
Submitted:
03 May 2023
Posted:
04 May 2023
You are already at the latest version
Abstract
Fluorescent hydrogels are promising candidate materials for portable biosensors to be used in point-of-care diagnosis because (1) they have a greater capacity for binding organic molecules than immunochromatographic test systems, determined by the immobilization of affinity labels within the three-dimensional hydrogel structure; (2) fluorescent detection is more sensitive than the colorimetric detection of gold nanoparticles or stained latex microparticles; (3) the properties of the gel matrix can be finely tuned for better compatibility and detection of different analytes; and (4) hydrogel biosensors can be made reusable and suitable for studying dynamic processes in real time. Water-soluble fluorescent nanocrystals are widely used for in vitro and in vivo biological imaging due to their unique optical properties, and hydrogels based on them allow preserving these properties in bulk composite macrostructures. Here we review the techniques for obtaining analyte-sensitive fluorescent hydrogels based on nanocrystals, the main methods used for detecting the fluorescent signal changes, and the approaches to the formation of inorganic fluorescent hydrogels via sol–gel phase transition using surface ligands of the nanocrystals.
Keywords:
inorganic fluorescent hydrogel
; fluorescent nanocrystal
; biosensor
1. Introduction
The current approach to engineering sensor diagnostic platforms is aimed at obtaining autonomous and versatile biosensors not requiring expensive equipment for analysis or time- consuming sample preparation, with the result available within minutes. This concept is referred to as point-of-care (PoC) or bedside diagnostics, but it is not restricted to medicine. Such sensors would be also useful for food testing, e.g., in rapid bacterial contamination tests, and for environment testing, e.g., in portable air or water contamination sensors. Many current miniature sensors were originally developed as the so-called labs-on-a-chip sensors used in more complex systems. Since fluids are the natural environment of most biological molecules, microfluidic flow cells and microfluidic chips are among the most common formats of these devices for the analysis of biological molecules. Macroscale analogs of these miniature devices include liquid chromatographs, flow cytometers, suspension chip systems, and classical enzyme immunoassay kits. Miniaturization offers many advantages, including a high throughput ensured by parallel assays, short time of analysis, low manufacturing costs, and small volumes of both the sample and the reagents, which is important in the case of expensive or hazardous reagents [1,2]. However, miniaturization also entails some limitations compared to traditional methods. For example, enzyme-linked immunosorbent assay (ELISA) uses a larger sample volume and a greater number of detection molecules, which ensures a more intense absolute signal than microfluidic-based sensors. Another example of the case when larger is better is chromatographic separation or purification of the sample, whose efficiency is directly proportional to the physical size of the column. Thus, miniaturization alone is not sufficient for successful use of microfluidic detection systems; it is also necessary to search for the ways of enhancing the sensitivity and increasing the effective interaction volume between the detecting molecules and the analyte. Fluorescence detection is the most sensitive of all optical detection methods [3,4], provided that the optimal type of the fluorescence label, e.g., organic dyes, fluorescent proteins, or fluorescent nanocrystals (NCs), has been selected for each specific task. The problem of increasing the effective interaction volume in microsystems is particularly urgent, because miniaturization reduces the overall number of detecting and reporter molecules. While, e.g., in chromatography, branched-surface resins functionalized with selective molecules are used to increase the effective interaction volume, this is not always applicable in microfluidic systems, because opaque media impede fluorescence detection. In addition, the particles of such medium can move with the fluid flow, which complicates detection and may lead to blocking of microfluidic channels or media washout from the microfluidic chip, thus compromising its analytical properties.
One of the most promising trends in the development of microfluidic sensors is the use of fluorescent labels based on inorganic NCs incorporated in a three-dimensional (3D) optically transparent polymeric matrix permeable for fluid flow. There are two most common types of such matrices: (1) matrices based on organic polymers with embedded fluorescent NCs [5,6] and (2) composite hydrogels formed from colloidal NCs via sol–gel phase transition [7]. These hydrogels have a porous branched structure, which is suitable for immobilization of sensory, affinity, and signal elements. In addition, hydrogels are hydrophilic, which is necessary for the unhindered passage of the analytes. The analyte can be specifically recognized by the structure of gel material itself [8]; otherwise, the functionalization of the hydrogel with affinity labels can be used for this purpose. The signal function can also be fulfilled by the hydrogel itself in some cases; e.g., there are gel materials that lose their structure when the pH is changed, either by dissolution or swelling [9]. However, the most common way is to incorporate signal molecules that generate fluorescent [10] or chromogenic [11] signals, or change their electrochemical [12] or conductive [13] properties.
In the next section we will discuss in detail the approaches to obtaining NC-based inorganic composite fluorescent hydrogels, as well as the principles of optical signal generation in hydrogel-based biosensors that rely on fluorescence detection.
2. Methods for Obtaining Fluorescent Gels
Fluorescent hydrogels constitute one of the main components of biosensors. In addition to the optical signal generation, they are responsible for the immobilization of affinity molecules and the maintenance of the 3D structure of the biosensor. In general, fluorescent gels are obtained by incorporating fluorescent labels, such as organic dyes [14], proteins/peptides [15], aromatic hydrocarbons [16], metal nanoparticles [17], or NCs [18], into the gel structure. The main requirements for fluorescent labels are the capability for multiplexed detection, high detection sensitivity and fluorescent signal intensity, photostability, and a reliable and convenient procedure for immobilizing fluorescent labels in the hydrogel. Multiplexed detection requires multiple labels with different fluorescence spectra but coinciding or overlapping absorption spectra, so that they could be excited simultaneously with a single source of radiation of a given wavelength. There are organic dyes and proteins that fluoresce in various spectral regions from visible to near-infrared. However, they have wider fluorescence spectra and narrower absorption spectra than fluorescent NCs, which makes it difficult to simultaneously excite and detect the fluorescent signal [19]. Fluorescent NCs exhibit the quantum size effect, allowing fluorescence spectra to be tailored to specific applications by selecting the NC size [20]. The NC photostability, i.e., the retention of their fluorescence properties upon exposure to light, is higher than that of organic fluorescent dyes [21], which makes it possible to increase the sensitivity through long-term signal accumulation, as well as to perform prolonged detection, e.g., in studying prolonged processes or sequentially analyzing several samples, without a decline in the signal intensity with time.
Fluorescent gels can be obtained using two fundamentally different approaches. The first approach is to embed fluorescent labels into the polymer structure, which is suitable for almost all types of fluorescent labels, both organic and inorganic. The second approach is based on the phenomenon of sol–gel phase transition and is traditionally used with inorganic fluorescent NCs (Figure 1). Below we will focus on the methods of inclusion of NC labels, because, as noted above, they have several advantages over other fluorescent labels and are the most promising for the use in this type of biosensors. In the first method of incorporation of fluorescent labels into a gel, the polymer and crosslinker components are mixed and let to polymerize. When the gel has been formed, it is incubated together with the fluorescent labels so that they diffuse into the resulting matrix (Figure 1a). Li et al. used this method to incorporate carbon dots into polymethacrylic acid gels [22]. Carbon dots were immobilized in the gel due to the formation of hydrogen bonds between the amino groups of the dot shell and the carboxyl groups of the polymer. However, this method does not guarantee permanent immobilization of the fluorescent labels, because further manipulations or pumping of the samples in polar solvents may cause disruption of hydrogen bonds and washout of the fluorescent labels from the gel. In the second method, the components for gel formation and fluorescent labels are mixed simultaneously, so that the labels become enclosed in the pores of the gel during its formation (Figure 1b). It should be noted that both methods require additional procedures to ensure uniform distribution of the labels within the gel, such as sonication [23]. The third method based on cross-linking of hydrogel monomers and NC surface chemical agents (Figure 1c) yields hydrogels containing uniformly distributed fluorescent NCs without additional treatment of the gel. For example, Xu et al. functionalized the ZnS NC surface with chemical agents that induced polymerization of methyl methacrylate or glycidyl methacrylate and acted as crosslinkers for the monomers [24]. Yang et al. described the formation of fluorescent gels based on acrylic acid and acrylamide containing CdS NCs with the use of gamma radiation as a polymerization initiator [25]. Coordination interaction between the Cd atoms on the NC surface and the N atoms of the polyacrylamide gel resulted in a homogeneous NC distribution in the gel and the absence of NC aggregation even if their concentration was increased. In addition, the polymer molecules can interact with heavy metal ions of not only the NCs, but also their precursors (Figure 1d). Jiang et al. powdered dried cellulose hydrogel and placed it into a solution of heavy metal precursors, cadmium chloride pentahydrate and sodium sulfide [26]. Positively charged Cd2+ ions were electrostatically and chemically absorbed on the gel surface, and their interaction with S2– ions gave rise to the growth of NCs steadily embedded in the gel structure. However, this method is disadvantageous compared to the above two ones in that it allows gel encapsulation of only core NCs lacking an epitaxial inorganic shell, and the latter is required to improve the stability and optical properties of NCs. In addition, this method imposes limitations on the NC synthesis procedure; e.g., it does not allow precise control of the NC size and, hence, their fluorescence spectrum. Figure 1e shows the method of obtaining fluorescent hydrogels via a sol–gel phase transition, when NC surface ligands are involved in gel formation. Gaponic et al. [27] have reported an example of the formation of such a gel formed of CdTe NCs whose surface is functionalized with compounds containing thiol groups. It is worth noting that this method of obtaining composite inorganic fluorescent hydrogels is the most versatile, because it allows using previously synthesized and characterized fluorescent core/shell NCs, ensures their uniform distribution in the hydrogel, and completely preserves the optical properties of the NCs by preventing their aggregation. Before reviewing the methods for modification of the NC surface and induction of gel formation, we will touch upon the principles of detection of target analytes in biosensors based on fluorescent gels.
