
Submitted:
01 May 2023
Posted:
02 May 2023
You are already at the latest version
Abstract
Japanese encephalitis virus (JEV) causes acute viral encephalitis in humans and reproductive disorders in pigs. JEV emerged during the 1870s in Japan and since that time, JEV has been transmitted exclusively throughout Asia, according to known reporting and sequencing records. A recent JEV outbreak occurred in Australia which affected commercial piggeries across different temperate southern Australian states and caused confirmed infections in humans. A total of 47 human cases and seven deaths were reported. The recent evolving situation of JEV needs to be reported due to its continuous circulation in endemic regions and spread to non-endemics areas. Here, we reconstructed the phylogeny and population dynamics of JEV using recent JEV isolates for the future perception of disease spread. Phylogenetic analysis shows the most recent common ancestor occurred about 3120 years ago (YA) (95% Highest posterior density [HPD], 2680 to 3715). Our results of the Bayesian skyline plot (BSP) demonstrates that JEV demography lacks fluctuations for the last two decades, but it shows that JEV genetic diversity has increased during the last ten years. This indicates the potential JEV replication in the reservoir host, which is helping it to maintain its genetic diversity, and to continue its dispersal into non-endemic areas. The continuous spread in Asia and recent detection from Australia further support these findings. Therefore, an enhanced surveillance system is needed along with precautionary measures such as regular vaccination and mosquito control to avoid future JEV outbreaks.
Keywords:
Japanese encephalitis virus
; Population dynamic
; Genetic diversity
1. Introduction
Japanese encephalitis (JE) is a vaccine-preventable disease, caused by the Japanese encephalitis virus (JEV) which is prevalent in Asian countries [1]. JEV has a positive-sense RNA genome belonging to the flavivirus genus within the flaviviridae family of five geographically and epidemiologically distinct genotypes (genotype I-V) [2]. Its genome contained 10,965 nucleotides and encoded polyprotein is further processed into three structural (capsid, membrane, envelope) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5). JEV genotype III (GIII) had been the most abundant genotype which led to several outbreaks in JEV endemic areas until 1990. However, recent data shows the emergence of genotype I (GI) as a dominant JEV genotype and it is gradually displacing GIII. The exact mechanism of this genotype displacement needs to be explored. In nature, the virus can circulate in invertebrate and vertebrate hosts. Invertebrates (mosquitoes) act as vectors and vertebrate hosts such as pigs and aquatic wading birds act as an amplifying/reservoir, and humans and equines are the dead-end hosts [3,4]. Approximately 300 million people live in Asia where JEV is circulating endemically, and they are at risk of JEV infection. Annually, it caused 68,000 clinical cases, and 10,000-15,000 associated deaths [1,5,6]. Recently, JEV cases have been controlled to a significant extent by the use of JEV vaccines all over Asian countries [7], and the demographic history of JEV has been reported in previous studies [2,8,9]. However, due to the continued spread of JEV to non-endemic areas such as Tibet, Xinjiang, Philippines, and Australia [10,11,12,13,14] and continuous detection from mosquitoes or vertebrate hosts of endemic regions [15,16,17,18,19,20,21], we felt that there is a need to reconstruct the molecular phylogeny and population dynamics of JEV using recent isolates (till December 2022) of JEV for the future perception of disease spread.
2. Methodology
All published and publicly available (n=160) complete JEV genomes (till December 2022) were retrieved from GenBank public database (Supplementary table 1). A MUSCLE based multiple sequence alignment (MSA) of the data set was generated using an online tool at EBI server (https://www.ebi.ac.uk/), which was then visualized in BioEdit software [22]. The model of evolution was tested by ModelFinder tool [23], which revealed GTR+F+I+G4 as the best-fit evolutionary model for the dataset judged by Akaike and Bayesian information criterions (AIC and BIC). Timeline phylogeny reconstructions were performed in a Bayesian framework with BEAST 2 [24] using Markov chain Monte Carlo (MCMC) algorithms [25]. The GTR site model with a strict molecular clock and a fixed rate of 1.002×10-4 mutations/site/year [18,26] was applied. The MCMC chain was run for 1 billion steps, with sampling of parameters every 2,000 steps. Tracer v1.7.1 was used to assess the MCMC generated results, and an ESS value of >200 was considered as acceptable for all parameters of interest. The maximum clade credibility tree was extracted by TreeAnnotator and it was then visualized and finished in FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
We reconstructed a Bayesian skyline plot (BSP) [27] using BEAST 2 [24]. The BSP was generated with a strict molecular clock and a fixed rate of 1.002×10-4 mutations/site/year [18]. The MCMC chain was run for 1 billion steps, with sampling of parameters every 2,000 steps. The ESS for all parameters of interest remained >200 as analyzed in Tracer. Finally, the BSP was visualized and extracted using Tracer v1.7.1 [28].
