic hydroxyproline content. The numbers in parentheses are the number of tested mice at each feeding period. Scale bars = 100 𝛍m. Data are means ± SEM (n = 113). *P < 0.05, **P < 0.01 and ***P < 0.001 between AIN93M diet group and OYC-NASH2 diet group.
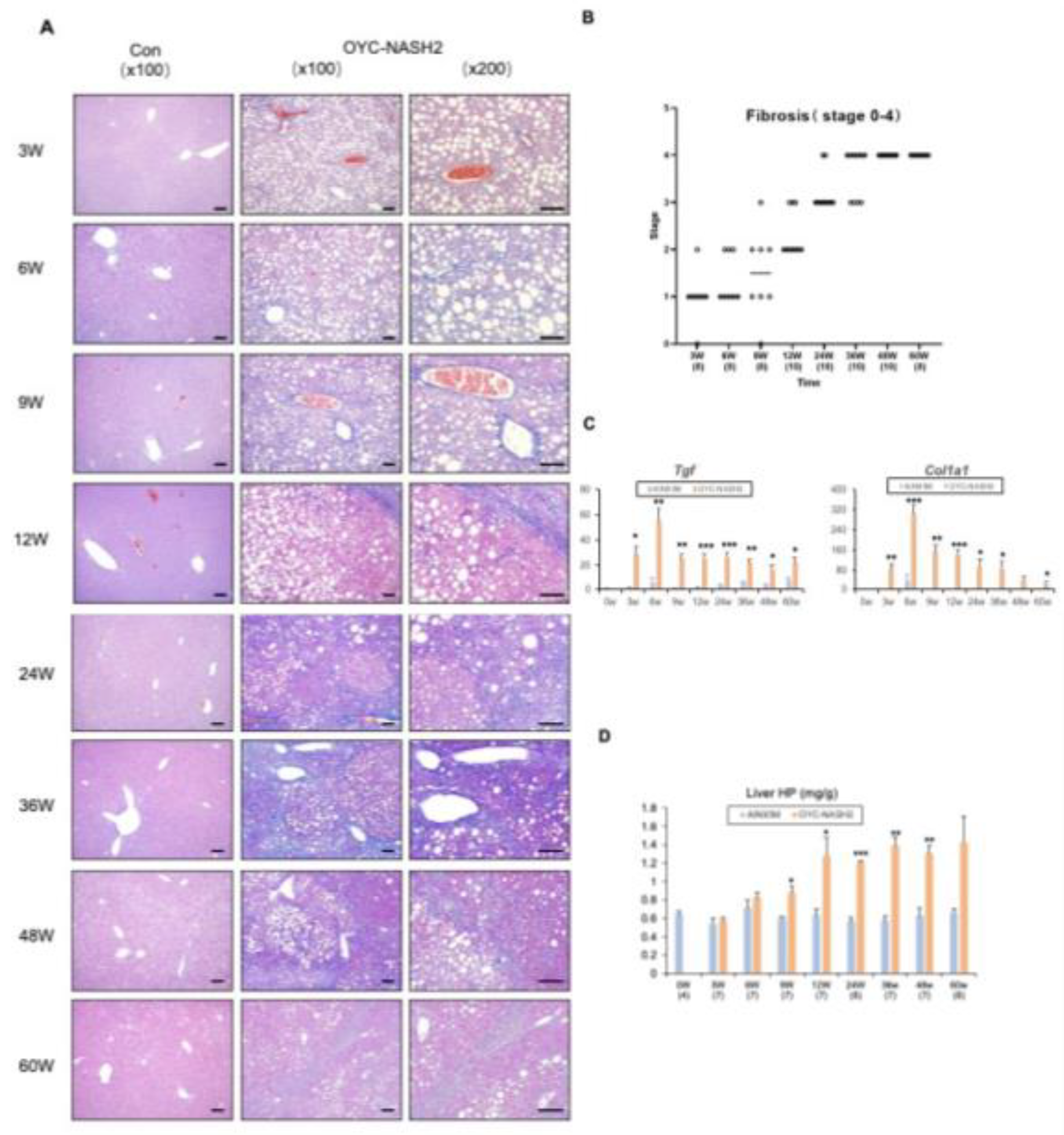
Figure 4.
Time course of biochemical parameters in the serum and liver tissues of mice fed the AIN93M diet or the OYC-NASH2 diet for 60 weeks. (A) Serum biochemical parameters of mice fed the AIN93M diet or the OYC-NASH2 diet for 60 weeks. (B) Liver tissue biochemical parameters of mice fed the AIN93M diet or the OYC-NASH2 diet for 60 weeks. Data are means ± SEM (n = 113). *P < 0.05, **P < 0.01 and ***P < 0.001 between AIN93M diet group and OYC-NASH2 diet group.
Figure 4.
Time course of biochemical parameters in the serum and liver tissues of mice fed the AIN93M diet or the OYC-NASH2 diet for 60 weeks. (A) Serum biochemical parameters of mice fed the AIN93M diet or the OYC-NASH2 diet for 60 weeks. (B) Liver tissue biochemical parameters of mice fed the AIN93M diet or the OYC-NASH2 diet for 60 weeks. Data are means ± SEM (n = 113). *P < 0.05, **P < 0.01 and ***P < 0.001 between AIN93M diet group and OYC-NASH2 diet group.