3. Methods of Analyte Detection Using Fluorescent Gels
Biosensors based on fluorescent gels employ different approaches to analyte detection, but all of them involve the recording of changes in the fluorescent properties of the gel. Depending on the analyte detected and the design of the biosensor, the changes in the optical properties may be of two types: first, quenching of NC fluorescence due to energy transfer (e.g., via the Förster resonance energy transfer, FRET), structural degradation of the fluorescent NCs, or their aggregation due to gel structure disruption; second, changes in the fluorescence spectrum due to the activation of new fluorescent labels. The specific mechanism of the change in the optical properties often depends not only on the properties of the fluorescent NCs and the gel, but also on the characteristics of the analyte and additional sensory molecules contained in the biosensor. In some cases, specific binding of the analyte is ensured by NC functionalization with surface ligands, such as cyclodextrins [28] or (2-hydroxyethyl) dithiocarbamate [29]. However, this is applicable almost exclusively to relatively simple analytes, such as metal ions [30], polyaromatic molecules [31], and NO2– [32]. Specific recognition of more complex organic molecules, such as antibodies or other proteins, requires the use of affinity molecules, e.g., antibodies. The main principles of detection used in fluorescent hydrogel biosensors are shown in Figure 2. One of the simplest detection principles (Figure 2a) is used with analytes that disrupt the gel structure upon contact, which results in NC aggregation and decreased fluorescence intensity. This detection principle was used by Bhattacharya et al. [33] for the detection of bacteria. They made a fluorescent gel from 6-O-(O-O′-dilauroyltharyl)-D-glucose and carbon NCs and, after its polymerization, added cultures of Bacillus and Staphylococcus strains. During the growth of the culture, the bacterial cells secreted esterases catalyzing the ester cleavage reaction. As a result, the fluorescence signal decreased, depending on the number of added bacteria, due to the degradation of the gel matrix and aggregation of the NCs. It should be noted that this approach entails irreversible destruction of the biosensor, which makes biosensors of this type hardly practicable. The detection principle shown in Figure 2b is applicable to analytes that either cause specific quenching of NC fluorescence via various excitation relaxation mechanisms or fluoresce themselves (i.e., they are optically active). For example, metal ions can act as electron acceptors upon their reduction, thereby decreasing the NC fluorescence intensity [34]. Many organic compounds can act as electron acceptors in the Förster resonance energy transfer, which also reduces the NC fluorescence intensity or leads to the formation of an additional fluorescence peak [35]. This principle is often used for the detection of metal ions, the selectivity being ensured by functionalization of the NC surface with a ligand that selectively interacts with a specific ion, e.g., Fe3+ [36], Cr6+ [37], or Cu2+ [38]. Figure 2c shows the scheme for detecting optically inactive analytes. In this case, auxiliary optically active labels are used that specifically bind the analyte and either quench the optical signal or fluoresce in a different spectral region. Antibodies or oligonucleotides can be used as capture molecules [39], and organic dyes [40], plasmonic nanoparticles [41], graphene [42], or other compounds causing changes in the fluorescence of hydrogels [43] are used as optically active labels. In the fourth detection method (Figure 2d), two enzymes, oxidase and horseradish peroxidase, are introduced into the pores of the fluorescent gel. The former specifically oxidizes the analyte to form hydrogen peroxide, and the latter uses hydrogen peroxide as a cofactor to catalyze the formation of OH– radicals, which weaken the hydrogel fluorescence [44], and the oxidation of the chromogenic dye, which shifts the spectral maximum of the absorption signal, also contributing to the quenching of the gel fluorescence [45]. For example, Cho et al. [44] developed a glucose biosensor operating on this principle. They immobilized fluorescent carbon NCs, rhodamine 6G, glucose oxidase, and horseradish peroxidase in a hydrogel. Upon excitation at a wavelength of 360 nm, the blue fluorescence of the carbon NCs was quenched by the bienzymatic reaction with glucose and the formation of OH– radicals, while the fluorescence of rhodamine 6G was used for calibration because it did not depend on the amount of glucose.
The miniature size of the biosensors imposes significant limitations on the area available for immobilization of sensor or affinity labels recognizing specific analytes. Switching from the 2D detection model to the 3D one by using 3D polymer matrices not only increases the capacity for binding the sensor and affinity labels, but also preserves the structure of the molecules, which could denature upon immobilization on a flat surface [46]. This prevents the deterioration of the properties necessary for sensing, such as specificity and sensitivity [47]. All this makes it possible to increase the detection sensitivity of microfluidic biosensors compared to their full-size counterparts. When the signal or recognition labels are immobilized in the 3D structure of the hydrogel, their orientation becomes irrelevant, because the analyte passes on all sides of them in any case, which increases the probability of the detection. For example, Gao et al. [48] have shown that the efficiency of DNA hybridization on a surface is 20–40 times lower than in a solution. When antibodies are immobilized on a flat surface, they can also lose specificity and affinity for their antigens, which is confirmed by numerous examples reviewed by Welch et al. [49]. Feng et al. [50] have shown that 3D arrangement of antibodies even on a flat surface can increase the detection sensitivity by a factor of 64. In addition, the hydrogel structure itself can perform a sensory function. For example, the selectivity for the analyte size or selectivity of analyte–hydrogel interaction can be increased, or nonspecific binding of the analyte decreased, by varying the pore size of the 3D matrix or by selecting materials with different physical and chemical properties for its formation. Yuan et al. [51] developed NC hydrogels with encapsulated tyrosinase (TYR)for biosensing of dopamine (Figure 3). The use of a neutral phosphate-buffered saline to dissolve the precipitated CdTe NCs capped with mercaptosuccinic acid (MSA) sufficiently shortened the NC gelation time (to several days), and the sol–gel transition was also observed in the as-prepared NC gels. The resulting gels had pores ranging from 10 to 50 nm in diameter, and the enzyme could be encapsulated in the mesopores of the gel network during the gelation. The encapsulation of TYR was confirmed by atomic force microscopy and the test reaction of tyrosinase-catalyzed oxidation of catechol. The enzymatic activity of TYR in the hydrogel was preserved even after immersion in a potassium buffer solution for at least one week. Tyrosinase catalyzes the oxidation of dopamine to dopamine-o-quinone, which quenches NC fluorescence. Addition of dopamine quenched the NC fluorescence in the tyrosinase-embedded hydrogel, but it had no obvious effect on the pure NC hydrogel in the absence of tyrosinase. The detection limit for dopamine was found to be 5.0×10−8 mol L−1, the practicable range of detection being from 5.0×10−5 to 1.0×10−3 mol L−1. This dopamine detection limit was four times lower than that determined for the CdTe NC sol [52] and comparable with those of electrochemical assays [53].
