
Submitted:
02 May 2023
Posted:
03 May 2023
You are already at the latest version
Abstract
Keywords: Obesity; hypothalamus; appetite; glucose homeostasis; weight-loss drugs; AGRP; POMC; NTS; incretins.
Keywords:
Obesity
; hypothalamus
; appetite
; glucose homeostasis
; weight-loss drugs
; AGRP
; POMC
; NTS
; incretins
1. Introduction
Extensive evidence has unequivocally confirmed the importance of the brain in metabolic disorders and obesity [1]. Research identifying its pathophysiological role has spanned over several decades. From its humble beginnings of employing rodents with hypothalamic lesions, which aided in identifying the role of distinct brain regions in appetite/satiety regulation, to the use of more sophisticated approaches such as chemogenetics and GWAS to identify novel therapeutic targets/pathways in the brain, the central nervous system (CNS) has now been firmly established as a critical component that is dysregulated in the development of obesity [2,3,4] . More recently, research on the metabolic role of the CNS has also paved the way for the identification of drug targets in metabolic disorders such as Type 2 diabetes and obesity. Hence a comprehensive understanding of how the CNS fine-tunes metabolic functions could aid in the further development of therapeutics for various metabolic disorders. A key aspect of current metabolic research is focused on understanding the contributions of the hypothalamic and the brainstem circuitry in the regulation of appetite and energy homeostasis. By manipulating neuronal populations located in these regions, research has uncovered several major neural circuits that exerts control over appetite and metabolic functions.
2. Hypothalamic and brainstem nuclei in appetite control and energy balance
2.1. Hypothalamus
The hypothalamus is one of the most well-studied brain regions in metabolism. Apart from regulating a broad range of thermoregulatory, reproductive and cardiovascular functions, it also exerts tremendous influence on several aspects of energy balance. The hypothalamus is composed of multiple nuclei located adjacent to the third ventricle. These nuclei comprise of a distinct subpopulation of neurons, capable of altering energy intake and/or energy expenditure via anabolic or catabolic functions. The complex nexus of hypothalamic neuronal interconnections can integrate responses from peripheral signals (hormones, nutrients and metabolites), to modulate appetite centrally, and to influence lipid and glucose metabolism, peripherally. Additionally, they also have reciprocal projections to and from extrahypothalamic nuclei located in the brainstem, midbrain and forebrain which can also alter synaptic activity in the hypothalamic metabolic circuits. Consequently, via integration and coordination of responses from the brain and periphery, hypothalamic nuclei are key regulators of energy homeostasis.
2.1.1. Arcuate nucleus (ARC)
The ARC is considered as one of most important brain regions involved in the regulation of appetite and energy expenditure. Located near the median eminence, a region enriched in fenestrated capillaries, the ARC is accessible to circulating hormones, nutrients and metabolites, thus, serving as an ideal relay center to communicate circulating peripheral signals to the brain. The ARC comprises of two distinct neuronal subpopulations that have opposing roles in energy homeostasis, the anabolic NPY/AGRP neurons and the catabolic, POMC/CART neurons (referred to as AGRP and POMC neurons henceforth respectively). Both these neurons are first order neurons, which have glucose and nutrient sensing capabilities, in addition to receiving input from circulating hormones and satiety signals [5,6,7,8]. These counterregulatory neuronal populations are modulated by energy status. Food deprivation rapidly activates AGRP neurons and inhibits POMC neurons [9,10,11]. AGRP neurons release both NPY, which is an agonist for the Y1-5 receptors, and AGRP, an inverse agonist for melanocortin receptors [12,13]. Ablation of AGRP neurons results in a dramatic reduction in feeding, while acute activation results in a robust increase in food intake, weight gain and altered autonomic outflow to several organs and tissues [14,15,16,17]. NPY was one of the first orexigenic neuropeptides to be identified, and subsequent functional studies revealed a potent, albeit fleeting, appetite-stimulating effect [18]. More recently, it has been revealed that NPY-mediated effects on feeding is mediated via the Y1 receptor, while its effects on energy expenditure are driven via the Y2 receptor [19]. Although both of these orexigenic neuropeptides, NPY and AGRP, have complimentary roles in triggering a hyperphagic response and reducing energy expenditure, the longer-lasting or sustained effect of these neurons on food intake is dependent on AGRP release, while the more rapid effect on food intake is dependent on NPY secretion [17,18,20]. Furthermore, AGRP neuronal activation results in an increase in adiposity and altered substrate utilization [21]. In stark contrast to AGRP neurons, POMC neurons have a pronounced catabolic effect due to their ability to release the anorectic neuropeptide, α-melanocyte-stimulating hormone (α-MSH), a major satiety neuropeptide which is an agonist of melanocortin receptors [22]. Ablation of POMC neurons results in a mild obesity phenotype characterized by reduced food intake [23]. Interestingly, only chronic, but not acute chemogenetic activation of these neurons results in suppression of food intake, suggesting a role for POMC in maintaining long-term energy homeostasis [24]. POMC neurons have been reported to exhibit functional and spatial heterogeneity characterized by differences in both molecular architecture and anatomical projections to distinct brain regions, suggesting a more complex neural network involved in metabolic control [25,26,27].
Both AGRP and POMC neurons also express receptors for insulin (IR) and leptin (LepR). Leptin depolarizes and increases firing frequency of POMC neurons, while hyperpolarizing and inhibiting AGRP/NPY neuronal activity and neuropeptide release [28,29,30,31,32]. Mechanistic studies revealed that deletion of Rho-kinase 1, a protein kinase activated by Gq/11 and G12/13 family of heterotrimeric G proteins, in both AGRP and POMC neuronal populations resulted in leptin resistance and obesity [33,34]. Collectively, these data point to a crucial central mechanism by which leptin can induce a negative energy balance. Studies investigating the role of insulin signaling in both AGRP and POMC neurons on appetite regulation have yielded contradictory results. While some studies reported on little-to-no effect on appetite and body weight change with IR deletion in AGRP neurons, others have described a more nuanced role of AGRP-specific insulin signaling on regulating meal size [35,36]. A context-dependent appetite suppression role is reported for insulin signaling in the AGRP neurons, which is characterized by acute repression of feeding bouts without altering total calorie intake, and suppression of highly palatable high fat diet food over standard chow [36]. In the case of POMC neurons, while deletion of LepR results in mild obesity, knockout of IR in these neurons had no significant effect on body weight [37,38]. Furthermore, both AGRP and POMC neurons are modulated by postprandial signals, such as ghrelin, incretins and amylin, to regulate food intake [39,40,41,42,43]. Apart from having integral roles in appetite and satiety regulation, these neuronal populations are also involved in maintaining glucose homeostasis as chemogenetic activation of AGRP and POMC neurons revealed distinct roles of G protein activation on food intake and glycemic control [15,17,24,44]. The mechanisms through which both these neuronal populations regulate glucose homeostasis will be discussed in later sections of this review.
2.1.2. Paraventricular nucleus (PVH)
The PVH serves as an important convergence/termination point for orexigenic and anorexigenic projections arising from the ARC and other hypothalamic regions. Neurons present in this region express two different types of melanocortin receptors subtypes (MC3R and MC4R) that can be activated by the melanocortin peptide, α-MSH [45,46]. α-MSH and AGRP, released from the ARC projections, can modulate PVH neuronal activity by either activating or antagonizing the melanocortin receptors respectively [13,46,47,48]. Thus, these neurons provide counterregulatory inputs to fine tune energy balance in response to changes in the levels of circulating signals. PVH neurons express single-minded 1 (Sim1), a transcriptional factor required for PVH development and the maintenance of energy homeostasis [49,50]. Sim1 neurons have pronounced effects on satiety and energy homeostasis as both sim1 heterozygous mice, and inducible Sim1-deficient mice, exhibit hyperphagia leading to obesity [50,51]. A major subset of Sim1 neurons in the PVH express MC4R [37,46] . Mutations in the MC4R gene are a leading cause of monogenic forms of obesity, and MC4R variants have been linked to increased obesity in certain populations [52,53,54,55]. The MC4R/Sim1 neurons, located in the PVH, together with the POMC neuronal projections, arising from the ARC, form the melanocortin pathway in the hypothalamus. Stimulation of MC4R neurons in the PVH results in pronounced satiation effects and thereby can induce a negative energy balance and confer protection against obesity [37,56,57,58] . Interestingly, short-term administration of MC4R agonists can also increase resting energy expenditure and shift substrate utilization towards increased fat oxidation in obese individuals suggesting additional mechanisms through which the melanocortin pathway and Sim1 neurons induce a negative energy balance [59]. Knockdown of MC4R results in potential disruption of synaptic plasticity and attenuation of long-term potentiation in the PVH [60]. Perturbation of MC4R signaling in the PVH alone, or in both PVH and DMV results in hyperphagic obesity with reduced energy expenditure and defects in insulin sensitivity [56,61]. It is to be noted that MC4R-expressing neurons are not just located in the hypothalamic nuclei, but also located in the brainstem, intermediolateral cell column of the spinal cord, and autonomic neurons where they not only exert prominent cardiovascular effects, but also regulate metabolic functions including thermogenesis, glucose homeostasis and energy expenditure [62,63,64,65]. Interestingly, PVH not only comprises of MC4R neurons, but also contains other neuronal populations such as prodynorphin-expressing neurons, which lack MC4R. These neuronal populations have comparable effects to the PVH-MC4R expressing neurons on regulating satiety [66]. Several such anatomically distinct neuronal populations have been identified in the PVH as having appetite-regulatory roles, which further highlights the complexity of this nucleus [67,68,69,70]. For a more detailed review on the pathophysiological roles of MC4R neurons, readers can refer to excellent reviews on this topic [71,72].
2.1.3. Ventromedial nucleus of the hypothalamus (VMH)
Despite having an inauspicious history in metabolism research, the VMH is still appreciated as one of the principal satiety centers in the brain [73]. Early studies have highlighted an important role of VMH in suppressing appetite [74,75]. Apart from regulating food intake, VMH neurons have also been associated with improvements in several metabolic parameters and conferring protection against obesity [76,77]. A major subset of VMH neurons express steroidogenic factor 1 (SF1), often serving as a biomarker to distinguish VMH from other hypothalamic nuclei. Similar in function to the POMC neurons, activated SF1 neurons elicit pronounced anorexigenic effects with increased energy expenditure [78,79]. These neurons not only provide excitatory input directly onto the POMC neurons, but also project to the paraventricular thalamus to induce an aversive effect and suppress appetite [80,81]. Deletion of LepR from SF1 neurons also resulted in similar degree of weight gain in mice when compared to LepR-specific KO in POMC neurons, suggesting important roles of leptin signaling in both sets of neuronal populations [37,82]. Moreover, SF1 neurons have distinct projections to other regions of the brain involved in negating insulin-induced hypoglycemia [83]. This will be covered in a later section. For a more detailed review on the role of SF1 neurons in metabolic disorders, the readers can refer to the review by Fosch et al [84] .
2.1.4. Dorsomedial hypothalamus (DMH)
Another hypothalamic nucleus that affects feeding response is the DMH. DMH lesion in both young and older rats produced a hypophagic response with reduced body weight [85]. Interestingly, the DMH expresses NPY, which shows altered levels in various models of obesity [86,87,88]. Overexpression of NPY in the DMH results in an increase in food intake, weight gain and an obese phenotype under high fat diet conditions, while knockdown of NPY ameliorated these effects in obese mice [89]. Inhibitory GABAergic neurons projecting to the PVH have been proposed as a key mechanism for eliciting a DMH-mediated orexigenic response [90]. Additionally, DMH neurons project to the ARC where they inhibit POMC neurons during fasting suggesting parallel neural circuits from DMH to regulate appetite [11]. The DMH may also be involved in the regulation of food intake by other hormones and peptides, as intra-DMH administration of the appetite-suppressing hormone, cholecystokinin (CCK), resulted in a suppression of food intake [91,92]. Interestingly, under refeeding conditions, excitatory glutamatergic projections are also activated by a subset of DMH glutamatergic neurons leading to reduced food intake [93]. A recently published study reported on DMH having bidirectional effects on food intake, which receive key leptin-responsive projections from the AGRP neurons [94]. Thus, it is likely that DMH projections could participate in the fine tuning of energy intake by activating distinct inhibitory and excitatory projections to other hypothalamic nuclei.
2.2. Brainstem
The brainstem exerts significant control over autonomous biological functions. The medulla is a key brainstem structure which has prominent cardioregulatory and metabolic functions, via specialized cardiovascular and satiety centers respectively. The medullary cardiovascular centers have well-established roles in the homeostatic regulation of blood pressure via the baroreflex [95]. Although the brainstem is not as well-investigated as their counterpart, the hypothalamus, in metabolism, studies dating back to the 1970s highlighted the importance of the caudal brainstem in mediating satiety and glucoregulatory responses [96,97]. Importantly, specialized medullary regions serve as crucial integration points between the CNS and the digestive tract. They receive visceral afferent input from gastrointestinal sensory neurons, the latter conveying satiety signals in response to a meal. Additionally, the brainstem also comprises of a circumventricular organ, area postrema (AP), which allows access to satiety signals. These signals in turn can modulate adjacent and supra-adjacent neuronal populations located in the brainstem. These proximally located neuronal populations in the caudal brainstem, in conjunction with the AP, are key structures in mediating postprandial satiety [98,99].
2.2.1. Dorsal vagal complex (DVC)
The caudal brainstem not only expresses receptors for circulating pressor peptides, but it can also be modulated by metabolic cues and thus exerts control over energy homeostasis [100,101,102,103,104]. The DVC located in the hindbrain is designated as the brainstem satiety center. The DVC comprises of the AP, the nucleus of the solitary tract (NTS), and the dorsal motor nucleus of the vagus nerve (DMV). The NTS serves as the primary hub for ascending neural signals from the nodose ganglia, which contains cell bodies for several vagal afferents that densely innervate the gastrointestinal tract (GIT) [105]. The sensory vagal nerve terminals in the GIT are heterogenous in nature conveying both chemosensory, from nutrients and gut hormones, and mechanosensory signals to the brainstem [105,106]. Postprandial gut hormones and nutrients suppress food intake by transmitting information via the sensory vagal afferent terminals to the NTS, a crucial entry point in the brain for visceral information [107,108]. CCK, one of the first gut peptides to be identified to mediate satiety, elicits its actions by acting on the CCK-A receptors that are abundantly expressed on the vagal afferents and the cell bodies of the nodose ganglia [107,109,110,111]. Additionally other receptors involved in regulating satiety, such as the LepR, are also expressed on these cell bodies [112]. As a result, circulating signals such as leptin can also act along with CCK on the nodose ganglia, to synergistically suppress food intake [113,114]. Another class of gut hormones, the incretins, also exert prominent effects on satiety and glucose homeostasis. The incretins, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), are released in response to a meal, and they act on their corresponding receptors (GLP1R and GIPR) located in pancreatic islets where they promote insulin release. These receptors are also expressed in non-islet cells, where they exert prominent metabolic actions independent of direct effects on pancreatic insulin secretion [115]. In the GIT, the GLP1R is expressed on mechanosensitive vagal sensory neurons, and its activation results in pronounced inhibition of food intake, while its knockdown is associated with an increased meal size [116,117].