3. Results
Recently, the epidemiology of JEV is changing and it has been expanded from Asia to other regions of the world such as Papua New Guinea, Australia. The rest of the world such as Europe, South and North America, and Pacific Islands are also receptive due to the presence of JEV competent mosquito vectors. To determine the continuously evolving situation of JEV, we downloaded the available complete genome sequences (n=160) and constructed phylogeny and population dynamics to report the possible threat of JE disease. We constructed a Bayesian molecular timeline phylogenetic tree to investigate the most recent common ancestor (tMRCA) for all genotypes using. Figure 1 explained that the tMRCA occurred about 3120 years ago (YA) (95% Highest posterior density [HPD], 2680 to 3715). The branching of the lineages occurred in the following order: genotype V, at the root of the tree; genotype IV, about 1589 YA (95% HPD, 1375 to 1754); genotype I, II, & III shared their MRCA about 824 YA (95% HPD, 730 to 1093); MRCA of genotype I & II was about 732 YA (95% HPD, 644 to 867). The mean rate of nucleotide substitution for all available (till December 2022) JEV strains isolated from a variety of hosts worldwide, estimated using a Bayesian MCMC approach, was 1.0628E-4 nucleotide substitutions per site per year (95% HPD values 8.7795E-5, 1.2564E-4).
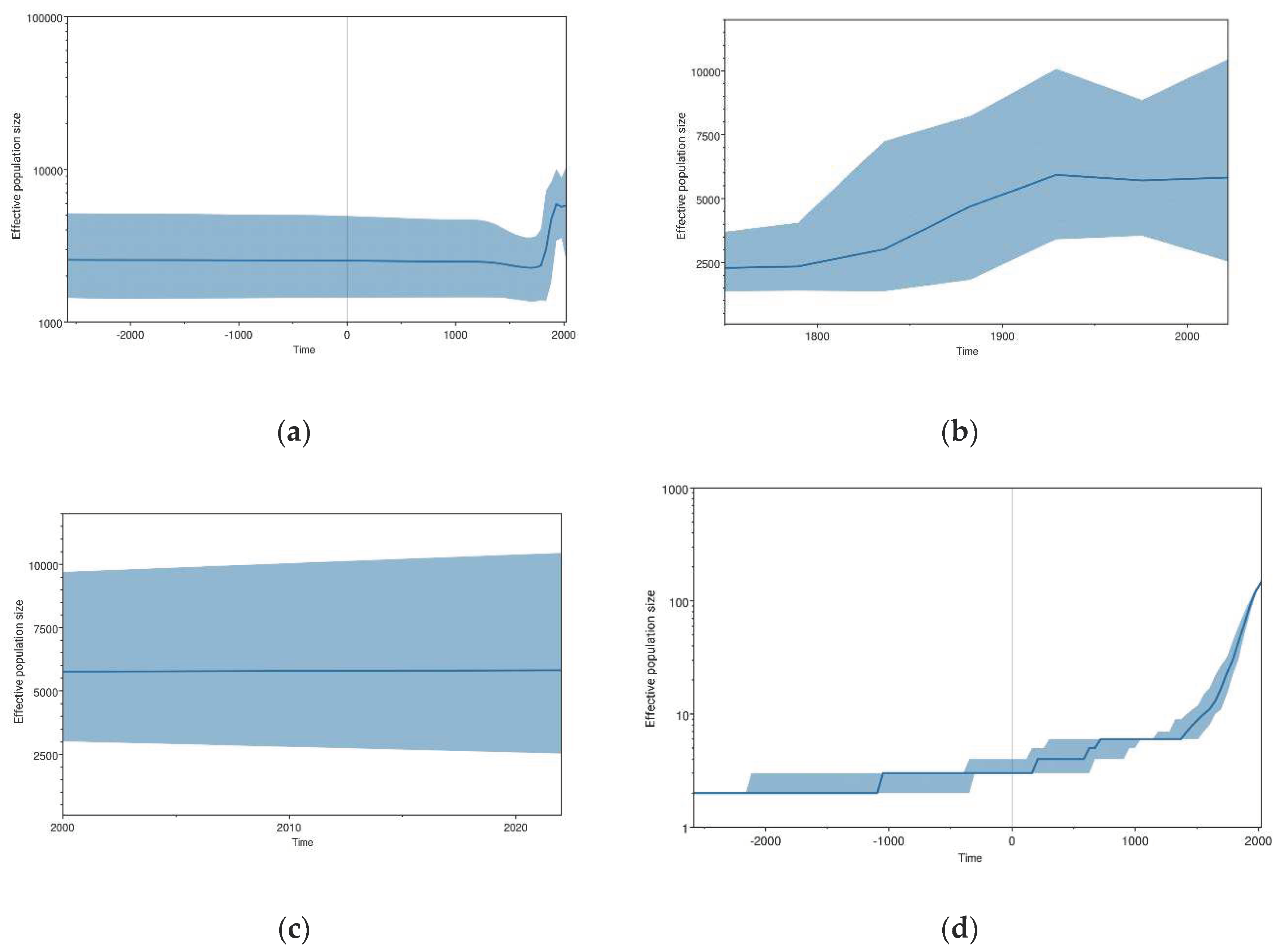
Figure 2a–c, and d illustrate the population dynamic of JEV. The skyline plot showed that the JEV population experienced complicated changes during the process of evolution after the 18th century. Initially, JEV remained relatively stable after its emergence (Figure 2 a). However, a fluctuation was observed in the late 18th century as shown in Figure 2 b. There was a gradual rise in the JEV population observed from 1790 to 1870. It was a time when the first recurrent epidemics of JEV occurred in Japan from 1871 onwards [29]. After 1870 it continued to rise and peaked during 1920 to 1930 and a little fluctuation was seen between 1975 to 1985 as presented in Figure 2 b. After that JEV population remained high (Figure 2 b). Figure 2 c explains that the JEV population is increasing slightly after 2010 to 2022, and genetic diversity has also increased slightly. The