Thus, the use of a 3D matrix for immobilizing the labels and detecting the analytes could significantly increase the detection sensitivity due to both increased numbers of fluorescent and affinity labels and an increased efficiency of binding the analyte by the affinity labels.
3. Gel Formation from Fluorescent Nanocrystals through Sol–Gel Phase Transition
Fluorescent NCs are semiconductor structures several nanometers, or sometimes several tens of nanometers, in size. Their optical properties, such as fluorescence and absorption spectra, depend on the NC size, structure, and composition. NC cores most commonly consist of elements of groups III–V, II–VI or groups IV–VI of the periodic system. The core may be coated with an outer epitaxial inorganic shell, which improves the NC optical properties and makes them more resistant to environmental factors [54]. The epitaxial shell made of a wide-gap material (ZnS or CdS) reduces the nonradiative recombination of charge carrier pairs on the surface defects of the core and improves fluorescence quantum yield. The NC composition determines not only their optical properties, but also their toxicity. CdSe, CdT (elements of groups II–VI), PbS, and PbSe (elements of groups IV–VI) NCs are highly toxic materials because of the heavy metals, which not only makes these NCs hazardous to synthesize and use, but also cause problems with their disposal. InP (elements of groups III–V) NCs do not contain heavy metals, but their optical properties are typically worse than those of their toxic counterparts [55]. Ternary NCs with cores containing three elements of groups I–III–VI, e.g., CuInS2 (CIS) and AgInS2 (AIS) NCs, constitute a better alternative. In order to increase fluorescence quantum yield, epitaxial ZnS or ZnSх/Se1-х shells are applied onto the CIS and AIS cores, which reduces charge recombination on the core surface [56].
The modification of the NC shell surface with ligands is necessary for rendering them hydrophilic and more biocompatible and for functionalizing them with various organic molecules, as well as for forming hydrogels through the sol–gel phase transition. Colloidal stability of NCs is typically provided by repulsive forces between NCs due to ligands bound to their surfaces after synthesis or postsynthetic ligand exchange. Gelation can involve either weakening of these stabilizing forces or creation of attraction forces between NCs. In some cases, gels can form spontaneously through aggregation of colloidally unstable nanoparticles [57], but only the formation of gels from stable colloidal NC dispersions allows assembling fluorescent NC gels with desired properties. Destabilization via ligand removal weakens the short-range interactions that hold the NCs apart and induce aggregation due to van der Waals attraction. However, this can lead to the formation of aggregates in the gel and loss of fluorescence properties of the NCs [58,59]. The sol–gel transition is usually initiated by optical irradiation, thermal treatment, or chemical destabilization of the sol, either spontaneous or caused by chemical agents. Gaponik et al. [27] found that storage of aqueous colloidal solutions of CdTe NCs coated with thioglycolic and mercaptopropionic acids during the synthesis eventually leads to spontaneous transition into a gel capable of maintaining its shape and integrity when flushed with buffer solutions. In this case, the gel was spontaneously formed due to oxidation of the stabilizing agents or partial hydrolysis of the NC ligands. This resulted in the formation of a structure several millimeters in size, and the NCs contained in it retained the optical properties characteristic of the NC solution. In addition, some parts of the gel had a red or yellow–green color. This indicated hierarchical organization of individual clusters, which were formed at different rates from NCs of different sizes. In practice, however, spontaneous gel formation due to aging is rarely used, because it does not allow obtaining reproducible results.
The formation of NC hydrogels induced by chemical factors that destabilize colloidal solutions was described, e.g., by Brock et al. [60]. In that case, CdSe/ZnS NCs coated with mercaptoundecanoic acid formed hydrogels upon addition of the oxidizing agent tetranitromethane (TNM). The authors noted that the optical properties of the core/shell NCs were better preserved compared with core NCs without a shell. It should also be noted that a strong oxidizing agent, such as TNM or hydrogen peroxide, can degrade the surface of core NCs, thereby decreasing the quality of the resultant fluorescent gels. Metal ions are an example of less aggressive agents that destabilize colloidal solutions. Hewa-Rehinduwage et al. [61] described the effects of Ni2+, Co2+, Ag+, and Zn2+ ions on the gel formation from CdS and CdSe NCs functionalized with thioglycolic acid and mercaptoundecanoic acid. The method proposed by these authors is also suitable for the formation of fluorescent gels on the surface of electrodes, which is relevant for the development of biosensors of various types. Gel formation can also be induced by adding an antisolvent or changing the dielectric properties of the buffer solution [62]. Lesnyak et al. [63] described the gel formation from NCs coated with 5-mercaptomethyltetrazole, with cadmium acetate as the gelation initiator. The gelation process itself took from several seconds to two or three weeks, depending on the amount of cadmium acetate added. This method yielded reproducible results, the synthesized gels remained transparent and their constituent NCs retained their optical properties, as evidenced by the small, if any, difference between the positions of the absorption maxima of the colloidal NC solution and the gel. At the same time, the fluorescence maximum was shifted by about 15 nm towards longer wavelengths, which was probably due to the energy transfer from smaller NCs to larger ones. This hypothesis was also supported by the shorter fluorescence decay in colloidal NCs than in gel-like ones, in which additional channels for nonradiative energy transfer were formed. These authors also tested the reversibility of chemically induced gelation. For this purpose, ethylenediaminetetraacetic acid (EDTA), which effectively chelates metal ions, was added to the resulting hydrogel. When EDTA was added in an amount equimolar to cadmium acetate, which was used to initiate gelation, the gel structure was destroyed, the solution became transparent, the NC fluorescence intensity was restored by 80%, and the fluorescence spectrum returned to its original shape. Subsequent increase in the cadmium acetate concentration again led to gel formation. Thiol-containing ligands are not the only ones used to form hydrogels. For example, Yao et al. showed the formation of gels from colloidal solutions of CdTe NCs coated with 16-mercaptohexadecanoic acid (MHA) or trioctylphosphine oxide (TOPO) ligands [64]. In both cases, gelation was chemically induced by the addition of TNM, with gelation of the MHA-coated NCs starting after 3–4 h, and that of the TOPO-coated NCs, after 3–4 days. The gelation of MHA-coated NCs could also be induced using a photochemical method, e.g., by irradiating the NCs with a mercury lamp for 9 h. Gacoin et al. [65] used H2O2 to obtain gels from CdS NCs functionalized with 4-fluorophenylthiol (4FPT) ligands via chemical oxidation of 4FPT. Figure 4 shows the time evolution of CdS NC colloids after oxidation by H2O2 as dependent on the ratio (X) between the added H2O2 and the amount of the 4FPT ligands on the NC surface. The addition of a small amount of H2O2 did not induce any noticeable change in the solution until the threshold value (Xmin) was reached, after which gelation was observed. At X ≥ Xmin, oxidized thiolate caused the gelation of the sol after some time as a consequence of the shrinkage of the solid network related to the solvent expulsion from the pores. In the case of very high amounts of the oxidant added (typically, 5Xmin), gelation was not observed, and the particles only precipitated. At even higher concentrations of the oxidant, the particles were partly dissolved as a result of the oxidation of the sulfide ions of CdS.
Fluorescent gels can also be obtained by using cross-coupling molecules that bind with the surface of the NCs. This provides additional control over the assembly of the gel structure. Jun-Jie et al. [66] proposed a hyperbranched macromolecule that could serve not only as an excellent ligand to stabilize CdSe/ZnS and CdSe NCs, but also as a good gelator inducing the formation of a reversible and multiresponsive fluorescent gel. Coupling molecules can act as bridges in the formation of NC gels, but non-adsorbing molecules can have a similar effect due to depletion interactions. Saez Cabezas et al. [67] reported a gelation method based on physical interactions between NCs, namely, short-range depletion attraction balanced by long-range electrostatic repulsion. The latter occurred upon removal of the native organic ligands that passivate tin-doped indium oxide NCs, while the former was induced by mixing with small polyethylene glycol chains. An increase in the polyethylene glycol concentration resulted in two gelation windows, the first arising from bridging effects and the second being attributed to depletion attraction according to phase behavior predicted by a unified theoretical model.