Interestingly, profiling of G-protein coupled receptors in the GIT revealed a lack of receptor expression for GIP and ghrelin on vagal afferents, potentially highlighting other CNS-dependent mechanisms to alter food intake [118]. Similar to the ARC, the AP lacks a well-defined blood brain barrier, as a result is accessible to various satiety signals and circulating hormones. Since the NTS is located in close proximity to the fourth ventricle, it serves as a crucial node for integrating signals from the gut and circulation. Satiety signals, such as CCK, GLP-1 and their analogs, have been shown to inhibit appetite by acting on their corresponding receptors localized to the brainstem neurons in the satiety center [119,120,121]. The GLP1R is highly expressed in the NTS, and knockdown of preproglucagon, a precursor for GLP1, in the brainstem results in hyperphagia and increased adiposity, suggesting a crucial role for central GLP1R in mediating satiety [122]. Interestingly, GIPR agonism not only enhances the anorectic effect of GLP1R agonism, but recent studies suggest that it improves tolerability of PYY analogs by modulating the brainstem neural circuits and blocking its anorectic effect [123,124,125]. Thereby, understanding of incretins-mediated modulation of the brainstem neural circuitry has significant implications for the development of weight loss drugs with an improved side effect profile. The role of incretins in the hypothalamic and brainstem neural circuitry in the regulation of energy homeostasis will be discussed in a later section. Other pancreatic and gut-derived postprandial signals, such as amylin and PYY, also act on neuronal populations in the AP and NTS to promote satiety [126,127,128,129,130]. Additionally, leptin-mediated signaling in the NTS also activates the satiation neural circuitry to suppress food intake and regulate energy balance [131,132,133].
Projections from the NTS extend to other brain regions involved in appetite control and food aversion behaviors, where they suppress appetite by triggering either a positive or negative valence [134,135]. The latter may well be dependent on both the molecular architecture of the neural circuit, and the brain regions innervated by it. For instance, NTS projections to calcitonin gene-related protein (CGRP) expressing neurons located in higher brain regions, are strongly involved in mediating anorexia and reducing body weight [136,137]. However, they can exert opposing motivational valences, since projections to specific brain regions can generate both a positive valence (NTS to PVH projection) and a negative valence (NTS to PBN projection); the latter aversive response triggered by the activation of CGRP neurons in the PBN [138,139,140]. In stark contrast to the CCK neurons, calcitonin receptor expressing neurons from the NTS do not activate CGRP neurons, and hence produce a non-aversive suppression of food intake despite projecting to the PBN [140]. Other neuronal populations such as GLP-1 expressing neurons, which are primarily located in the caudal NTS, have projections to the VTA where they regulate intake of highly-palatable food [141,142].
The NTS neurons also comprises of a small, but metabolically relevant, population of POMC expressing neurons, accounting for about 10% of the total POMC neuronal population [143,144]. Interestingly, while they are activated by postprandial visceral afferents from the gut, they do not co-express several of the other neuropeptide markers observed in the NTS, suggesting a distinct hub of neurons involved in mediating satiety [145,146]. These neuronal populations are functionally similar to the POMC neurons in the ARC, but they exhibit different kinetics in terms of suppression of food intake. ARC-POMC neurons are involved in long-term suppression, while the NTS-POMC neurons mediate short-term feeding responses [24]. The latter neurons potentially involved in a more rapid feeding suppression via circulating satiety signals. NTS-POMC neurons have been shown to be crucial for the acute appetite-suppressing effect of lorcaserin, indicative of their clinical relevance [147]. More recently, this effect of lorcaserin was also shown to be meditated via the GLP-1 neurons in the brainstem, in addition to the NTS-POMC neurons [148].
In addition to the regulation of food intake, the hindbrain circuitry also has important roles in glucose sensing and modulation of systemic glucose via vagal efferents [149,150]. Neuropeptide FF (NPFF), a key analgesic peptide which has been demonstrated to have a role in substrate utilization and regulation of energy balance, is strongly expressed in the caudal brainstem, mainly localized in the DVC [151]. More recently, a study reported on impairments in glucose homeostasis in mice deficient in NPFF, further highlighting the glucoregulatory role of the DVC [152]. The role of the various satiety signals in regulating glucose and lipid metabolism via brainstem circuits will be covered in more detail in later sections.
The sections so far highlight the pivotal roles of hypothalamic orexigenic and anorexigenic neuronal populations, along with the brainstem satiety center, in the regulation of energy intake and expenditure. While the NTS integrates multiple metabolic cues to promote satiation, the ARC neuronal populations are able exert both short- and long-term effects on energy homeostasis in response to energy demands.
3. Crosstalk between hypothalamic and brainstem nuclei with metabolic organs to regulate energy and glucose homeostasis
Autonomic dysfunction is associated with an elevated risk of developing metabolic syndrome and cardiovascular diseases [95,153]. In the case of metabolic disorders, this is due to an augmentation of sympathetic activity resulting in a breakdown of the glucose homeostatic processes [154]. The end result is a chronic elevation in blood glucose due to an imbalance between glucose production and glucose clearance from the blood by insulin-sensitive organs. The resulting hyperglycemic condition is known to result in extensive vascular complications due to endothelial damage, and hence serves as an independent risk factor for cardiovascular diseases [155,156]. Understanding the potential mechanisms involved in the maintenance of glucose homeostasis is therefore of high clinical relevance. Hypothalamic and brainstem nuclei are key components of a central network that help maintain a balance between sympathetic and parasympathetic nerve activity to the endocrine organs, resulting in exerting significant control over glucose metabolism. Circulating signals act on these neurons to recalibrate autonomic efferents to the peripheral metabolic organs. This section focusses on the crosstalk between the CNS and some of the important metabolic organs involved in maintenance of glucose and energy homeostasis.
3.1. Brain-pancreas axis and the role of central insulin signaling
Multiple brain regions, including several hypothalamic and brainstem nuclei, contain neural networks that regulate pancreatic islet function via autonomic efferents [157,158,159]. Functional validation of these circuits revealed important roles for several hypothalamic nuclei in pancreatic insulin release [159]. These hypothalamic nuclei exhibit bidirectional control over insulin release. Stimulation of a subpopulation of oxytocin neurons in the PVH suppressed insulin secretion, whereas increased glucokinase activity in the ARC augmented glucose-stimulated insulin secretion and improved glucose tolerance [160,161]. In addition to the hypothalamic nuclei, pancreas-projecting DMV neurons were reported to be excited by GLP1, which can then potentially increase insulin release by a vagal efferent pathway [162,163].
The IR is widely expressed in the brain, which enables circulating insulin to modulate neuronal populations that are integral to metabolism [164]. The hypothalamus represents a crucial insulin-responsive brain region involved in maintaining euglycemia [165]. The hypothalamic IR signaling and a downstream target of IR, the K-ATP channels, have both been reported to have essential roles in the regulation of endogenous glucose production [166,167]. ICV infusion of insulin into the third ventricle suppresses endogenous glucose production, while perturbation of central insulin signaling impaired both glucose and lipid metabolism [35,168]. Interestingly, the divergent mechanisms through which central insulin signaling regulates glucose and lipid homeostasis may be attributed to differing outcomes of IR activation in AGRP versus the POMC neurons. Insulin effects on AGRP neurons results in improved glucose homeostasis, while its action on POMC results in changes in lipid metabolism [169]. However, these effects may be more nuanced, as insulin receptor signaling in the POMC neurons has been shown to regulate glucose homeostasis which is shown to depend on the nutritional (fed vs fasted) and the pathophysiological status (obese vs lean) of the mice [170]. A similar subtle, yet significant, effect was observed for insulin-mediated feeding suppression via the AGRP neurons, which is described in an earlier section. More recently, the antihyperglycemic effect of hypothalamic insulin signaling was shown to be dependent on the neuropeptidegric system, 26Rfa and its receptor GPR103 [171]. ICV administration of both insulin and 26Rfa greatly augmented glucose-mediated insulin release, and GPR103 blockade greatly suppressed both their effects on glucose homeostasis, suggesting a potential key mechanism of central insulin signaling [171].
In addition to the ARC, insulin signaling in the brainstem also has been investigated. IR activation in the brainstem nuclei modulates both food intake and glucose production via distinct intracellular signaling complexes [172,173]. Furthermore, insulin has been shown to decrease synaptic activity in the DVC by hyperpolarization, to potentially alter gastric function [174,175]. While insulin mostly suppresses excitatory neuronal activity in the DMV, under certain conditions of elevated cAMP levels, it was able to suppress inhibitory neurotransmission in only normoglycemic, but not hyperglycemic mice, suggesting a mechanism of potential pathophysiological relevance [176]. Apart from circulating insulin being able to modulate hypothalamic and brainstem neuronal activity, there is evidence of insulin being produced locally in the brain as well [177,178]. While insulin producing hypothalamic neurons were shown to have both anabolic and catabolic roles, in the case of brainstem a recent study highlighted an anabolic role for them [179,180,181]. All of the aforementioned studies highlight the importance and diversity of hypothalamic and brainstem insulin signaling in metabolic control.
3.2. Brain-liver axis
The liver is a major site of glucose metabolism, by promoting both glucose production via gluconeogenesis and glycogenolysis, and stimulating storage via glycogenesis. These hepatic metabolic pathways are under the strict control of circulating hormones and hepatic autonomic efferents. While sympathetic innervation enhances glucose production, the vagal branch has been shown to inhibit glucose production and promote storage [182,183].
As described earlier, central insulin signaling plays a key role in improving glucose homeostasis. To a large extent, insulin modulates the hypothalamic neural circuitry to regulate autonomic efferents to the liver. Insulin activates the hypothalamic K-ATP potassium channels resulting in diminished hepatic gluconeogenesis via modulation of vagal efferent activity [184]. Activation of K-ATP channels is known to result in neuronal hyperpolarization and subsequent reduction in the release of neuropeptides [185]. In line with these findings, abrogation of neuropeptide release from AGRP neurons (both NPY and AGRP) has been described as a crucial mechanism by which insulin markedly alters hepatic efferents, both sympathetic and parasympathetic, to suppress hepatic glucose production [35,38,186,187].
In addition to AGRP, the role of POMC neurons in hepatic glucose control via both insulin and leptin signaling has been explored in the brain. Conflicting reports have emerged regarding the role of POMC-specific insulin signaling on altering hepatic gluconeogenesis. While some studies have reported that hepatic gluconeogenesis is mostly under the control of AGRP-specific and not POMC-specific insulin signaling [38,169], other investigators have concluded that POMC-insulin signaling plays a key role in suppressing hepatic glucose production [170]. The differences noted by various research groups may be attributed to the POMC neuronal heterogeneity which is described in an earlier section. Interestingly, both insulin and leptin can depolarize and also hyperpolarize a subset of POMC neuronal population, which could also contribute to the differences observed in the glucoregulatory outcomes observed with central insulin signaling [188,189,190]. POMC-specific leptin signaling has been demonstrated to improve glucose homeostasis, independent of its effects on food intake and appetite, via improvements in hepatic insulin sensitivity [191,192]. Interestingly, ICV leptin infusion does not alter glucose production in the liver, but triggers striking alterations in hepatic glucose fluxes [193,194]. Other hypothalamic nuclei, such as the SF1 neurons in VMH, also contributes to the regulation of hepatic glucose production. Stimulation of SF1 neuronal projections to specialized basal forebrain structures counteracts hypoglycemia by increasing blood glucose [83,195]. The VMH neurons maintain euglycemia in energy deprived states by regulating the expression and activity of hepatic gluconeogenic and glycogenolytic genes [79,196]. Therefore, VMH neurons may activate distinct neural circuits under glucopenic conditions to elevate endogenous glucose production. It is to be noted that VMH neurons exhibit neuronal heterogeneity and activation of SF1 neurons has been linked to hyperglycemic responses characterized by insulin resistance [81,197]. Apart from the VMH, hyperactivity of liver-projecting PVH neurons has also been reported in a diabetic mouse model [198].
The hypothalamic neural circuitry does not work in isolation to regulate endogenous glucose production. Glucoregulatory neural circuits between the hypothalamus and brainstem have been reported to regulate hepatic glucose production under both hyperglycemic and hypoglycemic conditions [199,200]. As mentioned earlier, neurons located in the DVC serve as an important integration point for ascending signals from the gut, as well as signals from other regions of the brain. Activation of NMDA receptors in the DVC lowered glucose production via hepatic vagal efferents [201]. Furthermore, administration of an NMDA blocker into the NTS blocked the hepatic glucose lowering effects of intestinal lipids [202]. This suggests an integral role of the brainstem neurons in nutrient-mediated changes in glucose homeostasis by regulating hepatic glucose production. A more detailed understanding of the hypothalamic and brainstem neural circuits in the regulation of hepatic efferents under both physiological and pathological conditions could aid in better understanding the mechanisms underlying impaired glucose homeostasis in metabolic disorders.
3.3. Brain-adipose tissue axis
A key metabolic tissue that has prominent roles in glucose and whole-body energy homeostasis is the adipose tissue. Chemogenetic and optogenetic modulation of adipose tissue has dramatic metabolic effects in both lean and obese conditions [203,204,205,206]. Hypothalamic and brainstem neuronal populations regulate key autonomic projections (mainly sympathetic) from the CNS, to both white and brown adipose tissues (WAT and BAT respectively) [207,208,209,210]. The best-defined neural circuits for adipose tissue regulation involves the AGRP and POMC neurons of the ARC. AGRP neuronal stimulation not only modulates hepatic glucose production, but also contributes to insulin resistance by altering glucose uptake from BAT [211]. AGRP stimulation also alters substrate utilization in adipose tissue shifting its energy source towards carbohydrates and away from lipids. This effect involves decreasing fat oxidation and increasing lipogenesis resulting in increased adiposity [212]. Interestingly, the role of orexigenic neuronal populations in the regulation of adipose tissue function is not limited to the ARC. NPY knockdown in the DMH resulted in a favorable metabolic profile characterized by an increased BAT mass and augmented beiging of WAT, leading to increased thermogenesis and energy expenditure [213]. In contrast to the AGRP neurons, perturbation of POMC neuronal activity by either knocking out IR or by genetic inactivation of key mitochondrial proteins, results in an altered adipose tissue lipolytic profile, which potentially contributes to high fat diet-induced metabolic impairments [169,214]. In addition to the ARC, other hypothalamic nuclei could also be involved in regulating lipid metabolism. There is evidence that hypothalamic AMPK, a metabolic regulator activated by low energy states, plays a role in glucose homeostasis via modulation of sympathetic outflow to adipose depots [215]. Specifically, AMPK in VMH has been linked to thermogenesis and beiging of WAT [216,217].
Leptin is a key adipokine released by WAT, and acts on the LepR expressed in the CNS to induce a negative energy balance. The LepR which is widely expressed in the CNS, plays a key role in modulating the neural circuits involved in regulating autonomic outflow to adipose tissue, thus modulating lipid metabolism in the adipose depots [218,219]. Potentiation of leptin and insulin signaling in POMC neurons confers protection against diet-induced obesity by increased WAT browning and decreased adiposity [220]. While activating LepR in key brainstem nuclei regulates sympathetic outflow to kidney [221,222], activation of the hypothalamic LepR alters metabolism via regulation of sympathetic outflow [218,223]. Furthermore, pancreatic peptides have also been shown to act in the brain to alter adipose tissue function. For instance, perturbation of amylin/calcitonin signaling in POMC neurons results in increased adiposity and decreased UCP1 in BAT, resulting in impaired glucose tolerance [224]. Therefore, via the hypothalamic and brainstem neural circuits, various aspects of adipose tissue functionality could be fine-tuned to have a sizable impact on whole-body energy homeostasis.