overall-JEV-lineage-through-time analysis (Figure 2 d) depicted a similar pattern of JEV population dynamics to that of BSP. It showed that the JEV population remained constant in the beginning, then increased stepwise, and finally attained a sharp increase. Whereas, after the sharp increase it makes a plateau-like pattern and maintains it to date (Figure 2 b&c).
4. Discussion
In the present study, we performed bioinformatic analysis of the available JEV genome sequences (n=160) to construct a timeline phylogenetic tree to determine the tMRCA and also determined the population dynamics of JEV. Our analysis showed that tMRCA occurred about 3120 years ago (YA) (95% Highest posterior density [HPD], 2680 to 3715). Furthermore, population dynamics analysis showed that JEV genetic diversity has been increasing since 2010. Therefore, we need to monitor JEV spread and update vaccines accordingly.
Recently, JEV cases have decreased due to the development and wide-scale application of JEV vaccines [30,31]. Although the JEV outbreak has been controlled, from this molecular data, we can conclude that disease threat still exists. Because a plateau-like pattern at high population diversity demonstrates the high genetic diversity of JEV, which infers that JEV is potentially replicating in the reservoir hosts, and it is a potential threat for future outbreaks (Figure 2 c). Our skyline data show that a fluctuation was observed after 1790 and gradual rise in the JEV population observed from 1790 to 1870 (Figure 2 b). It was a time when the first recurrent epidemics of JEV occurred in Japan from 1871 onwards [29], which is a good interpretation of our data. However, this data is not in line with previous reports where they observed the first rise in JEV population after 1930 [8]. This difference might come due selection of different models for analysis. However, Figure 2 c illustrates that after 2000, a similar trend was observed in previous and present JEV evolutionary studies [8,9,32]. In addition, despite the continuous application of different JEV vaccines, a high level of genetic diversity infers that the virus is replicating potentially and there are chances of gaining mutations that can lead to the emergence of new and highly pathogenic strains.