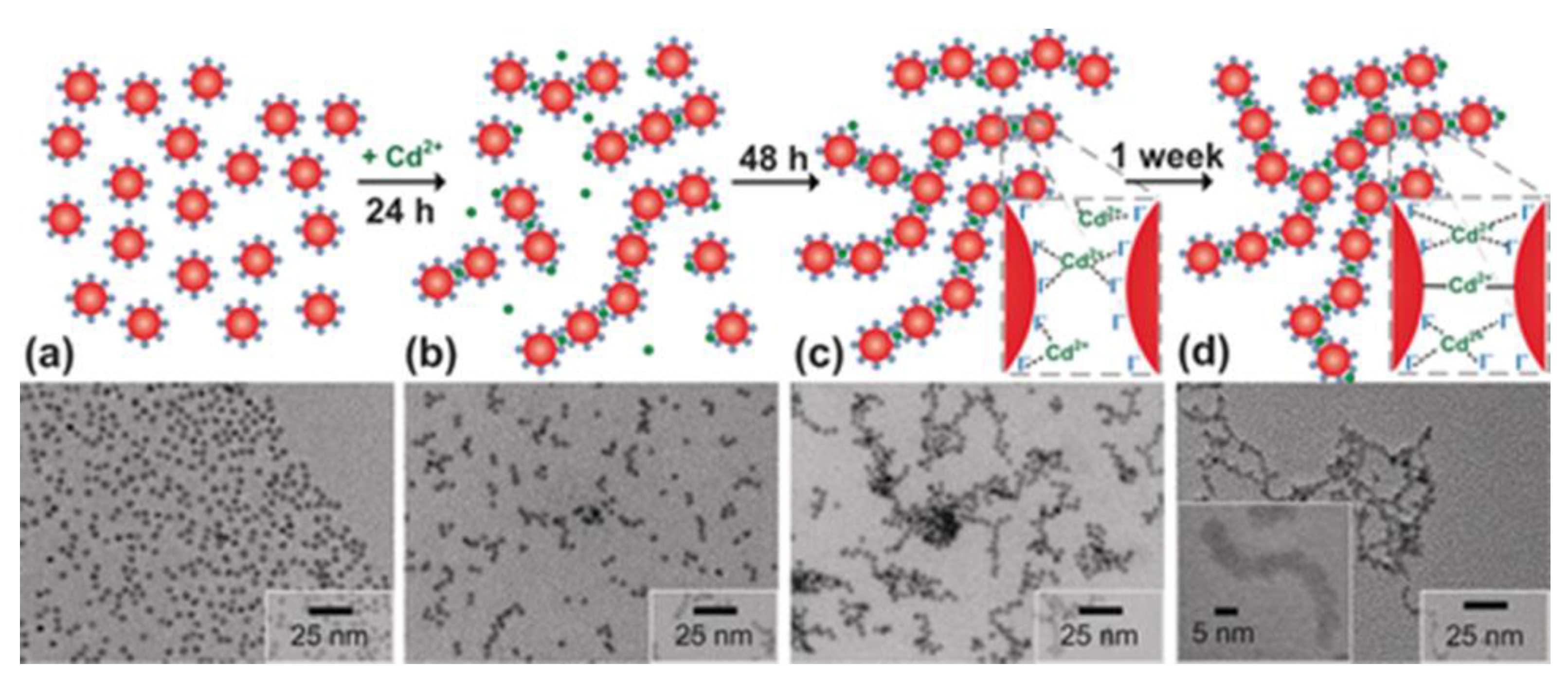
In addition, NC gels can be obtained by adding linkers that form bonds between NC surface ligands. This ensures controlled gelation process and allows new functional and sensor groups to be introduced into fluorescent gels. Sayevich et al. [68] demonstrated the assembly of all-inorganic colloidal NCs into gels mediated by NC linking with appropriate ions. They synthesized CdSe NCs with diameters of about 4.5 nm and replaced the surface ligands with NH4I, which resulted in I‒-capped NCs. The gelation was induced by adding cadmium acetate in N-methylformamide. The time course of gelation is schematically shown in Figure 5. The authors assumed that the aging process included the formation of strong covalent bonds, including I−Cd−I and Se−Cd−Se, accompanied by interfusion of the NCs, which determined the rigid framework of the gel structure. The lack of reversibility in the case of using strong complexation agents (EDTA) additionally confirmed the formation of strong bonds between the NCs. Transmission electron microscopy (TEM) showed the aggregation of CdSe(I−) NCs at early stages of gelation (Figure 5). First after 24 h, chains is formed; later up to 48h, they branched to give rise to more complex fractal aggregates, which acted as building blocks for the construction of a well-connected gel network after 1week.
Another method of initiating gelation is the irradiation of a colloidal NC solution with light. It should be noted that, in this method, it is important to control the radiation power and wavelength range so as to avoid NC damage because of the photoinduced etching of the surface [69]. High-power radiation causes degradation of NCs and the formation of a dispersed powdery precipitate. Irradiation in the infrared range also impairs the optical properties of the gels because of excessive heating. On the other hand, short-wavelength radiation is effectively absorbed by surface organic ligands, which interferes with uniform gel formation. The photoinduced gelation method can be used to form gels from NCs whose surface is functionalized with thiol-containing ligands, such as thioglycolic or mercaptopropionic acid [27]. When irradiated with a 150 W xenon lamp, the first signs of gelation appear within 5–10 h, and centrifugation of the solution for several hours after the start of gelation at low rates stimulates the gelation and allows the gel to be purified from the solvent. Note that the fluorescence intensity only slightly decreases during gelation. This decrease may be caused by aggregation of NCs due to the disruption of the outer organic shell. Minor changes in the position of the fluorescence peak may be related to changes in the size of NCs due to their degradation or, conversely, growth in response to irradiation.
Hewa-Rehinduwage et al. [70] studied the reversible electrochemical gelation of CdS, ZnS, and CdSe NCs. In the case of the CdS NCs, gelation occurs via electrochemical oxidation of surface-bound thiolate ligands in the form of dithiolates and solvation of Cd ions, after which the surface chalcogenides are oxidized and dichalcogenide bonds are formed between the NCs. The redox nature of the dichalcogenide bonds allows the gel network to be disassembled by reducing the NC surface to obtain chalcogenides at negative potentials. ZnS and CdSe NC hydrogels are formed in a similar way. The choice of ligands and specific modes of gel formation also depend on the parameters of the initial colloidal solution, such as the NC concentration and size [71,72]. The ligands required for gelation can be either added to NCs at the stage of synthesis [63] or, if this is impossible, replaced postsynthetically via surface ligand exchange [73].
5. Conclusions
Biosensors based on composite inorganic fluorescent hydrogels offer a number of advantages over the state-of-the-art microfluidic and immunochromatographic tests, which are often used in PoC diagnosis. In contrast to immunochromatographic tests, biosensors based on composite inorganic fluorescent hydrogels have a 3D structure, which allows immobilizing a larger number of capture molecules, preserving their structure, and orienting all analyte-binding sites towards the solution to ensure the maximal avidity of capture molecules. In addition, the use of a fluorescent signal increases the sensitivity of analyte detection compared to chromogenic staining methods. The change in the fluorescence signal level being reversible, biosensors based on inorganic fluorescent hydrogels can be operated in the flow mode for continuous monitoring or for sequential examination of several samples. Microfluidic systems have several advantages over their full-size counterparts, including a high throughput, a low manufacturing cost, the use of small volumes of the sample, and a short time of analysis. The most commonly used polymeric microfluidic chips are made of thermoset plastics, such as phenol formaldehyde and epoxy resins, or thermoplastic materials, such as polystyrene, polyetherketones, polyvinyl chloride, polycarbonate, and polydimethylsiloxane. Polymeric microfluidic chips are resistant to chemical agents, easy to chemically modify, and cheap to manufacture. Inorganic fluorescent gels can be immobilized inside microfluidic systems by two methods. The first one is based on gelation inside the assembled microfluidic chip containing a special cavity, into which a colloidal solution of NCs and affinity molecules is fed, and then gelation is initiated by either adding a gel-forming chemical agent or irradiating the chip with light. The second method is to apply the gel onto the surface of the plate immediately after its fabrication, before closing the microfluidic channels, and then to complete the assembly of the microfluidic system [74]. In the latter case, the colloidal solution is applied using a method similar to inkjet printing, except that the colloidal solution of NCs and affinity molecules is used instead of ink [75].