3.4. Gut-brain axis and the role of incretins
Neuronal populations located in the hypothalamus and brainstem express receptors for gut hormones. As discussed in earlier sections, the enteroendocrine system of the gut is responsible for chemosensing, and thus regulates the release of gut hormones in response to a meal to trigger a satiation response [225]. The incretins, GIP and GLP1, are one such class of gut hormones that are not only capable of suppressing appetite, but also have significant effects on maintenance of postprandial glucose levels. They exert prominent metabolic and glucoregulatory roles via their receptors expressed both in the periphery and the CNS [226]. In response to a meal, both GIP and GLP1 levels are elevated in the blood, which affects appetite and energy homeostasis by modulating neural activity in key hypothalamic and brainstem nuclei. The incretin receptors, GLP1R and GIPR, are highly expressed in the ARC and DMV, thereby can be activated by circulating incretins and their analogs to regulate food intake and energy balance [227,228,229,230,231,232]. Multiple studies have highlighted a crucial role of the ARC in mediating the appetite suppressing effects of incretins [233,234,235,236]. The appetite-reducing mechanism of the GLP1 analog, liraglutide, involves activation of GLP1R in ARC, specifically activation of the anorectic POMC neurons [229]. Additionally, CCK neurons in the NTS also play crucial roles in mediating the full anorectic effect of GLP1R agonists, but not GIPR agonists [237]. In the case of GIP, hypothalamic GIPR-expressing neurons are reported to have essential roles in mediating the effects of GIP on feeding and energy homeostasis [230,238]. Chemogenetic activation of GIPR in the hypothalamus, and modulation of hypothalamic neuronal activity by peripheral GIPR agonists resulted in an inhibition of food intake and improved glucose handling [230,231]. Studies have also reported on beneficial role of activating, and not antagonizing, GIPR in promoting weight loss in diet-induced obesity and improved glucose homeostasis [239,240]. More recently, acute administration of a long acting GIPR agonist, GIPFA-085, acted via the ARC POMC neurons to suppress feeding and increase lipid utilization, while subchronic administration was shown to reduce body weight in diet-induced obesity mice [241]. However there is ambiguity on whether activation or inhibition of GIPR has beneficial effects on obesity. There is evidence highlighting a positive correlation between elevated GIP levels and high-fat diet feeding [242,243]. Furthermore, both global and CNS-specific GIPR deletion resulted in protection against obesity, suggesting that they have essential roles in induction of weight gain and adiposity [231,239]. While it is apparent that central GIPR has a role in energy homeostasis, the relative contributions of the various neuronal populations in mediating the metabolic effects of GIPR are yet to be fully mapped out.
It is well-established that incretins improve glucose homeostasis by augmenting insulin secretion, following activation of their receptors on the beta cells of the pancreatic islets [244,245]. However, incretins are rapidly degraded once released into the GIT and the blood stream, suggesting the existence of a vagal afferent neural pathway as an intermediary mechanism to mediate low dose effects of incretins on regulating glucose homeostasis [116,246,247]. In agreement with this concept, nutrients and other gut hormones have also been shown to trigger the gut-brain axis to regulate glucose and energy homeostasis [202,248,249]. The incretin receptors in the hypothalamus and brainstem have been shown to have important roles in improving glucose homeostasis. Antagonizing the GLP1R located in the ARC resulted in worsening of glucose tolerance, while direct administration of GLP1 into the ARC reduced glucose production [250]. Furthermore, central GLP1R-mediated improvement in glucose homeostasis is preserved under high fat feeding conditions [251]. GLP1R-expressing neurons are also present in other hypothalamic nuclei. Stimulation of DMH-GLP1R resulted in a reduction of blood glucose via descending input to the DMV, which inturn augments pancreatic insulin release [252]. As discussed earlier, modulation of GLP1R activity in the DMV alone also regulates pancreatic autonomic efferents, and exerts influence over insulin secretion [162,163]. These studies highlight the fact that both peripheral and central incretin receptors, act in concert to trigger metabolic improvements observed with the incretins and their analogs. However, while glucose lowering ability of central GLP1R has been mostly reported, there have been reports of hyperglycemic responses by high doses of GLP1R agonist, exendin-4 [253]. In line with this, ICV administration of GLP1 also paradoxically reduced glucose-stimulated insulin secretion and caused mild glucose intolerance [254]. However, both these effects are a consequence of the sympathetic nerve activity activation, and could be due to the activation of GLP1R in several distinct neuronal populations present in the various hypothalamic nuclei. This could also suggest a negative feedback mechanism by which central GLP1R localized on distinct neuronal populations limits insulin release. Further investigation is needed to explore this aspect. Apart from incretins, other postprandial signals have also been involved in regulating glucose and lipid metabolism via the CNS. For instance, FGF19, a postprandial enterokine that has hypoglycemic effects, elicits its effects by modulating both hypothalamic and brainstem neuronal populations to improve glucose homeostasis [255,256,257,258]. Amylin’s effects on food intake and body weight were demonstrated to be depended on modulation of brainstem neuronal signaling, specifically lateral dorsal tegmental nucleus, resulting in increased SNS activity to BAT [259].
The CNS, specifically the hypothalamic and brainstem neural circuits, regulates several different facets of energy balance by altering autonomic outflow to multiple metabolic tissues and endocrine glands. A schematic summarizing some of the important complimentary and distinct metabolic roles of the neural pathways discussed in this review paper, is shown in Figure 1.
Green dotted lines/arrows represent modulation of hypothalamic neural circuits. Red dotted lines/arrows represent modulation of brainstem neural circuits.
4. Clinical implications
Multiple clinical studies have underscored the importance of the appetite/satiety centers in the brain for mediating the effects of gut hormones and circulating peptides on energy homeostasis [260,261]. Weight-loss drugs act by inducing a negative energy balance by either central or peripheral mechanisms, or a combination of both. This mainly includes appetite reduction via the melanocortin system, increased energy expenditure via both central and peripheral mechanisms, or restriction of calorie absorption from the intestine by acting on the intestinal enzymes [262]. Several of the anti-obesity drugs in the past were effective in inducing significant weight loss, however they were discontinued due to adverse effects. For instance, rimonabant improved several metabolic parameters along with promoting anorexia in several clinical studies, but it was associated with high neuropsychiatric adverse effects leading it to be withdrawn from the market [263]. Other drugs such as aminorex and sibutramine have been discontinued due to severe cardiovascular events [262,264]. The currently approved anti-obesity drugs demonstrate not only equivalent therapeutic efficacy, but also have favorable cardiovascular and neurological profile [262]. Centrally-acting drug combinations such as naltrexone + bupropion, which act on the melanocortin system via the opioid receptors, have been shown to be efficacious in reducing body weight without any CNS adverse effects [265]. Drugs such as orlistat act exclusively by inhibiting fatty acid absorption from the gut [266], while GLP1 analogs such as semaglutide act via multiple mechanisms which encompass both central and peripheral mechanisms to suppress appetite, improve glucose and lipid metabolism, and delay gastric emptying [267,268]. While these drugs have a much-improved cardiometabolic risk profile compared to the previous generations of weight loss medications, the long-term risk profile remains an outstanding question [269]. Additionally, gastrointestinal side effects such as nausea and diarrhea are commonly observed, and may diminish patient compliance which further limits their long-term efficacy [262]. Future research should be geared towards evaluating long term risk-benefit profile, using combination therapy with reduced doses to avoid GIT side effects. Deciphering the neural circuits that suppress appetite without triggering aversive responses could also aid in developing drugs with a favorable risk-benefit profile. Interestingly, some neuronal populations exhibit sexual dimorphism in metabolism and glucoregulation [270,271]. A better understanding of gender differences in energy and glucose homeostasis should aid in developing tailored therapeutic strategies for the treatment of obesity [272].
5. Conclusions
Obesity has been long considered to be at epidemic proportions globally, and is both a significant health and economic burden [273,274]. It is now evident that the brain is at the apex of the whole-body energy homeostatic machinery. Our understanding of the hypothalamic and brainstem neuronal circuits has already aided in the development of highly efficacious anti-obesity drugs. While several of these neuronal populations exhibit overlapping metabolic roles, recent studies have brought to light distinct and contrasting mechanisms, thereby enabling the CNS to fine tune metabolic functions under physiological conditions. It is important to note that metabolic disorders are highly heterogenous with distinct metabolic profiles [275,276]. A deeper understanding of the molecular architecture of the neuronal populations could aid in exploring multiple drug targets, potentially even tailor-made for the treatment of a specific metabolic profile. Such personalized therapies are already employed for several other pathological conditions, and hence feasible that this goal can be achieved for the future treatment of obesity and related metabolic disorders [277].
Author Contributions
All authors contributed to the conceptualization, literature search, writing and editing of the review paper.
Funding
The authors’ research is supported by the Intramural Research Program of the NIH, NIDDK, Bethesda, Maryland, USA
Acknowledgments
Artwork obtained from Servier Medical Art was used to construct aspects of the figure (www.servier.com). The authors would like to thank Drs. Jurgen Wess and Oksana Gavrilova for their many helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ahima, R.S.; Antwi, D.A. Brain Regulation of Appetite and Satiety. Endocrinol Metab Clin North Am 2008, 37, 811. [Google Scholar] [CrossRef]
- Hetherington, A.W.; Ranson, S.W. Hypothalamic Lesions and Adiposity in the Rat. Anat Rec 1940, 78, 149–172. [Google Scholar] [CrossRef]
- Wess, J.; Nakajima, K.; Jain, S. Novel Designer Receptors to Probe GPCR Signaling and Physiology. Trends Pharmacol Sci 2013, 34, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose Sensing by POMC Neurons Regulates Glucose Homeostasis and Is Impaired in Obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Okamura, T.; Brüning, J.C. Hormone and Glucose Signalling in POMC and AgRP Neurons. J Physiol 2009, 587, 5305. [Google Scholar] [CrossRef] [PubMed]
- Roh, E.; Song, D.K.; Kim, M.S. Emerging Role of the Brain in the Homeostatic Regulation of Energy and Glucose Metabolism. Experimental & Molecular Medicine 2016 48:3 2016, 48, e216–e216. [Google Scholar] [CrossRef]
- Yoon, N.A.; Diano, S. Hypothalamic Glucose-Sensing Mechanisms. Diabetologia 2021, 64, 985. [Google Scholar] [CrossRef]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger States Switch a Flip-Flop Memory Circuit via a Synaptic AMPK-Dependent Positive Feedback Loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef]
- Liu, T.; Kong, D.; Shah, B.P.; Ye, C.; Koda, S.; Saunders, A.; Ding, J.B.; Yang, Z.; Sabatini, B.L.; Lowell, B.B. Fasting Activation of AgRP Neurons Requires NMDA Receptors and Involves Spinogenesis and Increased Excitatory Tone. Neuron 2012, 73, 511. [Google Scholar] [CrossRef]
- Rau, A.R.; Hentges, S.T. GABAergic Inputs to POMC Neurons Originating from the Dorsomedial Hypothalamus Are Regulated by Energy State. Journal of Neuroscience 2019, 39, 6449–6459. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, D.; Morgan, D.G.A.; Meeran, K.; Edwards, C.M.B.; Turton, M.D.; Choi, S.J.; Heath, M.M.; Gunn, I.; Taylor, G.M.; Howard, J.K.; et al. Neuropeptide Y Induced Feeding in the Rat Is Mediated by a Novel Receptor. Endocrinology 1997, 138, 196–202. [Google Scholar] [CrossRef]