Flaviviruses such as West Nile virus (WNV), yellow fever virus (YFV), dengue virus (DENV), tick-borne encephalitis virus (TBEV), Zika virus (ZIKV), and JEV are the most abundant mosquito borne viruses which are causing outbreaks in different regions [33,34]. The evolutionary potential of viruses is determined by the nucleotide substitution rate [35]. In the present study, the mean rate of nucleotide substitution for JEV strains was 1.0628E-4 nucleotide substitutions per site per year (95% HPD values 8.7795E-5, 1.2564E-4) which is comparable with WNV 5.06E-4, TBEV 2.104E-4, and YFV 4.2E-4 substitutions per site per year [36,37,38] and shows the JEV evolutionary potential among flaviviruses. Recently, JEV GIV caused an outbreak in Australia which highlights its potential to spread to different regions of the world and cause epidemics. JEV GIV was also detected from Indonesia [39,40]. There are multiple factors which might play role in JEV spread to non-endemic areas such as infected mosquitoes transfer through wind-blown, harboring on planes, local mosquitoes can get JEV from infected migratory birds, adaptive changes in virus to produce long time viremia in vertebrate hosts, etc. Previous studies show the role of birds in the JEV zoonotic transmission as a natural reservoir and amplifying host [4,41]. Migratory birds can move hundreds to thousands of kilometers long and can extend across political boundaries [42]. Bird population is increasing because of development in agriculture. A recent study from Korea, reported that the distribution and density of migratory birds are correlated with JE cases in cities and they might be highly potential hosts contributing to transmit JEV in metropolitan areas [43]. JEV GIV infected birds’ migration from endemic areas to non-endemic regions might lead to JEV spread to Australia which requires intensive investigation.
Overall, our analysis shows that JEV genetic diversity has increased during the last five to ten years which indicates the potential JEV replication in the reservoir host, which is helping it to maintain its genetic diversity, and to continue its dispersal into non-endemic areas. Therefore, an enhanced surveillance system is needed along with precautionary measures such as regular vaccination and mosquito control to avoid future JEV outbreaks.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: JEV isolates analyzed in this study.
Funding
This communication was supported by the Natural Science Foundation of Hebei Province (No: C2020402050).
Conflicts of Interest
All authors declare no conflict of interest.
Ethical Statement
This communication does not contain any studies with human or animal subjects performed by any of the authors.
References
- Campbell GL, Hills SL, Fischer M, Jacobson JA, Hoke CH, Hombach JM, et al. Estimated global incidence of Japanese encephalitis: a systematic review. Bulletin of the World Health Organization. 2011;89:766-74.