The fluorescent hydrogel biosensors are being intensely developed by a number of research groups. To date, they can already be used to detect metal ions, enzymes, reactive oxygen species, some biologically active molecules, and markers of diseases and pathological conditions, as well as hazardous contaminants in food and the environment. Many studies are aimed at increasing the specificity of detection and extending the list of detectable analytes, which will increase the demand for biosensors based on composite inorganic gels in the PoC diagnosis segment.
Author Contributions
Conceptualization, I.N. and A.S.; methodology, P.Sam. and P.Sok.; formal analysis, P.Sok. and I.N.; writing—original draft preparation, P.Sok.; writing—review and editing, P.Sam., A.S., I.N.; supervision, I.N.; funding acquisition, I.N. and P.Sam. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation through grant no. 075-15-2021-935 (P.Sam. and P.Sok). I.N. and A.S. acknowledge support from the French National Research Agency (grant no. ANR-20-CE19-009-02) and from the Université de Reims Champagne-Ardenne.
Acknowledgments
The authors are grateful to Vladimir Ushakov for invaluable technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tu, R.; Zhang, Y.; Hua, E.; Bai, L.; Huang, H.; Yun, K.; Wang, M. Droplet-based microfluidic platform for high-throughput screening of Streptomyces. Commun. Biol. 2021, 4, 647. [Google Scholar] [CrossRef] [PubMed]
- Annabestani, M.; Esmaeili-Dokht, P.; Fardmanesh, M. A novel, low cost, and accessible method for rapid fabrication of the modifiable microfluidic devices. Sci. Rep. 2020, 10, 16513. [Google Scholar] [CrossRef]
- Troy, T.; Jekic-McMullen, D.; Sambucetti, L.; Rice, B. Quantitative comparison of the sensitivity of detection of fluorescent and bioluminescent reporters in animal models. Mol. Imaging 2004, 3, 9–23. [Google Scholar] [CrossRef] [PubMed]
- You, P.Y.; Li, F.C.; Liu, M.H.; Chan, Y.H. Colorimetric and fluorescent dual-mode immunoassay based on plasmon-enhanced fluorescence of polymer dots for detection of PSA in whole blood. ACS Appl. Mater. Interfaces 2019, 11, 9841–9849. [Google Scholar] [CrossRef] [PubMed]
- Abdullah Issa, M.; Abidin, Z. Sustainable development of enhanced luminescence polymer-carbon dots composite film for rapid Cd2+ removal from wastewater. Molecules 2020, 25, 3541. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, Z.-Z.; Weng, Y.; Tan, H. Pyrophosphate ion-responsive alginate hydrogel as an effective fluorescent sensing platform for alkaline phosphatase detection. Chem. Commun. 2019, 55, 11450–11453. [Google Scholar] [CrossRef]
- Guglielmi, M.; Martucci, A. Chapter 9: Semiconductor quantum dot-doped sol–gel materials. In Sol-Gel Derived Optical and Photonic Materials, Martucci, A., Santos, L., Estefanía Rojas Hernández, R., Almeida, R., Eds.; Woodhead Publishing: 2020; pp. 209–226.
- Richter, A.; Paschew, G.; Klatt, S.; Lienig, J.; Arndt, K.-F.; Adler, H.-J.P. Review on hydrogel-based pH sensors and microsensors. Sensors 2008, 8. [Google Scholar] [CrossRef]
- Zhang, Z.; He, C.; Chen, X. Hydrogels based on pH-responsive reversible carbon–nitrogen double-bond linkages for biomedical applications. Mater. Chem. Front. 2018, 2, 1765–1778. [Google Scholar] [CrossRef]
- Hashim, H.; Maruyama, H.; Akita, Y.; Arai, F. Hydrogel Fluorescence microsensor with fluorescence recovery for prolonged stable temperature measurements. Sensors 2019, 19, 5247. [Google Scholar] [CrossRef]
- Jia, Z.; Sukker, I.; Müller, M.; Schönherr, H. Selective discrimination of key enzymes of pathogenic and nonpathogenic bacteria on autonomously reporting shape-encoded hydrogel patterns. ACS Appl. Mater. Interfaces 2018, 10, 5175–5184. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, J.; Wu, C.; Hu, X.; Lu, Y.; Wang, G.; Yu, F.; Zhang, X.; Wang, Y. Flexible and self-healing electrochemical hydrogel sensor with high efficiency toward glucose monitoring. Biosens. Bioelectr. 2020, 155, 112105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, Y.; Hedenqvist, M.S.; Chen, C.; Cai, C.; Li, H.; Liu, H.; Fu, J. Multifunctional conductive hydrogels and their applications as smart wearable devices. J. Mater. Chem. B 2021, 9, 2561–2583. [Google Scholar] [CrossRef] [PubMed]
- Missirlis, D.; Baños, M.; Lussier, F.; Spatz, J.P. Facile and Versatile method for micropatterning poly(acrylamide) hydrogels using photocleavable comonomers. ACS Appl. Mater. Interfaces 2022, 14, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Xue, B.; Qin, M.; Cao, Y.; Li, Y.; Wang, W. Printable fluorescent hydrogels based on self-assembling peptides. Sci. Rep. 2017, 7, 9691. [Google Scholar] [CrossRef] [PubMed]
- Kar, T.; Patra, N. Pyrene-based fluorescent supramolecular hydrogel: scaffold for nanoparticle synthesis. J. Phys. Org. Chem. 2020, 33, e4026. [Google Scholar] [CrossRef]
- Wu, Y.; Jin, X.; Ashrafzadeh Afshar, E.; Taher, M.A.; Xia, C.; Joo, S.-W.; Mashifana, T.; Vasseghian, Y. Simple turn-off fluorescence sensor for determination of raloxifene using gold nanoparticles stabilized by chitosan hydrogel. Chemosphere 2022, 305, 135392. [Google Scholar] [CrossRef]
- Liu, C.; Li, Q.; Wang, H.; Wang, G.; Shen, H. Quantum dots-loaded self-healing gels for versatile fluorescent assembly. Nanomaterials 2022, 12. [Google Scholar] [CrossRef]
- Pisanic, T.R., 2nd; Zhang, Y.; Wang, T.H. Quantum dots in diagnostics and detection: principles and paradigms. Analyst 2014, 139, 2968–2981. [Google Scholar] [CrossRef]
- Dey, S.C.; Nath, S.S. Size-dependent fluorescence in CdSe quantum dots. Emerg. Mater. Res. 2012, 1, 117–120. [Google Scholar] [CrossRef]
- Montón, H.; Nogués, C.; Rossinyol, E.; Castell, O.; Roldán, M. QDs versus Alexa: Reality of promising tools for immunocytochemistry. J. Nanobiotechnol. 2009, 7, 4. [Google Scholar] [CrossRef]
- Li, C.Y.; Zheng, S.Y.; Du, C.; Ling, J.; Zhu, C.N.; Wang, Y.J.; Wu, Z.L.; Zheng, Q. Carbon dot/poly(methylacrylic acid) nanocomposite hydrogels with high toughness and strong fluorescence. ACS Appl. Polym. Mater. 2020, 2, 1043–1052. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Liao, Q.; Xu, Z.; Li, H.; Zheng, L.; Fu, H. Embedding perovskite nanocrystals into a polymer matrix for tunable luminescence probes in cell imaging. Adv. Funct. Mater. 2017, 27, 1604382. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Zhu, W.; Cui, Y. Synthesis of Polymeric nanocomposite hydrogels containing the pendant ZnS nanoparticles: approach to higher refractive index optical polymeric nanocomposites. Macromolecules 2018, 51, 2672–2681. [Google Scholar] [CrossRef]