- Nijenhuis, W.A.J.; Oosterom, J.; Adan, R.A.H. AgRP(83-132) Acts as an Inverse Agonist on the Human-Melanocortin-4 Receptor. Mol Endocrinol 2001, 15, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Perez, F.A.; Hnasko, T.S.; Palmiter, R.D. NPY/AgRP Neurons Are Essential for Feeding in Adult Mice but Can Be Ablated in Neonates. Science 2005, 310, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Krashes, M.J.; Koda, S.; Ye, C.P.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, Reversible Activation of AgRP Neurons Drives Feeding Behavior in Mice. J Clin Invest 2011, 121, 1424–1428. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Denis, R.G.P.; Castel, J.; Lacombe, A.; Cansell, C.; Rouch, C.; Kassis, N.; Dairou, J.; Cani, P.D.; Ventura-Clapier, R.; et al. Hypothalamic AgRP-Neurons Control Peripheral Substrate Utilization and Nutrient Partitioning. EMBO J 2012, 31, 4276–4288. [Google Scholar] [CrossRef]
- Nakajima, K.I.; Cui, Z.; Li, C.; Meister, J.; Cui, Y.; Fu, O.; Smith, A.S.; Jain, S.; Lowell, B.B.; Krashes, M.J.; et al. Gs-Coupled GPCR Signalling in AgRP Neurons Triggers Sustained Increase in Food Intake. Nat Commun 2016, 7. [Google Scholar] [CrossRef]
- Krashes, M.J.; Shah, B.P.; Koda, S.; Lowell, B.B. Rapid versus Delayed Stimulation of Feeding by the Endogenously Released AgRP Neuron Mediators GABA, NPY, and AgRP. Cell Metab 2013, 18, 588–595. [Google Scholar] [CrossRef]
- Qi, Y.; Lee, N.J.; Ip, C.K.; Enriquez, R.; Tasan, R.; Zhang, L.; Herzog, H. NPY Derived from AGRP Neurons Controls Feeding via Y1 and Energy Expenditure and Food Foraging Behaviour via Y2 Signalling. Mol Metab 2022, 59, 101455. [Google Scholar] [CrossRef]
- Luo, N.; Marcelin, G.; Liu, S.M.; Schwartz, G.; Chua, S. Neuropeptide Y and Agouti-Related Peptide Mediate Complementary Functions of Hyperphagia and Reduced Energy Expenditure in Leptin Receptor Deficiency. Endocrinology 2011, 152, 883. [Google Scholar] [CrossRef]
- Cavalcanti-de-Albuquerque, J.P.; Bober, J.; Zimmer, M.R.; Dietrich, M.O. Regulation of Substrate Utilization and Adiposity by Agrp Neurons. Nat Commun 2019, 10. [Google Scholar] [CrossRef]
- D’agostino, G.; Diano, S. Alpha-Melanocyte Stimulating Hormone: Production and Degradation. J Mol Med (Berl) 2010, 88, 1195. [Google Scholar] [CrossRef]
- Greenman, Y.; Kuperman, Y.; Drori, Y.; Asa, S.L.; Navon, I.; Forkosh, O.; Gil, S.; Stern, N.; Chen, A. Postnatal Ablation of POMC Neurons Induces an Obese Phenotype Characterized by Decreased Food Intake and Enhanced Anxiety-like Behavior. Mol Endocrinol 2013, 27, 1091–1102. [Google Scholar] [CrossRef]
- Zhan, C.; Zhou, J.; Feng, Q.; Zhang, J. en; Lin, S.; Bao, J.; Wu, P.; Luo, M. Acute and Long-Term Suppression of Feeding Behavior by POMC Neurons in the Brainstem and Hypothalamus, Respectively. J Neurosci 2013, 33, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Toda, C.; Santoro, A.; Kim, J.D.; Diano, S. POMC Neurons: From Birth to Death. Annu Rev Physiol 2017, 79, 209–236. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC Neuronal Heterogeneity in Energy Balance and beyond: An Integrated View. Nat Metab 2021, 3, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Saucisse, N.; Mazier, W.; Simon, V.; Binder, E.; Catania, C.; Bellocchio, L.; Romanov, R.A.; Léon, S.; Matias, I.; Zizzari, P.; et al. Functional Heterogeneity of POMC Neurons Relies on MTORC1 Signaling. Cell Rep 2021, 37, 109800. [Google Scholar] [CrossRef] [PubMed]
- Elias, C.F.; Kelly, J.F.; Lee, C.E.; Ahima, R.S.; Drucker, D.J.; Saper, C.B.; Elmquist, J.K. Chemical Characterization of Leptin-Activated Neurons in the Rat Brain. J Comp Neurol 2000, 423, 261–281. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin Activates Anorexigenic POMC Neurons through a Neural Network in the Arcuate Nucleus. Nature 2001 411:6836 2001, 411, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Korner, J.; Savontaus, E.; Chua, S.C.; Leibel, R.L.; Wardlaw, S.L. Leptin Regulation of Agrp and Npy MRNA in the Rat Hypothalamus. J Neuroendocrinol 2001, 13, 959–966. [Google Scholar] [CrossRef]
- Takahashi, K.A.; Cone, R.D. Fasting Induces a Large, Leptin-Dependent Increase in the Intrinsic Action Potential Frequency of Orexigenic Arcuate Nucleus Neuropeptide Y/Agouti-Related Protein Neurons. Endocrinology 2005, 146, 1043–1047. [Google Scholar] [CrossRef]
- Baver, S.B.; Hope, K.; Guyot, S.; Bjørbaek, C.; Kaczorowski, C.; O’Connell, K.M.S. Leptin Modulates the Intrinsic Excitability of AgRP/NPY Neurons in the Arcuate Nucleus of the Hypothalamus. The Journal of Neuroscience 2014, 34, 5486. [Google Scholar] [CrossRef]
- Huang, H.; Kong, D.; Byun, K.H.; Ye, C.; Koda, S.; Lee, D.H.; Oh, B.C.; Lee, S.W.; Lee, B.; Zabolotny, J.M.; et al. Rho-Kinase Regulates Energy Balance by Targeting Hypothalamic Leptin Receptor Signaling. Nat Neurosci 2012, 15, 1391–1398. [Google Scholar] [CrossRef]
- Huang, H.; Lee, S.H.; Ye, C.; Lima, I.S.; Oh, B.C.; Lowell, B.B.; Zabolotny, J.M.; Kim, Y.B. ROCK1 in AgRP Neurons Regulates Energy Expenditure and Locomotor Activity in Male Mice. Endocrinology 2013, 154, 3660. [Google Scholar] [CrossRef] [PubMed]
- Koch, L.; Wunderlich, F.T.; Seibler, J.; Könner, A.C.; Hampel, B.; Irlenbusch, S.; Brabant, G.; Kahn, C.R.; Schwenk, F.; Brüning, J.C. Central Insulin Action Regulates Peripheral Glucose and Fat Metabolism in Mice. J Clin Invest 2008, 118, 2132–2147. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Kim, S.J.; Méquinion, M.; Xirouchaki, C.E.; Brüning, J.C.; Andrews, Z.B.; Tiganis, T. Insulin Signaling in AgRP Neurons Regulates Meal Size to Limit Glucose Excursions and Insulin Resistance. Sci Adv 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef]
- Könner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab 2007, 5, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Liu, Z.W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 Mediates Ghrelin’s Action on NPY/AgRP Neurons by Lowering Free Radicals. Nature 2008, 454, 846. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Shi, X.; Li, X.; Chang, B.; Wang, Y.; Li, D.; Chan, L. GLP-2 Receptor in POMC Neurons Suppresses Feeding Behavior and Gastric Motility. Am J Physiol Endocrinol Metab 2012, 303, E853. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Alhadeff, A.L.; Betley, J.N. Nutritive, Post-Ingestive Signals Are the Primary Regulators of AgRP Neuron Activity. Cell Rep 2017, 21, 2724. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Coester, B.; Whiting, L.; Dunn-Meynell, A.A.; Boyle, C.N.; Bouret, S.G.; Levin, B.E.; Le Foll, C. Amylin Selectively Signals onto POMC Neurons in the Arcuate Nucleus of the Hypothalamus. Diabetes 2018, 67, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Nuzzaci, D.; Cansell, C.; Liénard, F.; Nédélec, E.; Ben Fradj, S.; Castel, J.; Foppen, E.; Denis, R.; Grouselle, D.; Laderrière, A.; et al. Postprandial Hyperglycemia Stimulates Neuroglial Plasticity in Hypothalamic POMC Neurons after a Balanced Meal. Cell Rep 2020, 30, 3067–3078.e5. [Google Scholar] [CrossRef] [PubMed]
- Üner, A.G.; Keçik, O.; Quaresma, P.G.F.; De Araujo, T.M.; Lee, H.; Li, W.; Kim, H.J.; Chung, M.; Bjørbæk, C.; Kim, Y.B. Role of POMC and AgRP Neuronal Activities on Glycaemia in Mice. Sci Rep 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Jacobowitz, D.M.; O’Donohue, T.L. α-Melanocyte Stimulating Hormone: Immunohistochemical Identification and Mapping in Neurons of Rat Brain. Proc Natl Acad Sci U S A 1978, 75, 6300–6304. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G.; Mortrud, M.T.; Low, M.J.; Simerly, R.B.; Cone, R.D. Localization of the Melanocortin-4 Receptor (MC4-R) in Neuroendocrine and Autonomic Control Circuits in the Brain. Mol Endocrinol 1994, 8, 1298–1308. [Google Scholar] [CrossRef]
- Lu, D.; Willard, D.; Patel, I.R.; Kadwell, S.; Overton, L.; Kost, T.; Luther, M.; Chen, W.; Woychik, R.P.; Wilkison, W.O.; et al. Agouti Protein Is an Antagonist of the Melanocyte-Stimulating-Hormone Receptor. Nature 1994, 371, 799–802. [Google Scholar] [CrossRef]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science (1979) 1997, 278, 135–138. [Google Scholar] [CrossRef]
- Michaud, J.L.; Rosenquist, T.; May, N.R.; Fan, C.M. Development of Neuroendocrine Lineages Requires the BHLH-PAS Transcription Factor SIM1. Genes Dev 1998, 12, 3264–3275. [Google Scholar] [CrossRef]
- Michaud, J.L.; Boucher, F.; Melnyk, A.; Gauthier, F.; Goshu, E.; Lévy, E.; Mitchell, G.A.; Himms-Hagen, J.; Fan, C.M. Sim1 Haploinsufficiency Causes Hyperphagia, Obesity and Reduction of the Paraventricular Nucleus of the Hypothalamus. Hum Mol Genet 2001, 10, 1465–1473. [Google Scholar] [CrossRef]
- Tolson, K.P.; Gemelli, T.; Meyer, D.; Yazdani, U.; Kozlitina, J.; Zinn, A.R. Inducible Neuronal Inactivation of Sim1 in Adult Mice Causes Hyperphagic Obesity. Endocrinology 2014, 155, 2436–2444. [Google Scholar] [CrossRef]
- Hinney, A.; Schmidt, A.; Nottebom, K.; Heibült, O.; Becker, I.; Ziegler, A.; Gerber, G.; Sina, M.; Görg, T.; Mayer, H.; et al. Several Mutations in the Melanocortin-4 Receptor Gene Including a Nonsense and a Frameshift Mutation Associated with Dominantly Inherited Obesity in Humans. J Clin Endocrinol Metab 1999, 84, 1483–1486. [Google Scholar] [CrossRef]
- Vaisse, C.; Clement, K.; Durand, E.; Hercberg, S.; Guy-Grand, B.; Froguel, P. Melanocortin-4 Receptor Mutations Are a Frequent and Heterogeneous Cause of Morbid Obesity. J Clin Invest 2000, 106, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.H.; Lank, E.J.; Cheetham, T.; O’Rahilly, S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N Engl J Med 2003, 348, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J.F.; Lindgren, C.M.; Li, S.; Wheeler, E.; Hua Zhao, J.; Prokopenko, I.; Inouye, M.; Freathy, R.M.; Attwood, A.P.; Beckmann, J.S.; et al. Common Variants near MC4R Are Associated with Fat Mass, Weight and Risk of Obesity. Nat Genet 2008, 40, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Shrestha, Y.; Pandey, M.; Chen, M.; Kablan, A.; Gavrilova, O.; Offermanns, S.; Weinstein, L.S. Gq/11α and Gsα Mediate Distinct Physiological Responses to Central Melanocortins. J Clin Invest 2016, 126, 40. [Google Scholar] [CrossRef]
- Garfield, A.S.; Li, C.; Madara, J.C.; Shah, B.P.; Webber, E.; Steger, J.S.; Campbell, J.N.; Gavrilova, O.; Lee, C.E.; Olson, D.P.; et al. A Neural Basis for Melanocortin-4 Receptor Regulated Appetite. Nat Neurosci 2015, 18, 863. [Google Scholar] [CrossRef]
- Matsumura, S.; Miyakita, M.; Miyamori, H.; Kyo, S.; Shima, D.; Yokokawa, T.; Ishikawa, F.; Sasaki, T.; Jinno, T.; Tanaka, J.; et al. Stimulation of GSsignaling in MC4R Cells by DREADD Increases Energy Expenditure, Suppresses Food Intake, and Increases Locomotor Activity in Mice. Am J Physiol Endocrinol Metab 2022, 322, E436–E445. [Google Scholar] [CrossRef]
- Chen, K.Y.; Muniyappa, R.; Abel, B.S.; Mullins, K.P.; Staker, P.; Brychta, R.J.; Zhao, X.; Ring, M.; Psota, T.L.; Cone, R.D.; et al. RM-493, a Melanocortin-4 Receptor (MC4R) Agonist, Increases Resting Energy Expenditure in Obese Individuals. J Clin Endocrinol Metab 2015, 100, 1639–1645. [Google Scholar] [CrossRef]
- Wang, X.; Cui, X.; Li, Y.; Li, F.; Li, Y.; Dai, J.; Hu, H.; Wang, X.; Sun, J.; Yang, Y.; et al. MC4R Deficiency Causes Dysregulation of Postsynaptic Excitatory Synaptic Transmission as a Crucial Culprit for Obesity. Diabetes 2022, 71, 2331–2343. [Google Scholar] [CrossRef]
- Podyma, B.; Sun, H.; Wilson, E.A.; Carlson, B.; Pritikin, E.; Gavrilova, O.; Weinstein, L.S.; Chen, M. The Stimulatory G Protein Gsα Is Required in Melanocortin 4 Receptor–Expressing Cells for Normal Energy Balance, Thermogenesis, and Glucose Metabolism. J Biol Chem 2018, 293, 10993. [Google Scholar] [CrossRef]
- Kuo, J.J.; Silva, A.A.; Hall, J.E. Hypothalamic Melanocortin Receptors and Chronic Regulation of Arterial Pressure and Renal Function. Hypertension 2003, 41, 768–774. [Google Scholar] [CrossRef]
- Sohn, J.W.; Harris, L.E.; Berglund, E.D.; Liu, T.; Vong, L.; Lowell, B.B.; Balthasar, N.; Williams, K.W.; Elmquist, J.K. Melanocortin 4 Receptors Reciprocally Regulate Sympathetic and Parasympathetic Preganglionic Neurons. Cell 2013, 152, 612–619. [Google Scholar] [CrossRef]
- Iwasa, M.; Kawabe, K.; Sapru, H.N. Activation of Melanocortin Receptors in the Intermediolateral Cell Column of the Upper Thoracic Cord Elicits Tachycardia in the Rat. Am J Physiol Heart Circ Physiol 2013, 305, H885. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Liu, T.; Kong, X.; Sohn, J.W.; Vong, L.; Deng, Z.; Lee, C.E.; Lee, S.; Williams, K.W.; Olson, D.P.; et al. Melanocortin 4 Receptors in Autonomic Neurons Regulate Thermogenesis and Glycemia. Nat Neurosci 2014, 17, 911–913. [Google Scholar] [CrossRef]