- Schuh AJ, Ward MJ, Brown AJL, Barrett AD. Phylogeography of Japanese encephalitis virus: genotype is associated with climate. PLoS neglected tropical diseases. 2013;7(8):e2411. [CrossRef]
- Hameed M, Liu K, Anwar MN, Wahaab A, Safdar A, Di D, et al. The emerged genotype I of Japanese encephalitis virus shows an infectivity similar to genotype III in Culex pipiens mosquitoes from China. PLoS neglected tropical diseases. 2019;13(9):e0007716. Epub 2019/09/27. PubMed PMID: 31557156; PubMed Central PMCID: PMCPMC6762057. [CrossRef]
- Hameed M, Wahaab A, Nawaz M, Khan S, Nazir J, Liu K, et al. Potential Role of Birds in Japanese Encephalitis Virus Zoonotic Transmission and Genotype Shift. Viruses. 2021;13(3). Epub 20210224. PubMed PMID: 33668224; PubMed Central PMCID: PMCPMC7996159. [CrossRef]
- Gould E, Solomon T. Pathogenic flaviviruses. The Lancet. 2008;371(9611):500-9. [CrossRef]
- Misra UK, Kalita J. Overview: japanese encephalitis. Progress in neurobiology. 2010;91(2):108-20. [CrossRef]
- Heffelfinger JD, Li X, Batmunkh N, Grabovac V, Diorditsa S, Liyanage JB, et al. Japanese Encephalitis Surveillance and Immunization - Asia and Western Pacific Regions, 2016. MMWR Morbidity and mortality weekly report. 2017;66(22):579-83. Epub 2017/06/09. PubMed PMID: 28594790; PubMed Central PMCID: PMCPMC5720240. [CrossRef]
- Gao X, Liu H, Li M, Fu S, Liang G. Insights into the evolutionary history of Japanese encephalitis virus (JEV) based on whole-genome sequences comprising the five genotypes. Virol J. 2015;12:43. Epub 2015/04/18. PubMed PMID: 25884184; PubMed Central PMCID: PMCPMC4369081. [CrossRef]
- Xu G, Gao T, Wang Z, Zhang J, Cui B, Shen X, et al. Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution. Viruses. 2023;15(3). Epub 20230224. PubMed PMID: 36992335; PubMed Central PMCID: PMCPMC10054483. [CrossRef]
- Zhang H, Rehman MU, Li K, Luo H, Lan Y, Nabi F, et al. Epidemiologic Survey of Japanese Encephalitis Virus Infection, Tibet, China, 2015. Emerging infectious diseases. 2017;23(6):1023-4. Epub 2017/05/19. PubMed PMID: 28518046; PubMed Central PMCID: PMCPMC5443422. [CrossRef]
- Zhang H, Luo H, Ur Rehman M, Nabi F, Li K, Lan Y, et al. Evidence of JEV in Culex tritaeniorhynchus and pigs from high altitude regions of Tibet, China. Journal of vector borne diseases. 2017;54(1):69-73. Epub 2017/03/30. PubMed PMID: 28352048.
- Aure WE, Sayama Y, Saito-Obata M, Salazar NP, Malbas FF, Jr., Galang HO, et al. Japanese encephalitis virus genotype III from mosquitoes in Tarlac, Philippines. IJID Reg. 2022;4:59-65. Epub 20220516. PubMed PMID: 36093364; PubMed Central PMCID: PMCPMC9453045. [CrossRef]
- Howard-Jones AR, Pham D, Jeoffreys N, Eden JS, Hueston L, Kesson AM, et al. Emerging Genotype IV Japanese Encephalitis Virus Outbreak in New South Wales, Australia. Viruses. 2022;14(9). Epub 20220824. PubMed PMID: 36146660; PubMed Central PMCID: PMCPMC9505215. [CrossRef]
- Waller C, Tiemensma M, Currie BJ, Williams DT, Baird RW, Krause VL. Japanese Encephalitis in Australia - A Sentinel Case. N Engl J Med. 2022;387(7):661-2. PubMed PMID: 36070717. [CrossRef]
- Liu W, Fu S, Ma X, Chen X, Wu D, Zhou L, et al. An outbreak of Japanese encephalitis caused by genotype Ib Japanese encephalitis virus in China, 2018: A laboratory and field investigation. PLoS neglected tropical diseases. 2020;14(5):e0008312. Epub 2020/05/27. PubMed PMID: 32453787; PubMed Central PMCID: PMCPMC7274457. [CrossRef]
- Fang Y, Li XS, Zhang W, Xue JB, Wang JZ, Yin SQ, et al. Molecular epidemiology of mosquito-borne viruses at the China-Myanmar border: discovery of a potential epidemic focus of Japanese encephalitis. Infectious diseases of poverty. 2021;10(1):57. Epub 2021/04/28. PubMed PMID: 33902684; PubMed Central PMCID: PMCPMC8073957. [CrossRef]
- Hameed M, Wahaab A, Shan T, Wang X, Khan S, Di D, et al. A Metagenomic Analysis of Mosquito Virome Collected From Different Animal Farms at Yunnan-Myanmar Border of China. Frontiers in microbiology. 2020;11:591478. Epub 2021/02/26. PubMed PMID: 33628201; PubMed Central PMCID: PMCPMC7898981. [CrossRef]
- Hameed M, Khan S, Xu J, Zhang J, Wang X, Di D, et al. Detection of Japanese encephalitis virus in mosquitoes from Xinjiang during next-generation sequencing arboviral surveillance. Transbound Emerg Dis. 2021;68(2):467-76. Epub 20200815. PubMed PMID: 32614516. [CrossRef]
- Sanborn MA, Wuertz KM, Heung-Chul K, Yang Y, Li T, Pollett SD, et al. Identification of Japanese Encephalitis Virus Genotype V and Other Mosquito-borne Viruses in Camp Humphreys, Republic of Korea, using Metagenomic Analysis. bioRxiv. 2021.