- Yang, T.; Li, Q.; Wen, W.; Hu, L.; He, W.; Liu, H. Cadmium sulfide quantum dots/poly(acrylic acid-co-acrylic amide) composite hydrogel synthesized by gamma irradiation. Rad. Phys. Chem. 2018, 145, 130–134. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, X.; Yang, G.; Yuan, Z.; Ji, X.; Kong, F.; Huang, B.; Dionysiou, D.D.; Chen, J. Hydrogel as a miniature hydrogen production reactor to enhance photocatalytic hydrogen evolution activities of CdS and ZnS quantum dots derived from modified gel crystal growth method. Chem. Eng. J. 2019, 373, 814–820. [Google Scholar] [CrossRef]
- Gaponik, N.; Wolf, A.; Marx, R.; Lesnyak, V.; Schilling, K.; Eychmüller, A. Three-dimensional self-assembly of thiol-capped CdTe nanocrystals: Gels and aerogels as building blocks for nanotechnology. Adv. Mater. 2008, 20, 4257–4262. [Google Scholar] [CrossRef]
- Wei, Y.; Li, H.; Hao, H.; Chen, Y.; Dong, C.; Wang, G. β-Cyclodextrin functionalized Mn-doped ZnS quantum dots for the chiral sensing of tryptophan enantiomers11Electronic supplementary information (ESI) available: pH effect on the PL intensity of the as-synthesized β-CD-Mn-ZnS QDs, PL spectra of β-CD-Mn-ZnS QDs/Trp enantiomers at different equilibration time and UV-vis spectra of the mixture of Trp enantiomers and β-CD at various standing time. See doi: 10.1039/C4PY00618F. Polym. Chem. 2015, 6, 591–598. [CrossRef]
- Yuan, C.; Zhang, K.; Zhang, Z.; Wang, S. Highly selective and sensitive detection of mercuric ion based on a visual fluorescence method. Anal. Chem. 2012, 84, 9792–9801. [Google Scholar] [CrossRef] [PubMed]
- Cayuela, A.; Soriano, M.L.; Kennedy, S.R.; Steed, J.W.; Valcárcel, M. Fluorescent carbon quantum dot hydrogels for direct determination of silver ions. Talanta 2016, 151, 100–105. [Google Scholar] [CrossRef]
- Martín-Pacheco, A.; Del Río Castillo, A.E.; Martín, C.; Herrero, M.A.; Merino, S.; García Fierro, J.L.; Díez-Barra, E.; Vázquez, E. Graphene quantum dot–aerogel: From nanoscopic to macroscopic fluorescent materials. Sensing polyaromatic compounds in water. ACS Appl. Mater. Interfaces 2018, 10, 18192–18201. [Google Scholar] [CrossRef]
- Zhan, Y.; Zeng, Y.; Li, L.; Luo, F.; Qiu, B.; Lin, Z.; Guo, L. Ratiometric fluorescent hydrogel test kit for on-spot visual detection of nitrite. ACS Sens. 2019, 4, 1252–1260. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Nandi, S.; Jelinek, R. Carbon-dot–hydrogel for enzyme-mediated bacterial detection. RSC Adv. 2017, 7, 588–594. [Google Scholar] [CrossRef]
- Chen, Y.; Rosenzweig, Z. Luminescent CdS quantum dots as selective ion probes. Ana. Chem. 2002, 74, 5132–5138. [Google Scholar] [CrossRef] [PubMed]
- Surana, K.; Bhattacharya, B. Fluorescence quenching by Förster resonance energy transfer in carbon–cadmium sulfide core-shell quantum dots. ACS Omega 2021, 6, 32749–32753. [Google Scholar] [CrossRef]
- Cheng, C.; Xing, M.; Wu, Q. Green synthesis of fluorescent carbon dots/hydrogel nanocomposite with stable Fe3+ sensing capability. J. Alloys Comp. 2019, 790, 221–227. [Google Scholar] [CrossRef]
- Truskewycz, A.; Beker, S.A.; Ball, A.S.; Murdoch, B.; Cole, I. Incorporation of quantum carbon dots into a PVP/ZnO hydrogel for use as an effective hexavalent chromium sensing platform. Anal. Chim. Acta 2020, 1099, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Mandani, S.; Thakur, N.; Sarma, T.K. Cd(II)–nucleobase supramolecular metallo-hydrogels for in situ growth of color tunable CdS quantum dots. Soft Matter 2018, 14, 5715–5720. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Kaufmann, L.; Dey, P.; Haag, R.; Schedler, U. Bioorthogonal in situ hydrogels based on polyether polyols for new biosensor materials with high sensitivity. ACS Appl. Mater. Interfaces 2018, 10, 11382–11390. [Google Scholar] [CrossRef] [PubMed]
- Aloraefy, M.; Pfefer, T.J.; Ramella-Roman, J.C.; Sapsford, K.E. In vitro evaluation of fluorescence glucose biosensor response. Sensors 2014, 14, 12127–12148. [Google Scholar] [CrossRef]
- Pourreza, N.; Ghomi, M. A novel metal enhanced fluorescence bio probe for insulin sensing based on poly vinyl alcohol-borax hydrogel functionalized by Ag dots. Sens. Actuators B Chem. 2017, 251, 609–616. [Google Scholar] [CrossRef]
- Sun, L.; Hu, N.; Peng, J.; Chen, L.; Weng, J. Ultrasensitive detection of mitochondrial DNA mutation by graphene oxide/DNA hydrogel electrode. Adv. Funct. Mater. 2014, 24, 6905–6913. [Google Scholar] [CrossRef]
- Chen, M.; Grazon, C.; Sensharma, P.; Nguyen, T.T.; Feng, Y.; Chern, M.; Baer, R.C.; Varongchayakul, N.; Cook, K.; Lecommandoux, S.; et al. Hydrogel-Embedded Quantum Dot-Transcription Factor Sensors for Quantitative Progesterone Detection. ACS Appl. Mater. Interfaces 2020, 12, 43513–43521. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.-J.; Park, S.-Y. Carbon-dot-based ratiometric fluorescence glucose biosensor. Sens. Actuators B Chem. 2019, 282, 719–729. [Google Scholar] [CrossRef]
- Dong, B.; Li, H.; Mujtaba Mari, G.; Yu, X.; Yu, W.; Wen, K.; Ke, Y.; Shen, J.; Wang, Z. Fluorescence immunoassay based on the inner-filter effect of carbon dots for highly sensitive amantadine detection in foodstuffs. Food Chem. 2019, 294, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Ekblad, T.; Andersson, O.; Liedberg, B. Photografted poly(ethylene glycol) matrix for affinity interaction studies. Biomacromolecules 2007, 8, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.Y.; Kim, J.S.; Choi, B.R.; Lee, K.; Lee, H. Hydrogel based biosensors for in vitro diagnostics of biochemicals, proteins, and genes. Adv. Healthcare Mater. 2017, 6, 1601475. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wolf, L.K.; Georgiadis, R.M. Secondary structure effects on DNA hybridization kinetics: a solution versus surface comparison. Nucleic Acids Res. 2006, 34, 3370–3377. [Google Scholar] [CrossRef] [PubMed]
- Welch, N.G.; Scoble, J.A.; Muir, B.W.; Pigram, P.J. Orientation and characterization of immobilized antibodies for improved immunoassays (Review). Biointerphases 2017, 12, 02D301. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Huang, S.; Ge, F.; Luo, Y.; Jia, D.; Dai, Y. 3D antibody immobilization on a planar matrix surface. Biosens. Bioelectron. 2011, 28, 91–96. [Google Scholar] [CrossRef]