- Li, M.M.; Madara, J.C.; Steger, J.S.; Krashes, M.J.; Balthasar, N.; Campbell, J.N.; Resch, J.M.; Conley, N.J.; Garfield, A.S.; Lowell, B.B. The Paraventricular Hypothalamus Regulates Satiety and Prevents Obesity via Two Genetically Distinct Circuits. Neuron 2019, 102, 653. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Liao, G.Y.; Kinney, C.E.; Sahibzada, N.; Xu, B. Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab 2015, 22, 175–188. [Google Scholar] [CrossRef]
- Li, C.; Navarrete, J.; Liang-Guallpa, J.; Lu, C.; Funderburk, S.C.; Chang, R.B.; Liberles, S.D.; Olson, D.P.; Krashes, M.J. Defined Paraventricular Hypothalamic Populations Exhibit Differential Responses to Food Contingent on Caloric State. Cell Metab 2019, 29, 681–694.e5. [Google Scholar] [CrossRef]
- Varela, L.; Horvath, T.L. Parallel Paths in PVH Control of Feeding. Neuron 2019, 102, 514–516. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Kinney, C.E.; Tan, J.W.; Liao, G.Y.; Kremer, E.J.; Xu, B. TrkB-Expressing Paraventricular Hypothalamic Neurons Suppress Appetite through Multiple Neurocircuits. Nature Communications 2020 11:1 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Krashes, M.J.; Lowell, B.B.; Garfield, A.S. Melanocortin-4 Receptor–Regulated Energy Homeostasis. Nature Neuroscience 2016 19:2 2016, 19, 206–219. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The Melanocortin Pathway and Control of Appetite-Progress and Therapeutic Implications. J Endocrinol 2019, 241, R1–R33. [Google Scholar] [CrossRef]
- King, B.M. The Rise, Fall, and Resurrection of the Ventromedial Hypothalamus in the Regulation of Feeding Behavior and Body Weight. Physiol Behav 2006, 87, 221–244. [Google Scholar] [CrossRef]
- Becker, E.E.; Kissileff, H.R. Inhibitory Controls of Feeding by the Ventromedial Hypothalamus. Am J Physiol 1974, 226, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Maes, H. Time Course of Feeding Induced by Pentobarbital-Injections into the Rat’s VMH. Physiol Behav 1980, 24, 1107–1114. [Google Scholar] [CrossRef]
- Gaur, A.; Pal, G.K.; Ananthanarayanan, P.H.; Pal, P. Role of Ventromedial Hypothalamus in High Fat Diet Induced Obesity in Male Rats: Association with Lipid Profile, Thyroid Profile and Insulin Resistance. Ann Neurosci 2014, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, B.; Stutz, B.; Liu, Z.W.; Horvath, T.L.; Yang, X. Ventromedial Hypothalamic OGT Drives Adipose Tissue Lipolysis and Curbs Obesity. Sci Adv 2022, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Viskaitis, P.; Irvine, E.E.; Smith, M.A.; Choudhury, A.I.; Alvarez-Curto, E.; Glegola, J.A.; Hardy, D.G.; Pedroni, S.M.A.; Paiva Pessoa, M.R.; Fernando, A.B.P.; et al. Modulation of SF1 Neuron Activity Coordinately Regulates Both Feeding Behavior and Associated Emotional States. Cell Rep 2017, 21, 3559. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.A.; Okamoto, S.; Ishikawa, A.W.; Yokota, S.; Wada, N.; Hirabayashi, T.; Saito, K.; Sato, T.; Takagi, K.; Wang, C.C.; et al. Activation of SF1 Neurons in the Ventromedial Hypothalamus by DREADD Technology Increases Insulin Sensitivity in Peripheral Tissues. Diabetes 2017, 66, 2372–2386. [Google Scholar] [CrossRef] [PubMed]
- Sternson, S.M.; Shepherd, G.M.G.; Friedman, J.M. Topographic Mapping of VMH → Arcuate Nucleus Microcircuits and Their Reorganization by Fasting. Nature Neuroscience 2005 8:10 2005, 8, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, D.; Sweeney, P.; Yang, Y. An Excitatory Ventromedial Hypothalamus to Paraventricular Thalamus Circuit That Suppresses Food Intake. Nature Communications 2020 11:1 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin Directly Activates SF1 Neurons in the VMH, and This Action by Leptin Is Required for Normal Body-Weight Homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Meek, T.H.; Nelson, J.T.; Matsen, M.E.; Dorfman, M.D.; Guyenet, S.J.; Damian, V.; Allison, M.B.; Scarlett, J.M.; Nguyen, H.T.; Thaler, J.P.; et al. Functional Identification of a Neurocircuit Regulating Blood Glucose. Proc Natl Acad Sci U S A 2016, 113, E2073–E2082. [Google Scholar] [CrossRef] [PubMed]
- Fosch, A.; Zagmutt, S.; Casals, N.; Rodríguez-Rodríguez, R. New Insights of SF1 Neurons in Hypothalamic Regulation of Obesity and Diabetes. Int J Mol Sci 2021, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, L.L.; Bernardis, L.L. The Dorsomedial Hypothalamic Nucleus and Its Role in Ingestive Behavior and Body Weight Regulation: Lessons Learned from Lesioning Studies. Physiol Behav 2002, 76, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Kesterson, R.A.; Huszar, D.; Lynch, C.A.; Simerly, R.B.; Cone, R.D. Induction of Neuropeptide Y Gene Expression in the Dorsal Medial Hypothalamic Nucleus in Two Models of the Agouti Obesity Syndrome. Mol Endocrinol 1997, 11, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.M.; Yu, H.; Van Der Ploeg, L.H.T. Evidence of Altered Hypothalamic Pro-Opiomelanocortin/Neuropeptide Y MRNA Expression in Tubby Mice. Molecular Brain Research 1998, 59, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Ladenheim, E.E.; Schwartz, G.J.; Moran, T.H. A Role for NPY Overexpression in the Dorsomedial Hypothalamus in Hyperphagia and Obesity of OLETF Rats. Am J Physiol Regul Integr Comp Physiol 2001, 281. [Google Scholar] [CrossRef]
- Yang, L.; Scott, K.A.; Hyun, J.; Tamashiro, K.L.; Tray, N.; Moran, T.H.; Bi, S. Role of Dorsomedial Hypothalamic Neuropeptide Y in Modulating Food Intake and Energy Balance. J Neurosci 2009, 29, 179–190. [Google Scholar] [CrossRef]
- Otgon-Uul, Z.; Suyama, S.; Onodera, H.; Yada, T. Optogenetic Activation of Leptin- and Glucose-Regulated GABAergic Neurons in Dorsomedial Hypothalamus Promotes Food Intake via Inhibitory Synaptic Transmission to Paraventricular Nucleus of Hypothalamus. Mol Metab 2016, 5, 709–715. [Google Scholar] [CrossRef]
- Chen, J.; Scott, K.A.; Zhao, Z.; Moran, T.H.; Bi, S. Characterization of the Feeding Inhibition and Neural Activation Produced by Dorsomedial Hypothalamic Cholecystokinin Administration. Neuroscience 2008, 152, 178. [Google Scholar] [CrossRef] [PubMed]
- Rust, V.A.; Crosby, K.M. Cholecystokinin Acts in the Dorsomedial Hypothalamus of Young Male Rats to Suppress Appetite in a Nitric Oxide-Dependent Manner. Neurosci Lett 2021, 764. [Google Scholar] [CrossRef]
- Imoto, D.; Yamamoto, I.; Matsunaga, H.; Yonekura, T.; Lee, M.L.; Kato, K.X.; Yamasaki, T.; Xu, S.; Ishimoto, T.; Yamagata, S.; et al. Refeeding Activates Neurons in the Dorsomedial Hypothalamus to Inhibit Food Intake and Promote Positive Valence. Mol Metab 2021, 54, 101366. [Google Scholar] [CrossRef]
- Han, Y.; He, Y.; Harris, L.; Xu, Y.; Wu, Q. Identification of a GABAergic Neural Circuit Governing Leptin Signaling Deficiency-Induced Obesity. Elife 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Clark, M.A. Neuroinflammation and Sympathetic Overactivity: Mechanisms and Implications in Hypertension. Autonomic Neuroscience 2018. [Google Scholar] [CrossRef]
- Grill, H.J.; Norgren, R. Chronically Decerebrate Rats Demonstrate Satiation but Not Bait Shyness. Science 1978, 201, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Dirocco, R.J.; Grill, H.J. The Forebrain Is Not Essential for Sympathoadrenal Hyperglycemic Response to Glucoprivation. Science (1979) 1979, 204, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.K.; Dow, S.A.; Young, C.N. Sensory Circumventricular Organs, Neuroendocrine Control, and Metabolic Regulation. Metabolites 2021, 11. [Google Scholar] [CrossRef]
- Watts, A.G.; Kanoski, S.E.; Sanchez-Watts, G.; Langhans, W. The Physiological Control of Eating: Signals, Neurons, and Networks. Physiol Rev 2022, 102, 689–813. [Google Scholar] [CrossRef]
- Grill, H.J.; Hayes, M.R. The Nucleus Tractus Solitarius: A Portal for Visceral Afferent Signal Processing, Energy Status Assessment and Integration of Their Combined Effects on Food Intake. Int J Obes (Lond) 2009, 33 Suppl 1. [Google Scholar] [CrossRef]
- Kandalam, U.; Sarmiento, N.; Haspula, D.; Clark, M.A. Angiotensin III Induces Signal Transducer and Activator of Transcription 3 and Interleukin-6 MRNA Levels in Cultured Rat Astrocytes. Journal of the Renin-Angiotensin-Aldosterone System 2015, 16, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Clark, M.A. MAPK Activation Patterns of AT1R and CB1R in SHR versus Wistar Astrocytes: Evidence of CB1R Hypofunction and Crosstalk between AT1R and CB1R. Cell Signal 2017, 40, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Clark, M.A. Molecular Basis of the Brain Renin Angiotensin System in Cardiovascular and Neurologic Disorders: Uncovering a Key Role for the Astroglial Angiotensin Type 1 Receptor AT1R. Journal of Pharmacology and Experimental Therapeutics 2018, 366, 251–264. [Google Scholar] [CrossRef]
- O’Connor, A.T.; Haspula, D.; Alanazi, A.Z.; Clark, M.A. Roles of Angiotensin III in the Brain and Periphery. Peptides (N.Y.) 2022, 153, 170802. [Google Scholar] [CrossRef] [PubMed]
- Moura-Assis, A.; Friedman, J.M.; Velloso, L.A. Gut-to-Brain Signals in Feeding Control. Am J Physiol Endocrinol Metab 2021, 320, E326. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D. V. A Gut-Brain Neural Circuit for Nutrient Sensory Transduction. Science 2018, 361. [Google Scholar] [CrossRef]
- Owyang, C.; Heldsinger, A. Vagal Control of Satiety and Hormonal Regulation of Appetite. J Neurogastroenterol Motil 2011, 17, 338. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Vagal Mechanisms as Neuromodulatory Targets for the Treatment of Metabolic Disease. Ann N Y Acad Sci 2019, 1454, 42. [Google Scholar] [CrossRef]
- Gibbs, J.; Young, R.C.; Smith, G.P. Cholecystokinin Decreases Food Intake in Rats. J Comp Physiol Psychol 1973, 84, 488–495. [Google Scholar] [CrossRef]
- Widdop, R.E.; Krstew, E.; Mercer, L.D.; Carlsberg, M.; Beart, P.M.; Jarrott, B. Electrophysiological and Autoradiographical Evidence for Cholecystokinin A Receptors on Rat Isolated Nodose Ganglia. J Auton Nerv Syst 1994, 46, 65–73. [Google Scholar] [CrossRef]
- SCHWARTZ, G.J.; MORAN, T.H. CCK Elicits and Modulates Vagal Afferent Activity Arising from Gastric and Duodenal Sites. Ann N Y Acad Sci 1994, 713, 121–128. [Google Scholar] [CrossRef]
- Leon Mercado, L.; Caron, A.; Wang, Y.; Burton, M.; Gautron, L. Identification of Leptin Receptor–Expressing Cells in the Nodose Ganglion of Male Mice. Endocrinology 2019, 160, 1307. [Google Scholar] [CrossRef]
- Barrachina, M.D.; Martínez, V.; Wang, L.; Wei, J.Y.; Taché, Y. Synergistic Interaction between Leptin and Cholecystokinin to Reduce Short-Term Food Intake in Lean Mice. Proc Natl Acad Sci U S A 1997, 94, 10455. [Google Scholar] [CrossRef]
- Brierley, D.I.; de Lartigue, G. Reappraising the Role of the Vagus Nerve in GLP-1-Mediated Regulation of Eating. Br J Pharmacol 2022, 179, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab 2013, 17, 819–837. [Google Scholar] [CrossRef]
- Krieger, J.P.; Arnold, M.; Pettersen, K.G.; Lossel, P.; Langhans, W.; Lee, S.J. Knockdown of GLP-1 Receptors in Vagal Afferents Affects Normal Food Intake and Glycemia. Diabetes 2016, 65, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Mesgarzadeh, S.; Ramesh, K.S.; Huey, E.L.; Liu, Y.; Gray, L.A.; Aitken, T.J.; Chen, Y.; Beutler, L.R.; Ahn, J.S.; et al. Genetic Identification of Vagal Sensory Neurons That Control Feeding. Cell 2019, 179, 1129. [Google Scholar] [CrossRef]
- Egerod, K.L.; Petersen, N.; Timshel, P.N.; Rekling, J.C.; Wang, Y.; Liu, Q.; Schwartz, T.W.; Gautron, L. Profiling of G Protein-Coupled Receptors in Vagal Afferents Reveals Novel Gut-to-Brain Sensing Mechanisms. Mol Metab 2018, 12, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, I.; Pacheco-López, G.; Rüttimann, E.B.; Arnold, M.; Asarian, L.; Langhans, W.; Geary, N.; Hillebrand, J.J.G. Hepatic-Portal Vein Infusions of Glucagon-like Peptide-1 Reduce Meal Size and Increase c-Fos Expression in the Nucleus Tractus Solitarii, Area Postrema and Central Nucleus of the Amygdala in Rats. J Neuroendocrinol 2010, 22, 557–563. [Google Scholar] [CrossRef]
- Campos, C.A.; Wright, J.S.; Czaja, K.; Ritter, R.C. CCK-Induced Reduction of Food Intake and Hindbrain MAPK Signaling Are Mediated by NMDA Receptor Activation. Endocrinology 2012, 153, 2633–2646. [Google Scholar] [CrossRef]
- Punjabi, M.; Arnold, M.; Rüttimann, E.; Graber, M.; Geary, N.; Pacheco-López, G.; Langhans, W. Circulating Glucagon-like Peptide-1 (GLP-1) Inhibits Eating in Male Rats by Acting in the Hindbrain and Without Inducing Avoidance. Endocrinology 2014, 155, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Barrera, J.G.; Jones, K.R.; Herman, J.P.; D’Alessio, D.A.; Woods, S.C.; Seeley, R.J. Hyperphagia and Increased Fat Accumulation in Two Models of Chronic CNS Glucagon-Like Peptide-1 Loss of Function. The Journal of Neuroscience 2011, 31, 3904. [Google Scholar] [CrossRef] [PubMed]