- Mackenzie JS, Williams DT, van den Hurk AF, Smith DW, Currie BJ. Japanese Encephalitis Virus: The Emergence of Genotype IV in Australia and Its Potential Endemicity. Viruses. 2022;14(11). Epub 20221109. PubMed PMID: 36366578; PubMed Central PMCID: PMCPMC9698845. [CrossRef]
- van den Hurk AF, Skinner E, Ritchie SA, Mackenzie JS. The Emergence of Japanese Encephalitis Virus in Australia in 2022: Existing Knowledge of Mosquito Vectors. Viruses. 2022;14(6). Epub 20220602. PubMed PMID: 35746679; PubMed Central PMCID: PMCPMC9231386. [CrossRef]
- Hall TA, editor BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series; 1999: [London]: Information Retrieval Ltd., c1979-c2000.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods. 2017;14(6):587-9. [CrossRef]
- Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS computational biology. 2014;10(4):e1003537. Epub 2014/04/12. PubMed PMID: 24722319; PubMed Central PMCID: PMCPMC3985171. [CrossRef]
- Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics. 2002;161(3):1307-20. Epub 2002/07/24. PubMed PMID: 12136032; PubMed Central PMCID: PMCPMC1462188.
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of molecular evolution. 1985;22(2):160-74. Epub 1985/01/01. PubMed PMID: 3934395. [CrossRef]
- Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular biology and evolution. 2005;22(5):1185-92. Epub 2005/02/11. PubMed PMID: 15703244. [CrossRef]
- Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic biology. 2018;67(5):901-4. [CrossRef]
- Lewis L, Taylor HG, et al. Japanese B encephalitis; clinical observations in an outbreak on Okinawa Shima. Archives of neurology and psychiatry. 1947;57(4):430-63. Epub 1947/04/01. PubMed PMID: 20297184.
- Gao X, Nasci R, Liang G. The neglected arboviral infections in mainland China. PLoS neglected tropical diseases. 2010;4(4):e624. [CrossRef]
- Yu Y. Phenotypic and genotypic characteristics of Japanese encephalitis attenuated live vaccine virus SA14-14-2 and their stabilities. Vaccine. 2010;28(21):3635-41. [CrossRef]
- Pan XL, Liu H, Wang HY, Fu SH, Liu HZ, Zhang HL, et al. Emergence of genotype I of Japanese encephalitis virus as the dominant genotype in Asia. J Virol. 2011;85(19):9847-53. Epub 2011/06/24. PubMed PMID: 21697481; PubMed Central PMCID: PMCPMC3196406. [CrossRef]
- Huang YS, Higgs S, Vanlandingham DL. Emergence and re-emergence of mosquito-borne arboviruses. Curr Opin Virol. 2019;34:104-9. Epub 20190208. PubMed PMID: 30743191. [CrossRef]
- Peinado RDS, Eberle RJ, Arni RK, Coronado MA. A Review of Omics Studies on Arboviruses: Alphavirus, Orthobunyavirus and Phlebovirus. Viruses. 2022;14(10). Epub 20221005. PubMed PMID: 36298749; PubMed Central PMCID: PMCPMC9607206. [CrossRef]
- Duchêne S, Di Giallonardo F, Holmes EC. Substitution Model Adequacy and Assessing the Reliability of Estimates of Virus Evolutionary Rates and Time Scales. Mol Biol Evol. 2016;33(1):255-67. Epub 20150928. PubMed PMID: 26416981. [CrossRef]