- Yuan, J.; Wen, D.; Gaponik, N.; Eychmüller, A. Enzyme-encapsulating quantum dot hydrogels and xerogels as biosensors: multifunctional platforms for both biocatalysis and fluorescent probing. Angew. Chem. Int. Ed. 2013, 52, 976–979. [Google Scholar] [CrossRef]
- Yuan, J.; Guo, W.; Wang, E. Utilizing a CdTe quantum dots-enzyme hybrid system for the determination of both phenolic compounds and hydrogen peroxide. Anal. Chem. 2008, 80, 1141–1145. [Google Scholar] [CrossRef]
- Yu, D.; Zeng, Y.; Qi, Y.; Zhou, T.; Shi, G. A novel electrochemical sensor for determination of dopamine based on AuNPs@SiO2 core-shell imprinted composite. Biosens. Bioelectron. 2012, 38, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Linkov, P.A.; Vokhmintcev, K.V.; Samokhvalov, P.S.; Nabiev, I.R. Ultrasmall quantum dots for fluorescent bioimaging in vivo and in vitro. Optics Spectrosc. 2017, 122, 8–11. [Google Scholar] [CrossRef]
- Kandi, D.; Mansingh, S.; Behera, A.; Parida, K. Calculation of relative fluorescence quantum yield and Urbach energy of colloidal CdS QDs in various easily accessible solvents. J. Luminesc. 2021, 231, 117792. [Google Scholar] [CrossRef]
- Neo, D.C.J.; Goh, W.P.; Lau, H.H.; Shanmugam, J.; Chen, Y.F. CuInS2 Quantum Dots with Thick ZnSexS1–x Shells for a Luminescent Solar Concentrator. ACS Appl. Nano Mater. 2020, 3, 6489–6496. [Google Scholar] [CrossRef]
- Zhu, C.; Shi, Q.; Fu, S.; Song, J.; Xia, H.; Du, D.; Lin, Y. Efficient synthesis of MCu (M = Pd, Pt, and Au) aerogels with accelerated gelation kinetics and their high electrocatalytic activity. Adv. Mater. 2016, 28, 8779–8783. [Google Scholar] [CrossRef]
- Anupama, K.; Paul, T.; Ann Mary, K.A. Solid-state fluorescent selenium quantum dots by a solvothermal-assisted sol–gel route for curcumin sensing. ACS Omega 2021, 6, 21525–21533. [Google Scholar] [CrossRef] [PubMed]
- Arachchige, I.U.; Brock, S.L. Sol–Gel Methods for the assembly of metal chalcogenide quantum dots. Accounts Chem. Res. 2007, 40, 801–809. [Google Scholar] [CrossRef]
- Arachchige, I.U.; Brock, S.L. Highly luminescent quantum-dot monoliths. J. Am. Chem. Soc. 2007, 129, 1840–1841. [Google Scholar] [CrossRef]
- Hewa-Rahinduwage, C.C.; Silva, K.L.; Brock, S.L.; Luo, L. Quantum dot assembly driven by electrochemically generated metal-ion crosslinkers. Chem. Mater. 2021, 33, 4522–4528. [Google Scholar] [CrossRef]
- Matter, F.; Luna, A.L.; Niederberger, M. From colloidal dispersions to aerogels: How to master nanoparticle gelation. Nano Today 2020, 30, 100827. [Google Scholar] [CrossRef]
- Lesnyak, V.; Voitekhovich, S.V.; Gaponik, P.N.; Gaponik, N.; Eychmüller, A. CdTe nanocrystals capped with a tetrazolyl analogue of thioglycolic acid: Aqueous synthesis, characterization, and metal-assisted assembly. ACS Nano 2010, 4, 4090–4096. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Brock, S.L. Porous CdTe nanocrystal assemblies: ligation effects on the gelation process and the properties of resultant aerogels. Inorg. Chem. 2011, 50 20, 9985–9992. [Google Scholar] [CrossRef]
- Gacoin, T.; Lahlil, K.; Larregaray, P.; Boilot, J.P. Transformation of CdS colloids: sols, gels, and precipitates. J. Phys. Chem. B 2001, 105, 10228–10235. [Google Scholar] [CrossRef]
- Yan, J.-J.; Wang, H.; Zhou, Q.-H.; You, Y.-Z. Reversible and multisensitive quantum dot gels. Macromolecules 2011, 44, 4306–4312. [Google Scholar] [CrossRef]
- Saez Cabezas, C.A.; Ong, G.K.; Jadrich, R.B.; Lindquist, B.A.; Agrawal, A.; Truskett, T.M.; Milliron, D.J. Gelation of plasmonic metal oxide nanocrystals by polymer-induced depletion attractions. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 8925–8930. [Google Scholar] [CrossRef] [PubMed]
- Sayevich, V.; Cai, B.; Benad, A.; Haubold, D.; Sonntag, L.; Gaponik, N.; Lesnyak, V.; Eychmüller, A. 3D assembly of all-inorganic colloidal nanocrystals into gels and aerogels. Angew. Chem. Int. Ed. 2016, 55, 6334–6338. [Google Scholar] [CrossRef] [PubMed]
- Gaponik, N.; Talapin, D.V.; Rogach, A.L.; Hoppe, K.; Shevchenko, E.V.; Kornowski, A.; Eychmüller, A.; Weller, H. Thiol-capping of CdTe nanocrystals: An alternative to organometallic synthetic routes. J. Phys. Chem. B 2002, 106, 7177–7185. [Google Scholar] [CrossRef]
- Hewa-Rahinduwage, C.C.; Geng, X.; Silva, K.L.; Niu, X.; Zhang, L.; Brock, S.L.; Luo, L. Reversible electrochemical gelation of metal chalcogenide quantum dots. J. Am. Chem. Soc. 2020, 142, 12207–12215. [Google Scholar] [CrossRef]
- Mohanan, J.L.; Brock, S.L. CdS aerogels: Effect of concentration and primary particle size on surface area and opto-electronic properties. J. Sol-Gel Sci. Technol. 2006, 40, 341–350. [Google Scholar] [CrossRef]
- Lesnyak, V.; Wolf, A.; Dubavik, A.; Borchardt, L.; Voitekhovich, S.V.; Gaponik, N.; Kaskel, S.; Eychmüller, A. 3D assembly of semiconductor and metal nanocrystals: Hybrid CdTe/Au structures with controlled content. J. Am. Chem. Soc. 2011, 133, 13413–13420. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, Y.; Tang, J.; Tang, W. Surface ligands engineering of semiconductor quantum dots for chemosensory and biological applications. Mater. Today 2017, 20, 360–376. [Google Scholar] [CrossRef]
- Moya, M.L.; Alonzo, L.F.; George, S.C. Microfluidic device to culture 3D in vitro human capillary networks. In Biomimetics and Stem Cells: Methods and Protocols, Vunjak-Novakovic, G., Turksen, K., Eds.; Springer New York: New York, NY, 2014; pp. 21–27.
- Park, D.; Lee, J.; Lee, Y.; Son, K.; Choi, J.W.; Jeang, W.J.; Choi, H.; Hwang, Y.; Kim, H.-Y.; Jeon, N.L. Aspiration-mediated hydrogel micropatterning using rail-based open microfluidic devices for high-throughput 3D cell culture. Scientific Reports 2021, 11, 19986. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Methods for obtaining fluorescent gels. (a) post synthesis loading of gel with NC; (b) loading of gel with NC during gel formation; (c) direct incorporation of NC into gel; (d) synthesis of NC during gel formation; (e) sol-gel approach of fluorescent gel formation.
Figure 1.
Methods for obtaining fluorescent gels. (a) post synthesis loading of gel with NC; (b) loading of gel with NC during gel formation; (c) direct incorporation of NC into gel; (d) synthesis of NC during gel formation; (e) sol-gel approach of fluorescent gel formation.