- Finan, B.; Ma, T.; Ottaway, N.; Müller, T.D.; Habegger, K.M.; Heppner, K.M.; Kirchner, H.; Holland, J.; Hembree, J.; Raver, C.; et al. Unimolecular Dual Incretins Maximize Metabolic Benefits in Rodents, Monkeys, and Humans. Sci Transl Med 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Vincelette, L.K.; Reimann, F.; Liberles, S.D. A Brainstem Circuit for Nausea Suppression. Cell Rep 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Samms, R.J.; Cosgrove, R.; Snider, B.M.; Furber, E.C.; Droz, B.A.; Briere, D.A.; Dunbar, J.; Dogra, M.; Alsina-Fernandez, J.; Borner, T.; et al. GIPR Agonism Inhibits PYY-Induced Nausea-Like Behavior. Diabetes 2022, 71, 1410–1423. [Google Scholar] [CrossRef]
- Lutz, T.A.; Mollet, A.; Rushing, P.A.; Riediger, T.; Scharrer, E. The Anorectic Effect of a Chronic Peripheral Infusion of Amylin Is Abolished in Area Postrema/Nucleus of the Solitary Tract (AP/NTS) Lesioned Rats. Int J Obes Relat Metab Disord 2001, 25, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Halatchev, I.G.; Cone, R.D. Peripheral Administration of PYY(3-36) Produces Conditioned Taste Aversion in Mice. Cell Metab 2005, 1, 159–168. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic Signals Controlling Food Intake; Insulin, Glucagon and Amylin. Philosophical Transactions of the Royal Society B: Biological Sciences 2006, 361, 1219. [Google Scholar] [CrossRef]
- Braegger, F.E.; Asarian, L.; Dahl, K.; Lutz, T.A.; Boyle, C.N. The Role of the Area Postrema in the Anorectic Effects of Amylin and Salmon Calcitonin: Behavioral and Neuronal Phenotyping. Eur J Neurosci 2014, 40, 3055–3066. [Google Scholar] [CrossRef]
- Coester, B.; Foll, C. Le; Lutz, T.A. Viral Depletion of Calcitonin Receptors in the Area Postrema: A Proof-of-Concept Study. Physiol Behav 2020, 223, 112992. [Google Scholar] [CrossRef]
- Scott, M.M.; Williams, K.W.; Rossi, J.; Lee, C.E.; Elmquist, J.K. Leptin Receptor Expression in Hindbrain Glp-1 Neurons Regulates Food Intake and Energy Balance in Mice. J Clin Invest 2011, 121, 2413–2421. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Zhao, S.; Guarnieri, D.J.; DiLeone, R.J.; Yan, J.; De Jonghe, B.C.; Bence, K.K.; Hayes, M.R.; Grill, H.J. Endogenous Leptin Receptor Signaling in the Medial Nucleus Tractus Solitarius Affects Meal Size and Potentiates Intestinal Satiation Signals. Am J Physiol Endocrinol Metab 2012, 303. [Google Scholar] [CrossRef]
- Cheng, W.; Ndoka, E.; Hutch, C.; Roelofs, K.; MacKinnon, A.; Khoury, B.; Magrisso, J.; Kim, K.S.; Rhodes, C.J.; Olson, D.P.; et al. Leptin Receptor-Expressing Nucleus Tractus Solitarius Neurons Suppress Food Intake Independently of GLP1 in Mice. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; Edlow, B.L.; Immordino-Yang, M.H. The Brainstem in Emotion: A Review. Front Neuroanat 2017, 11, 15. [Google Scholar] [CrossRef]
- Vander Weele, C.M.; Siciliano, C.A.; Matthews, G.A.; Namburi, P.; Izadmehr, E.M.; Espinel, I.C.; Nieh, E.H.; Schut, E.H.S.; Padilla-Coreano, N.; Burgos-Robles, A.; et al. Dopamine Enhances Signal-to-Noise Ratio in Cortical-Brainstem Encoding of Aversive Stimuli. Nature 2018, 563, 397–401. [Google Scholar] [CrossRef]
- Wu, Q.; Clark, M.S.; Palmiter, R.D. Deciphering a Neuronal Circuit That Mediates Appetite. Nature 2012 483:7391 2012, 483, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Roman, C.W.; Derkach, V.A.; Palmiter, R.D. Genetically and Functionally Defined NTS to PBN Brain Circuits Mediating Anorexia. Nature Communications 2016 7:1 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Lyons, D.J.; Cristiano, C.; Burke, L.K.; Madara, J.C.; Campbell, J.N.; Garcia, A.P.; Land, B.B.; Lowell, B.B.; Dileone, R.J.; et al. Appetite Controlled by a Cholecystokinin Nucleus of the Solitary Tract to Hypothalamus Neurocircuit. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Roman, C.W.; Sloat, S.R.; Palmiter, R.D. A Tale of Two Circuits: CCKNTS Neuron Stimulation Controls Appetite and Induces Opposing Motivational States by Projections to Distinct Brain Regions. Neuroscience 2017, 358, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Gonzalez, I.; Pan, W.; Tsang, A.H.; Adams, J.; Ndoka, E.; Gordian, D.; Khoury, B.; Roelofs, K.; Evers, S.S.; et al. Calcitonin Receptor Neurons in the Mouse Nucleus Tractus Solitarius Control Energy Balance via the Non-Aversive Suppression of Feeding. Cell Metab 2020, 31, 301–312.e5. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.J.; Tang-Christensen, M.; Holst, J.J.; Ørskov, C. Distribution of Glucagon-like Peptide-1 and Other Preproglucagon-Derived Peptides in the Rat Hypothalamus and Brainstem. Neuroscience 1997, 77, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Alhadeff, A.L.; Rupprecht, L.E.; Hayes, M.R. GLP-1 Neurons in the Nucleus of the Solitary Tract Project Directly to the Ventral Tegmental Area and Nucleus Accumbens to Control for Food Intake. Endocrinology 2012, 153, 647. [Google Scholar] [CrossRef] [PubMed]
- Palkovits, M.; Eskay, R.L. Distribution and Possible Origin of β-Endorphin and ACTH in Discrete Brainstem Nuclei of Rats. Neuropeptides 1987, 9, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, D.M.; Schafer, M.K.H.; Watson, S.J.; Akil, H. Evidence That Beta-Endorphin Is Synthesized in Cells in the Nucleus Tractus Solitarius: Detection of POMC MRNA. Brain Res 1992, 587, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Appleyard, S.M.; Bailey, T.W.; Doyle, M.W.; Jin, Y.H.; Smart, J.L.; Low, M.J.; Andresen, M.C. Proopiomelanocortin Neurons in Nucleus Tractus Solitarius Are Activated by Visceral Afferents: Regulation by Cholecystokinin and Opioids. J Neurosci 2005, 25, 3578–3585. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, T.; Lyons, D.; Doslikova, B.; Garcia, A.P.; Marston, O.; Burke, L.K.; Chianese, R.; Lam, B.Y.H.; Yeo, G.S.H.; Rochford, J.J.; et al. Neurochemical Characterization of Brainstem Pro-Opiomelanocortin Cells. Endocrinology 2020, 161, 1–13. [Google Scholar] [CrossRef]
- D’Agostino, G.; Lyons, D.; Cristiano, C.; Lettieri, M.; Olarte-Sanchez, C.; Burke, L.K.; Greenwald-Yarnell, M.; Cansell, C.; Doslikova, B.; Georgescu, T.; et al. Nucleus of the Solitary Tract Serotonin 5-HT2C Receptors Modulate Food Intake. Cell Metab 2018, 28, 619–630.e5. [Google Scholar] [CrossRef]
- Wagner, S.; Brierley, D.I.; Leeson-Payne, A.; Jiang, W.; Chianese, R.; Lam, B.Y.H.; Dowsett, G.K.C.; Cristiano, C.; Lyons, D.; Reimann, F.; et al. Obesity Medication Lorcaserin Activates Brainstem GLP-1 Neurons to Reduce Food Intake and Augments GLP-1 Receptor Agonist Induced Appetite Suppression. Mol Metab 2023, 68, 101665. [Google Scholar] [CrossRef]
- Ritter, S.; Dinh, T.T.; Zhang, Y. Localization of Hindbrain Glucoreceptive Sites Controlling Food Intake and Blood Glucose. Brain Res 2000, 856, 37–47. [Google Scholar] [CrossRef]
- Boychuk, C.R.; Smith, K.C.; Peterson, L.E.; Boychuk, J.A.; Butler, C.R.; Derera, I.D.; McCarthy, J.J.; Smith, B.N. A Hindbrain Inhibitory Microcircuit Mediates Vagally-Coordinated Glucose Regulation. Scientific Reports 2019 9:1 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Zhang, L.; Koller, J.; Ip, C.K.; Gopalasingam, G.; Bajaj, N.; Lee, N.J.; Enriquez, R.F.; Herzog, H. Lack of Neuropeptide FF Signalling in Mice Leads to Reduced Repetitive Behavior, Altered Drinking Behavior, and Fuel Type Selection. The FASEB Journal 2021, 35, e21980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Koller, J.; Gopalasingam, G.; Qi, Y.; Herzog, H. Central NPFF Signalling Is Critical in the Regulation of Glucose Homeostasis. Mol Metab 2022, 62, 101525. [Google Scholar] [CrossRef] [PubMed]
- Wulsin, L.R.; Horn, P.S.; Perry, J.L.; Massaro, J.M.; D’Agostino, R.B. Autonomic Imbalance as a Predictor of Metabolic Risks, Cardiovascular Disease, Diabetes, and Mortality. J Clin Endocrinol Metab 2015, 100, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Licht, C.M.M.; Vreeburg, S.A.; Van Reedt Dortland, A.K.B.; Giltay, E.J.; Hoogendijk, W.J.G.; DeRijk, R.H.; Vogelzangs, N.; Zitman, F.G.; De Geus, E.J.C.; Penninx, B.W.J.H. Increased Sympathetic and Decreased Parasympathetic Activity Rather than Changes in Hypothalamic-Pituitary-Adrenal Axis Activity Is Associated with Metabolic Abnormalities. J Clin Endocrinol Metab 2010, 95, 2458–2466. [Google Scholar] [CrossRef]
- Aronson, D.; Rayfield, E.J. How Hyperglycemia Promotes Atherosclerosis: Molecular Mechanisms. Cardiovasc Diabetol 2002, 1, 1–10. [Google Scholar] [CrossRef]
- Haspula, D.; Vallejos, A.K.; Moore, T.M.; Tomar, N.; Dash, R.K.; Hoffmann, B.R. Influence of a Hyperglycemic Microenvironment on a Diabetic versus Healthy Rat Vascular Endothelium Reveals Distinguishable Mechanistic and Phenotypic Responses. Front Physiol 2019, 10, 558. [Google Scholar] [CrossRef]
- Jansen, A.S.P.; Hoffman, J.L.; Loewy, A.D. CNS Sites Involved in Sympathetic and Parasympathetic Control of the Pancreas: A Viral Tracing Study. Brain Res 1997, 766, 29–38. [Google Scholar] [CrossRef]
- Buijs, R.M.; La Fleur, S.E.; Wortel, J.; Van Heyningen, C.; Zuiddam, L.; Mettenleiter, T.C.; Kalsbeek, A.; Nagai, K.; Niijima, A. The Suprachiasmatic Nucleus Balances Sympathetic and Parasympathetic Output to Peripheral Organs through Separate Preautonomic Neurons. Journal of Comparative Neurology 2003, 464, 36–48. [Google Scholar] [CrossRef]
- Rosario, W.; Singh, I.; Wautlet, A.; Patterson, C.; Flak, J.; Becker, T.C.; Ali, A.; Tamarina, N.; Philipson, L.H.; Enquist, L.W.; et al. The Brain-to-Pancreatic Islet Neuronal Map Reveals Differential Glucose Regulation From Distinct Hypothalamic Regions. Diabetes 2016, 65, 2711–2723. [Google Scholar] [CrossRef]
- Ma, Y.; Ratnasabapathy, R.; Izzi-Engbeaya, C.; Nguyen-Tu, M.S.; Richardson, E.; Hussain, S.; De Backer, I.; Holton, C.; Norton, M.; Carrat, G.; et al. Hypothalamic Arcuate Nucleus Glucokinase Regulates Insulin Secretion and Glucose Homeostasis. Diabetes Obes Metab 2018, 20, 2246. [Google Scholar] [CrossRef]
- Papazoglou, I.; Lee, J.-H.; Cui, Z.; Enquist, L.W.; Krashes, M.J.; Rane, S.G. A Distinct Hypothalamus-to-β Cell Circuit Modulates Insulin Secretion. Cell Metab 2022, 34, 285–298.e7. [Google Scholar] [CrossRef]
- Wan, S.; Coleman, F.H.; Travagli, R.A. Glucagon-like Peptide-1 Excites Pancreas-Projecting Preganglionic Vagal Motoneurons. Am J Physiol Gastrointest Liver Physiol 2007, 292. [Google Scholar] [CrossRef]
- Wan, S.; Browning, K.N.; Travagli, R.A. Glucagon-like Peptide-1 Modulates Synaptic Transmission to Identified Pancreas-Projecting Vagal Motoneurons. Peptides (N.Y.) 2007, 28, 2184–2191. [Google Scholar] [CrossRef]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science (1979) 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Dampney, R.A.L. Arcuate Nucleus – a Gateway for Insulin’s Action on Sympathetic Activity. J Physiol 2011, 589, 2109. [Google Scholar] [CrossRef]
- Gelling, R.W.; Morton, G.J.; Morrison, C.D.; Niswender, K.D.; Myers, M.G.; Rhodes, C.J.; Schwartz, M.W. Insulin Action in the Brain Contributes to Glucose Lowering during Insulin Treatment of Diabetes. Cell Metab 2006, 3, 67–73. [Google Scholar] [CrossRef]
- Kishore, P.; Boucai, L.; Zhang, K.; Li, W.; Koppaka, S.; Kehlenbrink, S.; Schiwek, A.; Esterson, Y.B.; Mehta, D.; Bursheh, S.; et al. Activation of K(ATP) Channels Suppresses Glucose Production in Humans. J Clin Invest 2011, 121, 4916–4920. [Google Scholar] [CrossRef] [PubMed]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic Insulin Signaling Is Required for Inhibition of Glucose Production. Nat Med 2002, 8, 1376–1382. [Google Scholar] [CrossRef]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 2017, 66, 1560. [Google Scholar] [CrossRef]
- Dodd, G.T.; Michael, N.J.; Lee-Young, R.S.; Mangiafico, S.P.; Pryor, J.T.; Munder, A.C.; Simonds, S.E.; Brüning, J.C.; Zhang, Z.Y.; Cowley, M.A.; et al. Insulin Regulates POMC Neuronal Plasticity to Control Glucose Metabolism. Elife 2018, 7. [Google Scholar] [CrossRef]
- El Mehdi, M.; Takhlidjt, S.; Devère, M.; Arabo, A.; Le Solliec, M.A.; Maucotel, J.; Bénani, A.; Nedelec, E.; Duparc, C.; Lefranc, B.; et al. The 26RFa (QRFP)/GPR103 Neuropeptidergic System in Mice Relays Insulin Signalling into the Brain to Regulate Glucose Homeostasis. Diabetologia 2022, 65, 1198–1211. [Google Scholar] [CrossRef]
- Niu, S.N.; Huang, Z.B.; Wang, H.; Rao, X.R.; Kong, H.; Xu, J.; Li, X.J.; Yang, C.; Sheng, G.Q. Brainstem Hap1-Ahi1 Is Involved in Insulin-Mediated Feeding Control. FEBS Lett 2011, 585, 85–91. [Google Scholar] [CrossRef]
- Filippi, B.M.; Yang, C.S.; Tang, C.; Lam, T.K.T. Insulin Activates Erk1/2 Signaling in the Dorsal Vagal Complex to Inhibit Glucose Production. Cell Metab 2012, 16, 500–510. [Google Scholar] [CrossRef]
- Krowicki, Z.K.; Nathan, N.A.; Hornby, P.J. Gastric Motor and Cardiovascular Effects of Insulin in Dorsal Vagal Complex of the Rat. Am J Physiol 1998, 275. [Google Scholar] [CrossRef]