- Añez G, Grinev A, Chancey C, Ball C, Akolkar N, Land KJ, et al. Evolutionary dynamics of West Nile virus in the United States, 1999-2011: phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl Trop Dis. 2013;7(5):e2245. Epub 20130530. PubMed PMID: 23738027; PubMed Central PMCID: PMCPMC3667762. [CrossRef]
- Egyed L, Rónai Z, Dán Á. Hungarian tick-borne encephalitis viruses isolated from a 0.5-ha focus are closely related to Finnish strains. Ticks Tick Borne Dis. 2018;9(5):1064-8. Epub 20180407. PubMed PMID: 29655579. [CrossRef]
- Bryant JE, Holmes EC, Barrett AD. Out of Africa: a molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 2007;3(5):e75. PubMed PMID: 17511518; PubMed Central PMCID: PMCPMC1868956. [CrossRef]
- Garjito TA, Widiarti, Anggraeni YM, Alfiah S, Tunggul Satoto TB, Farchanny A, et al. Japanese encephalitis in Indonesia: An update on epidemiology and transmission ecology. Acta Trop. 2018;187:240-7. Epub 20180815. PubMed PMID: 30118700. [CrossRef]
- Solomon T, Ni H, Beasley DW, Ekkelenkamp M, Cardosa MJ, Barrett AD. Origin and evolution of Japanese encephalitis virus in southeast Asia. J Virol. 2003;77(5):3091-8. PubMed PMID: 12584335; PubMed Central PMCID: PMCPMC149749. [CrossRef]
- Xiao C, Li C, Di D, Cappelle J, Liu L, Wang X, et al. Differential replication efficiencies between Japanese encephalitis virus genotype I and III in avian cultured cells and young domestic ducklings. PLoS neglected tropical diseases. 2018;12(12):e0007046. [CrossRef]
- Palm EC, Newman SH, Prosser DJ, Xiao X, Ze L, Batbayar N, et al. Mapping migratory flyways in Asia using dynamic Brownian bridge movement models. Movement ecology. 2015;3(1):3. Epub 2015/02/25. PubMed PMID: 25709838; PubMed Central PMCID: PMCPMC4337761. [CrossRef]
- Bae W, Kim JH, Kim J, Lee J, Hwang ES. Changes of epidemiological characteristics of Japanese encephalitis viral infection and birds as a potential viral transmitter in Korea. Journal of Korean medical science. 2018;33(9). [CrossRef]
Figure 1.
Bayesian inference based phylogenetic tree for all available JEV complete genomes till 2022 was reconstructed in the present study. BEAST Bayesian inference-based chronograms present the divergence time estimates, and phylogenetic relationship among previously reported JEV genomes. Tree was constructed based on C, E, NS2B and NS5 protein coding sequences of all publicly available JEV sequences.
Figure 1.
Bayesian inference based phylogenetic tree for all available JEV complete genomes till 2022 was reconstructed in the present study. BEAST Bayesian inference-based chronograms present the divergence time estimates, and phylogenetic relationship among previously reported JEV genomes. Tree was constructed based on C, E, NS2B and NS5 protein coding sequences of all publicly available JEV sequences.