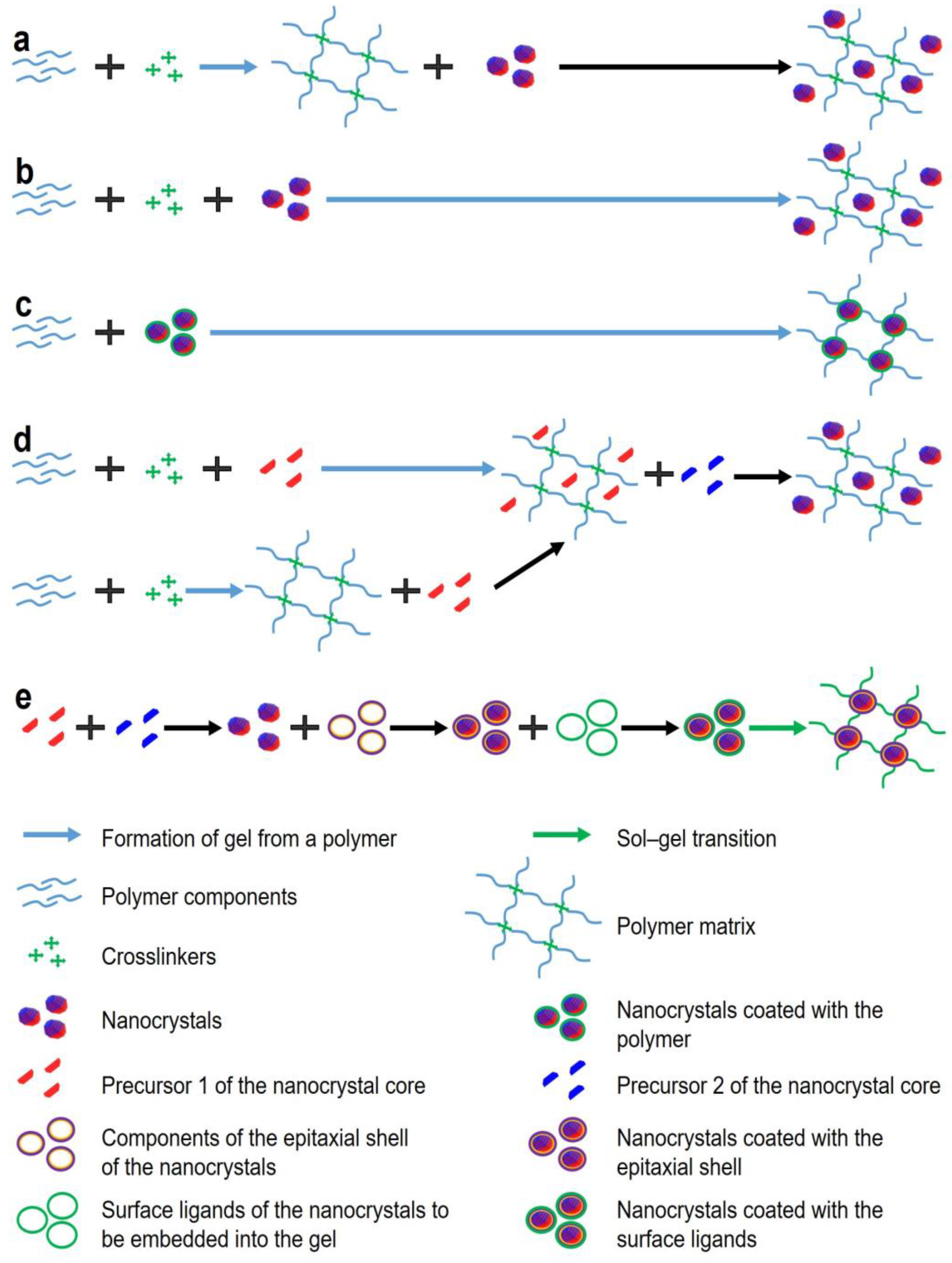
Figure 2.
Variants of detection of target analyte molecules in fluorescent gels. (a) target analyte disrupt gel structure; (b) analyte is optically active; (c) analyte is optically inactive; (d) enzyme dependent fluorescence quenching.
Figure 2.
Variants of detection of target analyte molecules in fluorescent gels. (a) target analyte disrupt gel structure; (b) analyte is optically active; (c) analyte is optically inactive; (d) enzyme dependent fluorescence quenching.
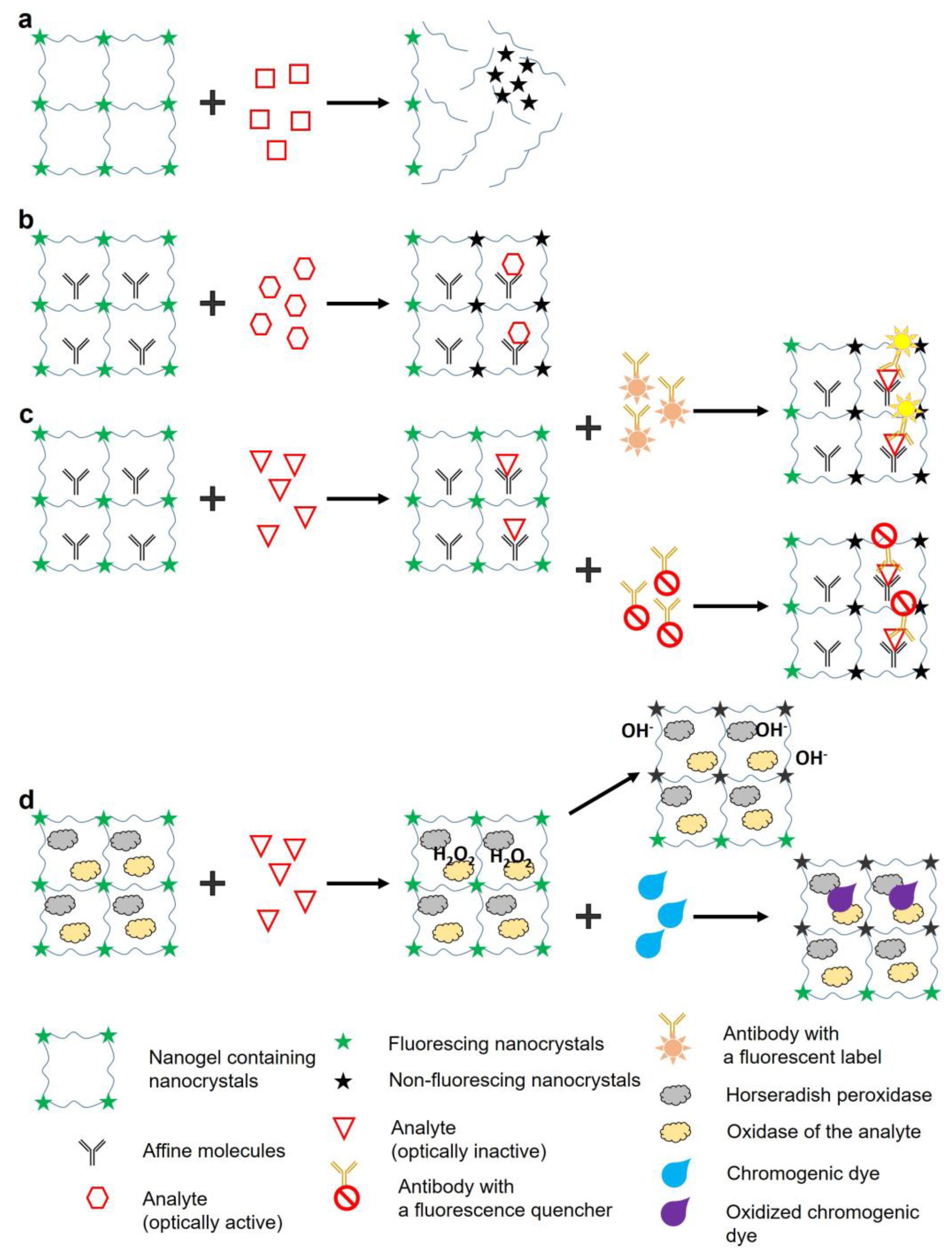
Figure 3.
Scheme of the encapsulation of enzymes in a NC gel using sol–gel switching process of MSA-capped CdTe NCs. The Scheme is adapted from Ref. [51].
Figure 3.
Scheme of the encapsulation of enzymes in a NC gel using sol–gel switching process of MSA-capped CdTe NCs. The Scheme is adapted from Ref. [51].
Figure 4.
Schematic representation of the time evolution of CdS colloids after oxidation by aqueous hydrogen peroxide solutions depending on the X, molar ratio between H2O2 and the grafted thiolate in the colloid. Reprinted with permission from Gacoin et al. [65]. Copyright {2023} American Chemical Society.
Figure 4.
Schematic representation of the time evolution of CdS colloids after oxidation by aqueous hydrogen peroxide solutions depending on the X, molar ratio between H2O2 and the grafted thiolate in the colloid. Reprinted with permission from Gacoin et al. [65]. Copyright {2023} American Chemical Society.

Figure 5.
Schematic representation of the gelation process and TEM images showing the gelation of CdSe(I–) NCs (a) 24 h (b), 48 h (c), and 1 week (d) after the addition of Cd2+ linker. Reprinted with permission from Eychmüller et al. [68]. Copyright {2016} John Wiley and Sons.
Figure 5.
Schematic representation of the gelation process and TEM images showing the gelation of CdSe(I–) NCs (a) 24 h (b), 48 h (c), and 1 week (d) after the addition of Cd2+ linker. Reprinted with permission from Eychmüller et al. [68]. Copyright {2016} John Wiley and Sons.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