- Blake, C.B.; Smith, B.N. Insulin Reduces Excitation in Gastric-Related Neurons of the Dorsal Motor Nucleus of the Vagus. Am J Physiol Regul Integr Comp Physiol 2012, 303, R807. [Google Scholar] [CrossRef]
- Blake, C.B.; Smith, B.N. CAMP-Dependent Insulin Modulation of Synaptic Inhibition in Neurons of the Dorsal Motor Nucleus of the Vagus Is Altered in Diabetic Mice. Am J Physiol Regul Integr Comp Physiol 2014, 307, R711–R720. [Google Scholar] [CrossRef]
- Schechter, R.; Holtzclaw, L.; Sadiq, F.; Kahn, A.; Devaskar, S. Insulin Synthesis by Isolated Rabbit Neurons. Endocrinology 1988, 123, 505–513. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Singh, B.S.; Carnaghi, L.R.; Rajakumar, P.A.; Giddings, S.J. Insulin II Gene Expression in Rat Central Nervous System. Regul Pept 1993, 48, 55–63. [Google Scholar] [CrossRef]
- Kong, D.; Tong, Q.; Ye, C.; Koda, S.; Fuller, P.M.; Krashes, M.J.; Vong, L.; Ray, R.S.; Olson, D.P.; Lowell, B.B. GABAergic RIP-Cre Neurons in the Arcuate Nucleus Selectively Regulate Energy Expenditure. Cell 2012, 151, 645–657. [Google Scholar] [CrossRef]
- Rother, E.; Belgardt, B.F.; Tsaousidou, E.; Hampel, B.; Waisman, A.; Myers, M.G.; Brüning, J.C. Acute Selective Ablation of Rat Insulin Promoter-Expressing (RIP HER) Neurons Defines Their Orexigenic Nature. Proc Natl Acad Sci U S A 2012, 109, 18132–18137. [Google Scholar] [CrossRef] [PubMed]
- Eerola, K.; Longo, F.; Reinbothe, T.M.; Richard, J.E.; Shevchouk, O.T.; López-Ferreras, L.; Mishra, D.; Asker, M.; Tolö, J.; Miranda, C.; et al. Hindbrain Insulin Controls Feeding Behavior. Mol Metab 2022, 66, 101614. [Google Scholar] [CrossRef]
- Pu¨schel, G.P.; Pu¨schel, P.; Pü, G.P. Control of Hepatocyte Metabolism by Sympathetic and Parasympathetic Hepatic Nerves. Anat Rec A Discov Mol Cell Evol Biol 2004, 280A, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.X.; la Fleur, S.E.; Fliers, E.; Kalsbeek, A. The Role of the Autonomic Nervous Liver Innervation in the Control of Energy Metabolism. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2010, 1802, 416–431. [Google Scholar] [CrossRef]
- Pocal, A.; Lam, T.K.T.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) Channels Control Hepatic Glucose Production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef]
- Liss, B.; Roeper, J. Molecular Physiology of Neuronal K-ATP Channels. Mol Membr Biol 2001, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Van Den Hoek, A.M.; Van Heijningen, C.; Schröder-van Der Elst, J.P.; Ouwens, D.M.; Havekes, L.M.; Romijn, J.A.; Kalsbeek, A.; Pijl, H. Intracerebroventricular Administration of Neuropeptide Y Induces Hepatic Insulin Resistance via Sympathetic Innervation. Diabetes 2008, 57, 2304. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Tanida, M.; Nagata, N.; Inaba, Y.; Watanabe, H.; Nagashimada, M.; Ota, T.; Asahara, S.I.; Kido, Y.; Matsumoto, M.; et al. Central Insulin Action Activates Kupffer Cells by Suppressing Hepatic Vagal Activation via the Nicotinic Alpha 7 Acetylcholine Receptor. Cell Rep 2016, 14, 2362–2374. [Google Scholar] [CrossRef]
- Williams, K.W.; Margatho, L.O.; Lee, C.E.; Choi, M.; Lee, S.; Scott, M.M.; Elias, C.F.; Elmquist, J.K. Segregation of Acute Leptin and Insulin Effects in Distinct Populations of Arcuate Proopiomelanocortin Neurons. The Journal of Neuroscience 2010, 30, 2472. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin Excites Anorexigenic Proopiomelanocortin Neurons via Activation of Canonical Transient Receptor Potential Channels. Cell Metab 2014, 19, 682–693. [Google Scholar] [CrossRef]
- Qiu, J.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin and Leptin Excite Anorexigenic Pro-Opiomelanocortin Neurones via Activation of TRPC5 Channels. J Neuroendocrinol 2018, 30. [Google Scholar] [CrossRef]
- Huo, L.; Gamber, K.; Greeley, S.; Silva, J.; Huntoon, N.; Leng, X.H.; Bjørbæk, C. Leptin-Dependent Control of Glucose Balance and Locomotor Activity by POMC Neurons. Cell Metab 2009, 9, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Vianna, C.R.; Donato, J.; Kim, M.H.; Chuang, J.C.; Lee, C.E.; Lauzon, D.A.; Lin, P.; Brule, L.J.; Scott, M.M.; et al. Direct Leptin Action on POMC Neurons Regulates Glucose Homeostasis and Hepatic Insulin Sensitivity in Mice. J Clin Invest 2012, 122, 1000. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. Sen; Karkanias, G.B.; Morales, J.C.; Hawkins, M.; Barzilai, N.; Wang, J.; Rossetti, L. Intracerebroventricular Leptin Regulates Hepatic but Not Peripheral Glucose Fluxes. Journal of Biological Chemistry 1998, 273, 31160–31167. [Google Scholar] [CrossRef]
- Gutiérrez-Juárez, R.; Obici, S.; Rossetti, L. Melanocortin-Independent Effects of Leptin on Hepatic Glucose Fluxes. J Biol Chem 2004, 279, 49704–49715. [Google Scholar] [CrossRef]
- Tong, Q.; Ye, C.P.; McCrimmon, R.J.; Dhillon, H.; Choi, B.; Kramer, M.D.; Yu, J.; Yang, Z.; Christiansen, L.M.; Lee, C.E.; et al. Synaptic Glutamate Release by Ventromedial Hypothalamic Neurons Is Part of the Neurocircuitry That Prevents Hypoglycemia. Cell Metab 2007, 5, 383–393. [Google Scholar] [CrossRef]
- Shimazu, T.; Ogasawara, S. Effects of Hypothalamic Stimulation on Gluconeogenesis and Glycolysis in Rat Liver. Am J Physiol 1975, 228, 1787–1793. [Google Scholar] [CrossRef]
- Tu, L.; Fukuda, M.; Tong, Q.; Xu, Y. The Ventromedial Hypothalamic Nucleus: Watchdog of Whole-Body Glucose Homeostasis. Cell & Bioscience 2022 12:1 2022, 12, 1–17. [Google Scholar] [CrossRef]
- Gao, H.; Molinas, A.J.R.; Miyata, K.; Qiao, X.; Zsombok, A. Overactivity of Liver-Related Neurons in the Paraventricular Nucleus of the Hypothalamus: Electrophysiological Findings in Db/Db Mice. The Journal of Neuroscience 2017, 37, 11140. [Google Scholar] [CrossRef]
- Pocai, A.; Obici, S.; Schwartz, G.J.; Rossetti, L. A Brain-Liver Circuit Regulates Glucose Homeostasis. Cell Metab 2005, 1, 53–61. [Google Scholar] [CrossRef]
- Kwon, E.; Joung, H.Y.; Liu, S.M.; Chua, S.C.; Schwartz, G.J.; Jo, Y.H. Optogenetic Stimulation of the Liver-Projecting Melanocortinergic Pathway Promotes Hepatic Glucose Production. Nature Communications 2020 11:1 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Lam, C.K.L.; Chari, M.; Su, B.B.; Cheung, G.W.C.; Kokorovic, A.; Yang, C.S.; Wang, P.Y.T.; Lai, T.Y.Y.; Lam, T.K.T. Activation of N-Methyl-d-Aspartate (NMDA) Receptors in the Dorsal Vagal Complex Lowers Glucose Production. J Biol Chem 2010, 285, 21913. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.T.; Caspi, L.; Lam, C.K.L.; Chari, M.; Li, X.; Light, P.E.; Gutierrez-Juarez, R.; Ang, M.; Schwartz, G.J.; Lam, T.K.T. Upper Intestinal Lipids Trigger a Gut-Brain-Liver Axis to Regulate Glucose Production. Nature 2008, 452, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pydi, S.P.; Zhu, L.; Barella, L.F.; Cui, Y.; Gavrilova, O.; Bence, K.K.; Vernochet, C.; Wess, J. Adipocyte Gi Signaling Is Essential for Maintaining Whole-Body Glucose Homeostasis and Insulin Sensitivity. Nature Communications 2020 11:1 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Ikeda, K.; Tanabe, Y.; Thomson, E.A.; Yoneshiro, T.; Oguri, Y.; Ferro, M.D.; Poon, A.S.Y.; Kajimura, S. Wireless Optogenetics Protects against Obesity via Stimulation of Non-Canonical Fat Thermogenesis. Nat Commun 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Lyons, C.E.; Razzoli, M.; Larson, E.; Svedberg, D.; Frontini, A.; Cinti, S.; Vulchanova, L.; Sanders, M.; Thomas, M.; Bartolomucci, A. Optogenetic-Induced Sympathetic Neuromodulation of Brown Adipose Tissue Thermogenesis. FASEB J 2020, 34, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Pydi, S.P.; Wang, L.; Haspula, D.; Cui, Y.; Lu, H.; König, G.M.; Kostenis, E.; Steinberg, G.R.; Gavrilova, O.; et al. Adipocyte Gq Signaling Is a Regulator of Glucose and Lipid Homeostasis in Mice. Nat Commun 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Aoki, V.T.; Adkison, M.G.; Warren, W.S.; Bartness, T.J. Central Nervous System Origins of the Sympathetic Nervous System Outflow to White Adipose Tissue. Am J Physiol 1998, 275. [Google Scholar] [CrossRef]
- Bartness, T.J.; Song, C.K. Brain-Adipose Tissue Neural Crosstalk. Physiol Behav 2007, 91, 343. [Google Scholar] [CrossRef]
- Stanley, S.; Pinto, S.; Segal, J.; Pérez, C.A.; Viale, A.; DeFalco, J.; Cai, X.; Heisler, L.K.; Friedman, J.M. Identification of Neuronal Subpopulations That Project from Hypothalamus to Both Liver and Adipose Tissue Polysynaptically. Proc Natl Acad Sci U S A 2010, 107, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Ryu, V.; Bartness, T.J. Short and Long Sympathetic-Sensory Feedback Loops in White Fat. Am J Physiol Regul Integr Comp Physiol 2014, 306. [Google Scholar] [CrossRef]
- Steculorum, S.M.; Ruud, J.; Karakasilioti, I.; Backes, H.; Engström Ruud, L.; Timper, K.; Hess, M.E.; Tsaousidou, E.; Mauer, J.; Vogt, M.C.; et al. AgRP Neurons Control Systemic Insulin Sensitivity via Myostatin Expression in Brown Adipose Tissue. Cell 2016, 165, 125. [Google Scholar] [CrossRef]
- Cavalcanti-de-Albuquerque, J.P.; Bober, J.; Zimmer, M.R.; Dietrich, M.O. Regulation of Substrate Utilization and Adiposity by Agrp Neurons. Nature Communications 2019 10:1 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Chao, P.T.; Yang, L.; Aja, S.; Moran, T.H.; Bi, S. Knockdown of NPY Expression in the Dorsomedial Hypothalamus Promotes Development of Brown Adipocytes and Prevents Diet-Induced Obesity. Cell Metab 2011, 13, 573. [Google Scholar] [CrossRef]
- Gómez-Valadés, A.G.; Pozo, M.; Varela, L.; Boudjadja, M.B.; Ramírez, S.; Chivite, I.; Eyre, E.; Haddad-Tóvolli, R.; Obri, A.; Milà-Guasch, M.; et al. Mitochondrial Cristae-Remodeling Protein OPA1 in POMC Neurons Couples Ca2+ Homeostasis with Adipose Tissue Lipolysis. Cell Metab 2021, 33, 1820. [Google Scholar] [CrossRef]
- López, M. Hypothalamic AMPK and Energy Balance. Eur J Clin Invest 2018, 48, 12996. [Google Scholar] [CrossRef]
- Martínez De Morentin, P.B.; González-García, I.; Martins, L.; Lage, R.; Fernández-Mallo, D.; Martínez-Sánchez, N.; Ruíz-Pino, F.; Liu, J.; Morgan, D.A.; Pinilla, L.; et al. Estradiol Regulates Brown Adipose Tissue Thermogenesis via Hypothalamic AMPK. Cell Metab 2014, 20, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Seoane-Collazo, P.; Roa, J.; Rial-Pensado, E.; Liñares-Pose, L.; Beiroa, D.; Ruíz-Pino, F.; López-González, T.; Morgan, D.A.; Pardavila, J.Á.; Sánchez-Tapia, M.J.; et al. SF1-Specific AMPKa1 Deletion Protects against Diet-Induced Obesity. Diabetes 2018, 67, 2213–2226. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Muse, E.D.; Cheng, A.; Chen, L.; Scherer, T.; Pocai, A.; Su, K.; Cheng, B.; Li, X.; Harvey-White, J.; et al. Leptin Controls Adipose Tissue Lipogenesis via Central, STAT3-Independent Mechanisms. Nat Med 2008, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.A.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic Neuro-Adipose Connections Mediate Leptin-Driven Lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and Insulin Act on POMC Neurons to Promote the Browning of White Fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef]
- Mark, A.L.; Agassandian, K.; Morgan, D.A.; Liu, X.; Cassell, M.D.; Rahmouni, K. Leptin Signaling in the Nucleus Tractus Solitarii Increases Sympathetic Nerve Activity to the Kidney. Hypertension 2009, 53, 375–380. [Google Scholar] [CrossRef]
- Barnes, M.J.; McDougal, D.H. Leptin into the Rostral Ventral Lateral Medulla (RVLM) Augments Renal Sympathetic Nerve Activity and Blood Pressure. Front Neurosci 2014, 8. [Google Scholar] [CrossRef]
- Shi, Z.; Pelletier, N.E.; Wong, J.; Li, B.; Sdrulla, A.D.; Madden, C.J.; Marks, D.L.; Brooks, V.L. Leptin Increases Sympathetic Nerve Activity via Induction of Its Own Receptor in the Paraventricular Nucleus. Elife 2020, 9, 1–34. [Google Scholar] [CrossRef]
- Coester, B.; Koester-Hegmann, C.; Lutz, T.A.; Foll, C. Le Amylin/Calcitonin Receptor–Mediated Signaling in POMC Neurons Influences Energy Balance and Locomotor Activity in Chow-Fed Male Mice. Diabetes 2020, 69, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Psichas, A.; Reimann, F.; Gribble, F.M. Gut Chemosensing Mechanisms. J Clin Invest 2015, 125, 908. [Google Scholar] [CrossRef]
- Kim, W.; Egan, J.M. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol Rev 2008, 60, 470. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, C.; Poulsen, S.S.; Møller, M.; Holst, J.J. Glucagon-like Peptide I Receptors in the Subfornical Organ and the Area Postrema Are Accessible to Circulating Glucagon-like Peptide I. Diabetes 1996, 45, 832–835. [Google Scholar] [CrossRef]
- Gu, G.; Roland, B.; Tomaselli, K.; Dolman, C.S.; Lowe, C.; Heilig, J.S. Glucagon-like Peptide-1 in the Rat Brain: Distribution of Expression and Functional Implication. J Comp Neurol 2013, 521, 2235–2261. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The Arcuate Nucleus Mediates GLP-1 Receptor Agonist Liraglutide-Dependent Loss. J Clin Invest 2014, 124, 4473. [Google Scholar] [CrossRef]