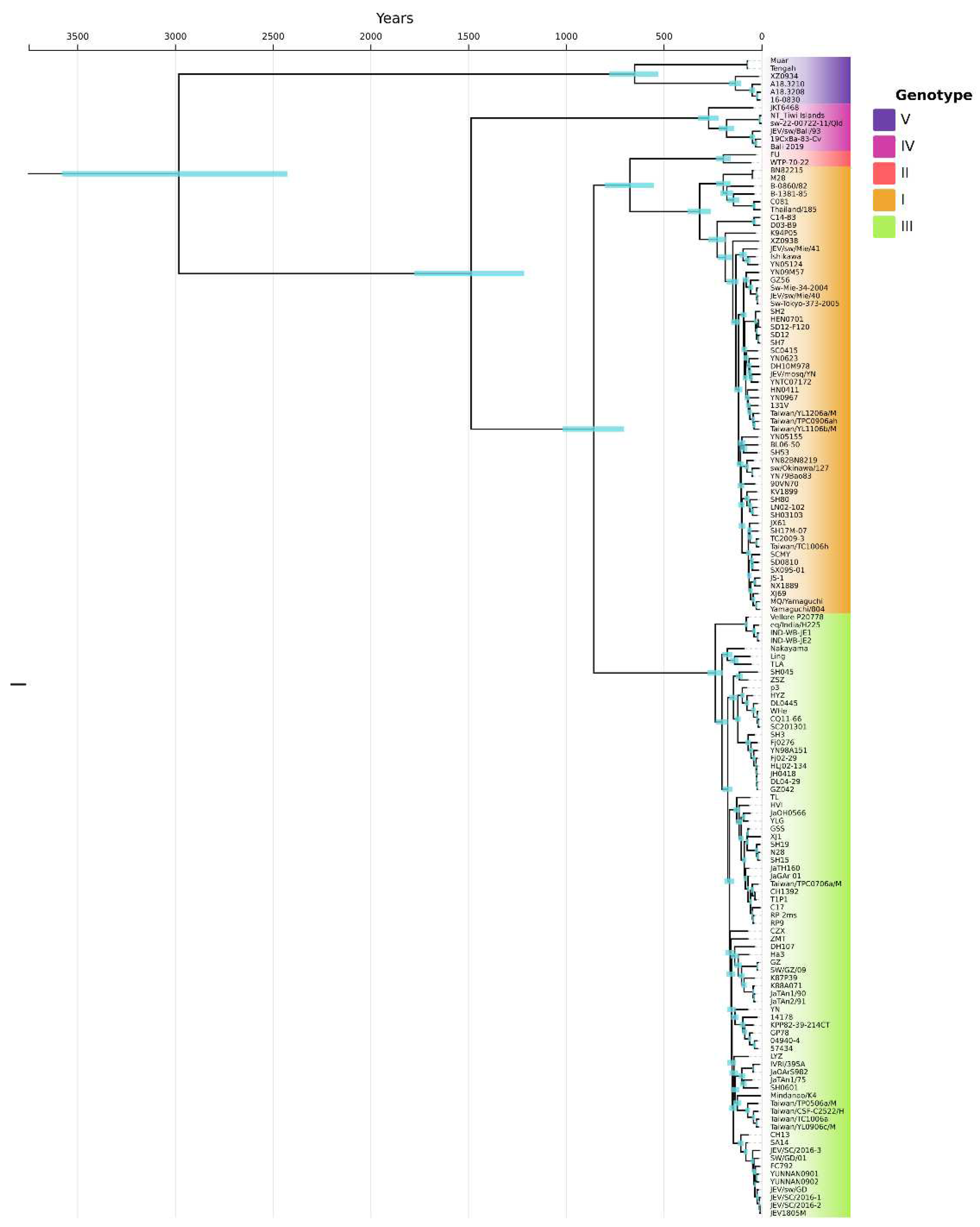
Figure 2.
Bayesian skyline plots representing the demographic history of JEV. The central solid blue line represents the median posterior value, and the shaded area represents the 95% HPD intervals. The x-axis corresponds to time (years), while the y-axis represents the effective population size. (a) Population modeling during the whole evolutionary history; (b) Population trends during 1750-2022, (c) Population dynamics from 2000 to 2022, (d) JEV lineage through time.
Figure 2.
Bayesian skyline plots representing the demographic history of JEV. The central solid blue line represents the median posterior value, and the shaded area represents the 95% HPD intervals. The x-axis corresponds to time (years), while the y-axis represents the effective population size. (a) Population modeling during the whole evolutionary history; (b) Population trends during 1750-2022, (c) Population dynamics from 2000 to 2022, (d) JEV lineage through time.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