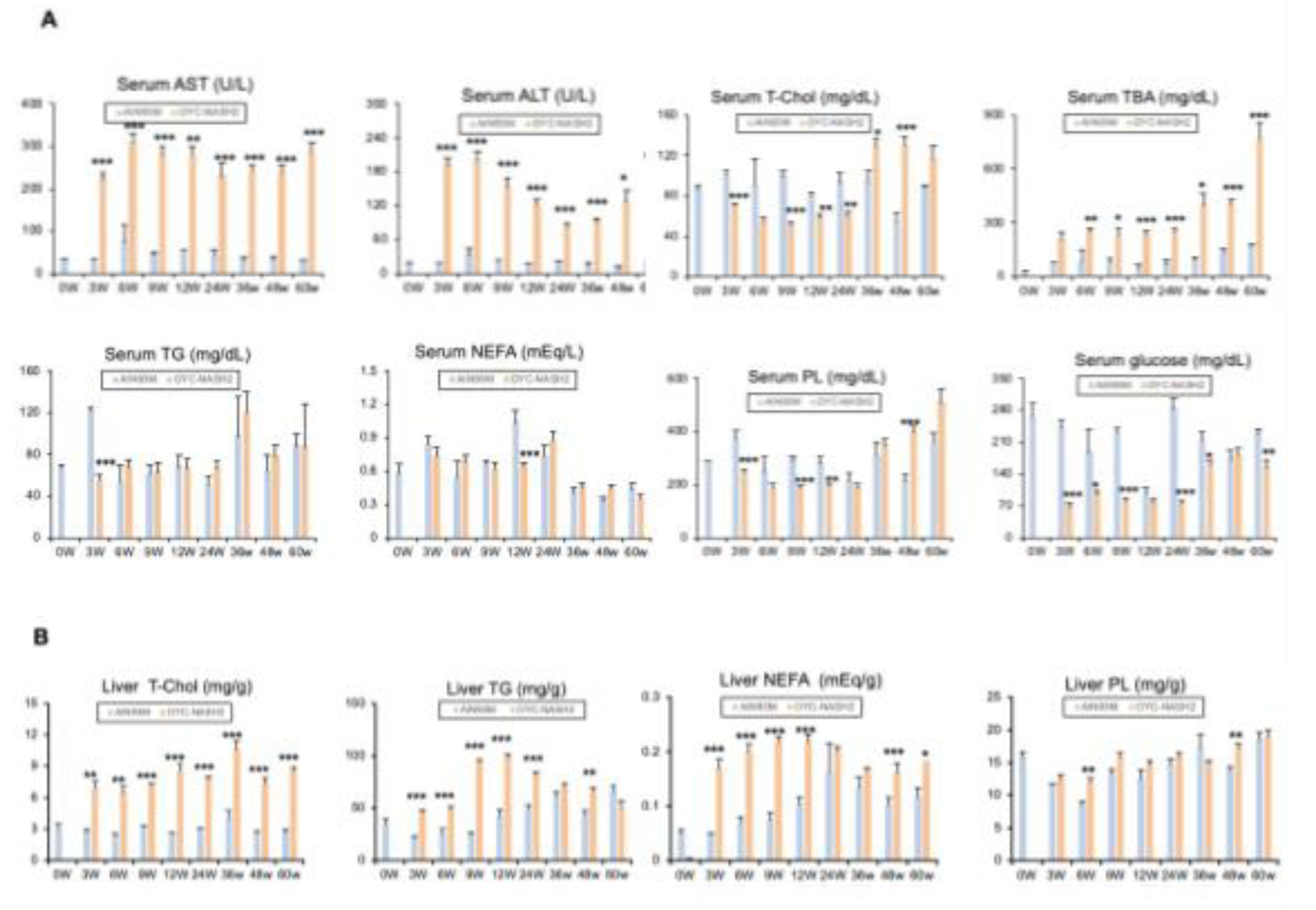
Figure 5.
Changes in gene expression. (A) Assessment of mRNA expression levels of the indicated genes associated with inflammation, ER stress and oxidative stress in the nodule-free areas of liver during feeding period by qPCR. (B) Expression of oncofetal markers (Myc, Ccnd1, Cdk4, Cdkn2a). All mRNA levels were normalized to 18S ribosomal RNA levels and subsequently expressed as values relative to those of control diet mice at 0 weeks. Data are means ± SEM (n = 113). *P < 0.05, **P < 0.01 and ***P < 0.001 between AIN93M diet group and OYC-NASH2 diet group.
Figure 5.
Changes in gene expression. (A) Assessment of mRNA expression levels of the indicated genes associated with inflammation, ER stress and oxidative stress in the nodule-free areas of liver during feeding period by qPCR. (B) Expression of oncofetal markers (Myc, Ccnd1, Cdk4, Cdkn2a). All mRNA levels were normalized to 18S ribosomal RNA levels and subsequently expressed as values relative to those of control diet mice at 0 weeks. Data are means ± SEM (n = 113). *P < 0.05, **P < 0.01 and ***P < 0.001 between AIN93M diet group and OYC-NASH2 diet group.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