- Adriaenssens, A.E.; Biggs, E.K.; Darwish, T.; Tadross, J.; Sukthankar, T.; Girish, M.; Polex-Wolf, J.; Lam, B.Y.; Zvetkova, I.; Pan, W.; et al. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab 2019, 30, 987–996.e6. [Google Scholar] [CrossRef]
- Zhang, Q.; Delessa, C.T.; Augustin, R.; Bakhti, M.; Colldén, G.; Drucker, D.J.; Feuchtinger, A.; Caceres, C.G.; Grandl, G.; Harger, A.; et al. The Glucose-Dependent Insulinotropic Polypeptide (GIP) Regulates Body Weight and Food Intake via CNS-GIPR Signaling. Cell Metab 2021, 33, 833–844.e5. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M. The Role of GIP Receptor in the CNS for the Pathogenesis of Obesity. Diabetes 2021, 70, 1929. [Google Scholar] [CrossRef] [PubMed]
- Turton, M.D.; O’Shea, D.; Gunn, I.; Beak, S.A.; Edwards, C.M.B.; Meeran, K.; Choi, S.J.; Taylor, G.M.; Heath, M.M.; Lambert, P.D.; et al. A Role for Glucagon-like Peptide-1 in the Central Regulation of Feeding. Nature 1996, 379, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Meeran, K.; O’Shea, D.; Edwards, C.M.B.; Turton, M.D.; Heath, M.M.; Gunn, I.; Abusnana, S.; Rossi, M.; Small, C.J.; Goldstone, A.P.; et al. Repeated Intracerebroventricular Administration of Glucagon-Like Peptide-1-(7–36) Amide or Exendin-(9–39) Alters Body Weight in the Rat*This Work Was Supported by the United Kingdom Medical Research Council. Endocrinology 1999, 140, 244–250. [Google Scholar] [CrossRef]
- Heppner, K.M.; Kirigiti, M.; Secher, A.; Paulsen, S.J.; Buckingham, R.; Pyke, C.; Knudsen, L.B.; Vrang, N.; Grove, K.L. Expression and Distribution of Glucagon-like Peptide-1 Receptor MRNA, Protein and Binding in the Male Nonhuman Primate (Macaca Mulatta) Brain. Endocrinology 2015, 156, 255–267. [Google Scholar] [CrossRef]
- NamKoong, C.; Kim, M.S.; Jang, B.T.; Lee, Y.H.; Cho, Y.M.; Choi, H.J. Central Administration of GLP-1 and GIP Decreases Feeding in Mice. Biochem Biophys Res Commun 2017, 490, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Ai, M.; Nunn, N.; Culotta, I.; Hunter, J.; Boudjadja, M.B.; Valencia-Torres, L.; Aviello, G.; Hodson, D.J.; Snider, B.M.; et al. Anorectic and Aversive Effects of GLP-1 Receptor Agonism Are Mediated by Brainstem Cholecystokinin Neurons, and Modulated by GIP Receptor Activation. Mol Metab 2022, 55, 101407. [Google Scholar] [CrossRef]
- Samms, R.J.; Sloop, K.W.; Gribble, F.M.; Reimann, F.; Adriaenssens, A.E. GIPR Function in the Central Nervous System: Implications and Novel Perspectives for GIP-Basedz Therapies in Treating Metabolic Disorders. Diabetes 2021, 70, 1938. [Google Scholar] [CrossRef]
- Kim, S.J.; Nian, C.; Karunakaran, S.; Clee, S.M.; Isales, C.M.; McIntosh, C.H.S. GIP-Overexpressing Mice Demonstrate Reduced Diet-Induced Obesity and Steatosis, and Improved Glucose Homeostasis. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Mroz, P.A.; Finan, B.; Gelfanov, V.; Yang, B.; Tschöp, M.H.; DiMarchi, R.D.; Perez-Tilve, D. Optimized GIP Analogs Promote Body Weight Lowering in Mice through GIPR Agonism Not Antagonism. Mol Metab 2019, 20, 51–62. [Google Scholar] [CrossRef]
- Han, W.; Wang, L.; Ohbayashi, K.; Takeuchi, M.; O’Farrell, L.; Coskun, T.; Rakhat, Y.; Yabe, D.; Iwasaki, Y.; Seino, Y.; et al. Glucose-Dependent Insulinotropic Polypeptide Counteracts Diet-Induced Obesity along with Reduced Feeding, Elevated Plasma Leptin and Activation of Leptin-Responsive and Proopiomelanocortin Neurons in the Arcuate Nucleus. Diabetes Obes Metab 2023. [Google Scholar] [CrossRef]
- Falko, J.M.; Crockett, S.E.; Cataland, S.; Mazzaferri, E.L. Gastric Inhibitory Polypeptide (GIP) Stimulated by Fat Ingestion in Man. J Clin Endocrinol Metab 1975, 41, 260–265. [Google Scholar] [CrossRef]
- Brøns, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of Short-Term High-Fat Feeding on Glucose and Insulin Metabolism in Young Healthy Men. J Physiol 2009, 587, 2387. [Google Scholar] [CrossRef] [PubMed]
- Szecowka, J.; Grill, V.; Sandberg, E.; Efendic, S. Effect of GIP on the Secretion of Insulin and Somatostatin and the Accumulation of Cyclic AMP in Vitro in the Rat. Acta Endocrinol (Copenh) 1982, 99, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like Peptide I Stimulates Insulin Gene Expression and Increases Cyclic AMP Levels in a Rat Islet Cell Line. Proc Natl Acad Sci U S A 1987, 84, 3434–3438. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B. Sensory Nerves Contribute to Insulin Secretion by Glucagon-like Peptide-1 in Mice. Am J Physiol Regul Integr Comp Physiol 2004, 286. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.W.C.; Kokorovic, A.; Lam, C.K.L.; Chari, M.; Lam, T.K.T. Intestinal Cholecystokinin Controls Glucose Production through a Neuronal Network. Cell Metab 2009, 10, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Schwartz, G.J. Duodenal Lipid Sensing Activates Vagal Afferents to Regulate Non-Shivering Brown Fat Thermogenesis in Rats. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Sandoval, D.A.; Bagnol, D.; Woods, S.C.; D’Alessio, D.A.; Seeley, R.J. Arcuate Glucagon-like Peptide 1 Receptors Regulate Glucose Homeostasis but Not Food Intake. Diabetes 2008, 57, 2046–2054. [Google Scholar] [CrossRef]
- Burmeister, M.A.; Ferre, T.; Ayala, J.E.; King, E.M.; Holt, R.M.; Ayala, J.E. Acute Activation of Central GLP-1 Receptors Enhances Hepatic Insulin Action and Insulin Secretion in High-Fat-Fed, Insulin Resistant Mice. Am J Physiol Endocrinol Metab 2012, 302, 334–343. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, L.; Zhang, J.; Conde, K.; Phansalkar, J.; Li, Z.; Yao, L.; Xu, Z.; Wang, W.; Zhou, J.; et al. Glucose-Sensing Glucagon-like Peptide-1 Receptor Neurons in the Dorsomedial Hypothalamus Regulate Glucose Metabolism. Sci Adv 2022, 8, 5345. [Google Scholar] [CrossRef]
- Pérez-Tilve, D.; González-Matías, L.; Aulinger, B.A.; Alvarez-Crespo, M.; Gil-Lozano, M.; Alvarez, E.; Andrade-Olivie, A.M.; Tschöp, M.H.; D’Alessio, D.A.; Mallo, F. Exendin-4 Increases Blood Glucose Levels Acutely in Rats by Activation of the Sympathetic Nervous System. Am J Physiol Endocrinol Metab 2010, 298. [Google Scholar] [CrossRef]
- Jessen, L.; Smith, E.P.; Ulrich-Lai, Y.; Herman, J.P.; Seeley, R.J.; Sandoval, D.; D’Alessio, D. Central Nervous System GLP-1 Receptors Regulate Islet Hormone Secretion and Glucose Homeostasis in Male Rats. Endocrinology 2017, 158, 2124. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S. Central Action of FGF19 Reduces Hypothalamic AGRP/NPY Neuron Activity and Improves Glucose Metabolism. Mol Metab 2013, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Marcelin, G.; Blouet, C.; Jeong, J.H.; Jo, Y.H.; Schwartz, G.J.; Chua, S. A Gut-Brain Axis Regulating Glucose Metabolism Mediated by Bile Acids and Competitive Fibroblast Growth Factor Actions at the Hypothalamus. Mol Metab 2018, 8, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-M.; Ifebi, B.; Johnson III, F.; Xu, A.; Ho, J.; Yang, Y.; Schwartz, G.J.; Jo, Y.-H.; Chua, S. The Gut Signals to AGRP-Expressing Cells of the Pituitary to Control Glucose Homeostasis. J Clin Invest 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Wean, J.B.; Smith, B.N. FGF19 in the Hindbrain Lowers Blood Glucose and Alters Excitability of Vagal Motor Neurons in Hyperglycemic Mice. Endocrinology 2021, 162, 1–14. [Google Scholar] [CrossRef]
- Fan, K.; Li, Q.; Pan, D.; Liu, H.; Li, P.; Hai, R.; Du, C. Effects of Amylin on Food Intake and Body Weight via Sympathetic Innervation of the Interscapular Brown Adipose Tissue. 2020, 25, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Batterham, R.L.; Ffytche, D.H.; Rosenthal, J.M.; Zelaya, F.O.; Barker, G.J.; Withers, D.J.; Williams, S.C.R. PYY Modulation of Cortical and Hypothalamic Brain Areas Predicts Feeding Behaviour in Humans. Nature 2007, 450, 106–109. [Google Scholar] [CrossRef]
- De Silva, A.; Salem, V.; Long, C.J.; Makwana, A.; Newbould, R.D.; Rabiner, E.A.; Ghatei, M.A.; Bloom, S.R.; Matthews, P.M.; Beaver, J.D.; et al. The Gut Hormones PYY 3-36 and GLP-1 7-36 Amide Reduce Food Intake and Modulate Brain Activity in Appetite Centers in Humans. Cell Metab 2011, 14, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Tak, Y.J.; Lee, S.Y. Long-Term Efficacy and Safety of Anti-Obesity Treatment: Where Do We Stand? Curr Obes Rep 2021, 10, 14–30. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int J Mol Sci 2020, 21, 1–65. [Google Scholar] [CrossRef] [PubMed]
- Coulter, A.A.; Rebello, C.J.; Greenway, F.L. Centrally Acting Agents for Obesity: Past, Present, and Future. Drugs 2018, 78, 1113–1132. [Google Scholar] [CrossRef] [PubMed]
- Greenway, F.L.; Fujioka, K.; Plodkowski, R.A.; Mudaliar, S.; Guttadauria, M.; Erickson, J.; Kim, D.D.; Dunayevich, E. Effect of Naltrexone plus Bupropion on Weight Loss in Overweight and Obese Adults (COR-I): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2010, 376, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Padwal, R.S.; Majumdar, S.R. Drug Treatments for Obesity: Orlistat, Sibutramine, and Rimonabant. Lancet 2007, 369, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.; Finlayson, G.; Axelsen, M.; Flint, A.; Gibbons, C.; Kvist, T.; Hjerpsted, J.B. Effects of Once-Weekly Semaglutide on Appetite, Energy Intake, Control of Eating, Food Preference and Body Weight in Subjects with Obesity. Diabetes Obes Metab 2017, 19, 1242–1251. [Google Scholar] [CrossRef]
- Hjerpsted, J.B.; Flint, A.; Brooks, A.; Axelsen, M.B.; Kvist, T.; Blundell, J. Semaglutide Improves Postprandial Glucose and Lipid Metabolism, and Delays First-Hour Gastric Emptying in Subjects with Obesity. Diabetes Obes Metab 2018, 20, 610–619. [Google Scholar] [CrossRef]
- Chao, A.M.; Wadden, T.A.; Berkowitz, R.I.; Quigley, K.; Silvestry, F. The Risk of Cardiovascular Complications with Current Obesity Drugs. Expert Opin Drug Saf 2020, 19, 1095. [Google Scholar] [CrossRef]
- Wang, C.; He, Y.; Xu, P.; Yang, Y.; Saito, K.; Xia, Y.; Yan, X.; Hinton, A.; Yan, C.; Ding, H.; et al. TAp63 Contributes to Sexual Dimorphism in POMC Neuron Functions and Energy Homeostasis. Nat Commun 2018, 9. [Google Scholar] [CrossRef]
- JOSIPOVIC, M.; STARICOFF, E.O.; RICHES, C.H.; SHEPPARD, L.; EVANS, M. 1311-P: Glucokinase in AGRP Neurons Is Required for Normal Insulin Secretion and Action in Female Mice. Diabetes 2022, 71. [Google Scholar] [CrossRef]
- Kroll, D.S.; Feldman, D.E.; Biesecker, C.L.; McPherson, K.L.; Manza, P.; Joseph, P.V.; Volkow, N.D.; Wang, G.J. Neuroimaging of Sex/Gender Differences in Obesity: A Review of Structure, Function, and Neurotransmission. Nutrients 2020, 12, 1–21. [Google Scholar] [CrossRef]
- Pi-Sunyer, X. The Medical Risks of Obesity. Postgrad Med 2009, 121, 21. [Google Scholar] [CrossRef] [PubMed]
- Haththotuwa, R.N.; Wijeyaratne, C.N.; Senarath, U. Worldwide Epidemic of Obesity. Obesity and Obstetrics 2020, 3–8. [Google Scholar] [CrossRef]
- Karalliedde, J.; Gnudi, L. Diabetes Mellitus, a Complex and Heterogeneous Disease, and the Role of Insulin Resistance as a Determinant of Diabetic Kidney Disease. Nephrology Dialysis Transplantation 2016, 31, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Poirier, P.; Després, J.P. The Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation 2018, 137, 1391. [Google Scholar] [CrossRef]
- Rodgers, R.J.; Tschöp, M.H.; Wilding, J.P.H. Anti-Obesity Drugs: Past, Present and Future. Dis Model Mech 2012, 5, 621. [Google Scholar] [CrossRef]
Figure 1.
Overview of hypothalamic and brainstem control of energy, glucose and lipid homeostasis: Both hypothalamic and brainstem neuronal populations exert prominent homeostatic control over energy intake and energy expenditure. Circulating signals can also activate/inhibit distinct neural pathways to recalibrate autonomic outflow to various metabolic organs and endocrine glands. This crosstalk between the CNS and the periphery is essential for the maintenance of glucose and lipid homeostasis. It is to be noted, that figure highlights only a limited number of metabolically relevant pathways due to space restrictions. Please see text for details.
Figure 1.
Overview of hypothalamic and brainstem control of energy, glucose and lipid homeostasis: Both hypothalamic and brainstem neuronal populations exert prominent homeostatic control over energy intake and energy expenditure. Circulating signals can also activate/inhibit distinct neural pathways to recalibrate autonomic outflow to various metabolic organs and endocrine glands. This crosstalk between the CNS and the periphery is essential for the maintenance of glucose and lipid homeostasis. It is to be noted, that figure highlights only a limited number of metabolically relevant pathways due to space restrictions. Please see text for details.
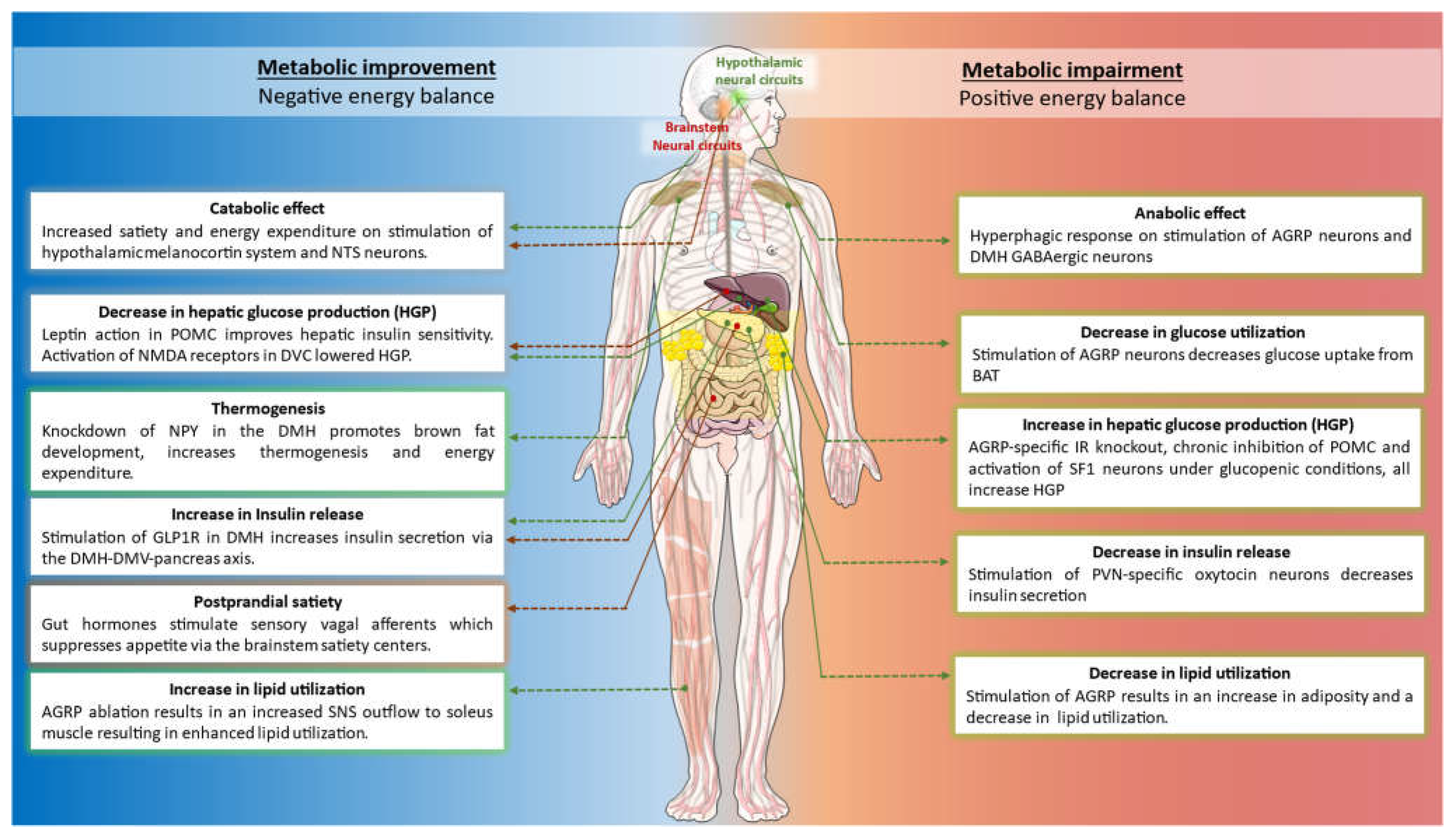
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



